Detection of a G-Quadruplex as a Regulatory Element in Thymidylate synthase for Gene Silencing Using Polypurine Reverse Hoogsteen Hairpins

 , ,
, ,  ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Results
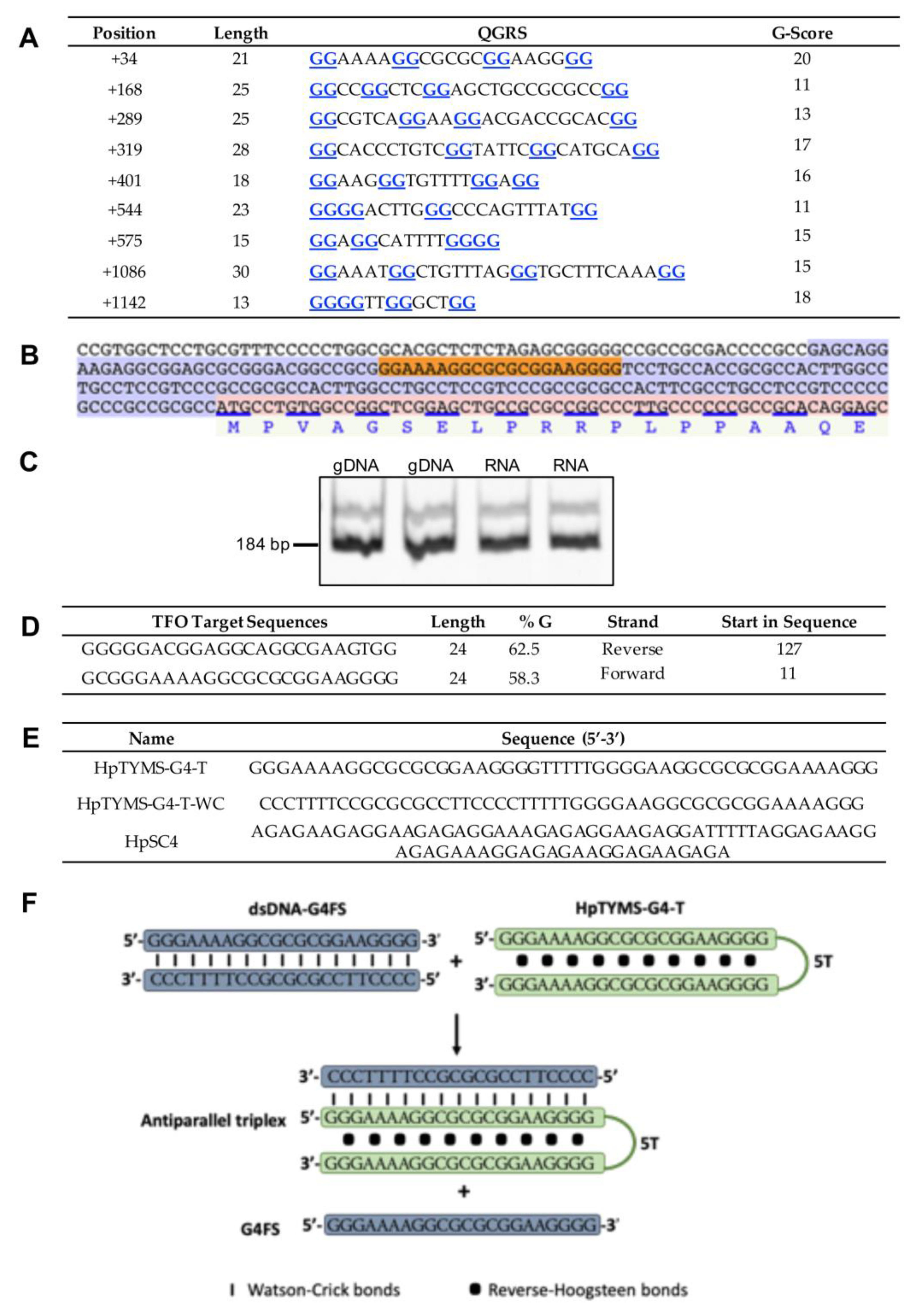
2.1. Detection of a G4 Structure in the 5’UTR of TYMS
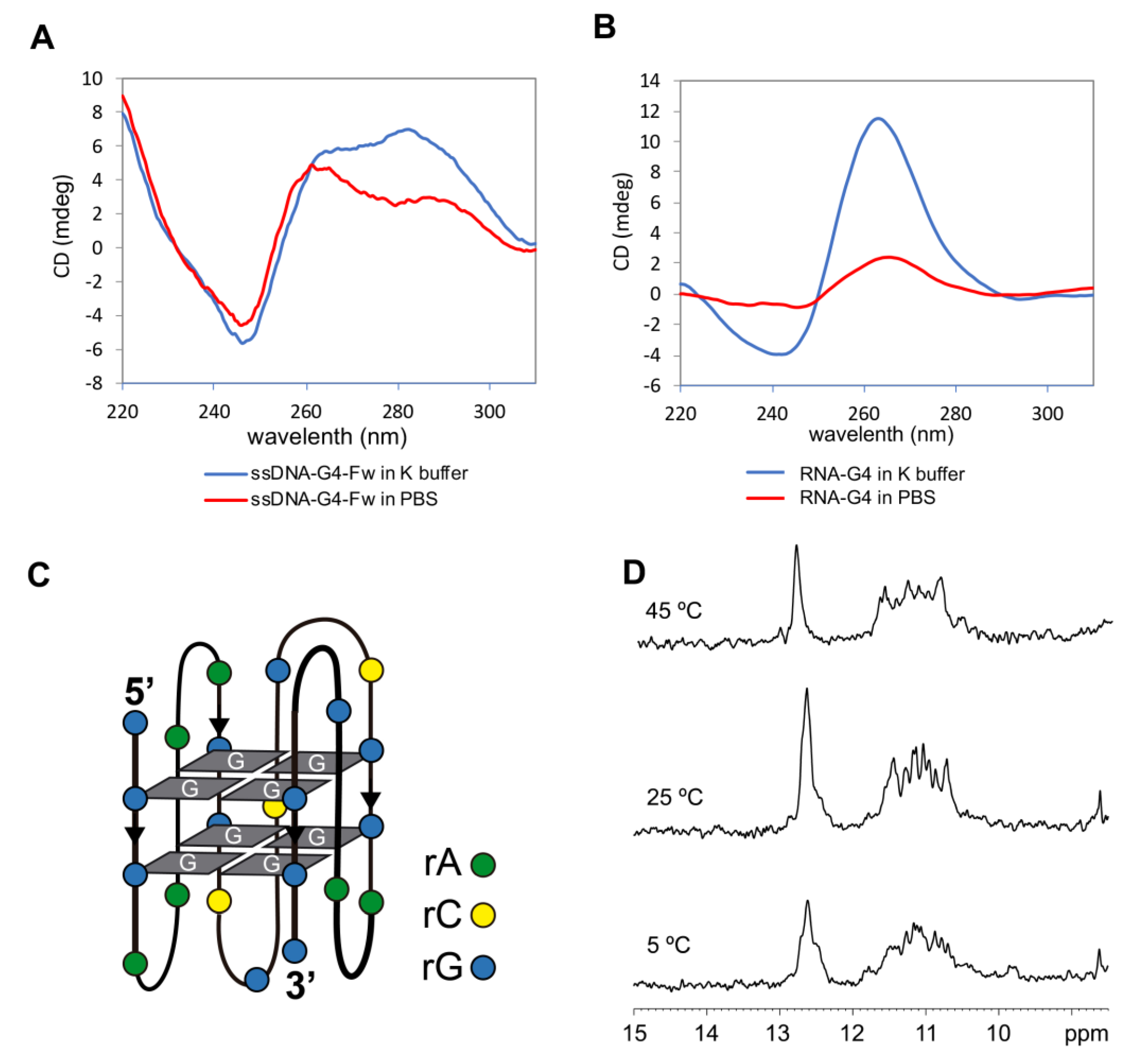
2.2. G4 Structure Confirmation in Both RNA and DNA
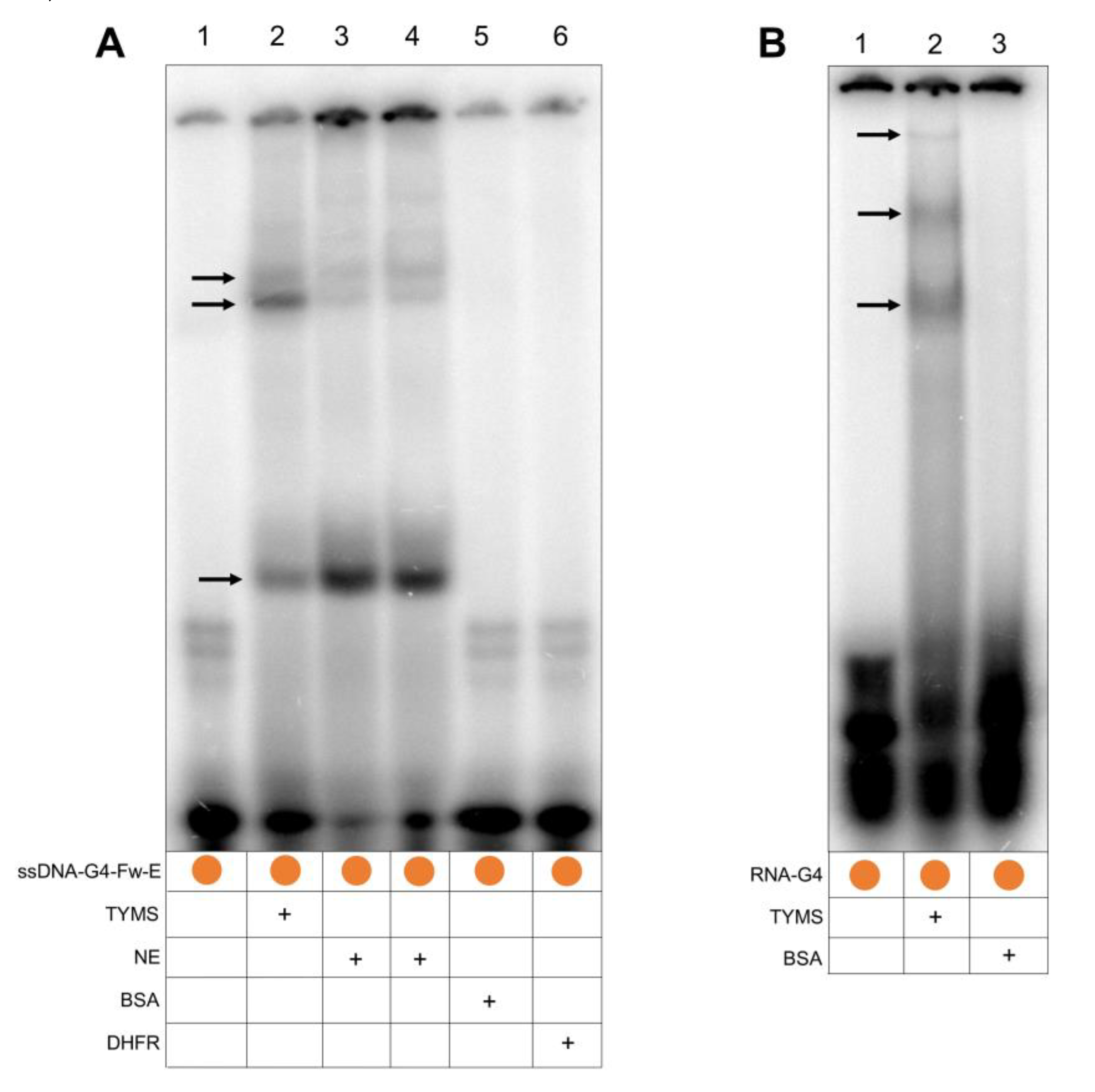
2.3. TYMS Protein Binds to the G4FS
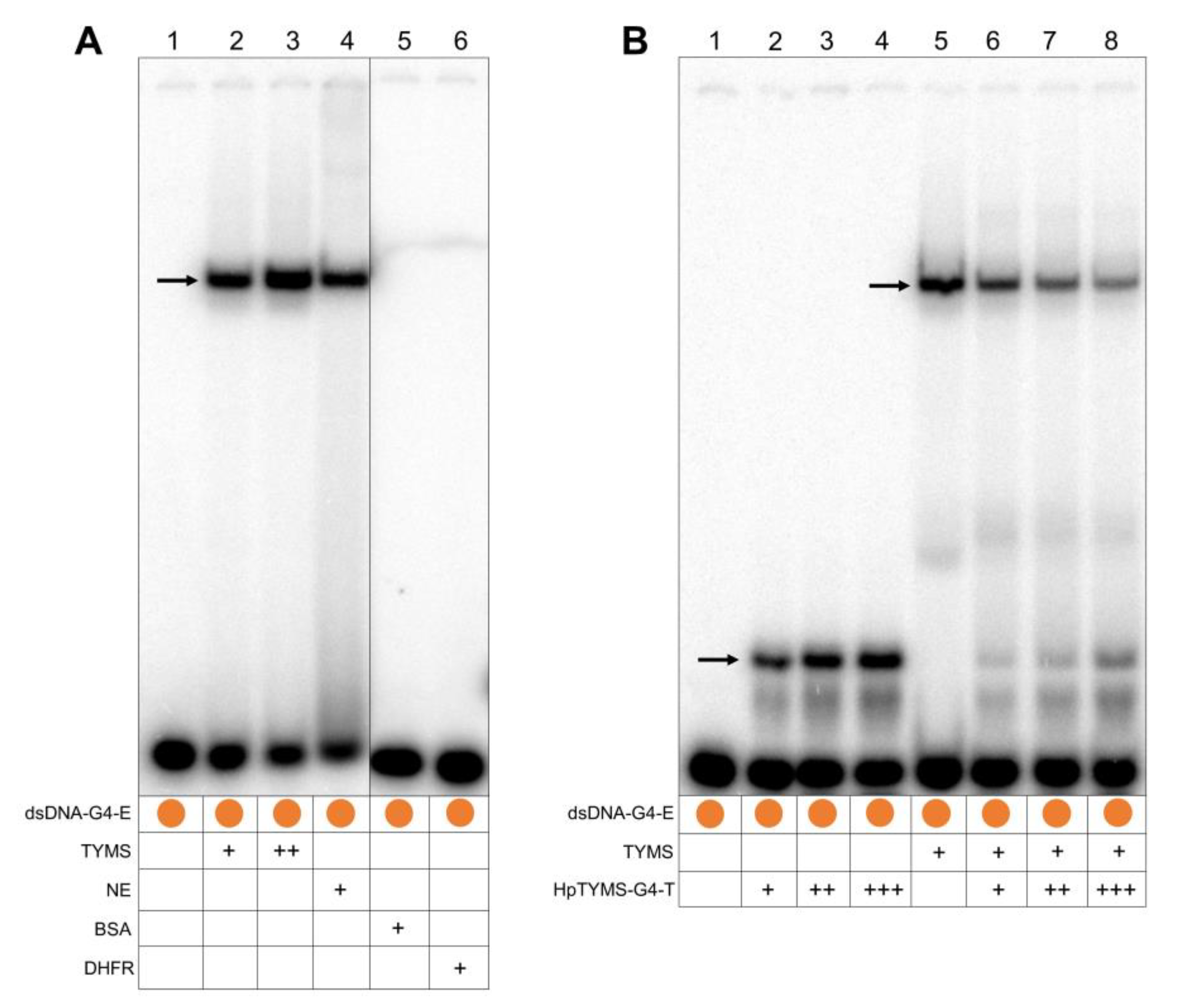
2.4. TYMS Protein and HpTYMS-G4-T Compete to Bind to the G4FS in the dsDNA
2.5. HpTYMS-G4-T Promotes the Formation of the G4 Structure
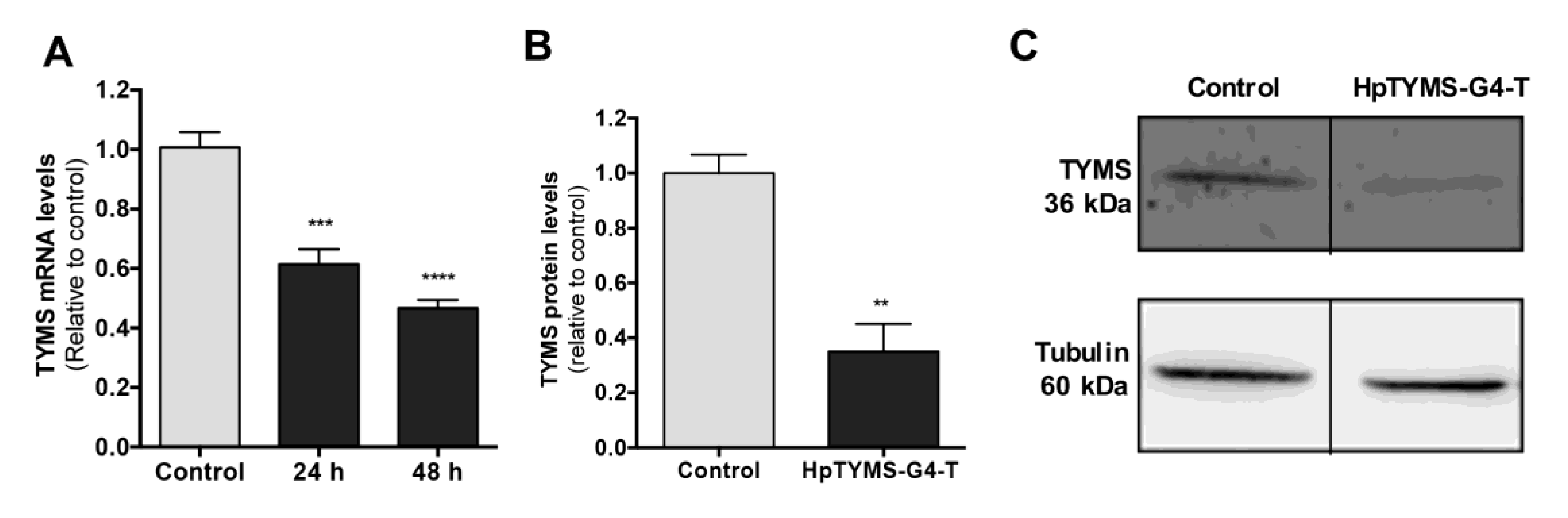
2.6. Effect of HpTYMS-G4-T on the Levels of TYMS mRNA and Protein
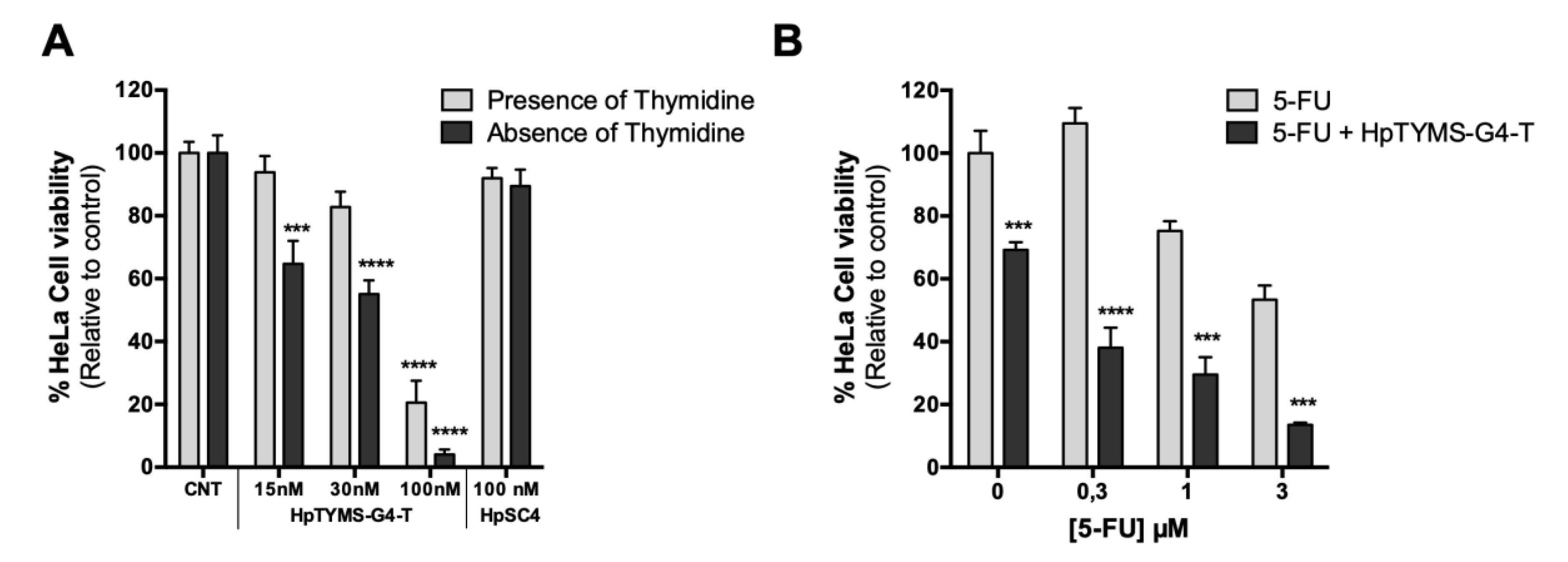
2.7. Effect of HpTYMS-G4-T as a Single Agent or Combined with 5-FU on HeLa Cell Viability
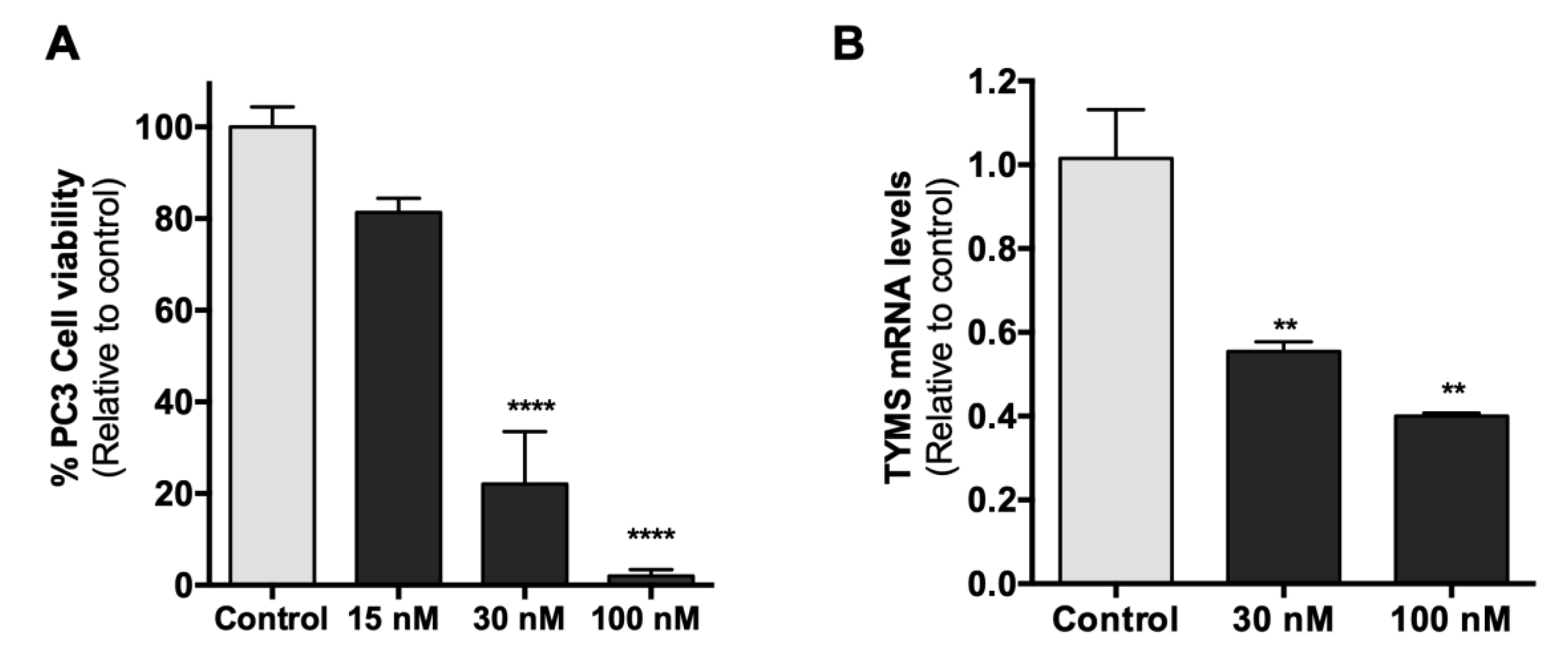
2.8. Effect of HpTYMS-G4-T on PC-3 Cell Viability and mRNA Levels
3. Discussion
4. Materials and Methods
4.1. Bioinformatic Detection of G4-Forming Sequences
4.2. Design of Polypurine Reverse Hoogsteen Hairpins
4.3. RNA and gDNA Extraction
4.4. Reverse Transcription
4.5. PCR
4.6. CD and UV Absorbance Spectroscopy
4.7. Thioflavin T Fluorescence Spectroscopy
4.8. Nuclear Magnetic Resonance
4.9. RT-qPCR
4.10. Electrophoretic Mobility Shift Assay
4.11. Detection of G4 Structures with ThT upon DNA Binding Assays
4.12. Cell Culture
4.13. Transfection of the PPRH
4.14. Western Blot Analyses
4.15. Combination Treatment of 5-FU plus HpTYMS-G4-T
4.16. MTT Assay
4.17. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| TYMS | Thymidylate synthase |
| 5-FU | 5-fluorouracil |
| ASO | Antisense oligodeoxynucleotides |
| G4 | G-quadruplex structures |
| G4FS | G4 forming sequence |
| PPRH | Polypurine reverse Hoogsteen hairpins |
| ThT | Thioflavin T |
| DOTAP | N-(1-(1,2-Di-(9Z-octadecenoyl)-3-trimethylammoniumpropane methyl sulfate |
References
- Carreras, C.W.; Santi, D.V. The Catalytic Mechanism and Structure of Thymidylate Synthase. Annu. Rev. Biochem. 1995, 64, 721–762. [Google Scholar] [CrossRef] [PubMed]
- Rose, M.G.; Farrell, M.P.; Schmitz, J.C. Thymidylate synthase: A critical target for cancer chemotherapy. Clin. Colorectal Cancer 2002, 1, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Gmeiner, W. Novel Chemical Strategies for Thymidylate Synthase Inhibition. Curr. Med. Chem. 2012, 12, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-Fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer. 2003, 3, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Yin, Y.; Xu, S.J.; Chen, W.S. 5-Fluorouracil: Mechanisms of resistance and reversal strategies. Molecules 2008, 13, 1551–1569. [Google Scholar] [CrossRef] [Green Version]
- Berger, S.H.; Jenh, C.H.; Johnson, L.F.; Berger, F.G. Thymidylate synthase overproduction and gene amplification in fluorodeoxyuridine-resistant human cells. Mol. Pharmacol. 1985, 28, 461–467. [Google Scholar]
- Rooney, P.H.; Stevenson, D.A.J.; Marsh, S.; Johnston, P.G.; Haites, N.E.; Cassidy, J.; McLeod, H.L. Comparative genomic hybridization analysis of chromosomal alterations induced by the development of resistance to thymidylate synthase inhibitors. Cancer Res. 1998, 58, 5042–5045. [Google Scholar]
- Chu, E.; Koeller, D.M.; Casey, J.L.; Drake, J.C.; Chabner, B.A.; Elwood, P.C.; Zinn, S.; Allegra, C.J. Autoregulation of human thymidylate synthase messenger RNA translation by thymidylate synthase. Proc. Natl. Acad. Sci. USA 1991, 88, 8977–8981. [Google Scholar] [CrossRef] [Green Version]
- Chu, E.; Voeller, D.; Koeller, D.M.; Drake, J.C.; Takimoto, C.H.; Maley, G.F.; Maley, F.; Allegra, C.J. Identification of an RNA binding site for human thymidylate synthase. Proc. Natl. Acad. Sci. USA 1993, 90, 517–521. [Google Scholar] [CrossRef]
- Garg, D.; Henrich, S.; Salo-Ahen, O.M.H.; Myllykallio, H.; Costi, M.P.; Wade, R.C. Novel approaches for targeting thymidylate synthase to overcome the resistance and toxicity of anticancer drugs. J. Med. Chem. 2010, 53, 6539–6549. [Google Scholar] [CrossRef]
- Brunn, N.D.; Dibrov, S.M.; Kao, M.B.; Ghassemian, M.; Hermann, T. Analysis of mRNA recognition by human thymidylate synthase. Biosci. Rep. 2014, 34, 905–913. [Google Scholar] [CrossRef]
- Tian, T.; Chen, Y.Q.; Wang, S.R.; Zhou, X. G-Quadruplex: A Regulator of Gene Expression and Its Chemical Targeting. Chem 2018, 4, 1314–1344. [Google Scholar] [CrossRef] [Green Version]
- Gellert, M.; Lipsett, M.N.; Davies, D.R. Helix formation by guanylic acid. Proc. Natl. Acad. Sci. USA 1962, 48, 2013–2018. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.T. G-Quartets 40 Years Later: From 5′-GMP to Molecular Biology and Supramolecular Chemistry. Angew. Chem. Int. Ed. 2004, 43, 668–698. [Google Scholar] [CrossRef] [PubMed]
- Lane, A.N.; Chaires, J.B.; Gray, R.D.; Trent, J.O. Stability and kinetics of G-quadruplex structures. Nucleic Acids Res. 2008, 36, 5482–5515. [Google Scholar] [CrossRef]
- Hänsel-Hertsch, R.; di Antonio, M.; Balasubramanian, S. DNA G-quadruplexes in the human genome: Detection, functions and therapeutic potential. Nat. Rev. Mol. Cell Biol. 2017, 18, 279–284. [Google Scholar] [CrossRef]
- Rhodes, D.; Lipps, H.J. Survey and summary G-quadruplexes and their regulatory roles in biology. Nucleic Acids Res. 2015, 43, 8627–8637. [Google Scholar] [CrossRef] [Green Version]
- Bugaut, A.; Balasubramanian, S. 5′-UTR RNA G-quadruplexes: Translation regulation and targeting. Nucleic Acids Res. 2012, 40, 4727–4741. [Google Scholar] [CrossRef]
- Song, J.; Perreault, J.-P.; Topisirovic, I.; Richard, S. RNA G-quadruplexes and their potential regulatory roles in translation. Translation 2016, 4, e1244031. [Google Scholar] [CrossRef] [Green Version]
- Chambers, V.S.; Marsico, G.; Boutell, J.M.; di Antonio, M.; Smith, G.P.; Balasubramanian, S. High-throughput sequencing of DNA G-quadruplex structures in the human genome. Nat. Biotechnol. 2015, 33, 877–881. [Google Scholar] [CrossRef] [Green Version]
- Ciudad, C.J.; Rodríguez, L.; Villalobos, X.; Félix, A.J.; Noé, V. Polypurine Reverse Hoogsteen Hairpins as a Gene Silencing Tool for Cancer. Curr. Med. Chem. 2017, 24, 2809–2826. [Google Scholar] [CrossRef] [PubMed]
- De Almagro, M.C.; Coma, S.; Noé, V.; Ciudad, C.J. Polypurine hairpins directed against the template strand of DNA knock down the expression of mammalian genes. J. Biol. Chem. 2009, 284, 11579–11589. [Google Scholar] [CrossRef] [PubMed]
- Goñi, J.R.; de la Cruz, X.; Orozco, M. Triplex-forming oligonucleotide target sequences in the human genome. Nucleic Acids Res. 2004, 32, 354–360. [Google Scholar] [CrossRef]
- Rodríguez, L.; Villalobos, X.; Dakhel, S.; Padilla, L.; Hervas, R.; Hernández, J.L.; Ciudad, C.J.; Noé, V. Polypurine reverse Hoogsteen hairpins as a gene therapy tool against survivin in human prostate cancer PC3 cells in vitro and in vivo. Biochem. Pharmacol. 2013, 86, 1541–1554. [Google Scholar] [CrossRef]
- De Almagro, M.C.; Mencia, N.; Noé, V.; Ciudad, C.J. Coding polypurine hairpins cause target-induced cell death in breast cancer cells. Hum. Gene Ther. 2011, 22, 451–463. [Google Scholar] [CrossRef]
- Villalobos, X.; Rodríguez, L.; Solé, A.; Lliberós, C.; Mencia, N.; Ciudad, C.J.; Noé, V. Effect of polypurine reverse hoogsteen hairpins on relevant cancer target genes in different human cell lines. Nucleic Acid Ther. 2015, 25, 198–208. [Google Scholar] [CrossRef]
- Aubets, E.; Noé, V.; Ciudad, C.J. Targeting replication stress response using polypurine reverse hoogsteen hairpins directed against WEE1 and CHK1 genes in human cancer cells. Biochem. Pharmacol. 2020, 175, 113911. [Google Scholar] [CrossRef] [PubMed]
- Bener, G.; Félix, A.J.; de Diego, C.S.; Fabregat, I.P.; Ciudad, C.J.; Noé, V. Silencing of CD47 and SIRPα by Polypurine reverse Hoogsteen hairpins to promote MCF-7 breast cancer cells death by PMA-differentiated THP-1 cells. BMC Immunol. 2016, 17, 32. [Google Scholar] [CrossRef]
- Ciudad, C.J.; Enriquez, M.M.M.; Félix, A.J.; Bener, G.; Noé, V. Silencing PD-1 and PD-L1: The potential of PolyPurine Reverse Hoogsteen hairpins for the elimination of tumor cells. Immunotherapy 2019, 11, 369–372. [Google Scholar] [CrossRef]
- Enríquez, M.M.M.; Félix, A.J.; Ciudad, C.J.; Noé, V. Cancer immunotherapy using PolyPurine Reverse Hoogsteen hairpins targeting the PD-1/PD-L1 pathway in human tumor cells. PLoS ONE. 2018, 13, e0206818. [Google Scholar]
- Mencia, N.; Selga, E.; Noé, V.; Ciudad, C.J. Underexpression of miR-224 in methotrexate resistant human colon cancer cells. Biochem. Pharm. 2011, 82, 1572–1582. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, S.; Nalbantoglu, J.; Takeishi, K.; Shimizu, K.; Gotoh, O.; Seno, T.; Ayusawa, D. Structural and functional analysis of the human thymidylate synthase gene. J. Biol. Chem. 1990, 265, 20277–20284. [Google Scholar]
- Gaddis, S.S.; Wu, Q.; Thames, H.D.; Digiovanni, J.; Walborg, E.F.; Macleod, M.C.; Vasquez, K.M. A web-based search engine for triplex-forming oligonucleotide target sequences. Oligonucleotides 2006, 16, 196–201. [Google Scholar] [CrossRef]
- Mergny, J.-L.; Phan, A.-T.; Lacroix, L. Following G-quartet formation by UV-spectroscopy. FEBS Lett. 1998, 435, 74–78. [Google Scholar] [CrossRef]
- Paramasivan, S.; Rujan, I.; Bolton, P.H. Circular dichroism of quadruplex DNAs: Applications to structure, cation effects and ligand binding. Methods 2007, 43, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Mergny, J.-L.; Li, J.; Lacroix, L.; Amrane, S.; Chaires, J.B. Thermal difference spectra: A specific signature for nucleic acid structures. Nucleic Acids Res. 2005, 33, e138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kettani, A.; Kumar, R.A.; Patel, D.J. Solution structure of a DNA quadruplex containing the fragile X syndrome triplet repeat. J. Mol. Biol. 1995, 254, 638–656. [Google Scholar] [CrossRef]
- Mohanty, J.; Barooah, N.; Dhamodharan, V.; Harikrishna, S.; Pradeepkumar, P.I.; Bhasikuttan, A.C. Thioflavin T as an Efficient Inducer and Selective Fluorescent Sensor for the Human Telomeric G-Quadruplex DNA. J. Am. Chem. Soc. 2013, 135, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Maiti, S.; Chaudhury, N.K.; Chowdhury, S. Hoechst 33258 binds to G-quadruplex in the promoter region of human c-myc. Biochem. Biophys. Res. Commun. 2003, 310, 505–512. [Google Scholar] [CrossRef]
- De la Faverie, A.R.; Guédin, A.; Bedrat, A.; Yatsunyk, L.A.; Mergny, J.L. Thioflavin T as a fluorescence light-up probe for G4 formation. Nucleic Acids Res. 2014, 42, e65. [Google Scholar] [CrossRef]
- Solé, A.; Delagoutte, E.; Ciudad, C.J.; Noé, V.; Alberti, P. Polypurine reverse-Hoogsteen (PPRH) oligonucleotides can form triplexes with their target sequences even under conditions where they fold into G-quadruplexes. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Chou, T.-C. Drug Combination Studies and Their Synergy Quantification Using the Chou-Talalay Method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Ligabue, A.; Marverti, G.; Liebl, U.; Myllykallio, H. Transcriptional Activation and Cell Cycle Block Are the Keys for 5-Fluorouracil Induced Up-Regulation of Human Thymidylate Synthase Expression. PLoS ONE 2012, 7, e47318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, G.J.; Backus, H.H.J.; Freemantle, S.; van Triest, B.; Codacci-Pisanelli, G.; van der Wilt, C.L.; Smid, K.; Lunec, J.; Calvert, A.H.; Marsh, S.; et al. Induction of thymidylate synthase as a 5-fluorouracil resistance mechanism. Biochim. Biophys. Acta Mol. Basis Dis. 2002, 1587, 194–205. [Google Scholar] [CrossRef]
- Peters, G.J.; van Triest, B.; Backus, H.H.J.; Kuiper, C.M.; van der Wilt, C.L.; Pinedo, H.M. Molecular downstream events and induction of thymidylate synthase in mutant and wild-type p53 colon cancer cell lines after treatment with 5-fluorouracil and the thymidylate synthase inhibitor raltitrexed. Eur. J. Cancer 2000, 36, 916–924. [Google Scholar] [CrossRef]
- Kikin, O.; D’Antonio, L.; Bagga, P.S. QGRS Mapper: A web-based server for predicting G-quadruplexes in nucleotide sequences. Nucleic Acids Res. 2006, 34, W676–W682. [Google Scholar] [CrossRef]
- Chu, E.; Takimoto, C.H.; Voeller, D.; Grem, J.L.; Allegra, C.J. Specific Binding of Human Dihydrofolate Reductase Protein to Dihydrofolate Reductase Messenger RNA in Vitro. Biochemistry 1993, 32, 4756–4760. [Google Scholar] [CrossRef]
- Bhardwaj, A.; Wilkinson, M.F. A metabolic enzyme doing double duty as a transcription factor. BioEssays 2005, 27, 467–471. [Google Scholar] [CrossRef]
- Zheng, L.; Roeder, R.G.; Luo, Y. S Phase Activation of the Histone H2B Promoter by OCA-S, a Coactivator Complex that Contains GAPDH as a Key Component. Cell 2003, 114, 255–266. [Google Scholar] [CrossRef] [Green Version]
- Chu, E.; Takechi, T.; Jones, K.L.; Voeller, D.M.; Copur, S.M.; Maley, G.F.; Maley, F.; Segal, S.; Allegra, C.J. Thymidylate synthase binds to c-myc RNA in human colon cancer cells and in vitro. Mol. Cell. Biol. 1995, 15, 179–185. [Google Scholar] [CrossRef] [Green Version]
- Ju, J.; Pedersen-Lane, J.; Maley, F.; Chu, E. Regulation of p53 expression by thymidylate synthase. Proc. Natl. Acad. Sci. USA 1999, 96, 3769–3774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, E.; Copur, S.M.; Ju, J.; Chen, T.; Khleif, S.; Voeller, D.M.; Mizunuma, N.; Patel, M.; Maley, G.F.; Maley, F.; et al. Thymidylate Synthase Protein and p53 mRNA Form an In Vivo Ribonucleoprotein Complex. Mol. Cell. Biol. 1999, 19, 1582–1594. [Google Scholar] [CrossRef]
- Chu, E. Identification of in vivo target RNA sequences bound by thymidylate synthase. Nucleic Acids Res. 1996, 24, 3222–3228. [Google Scholar] [CrossRef] [Green Version]
- Cogoi, S.; Shchekotikhin, A.E.; Xodo, L.E. HRAS is silenced by two neighboring G-quadruplexes and activated by MAZ, a zinc-finger transcription factor with DNA unfolding property. Nucleic Acids Res. 2014, 42, 8379–8388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Su, L.; Qiu, J.; Xiao, N.; Lin, J.; Tan, J.H.; Ou, T.M.; Gu, L.Q.; Huang, Z.S.; Li, D. Mechanistic studies for the role of cellular nucleic-acid-binding protein (CNBP) in regulation of c-myc transcription. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 4769–4777. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Bugaut, A.; Huppert, J.L.; Balasubramanian, S. An RNA G-quadruplex in the 5′ UTR of the NRAS proto-oncogene modulates translation. Nat. Chem. Biol. 2007, 3, 218–221. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.Y.; Vankayalapati, H.; Shin-Ya, K.; Wierzba, K.; Hurley, L.H. Telomestatin, a potent telomerase inhibitor that interacts quite specifically with the human telomeric intramolecular G-quadruplex. J. Am. Chem. Soc. 2002, 124, 2098–2099. [Google Scholar] [CrossRef]
- Zahler, A.M.; Williamson, J.R.; Cech, T.R.; Prescott, D.M. Inhibition of telomerase by G-quartet DMA structures. Nature 1991, 350, 718–720. [Google Scholar] [CrossRef]
- Asamitsu, S.; Obata, S.; Yu, Z.; Bando, T.; Sugiyama, H. Recent progress of targeted G-quadruplex-preferred ligands toward cancer therapy. Molecules 2019, 24, 429. [Google Scholar] [CrossRef]
- Siddiqui-Jain, A.; Grand, C.L.; Bearss, D.J.; Hurley, L.H. Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc. Natl. Acad. Sci. USA 2002, 99, 11593–11598. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.K.; Wu, Y.; Wang, X.Q.; Kuang, G.T.; Zhang, Q.; Lin, S.L.; Liu, H.Y.; Tan, J.H.; Huang, Z.S.; Ou, T.M. Discovery of Small Molecules for Repressing Cap-Independent Translation of Human Vascular Endothelial Growth Factor (hVEGF) as Novel Antitumor Agents. J. Med. Chem. 2017, 60, 5306–5319. [Google Scholar] [CrossRef]
- Beaudoin, J.D.; Perreault, J.P. Exploring mRNA 3′-UTR G-quadruplexes: Evidence of roles in both alternative polyadenylation and mRNA shortening. Nucleic Acids Res. 2013, 41, 5898–5911. [Google Scholar] [CrossRef] [PubMed]
- Che, T.; Wang, Y.Q.; Huang, Z.L.; Tan, J.H.; Huang, Z.S.; Chen, S.B. Natural alkaloids and heterocycles as G-quadruplex ligands and potential anticancer agents. Molecules 2018, 23, 493. [Google Scholar]
- Xu, H.; di Antonio, M.; McKinney, S.; Mathew, V.; Ho, B.; O’Neil, N.J.; Santos, N.D.; Silvester, J.; Wei, V.; Garcia, J.; et al. CX-5461 is a DNA G-quadruplex stabilizer with selective lethality in BRCA1/2 deficient tumours. Nat. Commun. 2017, 8, 14432. [Google Scholar] [CrossRef]
- Khot, A.; Brajanovski, N.; Cameron, D.P.; Hein, N.; Maclachlan, K.H.; Sanij, E.; Lim, J.; Soong, J.; Link, E.; Blombery, P.; et al. First-in-human RNA polymerase I transcription inhibitor CX-5461 in patients with advanced hematologic cancers: Results of a phase I dose-escalation study. Cancer Discov. 2019, 9, 1036–1049. [Google Scholar] [CrossRef]
- Drygin, D.; Siddiqui-Jain, A.; O’Brien, S.; Schwaebe, M.; Lin, A.; Bliesath, J.; Ho, C.B.; Proffitt, C.; Trent, K.; Whitten, J.P.; et al. Anticancer activity of CX-3543: A direct inhibitor of rRNA biogenesis. Cancer Res. 2009, 69, 7653–7661. [Google Scholar] [CrossRef] [PubMed]
- Local, A.; Zhang, H.; Benbatoul, K.D.; Folger, P.; Sheng, X.; Tsai, C.Y.; Howell, S.B.; Rice, W.G. APTO-253 stabilizes G-quadruplex DNA, inhibits MYC expression, and induces DNA damage in acute myeloid leukemia cells. Mol. Cancer Ther. 2018, 17, 1177–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huppert, J.L.; Balasubramanian, S. Prevalence of quadruplexes in the human genome. Nucleic Acids Res. 2005, 33, 2908–2916. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, J.C.; Chen, T.M.; Chu, E. Small Interfering Double-Stranded RNAs as Therapeutic Molecules to Restore Chemosensitivity to Thymidylate Synthase Inhibitor Compounds. Cancer Res. 2004, 64, 1431–1435. [Google Scholar] [CrossRef] [Green Version]
- Jason, T.L.H.; Berg, R.W.; Vincent, M.D.; Koropatnick, J. Antisense targeting of Thymidylate Synthase (TS) mRNA increases TS gene transcription and TS protein: Effects on human tumor cell sensitivity to TS enzyme-inhibiting drugs. Gene Expr. 2007, 13, 227–239. [Google Scholar] [CrossRef]
- Ferguson, P.J.; Collins, O.; Dean, N.M.; DeMoor, J.; Sha-Li, C.; Vincent, M.D.; Koropatnick, J. Antisense down-regulation of thymidylate synthase to suppress growth and enhance cytotoxicity of 5-FUdR, 5-FU and Tomudex in HeLa cells. Br. J. Pharmacol. 1999, 127, 1777–1786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berg, R.W.; Ferguson, P.J.; Vincent, M.D.; Koropatnick, D.J. A “combination oligonucleotide” antisense strategy to downregulate thymidylate synthase and decrease tumor cell growth and drug resistance. Cancer Gene Ther. 2003, 10, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Niu, R.L.; Wang, Z.; Lin, X.K. In vitro selected peptides bind with thymidylate synthase mRNA and inhibit its translation. Sci. China Ser. C Life Sci. 2007, 50, 630–636. [Google Scholar] [CrossRef]
- Villalobos, X.; Rodríguez, L.; Prévot, J.; Oleaga, C.; Ciudad, C.J.; Noé, V. Stability and immunogenicity properties of the gene-silencing polypurine reverse hoogsteen hairpins. Mol. Pharm. 2014, 11, 254–264. [Google Scholar] [CrossRef]
- Félix, A.J.; Ciudad, C.J.; Noé, V. Functional pharmacogenomics and toxicity of PolyPurine Reverse Hoogsteen hairpins directed against survivin in human cells. Biochem. Pharmacol. 2018, 155, 8–20. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence (5’-3’) | Length | Assay |
|---|---|---|---|
| 5’UTR-TYMS-Fw | GAGCAGGAAGAGGCGGAGCG | 20 | PCR |
| 5’UTR-TYMS-Rv | GCAGCTCCGAGCCGGCCACAGG | 22 | PCR |
| APRT-Ex1-Fw | CACCCCAGGCGTGGTATTCA | 20 | PCR |
| APRT-Ex2-Rv | CTGCGATGTAGTCGATGCGG | 20 | PCR |
| TYMS-Fw | CCTCGGTGTGCCTTTCAACATC | 22 | qPCR |
| TYMS-Rv | GGTCTGGGTTCTCGCTGAAGC | 21 | qPCR |
| PPIB-Fw | GGAGATGGCACAGGAGGAAA | 20 | qPCR |
| PPIB-Rv | CGTAGTGCTTCAGTTTGAAGTTCTCA | 26 | qPCR |
| RNA-G4 | GGGAAAAGGCGCGCGGAAGGGG | 22 | EMSA, CD, UV, Flu |
| ssDNA-G4-Fw-E | GGGAAAAGGCGCGCGGAAGGGG | 22 | EMSA |
| ssDNA-G4-Rv-E | CCCCTTCCGCGCGCCTTTTCCC | 22 | EMSA |
| ssDNA-G4-Fw | CGGGAAAAGGCGCGCGGAAGGGGT | 24 | TGS, CD, UV, Flu, NMR |
| ssDNA-G4-Rv | ACCCCTTCCGCGCGCCTTTTCCCG | 24 | TGS |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aubets, E.; J. Félix, A.; Garavís, M.; Reyes, L.; Aviñó, A.; Eritja, R.; Ciudad, C.J.; Noé, V. Detection of a G-Quadruplex as a Regulatory Element in Thymidylate synthase for Gene Silencing Using Polypurine Reverse Hoogsteen Hairpins. Int. J. Mol. Sci. 2020, 21, 5028. https://doi.org/10.3390/ijms21145028
Aubets E, J. Félix A, Garavís M, Reyes L, Aviñó A, Eritja R, Ciudad CJ, Noé V. Detection of a G-Quadruplex as a Regulatory Element in Thymidylate synthase for Gene Silencing Using Polypurine Reverse Hoogsteen Hairpins. International Journal of Molecular Sciences. 2020; 21(14):5028. https://doi.org/10.3390/ijms21145028
Chicago/Turabian StyleAubets, Eva, Alex J. Félix, Miguel Garavís, Laura Reyes, Anna Aviñó, Ramón Eritja, Carlos J. Ciudad, and Véronique Noé. 2020. "Detection of a G-Quadruplex as a Regulatory Element in Thymidylate synthase for Gene Silencing Using Polypurine Reverse Hoogsteen Hairpins" International Journal of Molecular Sciences 21, no. 14: 5028. https://doi.org/10.3390/ijms21145028