Lipotoxicity and Diabetic Nephropathy: Novel Mechanistic Insights and Therapeutic Opportunities

 , , ,
, , ,  and
and
Abstract
:1. Introduction
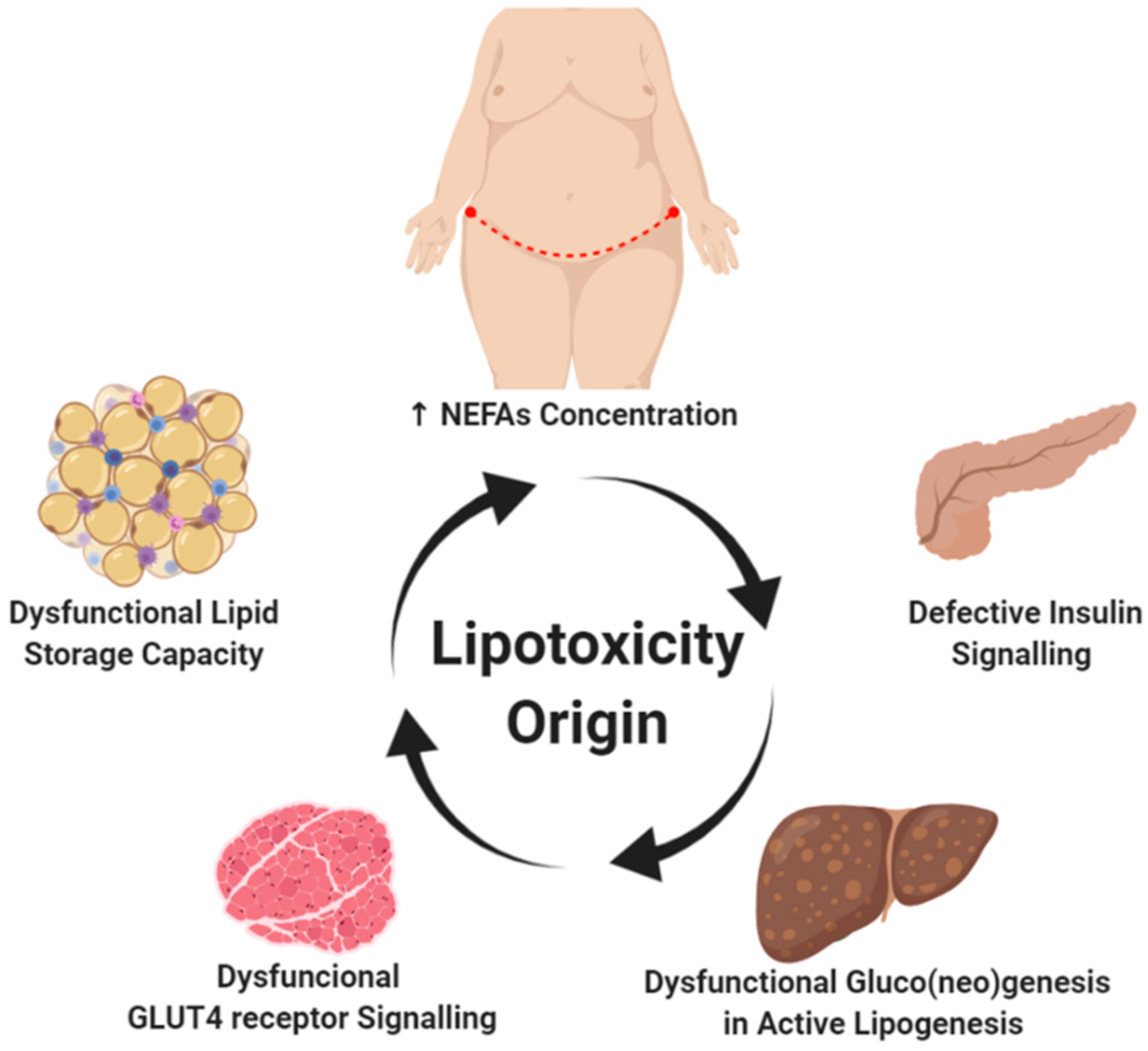
2. Lipotoxicity Origins
2.1. Subcutaneous Abdominal Fatty Deposits and High Plasma NEFA Levels
2.2. Dysfunctional Signaling in Adipose Tissue
2.3. Insulin Resistance and Lipid Accumulation
3. Glycogen versus Lipid Storage
4. Dyslipidemia in Diabetic Nephropathy
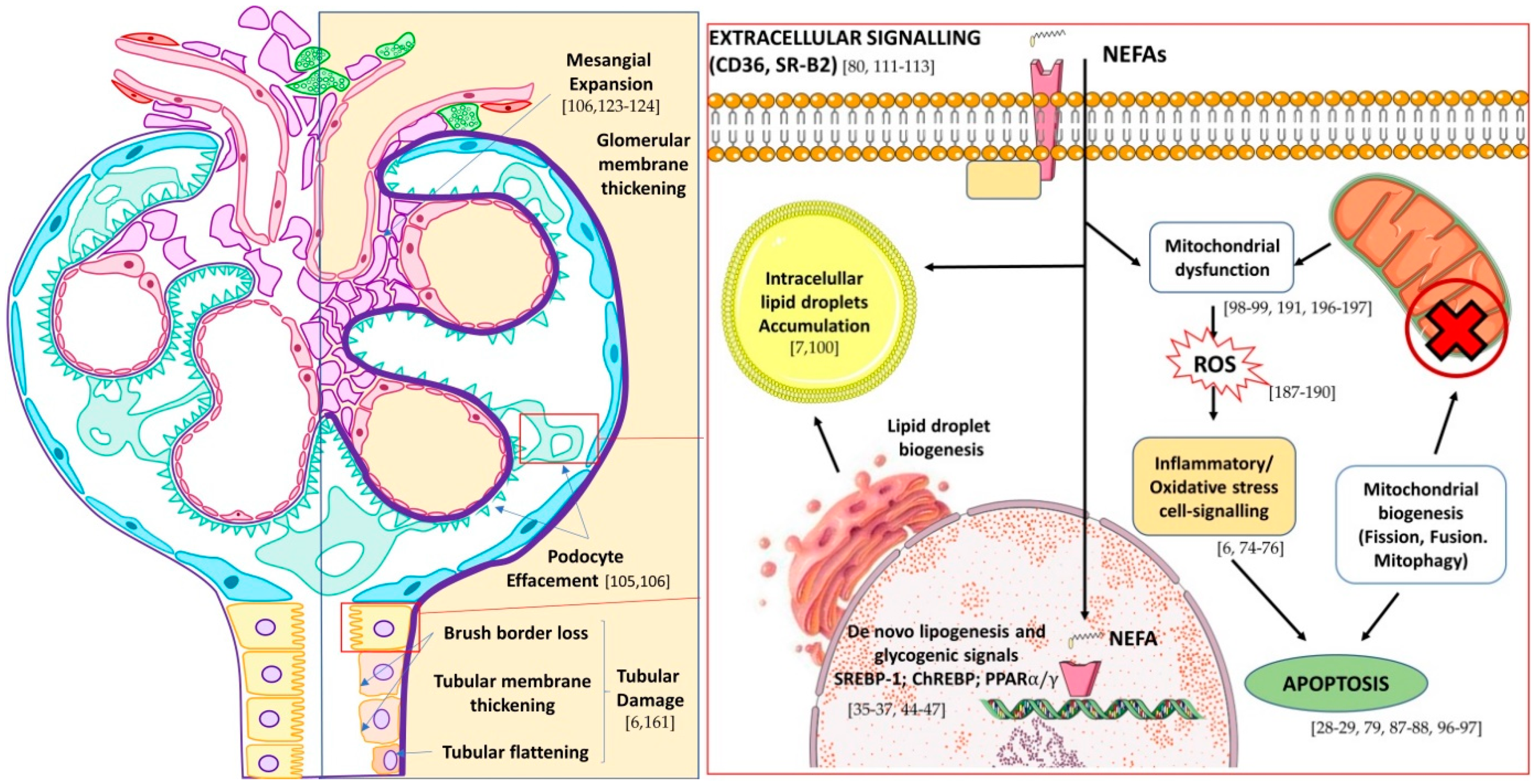
5. The Fatty Kidney in DN
Lipotoxicity in Renal Cells
6. Mitochondria as the Main Target of Lipotoxicity-Kidney Disease
7. Lipid Biomarkers as Predictors of DN
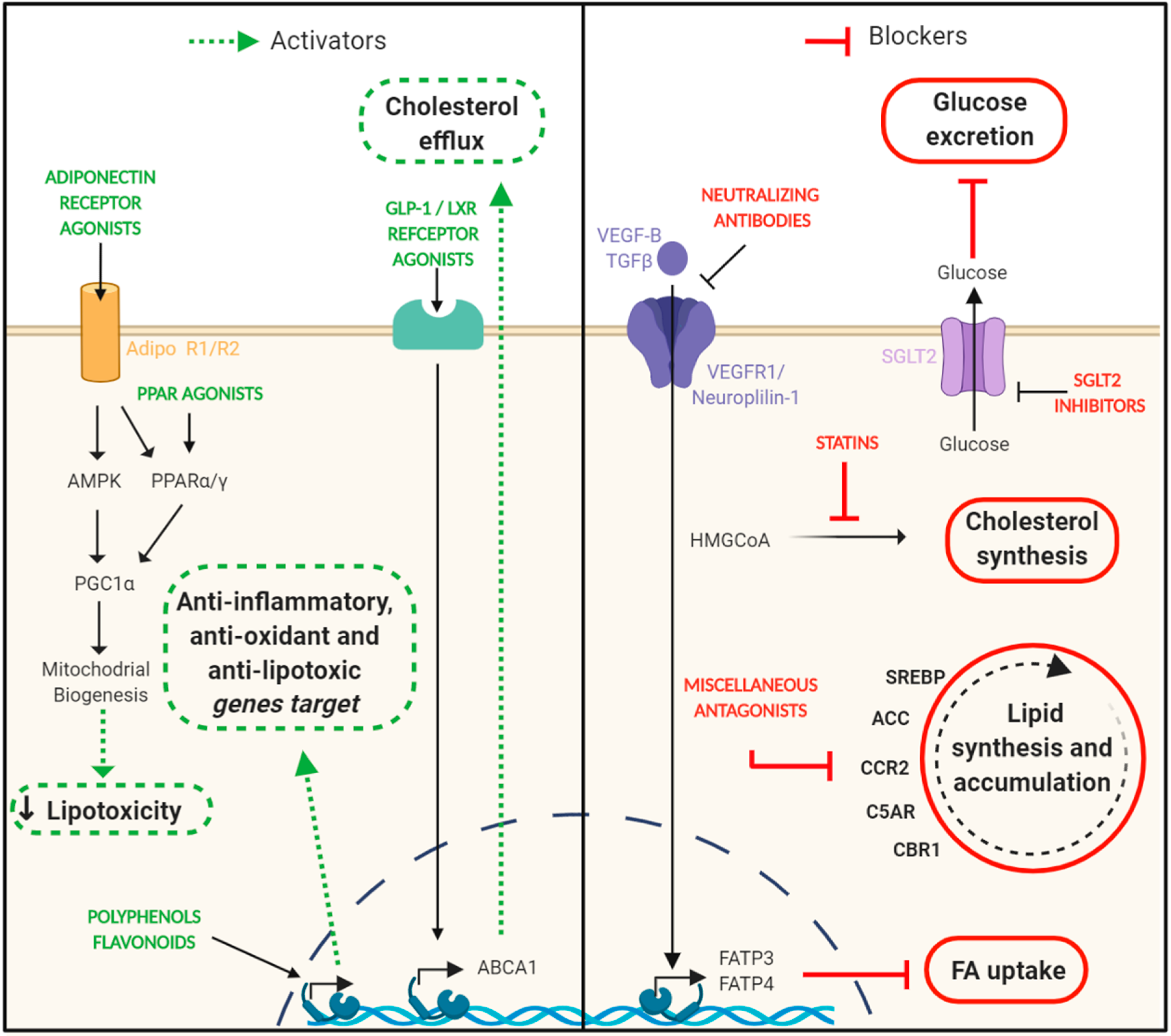
8. Targeting Lipotoxicity in DN. Is This Approach Feasible?
8.1. Statins
8.2. PPAR Agonists
8.3. Adiponectin Receptor Agonists
8.4. SGLT-2 Inhibitors
8.5. VEGF-B Signaling inhibition
8.6. Polyphenols, Flavonoids, and Nutraceuticals
8.7. Other Drugs Able to Impair Renal Lipid Deposition
8.8. Non-Pharmacological Approaches
9. Perspectives and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Verderio, C.; Gabrielli, M.; Giussani, P. Role of sphingolipids in the biogenesis and biological activity of extracellular vesicles. J. Lipid Res. 2018, 59, 1325–1340. [Google Scholar] [CrossRef] [Green Version]
- Vanni, S. Intracellular Lipid Droplets: From Structure to Function. Lipid Insights 2017, 10, 1178635317745518. [Google Scholar] [CrossRef] [Green Version]
- Olzmann, J.A.; Carvalho, P. Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell Biol. 2019, 20, 137–155. [Google Scholar] [CrossRef]
- Nishi, H.; Higashihara, T.; Inagi, R. Lipotoxicity in kidney, heart, and skeletal muscle dysfunction. Nutrients 2019, 11, 1664. [Google Scholar] [CrossRef] [Green Version]
- Walther, T.C.; Chung, J.; Farese, R.V. Lipid Droplet Biogenesis. Annu. Rev. Cell Dev. Biol. 2017, 33, 491–510. [Google Scholar] [CrossRef] [Green Version]
- Bobulescu, I.A. Renal lipid metabolism and lipotoxicity. Curr. Opin. Nephrol. Hypertens. 2010, 19, 393–402. [Google Scholar] [CrossRef] [Green Version]
- Herman-Edelstein, M.; Scherzer, P.; Tobar, A.; Levi, M.; Gafter, U. Altered renal lipid metabolism and renal lipid accumulation in human diabetic nephropathy. J. Lipid Res. 2014, 55, 561. [Google Scholar] [CrossRef] [Green Version]
- Rydén, M.; Arner, P. Subcutaneous Adipocyte Lipolysis Contributes to Circulating Lipid Levels. Arter. Thromb. Vasc. Biol. 2017, 37, 1782–1787. [Google Scholar] [CrossRef] [Green Version]
- Karpe, F.; Dickmann, J.R.; Frayn, K.N. Fatty acids, obesity, and insulin resistance: Time for a reevaluation. Diabetes 2011, 60, 2441–2449. [Google Scholar] [CrossRef] [Green Version]
- Koutsari, C.; Snozek, C.L.H.; Jensen, M.D. Plasma NEFA storage in adipose tissue in the postprandial state: Sex-related and regional differences. Diabetologia 2008, 51, 2041–2048. [Google Scholar] [CrossRef] [Green Version]
- Abrass, C.K. Lipid metabolism and renal disease. Contrib. Nephrol. 2006, 151, 106–121. [Google Scholar] [PubMed]
- Hellmuth, C.; Demmelmair, H.; Schmitt, I.; Peissner, W.; Blüher, M.; Koletzko, B. Association between Plasma Nonesterified Fatty Acids Species and Adipose Tissue Fatty Acid Composition. PLoS ONE 2013, 8, e74927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, M.J.; Skrtic, S.; Katsogiannos, P.; Abrahamsson, N.; Sidibeh, C.O.; Dahgam, S.; Månsson, M.; Risérus, U.; Kullberg, J.; Eriksson, J.W. Impaired adipose tissue lipid storage, but not altered lipolysis, contributes to elevated levels of NEFA in type 2 diabetes. Degree of hyperglycemia and adiposity are important factors. Metabolism 2016, 65, 1768–1780. [Google Scholar] [CrossRef] [PubMed]
- Utzschneider, K.M.; Kahn, S.E. The Role of Insulin Resistance in Nonalcoholic Fatty Liver Disease. J. Clin. Endocrinol. Metab. 2006, 91, 4753–4761. [Google Scholar] [CrossRef] [Green Version]
- Charles, M.A.; Eschwège, E.; Thibult, N.; Claude, J.R.; Warnet, J.M.; Rosselin, G.E.; Girard, J.; Balkau, B. The role of non-esterified fatty acids in the deterioration of glucose tolerance in Caucasian subjects: Results of the Paris Prospective Study. Diabetologia 1997, 40, 1101–1106. [Google Scholar] [CrossRef] [Green Version]
- Krssak, M.; Falk Petersen, K.; Dresner, A.; DiPietro, L.; Vogel, S.M.; Rothman, D.L.; Shulman, G.I.; Roden, M. Intramyocellular lipid concentrations are correlated with insulin sensitivity in humans: A 1H NMR spectroscopy study. Diabetologia 1999, 42, 113–116. [Google Scholar] [CrossRef] [Green Version]
- Nosadini, R.; Tonolo, G. Role of oxidized low density lipoproteins and free fatty acids in the pathogenesis of glomerulopathy and tubulointerstitial lesions in type 2 diabetes. Nutr. Metab. Cardiovasc. Dis. 2011, 21, 79–85. [Google Scholar] [CrossRef]
- Johnston, L.W.; Harris, S.B.; Retnakaran, R.; Giacca, A.; Liu, Z.; Bazinet, R.P.; Hanley, A.J. Association of NEFA composition with insulin sensitivity and beta cell function in the Prospective Metabolism and Islet Cell Evaluation (PROMISE) cohort. Diabetologia 2018, 61, 821–830. [Google Scholar] [CrossRef] [Green Version]
- Cnop, M.; Welsh, N.; Jonas, J.C.; Jörns, A.; Lenzen, S.; Eizirik, D.L. Mechanisms of pancreatic β-cell death in type 1 and type 2 diabetes: Many differences, few similarities. Diabetes 2005, 54, S97–S107. [Google Scholar] [CrossRef] [Green Version]
- Sharma, R.B.; Alonso, L.C. Lipotoxicity in the pancreatic beta cell: Not just survival and function, but proliferation as well? Curr. Diabetes Rep. 2014, 14, 492. [Google Scholar] [CrossRef] [Green Version]
- Oh, Y.S. Mechanistic insights into pancreatic beta-cell mass regulation by glucose and free fatty acids. Anat. Cell Biol. 2015, 48, 16–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, Y.S.; Bae, G.D.; Baek, D.J.; Park, E.Y.; Jun, H.S. Fatty acid-induced lipotoxicity in pancreatic beta-cells during development of type 2 diabetes. Front. Endocrinol. 2018, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, R.H.; Zerenturk, E.J.; Prakoso, D.; Calkin, A.C. Lipid metabolism and its implications for type 1 diabetes-associated cardiomyopathy. J. Mol. Endocrinol. 2017, 58, R225–R240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ertunc, M.E.; Hotamisligil, G.S. Lipid signaling and lipotoxicity in metaflammation: Indications for metabolic disease pathogenesis and treatment. J. Lipid Res. 2016, 57, 2099–2114. [Google Scholar] [CrossRef] [Green Version]
- Meex, R.C.R.; Blaak, E.E.; van Loon, L.J.C. Lipotoxicity plays a key role in the development of both insulin resistance and muscle atrophy in patients with type 2 diabetes. Obes. Rev. 2019, 20, 1205–1217. [Google Scholar] [CrossRef] [Green Version]
- Gai, Z.; Wang, T.; Visentin, M.; Kullak-Ublick, G.; Fu, X.; Wang, Z. Lipid Accumulation and Chronic Kidney Disease. Nutrients 2019, 11, 722. [Google Scholar] [CrossRef] [Green Version]
- Martínez-García, C.; Izquierdo-Lahuerta, A.; Vivas, Y.; Velasco, I.; Yeo, T.-K.; Chen, S.; Medina-Gomez, G. Renal Lipotoxicity-Associated Inflammation and Insulin Resistance Affects Actin Cytoskeleton Organization in Podocytes. PLoS ONE 2015, 10, e0142291. [Google Scholar] [CrossRef] [Green Version]
- Jaishy, B.; Abel, E.D. Lipids, lysosomes, and autophagy. J. Lipid Res. 2016, 57, 1619–1635. [Google Scholar] [CrossRef] [Green Version]
- Escasany, E.; Izquierdo-Lahuerta, A.; Medina-Gomez, G. Underlying Mechanisms of Renal Lipotoxicity in Obesity. Nephron 2019, 143, 28–32. [Google Scholar] [CrossRef]
- Edgerton, D.S.; Kraft, G.; Smith, M.; Farmer, B.; Williams, P.E.; Coate, K.C.; Printz, R.L.; O’Brien, R.M.; Cherrington, A.D. Insulin’s direct hepatic effect explains the inhibition of glucose production caused by insulin secretion. JCI Insight 2017, 2, e91863. [Google Scholar] [CrossRef] [Green Version]
- Guo, S. Insulin Signaling, Resistance, and the Metabolic Syndrome: Insights from Mouse Models to Disease Mechanisms. J. Endocrinol 2014, 220, T1–T23. [Google Scholar] [CrossRef] [PubMed]
- Minokoshi, Y.; Kahn, C.R.; Kahn, B.B. Tissue-specific Ablation of the GLUT4 Glucose Transporter or the Insulin Receptor Challenges Assumptions about Insulin Action and Glucose Homeostasis. J. Biol. Chem. 2003, 278, 33609–33612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perseghin, G.; Scifo, P.; De Cobelli, F.; Pagliato, E.; Battezzati, A.; Arcelloni, C.; Vanzulli, A.; Testolin, G.; Pozza, G.; Del Maschio, A.; et al. Intramyocellular triglyceride content is a determinant of in vivo insulin resistance in humans: A 1H-13C nuclear magnetic resonance spectroscopy assessment in offspring of type 2 diabetic parents. Diabetes 1999, 48, 1600–1606. [Google Scholar] [CrossRef] [PubMed]
- Manco, M. Insulin Resistance and NAFLD: A Dangerous Liaison beyond the Genetics. Children 2017, 4, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artunc, F.; Schleicher, E.; Weigert, C.; Fritsche, A.; Stefan, N.; Häring, H.U. The impact of insulin resistance on the kidney and vasculature. Nat. Rev. Nephrol. 2016, 12, 721–737. [Google Scholar] [CrossRef] [Green Version]
- Birkenfeld, A.L.; Shulman, G.I. Non Alcoholic Fatty Liver Disease, Hepatic Insulin Resistance and Type 2 Diabetes. Hepatology 2014, 59, 713–723. [Google Scholar] [CrossRef] [Green Version]
- Dresner, A.; Laurent, D.; Marcucci, M.; Griffin, M.E.; Dufour, S.; Cline, G.W.; Slezak, L.A.; Andersen, D.K.; Hundal, R.S.; Rothman, D.L.; et al. Effects of free fatty acids on glucose transport and IRS-1-associated phosphatidylinositol 3-kinase activity. J. Clin. Investig. 1999, 103, 253–259. [Google Scholar] [CrossRef] [Green Version]
- Milić, S.; Lulić, D.; Štimac, D. Non-alcoholic fatty liver disease and obesity: Biochemical, metabolic and clinical presentations. World J. Gastroenterol. 2014, 20, 9330–9337. [Google Scholar]
- Than, N.N.; Newsome, P.N. A concise review of non-alcoholic fatty liver disease. Atherosclerosis 2015, 239, 192–202. [Google Scholar] [CrossRef] [Green Version]
- Kolleritsch, S.; Kien, B.; Schoiswohl, G.; Diwoky, C.; Schreiber, R.; Heier, C.; Maresch, L.K.; Schweiger, M.; Eichmann, T.O.; Stryeck, S.; et al. Low cardiac lipolysis reduces mitochondrial fission and prevents lipotoxic heart dysfunction in Perilipin 5 mutant mice. Cardiovasc. Res. 2020, 116, 339–352. [Google Scholar] [CrossRef]
- Pecoits-Filho, R.; Abensur, H.; Betônico, C.C.R.; Machado, A.D.; Parente, E.B.; Queiroz, M.; Salles, J.E.N.; Titan, S.; Vencio, S. Interactions between kidney disease and diabetes: Dangerous liaisons. Diabetol. Metab. Syndr. 2016, 8, 50. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, K.; Soty, M.; Zitoun, C.; Duchampt, A.; Silva, M.; Philippe, E.; Gautier-Stein, A.; Rajas, F.; Mithieux, G. The role of kidney in the inter-organ coordination of endogenous glucose production during fasting. Mol. Metab. 2018, 16, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Rajas, F.; Labrune, P.; Mithieux, G. Glycogen storage disease type1 and diabetes: Learning by comparing and contrasting the two disorders. Diabetes Metab. 2013, 39, 377–387. [Google Scholar] [CrossRef]
- Mather, A.; Pollock, C. Glucose handling by the kidney. Kidney Int. 2011, 79, S1–S6. [Google Scholar] [CrossRef] [Green Version]
- Mittra, S.; Bansal, V.S.; Bhatnagar, P.K. From a glucocentric to a lipocentric approach towards metabolic syndrome. Drug Discov. Today 2008, 13, 211–218. [Google Scholar] [CrossRef]
- Mota, M.; Banini, B.A.; Cazanave, S.C.; Sanyal, A.J. Molecular mechanisms of lipotoxicity and glucotoxicity in nonalcoholic fatty liver disease. Metabolism 2016, 65, 1049–1061. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, M.A.; Forbes, J.M. Glucose and glycogen in the diabetic kidney: Heroes or villains? EBioMedicine 2019, 47, 590–597. [Google Scholar] [CrossRef] [Green Version]
- Musso, G.; Cassader, M.; Cohney, S.; De Michieli, F.; Pinach, S.; Saba, F.; Gambino, R. Fatty liver and chronic kidney disease: Novel mechanistic insights and therapeutic opportunities. Diabetes Care 2016, 39, 1830–1845. [Google Scholar] [CrossRef] [Green Version]
- Hager, M.R.; Narla, A.D.; Tannock, L.R. Dyslipidemia in patients with chronic kidney disease. Rev. Endocr. Metab. Disord. 2017, 18, 29–40. [Google Scholar] [CrossRef]
- Vaziri, N.D. Disorders of lipid metabolism in nephrotic syndrome: Mechanisms and consequences. Kidney Int. 2016, 90, 41–52. [Google Scholar] [CrossRef] [Green Version]
- Alicic, R.Z.; Rooney, M.T.; Tuttle, K.R. Diabetic kidney disease: Challenges, progress, and possibilities. Clin. J. Am. Soc. Nephrol. 2017, 12, 2032–2045. [Google Scholar] [CrossRef] [PubMed]
- Adiels, M.; Olofsson, S.-O.; Taskinen, M.-R.; Borén, J. Overproduction of very low-density lipoproteins is the hallmark of the dyslipidemia in the metabolic syndrome. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1225–1236. [Google Scholar] [CrossRef] [PubMed]
- Okubo, K.; Ikewaki, K.; Sakai, S.; Tada, N.; Kawaguchi, Y.; Mochizuki, S. Abnormal HDL Apolipoprotein A-I and A-II Kinetics in Hemodialysis Patients: A Stable Isotope Study. J. Am. Soc. Nephrol. 2004, 15, 1008–1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawanami, D.; Matoba, K.; Utsunomiya, K. Dyslipidemia in diabetic nephropathy. Ren. Replace. Ther. 2016, 2, 16. [Google Scholar] [CrossRef] [Green Version]
- Kramer, H.J.; Saranathan, A.; Luke, A.; Durazo-Arvizu, R.A.; Guichan, C.; Hou, S.; Cooper, R. Increasing body mass index and obesity in the incident ESRD population. J. Am. Soc. Nephrol. 2006, 17, 1453–1459. [Google Scholar] [CrossRef] [Green Version]
- Kaysen, G.A. Dyslipidemia in chronic kidney disease: Causes and consequences. Kidney Int. 2006, 70, S55–S58. [Google Scholar] [CrossRef] [Green Version]
- Kaysen, G.A. New insights into lipid metabolism in chronic kidney disease. J. Ren. Nutr. 2011, 21, 120–123. [Google Scholar] [CrossRef]
- Al-Shahrouri, H.Z.; Ramirez, P.; Fanti, P.; Abboud, H.; Lorenzo, C.; Haffner, S. NMR identifies atherogenic lipoprotein abnormalities in early diabetic nephropathy that are unrecognized by conventional analysis. Clin. Nephrol. 2010, 73, 180–189. [Google Scholar] [CrossRef]
- Bosmans, J.L.; Holvoet, P.; Dauwe, S.E.; Ysebaert, D.K.; Chapelle, T.; Jürgens, A.; Kovacic, V.; Van Marck, E.A.; De Broe, M.E.; Verpooten, G.A. Oxidative modification of low-density lipoproteins and the outcome of renal allografts at 1 1/2 years. Kidney Int. 2001, 59, 2346–2356. [Google Scholar] [CrossRef]
- Malle, E.; Woenckhaus, C.; Waeg, G.; Esterbauer, H.; Gröne, E.F.; Gröne, H.J. Immunological evidence for hypochlorite-modified proteins in human kidney. Am. J. Pathol. 1997, 150, 603–615. [Google Scholar]
- Scheuer, H.; Gwinner, W.; Hohbach, J.; Gröne, E.F.; Brandes, R.P.; Malle, E.; Olbricht, C.J.; Walli, A.K.; Gröne, H.J. Oxidant stress in hyperlipidemia-induced renal damage. Am. J. Physiol. Ren. Physiol. 2000, 278, F63–F74. [Google Scholar] [CrossRef] [PubMed]
- Bermúdez-López, M.; Arroyo, D.; Betriu, À.; Masana, L.; Fernández, E.; Valdivielso, J.M. New perspectives on CKD-induced dyslipidemia. Expert Opin. Ther. Targets 2017, 21, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Drüeke, T.B.; Khoa, T.N.; Massy, Z.A.; Witko-Sarsat, V.; Lacour, B.; Descamps-Latscha, B. Role of oxidized low-density lipoprotein in the atherosclerosis of uremia. Kidney Int. 2001, 59, S114–S119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.S. Oxidized LDL, glomerular mesangial cells and collagen. Diabetes Res. Clin. Pract. 1999, 45, 117–122. [Google Scholar] [CrossRef]
- Sastre, C.; Rubio-Navarro, A.; Buendía, I.; Gómez-Guerrero, C.; Blanco, J.; Mas, S.; Egido, J.; Blanco-Colio, L.M.; Ortiz, A.; Moreno, J.A. Hyperlipidemia-associated renal damage decreases Klotho expression in kidneys from ApoE knockout mice. PLoS ONE 2013, 8, e83713. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-García, B.; Moreno, J.A.; López-Franco, O.; Sanz, A.B.; Martín-Ventura, J.L.; Blanco, J.; Jakubowski, A.; Burkly, L.C.; Ortiz, A.; Egido, J.; et al. Tumor necrosis factor-like weak inducer of apoptosis (TWEAK) enhances vascular and renal damage induced by hyperlipidemic diet in ApoE-knockout mice. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 2061–2068. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.; Cabral, P.D.; Schilling, W.P.; Schmidt, Z.W.; Uddin, A.N.; Gingras, A.; Madhavan, S.M.; Garvin, J.L.; Schelling, J.R. Kidney Proximal Tubule Lipoapoptosis Is Regulated by Fatty Acid Transporter-2 (FATP2). J. Am. Soc. Nephrol. 2018, 29, 81–91. [Google Scholar] [CrossRef] [Green Version]
- Fineberg, D.; Jandeleit-Dahm, K.A.M.; Cooper, M.E. Diabetic nephropathy: Diagnosis and treatment. Nat. Rev. Endocrinol. 2013, 9, 713–723. [Google Scholar] [CrossRef]
- Ferrara, D.; Montecucco, F.; Dallegri, F.; Carbone, F. Impact of different ectopic fat depots on cardiovascular and metabolic diseases. J. Cell. Physiol. 2019, 234, 21630–21641. [Google Scholar] [CrossRef]
- De Vries, A.P.J.; Ruggenenti, P.; Ruan, X.Z.; Praga, M.; Cruzado, J.M.; Bajema, I.M.; D’Agati, V.D.; Lamb, H.J.; Barlovic, D.P.; Hojs, R.; et al. Fatty kidney: Emerging role of ectopic lipid in obesity-related renal disease. Lancet Diabetes Endocrinol. 2014, 2, 417–426. [Google Scholar] [CrossRef]
- DeZwaan-McCabe, D.; Sheldon, R.D.; Gorecki, M.C.; Guo, D.-F.; Gansemer, E.R.; Kaufman, R.J.; Rahmouni, K.; Gillum, M.P.; Taylor, E.B.; Teesch, L.M.; et al. ER Stress Inhibits Liver Fatty Acid Oxidation while Unmitigated Stress Leads to Anorexia-Induced Lipolysis and Both Liver and Kidney Steatosis. Cell Rep. 2017, 19, 1794–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonker, J.T.; de Heer, P.; Engelse, M.A.; van Rossenberg, E.H.; Klessens, C.Q.F.; Baelde, H.J.; Bajema, I.M.; Koopmans, S.J.; Coelho, P.G.; Streefland, T.C.M.; et al. Metabolic imaging of fatty kidney in diabesity: Validation and dietary intervention. Nephrol. Dial. Transpl. 2018, 33, 224–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Praga, M.; Morales, E. The Fatty Kidney: Obesity and Renal Disease. Nephron 2017, 136, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Foster, M.C.; Hwang, S.J.; Porter, S.A.; Massaro, J.M.; Hoffmann, U.; Fox, C.S. Fatty kidney, hypertension, and chronic kidney disease: The framingham heart study. Hypertension 2011, 58, 784–790. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, J.M. Lipotoxicity. Kidney Int. 2006, 70, 1560–1566. [Google Scholar] [CrossRef] [Green Version]
- Ruggiero, C.; Elks, C.M.; Kruger, C.; Cleland, E.; Addison, K.; Noland, R.C.; Stadler, K. Albumin-bound fatty acids but not albumin itself alter redox balance in tubular epithelial cells and induce a peroxide-mediated redox-sensitive apoptosis. Am. J. Physiol. Renal Physiol. 2014, 306, F896–F906. [Google Scholar] [CrossRef]
- Jao, T.-M.; Nangaku, M.; Wu, C.-H.; Sugahara, M.; Saito, H.; Maekawa, H.; Ishimoto, Y.; Aoe, M.; Inoue, T.; Tanaka, T.; et al. ATF6α downregulation of PPARα promotes lipotoxicity-induced tubulointerstitial fibrosis. Kidney Int. 2019, 95, 577–589. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Zhang, X.; Liu, P. Lipid droplet proteins and metabolic diseases. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1968–1983. [Google Scholar] [CrossRef]
- Krahmer, N.; Farese, R.V.; Walther, T.C.; Walther, T.C. Balancing the fat: Lipid droplets and human disease. EMBO Mol. Med. 2013, 5, 973–983. [Google Scholar] [CrossRef]
- Ashcroft, F.M.; Rohm, M.; Clark, A.; Brereton, M.F. Is Type 2 Diabetes a Glycogen Storage Disease of Pancreatic β Cells? Cell Metab. 2017, 26, 17–23. [Google Scholar] [CrossRef] [Green Version]
- Thongnak, L.; Pongchaidecha, A.; Lungkaphin, A. Renal Lipid Metabolism and Lipotoxicity in Diabetes. Am. J. Med. Sci. 2020, 359, 84–99. [Google Scholar] [CrossRef] [PubMed]
- Bandet, C.L.; Tan-Chen, S.; Bourron, O.; Le Stunff, H.; Hajduch, E. Sphingolipid metabolism: New insight into ceramide-induced lipotoxicity in muscle cells. Int. J. Mol. Sci. 2019, 20, 479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, M.C.; Shulman, G.I. Roles of Diacylglycerols and Ceramides in Hepatic Insulin Resistance. Trends Pharmacol. Sci. 2017, 38, 649–665. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.A.O.; Liebman, S.E.; Scott Lucia, M.; Li, J.; Levi, M. Role of altered renal lipid metabolism and the sterol regulatory element binding proteins in the pathogenesis of age-related renal disease. Kidney Int. 2005, 68, 2608–2620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gheith, O.A.; Sobh, M.A.K.; Mohamed, K.E.S.; El-Baz, M.A.; El-Husseini, F.; Gazarin, S.S.; Ahmed, H.A.E.H.; Rasem, M.W.; Amer, G.M. Impact of treatment of dyslipidemia on renal function, fat deposits and scarring in patients with persistent nephrotic syndrome. Nephron 2002, 91, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Li, X.; Zhu, C.; Wei, M. Glomerulosclerosis in adriamycin-induced nephrosis is accelerated by a lipid-rich diet. Pediatr. Nephrol. 2000, 15, 196–200. [Google Scholar] [CrossRef]
- Freedman, B.I.; Limou, S.; Ma, L.; Kopp, J.B. APOL1-Associated Nephropathy: A Key Contributor to Racial Disparities in CKD. Am. J. Kidney Dis. 2018, 72, S8–S16. [Google Scholar] [CrossRef] [Green Version]
- Couser, W.G. Primary Membranous Nephropathy. Clin. J. Am. Soc. Nephrol. 2017, 12, 983–997. [Google Scholar] [CrossRef]
- Diez-Sampedro, A.; Lenz, O.; Fornoni, A. Podocytopathy in Diabetes: A Metabolic and Endocrine Disorder. Am. J. Kidney Dis. 2011, 58, 637–646. [Google Scholar] [CrossRef] [Green Version]
- D’Agati, V.D.; Chagnac, A.; De Vries, A.P.J.; Levi, M.; Porrini, E.; Herman-Edelstein, M.; Praga, M. Obesity-related glomerulopathy: Clinical and pathologic characteristics and pathogenesis. Nat. Rev. Nephrol. 2016, 12, 453–471. [Google Scholar] [CrossRef]
- Hara, S.; Kobayashi, N.; Sakamoto, K.; Ueno, T.; Manabe, S.; Takashima, Y.; Hamada, J.; Pastan, I.; Fukamizu, A.; Matsusaka, T.; et al. Podocyte Injury-Driven Lipid Peroxidation Accelerates the Infiltration of Glomerular Foam Cells in Focal Segmental Glomerulosclerosis. Am. J. Pathol. 2015, 185, 2118–2131. [Google Scholar] [CrossRef] [PubMed]
- Merscher, S.; Pedigo, C.E.; Mendez, A.J. Metabolism, energetics, and lipid biology in the podocyte—Cellular cholesterol mediated glomerular injury. Front. Endocrinol. 2014, 5, 169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, C.; Kanter, J.E.; Bornfeldt, K.E.; Leboeuf, R.C.; Oram, J.F. Diabetes reduces the cholesterol exporter ABCA1 in mouse macrophages and kidneys. J. Lipid Res. 2010, 51, 1719–1728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proctor, G.; Jiang, T.; Iwahashi, M.; Wang, Z.; Li, J.; Levi, M. Regulation of renal fatty acid and cholesterol metabolism, inflammation, and fibrosis in Akita and OVE26 mice with type 1 diabetes. Diabetes 2006, 55, 2502–2509. [Google Scholar] [CrossRef] [Green Version]
- Tsun, J.G.S.; Yung, S.; Chau, M.K.M.; Shiu, S.W.M.; Chan, T.M.; Tan, K.C.B. Cellular cholesterol transport proteins in diabetic nephropathy. PLoS ONE 2014, 9, e105787. [Google Scholar] [CrossRef]
- Pedigo, C.E.; Ducasa, G.M.; Leclercq, F.; Sloan, A.; Mitrofanova, A.; Hashmi, T.; Molina-David, J.; Ge, M.; Lassenius, M.I.; Forsblom, C.; et al. Local TNF causes NFATc1-dependent cholesterol-mediated podocyte injury. J. Clin. Investig. 2016, 126, 3336–3350. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Collins, H.L.; Ranalletta, M.; Fuki, I.V.; Billheimer, J.T.; Rothblat, G.H.; Tall, A.R.; Rader, D.J. Macrophage ABCA1 and ABCG1, but not SR-BI, promote macrophage reverse cholesterol transport in vivo. J. Clin. Investig. 2007, 117, 2216–2224. [Google Scholar] [CrossRef] [Green Version]
- Hafiane, A.; Bielicki, J.K.; Johansson, J.O.; Genest, J. Novel apo E-derived ABCA1 agonist peptide (CS-6253) promotes reverse cholesterol transport and induces formation of preβ-1 HDL in vitro. PLoS ONE 2015, 10, e0131997. [Google Scholar] [CrossRef] [Green Version]
- Bielicki, J.K. ABCA1 agonist peptides for the treatment of disease. Curr. Opin. Lipidol. 2016, 27, 40–46. [Google Scholar] [CrossRef] [Green Version]
- Wahl, P.; Ducasa, G.M.; Fornoni, A. Systemic and renal lipids in kidney disease development and progression. Am. J. Physiol. Physiol. 2016, 310, F433–F445. [Google Scholar] [CrossRef] [Green Version]
- Merscher-Gomez, S.; Guzman, J.; Pedigo, C.E.; Lehto, M.; Aguillon-Prada, R.; Mendez, A.; Lassenius, M.I.; Forsblom, C.; Yoo, T.H.; Villarreal, R.; et al. Cyclodextrin protects podocytes in diabetic kidney disease. Diabetes 2013, 62, 3817–3827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tufro, A. Cholesterol accumulation in podocytes: A potential novel targetable pathway in diabetic nephropathy. Diabetes 2013, 62, 3661–3662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López, C.A.; de Vries, A.H.; Marrink, S.J. Molecular mechanism of cyclodextrin mediated cholesterol extraction. PLoS Comput. Biol. 2011, 7, e1002020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ducasa, G.M.; Fontanesi, F.; Fornoni, A.; Ducasa, G.M.; Mitrofanova, A.; Mallela, S.K.; Liu, X.; Molina, J.; Sloan, A.; Pedigo, C.E.; et al. ATP-binding cassette A1 deficiency causes cardiolipin-driven mitochondrial dysfunction in podocytes Find the latest version: ATP-binding cassette A1 deficiency causes cardiolipin-driven mitochondrial dysfunction in podocytes. J. Clin. Investig. 2019, 129, 3387–3400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitrofanova, A.; Sosa, M.A.; Fornoni, A. Lipid Mediators of Insulin Signaling in Diabetic Kidney Disease. Am. J. Physiol. Physiol. 2019, 317, F1241–F1252. [Google Scholar] [CrossRef]
- Mitrofanova, A.; Mallela, S.K.; Ducasa, G.M.; Yoo, T.H.; Rosenfeld-Gur, E.; Zelnik, I.D.; Molina, J.; Varona Santos, J.; Ge, M.; Sloan, A.; et al. SMPDL3b modulates insulin receptor signaling in diabetic kidney disease. Nat. Commun. 2019, 10, 2692. [Google Scholar] [CrossRef] [Green Version]
- Okada, M.; Takemura, T.; Yanagida, H.; Yoshioka, K. Response of mesangial cells to low-density lipoprotein and angiotensin II in diabetic (OLETF) rats. Kidney Int. 2002, 61, 113–124. [Google Scholar] [CrossRef] [Green Version]
- White, K.E.; Bilous, R.W.; Marshall, S.M.; El Nahas, M.; Remuzzl, G.; Piras, G.; De Cosmo, S.; Vlberti, G.C. Podocyte number in normotensive type 1 diabetic patients with albuminuria. Diabetes 2002, 51, 3083–3089. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Li, H.; Wen, Y.B.; Li, X.W. Very-low-density lipoprotein-induced triglyceride accumulation in human mesangial cells is mainly mediated by lipoprotein lipase. Nephron Physiol. 2008, 110, p1–p10. [Google Scholar] [CrossRef]
- Chen, G.; Wang, T.; Uttarwar, L.; van Krieken, R.; Li, R.; Chen, X.; Gao, B.; Ghayur, A.; Margetts, P.; Krepinsky, J.C. SREBP-1 is a novel mediator of TGFβ1 signaling in mesangial cells. J. Mol. Cell Biol. 2014, 6, 516–530. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.N.; Chen, X.; Li, R.; Gao, B.; Mohammed-Ali, Z.; Lu, C.; Yum, V.; Dickhout, J.G.; Krepinsky, J.C. SREBP-1 mediates angiotensin II-induced TGF-β1 upregulation and glomerular fibrosis. J. Am. Soc. Nephrol. 2015, 26, 1839–1854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uttarwar, L.; Gao, B.; Ingram, A.J.; Krepinsky, J.C. SREBP-1 activation by glucose mediates TGF-β upregulation in mesangial cells. Am. J. Physiol. Ren. Physiol. 2012, 302, F329–F341. [Google Scholar] [CrossRef] [PubMed]
- Park, M.J.; Han, H.J.; Kim, D. Il Lipotoxicity-induced PRMT1 exacerbates mesangial cell apoptosis via endoplasmic reticulum stress. Int. J. Mol. Sci. 2017, 18, 1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, R.; Simonson, M.S. Saturated free fatty acids and apoptosis in microvascular mesangial cells: Palmitate activates pro-apoptotic signaling involving caspase 9 and mitochondrial release of endonuclease G. Cardiovasc. Diabetol. 2005, 4, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Y.; Chen, Q.; Ma, K.; Ju, Y.; Ji, T.; Wang, Z.; Li, W.; Li, W. Astragaloside IV inhibits palmitate-mediated oxidative stress and fibrosis in human glomerular mesangial cells via downregulation of CD36 expression. Pharmacol. Reports 2019, 71, 319–329. [Google Scholar] [CrossRef]
- Lee, E.S.; Kwon, M.-H.; Kim, H.M.; Kim, N.; Kim, Y.M.; Kim, H.S.; Lee, E.Y.; Chung, C.H. Dibenzoylmethane ameliorates lipid-induced inflammation and oxidative injury in diabetic nephropathy. J. Endocrinol. 2019, 240, 169–179. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, Y.; Kume, S.; Araki, S.I.; Isshiki, K.; Chin-Kanasaki, M.; Sakaguchi, M.; Sugimoto, T.; Koya, D.; Haneda, M.; Kashiwagi, A.; et al. Fenofibrate, a PPARα agonist, has renoprotective effects in mice by enhancing renal lipolysis. Kidney Int. 2011, 79, 871–882. [Google Scholar] [CrossRef] [Green Version]
- Hagberg, C.E.; Falkevall, A.; Wang, X.; Larsson, E.; Huusko, J.; Nilsson, I.; Van Meeteren, L.A.; Samen, E.; Lu, L.; Vanwildemeersch, M.; et al. Vascular endothelial growth factor B controls endothelial fatty acid uptake. Nature 2010, 464, 917–921. [Google Scholar] [CrossRef] [Green Version]
- Muoio, D.M. Metabolism and vascular fatty acid transport. N. Engl. J. Med. 2010, 363, 291. [Google Scholar] [CrossRef]
- Hagberg, C.E.; Mehlem, A.; Falkevall, A.; Muhl, L.; Fam, B.C.; Ortsäter, H.; Scotney, P.; Nyqvist, D.; Samén, E.; Lu, L.; et al. Targeting VEGF-B as a novel treatment for insulin resistance and type 2 diabetes. Nature 2012, 490, 426–430. [Google Scholar] [CrossRef]
- Hagberg, C.; Mehlem, A.; Falkevall, A.; Muhl, L.; Eriksson, U. Endothelial fatty acid transport: Role of vascular endothelial growth factor B. Physiology 2013, 28, 125–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glass, C.K.; Olefsky, J.M. Inflammation and lipid signaling in the etiology of insulin resistance. Cell Metab. 2012, 15, 635–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Sun, S.-C. NF-κB in inflammation and renal diseases. Cell Biosci. 2015, 5, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Byrne, D.; Devaraj, S.; Islam, K.N.; Collazo, R.; McDonald, L.; Grundy, S.; Jialal, I. Low-density lipoprotein (LDL)-induced monocyte-endothelial cell adhesion, soluble cell adhesion molecules, and autoantibodies to oxidized-LDL in chronic renal failure patients on dialysis therapy. Metabolism 2001, 50, 207–215. [Google Scholar] [CrossRef]
- Tanaka, M.; Suzuki, Y.; Shirato, I.; Takahara, H.; Shibata, T.; Sugaya, T.; Shimamoto, K.; Horikoshi, S.; Tomino, Y. Tubular epithelial cells have the capacity to transdifferentiate into CD68-positive macrophage-like cells by oxidative stress. Inflamm. Res. 2008, 57, 593–600. [Google Scholar] [CrossRef]
- Prieur, X.; Roszer, T.; Ricote, M. Lipotoxicity in macrophages: Evidence from diseases associated with the metabolic syndrome. Biochim. Biophys. Acta 2010, 1801, 327–337. [Google Scholar] [CrossRef]
- Eom, M.; Hudkins, K.L.; Alpers, C.E. Foam cells and the pathogenesis of kidney disease. Curr. Opin. Nephrol. Hypertens. 2015, 24, 245–251. [Google Scholar] [CrossRef]
- Chaabane, C.; Coen, M.; Bochaton-Piallat, M.L. Smooth muscle cell phenotypic switch: Implications for foam cell formation. Curr. Opin. Lipidol. 2014, 25, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Zeller, I.; Srivastava, S. Macrophage functions in atherosclerosis. Circ. Res. 2014, 115, e83–e85. [Google Scholar] [CrossRef] [Green Version]
- Jones, N.L.; Reagan, J.W.; Willingham, M.C. The pathogenesis of foam cell formation: Modified LDL stimulates uptake of co-incubated LDL via macropinocytosis. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 773–781. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.H.; Fu, Y.C.; Zhang, D.W.; Yin, K.; Tang, C.K. Foam cells in atherosclerosis. Clin. Chim. Acta 2013, 424, 245–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, X.; Kushiyama, A.; Yoneda, M.; Nakatsu, Y.; Guo, Y.; Zhang, J.; Ono, H.; Kanna, M.; Sakoda, H.; Ono, H.; et al. Macrophage foam cell formation is augmented in serum from patients with diabetic angiopathy. Diabetes Res. Clin. Pract. 2010, 87, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.; Itabe, H.; Higashi, Y.; Fujimoto, Y.; Shiomi, M.; Yoshizumi, M.; Ouchi, Y.; Takano, T. Foam cell formation containing lipid droplets enriched with free cholesterol by hyperlipidemic serum. J. Lipid Res. 2001, 42, 1771–1781. [Google Scholar] [PubMed]
- Ruan, X.Z.; Varghese, Z.; Powis, S.H.; Moorhead, J.F. Dysregulation of LDL receptor under the influence of inflammatory cytokines: A new pathway for foam cell formation. Kidney Int. 2001, 60, 1716–1725. [Google Scholar] [CrossRef] [Green Version]
- Kanter, J.E.; Hsu, C.-C.; Bornfeldt, K.E. Monocytes and Macrophages as Protagonists in Vascular Complications of Diabetes. Front. Cardiovasc. Med. 2020, 7, 10. [Google Scholar] [CrossRef]
- Zhou, C.; Lei, H.; Chen, Y.; Liu, Q.; Li, L.C.; Moorhead, J.F.; Varghese, Z.; Ruan, X.Z. Enhanced SCAP Glycosylation by Inflammation Induces Macrophage Foam Cell Formation. PLoS ONE 2013, 8, e75650. [Google Scholar] [CrossRef]
- Yang, X.; Okamura, D.M.; Lu, X.; Chen, Y.; Moorhead, J.; Varghese, Z.; Ruan, X.Z. Cd36 in chronic kidney disease: Novel insights and therapeutic opportunities. Nat. Rev. Nephrol. 2017, 13, 769–781. [Google Scholar] [CrossRef]
- Glatz, J.F.C.; Luiken, J.J.F.P. Dynamic role of the transmembrane glycoprotein CD36 (SR-B2) in cellular fatty acid uptake and utilization. J. Lipid Res. 2018, 59, 1084–1093. [Google Scholar] [CrossRef] [Green Version]
- Cao, W.; Xu, J.; Zhou, Z.M.; Wang, G.B.; Hou, F.F.; Nie, J. Advanced oxidation protein products activate intrarenal renin-angiotensin system via a CD36-mediated, redox-dependent pathway. Antioxid. Redox Signal. 2013, 18, 19–35. [Google Scholar] [CrossRef] [Green Version]
- Usui, H.K.; Shikata, K.; Sasaki, M.; Okada, S.; Matsuda, M.; Shikata, Y.; Ogawa, D.; Kido, Y.; Nagase, R.; Yozai, K.; et al. Macrophage scavenger receptor-A-deficient mice are resistant against diabetic nephropathy through amelioration of microinflammation. Diabetes 2007, 56, 363–372. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhang, T.; Geng, J.; Wu, Z.; Xu, L.; Liu, J.; Tian, J.; Zhou, Z.; Nie, J.; Bai, X. Advanced Oxidation Protein Products Promote Lipotoxicity and Tubulointerstitial Fibrosis via CD36/β-Catenin Pathway in Diabetic Nephropathy. Antioxid. Redox Signal. 2019, 31, 521–538. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xu, L.; Hou, X.; Geng, J.; Tian, J.; Liu, X.; Bai, X. Advanced Oxidation Protein Products Aggravate Tubulointerstitial Fibrosis Through Protein Kinase C-Dependent Mitochondrial Injury in Early Diabetic Nephropathy. Antioxid. Redox Signal. 2019, 30, 1162–1185. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.-A.; Han, S.H.; Chinga, F.; Park, A.S.D.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef]
- Long, K.R.; Rbaibi, Y.; Gliozzi, M.L.; Ren, Q.; Weisz, O.A. Differential kidney proximal tubule cell responses to protein overload by albumin and its ligands. Am. J. Physiol. Physiol. 2020, 318, F851–F859. [Google Scholar] [CrossRef] [PubMed]
- Mesilati-Stahy, R.; Argov-Argaman, N. Changes in lipid droplets morphometric features in mammary epithelial cells upon exposure to non-esterified free fatty acids compared with VLDL. PLoS ONE 2018, 13, e0209565. [Google Scholar] [CrossRef]
- Jarad, G.; Knutsen, R.H.; Mecham, R.P.; Miner, J.H. Albumin contributes to kidney disease progression in alport syndrome. Am. J. Physiol. Ren. Physiol. 2016, 311, F120–F130. [Google Scholar] [CrossRef] [Green Version]
- Katsoulieris, E.; Mabley, J.G.; Samai, M.; Sharpe, M.A.; Green, I.C.; Chatterjee, P.K. Lipotoxicity in renal proximal tubular cells: Relationship between endoplasmic reticulum stress and oxidative stress pathways. Free Radic. Biol. Med. 2010, 48, 1654–1662. [Google Scholar] [CrossRef]
- Iwai, T.; Kume, S.; Chin-Kanasaki, M.; Kuwagata, S.; Araki, H.; Takeda, N.; Sugaya, T.; Uzu, T.; Maegawa, H.; Araki, S.-I. Stearoyl-CoA Desaturase-1 Protects Cells against Lipotoxicity-Mediated Apoptosis in Proximal Tubular Cells. Int. J. Mol. Sci. 2016, 17, 1868. [Google Scholar] [CrossRef] [Green Version]
- Sieber, J.; Weins, A.; Kampe, K.; Gruber, S.; Lindenmeyer, M.T.; Cohen, C.D.; Orellana, J.M.; Mundel, P.; Jehle, A.W. Susceptibility of podocytes to palmitic acid is regulated by stearoyl-CoA desaturases 1 and 2. Am. J. Pathol. 2013, 183, 735–744. [Google Scholar] [CrossRef] [Green Version]
- Sieber, J.; Jehle, A.W. Free Fatty Acids and Their Metabolism Affect Function and Survival of Podocytes. Front. Endocrinol. 2014, 5, 186. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Liu, S.; Zhai, A.; Zhang, B.; Tian, G. AMPK-mediated regulation of lipid metabolism by phosphorylation. Biol. Pharm. Bull. 2018, 41, 985–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, M.R.; Tran, M.T.; Parikh, S.M. PGC1α in the kidney. Am. J. Physiol. Ren. Physiol. 2018, 314, F1–F8. [Google Scholar] [CrossRef] [PubMed]
- Guo, K.; Lu, J.; Huang, Y.; Wu, M.; Zhang, L.; Yu, H.; Zhang, M.; Bao, Y.; He, J.C.; Chen, H.; et al. Protective role of PGC-1α in diabetic nephropathy is associated with the inhibition of ROS through mitochondrial dynamic remodeling. PLoS ONE 2015, 10, e0125176. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.A.; Lim, J.H.; Kim, M.Y.; Kim, T.W.; Kim, Y.; Yang, K.S.; Park, H.S.; Choi, S.R.; Chung, S.; Kim, H.W.; et al. Fenofibrate improves renal lipotoxicity through activation of AMPK-PGC-1α in db/db mice. PLoS ONE 2014, 9, e96147. [Google Scholar] [CrossRef]
- Mehlem, A.; Palombo, I.; Wang, X.; Hagberg, C.E.; Eriksson, U.; Falkevall, A. PGC-1α coordinates mitochondrial respiratory capacity and muscular fatty acid uptake via regulation of VEGF-B. Diabetes 2016, 65, 861–873. [Google Scholar] [CrossRef] [Green Version]
- Forbes, J.M.; Thorburn, D.R. Mitochondrial dysfunction in diabetic kidney disease. Nat. Rev. Nephrol. 2018, 14, 291–312. [Google Scholar] [CrossRef]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 2017, 13, 629–646. [Google Scholar] [CrossRef]
- Szeto, H.H.; Liu, S.; Soong, Y.; Alam, N.; Prusky, G.T.; Seshan, S.V. Protection of mitochondria prevents high-fat diet–induced glomerulopathy and proximal tubular injury. Kidney Int. 2016, 90, 997–1011. [Google Scholar] [CrossRef] [Green Version]
- Wei, P.Z.; Szeto, C.C. Mitochondrial dysfunction in diabetic kidney disease. Clin. Chim. Acta 2019, 496, 108–116. [Google Scholar] [CrossRef]
- Zhan, M.; Usman, I.; Yu, J.; Ruan, L.; Bian, X.; Yang, J.; Yang, S.; Sun, L.; Kanwar, Y.S. Perturbations in mitochondrial dynamics by p66Shc lead to renal tubular oxidative injury in human diabetic nephropathy. Clin. Sci. 2018, 132, 1297–1314. [Google Scholar] [CrossRef]
- Jang, H.-S.; Noh, M.R.; Kim, J.; Padanilam, B.J. Defective Mitochondrial Fatty Acid Oxidation and Lipotoxicity in Kidney Diseases. Front. Med. 2020, 7, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afshinnia, F.; Nair, V.; Lin, J.; Rajendiran, T.M.; Soni, T.; Byun, J.; Sharma, K.; Fort, P.E.; Gardner, T.W.; Looker, H.C.; et al. Increased lipogenesis and impaired B-oxidation predict type 2 diabetic kidney disease progression in American Indians. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Afshinnia, F.; Rajendiran, T.M.; Soni, T.; Byun, J.; Wernisch, S.; Sas, K.M.; Hawkins, J.; Bellovich, K.; Gipson, D.; Michailidis, G.; et al. Impaired B-oxidation and altered complex lipid fatty acid partitioning with advancing CKD. J. Am. Soc. Nephrol. 2018, 29, 295–306. [Google Scholar] [CrossRef] [Green Version]
- Benador, I.Y.; Veliova, M.; Mahdaviani, K.; Petcherski, A.; Wikstrom, J.D.; Assali, E.A.; Acín-Pérez, R.; Shum, M.; Oliveira, M.F.; Cinti, S.; et al. Mitochondria Bound to Lipid Droplets Have Unique Bioenergetics, Composition, and Dynamics that Support Lipid Droplet Expansion. Cell Metab. 2018, 27, 869–885. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Soong, Y.; Seshan, S.V.; Szeto, H.H. Novel cardiolipin therapeutic protects endothelial mitochondria during renal ischemia and mitigates microvascular rarefaction, inflammation, and fibrosis. Am. J. Physiol. Ren. Physiol. 2014, 306, F970–F980. [Google Scholar] [CrossRef] [Green Version]
- Lindblom, R.; Higgins, G.; Coughlan, M.; De Haan, J.B. Targeting mitochondria-and reactive oxygen species-driven pathogenesis in diabetic nephropathy. Rev. Diabet. Stud. 2015, 12, 134–156. [Google Scholar] [CrossRef] [Green Version]
- Higgins, G.C.; Coughlan, M.T. Mitochondrial dysfunction and mitophagy: The beginning and end to diabetic nephropathy? Br. J. Pharmacol. 2014, 171, 1917–1942. [Google Scholar] [CrossRef] [Green Version]
- Takabatake, Y.; Yamamoto, T.; Isaka, Y. Stagnation of autophagy: A novel mechanism of renal lipotoxicity. Autophagy 2017, 13, 775–776. [Google Scholar] [CrossRef] [Green Version]
- Chevalier, R.L. The proximal tubule is the primary target of injury and progression of kidney disease: Role of the glomerulotubular junction. Am. J. Physiol. Physiol. 2016, 311, F145–F161. [Google Scholar] [CrossRef]
- Tan, S.M.; de Haan, J.B. Combating oxidative stress in diabetic complications with Nrf2 activators: How much is too much? Redox Rep. 2014, 19, 107–117. [Google Scholar] [CrossRef]
- Coughlan, M.T.; Sharma, K. Challenging the dogma of mitochondrial reactive oxygen species overproduction in diabetic kidney disease. Kidney Int. 2016, 90, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Darshi, M.; Sharma, K. The Warburg Effect in Diabetic Kidney Disease. Semin. Nephrol. 2018, 238, 111–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, H.G.; Overgaard, J.; Lajer, M.; Hubalek, F.; Hojrup, P.; Pedersen, L.; Tarnow, L.; Rossing, P.; Pociot, F.; McGuire, J.N. Finding diabetic nephropathy biomarkers in the plasma peptidome by high-throughput magnetic bead processing and MALDI-TOF-MS analysis. Proteom. Clin. Appl. 2010, 4, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Liao, W.L.; Chang, C.T.; Liao, H.Y.; Tsai, F.J. Urine proteome analysis by C18 plate–matrix-assisted laser desorption/ionization time-of-flight mass spectrometry allows noninvasive differential diagnosis and prediction of diabetic nephropathy. PLoS ONE 2018, 13, e0200945. [Google Scholar] [CrossRef] [Green Version]
- Tofte, N.; Suvitaival, T.; Ahonen, L.; Winther, S.A.; Theilade, S.; Frimodt-Møller, M.; Ahluwalia, T.S.; Rossing, P. Lipidomic analysis reveals sphingomyelin and phosphatidylcholine species associated with renal impairment and all-cause mortality in type 1 diabetes. Sci. Rep. 2019, 9, 16398. [Google Scholar] [CrossRef] [PubMed]
- Han, L.D.; Xia, J.F.; Liang, Q.L.; Wang, Y.; Wang, Y.M.; Hu, P.; Li, P.; Luo, G.A. Plasma esterified and non-esterified fatty acids metabolic profiling using gas chromatography-mass spectrometry and its application in the study of diabetic mellitus and diabetic nephropathy. Anal. Chim. Acta 2011, 689, 85–91. [Google Scholar] [CrossRef]
- Pena, M.J.; Lambers Heerspink, H.J.; Hellemons, M.E.; Friedrich, T.; Dallmann, G.; Lajer, M.; Bakker, S.J.L.; Gansevoort, R.T.; Rossing, P.; de Zeeuw, D.; et al. Urine and plasma metabolites predict the development of diabetic nephropathy in individuals with Type 2 diabetes mellitus. Diabet. Med. 2014, 31, 1138–1147. [Google Scholar] [CrossRef]
- Hirayama, A.; Nakashima, E.; Sugimoto, M.; Akiyama, S.I.; Sato, W.; Maruyama, S.; Matsuo, S.; Tomita, M.; Yuzawa, Y.; Soga, T. Metabolic profiling reveals new serum biomarkers for differentiating diabetic nephropathy. Anal. Bioanal. Chem. 2012, 404, 3101–3109. [Google Scholar] [CrossRef]
- Sberna, A.L.; Bouillet, B.; Rouland, A.; Brindisi, M.C.; Nguyen, A.; Mouillot, T.; Duvillard, L.; Denimal, D.; Loffroy, R.; Vergès, B.; et al. European Association for the Study of the Liver (EASL), European Association for the Study of Diabetes (EASD) and European Association for the Study of Obesity (EASO) clinical practice recommendations for the management of non-alcoholic fatty liver disease: Evaluation of their application in people with Type 2 diabetes. Diabet. Med. 2018, 35, 368–375. [Google Scholar]
- International Diabetes Federation. IDF Diabetes Atlas, 9th ed.; Brussels, Belgium, 2019; Available online: https://www.diabetesatlas.org/en/ (accessed on 9 April 2020).
- Neuschwander-Tetri, B.A. Therapeutic landscape for NAFLD in 2020. Gastroenterology 2020. [Google Scholar] [CrossRef]
- Barter, P.J.; Rye, K.-A. New Era of Lipid-Lowering Drugs. Pharmacol. Rev. 2016, 68, 458–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bussolati, B.; Deregibus, M.C.; Fonsato, V.; Doublier, S.; Spatola, T.; Procida, S.; Di Carlo, F.; Camussi, G. Statins prevent oxidized LDL-induced injury of glomerular podocytes by activating the phosphatidylinositol 3-kinase/AKT-signaling pathway. J. Am. Soc. Nephrol. 2005, 16, 1936–1947. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Park, C.W. Mechanisms of Adiponectin Action: Implication of Adiponectin Receptor Agonism in Diabetic Kidney Disease. Int. J. Mol. Sci. 2019, 20, 1782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonolo, G.; Melis, M.G.; Formato, M.; Angius, M.F.; Carboni, A.; Brizzi, P.; Ciccarese, M.; Cherchi, G.M.; Maioli, M. Additive effects of Simvastatin beyond its effects on LDL cholesterol in hypertensive type 2 diabetic patients. Eur. J. Clin. Investig. 2000, 30, 980–987. [Google Scholar] [CrossRef] [PubMed]
- Tonolo, G.; Velussi, M.; Brocco, E.; Abaterusso, C.; Carraro, A.; Morgia, G.; Satta, A.; Faedda, R.; Abhyankar, A.; Luthman, H.; et al. Simvastatin maintains steady patterns of GFR and improves AER and expression of slit diaphragm proteins in type II diabetes. Kidney Int. 2006, 70, 177–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Wu, P.; Zhang, J.; Wang, S.; Zhang, G. The effect of statins on microalbuminuria, proteinuria, progression of kidney function, and all-cause mortality in patients with non-end stage chronic kidney disease: A meta-analysis. Pharmacol. Res. 2016, 105, 74–83. [Google Scholar] [CrossRef]
- Summary of Revisions: Standards of Medical Care in Diabetes-2020. Diabetes Care 2020, 43, S4–S6. [CrossRef] [Green Version]
- Choi, S.R.; Lim, J.H.; Kim, M.Y.; Kim, E.N.; Kim, Y.; Choi, B.S.; Kim, Y.-S.; Kim, H.W.; Lim, K.-M.; Kim, M.J.; et al. Adiponectin receptor agonist AdipoRon decreased ceramide, and lipotoxicity, and ameliorated diabetic nephropathy. Metabolism 2018, 85, 348–360. [Google Scholar] [CrossRef]
- Kim, Y.; Lim, J.H.; Kim, M.Y.; Kim, E.N.; Yoon, H.E.; Shin, S.J.; Choi, B.S.; Kim, Y.-S.; Chang, Y.S.; Park, C.W. The Adiponectin Receptor Agonist AdipoRon Ameliorates Diabetic Nephropathy in a Model of Type 2 Diabetes. J. Am. Soc. Nephrol. 2018, 29, 1108–1127. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.X.; Levi, J.; Luo, Y.; Myakala, K.; Herman-Edelstein, M.; Qiu, L.; Wang, D.; Peng, Y.; Grenz, A.; Lucia, S.; et al. SGLT2 protein expression is increased in human diabetic nephropathy: SGLT2 protein inhibition decreases renal lipid accumulation, inflammation, and the development of nephropathy in diabetic mice. J. Biol. Chem. 2017, 292, 5335–5348. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, S.; Sugiura, Y.; Saito, H.; Sugahara, M.; Higashijima, Y.; Yamaguchi, J.; Inagi, R.; Suematsu, M.; Nangaku, M.; Tanaka, T. Sodium–glucose cotransporter 2 inhibition normalizes glucose metabolism and suppresses oxidative stress in the kidneys of diabetic mice. Kidney Int. 2018, 94, 912–925. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.-H.; Zhang, R.; Li, L.; Wang, Y.-T.; Liu, J.-P.; Zhang, J.; Bai, L.; Cheng, J.-Q.; Fu, P.; Liu, F. Exendin-4 Ameliorates Lipotoxicity-induced Glomerular Endothelial Cell Injury by Improving ABC Transporter A1-mediated Cholesterol Efflux in Diabetic apoE Knockout Mice. J. Biol. Chem. 2016, 291, 26487–26501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Li, L.; Liu, S.; Liao, G.; Li, L.; Chen, Y.; Cheng, J.; Lu, Y.; Liu, J. GLP-1 receptor agonist ameliorates obesity-induced chronic kidney injury via restoring renal metabolism homeostasis. PLoS ONE 2018, 13, e0193473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falkevall, A.; Mehlem, A.; Palombo, I.; Heller Sahlgren, B.; Ebarasi, L.; He, L.; Ytterberg, A.J.; Olauson, H.; Axelsson, J.; Sundelin, B.; et al. Reducing VEGF-B Signaling Ameliorates Renal Lipotoxicity and Protects against Diabetic Kidney Disease. Cell Metab. 2017, 25, 713–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayachandran, M.; Wu, Z.; Ganesan, K.; Khalid, S.; Chung, S.M.; Xu, B. Isoquercetin upregulates antioxidant genes, suppresses inflammatory cytokines and regulates AMPK pathway in streptozotocin-induced diabetic rats. Chem. Biol. Interact. 2019, 303, 62–69. [Google Scholar] [CrossRef]
- Jiang, X.; Yu, J.; Wang, X.; Ge, J.; Li, N. Quercetin improves lipid metabolism via SCAP-SREBP2-LDLr signaling pathway in early stage diabetic nephropathy. Diabetes. Metab. Syndr. Obes. 2019, 12, 827–839. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Pan, Y.; Zhang, Q.Y.; Wang, F.M.; Kong, L.D. Quercetin and allopurinol ameliorate kidney injury in STZ-treated rats with regulation of renal NLRP3 inflammasome activation and lipid accumulation. PLoS ONE 2012, 7, e38285. [Google Scholar] [CrossRef] [Green Version]
- Koh, E.S.; Lim, J.H.; Kim, M.Y.; Chung, S.; Shin, S.J.; Choi, B.S.; Kim, H.W.; Hwang, S.Y.; Kim, S.W.; Park, C.W.; et al. Anthocyanin-rich Seoritae extract ameliorates renal lipotoxicity via activation of AMP-activated protein kinase in diabetic mice. J. Transl. Med. 2015, 13, 203. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.Y.; Lim, J.H.; Youn, H.H.; Hong, Y.A.; Yang, K.S.; Park, H.S.; Chung, S.; Koh, S.H.; Shin, S.J.; Choi, B.S.; et al. Resveratrol prevents renal lipotoxicity and inhibits mesangial cell glucotoxicity in a manner dependent on the AMPK-SIRT1-PGC1α axis in db/db mice. Diabetologia 2013, 56, 204–217. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.H.; Lee, E.S.; Choi, R.; Nawaboot, J.; Lee, M.Y.; Lee, E.Y.; Kim, H.S.; Chung, C.H. Protective Effects of Curcumin on Renal Oxidative Stress and Lipid Metabolism in a Rat Model of Type 2 Diabetic Nephropathy. Yonsei Med. J. 2016, 57, 664–673. [Google Scholar] [CrossRef] [Green Version]
- Soetikno, V.; Sari, F.R.; Sukumaran, V.; Lakshmanan, A.P.; Harima, M.; Suzuki, K.; Kawachi, H.; Watanabe, K. Curcumin decreases renal triglyceride accumulation through AMPK-SREBP signaling pathway in streptozotocin-induced type 1 diabetic rats. J. Nutr. Biochem. 2013, 24, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Park, C.H.; Noh, J.S.; Fujii, H.; Roh, S.-S.; Song, Y.-O.; Choi, J.S.; Chung, H.Y.; Yokozawa, T. Oligonol, a low-molecular-weight polyphenol derived from lychee fruit, attenuates gluco-lipotoxicity-mediated renal disorder in type 2 diabetic db/db mice. Drug Discov. Ther. 2015, 9, 13–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhaskaragoud, G.; Geetha, V.; Sharanappa, T.; Kumar, A.S.M.; Kumar, C.H.; Suresh Kumar, G. Hypolipidemic and Antioxidant Properties of Oryzanol Concentrate in Reducing Diabetic Nephropathy via SREBP1 Downregulation Rather than β-Oxidation. Mol. Nutr. Food Res. 2018, 62, e1700511. [Google Scholar]
- Qin, X.; Zhao, Y.; Gong, J.; Huang, W.; Su, H.; Yuan, F.; Fang, K.; Wang, D.; Li, J.; Zou, X.; et al. Berberine Protects Glomerular Podocytes via Inhibiting Drp1-Mediated Mitochondrial Fission and Dysfunction. Theranostics 2019, 9, 1698–1713. [Google Scholar] [CrossRef]
- Liu, P.; Peng, L.; Zhang, H.; Tang, P.M.K.; Zhao, T.; Yan, M.; Zhao, H.; Huang, X.; Lan, H.; Li, P. Tangshen formula attenuates diabetic nephropathy by promoting ABCA1-mediated renal cholesterol efflux in db/db mice. Front. Physiol. 2018, 9, 343. [Google Scholar] [CrossRef] [Green Version]
- Saravanan, S.; Pari, L. Protective effect of thymol on high fat diet induced diabetic nephropathy in C57BL/6J mice. Chem. Biol. Interact. 2016, 245, 1–11. [Google Scholar] [CrossRef]
- Chin, H.J.; Fu, Y.Y.; Ahn, J.M.; Na, K.Y.; Kim, Y.S.; Kim, S.; Chae, D.-W. Omacor, n-3 polyunsaturated fatty acid, attenuated albuminuria and renal dysfunction with decrease of SREBP-1 expression and triglyceride amount in the kidney of type II diabetic animals. Nephrol. Dial. Transplant 2010, 25, 1450–1457. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Z.B.; Yin, W.D.; Li, Q.K.; Cai, M.B.; Yu, J.; Li, H.G.; Zhang, C.; Zu, X.H. Preventive effect of Ibrolipim on suppressing lipid accumulation and increasing lipoprotein lipase in the kidneys of diet-induced diabetic minipigs. Lipids Health Dis. 2011, 10, 117. [Google Scholar] [CrossRef] [Green Version]
- Gai, Z.; Gui, T.; Hiller, C.; Kullak-Ublick, G.A. Farnesoid X Receptor Protects against Kidney Injury in Uninephrectomized Obese Mice. J. Biol. Chem. 2016, 291, 2397–2411. [Google Scholar] [CrossRef] [Green Version]
- Kiss, E.; Kränzlin, B.; Wagenblaβ, K.; Bonrouhi, M.; Thiery, J.; Gröne, E.; Nordström, V.; Teupser, D.; Gretz, N.; Malle, E.; et al. Lipid droplet accumulation is associated with an increase in hyperglycemia-induced renal damage: Prevention by liver X receptors. Am. J. Pathol. 2013, 182, 727–741. [Google Scholar] [CrossRef]
- Wilson, P.G.; Thompson, J.C.; Yoder, M.H.; Charnigo, R.; Tannock, L.R. Prevention of renal apoB retention is protective against diabetic nephropathy: Role of TGF-β inhibition. J. Lipid Res. 2017, 58, 2264–2274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, J.; Wilson, P.; Brandewie, K.; Taneja, D.; Schaefer, L.; Mitchell, B.; Tannock, L.R. Renal accumulation of biglycan and lipid retention accelerates diabetic nephropathy. Am. J. Pathol. 2011, 179, 1179–1187. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Lee, M.H.; Nam, D.H.; Song, H.K.; Kang, Y.S.; Lee, J.E.; Kim, H.W.; Cha, J.J.; Hyun, Y.Y.; Han, S.Y.; et al. Celastrol, an NF-κB inhibitor, improves insulin resistance and attenuates renal injury in db/db mice. PLoS ONE 2013, 8, e62068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Shao, M.; Yang, H.; Chen, L.; Yu, L.; Cong, W.; Tian, H.; Zhang, F.; Cheng, P.; Jin, L.; et al. Attenuation of hyperlipidemia- and diabetes-induced early-stage apoptosis and late-stage renal dysfunction via administration of fibroblast growth factor-21 is associated with suppression of renal inflammation. PLoS ONE 2013, 8, e82275. [Google Scholar] [CrossRef] [Green Version]
- Ma, K.L.; Liu, L.; Zhang, Y.; Wang, G.H.; Hu, Z.B.; Chen, P.P.; Lu, J.; Lu, C.C.; Gong, T.K.; Gong, Y.X.; et al. Aspirin attenuates podocyte injury in diabetic rats through overriding cyclooxygenase-2-mediated dysregulation of LDL receptor pathway. Int. Urol. Nephrol. 2019, 51, 551–558. [Google Scholar] [CrossRef]
- Mori, J.; Patel, V.B.; Ramprasath, T.; Alrob, O.A.; DesAulniers, J.; Scholey, J.W.; Lopaschuk, G.D.; Oudit, G.Y. Angiotensin 1-7 mediates renoprotection against diabetic nephropathy by reducing oxidative stress, inflammation, and lipotoxicity. Am. J. Physiol. Renal Physiol. 2014, 306, F812–F821. [Google Scholar] [CrossRef]
- Kang, Y.S.; Lee, M.H.; Song, H.K.; Ko, G.J.; Kwon, O.S.; Lim, T.K.; Kim, S.H.; Han, S.Y.; Han, K.H.; Lee, J.E.; et al. CCR2 antagonism improves insulin resistance, lipid metabolism, and diabetic nephropathy in type 2 diabetic mice. Kidney Int. 2010, 78, 883–894. [Google Scholar] [CrossRef] [Green Version]
- Yiu, W.H.; Li, R.X.; Wong, D.W.L.; Wu, H.J.; Chan, K.W.; Chan, L.Y.Y.; Leung, J.C.K.; Lai, K.N.; Sacks, S.H.; Zhou, W.; et al. Complement C5a inhibition moderates lipid metabolism and reduces tubulointerstitial fibrosis in diabetic nephropathy. Nephrol. Dial. Transplant 2018, 33, 1323–1332. [Google Scholar] [CrossRef]
- Nam, D.H.; Lee, M.H.; Kim, J.E.; Song, H.K.; Kang, Y.S.; Lee, J.E.; Kim, H.W.; Cha, J.J.; Hyun, Y.Y.; Kim, S.H.; et al. Blockade of cannabinoid receptor 1 improves insulin resistance, lipid metabolism, and diabetic nephropathy in db/db mice. Endocrinology 2012, 153, 1387–1396. [Google Scholar] [CrossRef] [Green Version]
- Jia, Z.; Sun, Y.; Yang, G.; Zhang, A.; Huang, S.; Heiney, K.M.; Zhang, Y. New Insights into the PPAR γ Agonists for the Treatment of Diabetic Nephropathy. PPAR Res. 2014, 2014, 818530. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Zhang, D.; Li, J.; Zhang, X.; Fan, F.; Guan, Y. Role of PPARgamma in renoprotection in Type 2 diabetes: Molecular mechanisms and therapeutic potential. Clin. Sci. 2009, 116, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.M.; Doss, H.M.; Kim, K.S. Multifaceted Physiological Roles of Adiponectin in Inflammation and Diseases. Int. J. Mol. Sci. 2020, 21, 1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada-Iwabu, M.; Yamauchi, T.; Iwabu, M.; Honma, T.; Hamagami, K.I.; Matsuda, K.; Yamaguchi, M.; Tanabe, H.; Kimura-Someya, T.; Shirouzu, M.; et al. A small-molecule AdipoR agonist for type 2 diabetes and short life in obesity. Nature 2013, 503, 493–499. [Google Scholar] [CrossRef]
- Szendroedi, J.; Yoshimura, T.; Phielix, E.; Koliaki, C.; Marcucci, M.; Zhang, D.; Jelenik, T.; Müller, J.; Herder, C.; Nowotny, P.; et al. Role of diacylglycerol activation of PKCθ in lipid-induced muscle insulin resistance in humans. Proc. Natl. Acad. Sci. USA 2014, 111, 9597–9602. [Google Scholar] [CrossRef] [Green Version]
- Saxena, N.K.; Anania, F.A. Adipocytokines and Hepatic Fibrosis. Trends Endocrinol. Metab. 2015, 26, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Zha, D.; Wu, X.; Gao, P. Adiponectin and its receptors in diabetic kidney disease: Molecular mechanisms and clinical potential. Endocrinology 2017, 158, 2022–2034. [Google Scholar] [CrossRef] [Green Version]
- Yamakado, S.; Cho, H.; Inada, M.; Morikawa, M.; Jiang, Y.H.; Saito, K.; Nakaishi, K.; Watabe, S.; Takagi, H.; Kaneda, M.; et al. Urinary adiponectin as a new diagnostic index for chronic kidney disease due to diabetic nephropathy. BMJ Open Diabetes Res. Care 2019, 7, e000661. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Hu, F.B.; Curhan, G. Serum adiponectin and renal dysfunction in men with type 2 diabetes. Diabetes Care 2007, 30, 239–244. [Google Scholar] [CrossRef] [Green Version]
- Saraheimo, M.; Forsblom, C.; Fagerudd, J.; Teppo, A.M.; Pettersson-Fernholm, K.; Frystyk, J.; Flyvbjerg, A.; Groop, P.H. Serum adiponectin is increased in type 1 diabetic patients with nephropathy. Diabetes Care 2005, 28, 1410–1414. [Google Scholar] [CrossRef] [Green Version]
- Cherney, D.Z.I.; Zinman, B.; Inzucchi, S.E.; Koitka-Weber, A.; Mattheus, M.; von Eynatten, M.; Wanner, C. Effects of empagliflozin on the urinary albumin-to-creatinine ratio in patients with type 2 diabetes and established cardiovascular disease: An exploratory analysis from the EMPA-REG OUTCOME randomised, placebo-controlled trial. Lancet Diabetes Endocrinol. 2017, 5, 610–621. [Google Scholar] [CrossRef]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. Canagliflozin and renal outcomes in type 2 diabetes and nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheen, A.J. Beneficial effects of SGLT2 inhibitors on fatty liver in type 2 diabetes: A common comorbidity associated with severe complications. Diabetes Metab. 2019, 45, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Gnudi, L.; Coward, R.J.M.; Long, D.A. Diabetic Nephropathy: Perspective on Novel Molecular Mechanisms. Trends Endocrinol. Metab. 2016, 27, 820–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahl, S.; Gancheva, S.; Straßburger, K.; Herder, C.; Machann, J.; Katsuyama, H.; Kabisch, S.; Henkel, E.; Kopf, S.; Lagerpusch, M.; et al. Empagliflozin Effectively Lowers Liver Fat Content in Well-Controlled Type 2 Diabetes: A Randomized, Double-Blind, Phase 4, Placebo-Controlled Trial. Diabetes Care 2020, 43, 298–305. [Google Scholar] [CrossRef] [Green Version]
- Johansson, L.; Hockings, P.D.; Johnsson, E.; Dronamraju, N.; Maaske, J.; Garcia-Sanchez, R.; Wilding, J.P.H. Dapagliflozin plus saxagliptin add-on to metformin reduces liver fat and adipose tissue volume in patients with type 2 diabetes. Diabetes. Obes. Metab. 2020. [Google Scholar] [CrossRef] [PubMed]
- Gastaldelli, A.; Repetto, E.; Guja, C.; Hardy, E.; Han, J.; Jabbour, S.A.; Ferrannini, E. Exenatide and dapagliflozin combination improves markers of liver steatosis and fibrosis in patients with type 2 diabetes. Diabetes. Obes. Metab. 2020, 22, 393–403. [Google Scholar] [CrossRef]
- Hosokawa, K.; Takata, T.; Sugihara, T.; Matono, T.; Koda, M.; Kanda, T.; Taniguchi, S.; Ida, A.; Mae, Y.; Yamamoto, M.; et al. Ipragliflozin ameliorates endoplasmic reticulum stress and apoptosis through preventing ectopic lipid deposition in renal tubules. Int. J. Mol. Sci. 2020, 21, 190. [Google Scholar] [CrossRef] [Green Version]
- Fioretto, P.; Zambon, A.; Rossato, M.; Busetto, L.; Vettor, R. SGLT2 inhibitors and the diabetic kidney. Diabetes Care 2016, 39, S165–S171. [Google Scholar] [CrossRef] [Green Version]
- Nespoux, J.; Vallon, V. SGLT2 inhibition and kidney protection. Clin. Sci. 2018, 132, 1329–1339. [Google Scholar] [CrossRef]
- Caro-Ordieres, T.; Marín-Royo, G.; Opazo-Ríos, L.; Jiménez-Castilla, L.; Moreno, J.A.; Gómez-Guerrero, C.; Egido, J. The Coming Age of Flavonoids in the Treatment of Diabetic Complications. J. Clin. Med. 2020, 9, 346. [Google Scholar]
- Taylor, R.; Leslie, W.S.; Barnes, A.C.; Brosnahan, N.; Thom, G.; McCombie, L.; Sattar, N.; Welsh, P.; Peters, C.; Zhyzhneuskaya, S.; et al. Clinical and metabolic features of the randomised controlled Diabetes Remission Clinical Trial (DiRECT) cohort. Diabetologia 2018, 61, 589–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lean, M.E.J.; Leslie, W.S.; Barnes, A.C.; Brosnahan, N.; Thom, G.; McCombie, L.; Peters, C.; Zhyzhneuskaya, S.; Al-Mrabeh, A.; Hollingsworth, K.G.; et al. Durability of a primary care-led weight-management intervention for remission of type 2 diabetes: 2-year results of the DiRECT open-label, cluster-randomised trial. Lancet Diabetes Endocrinol. 2019, 7, 344–355. [Google Scholar] [CrossRef] [Green Version]
- Lean, M.E.; Leslie, W.S.; Barnes, A.C.; Brosnahan, N.; Thom, G.; McCombie, L.; Peters, C.; Zhyzhneuskaya, S.; Al-Mrabeh, A.; Hollingsworth, K.G.; et al. Primary care-led weight management for remission of type 2 diabetes (DiRECT): An open-label, cluster-randomised trial. Lancet 2018, 391, 541–551. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, M.D.; Taylor, R.; Lean, M.E.J. The DiRECT principles: Giving Type 2 diabetes remission programmes the best chance of success. Diabet. Med. 2019, 36, 1703–1704. [Google Scholar] [CrossRef] [Green Version]
- Taylor, R.; Al-Mrabeh, A.; Sattar, N. Understanding the mechanisms of reversal of type 2 diabetes. Lancet Diabetes Endocrinol. 2019, 7, 726–736. [Google Scholar] [CrossRef]
- Leehey, D.J.; Moinuddin, I.; Bast, J.P.; Qureshi, S.; Jelinek, C.S.; Cooper, C.; Edwards, L.C.; Smith, B.M.; Collins, E.G. Aerobic exercise in obese diabetic patients with chronic kidney disease: A randomized and controlled pilot study. Cardiovasc. Diabetol. 2009, 8, 62. [Google Scholar] [CrossRef] [Green Version]
- Dungey, M.; Hull, K.L.; Smith, A.C.; Burton, J.O.; Bishop, N.C. Inflammatory factors and exercise in chronic kidney disease. Int. J. Endocrinol. 2013, 2013, 569831. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.C.; Burton, J.O.; Hons, B.A.; Hons, M. Exercise in Kidney Disease and Diabetes: Time for Action. J. Ren. Care 2012, 52–58. [Google Scholar] [CrossRef]
- Golbidi, S.; Laher, I. Exercise induced adipokine changes and the metabolic syndrome. J. Diabetes Res. 2014, 2014, 726861. [Google Scholar] [CrossRef] [Green Version]
- Zacharewicz, E.; Hesselink, M.K.C.; Schrauwen, P. Exercise counteracts lipotoxicity by improving lipid turnover and lipid droplet quality. J. Intern. Med. 2018, 284, 505–518. [Google Scholar] [CrossRef] [Green Version]
- Gleeson, M.; Bishop, N.C.; Stensel, D.J.; Lindley, M.R.; Mastana, S.S.; Nimmo, M.A. The anti-inflammatory effects of exercise: Mechanisms and implications for the prevention and treatment of disease. Nat. Rev. Immunol. 2011, 11, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Van Craenenbroeck, A.H.; Van Craenenbroeck, E.M.; Van Ackeren, K.; Vrints, C.J.; Conraads, V.M.; Verpooten, G.A.; Kouidi, E.; Couttenye, M.M. Effect of Moderate Aerobic Exercise Training on Endothelial Function and Arterial Stiffness in CKD Stages 3–4: A Randomized Controlled Trial. Am. J. Kidney Dis. 2015, 66, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Codella, R.; Lanzoni, G.; Zoso, A.; Caumo, A.; Montesano, A.; Terruzzi, I.M.; Ricordi, C.; Luzi, L.; Inverardi, L. Moderate Intensity Training Impact on the Inflammatory Status and Glycemic Profiles in NOD Mice. J. Diabetes Res. 2015, 2015, 737586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ploeger, H.E.; Takken, T.; De Greef, M.H.G.; Timmons, B.W. The effects of acute and chronic exercise on inflammatory markers in children and adults with a chronic inflammatory disease: A systematic review. Exerc. Immunol. Rev. 2009, 15, 6–41. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Condition | Patients | Sample | Comparison | Disease-Associated Metabolites | Ref. |
|---|---|---|---|---|---|
| T2D | 92 (American Indians) | Serum | DN Progression | ↑Polyunsaturated triacylglycerols (TAGs) ↓C16–C20 acylcarnitines (ACs) | [163] |
| T1D | 669 | Serum | Combined renal end-point | ↓PC(O-34:2), PC(O-34:3), SM(d18:1/24:0), SM(d40:1), SM(d41:1). | [175] |
| All-cause mortality | ↓PC(O-34:3), SM(d40:1) and SM(d41:1). | ||||
| Albuminuria progression | ↓SM(d18:1/24:0) | ||||
| T2D + renal involvement | 150 | Plasma | Control-T2D | ↓EFAs ↑NEFAs | [176] |
| T2D-DN III | ↑EFAs =NEFAs | ||||
| DN III-DN IV | ↓EFAs ↓↓NEFAs | ||||
| DN IV-DN V | ↑EFAs ↑NEFAs | ||||
| T2D | 90 | Plasma | Δ UACR or Δ eGFR | ↓Histidine; ↑butenoylcarnitine | [177] |
| Urine | ↓Hexose, Glutamine, Tyrosine | ||||
| T2D | 78 | Serum | Albuminuria | ↑Creatinine, aspartic acid, γ-butyrobetaine, citrulline, symmetric dimethylarginine (SDMA), kynurenine, azelaic acid, galactaric acid | [178] |
| Drug | Category | Pathway | Experimental Model | Observed Effect | Ref. |
|---|---|---|---|---|---|
| Cyclosporin A2/hydroxypropyl-β-cyclodextrin | Calcineurin inhibitor/lipid chelator | TNF/NFAT/ABCA1/SOAT1 signaling | Podo-Abca1 KO, double, triple, and inducible KO mice | ↓ UACR; ↓ Histological changes; ↓ Inflammation/oxidative stress/apoptosis; ↓ Lipid accumulation | [96] |
| A30/Elamipretide | ABCA1inductor/Cardiolipin peroxidase inhibitor | Mitochondrial dysfunction pathway | Podo-Abca1 KO SOAT KO, db/db and BTBR ob/ob mice | ↓ UACR; ↓ Histological changes; ↓ Oxidative stress/Mitochondrial dysfunction; ↓ Lipid accumulation | [104] |
| Ceramide-1-Phosphate | Lipid supplementation | SMPDL3b/C1P/IR/Cav-1/Akt signaling | Podo-Smpdl3 KO, double KO, and db/db mice | ↓ UACR; ↓ Histological changes; ↓ Lipid accumulation | [106] |
| Fenofibrate | Fibrate | PPARα modulator | HFD | ↓ Albuminuria; ↓ Histological changes; ↓ Oxidative stress/fibrosis; ↓ Lipid accumulation | [117] |
| Fenofibrate | Fibrate | PPARα modulator/AMPK-PGC-1α axis | db/db mice | ↓ Albuminuria; ↓ Histological changes; ↓ Inflammation/oxidative stress; ↓ Apoptosis/fibrosis; ↓ Lipid accumulation | [154] |
| AdipoRon | Adiponectin agonist | AMPK/PPARα pathway | db/db mice | ↓UACR; ↓ Oxidative stress/apoptosis/fibrosis; ↓ Lipid accumulation | [189,190] |
| Ipraglifozin | SGLT2i | ER stress pathway | FTL ob/ob mice | ↓ Histological changes; ↓ ER stress/apoptosis/fibrosis; ↓ Lipid accumulation | [191] |
| JNJ-39933673 | SGLT2i | Glycogenic and lipogenic pathways | db/db mice | ↓ UACR; ↓ Histological changes; ↓ Inflammation/fibrosis; ↓ Lipid accumulation | [192] |
| Exendin-4 | GLP-1 RA | Cholesterol efflux pathway | ApoE KO HFD + STZ | ↓UACR; ↓ Lipid accumulation | [193] |
| Liraglutide | GLP-1 RA | AMPK/SIRT1/PGC-1α axis | SD rats + HFD | ↓UACR; ↓ Inflammation/fibrosis; ↓ Lipid accumulation | [194] |
| Neutralizing Monoclonal VEGF-B antibody | VEGF-B antagonism | VEGF-B signaling | Podo-VegfBKO; db/db mice; double KO; + HFD; + STZ | ↓ UACR; ↓ Histological changes; ↓ Inflammation; ↓ Lipid accumulation | [195] |
| Isoquercetin | Flavonoid | NF-κB-AMPK-NRF2 axis | Wistar rats + STZ | ↓ Inflammation/oxidative stress; ↓ Lipid accumulation | [196] |
| Quercetin | Flavonoid | SCAP-SREBP-2-LDLR pathway | db/db mice | ↓UACR; ↓ Histological changes; ↓ Lipid accumulation | [197] |
| Quercetin + Allopurinol | Flavonoid + uric acid inhibitor | NLRP3 Inflammasome pathway | SD rats + STZ | ↓ Albuminuria; ↓ Histological changes; ↓ Inflammation; ↓ Lipid accumulation | [198] |
| Anthocyanin-rich Seoritae extract | Flavonoid | AMPK/PGC-1α | db/db mice | ↓ Albuminuria; ↓ Oxidative stress; ↓ Apoptosis/fibrosis; ↓ Lipid accumulation | [199] |
| Resveratrol | Polyphenol | AMPK–SIRT1–PGC-1α axis | db/db mice | ↓ Albuminuria; ↓ Histological changes; ↓ Inflammation/oxidative stress; ↓Apoptosis/fibrosis; ↓ Lipid accumulation | [200] |
| Curcumin | Polyphenol | AMPK/NRF2 pathway | OLETF rats | ↓ Albuminuria; ↓ Histological changes; ↓ Inflammation/oxidative stress; ↓ Lipid accumulation | [201] |
| Curcumin | Polyphenol | AMPK/SREBP-1 pathway | SD rats + STZ | ↓ Albuminuria; ↓ Histological changes; ↓ Inflammation/fibrosis; ↓ Lipid accumulation | [202] |
| Oligonol-derived lychee fruit | Polyphenol | Adiponectin pathway | db/db mice | ↓ Histological changes; ↓ Inflammation ↓ Apoptosis/oxidative stress; ↓ Lipid accumulation | [203] |
| Oryzanol Concentrate | Rice bran oil | SREBP-1 pathway | Wistar rats + HFD + STZ | ↓ Albuminuria; ↓ Histological changes; ↓ Inflammation/oxidative stress/fibrosis; ↓ Lipid accumulation | [204] |
| Berberine | Flavonoid | Mitochondrial dysfunction pathway | db/db mice | ↓ UACR; ↓ Histological changes; ↓ Oxidative stress; ↓ Mitochondrial dysfunction; ↓ Lipid accumulation | [205] |
| Tangshen Formula | Traditional Chinese formulation | PGC-1α-LXR-ABCA1 pathway | db/db mice | ↓ UACR; ↓ Histological changes; ↓ Lipid accumulation | [206] |
| Thymol | Monoterpene phenolic compound | SREBP-1 pathway | HFD | ↓ Albuminuria; ↓ Histological changes; ↓ Oxidative stress/fibrosis; ↓Lipid accumulation | [207] |
| Omacor | n-3 polyunsaturated fatty acid | NF-κB and lipogenic pathway | db/db mice | ↓ Albuminuria; ↓ Histological changes; ↓ Inflammation/fibrosis; ↓ Lipid accumulation | [208] |
| Ibrolipim (NO-1886) | Renal lipoprotein lipase agonist | Activation renal lipoprotein lipase | CB minipigs + HSFD | ↓ UACR; ↓ Histological changes; ↓ Lipid accumulation | [209] |
| Obeticholic acid | FXR agonist | Glutathione metabolism pathway | HFD + UNX | ↓ UACR; ↓ Histological changes; ↓ Oxidative stress/apoptosis; ↓ Lipid accumulation | [210] |
| GW3965 | LXRα agonist | LXRαin macrophages | LDLR KO and transgenic mice; WD + STZ | ↓ Albuminuria; ↓ Histological changes; ↓ Inflammation/oxidative stress/fibrosis; ↓ Lipid accumulation | [211] |
| 1D11 | pan-TGFβ-neutralizing antibody | TGFβ-ApoB axis | Double KO STZ + CholD | ↓ UACR; ↓ Fibrosis; ↓ Lipid accumulation | [212,213] |
| Celastrol | NF-κB inhibitor | NF-κB pathway | db/db mice | ↓ UACR; ↓ Histological changes; ↓ Inflammation/oxidative stress/fibrosis; ↓ Lipid accumulation | [214] |
| Fibroblast growth factor-21 | Growth factor | TGFβ pathway | FGF21 KO mice + STZ; BSA–FFA | ↓ UACR; ↓ Inflammation/oxidative stress/apoptosis; ↓ Lipid accumulation | [215] |
| Aspirin | COX-2 inhibitor | COX-2/LDLR pathway | SD rats + STZ | ↓UACR; ↓ Histological changes; ↓ Inflammation; ↓ Lipid accumulation | [216] |
| Angiotensin 1–7 | ACEi | ACE2/Ang 1–7/Mas receptor axis | db/db mice | ↓ Albuminuria; ↓ Histological changes; ↓ Inflammation/oxidative stress/fibrosis; ↓ Lipid accumulation | [217] |
| RS504393 | CCR2 antagonist | CCL2/CCR2 axis | db/db mice | ↓ Albuminuria; ↓ Histological changes; ↓ Inflammation/fibrosis; ↓ Lipid accumulation | [218] |
| NOX-D21 | Complement C5a inhibitor | C5a/C5a receptor axis | db/db mice + UNX | ↓ UACR; ↓ Histological changes; ↓ Inflammation/fibrosis; ↓ Lipid accumulation | [219] |
| SR141716 | CB-1 receptor antagonist | CB-1 receptor pathway | db/db mice | ↓ UACR; ↓ Histological changes; ↓ Inflammation/oxidative stress/fibrosis; ↓ Lipid accumulation | [220] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Opazo-Ríos, L.; Mas, S.; Marín-Royo, G.; Mezzano, S.; Gómez-Guerrero, C.; Moreno, J.A.; Egido, J. Lipotoxicity and Diabetic Nephropathy: Novel Mechanistic Insights and Therapeutic Opportunities. Int. J. Mol. Sci. 2020, 21, 2632. https://doi.org/10.3390/ijms21072632
Opazo-Ríos L, Mas S, Marín-Royo G, Mezzano S, Gómez-Guerrero C, Moreno JA, Egido J. Lipotoxicity and Diabetic Nephropathy: Novel Mechanistic Insights and Therapeutic Opportunities. International Journal of Molecular Sciences. 2020; 21(7):2632. https://doi.org/10.3390/ijms21072632
Chicago/Turabian StyleOpazo-Ríos, Lucas, Sebastián Mas, Gema Marín-Royo, Sergio Mezzano, Carmen Gómez-Guerrero, Juan Antonio Moreno, and Jesús Egido. 2020. "Lipotoxicity and Diabetic Nephropathy: Novel Mechanistic Insights and Therapeutic Opportunities" International Journal of Molecular Sciences 21, no. 7: 2632. https://doi.org/10.3390/ijms21072632