Theoretical Insight into the Interaction between Chloramphenicol and Functional Monomer (Methacrylic Acid) in Molecularly Imprinted Polymers

Abstract
:1. Introduction
2. Results and Discussion
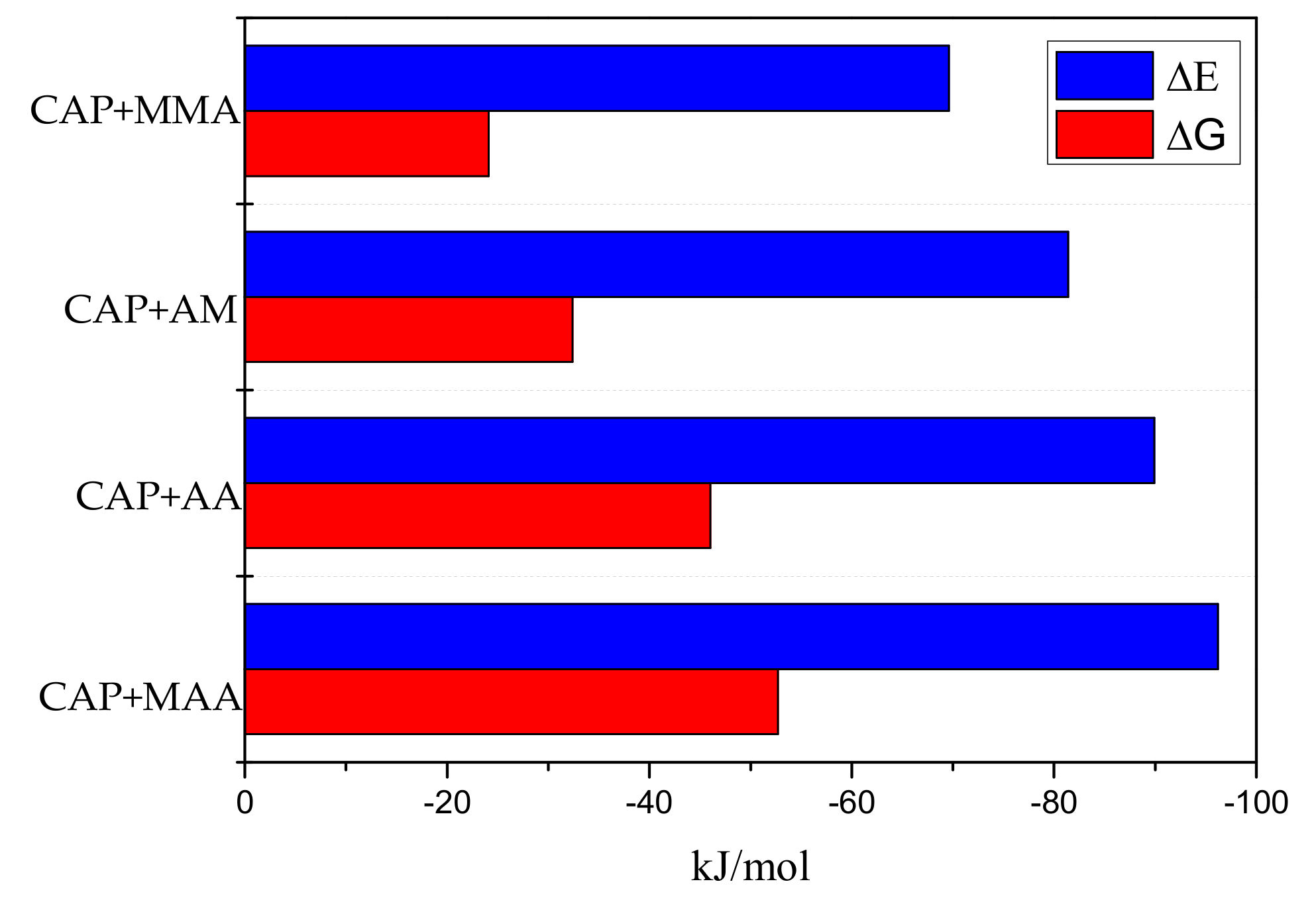
2.1. Theoretical Screening of Functional Monomers
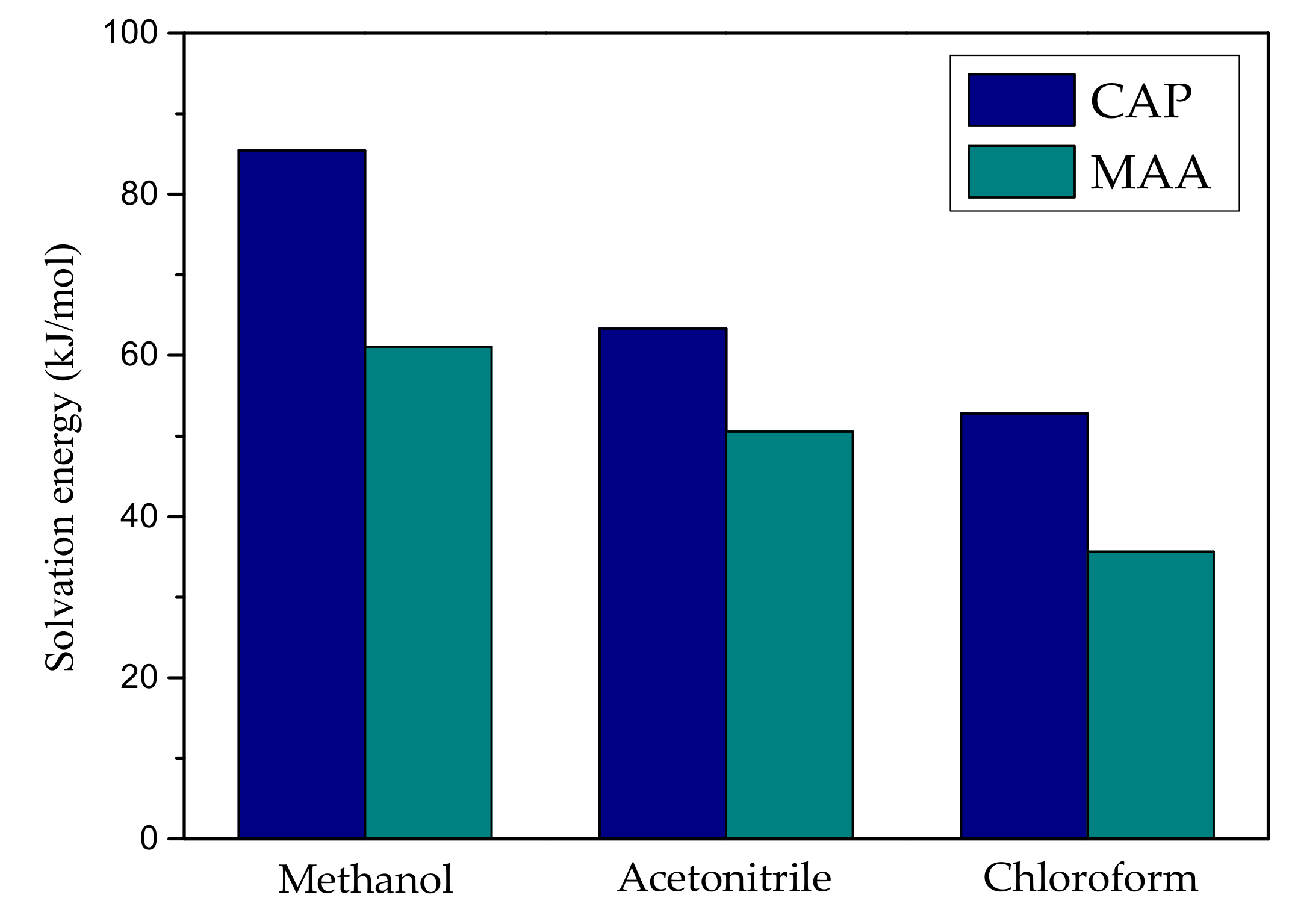
2.2. The Effect of Solvent
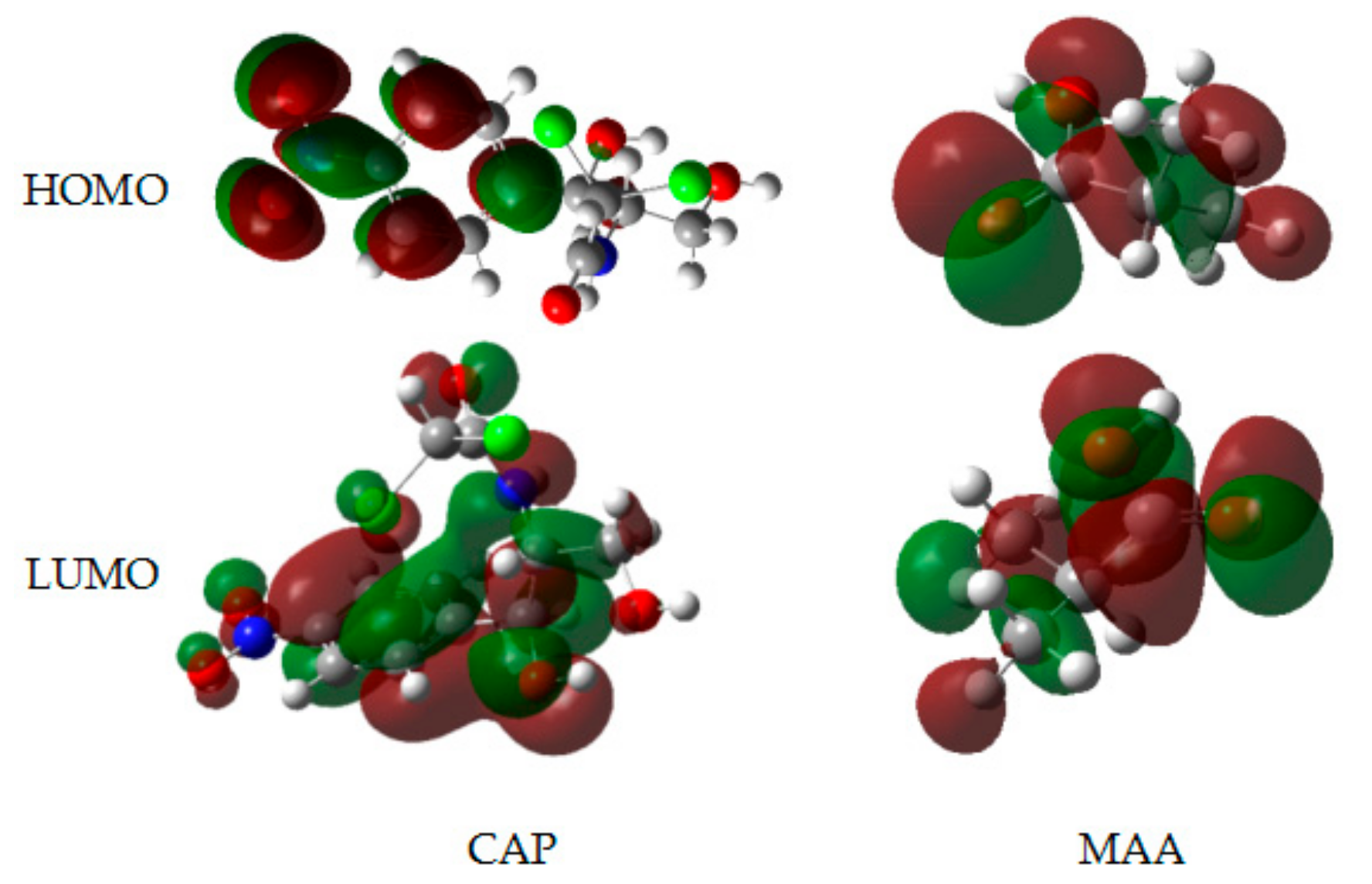
2.3. Analysis of FMOs
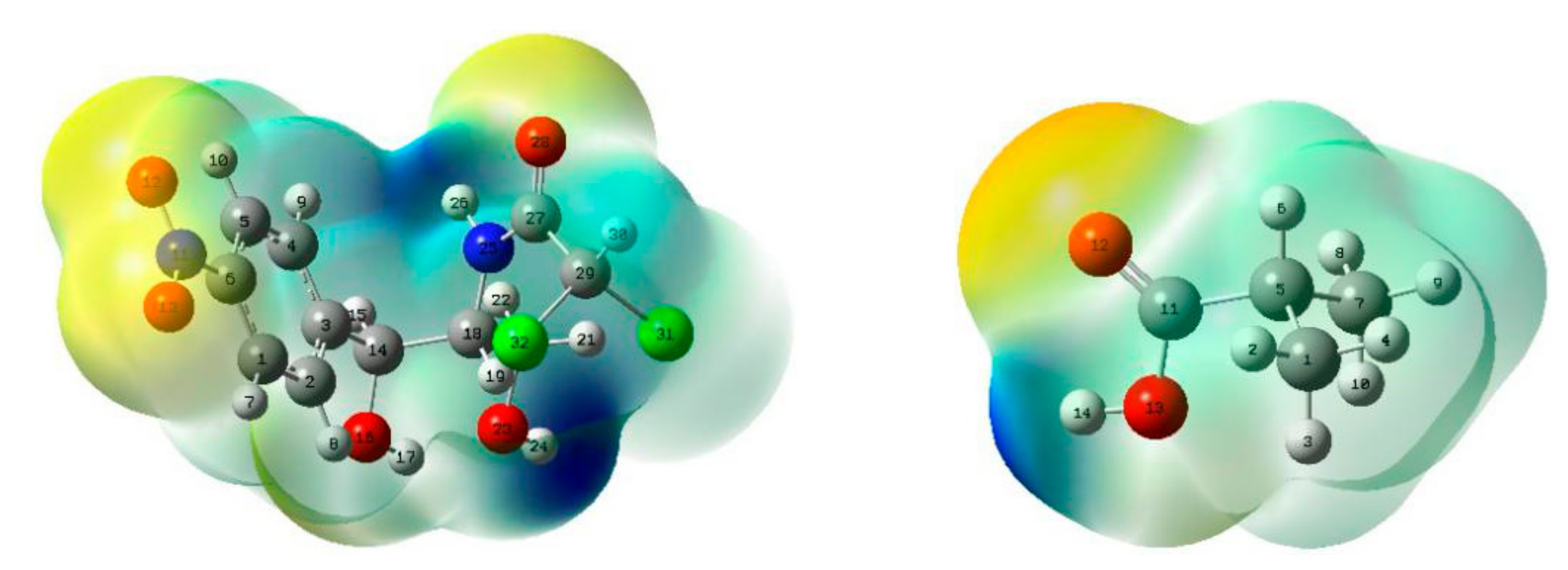
2.4. Analysis of MEPs
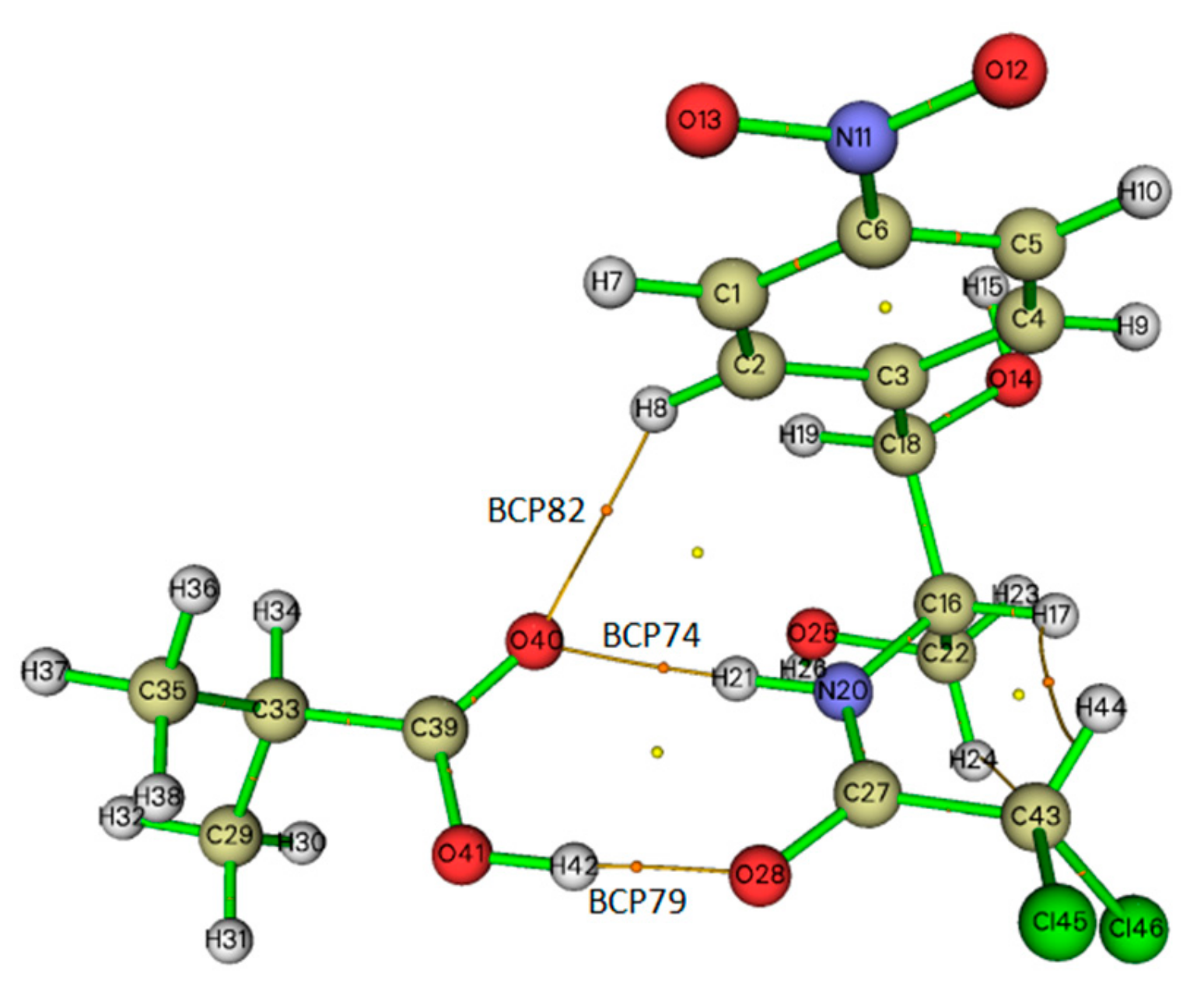
2.5. Topological Analysis
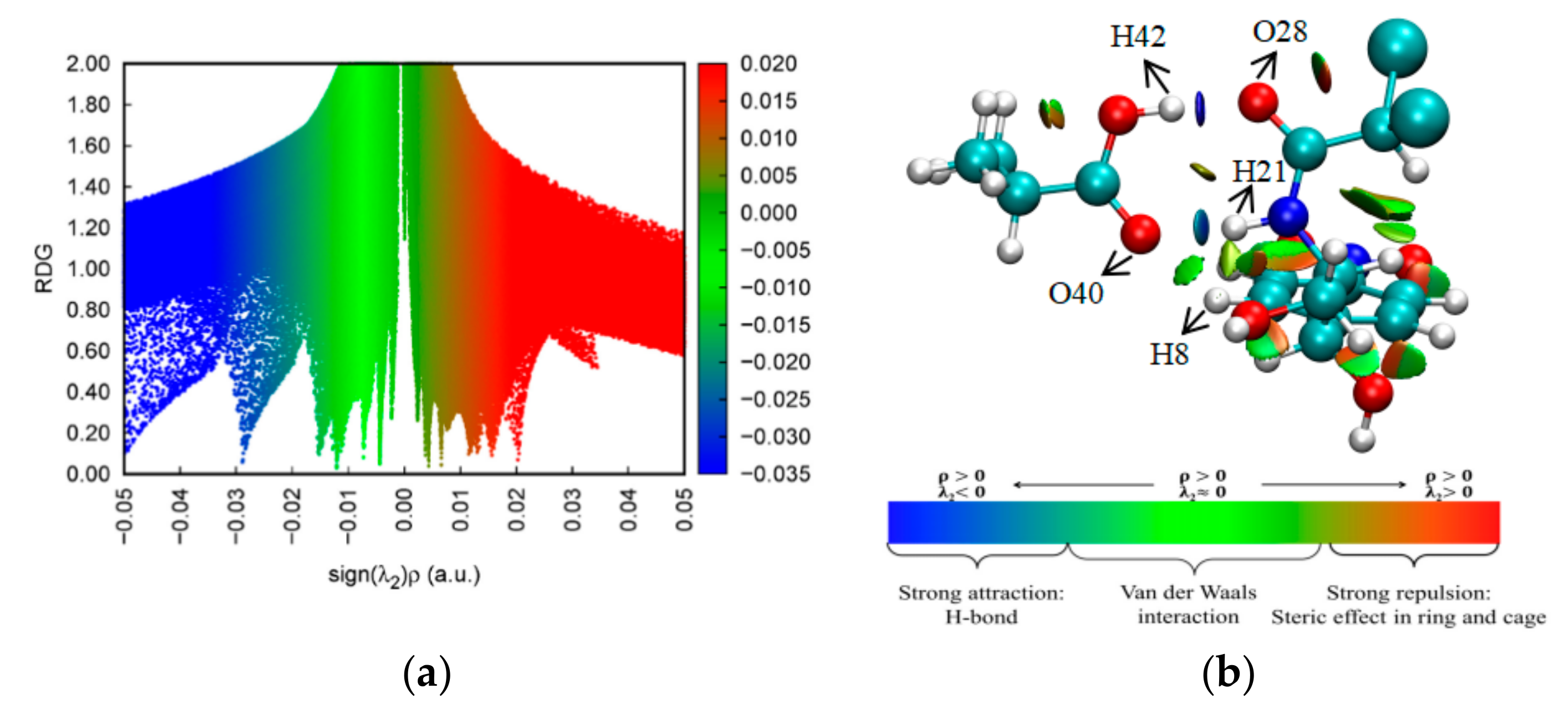
2.6. NCI-RDG Analysis
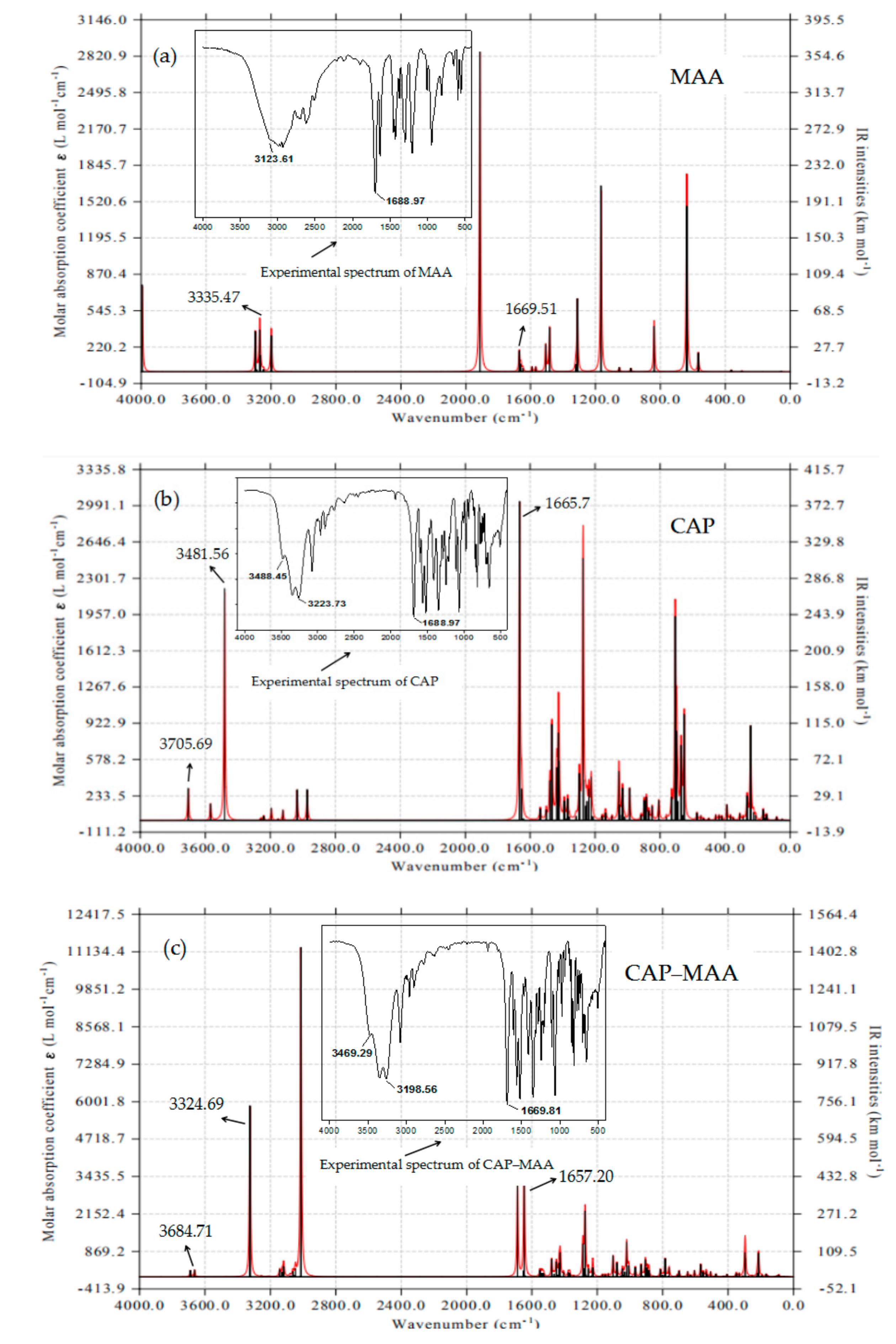
2.7. FTIR Analysis
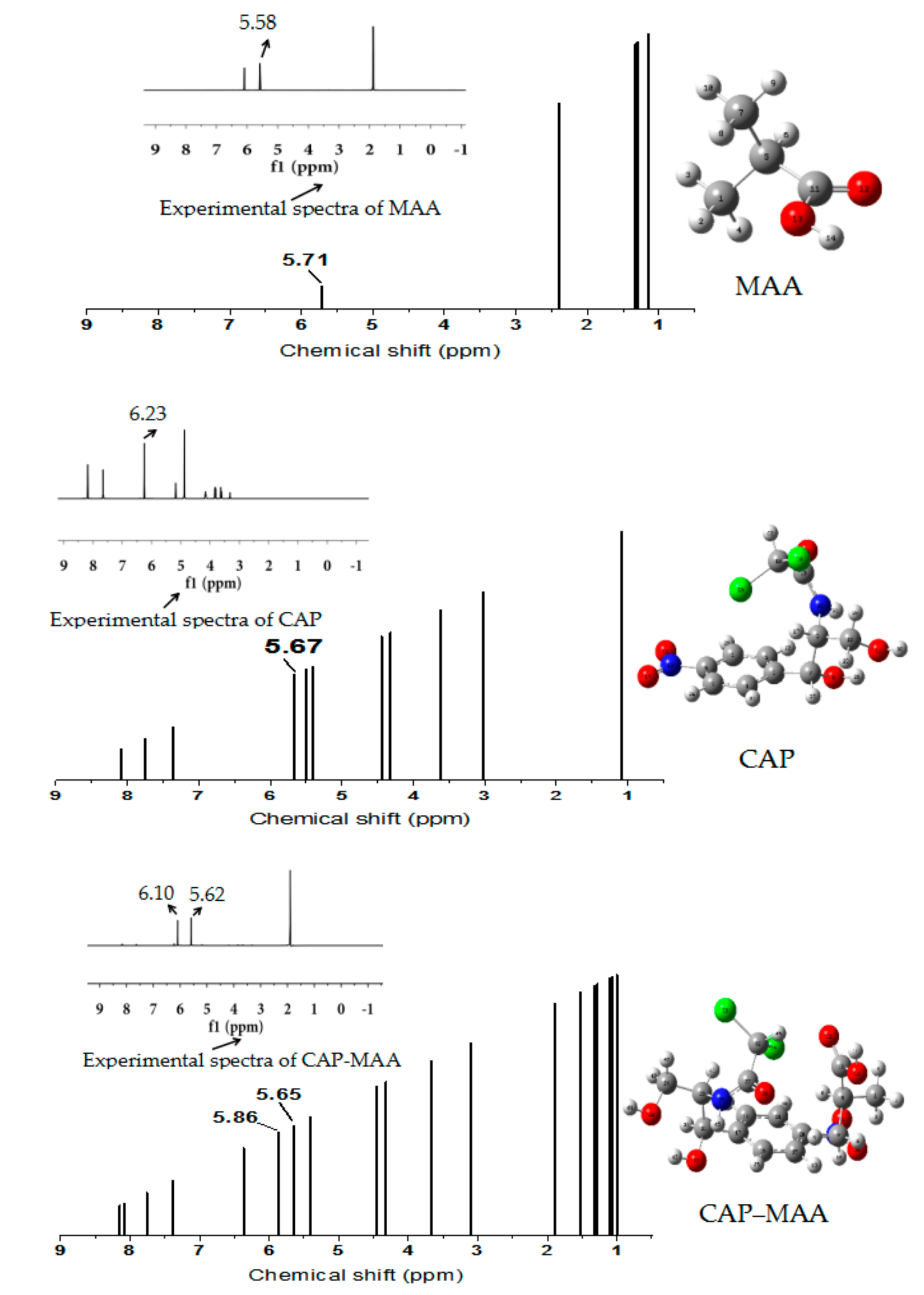
2.8. 1H-NMR Analysis
3. Materials and Methods
3.1. Materials
3.2. Computational Details
3.3. FTIR Analysis
3.4. 1H-NMR Analysis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chen, H.; Ying, J.; Chen, H.; Huang, J.L.; Liao, L. LC determination of chloramphenicol in honey using dispersive liquid-liquid microextraction. Chromatographia 2008, 68, 629–634. [Google Scholar] [CrossRef]
- Zhang, R.L.; Zhou, Z.P.; Xie, A.; Dai, J.D.; Cui, J.Y.; Lang, J.H.; Wei, M.B.; Dai, X.H.; Li, C.X.; Yan, Y.S. Preparation of hierarchical porous carbons from sodium carboxymethyl cellulose via halloysite template strategy coupled with KOH-activation for efficient removal of chloramphenicol. J. Taiwan Inst. Chem. Eng. 2017, 80, 424–433. [Google Scholar] [CrossRef]
- Qin, L.; Zhou, Z.P.; Dai, J.D.; Ma, P.; Zhao, H.B.; He, J.S.; Xie, A.; Li, C.X.; Yan, Y.S. Novel N-doped hierarchically porous carbons derived from sustainable shrimp shell for high-performance removal of sulfamethazine and chloramphenicol. J. Taiwan Inst. Chem. Eng. 2016, 62, 228–238. [Google Scholar] [CrossRef]
- Yusof, N.A.; Rahman, S.K.A.; Hussein, M.Z.; Ibrahim, N.A. Preparation and characterization of molecularly imprinted polymer as SPE sorbent for melamine isolation. Polymers 2013, 5, 1215–1228. [Google Scholar] [CrossRef] [Green Version]
- Douny, C.; Widart, J.; Pauw, d.E.; Maghuin-Rogister, G.; Scippo, M.L. Determination of chloramphenicol in honey, shrimp, and poultry meat with liquid chromatography–mass spectrometry: Validation of the method according to commission decision 2002/657/EC. Food Anal. Methods 2013, 6, 1458–1465. [Google Scholar] [CrossRef]
- Xu, L.H.; Pan, M.F.; Fang, G.Z.; Wang, S. Carbon dots embedded metal-organic framework@molecularly imprinted nanoparticles for highly sensitive and selective detection of quercetin. Sens. Actuators B 2019, 286, 321–327. [Google Scholar] [CrossRef]
- Gómez-Pineda, L.E.; Pina-Luis, G.E.; Cuán, Á.; García-Calzón, J.A.; Díaz-García, M.E. Physico-chemical characterization of flavonol molecularly imprinted polymers. React. Funct. Polym. 2011, 71, 402–408. [Google Scholar] [CrossRef]
- Whitcombe, M.J.; Kirsch, N.; Nicholls, I.A. Molecular imprinting science and technology: A survey of the literature for the years 2004–2011. J. Mol. Recognit. 2014, 27, 297–401. [Google Scholar] [PubMed] [Green Version]
- Jia, B.J.; He, X.; Cui, P.L.; Liu, J.X.; Wang, J.P. Detection of chloramphenicol in meat with a chemiluminescence resonance energy transfer platform based on molecularly imprinted graphene. Anal. Chim. Acta 2019, 1063, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.Y.; Li, S.J.; Lei, M.; Han, Z.H. Online enrichment combined with high performance liquid chromatography for quantitation of trace-level chloramphenicol in milk. Food Sci. Technol. Res. 2018, 24, 963–969. [Google Scholar] [CrossRef]
- Liu, J.B.; Wang, G.Y.; Tang, S.S.; Gao, Q.; Liang, D.D.; Jin, R.F. Theoretical and experimental research on self-assembly system of molecularly imprinted polymers formed via chloramphenicol and methacrylic acid. J. Sep. Sci. 2019, 42, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Xiong, M.Z.; Li, Y.; Li, L.; Xie, X.A.; Zheng, L.S. Molecular dynamics simulation on the pre-organization system of chloramphenicol molecularly imprinted polymers. Comput. Appl. Chem. 2012, 5, 597–600. [Google Scholar]
- Sánchez-González, J.; Peña-Gallego, A.; Sanmartín, J.; Bermejo, A.M.; Bermejo-Barrera, P.; Moreda-Piñeiro, A. NMR spectroscopy for assessing cocaine-functional monomer interactions when preparing molecularly imprinted polymers. Microchem. J. 2019, 147, 813–817. [Google Scholar] [CrossRef]
- Wu, H.; Tian, Q.; Zheng, W.; Jiang, Y.; Xu, J.C.; Li, X.; Zhang, W.C.; Qiu, F.X. Non-enzymatic glucose sensor based on molecularly imprinted polymer: A theoretical, strategy fabrication and application. J. Solid State Electrochem. 2019, 23, 1379–1388. [Google Scholar] [CrossRef]
- Zhong, M.; Wang, Y.H.; Wang, L.; Long, R.Q.; Chen, C.L. Preparation and application of magnetic molecularly imprinted polymers for the isolation of chelerythrine from Macleaya cordata. J. Sep. Sci. 2018, 41, 3318–3327. [Google Scholar] [CrossRef] [PubMed]
- Cowen, T.; Karim, K.; Piletsky, S. Computational approaches in the design of synthetic receptors—A review. Anal. Chim. Acta 2016, 936, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.C.; Fan, X.; Zhao, D.Y. Computer-aided design of molecularly imprinted polymers for simultaneous detection of clenbuterol and its metabolites. Polymers 2019, 11, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, L.; Li, D.Z.; Zhang, L.J. Theoretical insights into the hydrogen bonding interaction in the complexation of epinephrine with uracil. J. Mol. Model. 2019, 25, 252. [Google Scholar] [CrossRef] [PubMed]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Dong, W.; Yan, M.; Zhang, M.L.; Liu, Z.; Li, Y.M. A computational and experimental investigation of the interaction between the template molecule and the functional monomer used in the molecularly imprinted polymer. Anal. Chim. Acta 2005, 542, 186–192. [Google Scholar] [CrossRef]
- Wu, H.Y.; Cui, C.C.; Song, Q.J.; Wang, H.J.; Wu, A.P. Theoretical study of the peroxidation of chlorophenols in gas phase and aqueous solutions. J. Mol. Struct. 2009, 916, 86–90. [Google Scholar] [CrossRef]
- Wu, L.Q.; Sun, B.W.; Li, Y.Z.; Chang, W.B. Study properties of molecular imprinting polymer using a computational approach. Analyst 2003, 128, 944–949. [Google Scholar] [CrossRef]
- Halim, S.A.; Ibrahim, M.A. Synthesis, DFT calculations, electronic structure, electronic absorption spectra, natural bond orbital (NBO) and nonlinear optical (NLO) analysis of the novel 5-methyl-8H-benzo[h]chromeno[2,3-b][1,6] naphthyridine-6(5H),8-dione (MBCND). J. Mol. Struct. 2017, 1130, 543–558. [Google Scholar] [CrossRef]
- Putz, M.V.; Chattaraj, P.K. Electrophilicity kernel and its hierarchy through softness in conceptual density functional theory. Int. J. Quantum Chem. 2013, 113, 2163–2171. [Google Scholar] [CrossRef]
- Dai, Z.Q.; Liu, J.B.; Tang, S.S.; Wang, Y.; Wang, Y.M.; Jin, R.F. Optimization of enrofloxacin-imprinted polymers by computer-aided design. J. Mol. Model. 2015, 21, 290. [Google Scholar] [CrossRef] [PubMed]
- Vanasundari, K.; Balachandran, V.; Kavimani, M.; Narayana, B. Spectroscopic investigation, vibrational assignments, Fukui functions, HOMO-LUMO, MEP and molecular docking evaluation of 4-[(3,4-dichlorophenyl)amino]2-methylidene 4-oxo butanoic acid by DFT method. J. Mol. Struct. 2017, 1147, 136–147. [Google Scholar] [CrossRef]
- Ramalingam, S.; Babu, P.D.S.; Periandy, S.; Fereyduni, E. Vibrational investigation, molecular orbital studies and molecular electrostatic potential map analysis on 3-chlorobenzoic acid using hybrid computational calculations. Spectrochim. Acta Part A 2011, 84, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Prathap, K.N.C.; Lokanath, N. Three novel coumarin-benzenesulfonylhydrazide hybrids: Synthesis, characterization, crystal structure, Hirshfeld surface, DFT and NBO studies. J. Mol. Struct. 2018, 1171, 564–577. [Google Scholar] [CrossRef]
- Minaev, B.F.; Baryshnikov, G.V.; Minaeva, V.A. Electronic structure and spectral properties of the triarylamine-dithienosilole dyes for efficient organic solar cells. Dyes Pigm. 2012, 92, 531–536. [Google Scholar] [CrossRef]
- Bader, R.F.W. A bond path: A universal indicator of bonded interactions. J. Phys. Chem. A 1998, 102, 7314–7323. [Google Scholar] [CrossRef]
- Rozas, I.; Alkorta, I.; Elguero, J. Behavior of ylides containing N, O, and C atoms as hydrogen bond acceptors. J. Am. Chem. Soc. 2000, 122, 11154–11161. [Google Scholar] [CrossRef]
- Fuster, G.; Schuhmacher, M.; Domingo, J.L. Human exposure to dioxins and furans: Application of the substance flow analysis to health risk assessment. Environ. Sci. Pollut. Res. 2002, 9, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Koch, U.; Popelier, P.L.A. Characterization of CHO hydrogen bonds on the basis of the charge density. J. Phys. Chem. 1995, 99, 9747–9754. [Google Scholar] [CrossRef]
- Lipkowski, P.; Grabowski, S.J.; Robinson, T.L.; Leszczynski, J. Properties of the C-H···H dihydrogen bond: An ab initio and topological analysis. J. Phys. Chem. A 2004, 108, 10865–10872. [Google Scholar] [CrossRef]
- Venkataramanan, N.S.; Suvitha, A. Theoretical investigation of the binding of nucleobases to cucurbiturils by dispersion corrected DFT approaches. J. Phys. Chem. B 2017, 121, 4733–4744. [Google Scholar] [CrossRef] [PubMed]
- Venkataramanan, N.S.; Suvitha, A.; Kawazoe, Y. Unravelling the nature of binding of cubane and substituted cubanes within cucurbiturils: A DFT and NCI study. J. Mol. Liq. 2018, 260, 18–29. [Google Scholar] [CrossRef]
- Saloni, J.; Lipkowski, P.; Dasary, S.S.R.; Anjaneyulu, Y.; Yu, H.T.; Hill, G., Jr. Theoretical study of molecular interactions of TNT, acrylic acid, and ethylene glycol dimethacrylate—Elements of molecularly imprinted polymer modeling process. Polymer 2011, 52, 1206–1216. [Google Scholar] [CrossRef]
- She, Y.X.; Wang, M.; Shi, X.M.; Liu, J.J.; Lu, X.L.; Xiao, H.; Cao, W.Q.; Wang, J. Spectroscopy study of binding mechanisms and molecular recognition of class-specific molecularly imprinted polymer beads. Spectrosc. Spectral Anal. 2010, 30, 3052–3055. [Google Scholar]
- Dugo, G.; Rotondo, A.; Mallamace, D.; Cicero, N.; Salvo, A.; Rotondo, E.; Corsaro, C. Enhanced detection of aldehydes in Extra-Virgin Olive Oil by means of band selective NMR spectroscopy. Physica A 2015, 420, 258–264. [Google Scholar] [CrossRef]
- Zheng, Y.Z.; Wang, N.N.; Luo, J.J.; Zhou, Y.; Yu, Z.W. Hydrogen-bonding interactions between [BMIM][BF4] and acetonitrile. Phys. Chem. Chem. Phys. 2013, 15, 18055–18064. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, M.C.; Nascimento, C.S., Jr.; Borges, K.B. Theoretical investigation on functional monomer and solvent selection for molecular imprinting of tramadol. Chem. Phys. Lett. 2016, 645, 174–179. [Google Scholar] [CrossRef]
- Simon, S.; Duran, M.; Dannenberg, J.J. How does basis set superposition error change the potential surfaces for hydrogen-bonded dimers? J. Chem. Phys. 1996, 105, 11024–11103. [Google Scholar] [CrossRef] [Green Version]
- Boys, S.F.; Bernardi, F. Calculation of small molecular interactions by differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M. Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Pierens, G.K. 1H and 13C NMR scaling factors for the calculation of chemical shifts in commonly used solvents using density functional theory. J. Comput. Chem. 2014, 35, 1388–1394. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | EHOMO (eV) | ELUMO (eV) | ω |
|---|---|---|---|
| CAP | −2.283 | 1.377 | 0.038 |
| MAA | −2.335 | 0.824 | 0.189 |
| Complex | BCP | ρ(r) (a.u.) | ▽2ρ(r) (a.u.) | V(r) (a.u.) | H(r) (a.u.) |
|---|---|---|---|---|---|
| CAP-MAA | BCP82, H(8)···O(40) | 0.007 | 0.028 | −0.004 | 0.00112 |
| BCP74, H(21)···O(40) | 0.028 | 0.098 | −0.024 | −0.00005 | |
| BCP79, O(28)···H(42) | 0.050 | 0.134 | −0.045 | −0.00094 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, L.; Xiao, N.; Li, L.; Xie, X.; Li, Y. Theoretical Insight into the Interaction between Chloramphenicol and Functional Monomer (Methacrylic Acid) in Molecularly Imprinted Polymers. Int. J. Mol. Sci. 2020, 21, 4139. https://doi.org/10.3390/ijms21114139
Xie L, Xiao N, Li L, Xie X, Li Y. Theoretical Insight into the Interaction between Chloramphenicol and Functional Monomer (Methacrylic Acid) in Molecularly Imprinted Polymers. International Journal of Molecular Sciences. 2020; 21(11):4139. https://doi.org/10.3390/ijms21114139
Chicago/Turabian StyleXie, Lei, Nan Xiao, Lu Li, Xinan Xie, and Yan Li. 2020. "Theoretical Insight into the Interaction between Chloramphenicol and Functional Monomer (Methacrylic Acid) in Molecularly Imprinted Polymers" International Journal of Molecular Sciences 21, no. 11: 4139. https://doi.org/10.3390/ijms21114139