New Insights into the Role of Glutathione in the Mechanism of Fever

 , , and
, , and 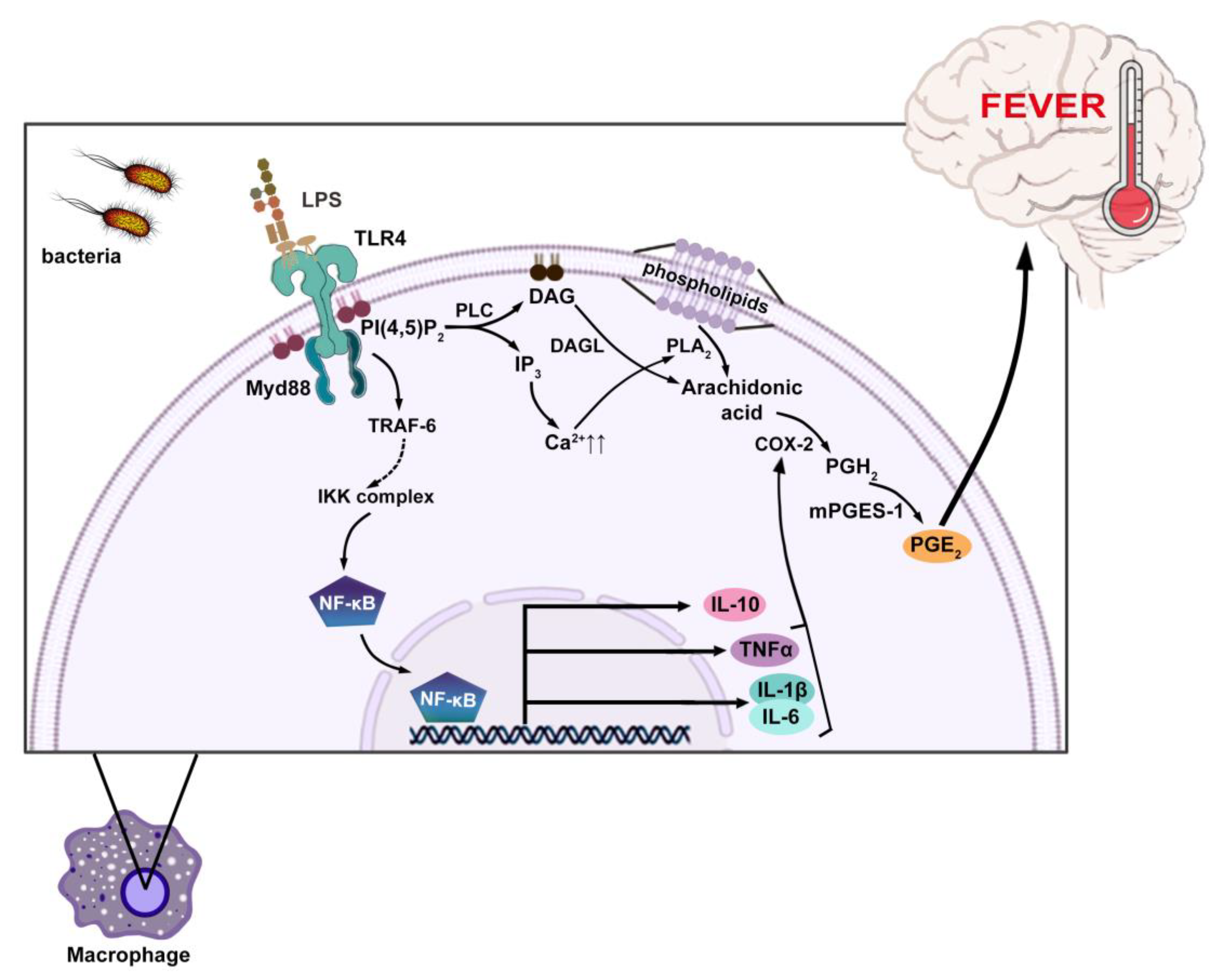{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Fever—A Brief Overview
2. Fever Induction and Antipyresis
3. Redox Status Modulates Thermal Response in Organisms
4. Glutathione
4.1. Glutathione Affects the Immune System and Signaling Molecules Involved in the Mechanism of Fever
4.2. Direct Evidence of the Effect of Modulation of Glutathione Level on the Mechanism of Fever
4.2.1. Fever in Rats Treated with N-acetyl-l-cysteine (NAC)
4.2.2. Fever in Organisms with Pharmacologically Decreased Glutathione Level
5. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Zhao, Z.-D.; Yang, W.Z.; Gao, C.; Fu, X.; Zhang, W.; Zhou, Q.; Chen, W.; Ni, X.; Lin, J.-K.; Yang, J.; et al. A hypothalamic circuit that controls body temperature. Proc. Natl. Acad. Sci. USA 2017, 114, 2042–2047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, A.; Wilhelms, D.B.; Mirrasekhian, E.; Jaarola, M.; Blomqvist, A.; Engblom, D. Inflammation-induced anorexia and fever are elicited by distinct prostaglandin dependent mechanisms, whereas conditioned taste aversion is prostaglandin independent. Brain Behav. Immun. 2017, 61, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Soszyński, D. “Sickness behavior”—Mechanisms of origin and significance. Postepy Hig. Med. Dosw. 2004, 58, 74–82. [Google Scholar]
- Cabanac, M.; Massonnet, B. Temperature regulation during fever: Change of set point or change of gain? A tentative answer from a behavioural study in man. J. Physiol. 1974, 238, 561–568. [Google Scholar] [CrossRef]
- Cannon, J.G. Perspective on fever: The basic science and conventional medicine. Complement. Ther. Med. 2013, 21 (Suppl. S1), S54–S60. [Google Scholar] [CrossRef]
- Ogoina, D. Fever, fever patterns and diseases called ‘fever’—A review. J. Infect. Public Health 2011, 4, 108–124. [Google Scholar] [CrossRef] [Green Version]
- Dinarello, C.A. Thermoregulation and the pathogenesis of fever. Infect. Dis. Clin. 1996, 10, 433–449. [Google Scholar] [CrossRef]
- Dal Nogare, A.R.; Sharma, S. Exogenous pyrogens. In Fever: Basic Mechanisms and Management, 2nd ed.; Mackowiak, P.A., Ed.; Lippincott-Raven Publishers: Philadelphia, PA, USA, 1997; pp. 79–85. [Google Scholar]
- Kozak, W.; Wrotek, S.; Kozak, A. Pyrogenicity of CpG-DNA in mice: Role of interleukin-6, cyclooxygenases, and nuclear factor-kappaB. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 290, R871–R880. [Google Scholar] [CrossRef] [Green Version]
- Proft, T.; Fraser, J.D. Bacterial superantigens. Clin. Exp. Immunol. 2003, 133, 299–306. [Google Scholar] [CrossRef]
- Kluger, M.J.; Kozak, W.; Conn, C.A.; Leon, L.R.; Soszynski, D. The adaptive value of fever. Infect. Dis. Clin. North Am. 1996, 10, 1–20. [Google Scholar] [CrossRef]
- Kluger, M.J.; Kozak, W.; Conn, C.A.; Leon, L.R.; Soszynski, D. Role of fever in disease. Ann. N. Y. Acad. Sci. 1998, 856, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Hoption Cann, S.A.; van Netten, J.P.; van Netten, C.; Glover, D.W. Spontaneous regression: A hidden treasure buried in time. Med. Hypotheses 2002, 58, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Hoption Cann, S.A.; van Netten, J.P.; van Netten, C. Acute infections as a means of cancer prevention: Opposing effects to chronic infections? Cancer Detect. Prev. 2006, 30, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Wrotek, S.; Jedrzejewski, T.; Potera-Kram, E.; Kozak, W. Antipyretic activity of N-acetylcysteine. J. Physiol. Pharmacol. 2011, 62, 669–675. [Google Scholar]
- Clint, E.; Fessler, D.M.T. Insurmountable Heat: The Evolution and Persistence of Defensive Hyperthermia. Q. Rev. Biol. 2016, 91, 25–46. [Google Scholar] [CrossRef] [Green Version]
- Day, J.D.; LeGrand, E.K. Synergy of local, regional, and systemic non-specific stressors for host defense against pathogens. J. Theor. Biol. 2015, 367, 39–48. [Google Scholar] [CrossRef]
- Evans, S.S.; Repasky, E.A.; Fisher, D.T. Fever and the thermal regulation of immunity: The immune system feels the heat. Nat. Rev. Immunol. 2015, 15, 335–349. [Google Scholar] [CrossRef]
- LeGrand, E.K.; Day, J.D. Self-harm to preferentially harm the pathogens within: Non-specific stressors in innate immunity. Proc. Biol. Sci. 2016, 283, 20160266. [Google Scholar] [CrossRef] [Green Version]
- Baumann, H.; Gauldie, J. The acute phase response. Immunol. Today 1994, 15, 74–80. [Google Scholar] [CrossRef]
- Roberts Jr, N.J. Impact of temperature elevation on immunologic defenses. Rev. Infect. Dis. 1991, 13, 462–472. [Google Scholar] [CrossRef]
- Blomqvist, A.; Engblom, D. Neural Mechanisms of Inflammation-Induced Fever. Neuroscientist 2018, 24, 381–399. [Google Scholar] [CrossRef] [PubMed]
- Harden, L.M.; Kent, S.; Pittman, Q.J.; Roth, J. Fever and sickness behavior: Friend or foe? Brain Behav. Immun. 2015, 50, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Young, P.J.; Saxena, M.; Beasley, R.; Bellomo, R.; Bailey, M.; Pilcher, D.; Finfer, S.; Harrison, D.; Myburgh, J.; Rowan, K. Early peak temperature and mortality in critically ill patients with or without infection. Intensive Care Med. 2012, 38, 437–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doran, T.F.; De Angelis, C.; Baumgardner, R.A.; Mellits, E.D. Acetaminophen: More harm than good for chickenpox? J. Pediatr. 1989, 114, 1045–1048. [Google Scholar] [CrossRef]
- Plaisance, K.I.; Kudaravalli, S.; Wasserman, S.S.; Levine, M.M.; Mackowiak, P.A. Effect of antipyretic therapy on the duration of illness in experimental influenza A, Shigella sonnei, and Rickettsia rickettsii infections. Pharmacotherapy 2000, 20, 1417–1422. [Google Scholar] [CrossRef]
- Grossarth-Maticek, R.; Frentzel-Beyme, R.; Kanazir, D.; Jankovic, M.; Vetter, H. Reported herpes-virus-infection, fever and cancer incidence in a prospective study. J. Chronic. Dis. 1987, 40, 967–976. [Google Scholar] [CrossRef]
- Kölmel, K.F.; Gefeller, O.; Haferkamp, B. Febrile infections and malignant melanoma: Results of a case-control study. Melanoma Res. 1992, 2, 207–211. [Google Scholar] [CrossRef]
- Wrotek, S.; Jędrzejewski, T.; Nowakowska, A.; Kozak, W. LPS alters pattern of sickness behavior but does not affect glutathione level in aged male rats. Biogerontology 2016, 17, 715–723. [Google Scholar] [CrossRef]
- Wrotek, S.; Kamecki, K.; Kwiatkowski, S.; Kozak, W. Cancer patients report a history of fewer fevers during infections than healthy controls. J. Pre-Clin Clin. Res. 2009, 3, 31–35. [Google Scholar]
- Challis, G.B.; Stam, H.J. The spontaneous regression of cancer. A review of cases from 1900 to 1987. Acta Oncol. 1990, 29, 545–550. [Google Scholar] [CrossRef]
- O’Regan, B.; Hirshberg, C. Spontaneous Remission: An Annotated Bibliography; Institute of Noetic Sciences: Sausalito, CA, USA, 1993; ISBN 978-0-943951-17-1. [Google Scholar]
- Køstner, A.H.; Johansen, R.F.; Schmidt, H.; Mølle, I. Regression in cancer following fever and acute infection. Acta Oncol. 2013, 52, 455–457. [Google Scholar] [CrossRef] [PubMed]
- Wrotek, S.; Brycht, Ł.; Wrotek, W.; Kozak, W. Fever as a factor contributing to long-term survival in a patient with metastatic melanoma: A case report. Complement. Ther. Med. 2018, 38, 7–10. [Google Scholar] [CrossRef] [PubMed]
- El-Radhi, A.S.M. Why is the evidence not affecting the practice of fever management? Arch. Dis. Child. 2008, 93, 918–920. [Google Scholar] [CrossRef] [PubMed]
- Atkins, E. Pathogenesis of fever. Physiol. Rev. 1960, 40, 580–646. [Google Scholar] [CrossRef]
- Wrotek, S.; Domagalski, K.; Jędrzejewski, T.; Dec, E.; Kozak, W. Buthionine sulfoximine, a glutathione depletor, attenuates endotoxic fever and reduces IL-1β and IL-6 level in rats. Cytokine 2017, 90, 31–37. [Google Scholar] [CrossRef]
- Wrotek, S.; Jędrzejewski, T.; Nowakowska, A.; Kozak, W. Glutathione deficiency attenuates endotoxic fever in rats. Int. J. Hyperth. 2015, 31, 793–799. [Google Scholar] [CrossRef]
- Zhang, Z.; La Placa, D.; Nguyen, T.; Kujawski, M.; Le, K.; Li, L.; Shively, J.E. CEACAM1 regulates the IL-6 mediated fever response to LPS through the RP105 receptor in murine monocytes. BMC Immunol. 2019, 20, 7. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-J.; Huang, W.-T.; Shao, D.-Z.; Liao, J.-F.; Lin, M.-T. Blocking NF-kappaB activation may be an effective strategy in the fever therapy. Jpn. J. Physiol. 2003, 53, 367–375. [Google Scholar] [CrossRef] [Green Version]
- Kluger, M.J.; Kozak, W.; Leon, L.R.; Soszynski, D.; Conn, C.A. Cytokines and fever. Neuroimmunomodulation 1995, 2, 216–223. [Google Scholar] [CrossRef]
- Milton, A.S. Thermoregulatory actions of eicosanoids in the central nervous system with particular regard to the pathogenesis of fever. Ann. N. Y. Acad. Sci. 1989, 559, 392–410. [Google Scholar] [CrossRef]
- Roth, J.; Blatteis, C.M. Mechanisms of fever production and lysis: Lessons from experimental LPS fever. Compr. Physiol. 2014, 4, 1563–1604. [Google Scholar]
- Roth, J.; De Souza, G.E. Fever induction pathways: Evidence from responses to systemic or local cytokine formation. Braz. J. Med. Biol. Res. 2001, 34, 301–314. [Google Scholar] [CrossRef]
- Leschner, J.; Ring, L.; Feierler, J.; Dinkel, K.; Jochum, M.; Faussner, A. Fever-like temperature modification differentially affects in vitro signaling of bradykinin B(1) and B(2) receptors. Biol. Chem. 2011, 392, 1021–1029. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Wellman, L.L.; Tang, X.; Sanford, L.D. Effects of corticotropin releasing factor (CRF) on sleep and body temperature following controllable footshock stress in mice. Physiol. Behav. 2011, 104, 886–892. [Google Scholar] [CrossRef] [Green Version]
- Kozak, W.; Kozak, A. Genetic Models in Applied Physiology. Differential role of nitric oxide synthase isoforms in fever of different etiologies: Studies using Nos gene-deficient mice. J. Appl. Physiol. 2003, 94, 2534–2544. [Google Scholar] [CrossRef] [Green Version]
- Miñano, F.J.; Sancibrian, M.; Myers, R.D. Fever induced by macrophage inflammatory protein-1 (MIP-1) in rats: Hypothalamic sites of action. Brain Res. Bull. 1991, 27, 701–706. [Google Scholar] [CrossRef]
- Fabricio, A.S.C.; Rae, G.A.; D’Orléans-Juste, P.; Souza, G.E.P. Endothelin-1 as a central mediator of LPS-induced fever in rats. Brain Res. 2005, 1066, 92–100. [Google Scholar] [CrossRef]
- Shepard, A.M.; Bharwani, A.; Durisko, Z.; Andrews, P.W. Reverse Engineering the Febrile System. Q. Rev. Biol. 2016, 91, 419–457. [Google Scholar] [CrossRef]
- Lee, J.J.; Simmons, D.L. Antipyretic therapy: Clinical pharmacology. Handb. Clin. Neurol. 2018, 157, 869–881. [Google Scholar]
- Sajadi, M.M.; Mackowiak, P.A. 55—Temperature Regulation and the Pathogenesis of Fever. In Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases; Bennett, J.E., Dolin, R., Blaser, M.J., Eds.; Content Repository Only: Philadelphia, PA, USA, 2015; pp. 708–720. ISBN 978-1-4557-4801-3. [Google Scholar]
- Kozak, W.; Kluger, M.J.; Kozak, A.; Wachulec, M.; Dokladny, K. Role of cytochrome P-450 in endogenous antipyresis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 279, R455–460. [Google Scholar] [CrossRef] [Green Version]
- El-Radhi, A.S.M. Fever management: Evidence vs current practice. World J. Clin. Pediatr. 2012, 1, 29–33. [Google Scholar] [CrossRef]
- Harirforoosh, S.; Asghar, W.; Jamali, F. Adverse effects of nonsteroidal antiinflammatory drugs: An update of gastrointestinal, cardiovascular and renal complications. J. Pharm. Pharm. Sci. 2013, 16, 821–847. [Google Scholar] [CrossRef] [Green Version]
- Plaisance, K.I. Toxicities of drugs used in the management of fever. Clin. Infect. Dis. 2000, 31 (Suppl. S5), S219–S223. [Google Scholar] [CrossRef] [Green Version]
- Zampronio, A.R.; Soares, D.M.; Souza, G.E.P. Central mediators involved in the febrile response: Effects of antipyretic drugs. Temperature 2015, 2, 506–521. [Google Scholar] [CrossRef] [Green Version]
- Pittman, Q.J.; Wilkinson, M.F. Central arginine vasopressin and endogenous antipyresis. Can. J. Physiol. Pharmacol. 1992, 70, 786–790. [Google Scholar] [CrossRef]
- Pittman, Q.J.; Poulin, P.; Wilkinson, M.F. Role of neurohypophysial hormones in temperature regulation. Ann. N. Y. Acad. Sci. 1993, 689, 375–381. [Google Scholar] [CrossRef]
- Morrow, L.E.; McClellan, J.L.; Conn, C.A.; Kluger, M.J. Glucocorticoids alter fever and IL-6 responses to psychological stress and to lipopolysaccharide. Am. J. Physiol. 1993, 264, R1010–1016. [Google Scholar] [CrossRef]
- Ledeboer, A.; Brevé, J.J.; Poole, S.; Tilders, F.J.; Van Dam, A.M. Interleukin-10, interleukin-4, and transforming growth factor-beta differentially regulate lipopolysaccharide-induced production of pro-inflammatory cytokines and nitric oxide in co-cultures of rat astroglial and microglial cells. Glia 2000, 30, 134–142. [Google Scholar] [CrossRef]
- Harden, L.M.; Rummel, C.; Laburn, H.P.; Damm, J.; Wiegand, F.; Poole, S.; Gerstberger, R.; Roth, J. Critical role for peripherally-derived interleukin-10 in mediating the thermoregulatory manifestations of fever and hypothermia in severe forms of lipopolysaccharide-induced inflammation. Pflugers Arch. 2014, 466, 1451–1466. [Google Scholar] [CrossRef]
- Riedel, W.; Maulik, G. Fever: An integrated response of the central nervous system to oxidative stress. Mol. Cell. Biochem. 1999, 196, 125–132. [Google Scholar] [CrossRef]
- Gomes, B.R.B.; Firmino, M.; Jorge, J.S.; Ferreira, M.L.O.; Rodovalho, T.M.; Weis, S.N.; Souza, G.E.P.; Morais, P.C.; Sousa, M.V.; Souza, P.E.N.; et al. Increase of reactive oxygen species in different tissues during lipopolysaccharide-induced fever and antipyresis: An electron paramagnetic resonance study. Free Radic. Res. 2018, 52, 351–361. [Google Scholar] [CrossRef]
- Riedel, W.; Lang, U.; Oetjen, U.; Schlapp, U.; Shibata, M. Inhibition of oxygen radical formation by methylene blue, aspirin, or alpha-lipoic acid, prevents bacterial-lipopolysaccharide-induced fever. Mol. Cell. Biochem. 2003, 247, 83–94. [Google Scholar] [CrossRef]
- Ettebong, E.O.; Nwafor, P.A. Antipyretic and antioxidant activities of Eleucine indica. J. Phytopharmacol. 2015, 4, 235–242. [Google Scholar]
- Desideri, E.; Ciccarone, F.; Ciriolo, M.R. Targeting Glutathione Metabolism: Partner in Crime in Anticancer Therapy. Nutrients 2019, 11, 1926. [Google Scholar] [CrossRef] [Green Version]
- Lv, H.; Zhen, C.; Liu, J.; Yang, P.; Hu, L.; Shang, P. Unraveling the Potential Role of Glutathione in Multiple Forms of Cell Death in Cancer Therapy. Oxid. Med. Cell. Longev. 2019, 2019, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Minich, D.M.; Brown, B.I. A Review of Dietary (Phyto)Nutrients for Glutathione Support. Nutrients 2019, 11, 2073. [Google Scholar] [CrossRef] [Green Version]
- Bajic, V.P.; Van Neste, C.; Obradovic, M.; Zafirovic, S.; Radak, D.; Bajic, V.B.; Essack, M.; Isenovic, E.R. Glutathione “Redox Homeostasis” and Its Relation to Cardiovascular Disease. Oxid. Med. Cell. Longev. 2019, 2019, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Adeoye, O.; Olawumi, J.; Opeyemi, A.; Christiania, O. Review on the role of glutathione on oxidative stress and infertility. JBRA Assist. Reprod. 2017, 22, 61. [Google Scholar] [CrossRef]
- Teskey, G.; Abrahem, R.; Cao, R.; Gyurjian, K.; Islamoglu, H.; Lucero, M.; Martinez, A.; Paredes, E.; Salaiz, O.; Robinson, B.; et al. Glutathione as a Marker for Human Disease. Adv. Clin. Chem. 2018, 87, 141–159. [Google Scholar]
- Wang, L.; Ahn, Y.J.; Asmis, R. Sexual dimorphism in glutathione metabolism and glutathione-dependent responses. Redox Biol. 2019. [Google Scholar] [CrossRef]
- Maciejczyk, M.; Zalewska, A.; Ładny, J.R. Salivary Antioxidant Barrier, Redox Status, and Oxidative Damage to Proteins and Lipids in Healthy Children, Adults, and the Elderly. Oxid. Med. Cell. Longev. 2019, 2019, 4393460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuppner, M.C.; Scharner, A.; Milani, V.; von Hesler, C.; Tschöp, K.E.; Heinz, O.; Issels, R.D. Ifosfamide impairs the allostimulatory capacity of human dendritic cells by intracellular glutathione depletion. Blood 2003, 102, 3668–3674. [Google Scholar] [CrossRef] [Green Version]
- Sido, B.; Braunstein, J.; Breitkreutz, R.; Herfarth, C.; Meuer, S.C. Thiol-Mediated Redox Regulation of Intestinal Lamina Propria T Lymphocytes. J. Exp. Med. 2000, 192, 907–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadzic, T.; Li, L.; Cheng, N.; Walsh, S.A.; Spitz, D.R.; Knudson, C.M. The Role of Low Molecular Weight Thiols in T Lymphocyte Proliferation and IL-2 Secretion. J. Immunol. 2005, 175, 7965–7972. [Google Scholar] [CrossRef] [Green Version]
- Arunachalam, B.; Phan, U.T.; Geuze, H.J.; Cresswell, P. Enzymatic reduction of disulfide bonds in lysosomes: Characterization of a Gamma-interferon-inducible lysosomal thiol reductase (GILT). Proc. Natl. Acad. Sci. USA 2000, 97, 745–750. [Google Scholar] [CrossRef] [Green Version]
- Short, S.; Merkel, B.J.; Caffrey, R.; McCoy, K.L. Defective antigen processing correlates with a low level of intracellular glutathione. Eur. J. Immunol. 1996, 26, 3015–3020. [Google Scholar] [CrossRef]
- Fraternale, A.; Brundu, S.; Magnani, M. Glutathione and glutathione derivatives in immunotherapy. Biol. Chem. 2017, 398, 261–275. [Google Scholar] [CrossRef]
- Mak, T.W.; Grusdat, M.; Duncan, G.S.; Dostert, C.; Nonnenmacher, Y.; Cox, M.; Binsfeld, C.; Hao, Z.; Brüstle, A.; Itsumi, M.; et al. Glutathione Primes T Cell Metabolism for Inflammation. Immunity 2017, 46, 675–689. [Google Scholar] [CrossRef] [Green Version]
- Murata, Y.; Ohteki, T.; Koyasu, S.; Hamuro, J. IFN-γ and pro-inflammatory cytokine production by antigen-presenting cells is dictated by intracellular thiol redox status regulated by oxygen tension. Eur. J. Immunol. 2002, 32, 2866–2873. [Google Scholar] [CrossRef]
- Dröge, W.; Breitkreutz, R. Glutathione and immune function. Proc. Nutr. Soc. 2000, 59, 595–600. [Google Scholar] [CrossRef]
- Kent, S.; Bluthé, R.M.; Kelley, K.W.; Dantzer, R. Sickness behavior as a new target for drug development. Trends Pharmacol. Sci. 1992, 13, 24–28. [Google Scholar] [CrossRef]
- Haddad, J.J.; Safieh-Garabedian, B.; Saadé, N.E.; Land, S.C. Thiol regulation of pro-inflammatory cytokines reveals a novel immunopharmacological potential of glutathione in the alveolar epithelium. J. Pharmacol. Exp. Ther. 2001, 296, 996–1005. [Google Scholar]
- Kudo, I.; Murakami, M. Prostaglandin E synthase, a terminal enzyme for prostaglandin E2 biosynthesis. J. Biochem. Mol. Biol. 2005, 38, 633–638. [Google Scholar] [CrossRef] [Green Version]
- Kil, I.S.; Kim, S.Y.; Park, J.-W. Glutathionylation regulates IkappaB. Biochem. Biophys. Res. Commun. 2008, 373, 169–173. [Google Scholar] [CrossRef]
- Seidel, P.; Merfort, I.; Hughes, J.M.; Oliver, B.G.G.; Tamm, M.; Roth, M. Dimethylfumarate inhibits NF-{kappa}B function at multiple levels to limit airway smooth muscle cell cytokine secretion. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 297, L326–339. [Google Scholar] [CrossRef]
- Oka, S.; Kamata, H.; Kamata, K.; Yagisawa, H.; Hirata, H. N-acetylcysteine suppresses TNF-induced NF-kappaB activation through inhibition of IkappaB kinases. FEBS Lett. 2000, 472, 196–202. [Google Scholar] [CrossRef] [Green Version]
- Lou, H.; Kaplowitz, N. Glutathione depletion down-regulates tumor necrosis factor alpha-induced NF-kappaB activity via IkappaB kinase-dependent and -independent mechanisms. J. Biol. Chem. 2007, 282, 29470–29481. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, A.J.; Krieg, J.-C.; Vedder, H. Interleukin-6 induces glutathione in hippocampal cells. Prog. Neuropsychopharmacol. Biol. Psychiatry 2005, 29, 321–326. [Google Scholar] [CrossRef]
- Murakami, M.; Naraba, H.; Tanioka, T.; Semmyo, N.; Nakatani, Y.; Kojima, F.; Ikeda, T.; Fueki, M.; Ueno, A.; Oh, S.; et al. Regulation of prostaglandin E2 biosynthesis by inducible membrane-associated prostaglandin E2 synthase that acts in concert with cyclooxygenase-2. J. Biol. Chem. 2000, 275, 32783–32792. [Google Scholar] [CrossRef] [Green Version]
- Engblom, D.; Saha, S.; Engström, L.; Westman, M.; Audoly, L.P.; Jakobsson, P.-J.; Blomqvist, A. Microsomal prostaglandin E synthase-1 is the central switch during immune-induced pyresis. Nat. Neurosci. 2003, 6, 1137–1138. [Google Scholar] [CrossRef]
- Ogawa, M. The Effect of Glutathione on Fever and Glycolysis. J. Agric. Chem. Soc. Jpn. 1939, 15, 775–782. [Google Scholar]
- Kolesnichenko, L.S.; Kulinsky, V.I.; Sotnikova, G.V.; Kovtun, V.Y. Influence of changes in glutathione concentration on body temperature and tolerance to cerebral ischemia. Biochem. Mosc. 2003, 68, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Costa, L.G.; Murphy, S.D. Effect of diethylmaleate and other glutathione depletors on protein synthesis. Biochem. Pharmacol. 1986, 35, 3383–3388. [Google Scholar] [CrossRef]
- Wrotek, S.; Jędrzejewski, T.; Piotrowski, J.; Kozak, W. N-Acetyl-l-cysteine exacerbates generation of IL-10 in cells stimulated with endotoxin in vitro and produces antipyresis via IL-10 dependent pathway in vivo. Immunol. Lett. 2016, 177, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Lou, Q.; Wang, F.; Li, E.; Sun, J.; Fang, H.; Xi, J.; Ju, L. N-acetylcysteine protects against liver injure induced by carbon tetrachloride via activation of the Nrf2/HO-1 pathway. Int. J. Clin. Exp. Pathol. 2015, 8, 8655–8662. [Google Scholar] [PubMed]
- Ortiz, M.S.; Forti, K.M.; Suárez Martinez, E.B.; Muñoz, L.G.; Husain, K.; Muñiz, W.H. Effects of Antioxidant N-acetylcysteine Against Paraquat-Induced Oxidative Stress in Vital Tissues of Mice. Int. J. Sci. Basic. Appl. Res. 2016, 26, 26–46. [Google Scholar]
- Pirabbasi, E.; Shahar, S.; Manaf, Z.A.; Rajab, N.F.; Manap, R.A. Efficacy of Ascorbic Acid (Vitamin C) and/N-Acetylcysteine (NAC) Supplementation on Nutritional and Antioxidant Status of Male Chronic Obstructive Pulmonary Disease (COPD) Patients. J. Nutr. Sci. Vitaminol. 2016, 62, 54–61. [Google Scholar] [CrossRef] [Green Version]
- Hamzeh, N.; Li, L.; Barkes, B.; Huang, J.; Canono, B.; Gillespie, M.; Maier, L.; Day, B. The effect of an oral anti-oxidant, N-Acetyl-cysteine, on inflammatory and oxidative markers in pulmonary sarcoidosis. Respir. Med. 2016, 112, 106–111. [Google Scholar] [CrossRef] [Green Version]
- Turkyilmaz, S.; Usta, A.; Cekic, A.B.; Alhan, E.; Kural, B.V.; Ercin, C. N-acetylcysteine amid reduces pancreatic damage in a rat model of acute necrotizing pancreatitis. J. Surg. Res. 2016, 203, 383–389. [Google Scholar] [CrossRef]
- Mikolka, P.; Kopincova, J.; Mikusiakova, L.T.; Kosutova, P.; Calkovska, A.; Mokra, D. Antiinflammatory Effect of N-Acetylcysteine Combined with Exogenous Surfactant in Meconium-Induced Lung Injury. Adv. Exp. Med. Biol. 2016, 934, 63–75. [Google Scholar]
- Cooper, A.L.; Brouwer, S.; Turnbull, A.V.; Luheshi, G.N.; Hopkins, S.J.; Kunkel, S.L.; Rothwell, N.J. Tumor necrosis factor-alpha and fever after peripheral inflammation in the rat. Am. J. Physiol. 1994, 267, R1431–R1436. [Google Scholar] [CrossRef] [PubMed]
- Kozak, W.; Poli, V.; Soszynski, D.; Conn, C.A.; Leon, L.R.; Kluger, M.J. Sickness behavior in mice deficient in interleukin-6 during turpentine abscess and influenza pneumonitis. Am. J. Physiol. 1997, 272, R621–630. [Google Scholar] [CrossRef] [PubMed]
- Kozak, W.; Kluger, M.J.; Soszynski, D.; Conn, C.A.; Rudolph, K.; Leon, L.R.; Zheng, H. IL-6 and IL-1 beta in fever. Studies using cytokine-deficient (knockout) mice. Ann. N. Y. Acad. Sci. 1998, 856, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.P.O.; Pasin, J.S.M.; Saraiva, A.L.L.; Ratzlaff, V.; Rossato, M.F.; Andrighetto, R.; Rubin, M.A.; Ferreira, J.; Mello, C.F. N-Acetylcysteine prevents baker’s-yeast-induced inflammation and fever. Inflamm. Res. 2012, 61, 103–112. [Google Scholar] [CrossRef]
- Sanchez-Alavez, M.; Bortell, N.; Galmozzi, A.; Conti, B.; Marcondes, M.C.G. Reactive oxygen species scavenger N-acetyl cysteine reduces methamphetamine-induced hyperthermia without affecting motor activity in mice. Temperature 2014, 1, 227–241. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.S.; Kim, S.J.; Kwon, D.Y.; Kim, Y.C. Comparison of the effects of buthioninesulfoximine and phorone on the metabolism of sulfur-containing amino acids in rat liver. Biochem. Biophys. Res. Commun. 2008, 368, 913–918. [Google Scholar] [CrossRef]
- Traber, J.; Suter, M.; Walter, P.; Richter, C. In vivo modulation of total and mitochondrial glutathione in rat liver. Depletion by phorone and rescue by N-acetylcysteine. Biochem. Pharmacol. 1992, 43, 961–964. [Google Scholar] [CrossRef]
- Haddad, J.J.; Land, S.C. Redox/ROS regulation of lipopolysaccharide-induced mitogen-activated protein kinase (MAPK) activation and MAPK-mediated TNF-α biosynthesis. Br. J. Pharmacol. 2002, 135, 520–536. [Google Scholar] [CrossRef] [Green Version]
- Griffith, O.W. Mechanism of action, metabolism, and toxicity of buthionine sulfoximine and its higher homologs, potent inhibitors of glutathione synthesis. J. Biol. Chem. 1982, 257, 13704–13712. [Google Scholar]
- Plummer, J.L.; Smith, B.R.; Sies, H.; Bend, J.R. Chemical depletion of glutathione in vivo. Meth. Enzymol. 1981, 77, 50–59. [Google Scholar]
- Sunahara, G.I.; Chiesa, A. Phorone (diisopropylidene acetone), a glutathione depletor, decreases rat glucocorticoid receptor binding in vivo. Carcinogenesis 1992, 13, 1083–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Housby, J.N.; Cahill, C.M.; Chu, B.; Prevelige, R.; Bickford, K.; Stevenson, M.A.; Calderwood, S.K. Non-steroidal anti-inflammatory drugs inhibit the expression of cytokines and induce HSP70 in human monocytes. Cytokine 1999, 11, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Page, T.H.; Turner, J.J.O.; Brown, A.C.; Timms, E.M.; Inglis, J.J.; Brennan, F.M.; Foxwell, B.M.J.; Ray, K.P.; Feldmann, M. Nonsteroidal anti-inflammatory drugs increase TNF production in rheumatoid synovial membrane cultures and whole blood. J. Immunol. 2010, 185, 3694–3701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbara, G.; Xing, Z.; Hogaboam, C.M.; Gauldie, J.; Collins, S.M. Interleukin 10 gene transfer prevents experimental colitis in rats. Gut 2000, 46, 344–349. [Google Scholar] [CrossRef] [Green Version]
- Rott, O.; Fleischer, B.; Cash, E. Interleukin-10 prevents experimental allergic encephalomyelitis in rats. Eur. J. Immunol. 1994, 24, 1434–1440. [Google Scholar] [CrossRef]
- Van Laethem, J.L.; Marchant, A.; Delvaux, A.; Goldman, M.; Robberecht, P.; Velu, T.; Devière, J. Interleukin 10 prevents necrosis in murine experimental acute pancreatitis. Gastroenterology 1995, 108, 1917–1922. [Google Scholar] [CrossRef]
- Pennline, K.J.; Roque-Gaffney, E.; Monahan, M. Recombinant human IL-10 prevents the onset of diabetes in the nonobese diabetic mouse. Clin. Immunol. Immunopathol. 1994, 71, 169–175. [Google Scholar] [CrossRef]
- Gérard, C.; Bruyns, C.; Marchant, A.; Abramowicz, D.; Vandenabeele, P.; Delvaux, A.; Fiers, W.; Goldman, M.; Velu, T. Interleukin 10 reduces the release of tumor necrosis factor and prevents lethality in experimental endotoxemia. J. Exp. Med. 1993, 177, 547–550. [Google Scholar] [CrossRef]
- Persson, S.; Mikulowska, A.; Narula, S.; O’Garra, A.; Holmdahl, R. Interleukin-10 suppresses the development of collagen type II-induced arthritis and ameliorates sustained arthritis in rats. Scand. J. Immunol. 1996, 44, 607–614. [Google Scholar] [CrossRef]
- Deml, E.; Oesterle, D. Histochemical demonstration of enhanced glutathione content in enzyme-altered islands induced by carcinogens in rat liver. Cancer Res. 1980, 40, 490–491. [Google Scholar]
- Awasthi, Y.C.; Sharma, R.; Yadav, S.; Dwivedi, S.; Sharma, A.; Awasthi, S. The non-ABC drug transporter RLIP76 (RALBP-1) plays a major role in the mechanisms of drug resistance. Curr. Drug Metab. 2007, 8, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Miura, M.; Sasaki, T. Role of glutathione in the intrinsic radioresistance of cell lines from a mouse squamous cell carcinoma. Radiat. Res. 1991, 126, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Traverso, N.; Ricciarelli, R.; Nitti, M.; Marengo, B.; Furfaro, A.L.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. Role of glutathione in cancer progression and chemoresistance. Oxid. Med. Cell. Longev. 2013, 2013, 972913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Dwyer, P.J.; Hamilton, T.C.; LaCreta, F.P.; Gallo, J.M.; Kilpatrick, D.; Halbherr, T.; Brennan, J.; Bookman, M.A.; Hoffman, J.; Young, R.C.; et al. Phase I trial of buthionine sulfoximine in combination with melphalan in patients with cancer. J. Clin. Oncol. 1996, 14, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Kinscherf, R.; Fischbach, T.; Mihm, S.; Roth, S.; Hohenhaus-Sievert, E.; Weiss, C.; Edler, L.; Bärtsch, P.; Dröge, W. Effect of glutathione depletion and oral N-acetyl-cysteine treatment on CD4+ and CD8+ cells. FASEB J. 1994, 8, 448–451. [Google Scholar] [CrossRef] [PubMed]
- Kabe, Y.; Ando, K.; Hirao, S.; Yoshida, M.; Handa, H. Redox Regulation of NF-κB Activation: Distinct Redox Regulation Between the Cytoplasm and the Nucleus. Antioxid. Redox Signal. 2005, 7, 395–403. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wrotek, S.; Sobocińska, J.; Kozłowski, H.M.; Pawlikowska, M.; Jędrzejewski, T.; Dzialuk, A. New Insights into the Role of Glutathione in the Mechanism of Fever. Int. J. Mol. Sci. 2020, 21, 1393. https://doi.org/10.3390/ijms21041393
Wrotek S, Sobocińska J, Kozłowski HM, Pawlikowska M, Jędrzejewski T, Dzialuk A. New Insights into the Role of Glutathione in the Mechanism of Fever. International Journal of Molecular Sciences. 2020; 21(4):1393. https://doi.org/10.3390/ijms21041393
Chicago/Turabian StyleWrotek, Sylwia, Justyna Sobocińska, Henryk M. Kozłowski, Małgorzata Pawlikowska, Tomasz Jędrzejewski, and Artur Dzialuk. 2020. "New Insights into the Role of Glutathione in the Mechanism of Fever" International Journal of Molecular Sciences 21, no. 4: 1393. https://doi.org/10.3390/ijms21041393