The PERK-Dependent Molecular Mechanisms as a Novel Therapeutic Target for Neurodegenerative Diseases

 ,
,
Abstract
:1. Introduction
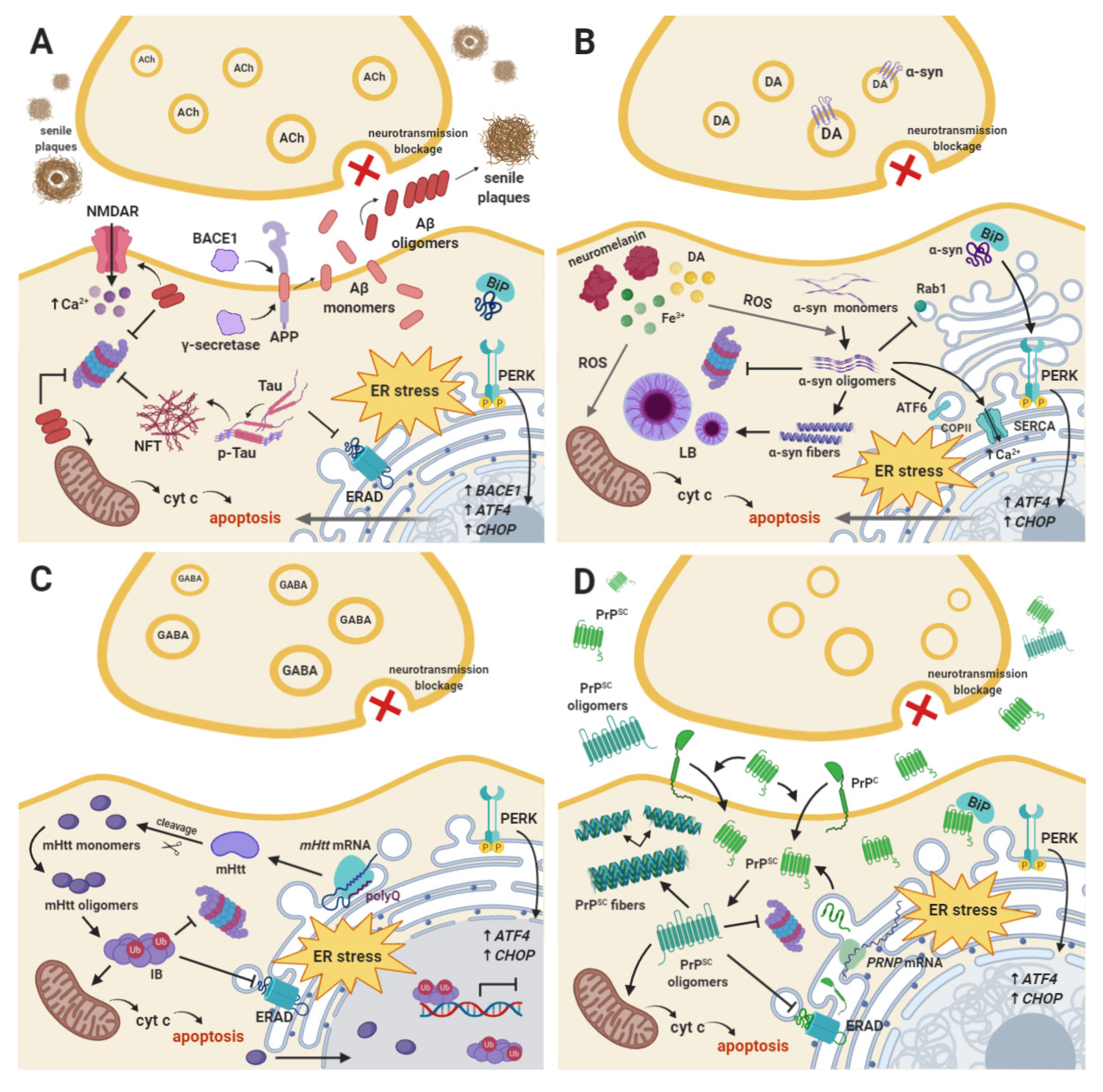
2. Alzheimer’s Disease
3. Parkinson’s Disease
4. Other Neurodegenerative Diseases
5. ER Stress and the UPR Signaling Branches
5.1. PERK-Dependent pro-Adaptive Branch of the UPR
5.2. PERK-Dependent pro-Apoptotic Branch of the UPR
6. Modulatory Compounds of the UPR Signaling Pathways
6.1. GSK2606414
6.2. GSK2656157
6.3. Salubrinal
6.4. ISRIB
6.5. Guanabenz
6.6. Sepin1
6.7. Trazodone Hydrochloride and Dibenzoylmethane
6.8. LDN-0060609
6.9. Oxyresveratrol
6.10. β-Asarone
6.11. Gastrodia Elata Derivatives
7. Summary and Perspective
Author Contributions
Funding
Conflicts of Interest
References
- Brown, R.C.; Lockwood, A.H.; Sonawane, B.R. Neurodegenerative diseases: An overview of environmental risk factors. Environ. Health Perspect. 2005, 113, 1250–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skoog, I.; Börjesson-Hanson, A.; Kern, S.; Johansson, L.; Falk, H.; Sigström, R.; Östling, S. Decreasing prevalence of dementia in 85-year olds examined 22 years apart: The influence of education and stroke. Sci. Rep. 2017, 7, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prince, M.; Bryce, R.; Albanese, E.; Wimo, A.; Ribeiro, W.; Ferri, C.P. The Global Prevalence of Dementia: A Systematic Review and Metaanalysis. Alzheimers Dement. 2013, 9, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Babusikova, E.; Evinova, A.; Jurecekova, J.; Jesenak, M.; Dobrot, D. Alzheimer’s Disease: Definition, Molecular and Genetic Factors. In Advanced Understanding of Neurodegenerative Diseases; IntechOpen: London, UK, 2011. [Google Scholar]
- Krantic, S. Editorial Thematic Issue: From Current Diagnostic Tools and Therapeutics for Alzheimer’s Disease towards Earlier Diagnostic Markers and Treatment Targets. Curr. Alzheimer Res. 2016, 14, 2–5. [Google Scholar] [CrossRef] [PubMed]
- Wortmann, M. Dementia: A global health priority—Highlights from an ADI and World Health Organization report. Alzheimer’s Res. Ther. 2012, 4, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Ham, T.J.; Breitling, R.; Swertz, M.A.; Nollen, E.A.A.A. Neurodegenerative diseases: Lessons from genome-wide screens in small model organisms. EMBO Mol. Med. 2009, 1, 360–370. [Google Scholar] [CrossRef]
- Brundin, P.; Melki, R.; Kopito, R. Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat. Rev. Mol. Cell Biol. 2010, 11, 301–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elharram, A.; Czegledy, N.M.; Golod, M.; Milne, G.L.; Pollock, E.; Bennett, B.M.; Shchepinov, M.S. Deuterium-reinforced polyunsaturated fatty acids improve cognition in a mouse model of sporadic Alzheimer’s disease. FEBS J. 2017, 284, 4083–4095. [Google Scholar] [CrossRef]
- Raefsky, S.M.; Furman, R.; Milne, G.; Pollock, E.; Axelsen, P.; Mattson, M.P.; Shchepinov, M.S. Deuterated polyunsaturated fatty acids reduce brain lipid peroxidation and hippocampal amyloid β-peptide levels, without discernable behavioral effects in an APP/PS1 mutant transgenic mouse model of Alzheimer’s disease. Neurobiol. Aging 2018, 66, 165–176. [Google Scholar] [CrossRef]
- Shchepinov, M.S.; Chou, V.P.; Pollock, E.; Langston, J.W.; Cantor, C.R.; Molinari, R.J.; Manning-Boĝ, A.B. Isotopic reinforcement of essential polyunsaturated fatty acids diminishes nigrostriatal degeneration in a mouse model of Parkinson’s disease. Toxicol. Lett. 2011, 207, 97–103. [Google Scholar] [CrossRef]
- Hatami, A.; Zhu, C.; Relaño-Gines, A.; Elias, C.; Galstyan, A.; Jun, M.; Milne, G.; Cantor, C.R.; Chesselet, M.F.; Shchepinov, M.S. Deuterium-reinforced linoleic acid lowers lipid peroxidation and mitigates cognitive impairment in the Q140 knock in mouse model of Huntington’s disease. FEBS J. 2018, 285, 3002–3012. [Google Scholar] [CrossRef] [PubMed]
- Halliday, M.; Mallucci, G.R. Review: Modulating the unfolded protein response to prevent neurodegeneration and enhance memory. Neuropathol. Appl. Neurobiol. 2015, 41, 414–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozpedek, W.; Markiewicz, L.; Diehl, J.; Pytel, D.; Majsterek, I. Unfolded Protein Response and PERK Kinase as a New Therapeutic Target in the Pathogenesis of Alzheimer’s Disease. Curr. Med. Chem. 2015, 22, 3169–3184. [Google Scholar] [CrossRef] [PubMed]
- Paraskevaidi, M.; Morais, C.L.M.; Lima, K.M.G.; Snowden, J.S.; Saxon, J.A.; Richardson, A.M.T.; Jones, M.; Mann, D.M.A.; Allsop, D.; Martin-Hirsch, P.L.; et al. Differential diagnosis of Alzheimer’s disease using spectrochemical analysis of blood. Proc. Natl. Acad. Sci. USA 2017, 114, E7929–E7938. [Google Scholar] [CrossRef] [Green Version]
- Rocca, W.A.; Petersen, R.C.; Knopman, D.S.; Hebert, L.E.; Evans, D.A.; Hall, K.S.; Gao, S.; Unverzagt, F.W.; Langa, K.M.; Larson, E.B.; et al. Trends in the incidence and prevalence of Alzheimer’s disease, dementia, and cognitive impairment in the United States. Alzheimer’s Dement. 2011, 7, 80–93. [Google Scholar] [CrossRef] [Green Version]
- Van Giau, V.; Pyun, J.M.; Suh, J.; Bagyinszky, E.; An, S.S.A.; Kim, S.Y. A pathogenic PSEN1 Trp165Cys mutation associated with early-onset Alzheimer’s disease. BMC Neurol. 2019, 19, 188. [Google Scholar] [CrossRef]
- Meraz-Ríos, M.A.; Franco-Bocanegra, D.; Toral Rios, D.; Campos-Peña, V. Early onset Alzheimer’s disease and oxidative stress. Oxidative Med. Cell. Longev. 2014, 2014. [Google Scholar] [CrossRef] [Green Version]
- Snowden, S.G.; Ebshiana, A.A.; Hye, A.; An, Y.; Pletnikova, O.; O’Brien, R.; Troncoso, J.; Legido-Quigley, C.; Thambisetty, M. Association between fatty acid metabolism in the brain and Alzheimer disease neuropathology and cognitive performance: A nontargeted metabolomic study. PLoS Med. 2017, 14, e1002266. [Google Scholar] [CrossRef] [Green Version]
- Forchetti, C.M. Treating patients with moderate to severe Alzheimer’s Disease: Implications of recent pharmacologic studies. Prim. Care Companion J. Clin. Psychiatry 2005, 7, 155–161. [Google Scholar] [CrossRef]
- Holtzman, D.M.; Morris, J.C.; Goate, A.M. Alzheimer’s disease: The challenge of the second century. Sci. Transl. Med. 2011, 3. [Google Scholar] [CrossRef] [Green Version]
- Ferris, S.H.; Farlow, M. Language impairment in alzheimer’s disease and benefits of acetylcholinesterase inhibitors. Clin. Interv. Aging 2013, 8, 1007–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nisbet, R.M.; Polanco, J.C.; Ittner, L.M.; Götz, J. Tau aggregation and its interplay with amyloid-β. Acta Neuropathol. 2015, 129, 207–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Qing, H.; Deng, Y. Biomarkers in Alzheimer’s disease analysis by mass spectrometry-based proteomics. Int. J. Mol. Sci. 2014, 15, 7865–7882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Lacosta, A.M.; Insua, D.; Badi, H.; Pesini, P.; Sarasa, M. Neurofibrillary Tangles of Aβx-40 in Alzheimer’s Disease Brains. J. Alzheimer’s Dis. 2017, 58, 661–667. [Google Scholar] [CrossRef]
- Donev, R.; Kolev, M.; Millet, B.; Thome, J. Neuronal death in Alzheimer’s disease and therapeutic opportunities. J. Cell. Mol. Med. 2009, 13, 4329–4348. [Google Scholar] [CrossRef] [Green Version]
- Jahn, H. Memory loss in alzheimer’s disease. Dialogues Clin. Neurosci. 2013, 15, 445–454. [Google Scholar]
- Salminen, A.; Kauppinen, A.; Suuronen, T.; Kaarniranta, K.; Ojala, J. ER stress in Alzheimer’s disease: A novel neuronal trigger for inflammation and Alzheimer’s pathology. J. Neuroinflamm. 2009, 6, 41. [Google Scholar] [CrossRef] [Green Version]
- Rahman, S.; Archana, A.; Jan, A.T.; Minakshi, R. Dissecting endoplasmic reticulum unfolded protein response (UPRER) in managing clandestine modus operandi of Alzheimer’s disease. Front. Aging Neurosci. 2018, 10, 30. [Google Scholar] [CrossRef] [Green Version]
- Glabe, C. Intracellular mechanisms of amyloid accumulation and pathogenesis in Alzheimer’s disease. J. Mol. Neurosci. 2001, 17, 137–145. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Swomley, A.M.; Sultana, R. Amyloid β-Peptide (1–42)-induced oxidative stress in alzheimer disease: Importance in disease pathogenesis and progression. Antioxid. Redox Signal. 2013, 19, 823–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunan, J.; Small, D.H. Regulation of APP cleavage by α-, β- and γ-secretases. FEBS Lett. 2000, 483, 6–10. [Google Scholar] [CrossRef] [Green Version]
- Khalifa, N.B.; Van Hees, J.; Tasiaux, B.; Huysseune, S.; Smith, S.O.; Constantinescu, S.N.; Octave, J.N.; Kienlen-Campard, P. What is the role of amyloid precursor protein dimerization? Cell Adhes. Migr. 2010, 4, 268–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, R.J.; Wong, P.C. Amyloid Precursor Protein Processing and Alzheimer’s Disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weggen, S.; Beher, D. Molecular consequences of amyloid precursor protein and presenilin mutations causing autosomal-dominant Alzheimer’s disease. Alzheimer’s Res. Ther. 2012, 4, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Strooper, B. Aph-1, Pen-2, and Nicastrin with Presenilin generate an active γ-Secretase complex. Neuron 2003, 38, 9–12. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, Y.; Matsuoka, M. A mutation protective against Alzheimer’s disease renders amyloid β precursor protein incapable of mediating neurotoxicity. J. Neurochem. 2014, 130, 291–300. [Google Scholar] [CrossRef] [Green Version]
- Portelius, E.; Price, E.; Brinkmalm, G.; Stiteler, M.; Olsson, M.; Persson, R.; Westman-Brinkmalm, A.; Zetterberg, H.; Simon, A.J.; Blennow, K. A novel pathway for amyloid precursor protein processing. Neurobiol. Aging 2011, 32, 1090–1098. [Google Scholar] [CrossRef]
- Miras-Portugal, M.T.; Diaz-Hernandez, J.I.; Gomez-Villafuertes, R.; Diaz-Hernandez, M.; Artalejo, A.R.; Gualix, J. Role of P2X7 and P2Y2 receptors on α-secretase-dependent APP processing: Control of amyloid plaques formation “in vivo” by P2X7 receptor. Comput. Struct. Biotechnol. J. 2015, 13, 176–181. [Google Scholar] [CrossRef] [Green Version]
- Hicks, D.A.; Nalivaeva, N.N.; Turner, A.J. Lipid rafts and Alzheimer’s disease: Protein-lipid interactions and perturbation of signaling. Front. Physiol. 2012, 3, 189. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Wang, Z.; Cai, F.; Zhang, M.; Wu, Y.; Zhang, J.; Song, W. BACE1 cleavage site selection critical for amyloidogenesis and Alzheimer’s pathogenesis. J. Neurosci. 2017, 37, 6915–6925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohandas, E.; Rajmohan, V.; Raghunath, B. Neurobiology of Alzheimer’s disease. Indian J. Psychiatry 2009, 51, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.B.; Lindholm, K.; Yan, R.; Citron, M.; Xia, W.; Yang, X.L.; Beach, T.; Sue, L.; Wong, P.; Price, D.; et al. Elevated β-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat. Med. 2003, 9, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Holsinger, R.M.D.; McLean, C.A.; Beyreuther, K.; Masters, C.L.; Evin, G. Increased expression of the amyloid precursor β-secretase in Alzheimer’s disease. Ann. Neurol. 2002, 51, 783–786. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, H.; Cheung, B.S.; Hyman, B.T.; Irizarry, M.C. β-secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch. Neurol. 2002, 59, 1381–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connor, T.; Sadleir, K.R.; Maus, E.; Velliquette, R.A.; Zhao, J.; Cole, S.L.; Eimer, W.A.; Hitt, B.; Bembinster, L.A.; Lammich, S.; et al. Phosphorylation of the Translation Initiation Factor eIF2α Increases BACE1 Levels and Promotes Amyloidogenesis. Neuron 2008, 60, 988–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoozemans, J.J.M.; Van Haastert, E.S.; Nijholt, D.A.T.; Rozemuller, A.J.M.; Eikelenboom, P.; Scheper, W. The unfolded protein response is activated in pretangle neurons in alzheimer’s disease hippocampus. Am. J. Pathol. 2009, 174, 1241–1251. [Google Scholar] [CrossRef] [Green Version]
- Stutzbach, L.D.; Xie, S.X.; Naj, A.C.; Albin, R.; Gilman, S.; Lee, V.M.Y.; Trojanowski, J.Q.; Devlin, B.; Schellenberg, G.D. The unfolded protein response is activated in disease-affected brain regions in progressive supranuclear palsy and Alzheimer’s disease. Acta Neuropathol. Commun. 2014, 2, 31. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Guo, Y.; Tang, J.; Jiang, J.; Chen, Z. New insights into the roles of CHOP-induced apoptosis in ER stress. Acta Biochim. Biophys. Sin. 2014, 46, 629–640. [Google Scholar] [CrossRef] [Green Version]
- Sadleir, K.R.; Popovic, J.; Vassar, R. ER stress is not elevated in the 5XFAD mouse model of Alzheimer’s disease. J. Biol. Chem. 2018, 293, 18434–18443. [Google Scholar] [CrossRef] [Green Version]
- Spatara, M.L.; Robinson, A.S. Transgenic mouse and cell culture models demonstrate a lack of mechanistic connection between endoplasmic reticulum stress and tau dysfunction. J. Neurosci. Res. 2010, 88, 1951–1961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitera, A.P.; Asuni, A.A.; O’Connor, V.; Deinhardt, K. Pathogenic tau does not drive activation of the unfolded protein response. J. Biol. Chem. 2019, 294, 9679–9688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohno, M. PERK as a hub of multiple pathogenic pathways leading to memory deficits and neurodegeneration in Alzheimer’s disease. Brain Res. Bull. 2018, 141, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Ray Dorsey, E.; Elbaz, A.; Nichols, E.; Abd-Allah, F.; Abdelalim, A.; Adsuar, J.C.; Ansha, M.G.; Brayne, C.; Choi, J.Y.J.; Collado-Mateo, D.; et al. Global, regional, and national burden of Parkinson’s disease, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 939–953. [Google Scholar] [CrossRef] [Green Version]
- Calabrese, V.P. Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology 2007, 68, 384–386. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Prim. 2017, 3, 1–21. [Google Scholar] [CrossRef]
- Mercado, G.; Castillo, V.; Vidal, R.; Hetz, C. ER proteostasis disturbances in Parkinson’s disease: Novel insights. Front. Aging Neurosci. 2015, 7, 39. [Google Scholar] [CrossRef]
- Valdés, P.; Mercado, G.; Vidal, R.L.; Molina, C.; Parsons, G.; Court, F.A.; Martinez, A.; Galleguillos, D.; Armentano, D.; Schneider, B.L.; et al. Control of dopaminergic neuron survival by the unfolded protein response transcription factor XBP1. Proc. Natl. Acad. Sci. USA 2014, 111, 6804–6809. [Google Scholar] [CrossRef] [Green Version]
- Driver, J.A.; Logroscino, G.; Gaziano, J.M.; Kurth, T. Incidence and remaining lifetime risk of Parkinson disease in advanced age. Neurology 2009, 72, 432–438. [Google Scholar] [CrossRef]
- Ascherio, A.; Schwarzschild, M.A. The epidemiology of Parkinson’s disease: Risk factors and prevention. Lancet Neurol. 2016, 15, 1257–1272. [Google Scholar] [CrossRef]
- Kim, C.Y.; Alcalay, R.N. Genetic Forms of Parkinson’s Disease. Semin. Neurol. 2017, 37, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Mercado, G.; Castillo, V.; Soto, P.; Sidhu, A. ER stress and Parkinson’s disease: Pathological inputs that converge into the secretory pathway. Brain Res. 2016, 1648, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Lehtonen, Š.; Sonninen, T.M.; Wojciechowski, S.; Goldsteins, G.; Koistinaho, J. Dysfunction of cellular proteostasis in Parkinson’s disease. Front. Neurosci. 2019, 13, 457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaya, J.; Aluf, Y.; Finberg, J.P.M. Oxidative Stress in Parkinson’s Disease. In Oxidative Stress and Free Radical Damage in Neurology; Humana Press: Totowa, NJ, USA, 2011; pp. 191–223. [Google Scholar]
- Puspita, L.; Chung, S.Y.; Shim, J.W. Oxidative stress and cellular pathologies in Parkinson’s disease. Mol. Brain 2017, 10, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cifu, D.X.; Eapen, B.C.; Janak, J.C.; Pugh, M.J.; Orman, J.A.L. Epidemiology of traumatic brain injury. In Traumatic Brain Injury Rehabilitation Medicine; Future Medicine Ltd.: London, UK, 2015; pp. 6–35. [Google Scholar]
- Gardner, R.C.; Byers, A.L.; Barnes, D.E.; Li, Y.; Boscardin, J.; Yaffe, K. Mild TBI and risk of Parkinson disease: A Chronic Effects of Neurotrauma Consortium Study. Neurology 2018, 90, E1771–E1779. [Google Scholar] [CrossRef] [PubMed]
- Pozo Devoto, V.M.; Falzone, T.L. Mitochondrial dynamics in Parkinson’s disease: A role for α-synuclein? DMM Dis. Model. Mech. 2017, 10, 1075–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grosch, J.; Winkler, J.; Kohl, Z. Early degeneration of both dopaminergic and serotonergic axons—A common mechanism in parkinson’s disease. Front. Cell. Neurosci. 2016, 10, 293. [Google Scholar] [CrossRef] [Green Version]
- Varrone, A.; Halldin, C. Molecular imaging of the dopamine transporter. J. Nucl. Med. 2010, 51, 1331–1334. [Google Scholar] [CrossRef] [Green Version]
- Fields, C.R.; Bengoa-Vergniory, N.; Wade-Martins, R. Targeting Alpha-Synuclein as a Therapy for Parkinson’s Disease. Front. Mol. Neurosci. 2019, 12, 299. [Google Scholar] [CrossRef] [Green Version]
- Perri, E.R.; Thomas, C.J.; Parakh, S.; Spencer, D.M.; Atkin, J.D. The unfolded protein response and the role of protein disulfide isomerase in neurodegeneration. Front. Cell Dev. Biol. 2016, 3, 80. [Google Scholar] [CrossRef] [Green Version]
- Doyle, K.M.; Kennedy, D.; Gorman, A.M.; Gupta, S.; Healy, S.J.M.; Samali, A. Unfolded proteins and endoplasmic reticulum stress in neurodegenerative disorders. J. Cell. Mol. Med. 2011, 15, 2025–2039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Pablo-Fernandez, E.; Tur, C.; Revesz, T.; Lees, A.J.; Holton, J.L.; Warner, T.T. Association of autonomic dysfunction with disease progression and survival in Parkinson disease. JAMA Neurol. 2017, 74, 970–976. [Google Scholar] [CrossRef]
- Bhardwaj, R.; Deshmukh, R. Parkinson’s Disease: An Insight into Mechanisms and Model Systems. Int. J. Med. Res. Health Sci. 2018, 7, 38–51. [Google Scholar]
- Volpicelli-Daley, L.; Brundin, P. Prion-like propagation of pathology in Parkinson disease. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2018; Volume 153, pp. 321–335. ISBN 9780444639455. [Google Scholar]
- Fullard, M.E.; Morley, J.F.; Duda, J.E. Olfactory Dysfunction as an Early Biomarker in Parkinson’s Disease. Neurosci. Bull. 2017, 33, 515–525. [Google Scholar] [CrossRef]
- Floor, E.; Wetzel, M.G. Increased Protein Oxidation in Human Substantia Nigra Pars Compacta in Comparison with Basal Ganglia and Prefrontal Cortex Measured with an Improved Dinitrophenylhydrazine Assay. J. Neurochem. 2002, 70, 268–275. [Google Scholar] [CrossRef]
- Nikolova, G. Oxidative stress and Parkinson disease. TRAKIA J. Sci. 2012, 10, 92–100. [Google Scholar]
- Bourdenx, M.; Koulakiotis, N.S.; Sanoudou, D.; Bezard, E.; Dehay, B.; Tsarbopoulos, A. Protein aggregation and neurodegeneration in prototypical neurodegenerative diseases: Examples of amyloidopathies, tauopathies and synucleinopathies. Prog. Neurobiol. 2017, 155, 171–193. [Google Scholar] [CrossRef]
- Shimozawa, A.; Ono, M.; Takahara, D.; Tarutani, A.; Imura, S.; Masuda-Suzukake, M.; Higuchi, M.; Yanai, K.; Hisanaga, S.I.; Hasegawa, M. Propagation of pathological α-synuclein in marmoset brain. Acta Neuropathol. Commun. 2017, 5, 12. [Google Scholar] [CrossRef] [Green Version]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.B.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. α-Synuclein Locus Triplication Causes Parkinson’s Disease. Science 2003, 302, 841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visanji, N.P.; Lang, A.E.; Kovacs, G.G. Beyond the synucleinopathies: Alpha synuclein as a driving force in neurodegenerative comorbidities. Transl. Neurodegener. 2019, 8, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, A.; Lopez, N.; Gonzalez, C.; Hetz, C. Targeting of the unfolded protein response (UPR) as therapy for Parkinson’s disease. Biol. Cell 2019, 111, 161–168. [Google Scholar] [CrossRef]
- Bertoncini, C.W.; Jung, Y.S.; Fernandez, C.O.; Hoyer, W.; Griesinger, C.; Jovin, T.M.; Zweckstetter, M. Release of long-range tertiary interactions potentiates aggregation of natively unstructured α-synuclein. Proc. Natl. Acad. Sci. USA 2005, 102, 1430–1435. [Google Scholar] [CrossRef] [Green Version]
- Stefani, C.I.; Wright, D.; Polizzi, M.K.; Kontoravdi, C. The Role of ER Stress-Induced Apoptosis in Neurodegeneration. Curr. Alzheimer Res. 2012, 9, 373–387. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.P.; Walker, D.E.; Goldstein, J.M.; De Laat, R.; Banducci, K.; Caccavello, R.J.; Barbour, R.; Huang, J.; Kling, K.; Lee, M.; et al. Phosphorylation of Ser-129 is the dominant pathological modification of α-synuclein in familial and sporadic lewy body disease. J. Biol. Chem. 2006, 281, 29739–29752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colla, E. Linking the endoplasmic reticulum to Parkinson’s disease and alpha-synucleinopathy. Front. Neurosci. 2019, 13, 560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, A.A.; Gitler, A.D.; Cashikar, A.; Haynes, C.M.; Hill, K.J.; Bhullar, B.; Liu, K.; Xu, K.; Strathearn, K.E.; Liu, F.; et al. α-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 2006, 313, 324–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzulli, J.R.; Zunke, F.; Isacson, O.; Studer, L.; Krainc, D. α-Synuclein-induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proc. Natl. Acad. Sci. USA 2016, 113, 1931–1936. [Google Scholar] [CrossRef] [Green Version]
- Credle, J.J.; Forcelli, P.A.; Delannoy, M.; Oaks, A.W.; Permaul, E.; Berry, D.L.; Duka, V.; Wills, J.; Sidhu, A. α-Synuclein-mediated inhibition of ATF6 processing into COPII vesicles disrupts UPR signaling in Parkinson’s disease. Neurobiol. Dis. 2015, 76, 112–125. [Google Scholar] [CrossRef]
- Betzer, C.; Lassen, L.B.; Olsen, A.; Kofoed, R.H.; Reimer, L.; Gregersen, E.; Zheng, J.; Calì, T.; Gai, W.; Chen, T.; et al. Alpha-synuclein aggregates activate calcium pump SERCA leading to calcium dysregulation. EMBO Rep. 2018, 19. [Google Scholar] [CrossRef]
- Paiva, I.; Jain, G.; Lázaro, D.F.; Jerčić, K.G.; Hentrich, T.; Kerimoglu, C.; Pinho, R.; Szegő, È.M.; Burkhardt, S.; Capece, V.; et al. Alpha-synuclein deregulates the expression of COL4A2 and impairs ER-Golgi function. Neurobiol. Dis. 2018, 119, 121–135. [Google Scholar] [CrossRef]
- Bellucci, A.; Navarria, L.; Zaltieri, M.; Falarti, E.; Bodei, S.; Sigala, S.; Battistin, L.; Spillantini, M.; Missale, C.; Spano, P. Induction of the unfolded protein response by α-synuclein in experimental models of Parkinson’s disease. J. Neurochem. 2011, 116, 588–605. [Google Scholar] [CrossRef] [PubMed]
- Bellani, S.; Mescola, A.; Ronzitti, G.; Tsushima, H.; Tilve, S.; Canale, C.; Valtorta, F.; Chieregatti, E. GRP78 clustering at the cell surface of neurons transduces the action of exogenous alpha-synuclein. Cell Death Differ. 2014, 21, 1971–1983. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Noh, J.Y.; Oh, Y.; Kim, Y.; Chang, J.W.; Chung, C.W.; Lee, S.T.; Kim, M.; Ryu, H.; Jung, Y.K. IRE1 plays an essential role in ER stress-mediated aggregation of mutant huntingtin via the inhibition of autophagy flux. Hum. Mol. Genet. 2012, 21, 101–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidal, R.; Caballero, B.; Couve, A.; Hetz, C. Converging Pathways in the Occurrence of Endoplasmic Reticulum (ER) Stress in Huntingtons Disease. Curr. Mol. Med. 2011, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Illarioshkin, S.N.; Klyushnikov, S.A.; Vigont, V.A.; Seliverstov, Y.A.; Kaznacheyeva, E.V. Molecular Pathogenesis in Huntington’s Disease. Biochemistry 2018, 83, 1030–1039. [Google Scholar] [CrossRef]
- Zuleta, A.; Vidal, R.L.; Armentano, D.; Parsons, G.; Hetz, C. AAV-mediated delivery of the transcription factor XBP1s into the striatum reduces mutant Huntingtin aggregation in a mouse model of Huntington’s disease. Biochem. Biophys. Res. Commun. 2012, 420, 558–563. [Google Scholar] [CrossRef] [PubMed]
- Xiang, C.; Wang, Y.; Zhang, H.; Han, F. The role of endoplasmic reticulum stress in neurodegenerative disease. Apoptosis 2017, 22, 1–26. [Google Scholar] [CrossRef]
- Margulis, J.; Finkbeiner, S. Proteostasis in striatal cells and selective neurodegeneration in Huntington’s disease. Front. Cell. Neurosci. 2014, 8, 218. [Google Scholar] [CrossRef] [Green Version]
- Shacham, T.; Sharma, N.; Lederkremer, G.Z. Protein misfolding and ER stress in Huntington’s disease. Front. Mol. Biosci. 2019, 6, 20. [Google Scholar] [CrossRef] [Green Version]
- Valionyte, E.; Yang, Y.; Roberts, S.L.; Kelly, J.; Lu, B.; Luo, S. Lowering Mutant Huntingtin Levels and Toxicity: Autophagy-Endolysosome Pathways in Huntington’s Disease. J. Mol. Biol. 2019. [Google Scholar] [CrossRef]
- Jiang, Y.; Chadwick, S.R.; Lajoie, P. Endoplasmic reticulum stress: The cause and solution to Huntington’s disease? Brain Res. 2016, 1648, 650–657. [Google Scholar] [CrossRef] [PubMed]
- Park, K.W.; Eun Kim, G.; Morales, R.; Moda, F.; Moreno-Gonzalez, I.; Concha-Marambio, L.; Lee, A.S.; Hetz, C.; Soto, C. The Endoplasmic Reticulum Chaperone GRP78/BiP Modulates Prion Propagation in vitro and in vivo. Sci. Rep. 2017, 7, 44723. [Google Scholar] [CrossRef] [Green Version]
- Scheper, W.; Hoozemans, J.J.M. The unfolded protein response in neurodegenerative diseases: A neuropathological perspective. Acta Neuropathol. 2015, 130, 315–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanemoto, S. Targeting the endoplasmic reticulum in prion disease treatment: Breakthroughs and challenges. Res. Rep. Biochem. 2015, 5, 31–38. [Google Scholar] [CrossRef] [Green Version]
- Wiersma, V.I.; van Hecke, W.; Scheper, W.; van Osch, M.A.J.; Hermsen, W.J.M.; Rozemuller, A.J.M.; Hoozemans, J.J.M. Activation of the unfolded protein response and granulovacuolar degeneration are not common features of human prion pathology. Acta Neuropathol. Commun. 2016, 4, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satterfield, T.; Pritchett, J.; Cruz, S.; Kemp, K. Prion disease and endoplasmic reticulum stress pathway correlations and treatment pursuits. Endoplasmic Reticulum Stress Dis. 2017, 4, 27–36. [Google Scholar] [CrossRef] [Green Version]
- Halliday, M.; Radford, H.; Mallucci, G.R. Prions: Generation and spread versus neurotoxicity. J. Biol. Chem. 2014, 289, 19862–19868. [Google Scholar] [CrossRef] [Green Version]
- Jaunmuktane, Z.; Brandner, S. Invited Review: The role of prion-like mechanisms in neurodegenerative diseases. Neuropathol. Appl. Neurobiol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Olsson, T.T.; Klementieva, O.; Gouras, G.K. Prion-like seeding and nucleation of intracellular amyloid-β. Neurobiol. Dis. 2018, 113, 1–10. [Google Scholar] [CrossRef]
- Ye, L.; Rasmussen, J.; Kaeser, S.A.; Marzesco, A.; Obermüller, U.; Mahler, J.; Schelle, J.; Odenthal, J.; Krüger, C.; Fritschi, S.K.; et al. Aβ seeding potency peaks in the early stages of cerebral β-amyloidosis. EMBO Rep. 2017, 18, 1536–1544. [Google Scholar] [CrossRef]
- Holmes, B.B.; Furman, J.L.; Mahan, T.E.; Yamasaki, T.R.; Mirbaha, H.; Eades, W.C.; Belaygorod, L.; Cairns, N.J.; Holtzman, D.M.; Diamond, M.I. Proteopathic tau seeding predicts tauopathy in vivo. Proc. Natl. Acad. Sci. USA 2014, 111, E4376–E4385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narasimhan, S.; Guo, J.L.; Changolkar, L.; Stieber, A.; McBride, J.D.; Silva, L.V.; He, Z.; Zhang, B.; Gathagan, R.J.; Trojanowski, J.Q.; et al. Pathological tau strains from human brains recapitulate the diversity of tauopathies in nontransgenic mouse brain. J. Neurosci. 2017, 37, 11406–11423. [Google Scholar] [CrossRef] [PubMed]
- Sanders, D.W.; Kaufman, S.K.; DeVos, S.L.; Sharma, A.M.; Mirbaha, H.; Li, A.; Barker, S.J.; Foley, A.C.; Thorpe, J.R.; Serpell, L.C.; et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 2014, 82, 1271–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clavaguera, F.; Akatsu, H.; Fraser, G.; Crowther, R.A.; Frank, S.; Hench, J.; Probst, A.; Winkler, D.T.; Reichwald, J.; Staufenbiel, M.; et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc. Natl. Acad. Sci. USA 2013, 110, 9535–9540. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, S.K.; Sanders, D.W.; Thomas, T.L.; Ruchinskas, A.J.; Vaquer-Alicea, J.; Sharma, A.M.; Miller, T.M.; Diamond, M.I. Tau Prion Strains Dictate Patterns of Cell Pathology, Progression Rate, and Regional Vulnerability In Vivo. Neuron 2016, 92, 796–812. [Google Scholar] [CrossRef] [Green Version]
- Prusiner, S.B.; Woerman, A.L.; Mordes, D.A.; Watts, J.C.; Rampersaud, R.; Berry, D.B.; Patel, S.; Oehler, A.; Lowe, J.K.; Kravitz, S.N.; et al. Evidence for α-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc. Natl. Acad. Sci. USA 2015, 112, E5308–E5317. [Google Scholar] [CrossRef] [Green Version]
- French, R.L.; Grese, Z.R.; Aligireddy, H.; Dhavale, D.D.; Reeb, A.N.; Kedia, N.; Kotzbauer, P.T.; Bieschke, J.; Ayala, Y.M. Detection of TAR DNA-binding protein 43 (TDP-43) oligomers as initial intermediate species during aggregate formation. J. Biol. Chem. 2019, 294, 6696–6709. [Google Scholar] [CrossRef]
- Fang, Y.S.; Tsai, K.J.; Chang, Y.J.; Kao, P.; Woods, R.; Kuo, P.H.; Wu, C.C.; Liao, J.Y.; Chou, S.C.; Lin, V.; et al. Full-length TDP-43 forms toxic amyloid oligomers that are present in frontotemporal lobar dementia-TDP patients. Nat. Commun. 2014, 5, 4824. [Google Scholar] [CrossRef]
- Kao, P.F.; Chen, Y.R.; Liu, X.B.; Decarli, C.; Seeley, W.W.; Jin, L.W. Detection of TDP-43 oligomers in frontotemporal lobar degeneration-TDP. Ann. Neurol. 2015, 78, 211–221. [Google Scholar] [CrossRef] [Green Version]
- Porta, S.; Xu, Y.; Restrepo, C.R.; Kwong, L.K.; Zhang, B.; Brown, H.J.; Lee, E.B.; Trojanowski, J.Q.; Lee, V.M.Y. Patient-derived frontotemporal lobar degeneration brain extracts induce formation and spreading of TDP-43 pathology in vivo. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Clavaguera, F.; Bolmont, T.; Crowther, R.A.; Abramowski, D.; Frank, S.; Probst, A.; Fraser, G.; Stalder, A.K.; Beibel, M.; Staufenbiel, M.; et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 2009, 11, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M.Y. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012, 338, 949–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watts, J.C.; Giles, K.; Oehler, A.; Middleton, L.; Dexter, D.T.; Gentleman, S.M.; DeArmond, S.J.; Prusiner, S.B. Transmission of multiple system atrophy prions to transgenic mice. Proc. Natl. Acad. Sci. USA 2013, 110, 19555–19560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaunmuktane, Z.; Mead, S.; Ellis, M.; Wadsworth, J.D.F.; Nicoll, A.J.; Kenny, J.; Launchbury, F.; Linehan, J.; Richard-Loendt, A.; Walker, A.S.; et al. Evidence for human transmission of amyloid-β pathology and cerebral amyloid angiopathy. Nature 2015, 525, 247–250. [Google Scholar] [CrossRef]
- Jaunmuktane, Z.; Quaegebeur, A.; Taipa, R.; Viana-Baptista, M.; Barbosa, R.; Koriath, C.; Sciot, R.; Mead, S.; Brandner, S. Evidence of amyloid-β cerebral amyloid angiopathy transmission through neurosurgery. Acta Neuropathol. 2018, 135, 671–679. [Google Scholar] [CrossRef] [Green Version]
- Frontzek, K.; Lutz, M.I.; Aguzzi, A.; Kovacs, G.G.; Budka, H. Amyloid-β pathology and cerebral amyloid angiopathy are frequent in iatrogenic Creutzfeldt-Jakob disease after dural grafting. Swiss Med. Wkly. 2016, 146, w14287. [Google Scholar] [CrossRef] [Green Version]
- Moreno, J.A.; Radford, H.; Peretti, D.; Steinert, J.R.; Verity, N.; Martin, M.G.; Halliday, M.; Morgan, J.; Dinsdale, D.; Ortori, C.A.; et al. Sustained translational repression by eIF2α-P mediates prion neurodegeneration. Nature 2012, 485, 507–511. [Google Scholar] [CrossRef] [Green Version]
- Unterberger, U.; Höftberger, R.; Gelpi, E.; Flicker, H.; Budka, H.; Voigtländer, T. Endoplasmic Reticulum Stress Features Are Prominent in Alzheimer Disease but Not in Prion Diseases In Vivo. J. Neuropathol. Exp. Neurol. 2006, 65, 348–357. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Lee, A.H.; Gonzalez-Romero, D.; Thielen, P.; Castilla, J.; Soto, C.; Glimcher, L.H. Unfolded protein response transcription factor XBP-1 does not influence prion replication or pathogenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 757–762. [Google Scholar] [CrossRef] [Green Version]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [Green Version]
- Bravo, R.; Parra, V.; Gatica, D.; Rodriguez, A.E.; Torrealba, N.; Paredes, F.; Wang, Z.V.; Zorzano, A.; Hill, J.A.; Jaimovich, E.; et al. Endoplasmic Reticulum and the Unfolded Protein Response. Dynamics and Metabolic Integration. In International Review of Cell and Molecular Biology; Elsevier: Amsterdam, The Netherlands, 2013; Volume 301, pp. 215–290. [Google Scholar]
- Chong, W.C.; Shastri, M.D.; Eri, R. Endoplasmic reticulum stress and oxidative stress: A vicious nexus implicated in bowel disease pathophysiology. Int. J. Mol. Sci. 2017, 18, 771. [Google Scholar] [CrossRef]
- Wojda, U.; Salinska, E.; Kuznicki, J. Calcium ions in neuronal degeneration. IUBMB Life 2008, 60, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Yang, J.; Li, L.; Shi, W.; Yuan, X.; Wu, L. The Natural Occurring Compounds Targeting Endoplasmic Reticulum Stress. Evid. Based Complement. Altern. Med. 2016, 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, S.S.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef] [PubMed]
- Erguler, K.; Pieri, M.; Deltas, C. A mathematical model of the unfolded protein stress response reveals the decision mechanism for recovery, adaptation and apoptosis. BMC Syst. Biol. 2013, 7, 16. [Google Scholar] [CrossRef] [Green Version]
- Urra, H.; Dufey, E.; Lisbona, F.; Rojas-Rivera, D.; Hetz, C. When ER stress reaches a dead end. Biochim. Biophys. Acta Mol. Cell Res. 2013, 1833, 3507–3517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindholm, D.; Korhonen, L.; Eriksson, O.; Kõks, S. Recent insights into the role of unfolded protein response in ER stress in health and disease. Front. Cell Dev. Biol. 2017, 5, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardner, B.M.; Pincus, D.; Gotthardt, K.; Gallagher, C.M.; Walter, P. Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb. Perspect. Biol. 2013, 5, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Rozpędek, W.; Pytel, D.; Nowak-Zduńczyk, A.; Lewko, D.; Wojtczak, R.; Diehl, J.A.; Majsterek, I. Breaking the DNA Damage Response via Serine/Threonine Kinase Inhibitors to Improve Cancer Treatment. Curr. Med. Chem. 2018, 26, 1425–1445. [Google Scholar] [CrossRef]
- Zhang, K.; Kaufman, R.J. Signaling the unfolded protein response from the endoplasmic reticulum. J. Biol. Chem. 2004, 279, 25935–25938. [Google Scholar] [CrossRef] [Green Version]
- Carrara, M.; Prischi, F.; Nowak, P.R.; Kopp, M.C.; Ali, M.M.U. Noncanonical binding of BiP ATPase domain to Ire1 and Perk is dissociated by unfolded protein CH1 to initiate ER stress signaling. Elife 2015, 4, e03522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.; Prywes, R. Dependence of site-2 protease cleavage of ATF6 on prior site-1 protease digestion is determined by the size of the luminal domain of ATF6. J. Biol. Chem. 2004, 279, 43046–43051. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.L.; Li, X.; Gan, L.; He, Y.Y.; Wang, L.L.; He, K.L. High-content screening identifies inhibitors of the nuclear translocation of ATF6. Int. J. Mol. Med. 2016, 37, 407–414. [Google Scholar] [CrossRef]
- Smith, H.L.; Mallucci, G.R. The unfolded protein response: Mechanisms and therapy of neurodegeneration. Brain 2016, 139, 2113–2121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Hendershot, L.M. The role of the unfolded protein response in tumour development: Friend or foe? Nat. Rev. Cancer 2004, 4, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Luo, B.; Lee, A.S. The critical roles of endoplasmic reticulum chaperones and unfolded protein response in tumorigenesis and anticancer therapies. Oncogene 2013, 32, 805–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.C.; Chan, N.; Labib, S.; Yu, J.; Cho, H.Y.; Hofman, F.M.; Schönthal, A.H. Induction of pro-apoptotic endoplasmic reticulum stress in multiple myeloma cells by NEO214, perillyl alcohol conjugated to rolipram. Int. J. Mol. Sci. 2018, 19, 277. [Google Scholar] [CrossRef] [Green Version]
- Cui, W.; Li, J.; Ron, D.; Sha, B. The structure of the PERK kinase domain suggests the mechanism for its activation. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 423–428. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Lv, Y.; Zhao, N.; Guan, G.; Wang, J. Protein kinase R-like ER kinase and its role in endoplasmic reticulum stress-decided cell fate. Cell Death Dis. 2015, 6, e1822. [Google Scholar] [CrossRef] [Green Version]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef]
- Wang, P.; Li, J.; Sha, B. The ER stress sensor PERK luminal domain functions as a molecular chaperone to interact with misfolded proteins. Acta Crystallogr. Sect. D Struct. Biol. 2016, 72, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Pytel, D.; Majsterek, I.; Diehl, J.A. Tumor progression and the different faces of the PERK kinase. Oncogene 2016, 35, 1207–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DuRose, J.B.; Scheuner, D.; Kaufman, R.J.; Rothblum, L.I.; Niwa, M. Phosphorylation of Eukaryotic Translation Initiation Factor 2 Coordinates rRNA Transcription and Translation Inhibition during Endoplasmic Reticulum Stress. Mol. Cell. Biol. 2009, 29, 4295–4307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozpedek, W.; Pytel, D.; Mucha, B.; Leszczynska, H.; Diehl, J.A.; Majsterek, I. The Role of the PERK/eIF2α/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress. Curr. Mol. Med. 2016, 16, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Devi, L.; Ohno, M. PERK mediates eIF2α phosphorylation responsible for BACE1 elevation, CREB dysfunction and neurodegeneration in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2014, 35, 2272–2281. [Google Scholar] [CrossRef] [Green Version]
- Fusakio, M.E.; Willy, J.A.; Wang, Y.; Mirek, E.T.; Baghdadi, R.J.T.A.; Adams, C.M.; Anthony, T.G.; Wek, R.C. Transcription factor ATF4 directs basal and stress-induced gene expression in the unfolded protein response and cholesterol metabolism in the liver. Mol. Biol. Cell 2016, 27, 1536–1551. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Grek, C.; Townsend, D.M. Protein Disulfide Isomerase Superfamily in Disease and the Regulation of Apoptosis. Endoplasmic Reticulum Stress Dis. 2013, 1, 4–17. [Google Scholar] [CrossRef]
- Devi, L.; Ohno, M. Mechanisms that lessen benefits of β-secretase reduction in a mouse model of Alzheimer’s disease. Transl. Psychiatry 2013, 3, e284. [Google Scholar] [CrossRef] [Green Version]
- Devi, L.; Ohno, M. Phospho-eIF2α Level Is Important for Determining Abilities of BACE1 Reduction to Rescue Cholinergic Neurodegeneration and Memory Defects in 5XFAD Mice. PLoS ONE 2010, 5, e12974. [Google Scholar] [CrossRef] [Green Version]
- Devi, L.; Ohno, M. Deletion of the eIF2α Kinase GCN2 Fails to Rescue the Memory Decline Associated with Alzheimer’s Disease. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadleir, K.R.; Eimer, W.A.; Kaufman, R.J.; Osten, P.; Vassar, R. Genetic Inhibition of Phosphorylation of the Translation Initiation Factor eIF2α Does Not Block Aβ-Dependent Elevation of BACE1 and APP Levels or Reduce Amyloid Pathology in a Mouse Model of Alzheimer’s Disease. PLoS ONE 2014, 9, e101643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, D.; Mallucci, G.R. The unfolded protein response in neurodegenerative disorders—Therapeutic modulation of the PERK pathway. FEBS J. 2019, 286, 342–355. [Google Scholar] [CrossRef] [PubMed]
- López, I.; Tournillon, A.S.; Martins, R.P.; Karakostis, K.; Malbert-Colas, L.; Nylander, K.; Fåhraeus, R. P53-mediated suppression of BiP triggers BIK-induced apoptosis during prolonged endoplasmic reticulum stress. Cell Death Differ. 2017, 24, 1717–1729. [Google Scholar] [CrossRef] [Green Version]
- Pasini, S.; Corona, C.; Liu, J.; Greene, L.A.; Shelanski, M.L. Specific downregulation of hippocampal ATF4 reveals a necessary role in synaptic plasticity and memory. Cell Rep. 2015, 11, 183–191. [Google Scholar] [CrossRef] [Green Version]
- Martínez, G.; Vidal, R.L.; Mardones, P.; Serrano, F.G.; Ardiles, A.O.; Wirth, C.; Valdés, P.; Thielen, P.; Schneider, B.L.; Kerr, B.; et al. Regulation of Memory Formation by the Transcription Factor XBP1. Cell Rep. 2016, 14, 1382–1394. [Google Scholar] [CrossRef] [Green Version]
- Yan, R.; Vassar, R. Targeting the β secretase BACE1 for Alzheimer’s disease therapy. Lancet Neurol. 2014, 13, 319–329. [Google Scholar] [CrossRef] [Green Version]
- Macleod, R.; Hillert, E.K.; Cameron, R.T.; Baillie, G.S. The role and therapeutic targeting of α-, β-and γ-secretase in Alzheimer’s disease. Future Sci. OA 2015, 1. [Google Scholar] [CrossRef] [Green Version]
- Pillai, S. Birth pangs: The stressful origins of lymphocytes. J. Clin. Investig. 2005, 115, 224–227. [Google Scholar] [CrossRef]
- Endres, K.; Reinhardt, S. ER-stress in Alzheimer’s disease: Turning the scale? Am. J. Neurodegener. Dis. 2013, 2, 247–265. [Google Scholar]
- Siwecka, N.; Rozpȩdek, W.; Pytel, D.; Wawrzynkiewicz, A.; Dziki, A.; Dziki, Ł.; Alan Diehl, J.; Majsterek, I.; Dziki, L.; Alan Diehl, J.; et al. Dual role of endoplasmic reticulum stress-mediated unfolded protein response signaling pathway in carcinogenesis. Int. J. Mol. Sci. 2019, 20, 4354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malhotra, J.D.; Kaufman, R.J. The endoplasmic reticulum and the unfolded protein response. Semin. Cell Dev. Biol. 2007, 18, 716–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinszner, H.; Kuroda, M.; Wang, X.Z.; Batchvarova, N.; Lightfoot, R.T.; Remotti, H.; Stevens, J.L.; Ron, D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998, 12, 982–995. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, D.; Rojas-Rivera, D.; Hetz, C. Integrating stress signals at the endoplasmic reticulum: The BCL-2 protein family rheostat. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 564–574. [Google Scholar] [CrossRef] [Green Version]
- O’neill, K.L.; Huang, K.; Zhang, J.; Chen, Y.; Luo, X. Inactivation of prosurvival Bcl-2 proteins activates Bax/Bak through the outer mitochondrial membrane. Genes Dev. 2016, 30, 973–988. [Google Scholar] [CrossRef] [Green Version]
- Wali, J.A.; Rondas, D.; McKenzie, M.D.; Zhao, Y.; Elkerbout, L.; Fynch, S.; Gurzov, E.N.; Akira, S.; Mathieu, C.; Kay, T.W.H.; et al. The proapoptotic BH3-only proteins Bim and Puma are downstream of endoplasmic reticulum and mitochondrial oxidative stress in pancreatic islets in response to glucotoxicity. Cell Death Dis. 2014, 5, e1124. [Google Scholar] [CrossRef] [Green Version]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef] [Green Version]
- Galehdar, Z.; Swan, P.; Fuerth, B.; Callaghan, S.M.; Park, D.S.; Cregan, S.P. Neuronal apoptosis induced by endoplasmic reticulum stress is regulated by ATF4-CHOP-mediated induction of the Bcl-2 homology 3-only member PUMA. J. Neurosci. 2010, 30, 16938–16948. [Google Scholar] [CrossRef]
- Bunk, E.C.; König, H.G.; Bernas, T.; Engel, T.; Henshall, D.C.; Kirby, B.P.; Prehn, J.H.M. BH3-only proteins BIM and PUMA in the regulation of survival and neuronal differentiation of newly generated cells in the adult mouse hippocampus. Cell Death Dis. 2010, 1, e15. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.P.; Klocke, B.J.; Ballestas, M.E.; Roth, K.A. CHOP potentially co-operates with FOXO3a in neuronal cells to regulate PUMA and BIM expression in response to ER stress. PLoS ONE 2012, 7, e39586. [Google Scholar] [CrossRef]
- Kudo, W.; Lee, H.G.P.; Smith, M.A.; Zhu, X.; Matsuyama, S.; Lee, H.G.P. Inhibition of Bax protects neuronal cells from oligomeric Aβ neurotoxicity. Cell Death Dis. 2012, 3, e309. [Google Scholar] [CrossRef] [PubMed]
- Rojas, M.; Vasconcelos, G.; Dever, T.E. An eIF2α-binding motif in protein phosphatase 1 subunit GADD34 and its viral orthologs is required to promote dephosphorylation of eIF2α. Proc. Natl. Acad. Sci. USA 2015, 112, E3466–E3475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novoa, I.; Zeng, H.; Harding, H.P.; Ron, D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2α. J. Cell Biol. 2001, 153, 1011–1021. [Google Scholar] [CrossRef] [Green Version]
- Hollander, M.C.; Zhan, Q.; Bae, I.; Fornace, A.J. Mammalian GADD34, an apoptosis and DNA damage-inducible gene. J. Biol. Chem. 1997, 272, 13731–13737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adler, H.T.; Chinery, R.; Wu, D.Y.; Kussick, S.J.; Payne, J.M.; Fornace, A.J.; Tkachuk, D.C. Leukemic HRX Fusion Proteins Inhibit GADD34-Induced Apoptosis and Associate with the GADD34 and hSNF5/INI1 Proteins. Mol. Cell. Biol. 1999, 19, 7050–7060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldman, D.E.; Chauhan, V.; Koong, A.C. The unfolded protein response: A novel component of the hypoxic stress response in tumors. Mol. Cancer Res. 2005, 3, 597–605. [Google Scholar] [CrossRef] [Green Version]
- Cornejo, V.H.; Hetz, C. The unfolded protein response in Alzheimer’s disease. Semin. Immunopathol. 2013, 35, 277–292. [Google Scholar] [CrossRef]
- Boyce, M.; Bryant, K.F.; Jousse, C.; Long, K.; Harding, H.P.; Scheuner, D.; Kaufman, R.J.; Ma, D.; Coen, D.M.; Ron, D.; et al. A selective inhibitor of elF2α dephosphorylation protects cells from ER stress. Science 2005, 307, 935–939. [Google Scholar] [CrossRef]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef]
- Kalaria, R.N. The role of cerebral ischemia in Alzheimer’s disease. Neurobiol. Aging 2000, 21, 321–330. [Google Scholar] [CrossRef]
- Lucke-Wold, B.P.; Turner, R.C.; Logsdon, A.F.; Simpkins, J.W.; Alkon, D.L.; Smith, K.E.; Chen, Y.W.; Tan, Z.; Huber, J.D.; Rosen, C.L. Common mechanisms of Alzheimer’s disease and ischemic stroke: The role of protein kinase C in the progression of age-related neurodegeneration. J. Alzheimer’s Dis. 2015, 43, 711–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pluta, R.; Jabłoński, M.; Ułamek-Kozioł, M.; Kocki, J.; Brzozowska, J.; Januszewski, S.; Furmaga-Jabłońska, W.; Bogucka-Kocka, A.; Maciejewski, R.; Czuczwar, S.J. Sporadic alzheimer’s disease begins as episodes of brain ischemia and ischemically dysregulated alzheimer’s disease genes. Mol. Neurobiol. 2013, 48, 500–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tajiri, S.; Oyadomari, S.; Yano, S.; Morioka, M.; Gotoh, T.; Hamada, J.I.; Ushio, Y.; Mori, M. Ischemia-induced neuronal cell death is mediated by the endoplasmic reticulum stress pathway involving CHOP. Cell Death Differ. 2004, 11, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Poone, G.; Hasseldam, H.; Munkholm, N.; Rasmussen, R.; Grønberg, N.; Johansen, F. The Hypothermic Influence on CHOP and Ero1-α in an Endoplasmic Reticulum Stress Model of Cerebral Ischemia. Brain Sci. 2015, 5, 178–187. [Google Scholar] [CrossRef] [Green Version]
- Scull, C.M.; Tabas, I. Mechanisms of ER stress-induced apoptosis in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2792–2797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeeshan, H.M.A.; Lee, G.H.; Kim, H.R.; Chae, H.J. Endoplasmic reticulum stress and associated ROS. Int. J. Mol. Sci. 2016, 17, 327. [Google Scholar] [CrossRef] [Green Version]
- Laurindo, F.R.M.; Araujo, T.L.S.; Abrahão, T.B. Nox NADPH oxidases and the endoplasmic reticulum. Antioxid. Redox Signal. 2014, 20, 2755–2775. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.Y.; Lee, K.-S.S.; Lee, H.J.; Kim, D.H.D.K.D.H.; Noh, Y.H.; Yu, K.; Jung, H.-Y.Y.; Lee, S.H.; Lee, J.Y.; Youn, Y.C.; et al. Activation of PERK signaling attenuates Aβ-mediated ER stress. PLoS ONE 2010, 5, e10489. [Google Scholar] [CrossRef]
- Plácido, A.I.; Oliveira, C.R.; Moreira, P.I.; Pereira, C.M.F. Enhanced Amyloidogenic Processing of Amyloid Precursor Protein and Cell Death Under Prolonged Endoplasmic Reticulum Stress in Brain Endothelial Cells. Mol. Neurobiol. 2015, 51, 571–590. [Google Scholar] [CrossRef]
- Fonseca, A.C.R.G.; Ferreiro, E.; Oliveira, C.R.; Cardoso, S.M.; Pereira, C.F. Activation of the endoplasmic reticulum stress response by the amyloid-beta 1–40 peptide in brain endothelial cells. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 2191–2203. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Chan, Y.; Wan, W.; Li, Y.; Zhang, C. Aβ1-42 induces cell damage via RAGE-dependent endoplasmic reticulum stress in bEnd.3 cells. Exp. Cell Res. 2018, 362, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Pan, X.D.; Zhang, J.; Zeng, Y.Q.; Zhou, M.; Yang, L.M.; Ye, B.; Dai, X.M.; Zhu, Y.G.; Chen, X.C. Endoplasmic reticulum stress induces the early appearance of pro-apoptotic and anti-apoptotic proteins in neurons of five familial alzheimer’s disease mice. Chin. Med. J. 2016, 129, 2845–2852. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Wang, S.; Wang, Z.; Wang, Z.; Sun, C.; Zhang, Y. Inhibition of PTEN Attenuates Endoplasmic Reticulum Stress and Apoptosis via Activation of PI3K/AKT Pathway in Alzheimer’s Disease. Neurochem. Res. 2017, 42, 3052–3060. [Google Scholar] [CrossRef] [PubMed]
- Alberdi, E.; Wyssenbach, A.; Alberdi, M.; Sánchez-Gómez, M.V.; Cavaliere, F.; Rodríguez, J.J.; Verkhratsky, A.; Matute, C. Ca2+ dependent endoplasmic reticulum stress correlates with astrogliosis in oligomeric amyloid β-treated astrocytes and in a model of Alzheimer’s disease. Aging Cell 2013, 12, 292–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soejima, N.; Ohyagi, Y.; Nakamura, N.; Himeno, E.; Iinuma, M.K.; Sakae, N.; Yamasaki, R.; Tabira, T.; Murakami, K.; Irie, K.; et al. Intracellular Accumulation of Toxic Turn Amyloid-β is Associated with Endoplasmic Reticulum Stress in Alzheimer’s Disease. Curr. Alzheimer Res. 2013, 10, 11–20. [Google Scholar] [PubMed]
- Katayama, T.; Imaizumi, K.; Sato, N.; Miyoshi, K.; Kudo, T.; Hitomi, J.; Morihara, T.; Yoneda, T.; Gomi, F.; Mori, Y.; et al. Presenilin-1 mutations downregulate the signalling pathway of the unfolded-protein response. Nat. Cell Biol. 1999, 1, 479–485. [Google Scholar] [CrossRef]
- Ho, Y.S.; Yang, X.; Lau, J.C.F.; Hung, C.H.L.; Wuwongse, S.; Zhang, Q.; Wang, J.; Baum, L.; So, K.F.; Chang, R.C.C. Endoplasmic reticulum stress induces tau pathology and forms a vicious cycle: Implication in Alzheimer’s disease pathogenesis. J. Alzheimer’s Dis. 2012, 28, 839–854. [Google Scholar] [CrossRef] [Green Version]
- Cheng, K.C.; Chiang, H.C. XBP1 and PERK Have Distinct Roles in Aβ-Induced Pathology. Mol. Neurobiol. 2018, 55, 7523–7532. [Google Scholar] [CrossRef]
- Köhler, C.; Dinekov, M.; Götz, J. Granulovacuolar degeneration and unfolded protein response in mouse models of tauopathy and Aβ amyloidosis. Neurobiol. Dis. 2014, 71, 169–179. [Google Scholar] [CrossRef]
- Mercado, G.; Hetz, C. Drug repurposing to target proteostasis and prevent neurodegeneration: Accelerating translational efforts. Brain 2017, 140, 1544–1547. [Google Scholar] [CrossRef]
- Ma, T.; Trinh, M.A.; Wexler, A.J.; Bourbon, C.; Gatti, E.; Pierre, P.; Cavener, D.R.; Klann, E. Suppression of eIF2α kinases alleviates Alzheimer’s disease-related plasticity and memory deficits. Nat. Neurosci. 2013, 16, 1299–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baleriola, J.; Walker, C.A.; Jean, Y.Y.; Crary, J.F.; Troy, C.M.; Nagy, P.L.; Hengst, U. Axonally synthesized ATF4 transmits a neurodegenerative signal across brain regions. Cell 2014, 158, 1159–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mamada, N.; Tanokashira, D.; Hosaka, A.; Kametani, F.; Tamaoka, A.; Araki, W. Amyloid β-protein oligomers upregulate the β-secretase, BACE1, through a post-translational mechanism involving its altered subcellular distribution in neurons. Mol. Brain 2015, 8, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segev, Y.; Barrera, I.; Ounallah-Saad, H.; Wibrand, K.; Sporild, I.; Livne, A.; Rosenberg, T.; David, O.; Mints, M.; Bramham, C.R.; et al. PKR inhibition rescues memory deficit and atf4 overexpression in apoe ε4 human replacement mice. J. Neurosci. 2015, 35, 12986–12993. [Google Scholar] [CrossRef] [Green Version]
- Zhong, N.; Ramaswamy, G.; Weisgraber, K.H. Apolipoprotein E4 domain interaction induces endoplasmic reticulum stress and impairs astrocyte function. J. Biol. Chem. 2009, 284, 27273–27280. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Won, S.M.; Suh, J.; Son, S.J.; Moon, G.J.; Park, U.J.; Gwag, B.J. Induction of the unfolded protein response and cell death pathway in alzheimer’s disease, but not in aged Tg2576 mice. Exp. Mol. Med. 2010, 42, 386–394. [Google Scholar] [CrossRef] [Green Version]
- Xu, N.; Xiao, Z.; Zou, T.; Huang, Z. Induction of GADD34 regulates the neurotoxicity of amyloid β. Am. J. Alzheimer’s. Dis. Other Dement. 2015, 30, 313–319. [Google Scholar] [CrossRef]
- Honjo, Y.; Ayaki, T.; Tomiyama, T.; Horibe, T.; Ito, H.; Mori, H.; Takahashi, R.; Kawakami, K. Increased GADD34 in oligodendrocytes in Alzheimer’s disease. Neurosci. Lett. 2015, 602, 50–55. [Google Scholar] [CrossRef]
- Chiu, C.C.; Lu, C.S.; Weng, Y.H.; Chen, Y.L.; Huang, Y.Z.; Chen, R.S.; Cheng, Y.C.; Huang, Y.C.; Liu, Y.C.; Lai, S.C.; et al. PARK14 (D331Y) PLA2G6 Causes Early-Onset Degeneration of Substantia Nigra Dopaminergic Neurons by Inducing Mitochondrial Dysfunction, ER Stress, Mitophagy Impairment and Transcriptional Dysregulation in a Knockin Mouse Model. Mol. Neurobiol. 2019, 56, 3835–3853. [Google Scholar] [CrossRef]
- Holtz, W.A.; O’Malley, K.L. Parkinsonian Mimetics Induce Aspects of Unfolded Protein Response in Death of Dopaminergic Neurons. J. Biol. Chem. 2003, 278, 19367–19377. [Google Scholar] [CrossRef] [Green Version]
- Yamamuro, A.; Yoshioka, Y.; Ogita, K.; Maeda, S. Involvement of Endoplasmic Reticulum Stress on the Cell Death Induced by 6-Hydroxydopamine in Human Neuroblastoma SH-SY5Y Cells. Neurochem. Res. 2006, 31, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Salganik, M.; Sergeyev, V.G.; Shinde, V.; Meyers, C.A.; Gorbatyuk, M.S.; Lin, J.H.; Zolotukhin, S.; Gorbatyuk, O.S. The loss of glucose-regulated protein 78 (GRP78) during normal aging or from siRNA knockdown augments human alpha-synuclein (α-syn) toxicity to rat nigral neurons. Neurobiol. Aging 2015, 36, 2213–2223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorbatyuk, M.S.; Shabashvili, A.; Chen, W.; Meyers, C.; Sullivan, L.F.; Salganik, M.; Lin, J.H.; Lewin, A.S.; Muzyczka, N.; Gorbatyuk, O.S. Glucose regulated protein 78 diminishes α-synuclein neurotoxicity in a rat model of parkinson disease. Mol. Ther. 2012, 20, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Baek, J.H.; Mamula, D.; Tingstam, B.; Pereira, M.; He, Y.; Svenningsson, P. GRP78 level is altered in the brain, but not in plasma or cerebrospinal fluid in Parkinson’s disease patients. Front. Neurosci. 2019, 13, 697. [Google Scholar] [CrossRef]
- Yang, J.; Kim, K.S.; Iyirhiaro, G.O.; Marcogliese, P.C.; Callaghan, S.M.; Qu, D.; Kim, W.J.; Slack, R.S.; Park, D.S. DJ-1 modulates the unfolded protein response and cell death via upregulation of ATF4 following ER stress. Cell Death Dis. 2019, 10, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Mercado, G.; Castillo, V.; Soto, P.; López, N.; Axten, J.M.; Sardi, S.P.; Hoozemans, J.J.M.; Hetz, C. Targeting PERK signaling with the small molecule GSK2606414 prevents neurodegeneration in a model of Parkinson’s disease. Neurobiol. Dis. 2018, 112, 136–148. [Google Scholar] [CrossRef]
- Amodio, G.; Moltedo, O.; Fasano, D.; Zerillo, L.; Oliveti, M.; Di Pietro, P.; Faraonio, R.; Barone, P.; Pellecchia, M.T.; De Rosa, A.; et al. PERK-mediated unfolded protein response activation and oxidative stress in PARK20 fibroblasts. Front. Neurosci. 2019, 13, 673. [Google Scholar] [CrossRef] [Green Version]
- Hashida, K.; Kitao, Y.; Sudo, H.; Awa, Y.; Maeda, S.; Mori, K.; Takahashi, R.; Iinuma, M.; Hori, O. ATF6alpha Promotes Astroglial Activation and Neuronal Survival in a Chronic Mouse Model of Parkinson’s Disease. PLoS ONE 2012, 7, e47950. [Google Scholar] [CrossRef] [PubMed]
- Celardo, I.; Costa, A.C.; Lehmann, S.; Jones, C.; Wood, N.; Mencacci, N.E.; Mallucci, G.R.; Loh, S.H.Y.; Martins, L.M. Mitofusin-mediated ER stress triggers neurodegeneration in pink1/parkin models of Parkinson’s disease. Cell Death Dis. 2016, 7, e2271. [Google Scholar] [CrossRef] [Green Version]
- Gully, J.C.; Sergeyev, V.G.; Bhootada, Y.; Mendez-Gomez, H.; Meyers, C.A.; Zolotukhin, S.; Gorbatyuk, M.S.; Gorbatyuk, O.S. Up-regulation of activating transcription factor 4 induces severe loss of dopamine nigral neurons in a rat model of Parkinson’s disease. Neurosci. Lett. 2016, 627, 36–41. [Google Scholar] [CrossRef]
- Reijonen, S.; Putkonen, N.; Nørremølle, A.; Lindholm, D.; Korhonen, L. Inhibition of endoplasmic reticulum stress counteracts neuronal cell death and protein aggregation caused by N-terminal mutant huntingtin proteins. Exp. Cell Res. 2008, 314, 950–960. [Google Scholar] [CrossRef] [PubMed]
- Waelter, S.; Boeddrich, A.; Lurz, R.; Scherzinger, E.; Lueder, G.; Lehrach, H.; Wanker, E.E. Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol. Biol. Cell 2001, 12, 1393–1407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishitoh, H.; Matsuzawa, A.; Tobiume, K.; Saegusa, K.; Takeda, K.; Inoue, K.; Hori, S.; Kakizuka, A.; Ichijo, H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002, 16, 1345–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Ahn, H.H.; Lee, W.J.; Oh, Y.; Choi, H.; Shim, S.M.; Shin, J.; Jung, Y.K. ENC1 Modulates the Aggregation and Neurotoxicity of Mutant Huntingtin Through p62 Under ER Stress. Mol. Neurobiol. 2016, 53, 6620–6634. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Lv, H.; Liao, M.; Xu, X.; Huang, S.; Tan, H.; Peng, T.; Zhang, Y.; Li, H. GRP78 counteracts cell death and protein aggregation caused by mutant huntingtin proteins. Neurosci. Lett. 2012, 516, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Lajoie, P.; Snapp, E.L. Changes in BiP availability reveal hypersensitivity to acute endoplasmic reticulum stress in cells expressing mutant huntingtin. J. Cell Sci. 2011, 124, 3332–3343. [Google Scholar] [CrossRef] [Green Version]
- Leitman, J.; Barak, B.; Benyair, R.; Shenkman, M.; Ashery, U.; Hartl, F.U.; Lederkremer, G.Z. ER Stress-Induced eIF2-alpha Phosphorylation Underlies Sensitivity of Striatal Neurons to Pathogenic Huntingtin. PLoS ONE 2014, 9, e90803. [Google Scholar] [CrossRef] [Green Version]
- Hyrskyluoto, A.; Bruelle, C.; Lundh, S.H.; Do, H.T.; Kivinen, J.; Rappou, E.; Reijonen, S.; Waltimo, T.; Petersén, Å.; Lindholm, D.; et al. Ubiquitin-specific protease-14 reduces cellular aggregates and protects against mutant huntingtin-induced cell degeneration: Involvement of the proteasome and ER stress-activated kinase IRE1α. Hum. Mol. Genet. 2014, 23, 5928–5939. [Google Scholar] [CrossRef] [Green Version]
- Sbodio, J.I.; Snyder, S.H.; Paul, B.D. Transcriptional control of amino acid homeostasis is disrupted in Huntington’s disease. Proc. Natl. Acad. Sci. USA 2016, 113, 8843–8848. [Google Scholar] [CrossRef] [Green Version]
- Fu, Z.; Liu, F.; Liu, C.; Jin, B.; Jiang, Y.; Tang, M.; Qi, X.; Guo, X. Mutant huntingtin inhibits the mitochondrial unfolded protein response by impairing ABCB10 mRNA stability. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1428–1435. [Google Scholar] [CrossRef]
- Hetz, C.; Castilla, J.; Soto, C. Perturbation of endoplasmic reticulum homeostasis facilitates prion replication. J. Biol. Chem. 2007, 282, 12725–12733. [Google Scholar] [CrossRef] [Green Version]
- Ferreiro, E.; Costa, R.; Marques, S.; Cardoso, S.M.; Oliveira, C.R.; Pereira, C.M.F. Involvement of mitochondria in endoplasmic reticulum stress-induced apoptotic cell death pathway triggered by the prion peptide PrP106–126. J. Neurochem. 2008, 104, 766–776. [Google Scholar] [CrossRef] [PubMed]
- Provansal, M.; Roche, S.; Pastore, M.; Casanova, D.; Belondrade, M.; Alais, S.; Leblanc, P.; Windl, O.; Lehmann, S. Proteomic consequences of expression and pathological conversion of the prion protein in inducible neuroblastoma N2a cells. Prion 2010, 4, 292–301. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.B.; Shi, Q.; Xu, Y.; Xie, W.L.; Zhang, J.; Tian, C.; Guo, Y.; Wang, K.; Zhang, B.Y.; Chen, C.; et al. Protein disulfide isomerase regulates endoplasmic reticulum stress and the apoptotic process during prion infection and PrP mutant-induced cytotoxicity. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Tang, Y.; Xiang, W.; Terry, L.; Kretzschmar, H.A.; Windl, O. Transcriptional analysis implicates endoplasmic reticulum stress in bovine spongiform encephalopathy. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, T.; Gu, Y.; Zanusso, G.; Sy, M.S.; Kumar, A.; Cohen, M.; Gambetti, P.; Singh, N. The chaperone protein BiP binds to a mutant prion protein and mediates its degradation by the proteasome. J. Biol. Chem. 2000, 275, 38699–38704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, Y.M.; Jeung, E.B. Calbindin-D28k in the Brain Influences the Expression of Cellular Prion Protein. Oxidative Med. Cell. Longev. 2018, 2018. [Google Scholar] [CrossRef] [Green Version]
- Peters, S.L.; Déry, M.A.; LeBlanc, A.C. Familial prion protein mutants inhibit Hrd1-mediated retrotranslocation of misfolded proteins by depleting misfolded protein sensor BiP. Hum. Mol. Genet. 2016, 25, 976–988. [Google Scholar] [CrossRef] [Green Version]
- Moreno, J.A.; Halliday, M.; Molloy, C.; Radford, H.; Verity, N.; Axten, J.M.; Ortori, C.A.; Willis, A.E.; Fischer, P.M.; Barrett, D.A.; et al. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef]
- Herrmann, U.S.; Sonati, T.; Falsig, J.; Reimann, R.R.; Dametto, P.; O’Connor, T. Correction: Prion Infections and Anti-PrP Antibodies Trigger Converging Neurotoxic Pathways. PLoS Pathog. 2015, 11. [Google Scholar] [CrossRef] [Green Version]
- Dametto, P.; Lakkaraju, A.K.K.; Bridel, C.; Villiger, L.; O’Connor, T.; Herrmann, U.S.; Pelczar, P.; Rülicke, T.; McHugh, D.; Adili, A.; et al. Neurodegeneration and unfolded-protein response in mice expressing a membrane-tethered flexible tail of PrP. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Shi, Q.; Xu, K.; Gao, C.; Chen, C.; Li, X.L.; Wang, G.R.; Tian, C.; Han, J.; Dong, X.P. Familial CJD associated PrP mutants within transmembrane region induced CTM-PrP retention in ER and Triggered apoptosis by ER stress in SH-SY5Y cells. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Klann, E. PERK: A novel therapeutic target for neurodegenerative diseases? Alzheimer’s Res. Ther. 2014, 6, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Axten, J.M.; Medina, J.R.; Feng, Y.; Shu, A.; Romeril, S.P.; Grant, S.W.; Li, W.H.H.; Heerding, D.A.; Minthorn, E.; Mencken, T.; et al. Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2, 3-dihydro-1H-indol-5-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J. Med. Chem. 2012, 55, 7193–7207. [Google Scholar] [CrossRef]
- Radford, H.; Moreno, J.A.; Verity, N.; Halliday, M.; Mallucci, G.R. PERK inhibition prevents tau-mediated neurodegeneration in a mouse model of frontotemporal dementia. Acta Neuropathol. 2015, 130, 633–642. [Google Scholar] [CrossRef] [Green Version]
- Ounallah-Saad, H.; Sharma, V.; Edry, E.; Rosenblum, K. Genetic or pharmacological reduction of PERK enhances cortical-dependent taste learning. J. Neurosci. 2014, 34, 14624–14632. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.; Henninger, K.; McGrath, B.C.; Cavener, D.R. PERK Regulates Working Memory and Protein Synthesis-Dependent Memory Flexibility. PLoS ONE 2016, 11, e0162766. [Google Scholar] [CrossRef]
- Sharma, V.; Ounallah-Saad, H.; Chakraborty, D.; Hleihil, M.; Sood, R.; Barrera, I.; Edry, E.; Chandran, S.K.; de Leon, S.B.T.; Kaphzan, H.; et al. Local inhibition of PERK enhances memory and reverses age-related deterioration of cognitive and neuronal properties. J. Neurosci. 2018, 38, 648–658. [Google Scholar] [CrossRef] [Green Version]
- Grande, V.; Ornaghi, F.; Comerio, L.; Restelli, E.; Masone, A.; Corbelli, A.; Tolomeo, D.; Capone, V.; Axten, J.M.; Laping, N.J.; et al. PERK inhibition delays neurodegeneration and improves motor function in a mouse model of Marinesco-Sjögren syndrome. Hum. Mol. Genet. 2018, 27, 2477–2489. [Google Scholar] [CrossRef]
- Rojas-Rivera, D.; Delvaeye, T.; Roelandt, R.; Nerinckx, W.; Augustyns, K.; Vandenabeele, P.; Bertrand, M.J.M. When PERK inhibitors turn out to be new potent RIPK1 inhibitors: Critical issues on the specificity and use of GSK2606414 and GSK2656157. Cell Death Differ. 2017, 24, 1100–1110. [Google Scholar] [CrossRef]
- Atkins, C.; Liu, Q.; Minthorn, E.; Zhang, S.Y.; Figueroa, D.J.; Moss, K.; Stanley, T.B.; Sanders, B.; Goetz, A.; Gaul, N.; et al. Characterization of a novel PERK kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res. 2013, 73, 1993–2002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Chen, Y.; Zhang, H.; Ma, Q.; Zhang, Y.W.; Xu, H. Salubrinal attenuates β-amyloid-induced neuronal death and microglial activation by inhibition of the NF-κB pathway. Neurobiol. Aging 2012, 33, e9–e1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colla, E.; Coune, P.; Liu, Y.; Pletnikova, O.; Troncoso, J.C.; Iwatsubo, T.; Schneider, B.L.; Lee, M.K. Endoplasmic reticulum stress is important for the manifestations of α-synucleinopathy in vivo. J. Neurosci. 2012, 32, 3306–3320. [Google Scholar] [CrossRef] [PubMed]
- Colla, E.; Jensen, P.H.; Pletnikova, O.; Troncoso, J.C.; Glabe, C.; Lee, M.K. Accumulation of toxic α-synuclein oligomer within endoplasmic reticulum occurs in α-synucleinopathy in vivo. J. Neurosci. 2012, 32, 3301–3305. [Google Scholar] [CrossRef] [Green Version]
- Smith, W.W.; Jiang, H.; Pei, Z.; Tanaka, Y.; Morita, H.; Sawa, A.; Dawson, V.L.; Dawson, T.M.; Ross, C.A. Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alpha-synuclein-induced toxicity. Hum. Mol. Genet. 2005, 14, 3801–3811. [Google Scholar] [CrossRef]
- Wu, L.; Luo, N.; Zhao, H.R.; Gao, Q.; Lu, J.; Pan, Y.; Shi, J.P.; Tian, Y.Y.; Zhang, Y.D. Salubrinal protects against rotenone-induced SH-SY5Y cell death via ATF4-parkin pathway. Brain Res. 2014, 1549, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Niso-Santano, M.; Bravo-San Pedro, J.M.; Gómez-Sánchez, R.; Climent, V.; Soler, G.; Fuentes, J.M.; González-Polo, R.A. ASK1 overexpression accelerates paraquat-induced autophagy via endoplasmic reticulum stress. Toxicol. Sci. 2011, 119, 156–168. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Tiffany-Castiglioni, E.; Koh, H.C.; Son, I.H. Paraquat activates the IRE1/ASK1/JNK cascade associated with apoptosis in human neuroblastoma SH-SY5Y cells. Toxicol. Lett. 2009, 191, 203–210. [Google Scholar] [CrossRef]
- Kim, J.S.; Heo, R.W.; Kim, H.; Yi, C.O.; Shin, H.J.; Han, J.W.; Roh, G.S. Salubrinal, ER stress inhibitor, attenuates kainic acid-induced hippocampal cell death. J. Neural Transm. 2014, 121, 1233–1243. [Google Scholar] [CrossRef]
- Sidrauski, C.; Acosta-Alvear, D.; Khoutorsky, A.; Vedantham, P.; Hearn, B.R.; Li, H.; Gamache, K.; Gallagher, C.M.; Ang, K.K.H.; Wilson, C.; et al. Pharmacological brake-release of mRNA translation enhances cognitive memory. Elife 2013, 2, e00498. [Google Scholar] [CrossRef]
- Sidrauski, C.; McGeachy, A.M.; Ingolia, N.T.; Walter, P. The small molecule ISRIB reverses the effects of eIF2α phosphorylation on translation and stress granule assembly. Elife 2015, 4, e05033. [Google Scholar] [CrossRef] [PubMed]
- Halliday, M.; Radford, H.; Sekine, Y.; Moreno, J.; Verity, N.; Le Quesne, J.; Ortori, C.A.; Barrett, D.A.; Fromont, C.; Fischer, P.M.; et al. Partial restoration of protein synthesis rates by the small molecule ISRIB prevents neurodegeneration without pancreatic toxicity. Cell Death Dis. 2015, 6, e1672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosoi, T.; Kakimoto, M.; Tanaka, K.; Nomura, J.; Ozawa, K. Unique pharmacological property of ISRIB in inhibition of Aβ-induced neuronal cell death. J. Pharmacol. Sci. 2016, 131, 292–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halliday, M.; Radford, H.; Zents, K.A.M.; Molloy, C.; Moreno, J.A.; Verity, N.C.; Smith, E.; Ortori, C.A.; Barrett, D.A.; Bushell, M.; et al. Repurposed drugs targeting eIF2α-P-mediated translational repression prevent neurodegeneration in mice. Brain 2017, 140, 1768–1783. [Google Scholar] [CrossRef] [Green Version]
- Tsaytler, P.; Harding, H.P.; Ron, D.; Bertolotti, A. Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science 2011, 332, 91–94. [Google Scholar] [CrossRef]
- Wang, L.; Popko, B.; Tixier, E.; Roos, R.P. Guanabenz, which enhances the unfolded protein response, ameliorates mutant SOD1-induced amyotrophic lateral sclerosis. Neurobiol. Dis. 2014, 71, 317–324. [Google Scholar] [CrossRef] [Green Version]
- Holmes, B.; Brogden, R.N.; Heel, R.C.; Speight, T.M.; Avery, G.S. Guanabenz: A Review of its Pharmacodynamic Properties and Therapeutic Efficacy in Hypertension. Drugs 1983, 26, 212–229. [Google Scholar] [CrossRef]
- McMahon, F.G.; Ryan, J.R.; Jain, A.K.; Vargas, R.; Vanov, S.K. Guanabenz in essential hypertension. Clin. Pharmacol. Ther. 1977, 21, 272–277. [Google Scholar] [CrossRef]
- Das, I.; Krzyzosiak, A.; Schneider, K.; Wrabetz, L.; D’Antonio, M.; Barry, N.; Sigurdardottir, A.; Bertolotti, A. Preventing proteostasis diseases by selective inhibition of a phosphatase regulatory subunit. Science 2015, 348, 239–242. [Google Scholar] [CrossRef] [Green Version]
- Crespillo-Casado, A.; Chambers, J.E.; Fischer, P.M.; Marciniak, S.J.; Ron, D. PPP1R15A-mediated dephosphorylation of eIF2a is unaffected by sephin1 or guanabenz. Elife 2017, 6, e26109. [Google Scholar] [CrossRef]
- Li, J.L. Trazodone. In The Essence of Analgesia and Analgesics; Cambridge University Press: Cambridge, UK, 2010; pp. 351–353. ISBN 9780511841378. [Google Scholar]
- Fagiolini, A.; Comandini, A.; Dell’Osso, M.C.; Kasper, S. Rediscovering trazodone for the treatment of major depressive disorder. CNS Drugs 2012, 26, 1033–1049. [Google Scholar] [CrossRef] [PubMed]
- Khouzam, H.R. A review of trazodone use in psychiatric and medical conditions. Postgrad. Med. 2017, 129, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Rozpędek, W.; Pytel, D.; Popławski, T.; Walczak, A.; Gradzik, K.; Wawrzynkiewicz, A.; Wojtczak, R.; Mucha, B.; Diehl, J.A.; Majsterek, I. Inhibition of the PERK-dependent unfolded protein response signaling pathway involved in the pathogenesis of Alzheimer’s disease. Curr. Alzheimer Res. 2019, 16, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Chao, J.; Yu, M.S.; Ho, Y.S.; Wang, M.; Chang, R.C.C. Dietary oxyresveratrol prevents parkinsonian mimetic 6-hydroxydopamine neurotoxicity. Free Radic. Biol. Med. 2008, 45, 1019–1026. [Google Scholar] [CrossRef]
- Choi, B.; Kim, S.; Jang, B.G.; Kim, M.J. Piceatannol, a natural analogue of resveratrol, effectively reduces beta-amyloid levels via activation of alpha-secretase and matrix metalloproteinase-9. J. Funct. Foods 2016, 23, 124–134. [Google Scholar] [CrossRef]
- Sangsen, Y.; Sooksawate, T.; Likhitwitayawuid, K.; Sritularak, B.; Wiwattanapatapee, R. A Self-Microemulsifying Formulation of Oxyresveratrol Prevents Amyloid Beta Protein-Induced Neurodegeneration in Mice. Planta Med. 2018, 84, 820–828. [Google Scholar] [CrossRef]
- Hyo, J.K.; Ki, W.L.; Hyong, J.L. Protective effects of piceatannol against beta-amyloid-induced neuronal cell death. Annals of the New York Academy of Sciences; Blackwell Publishing Inc.: Hoboken, NJ, USA, 2007; Volume 1095, pp. 473–482. [Google Scholar]
- Temsamani, H.; Krisa, S.; Decossas-Mendoza, M.; Lambert, O.; Mérillon, J.M.; Richard, T. Piceatannol and other wine stilbenes: A pool of inhibitors against α-synuclein aggregation and cytotoxicity. Nutrients 2016, 8, 367. [Google Scholar] [CrossRef] [Green Version]
- Shah, A.; Chao, J.; Legido-Quigley, C.; Chang, R.C.C. Oxyresveratrol exerts ATF4-and Grp78-mediated neuroprotection against endoplasmic reticulum stress in experimental Parkinson’s disease. Nutr. Neurosci. 2019, 1–16. [Google Scholar] [CrossRef]
- Yang, Y.; Xuan, L.; Chen, H.; Dai, S.; Ji, L.; Bao, Y.; Li, C. Neuroprotective Effects and Mechanism of β -Asarone against A β 1-42-Induced Injury in Astrocytes. Evid. Based Complement. Altern. Med. 2017, 2017, 8516518. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Xu, F.; Zhang, Q.; Li, X.; Guo, M.; Zhang, Y.; Wang, Z.; Wang, J.; Zhao, J.; Tian, Y.; et al. Effect of α-asarone on ethanol-induced learning and memory impairment in mice and its underlying mechanism. Life Sci. 2019, 238, 116898. [Google Scholar] [CrossRef]
- Chen, Y.; Gao, X.; Liu, Q.; Zeng, L.; Zhang, K.; Mu, K.; Zhang, D.; Zou, H.; Wu, N.; Ou, J.; et al. Alpha-asarone improves cognitive function of aged rats by alleviating neuronal excitotoxicity via GABAA receptors. Neuropharmacology 2020, 162, 107843. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.E.; Kim, N.; Yeo, J.Y.; Seo, D.G.; Kim, S.; Lee, J.S.E.; Hwang, K.W.; Park, S.Y. Anti-amyloidogenic effects of asarone derivatives from perilla frutescens leaves against beta-amyloid aggregation and nitric oxide production. Molecules 2019, 24, 4297. [Google Scholar] [CrossRef] [Green Version]
- Ning, B.; Deng, M.; Zhang, Q.; Wang, N.; Fang, Y. β-Asarone Inhibits IRE1/XBP1 Endoplasmic Reticulum Stress Pathway in 6-OHDA-Induced Parkinsonian Rats. Neurochem. Res. 2016, 41, 2097–2101. [Google Scholar] [CrossRef] [PubMed]
- Ning, B.; Zhang, Q.; Wang, N.; Deng, M.; Fang, Y. β-Asarone Regulates ER Stress and Autophagy Via Inhibition of the PERK/CHOP/Bcl-2/Beclin-1 Pathway in 6-OHDA-Induced Parkinsonian Rats. Neurochem. Res. 2019, 44, 1159–1166. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.S.; Zhou, S.F.; Wang, Q.; Guo, J.N.; Liang, H.M.; Deng, J.B.; He, W.Y. Gastrodin suppresses BACE1 expression under oxidative stress condition via inhibition of the PKR/eIF2α pathway in Alzheimer’s disease. Neuroscience 2016, 325, 1–9. [Google Scholar] [CrossRef]
- Lee, G.H.; Kim, H.R.; Han, S.Y.; Bhandary, B.; Kim, D.S.; Kim, M.G.; So, B.O.; Kim, S.Y.; Jo, K.S.; Lee, B.H.; et al. Gastrodia elata blume and its pure compounds protect BV-2 microglial-derived cell lines against β-amyloid: The involvement of GRP78 and CHOP. Biol. Res. 2012, 45, 403–410. [Google Scholar]
- Mou, Z.; Yuan, Y.H.; Lou, Y.X.; Heng, Y.; Huang, J.Y.; Xia, C.Y.; Gao, Y.; Zhu, C.G.; Chu, S.F.; Luo, P.; et al. Bibenzyl compound 20c protects against endoplasmic reticulum stress in tunicamycin-treated PC12 cells in vitro. Acta Pharmacol. Sin. 2016, 37, 1525–1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.Y.; Yuan, Y.H.; Yan, J.Q.; Wang, Y.N.; Chu, S.F.; Zhu, C.G.; Guo, Q.L.; Shi, J.G.; Chen, N.H. 20C, a bibenzyl compound isolated from Gastrodia elata, protects PC12 cells against rotenone-induced apoptosis via activation of the Nrf2/ARE/HO-1 signaling pathway. Acta Pharmacol. Sin. 2016, 37, 731–740. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Neurodegenerative Disease | UPR Markers | Experimental Model |
|---|---|---|
| Alzheimer’s disease | ↑ GRP78 | Aβ-treated SK-N-SH cells [203], RBE4 cells [204], Aβ1–40-treated RBE4 cells [205], Aβ1–42-treated bEnd.3 cells [206], 5XFAD mouse model [207], APP/PS1 transgenic mice [208], Aβ1–42-treated rat astrocytes, 3xTg-AD mice [209], mutant PS1 cells, 3xTg-AD mice [210]. |
| ↓ GRP78 | PS1 mutant SK-N-SH cells [211]. | |
| ↑ PERK | Hippocampus of human AD brains [48], 5XFAD mouse model [160,166], Aβ-treated SK-N-SH cells [203], JNPL3 mice, rat cortical neurons [212], Aβ42 transgenic flies [213], pR5 mice [214]. | |
| ↑ p-eIF2α | AD mouse model [215], hippocampus of human AD brains [48], 5XFAD mouse model [160,166], APP/PS1 mice [216], Aβ-treated SK-N-SH cells [203], JNPL3 mice, primary cultures of rat cortical neurons [212], Aβ1–42-treated rat embryonic hippocampal neurons [217], pR5 mice [214], Aβ1–42-treated rat primary cortical neurons [218], Aβ1–42-treated rat cerebral cortical astrocytes [209]. | |
| ↑ ATF4 | 5XFAD mouse model [160,166], Aβ1–40-treated RBE4 cells [205], human APOEe4 allele-expressing human and mouse AD models [219], Aβ1–42-treated rat embryonic hippocampal neurons, Aβ1–42-treated mice, human AD brains [217], Arg-61 APOE astrocytes [220]. | |
| ↑ CHOP | Aβ-treated SK-N-SH cells [203], primary rat cortical neurons [212], RBE4 cells [204], temporal cortex of human AD brains [221], Aβ1–40-treated RBE4 cell line [205], Aβ1–42-treated bEnd.3 cells [206], 5XFAD mouse model [207], APP/PS1 transgenic mice [208]. | |
| ↑ GADD34 | J20 mice [222], AD mouse model, human AD brains [223]. | |
| Parkinson’s disease | ↑ GRP78 | SYN120 mice, SH-SY5Y+ cells, HEK 293 cells [95], PARK14 knock-in mouse model [224], 6-OHDA-treated MN9D cells [225], 6-OHDA-treated SH-SY5Y cells [226]. |
| ↓ GRP78 | Rat PD model [227,228], human PD brains [229], DJ-1 KO neurons, MEFs and KD SH-SY5Y+ cells [230]. | |
| ↑ PERK | Human PD brains, rat PD models [231], PARK20 fibroblasts [232], mouse model of chronic MPTP/P injection [233], rat PD model [228], PARK14 mice [224], 6-OHDA- or MPP+-treated MN9D cells [225], Drosophila PINK1 and PARKIN mutants [234], DJ-1 KO MEFs [230]. | |
| ↑ eIF2α | PARK20 fibroblasts [232], mouse model of chronic MPTP/P injection [233], 6-OHDA- or MPP+-treated MN9D cells [225], DJ-1 KO MEFs [230]. | |
| ↑ ATF4 | Rat PD model [235], PD cellular models [95], mouse model of chronic MPTP/P injection [233], PARK20 fibroblasts [232]. | |
| ↓ ATF4 | DJ-1 KO neurons, MEFs and KD SH-SY5Y+ cells [230]. | |
| ↑ CHOP | 6-OHDA- or MPP+- treated MN9D cells [225], 6-OHDA-treated SH-SY5Y cells [226], PARK14 mice [224], rat PD model [228], PARK20 fibroblasts [232]. | |
| ↓ CHOP | DJ-1 KO neurons, MEFs and KD SH-SY5Y+ cells [230]. | |
| Huntington’s disease | ↑ GRP78 | PC6.3 cell [236], mHtt-expressing 293 Tet-Off cells [237], PC12-Q79 cells [238], HEK293T cells [239], Htt150Q-expressing N2a cells [240]. |
| ↓ GRP78 | mHtt-expressing mouse striatal cell lines [241]. | |
| ↑ PERK | Human and murine striatal cells, N171-82Q mice [242], PC12-Q79 cells [238]. | |
| ↑ eIF2α | Human and murine striatal cells, N171-82Q mice [242], 120Q-Htt-expressing PC6.3 cells [243]. | |
| ↑ ATF4 | Q7 cells [244]. | |
| ↓ ATF4 | Q111 cells [244]. | |
| ↑ CHOP | PC6.3 cell [236], PC12-Q79 cells [238], Q7 and Q111 cells [245], HEK293T cells [239]. | |
| Prion disease | ↑ GRP78 | PrP-expressing N2a cells [246], PrP(106-126)-treated NT2 rho0 cells and NT2 rho0+ cells [247], PrP-expressing N2a cells [248], 263K infected hamsters brain tissues, PrP-expressing 293-T cells [249], brains of BSE cattle [250], PrP-treated M17 cells [251]. |
| ↓ GRP78 | Prion infected GRP78+/− and GRP78+/+ mice, CAD5 cell line [106], CaBP-KO mice [252], PrP-treated CR7 cells [253]. | |
| ↑ PERK | Prion-infected mice [254], POM1-treated cultured organotypic cerebellar slices [255], FTgpi mice [256]. | |
| ↑ p-eIF2α | Prion-diseased mice [131,254], POM1-treated cultured organotypic cerebellar slices [255], FTgpi mice [256]. | |
| ↑ ATF4 | POM1-treated cultured organotypic cerebellar slices [255]. | |
| ↑ CHOP | FTgpi mice [256], PrP-expressing N2a cells [246], CaBP-KO mice [252], PrP-treated SH-SY5Y cells [257]. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rozpędek-Kamińska, W.; Siwecka, N.; Wawrzynkiewicz, A.; Wojtczak, R.; Pytel, D.; Diehl, J.A.; Majsterek, I. The PERK-Dependent Molecular Mechanisms as a Novel Therapeutic Target for Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 2108. https://doi.org/10.3390/ijms21062108
Rozpędek-Kamińska W, Siwecka N, Wawrzynkiewicz A, Wojtczak R, Pytel D, Diehl JA, Majsterek I. The PERK-Dependent Molecular Mechanisms as a Novel Therapeutic Target for Neurodegenerative Diseases. International Journal of Molecular Sciences. 2020; 21(6):2108. https://doi.org/10.3390/ijms21062108
Chicago/Turabian StyleRozpędek-Kamińska, Wioletta, Natalia Siwecka, Adam Wawrzynkiewicz, Radosław Wojtczak, Dariusz Pytel, J. Alan Diehl, and Ireneusz Majsterek. 2020. "The PERK-Dependent Molecular Mechanisms as a Novel Therapeutic Target for Neurodegenerative Diseases" International Journal of Molecular Sciences 21, no. 6: 2108. https://doi.org/10.3390/ijms21062108