Spinal Cord Injury: Pathophysiology, Multimolecular Interactions, and Underlying Recovery Mechanisms

 ,
,  , ,
, ,
Abstract
:
1. Introduction
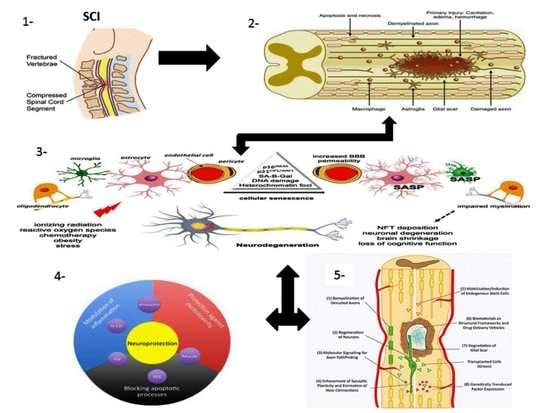
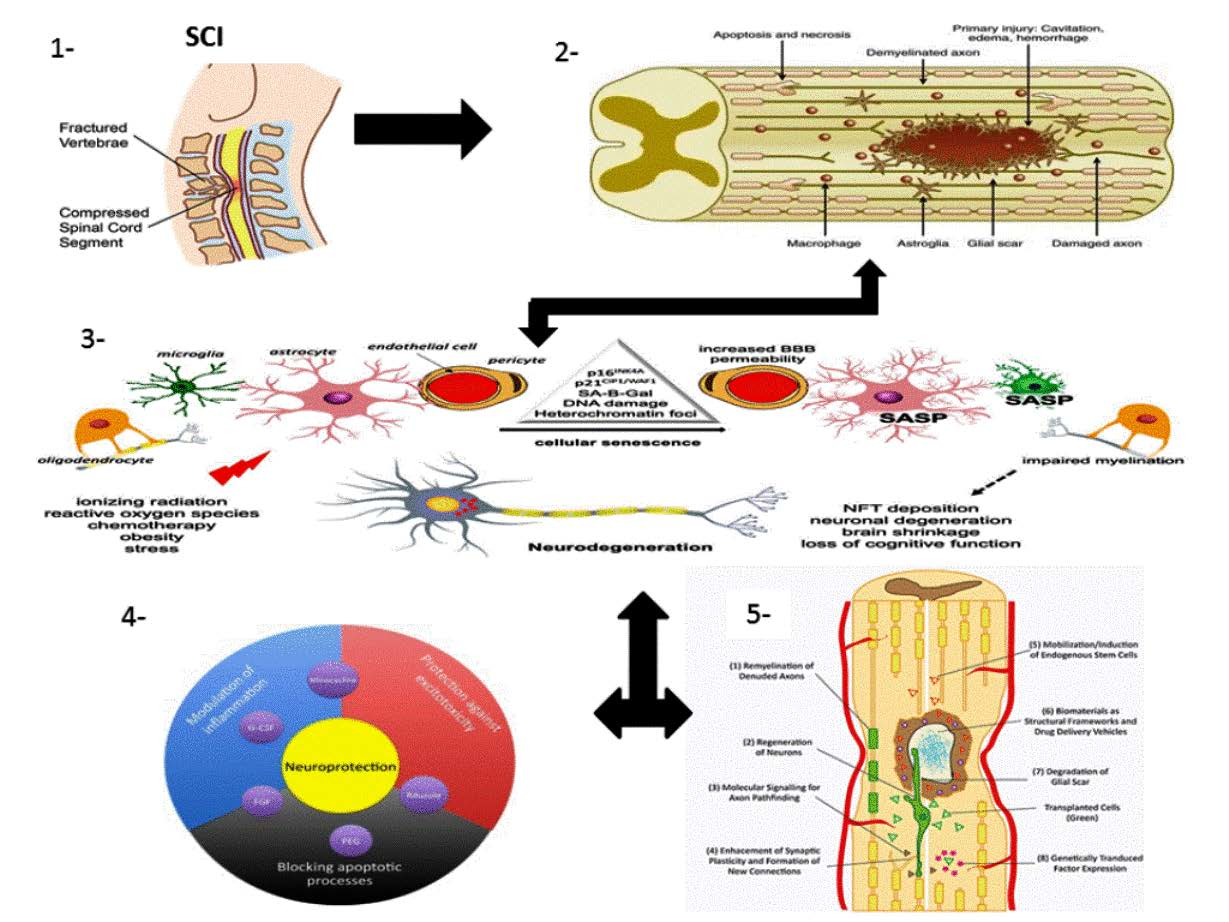
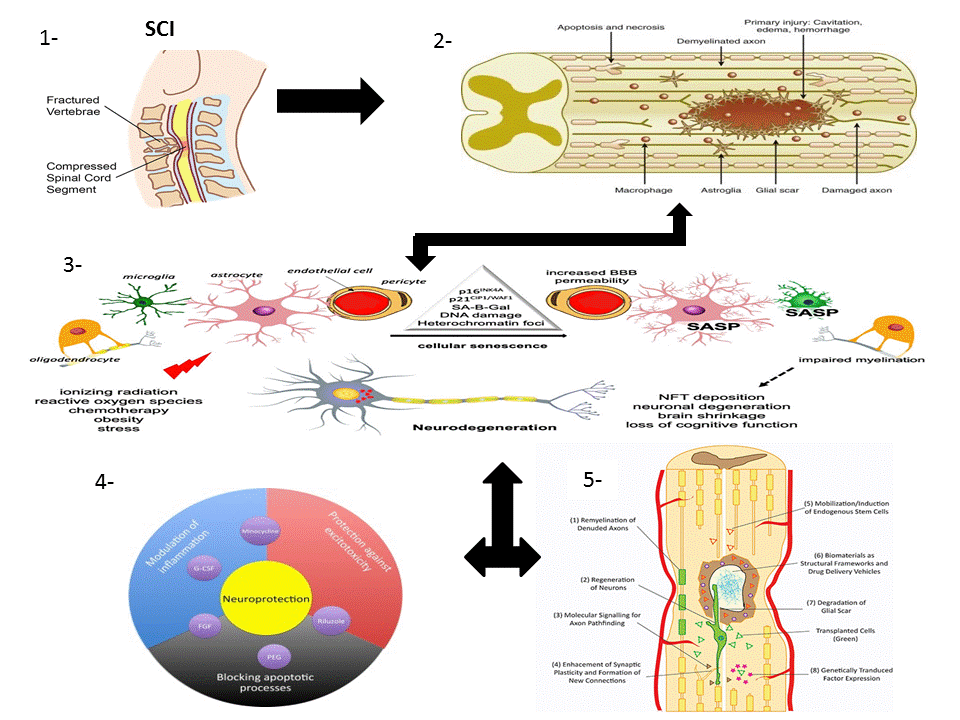
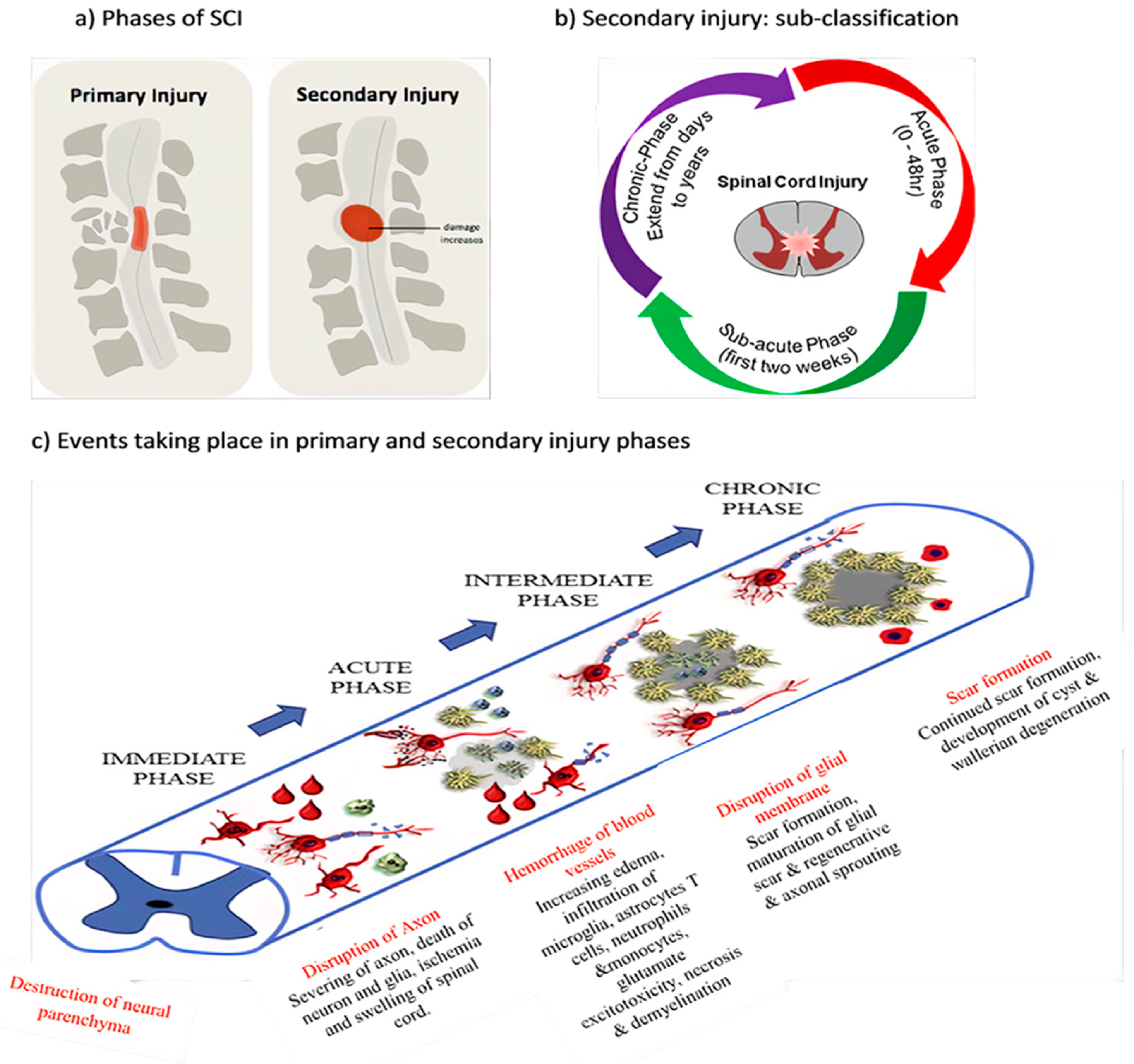
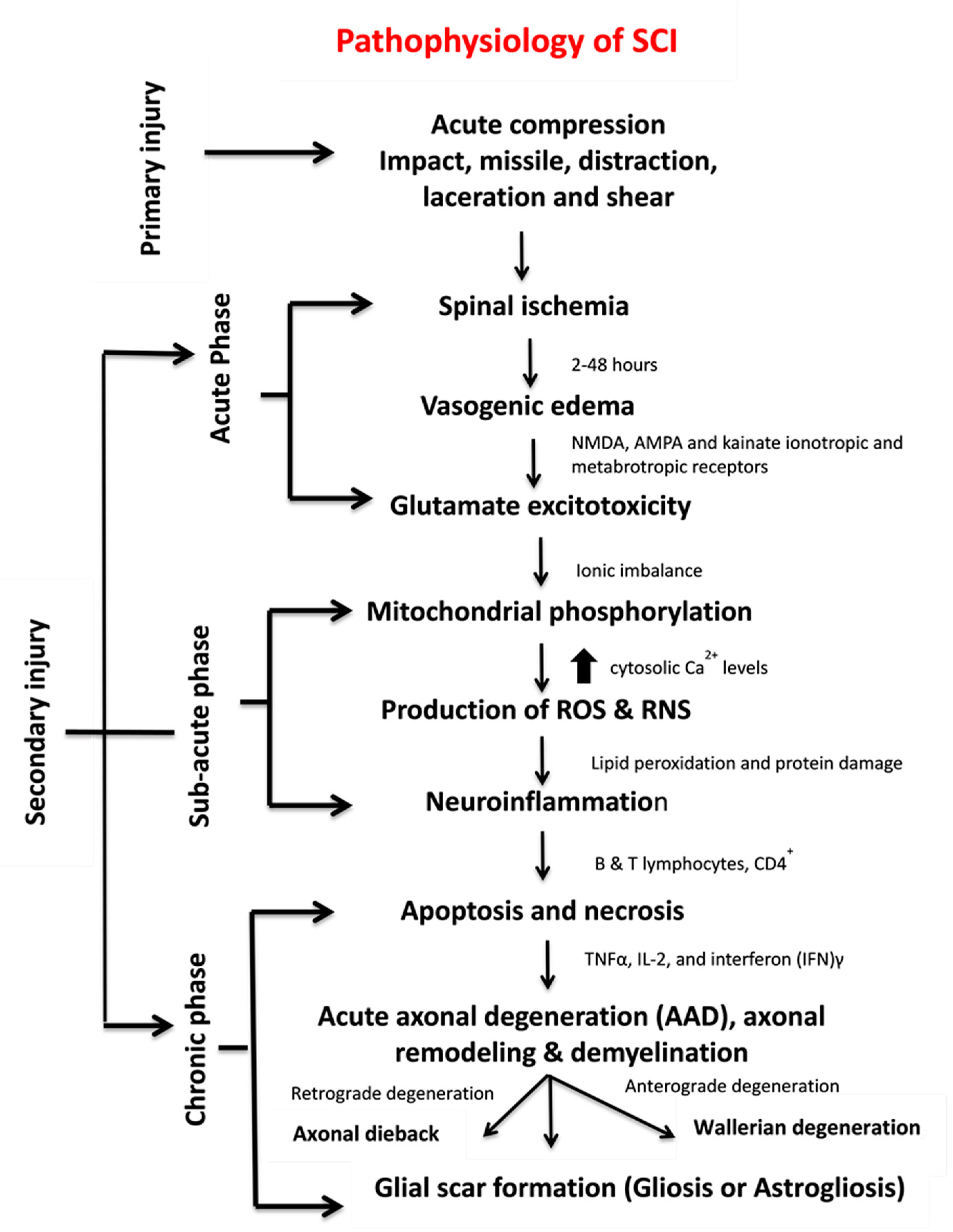
1.1. SCI Phases
1.1.1. Primary Injury
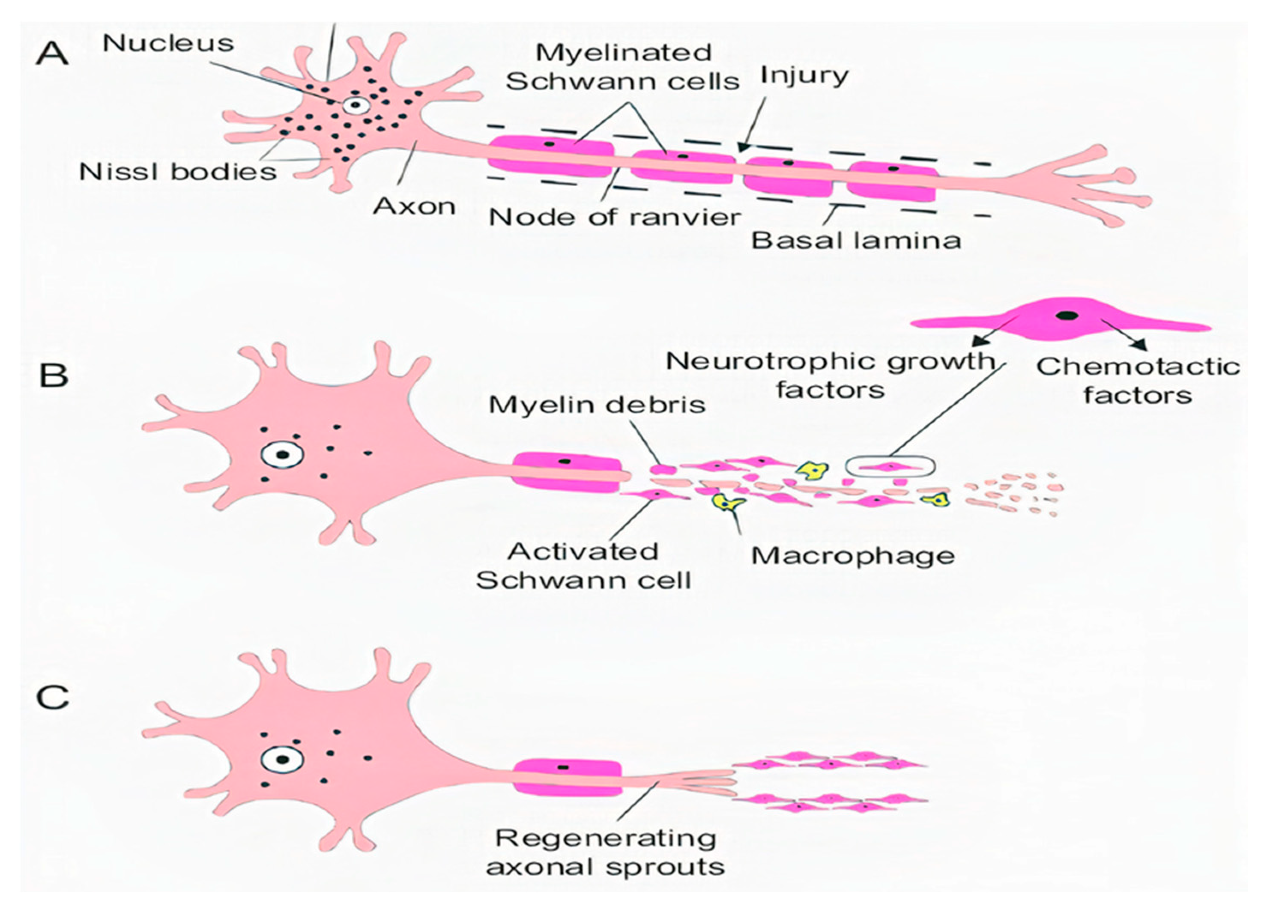
1.1.2. Secondary Injury
1.2. Pathophysiology of SCI
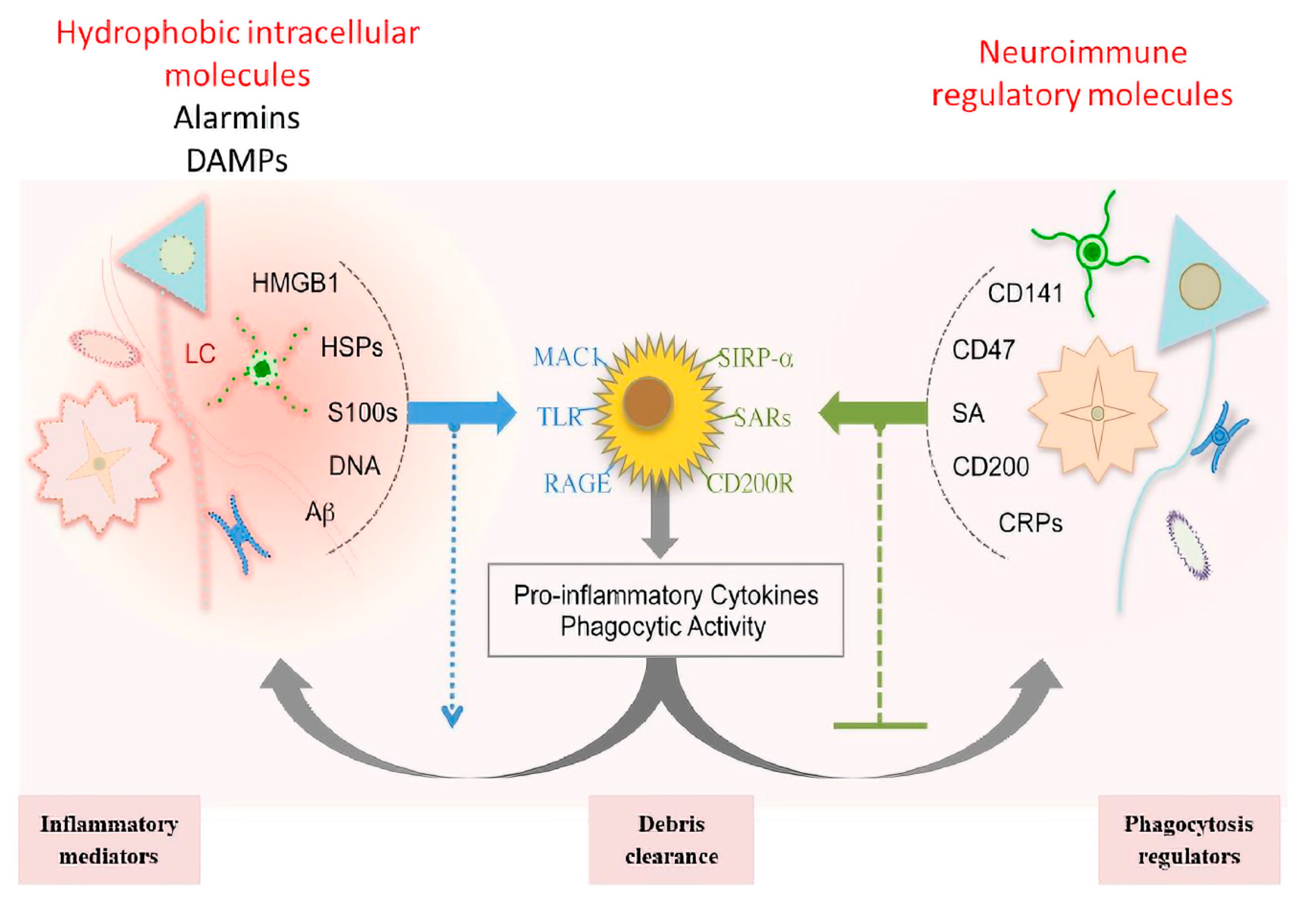
1.3. Multicellular and Multi-Molecular Interactions
1.3.1. Fibrotic Scar
1.3.2. Astroglial Scar
1.3.3. Perilesion Perimeters
1.4. Mechanism of Spinal Cord Recovery Pathways
1.4.1. Neuroprotective Pathways
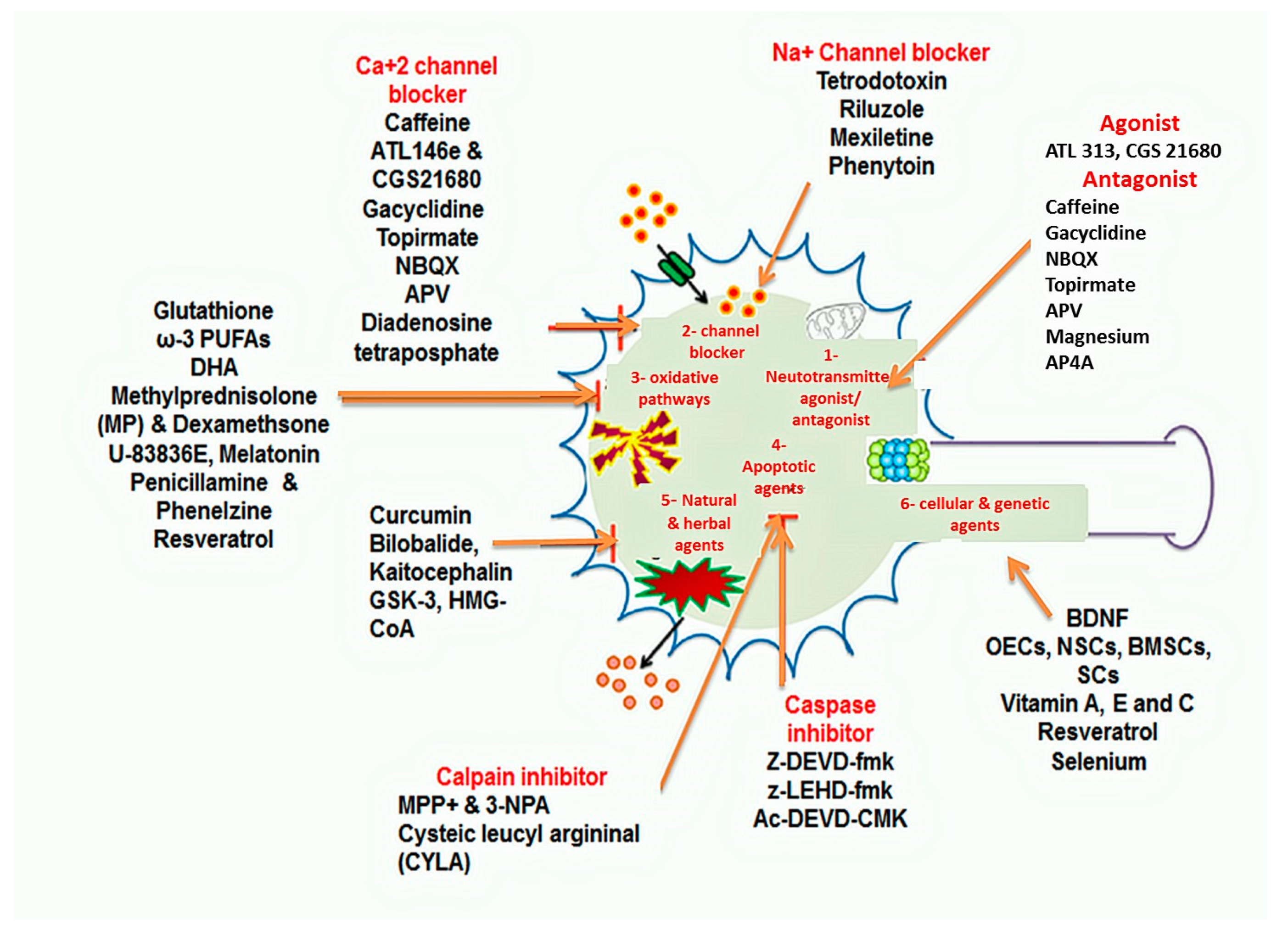
1.4.2. Pharmacological Approaches
Neurotransmitter Agonist and Receptor Antagonist
Channel Blockers
Anti-Oxidative Therapies
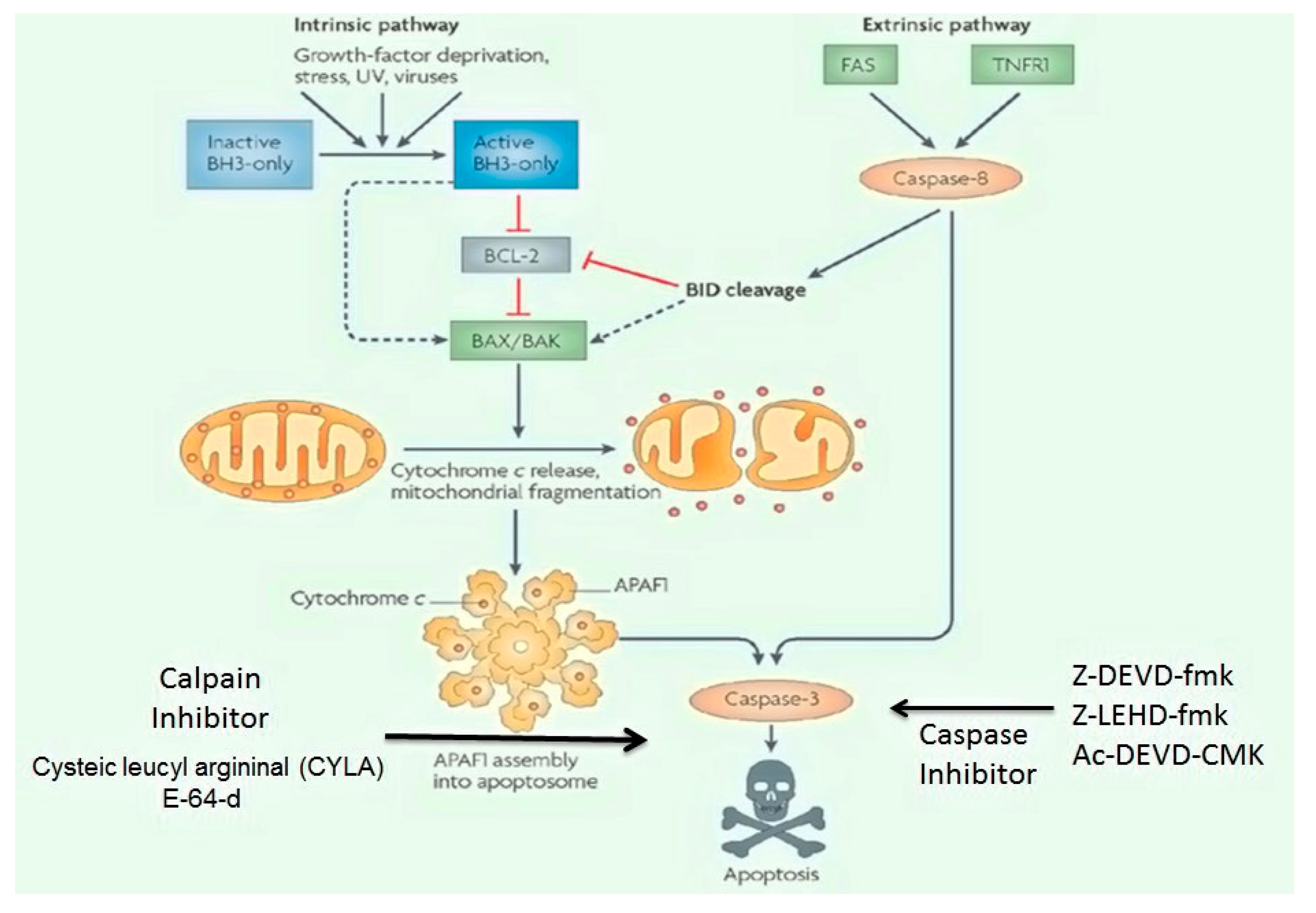
Apoptosis-Related Signaling Pathways Inhibitors
Herbal and Natural Agents
1.4.3. Non-Pharmacological Approaches
1.4.4. Cellular and Genetic Approaches
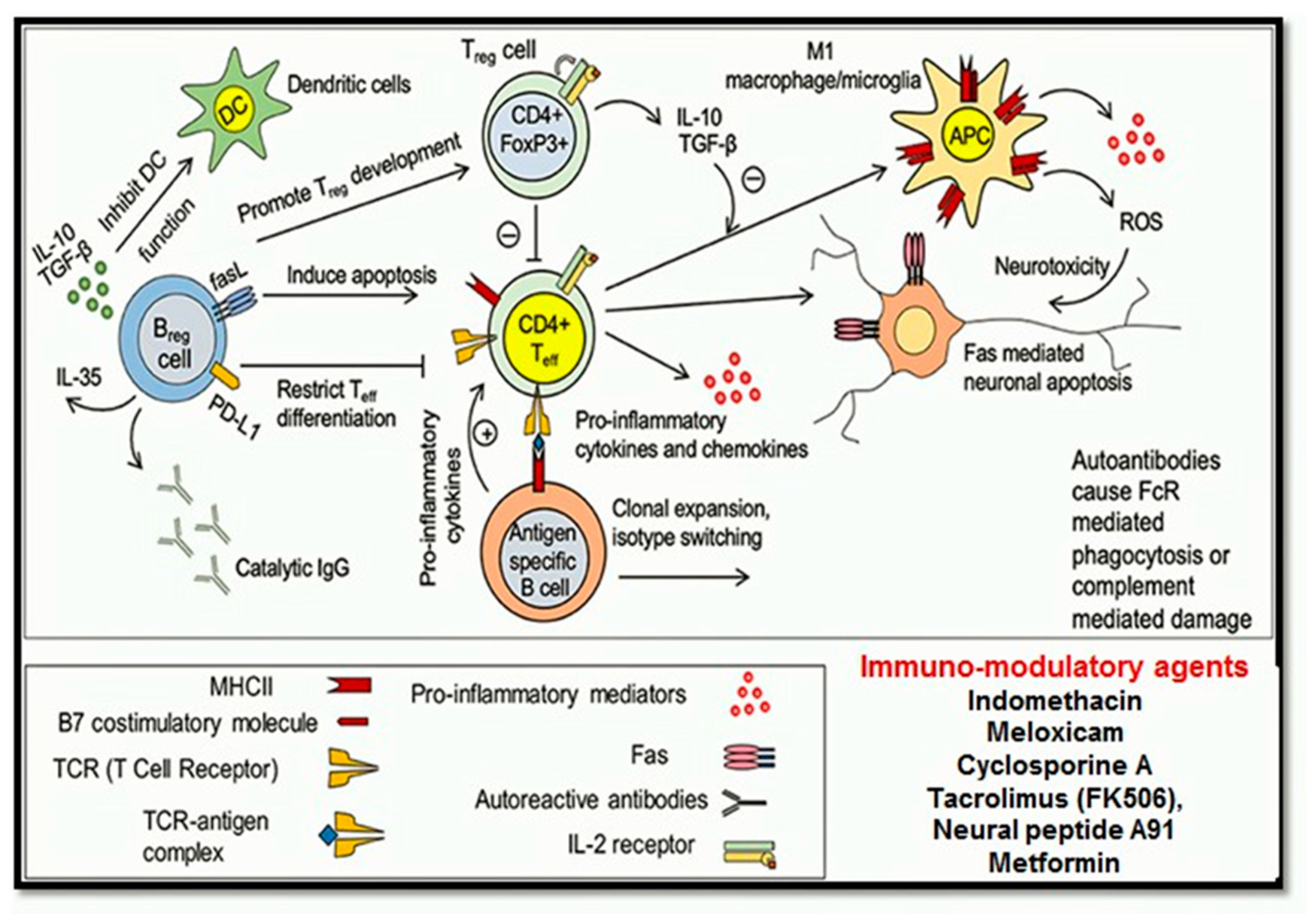
1.4.5. Immuno-Modulatory Pathways
Neuroinflammation
Immunosuppressive or Immunomodulatory Drugs
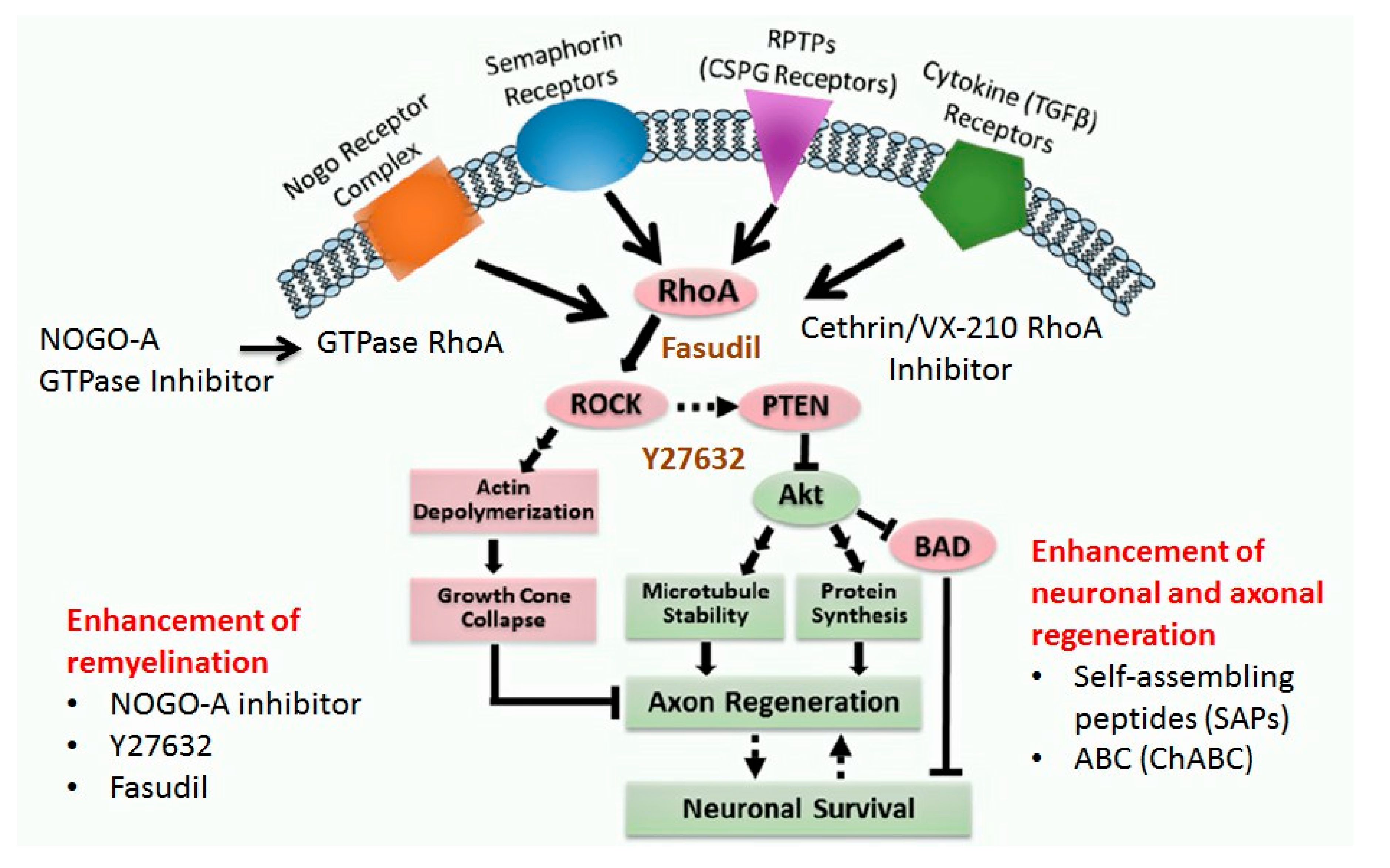
1.4.6. Neuro-Regenerative Pathways
RhoA-ROCK Kinase Pathway
1.4.7. Neuro-Regenerative Approaches
2. Discussion
3. Conclusions
Funding
Conflicts of Interest
References
- Khorasanizadeh, M.; Yousefifard, M.; Eskian, M.; Lu, Y.; Chalangari, M.; Harrop, J.S.; Rahimi-Movaghar, V. Neurological recovery following traumatic spinal cord injury: A systematic review and meta-analysis. J. Neurosurg. 2019, 30, 683–699. [Google Scholar] [CrossRef] [PubMed]
- Katoh, H.; Yokota, K.; Fehlings, M.G. Regeneration of spinal cord connectivity through stem cell transplantation and biomaterial scaffolds. Front. Cell. Neurosci. 2019, 13, 248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Htwe, O.; Hussain, R.I.; Naicker, A.S. Challenges in Managing Severe Lower Limb Spasticity Associated with Bilateral Hip Joints Subluxation. Eur. J. Gen. Med. 2016, 13, 165–167. [Google Scholar]
- Ohnmar, H.; Das, S.; Naicker, A.S. An interesting case of Autonomic Dysreflexia. Clin. Ter. 2009, 160, 371. [Google Scholar] [PubMed]
- O’Shea, T.M.; Burda, J.E.; Sofroniew, M.V. Cell biology of spinal cord injury and repair. J. Clin. Investig. 2017, 127, 3259–3270. [Google Scholar] [CrossRef]
- Couillard-Despres, S.; Bieler, L.; Vogl, M. Pathophysiology of traumatic spinal cord injury. Neurological Aspects of Spinal Cord Injury; Springer: Cham, Switzerland, 2017; pp. 503–528. [Google Scholar]
- Dimitrijevic, M.R.; Danner, S.M.; Mayr, W. Neurocontrol of movement in humans with spinal cord injury. Artif. Organs 2017, 39, 823–833. [Google Scholar] [CrossRef]
- Turtle, J.D.; Henwood, M.K.; Strain, M.M.; Huang, Y.J.; Miranda, R.C.; Grau, J.W. Engaging pain fibers after a spinal cord injury fosters hemorrhage and expands the area of secondary injury. Exp. Neurol. 2019, 311, 115–124. [Google Scholar] [CrossRef]
- Alizadeh, A.; Dyck, S.M.; Karimi-Abdolrezaee, S. Traumatic spinal cord injury: An overview of pathophysiology, models and acute injury mechanisms. Front. Neurol. 2019, 10, 282. [Google Scholar] [CrossRef] [Green Version]
- Tran, A.P.; Warren, P.M.; Silver, J. The biology of regeneration failure and success after spinal cord injury. Physiol. Rev. 2018, 98, 881–917. [Google Scholar] [CrossRef]
- Jha, R.M.; Kochanek, P.M.; Simard, J.M. Pathophysiology and treatment of cerebral edema in traumatic brain injury. Neuropharmacology 2019, 145, 230–246. [Google Scholar] [CrossRef]
- Vanzulli, I.; Butt, A.M. mGluR5 protect astrocytes from ischemic damage in postnatal CNS white matter. Cell Calcium 2015, 58, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Lv, G.; Wang, Y.S.; Fan, Z.K.; Bi, Y.L.; Zhao, L.; Guo, Z.P. Mitochondrial fusion and fission after spinal sacord injury in rats. Brain Res. 2013, 1522, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Pivovarova, N.B.; Andrews, S.B. Calcium-dependent mitochondrial function and dysfunction in neurons. FEBS J. 2010, 277, 3622–3636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, E.D. Antioxidant therapies for acute spinal cord injury. Neurotherapeutics 2011, 8, 152–167. [Google Scholar] [CrossRef] [Green Version]
- Golpich, M.; Amini, E.; Mohamed, Z.; Azman Ali, R.; Mohamed Ibrahim, N.; Ahmadiani, A. Mitochondrial dysfunction and biogenesis in neurodegenerative diseases: Pathogenesis and treatment. CNS Neurosci. Ther. 2017, 23, 5–22. [Google Scholar] [CrossRef]
- Hall, E.D.; Wang, J.A.; Bosken, J.M.; Singh, I.N. Lipid peroxidation in brain or spinal cord mitochondria after injury. J. Bioenerg. Biomembr. 2016, 48, 169–174. [Google Scholar] [CrossRef] [Green Version]
- Miron, V.E.; Franklin, R.J. Macrophages and CNS remyelination. J. Neurochem. 2014, 130, 165–171. [Google Scholar] [CrossRef]
- Jones, T.B. Lymphocytes and autoimmunity after spinal cord injury. Exp. Neurol. 2014, 258, 78–90. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012, 19, 107–120. [Google Scholar] [CrossRef]
- Dunai, Z.; Bauer, P.I.; Mihalik, R. Necroptosis: Biochemical, physiological and pathological aspects. Pathol. Oncol. Res. 2011, 17, 791–800. [Google Scholar] [CrossRef]
- Liu, S.; Li, Y.; Choi, H.M.C.; Sarkar, C.; Koh, E.Y.; Wu, J.; Lipinski, M.M. Lysosomal damage after spinal cord injury causes accumulation of RIPK1 and RIPK3 proteins and potentiation of necroptosis. Cell Death Dis. 2018, 9, 476. [Google Scholar] [CrossRef] [PubMed]
- Sobrido-Cameán, D.; Barreiro-Iglesias, A. Role of caspase-8 and Fas in cell death after spinal cord injury. Front. Mol. Neurosci. 2018, 11, 101. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.R.; Fehlings, M.G. Fas/FasL-mediated apoptosis and inflammation are key features of acute human spinal cord injury: Implications for translational, clinical application. Acta Neuropathol. 2011, 122, 747–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipinski, M.M.; Wu, J.; Faden, A.I.; Sarkar, C. Function and mechanisms of autophagy in brain and spinal cord trauma. Antioxid. Redox Signal. 2015, 23, 565–577. [Google Scholar] [CrossRef]
- Liu, M.; Wu, W.; Li, H.; Li, S.; Huang, L.T.; Yang, Y.Q.; Sun, Q.; Wang, C.X.; Yu, Z.; Hang, C.H. Necroptosis, a novel type of programmed cell death, contributes to early neural cells damage after spinal cord injury in adult mice. J. Spinal Cord Med. 2015, 38, 745–753. [Google Scholar] [CrossRef] [Green Version]
- Ginet, V.; Spiehlmann, A.; Rummel, C.; Rudinskiy, N.; Grishchuk, Y.; Luthi-Carter, R.; Clarke, P.G.; Truttmann, A.C.; Puyal, J. Involvement of autophagy in hypoxic-excitotoxic neuronal death. Autophagy 2014, 10, 846–860. [Google Scholar] [CrossRef] [Green Version]
- Williams, P.R.; Marincu, B.N.; Sorbara, C.D.; Mahler, C.F.; Schumacher, A.M.; Griesbeck, O.; Kerschensteiner, M.; Misgeld, T. A recoverable state of axon injury persists for hours after spinal cord contusion in vivo. Nat. Commun. 2014, 5, 1–11. [Google Scholar] [CrossRef]
- Wang, J.T.; Medress, Z.A.; Barres, B.A. Axon degeneration: Molecular mechanisms of a self-destruction pathway. J. Cell Biol. 2012, 196, 7–18. [Google Scholar] [CrossRef] [Green Version]
- Cohen-Adad, J.; El Mendili, M.M.; Lehéricy, S.; Pradat, P.F.; Blancho, S.; Rossignol, S.; Benali, H. Demyelination and degeneration in the injured human spinal cord detected with diffusion and magnetization transfer MRI. NeuroImage 2011, 55, 1024–1033. [Google Scholar] [CrossRef]
- Domingues, H.S.; Portugal, C.C.; Socodato, R.; Relvas, J.B. Oligodendrocyte, astrocyte, and microglia crosstalk in myelin development, damage, and repair. Front. Cell Dev. Biol. 2016, 4, 71. [Google Scholar]
- Almad, A.; Sahinkaya, F.R.; McTigue, D.M. Oligodendrocyte fate after spinal cord injury. Neurotherapeutics 2011, 8, 262–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mekhail, M.; Almazan, G.; Tabrizian, M. Oligodendrocyte-protection and remyelination post-spinal cord injuries: A review. Prog. Neurobiol. 2012, 96, 322–339. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.M.; He, C. The glial scar in spinal cord injury and repair. Neurosci. Bull. 2013, 29, 421–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billakanti, R.; Karimi-Abdolrezaee, S. Reactive astrogliosis after spinal cord injury-beneficial and detrimental effects. Mol. Neurobiol. 2012, 46, 251–264. [Google Scholar]
- Göritz, C.; Dias, D.O.; Tomilin, N.; Barbacid, M.; Shupliakov, O.; Frisén, J. A pericyte origin of spinal cord scar tissue. Science 2011, 333, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Zhang, K.; Shan, L.; Kuang, F.; Chen, K.; Zhu, K.; Ma, H.; Ju, G.; Wang, Y.Z. Reactive astrocytes undergo M1 microglia/macrohpages-induced necroptosis in spinal cord injury. Mol. Neurodegener. 2016, 11, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krebs, J.; Koch, H.G.; Hartmann, K.; Frotzler, A. The characteristics of posttraumatic syringomyelia. Spinal Cord 2016, 54, 463–466. [Google Scholar] [CrossRef] [Green Version]
- Kramer, J.L.; Minhas, N.K.; Jutzeler, C.R.; Erskine, E.L.; Liu, L.J.; Ramer, M.S. Neuropathic pain following traumatic spinal cord injury: Models, measurement, and mechanisms. J. Neurosci. Res. 2017, 95, 1295–1306. [Google Scholar] [CrossRef]
- Tuszynski, M.H.; Steward, O. Concepts and methods for the study of axonal regeneration in the CNS. Neuron 2012, 74, 777–791. [Google Scholar] [CrossRef] [Green Version]
- Burda, J.E.; Sofroniew, M.V. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron 2014, 81, 229–248. [Google Scholar] [CrossRef] [Green Version]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, E.G.; Kang, S.H.; Fukaya, M.; Bergles, D.E. Oligodendrocyte progenitors balance growth with self-repulsion to achieve homeostasis in the adult brain. Nat. Neurosci. 2013, 16, 668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soderblom, C.; Luo, X.; Blumenthal, E.; Bray, E.; Lyapichev, K.; Ramos, J.; Krishnan, V.; Lai-Hsu, C.; Park, K.K.; Tsoulfas, P.; et al. Perivascular fibroblasts form the fibrotic scar after contusive spinal cord injury. J. Neurosci. 2013, 33, 13882–13887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hesp, Z.C.; Yoseph, R.Y.; Suzuki, R.; Jukkola, P.; Wilson, C.; Nishiyama, A.; McTigue, D.M. Proliferating NG2-cell-dependent angiogenesis and scar formation alter axon growth and functional recovery after spinal cord injury in mice. J. Neurosci. 2018, 6, 1366–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kokaia, Z.; Martino, G.; Schwartz, M.; Lindvall, O. Cross-talk between neural stem cells and immune cells: The key to better brain repair? Nat. Neurosci. 2012, 15, 1078. [Google Scholar] [CrossRef]
- Wanner, I.B.; Anderson, M.A.; Song, B.; Levine, J.; Fernandez, A.; Gray-Thompson, Z.; Sofroniew, M.V. Glial scar borders are formed by newly proliferated, elongated astrocytes that interact to corral inflammatory and fibrotic cells via STAT3-dependent mechanisms after spinal cord injury. J. Neurosci. 2013, 33, 12870–12886. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, L.A.; Devitt, A. Current understanding of the mechanisms for clearance of apoptotic cells—A fine balances. J. Cell Death 2013, 6, JCD-S11037. [Google Scholar] [CrossRef] [Green Version]
- Cooper, J.G.; Jeong, S.J.; McGuire, T.; Sharma, S.; Wang, W.; Bhattacharyya, S.; Varga, J.; Kessler, J.A. Fibronectin EDA forms the chronic fibrotic scar after contusive spinal cord injury. Neurobiol. Dis. 2018, 116, 60–68. [Google Scholar] [CrossRef]
- Zhu, Y.; Soderblom, C.; Krishnan, V.; Ashbaugh, J.; Bethea, J.R.; Lee, J.K. Hematogenous macrophage depletion reduces the fibrotic scar and increases axonal growth after spinal cord injury. Neurobiol. Dis. 2015, 74, 114–125. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.J.; Nagarajah, R.; Banati, R.B.; Bennett, M.R. Glutamate induces directed chemotaxis of microglia. Eur. J. Neurosci. 2009, 29, 1108–1118. [Google Scholar] [CrossRef]
- Dosch, M.; Zindel, J.; Jebbawi, F.; Melin, N.; Sanchez-Taltavull, D.; Stroka, D.; Candinas, D.; Beldi, G. Connexin-43-dependent ATP release mediates macrophage activation during sepsis. eLife 2019, 8, 42670. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.K.; Roth, J.; Oppenheim, J.J.; Tracey, K.J.; Vogl, T.; Feldmann, M.; Horwood, N.; Nanchahal, J. Alarmins: Awaiting a clinical response. J. Clin. Investig. 2012, 122, 2711–2719. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.M.; Zhou, H.; Zhang, F.; Wilson, B.C.; Kam, W.; Hong, J.S. HMGB1 acts on microglia Mac1 to mediate chronic neuroinflammation that drives progressive neurodegeneration. J. Neurosci. 2011, 31, 1081–1092. [Google Scholar] [CrossRef] [PubMed]
- Ankeny, D.P.; Popovich, P.G. Mechanisms and implications of adaptive immune responses after traumatic spinal cord injury. Neuroscience 2009, 158, 1112–1121. [Google Scholar] [CrossRef] [Green Version]
- Zamanian, J.L.; Xu, L.; Foo, L.C.; Nouri, N.; Zhou, L.; Giffard, R.G.; Barres, B.A. Genomic analysis of reactive astrogliosis. J. Neurosci. 2012, 32, 6391–6410. [Google Scholar] [CrossRef] [Green Version]
- Seo, J.H.; Miyamoto, N.; Hayakawa, K.; Pham, L.D.; Maki, T.; Ayata, C.; Kim, K.W.; Lo, E.H.; Arai, K. Oligodendrocyte precursors induce early blood-brain barrier opening after white matter injury. J. Clin. Investig. 2013, 123, 782–786. [Google Scholar] [CrossRef] [Green Version]
- Beck, K.D.; Nguyen, H.X.; Galvan, M.D.; Salazar, D.L.; Woodruff, T.M.; Anderson, A.J. Quantitative analysis of cellular inflammation after traumatic spinal cord injury: Evidence for a multiphasic inflammatory response in the acute to chronic environment. Brain 2010, 133, 433–447. [Google Scholar] [CrossRef]
- Chang, R.C.C.; Ho, Y.S. Introductory Chapter: Concept of Neuroprotection—A New Perspective. In Neuroprotection; IntechOpen: London, UK, 2019. [Google Scholar]
- Gaidina, S.G.; Turovskayaa, M.V.; Mal’tsevaa, V.N.; Zinchenkoa, V.P.; Blinovab, E.V.; Turovskya, E.A. A complex neuroprotective effect of alpha-2-adrenergic receptor agonists in a model of cerebral ischemia–reoxygenation in vitro. Biochem. (Mosc.) Suppl. Ser. A Membr. Cell Biol. 2019, 13, 319–333. [Google Scholar] [CrossRef]
- Rivera-Oliver, M.; Díaz-Ríos, M. Using caffeine and other adenosine receptor antagonists and agonists as therapeutic tools against neurodegenerative diseases: A review. Life Sci. 2014, 101, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Aydoseli, A.; Can, H.; Aras, Y.; Sabanci, P.A.; Akcakaya, M.O.; Unal, O.F. Memantine and Q-VD-OPh treatments in experimental spinal cord injury: Combined inhibition of necrosis and apoptosis. Turk. Neurosurg. 2016, 26, 783–789. [Google Scholar] [CrossRef] [Green Version]
- Gerber, Y.N.; Privat, A.; Perrin, F.E. Gacyclidine improves the survival and reduces motor deficits in a mouse model of amyotrophic lateral sclerosis. Front. Cell. Neurosci. 2013, 7, 280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gensel, J.C.; Tovar, C.A.; Bresnahan, J.C.; Beattie, M.S. Topiramate treatment is neuroprotective and reduces oligodendrocyte loss after cervical spinal cord injury. PLoS ONE 2012, 7, e33519. [Google Scholar] [CrossRef] [PubMed]
- Vural, M.; Arslantaş, A.; Yazihan, N.; Köken, T.; Uzuner, K.; Arslantaş, D.; Özbek, Z. NMDA receptor blockage with 2-amino-5-phosphonovaleric acid improves oxidative stress after spinal cord trauma in rats. Spinal Cord 2010, 48, 285–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reigada, D.; Navarro-Ruiz, R.M.; Caballero-López, M.J.; Del Águila, Á.; Muñoz-Galdeano, T.; Maza, R.M.; Nieto-Díaz, M. Diadenosine tetraphosphate (Ap 4 A) inhibits ATP-induced excitotoxicity: A neuroprotective strategy for traumatic spinal cord injury treatment. Purinergic Signal. 2017, 13, 75–87. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Barrera, R.; Garibay-López, M.; Ibarra, A. Trends in Neuroprotective Strategies after Spinal Cord Injury: State of the Art. In Neuroprotection-New Approaches Prospect; IntechOpen: London, UK, 2019. [Google Scholar]
- Gurkoff, G.; Shahlaie, K.; Lyeth, B.; Berman, R. Voltage-gated calcium channel antagonists and traumatic brain injury. Pharmaceuticals 2013, 6, 788–812. [Google Scholar] [CrossRef] [Green Version]
- Wilson, J.R.; Fehlings, M.G. Riluzole for acute traumatic spinal cord injury: A promising neuroprotective treatment strategy. World Neurosurg. 2014, 81, 825–829. [Google Scholar] [CrossRef]
- Nagoshi, N.; Nakashima, H.; Fehlings, M.G. Riluzole as a neuroprotective drug for spinal cord injury: From bench to bedside. Molecules 2015, 20, 7775–7789. [Google Scholar] [CrossRef]
- Ates, O.; Cayli, S.R.; Gurses, I.; Turkoz, Y.; Tarim, O.; Cakir, C.O.; Kocak, A. Comparative neuroprotective effect of sodium channel blockers after experimental spinal cord injury. J. Clin. Neurosci. 2007, 14, 658–665. [Google Scholar] [CrossRef]
- Kopecky, B.J.; Liang, R.; Bao, J. T-type calcium channel blockers as neuroprotective agents. Pflügers Arch. Eur. J. Physiol. 2014, 466, 757–765. [Google Scholar] [CrossRef] [Green Version]
- Wildburger, N.C.; Lin-Ye, A.; Baird, M.A.; Lei, D.; Bao, J. Neuroprotective effects of blockers for T-type calcium channels. Mol. Neurodegener. 2009, 4, 44. [Google Scholar] [CrossRef] [Green Version]
- Teleanu, R.I.; Chircov, C.; Grumezescu, A.M.; Volceanov, A.; Teleanu, D.M. Antioxidant Therapies for Neuroprotection—A Review. J. Clin. Med. 2019, 8, 1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bains, M.; Hall, E.D. Antioxidant therapies in traumatic brain and spinal cord injury. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2012, 1822, 675–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mustafa, A.G.; Wang, J.A.; Carrico, K.M.; Hall, E.D. Pharmacological inhibition of lipid peroxidation attenuates calpain-mediated cytoskeletal degradation after traumatic brain injury. J. Neurochem. 2011, 117, 579–588. [Google Scholar] [CrossRef] [Green Version]
- Fehlings, M.G.; Wilson, J.R.; Tetreault, L.A.; Aarabi, B.; Anderson, P.; Arnold, P.M.; Brodke, D.S.; Burns, A.S.; Chiba, K.; Dettori, J.R.; et al. A clinical practice guideline for the management of patients with acute spinal cord injury: Recommendations on the use of methylprednisolone sodium succinate. Glob. Spine J. 2017, 203S–211S. [Google Scholar] [CrossRef]
- Mustafa, A.G.; Singh, I.N.; Wang, J.; Carrico, K.M.; Hall, E.D. Mitochondrial protection after traumatic brain injury by scavenging lipid peroxyl radicals. J. Neurochem. 2010, 114, 271–280. [Google Scholar] [CrossRef] [Green Version]
- Samantaray, S.; Das, A.; Thakore, N.P.; Matzelle, D.D.; Reiter, R.J.; Ray, S.K.; Banik, N.L. Therapeutic potential of melatonin in traumatic central nervous system injury. J. Pineal Res. 2009, 47, 134–142. [Google Scholar] [CrossRef]
- Vaishnav, R.A.; Singh, I.N.; Miller, D.M.; Hall, E.D. Lipid peroxidation-derived reactive aldehydes directly and differentially impair spinal cord and brain mitochondrial function. J. Neurotrauma 2010, 27, 1311–1320. [Google Scholar] [CrossRef]
- Xiong, Y.; Singh, I.N.; Hall, E.D. Tempol protection of spinal cord mitochondria from peroxynitrite-induced oxidative damage. Free Radic. Res. 2009, 43, 604–612. [Google Scholar] [CrossRef]
- Liu, C.; Shi, Z.; Fan, L.; Zhang, C.; Wang, K.; Wang, B. Resveratrol improves neuron protection and functional recovery in rat model of spinal cord injury. Brain Res. 2011, 1374, 100–109. [Google Scholar] [CrossRef]
- Petri, S.; Körner, S.; Kiaei, M. Nrf2/ARE signaling pathway: Key mediator in oxidative stress and potential therapeutic target in ALS. Neurol. Res. Int. 2012, 2012, 878030. [Google Scholar] [CrossRef]
- Chen, G.; Fang, Q.; Zhang, J.; Zhou, D.; Wang, Z. Role of the Nrf2-ARE pathway in early brain injury after experimental subarachnoid hemorrhage. J. Neurosci. Res. 2011, 89, 515–523. [Google Scholar] [CrossRef]
- Jin, W.; Kong, J.; Wang, H.; Wu, J.; Lu, T.; Jiang, J.; Ni, H.; Liang, W. Protective effect of tert-butylhydroquinone on cerebral inflammatory response following traumatic brain injury in mice. Injury 2011, 42, 714–718. [Google Scholar] [CrossRef]
- Galluzzi, L.; Blomgren, K.; Kroemer, G. Mitochondrial membrane permeabilization in neuronal injury. Nat. Rev. Neurosci. 2009, 10, 481–494. [Google Scholar] [CrossRef]
- Sureda, F.X.; Junyent, F.; Verdaguer, E.; Auladell, C.; Pelegri, C.; Vilaplana, J.; Folch, J.; Maria Canudas, A.; Beas Zarate, C.; Pallàs, M.; et al. Antiapoptotic drugs: A therapautic strategy for the prevention of neurodegenerative diseases. Curr. Pharm. Des. 2011, 17, 230–245. [Google Scholar] [CrossRef]
- Hisatomi, T.; Ishibashi, T.; Miller, J.W.; Kroemer, G. Pharmacological inhibition of mitochondrial membrane permeabilization for neuroprotection. Exp. Neurol. 2009, 218, 347–352. [Google Scholar] [CrossRef]
- Ma, B.; Shi, J.; Jia, L.; Yuan, W.; Wu, J.; Fu, Z.; Wang, Y.; Liu, N.; Guan, Z. Over-expression of PUMA correlates with the apoptosis of spinal cord cells in rat neuropathic intermittent claudication model. PLoS ONE 2013, 8, e56580. [Google Scholar] [CrossRef]
- Shiri, R.; Yari, F.; Ahmadinejad, M.; Vaeli, S.; Tabatabaei, M.R. The caspase-3 inhibitor (peptide Z-DEVD-FMK) affects the survival and function of platelets in platelet concentrate during storage. Blood Res. 2014, 49, 49–53. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Yan, H.; Chen, S.; Sabel, B.A. Caspase-3 inhibitor Z-DEVD-FMK enhances retinal ganglion cell survival and vision restoration after rabbit traumatic optic nerve injury. Restor. Neurol. Neurosci. 2015, 33, 205–220. [Google Scholar]
- Çolak, A.; Karaoǧlan, A.; Barut, Ş.; Köktürk, S.; Akyildiz, A.I.; Taşyürekli, M. Neuroprotection and functional recovery after application of the caspase-9 inhibitor z-LEHD-fmk in a rat model of traumatic spinal cord injury. J. Neurosurg. Spine 2005, 2, 327–334. [Google Scholar] [CrossRef]
- Glória, P.M.; Coutinho, I.; Gonçalves, L.M.; Baptista, C.; Soares, J.; Newton, A.S.; Moreira, R.; Saraiva, L.; Santos, M.M. Aspartic vinyl sulfones: Inhibitors of a caspase-3-dependent pathway. Eur. J. Med. Chem. 2011, 46, 2141–2146. [Google Scholar] [CrossRef]
- Camins, A.; Crespo-Biel, N.; Junyent, F.; Verdaguer, E.; Canudas, A.M.; Pallas, M. Calpains as a target for therapy of neurodegenerative diseases: Putative role of lithium. Curr. Drug Metab. 2009, 10, 433–447. [Google Scholar] [CrossRef]
- David, J.S.; Melamud, A.; Kesner, L.; Roth, S.; Rosenbaum, P.S.; Barone, F.C.; Popp, S.; Hassen, G.W.; Stracher, A.; Rosenbaum, D.M. A novel calpain inhibitor for treatment of transient retinal ischemia in the rat. Neuroreport 2011, 22, 633. [Google Scholar] [CrossRef]
- Zhang, Z.; Huang, Z.; Dai, H.; Wei, L.; Sun, S.; Gao, F. Therapeutic efficacy of E-64-d, a selective calpain inhibitor, in experimental acute spinal cord injury. BioMed Res. Int. 2015, 2015, 134242. [Google Scholar] [CrossRef] [Green Version]
- Lei, F.; He, W.; Tian, X.; Zhou, Q.; Zheng, L.; Kang, J.; Song, Y.; Feng, D. GSK-3 Inhibitor Promotes Neuronal Cell Regeneration and Functional Recovery in a Rat Model of Spinal Cord Injury. BioMed Res. Int. 2019, 2019, 9628065. [Google Scholar] [CrossRef] [Green Version]
- Koob, A.O.; Ubhi, K.; Paulsson, J.F.; Kelly, J.; Rockenstein, E.; Mante, M.; Adame, A.; Masliah, E. Lovastatin ameliorates α-synuclein accumulation and oxidation in transgenic mouse models of α-synucleinopathies. Exp. Neurol. 2010, 221, 267–274. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, C.; Tercero, I.; Pineda, A.; Burgos, J.S. Simvastatin is the statin that most efficiently protects against kainate-induced excitotoxicity and memory impairment. J. Alzheimer Dis. 2011, 24, 161–174. [Google Scholar] [CrossRef]
- Bagli, E.; Goussia, A.; Moschos, M.M.; Agnantis, N.; Kitsos, G. Natural compounds and neuroprotection: Mechanisms of action and novel delivery systems. In Vivo 2016, 30, 535–547. [Google Scholar]
- Puzi, N.N.; Lokanathan, Y.; Idrus, R.B.H. The Effect of Centella asiatica (L.) Urban on the Organotypic Model of Spinal Cord Injury. Sains Malays. 2018, 47, 2783–2788. [Google Scholar] [CrossRef]
- Kumar, R.; Htwe, O.; Baharudin, A.; Ariffin, M.H.; Rhani, S.A.; Ibrahim, K.; Rustam, A.; Gan, R. Spinal Cord Injury—Assessing Tolerability and Use of Combined Rehabilitation and NeuroAiD (SATURN Study): Protocol of An Exploratory Study In Assessing the Safety and Efficacy of NeuroAiD Amongst People Who Sustain Severe Spinal Cord Injury. JMIR Res. Protoc. 2016, 5, 230. [Google Scholar] [CrossRef]
- Fraunberger, E.A.; Scola, G.; Laliberté, V.L.; Duong, A.; Andreazza, A.C. Redox modulations, antioxidants, and neuropsychiatric disorders. Oxid. Med. Cell. Longev. 2016, 2016, 4729192. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, A.H.; Hamada, M.; Shinada, T.; Ohfune, Y.; Weerasinghe, L.; Garner, P.P.; Oswald, R.E. The Structure of (−)-Kaitocephalin Bound to the Ligand Binding Domain of the (S)-α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic Acid (AMPA)/Glutamate Receptor, GluA2. J. Biol. Chem. 2012, 287, 41007–41013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimmyo, Y.; Kihara, T.; Akaike, A.; Niidome, T.; Sugimoto, H. Three distinct neuroprotective functions of myricetin against glutamate-induced neuronal cell death: Involvement of direct inhibition of caspase-3. J. Neurosci. Res. 2008, 86, 1836–1845. [Google Scholar] [CrossRef] [PubMed]
- Lalkovičová, M.; Danielisová, V. Neuroprotection and antioxidants. Neural Regen. Res. 2016, 11, 865. [Google Scholar] [CrossRef] [PubMed]
- Kieliszek, M.; Błażejak, S. Selenium: Significance, and outlook for supplementation. Nutrition 2013, 29, 713–718. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, C.S.; Fehlings, M. Concise review: Bridging the gap: Novel neuroregenerative and neuroprotective strategies in spinal cord injury. Stem Cells Transl. Med. 2016, 5, 914–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallner, S.; Peters, S.; Pitzer, C.; Resch, H.; Bogdahn, U.; Schneider, A. The granulocyte-colony stimulating factor has a dual role in neuronal and vascular plasticity. Front. Cell Dev. Biol. 2015, 3, 48. [Google Scholar] [CrossRef] [Green Version]
- Ji, X.C.; Dang, Y.Y.; Gao, H.Y.; Wang, Z.T.; Gao, M.; Yang, Y.; Zhang, H.-T.; Xu, R.X. Local injection of Lenti–BDNF at the lesion site promotes M2 macrophage polarization and inhibits inflammatory response after spinal cord injury in mice. Cell. Mol. Neurobiol. 2015, 35, 881–890. [Google Scholar] [CrossRef]
- Kohta, M.; Kohmura, E.; Yamashita, T. Inhibition of TGF-β1 promotes functional recovery after spinal cord injury. Neurosci. Res. 2009, 65, 393–401. [Google Scholar] [CrossRef]
- Rong, Y.; Liu, W.; Wang, J.; Fan, J.; Luo, Y.; Li, L.; Kong, F.; Chen, J.; Tang, P.; Cai, W. Neural stem cell-derived small extracellular vesicles attenuate apoptosis and neuroinflammation after traumatic spinal cord injury by activating autophagy. Cell Death Dis. 2019, 10, 1–18. [Google Scholar] [CrossRef]
- Ankeny, D.P.; McTigue, D.M.; Jakeman, L.B. Bone marrow transplants provide tissue protection and directional guidance for axons after contusive spinal cord injury in rats. Exp. Neurol. 2004, 190, 17–31. [Google Scholar] [CrossRef]
- Wu, S.; Cui, G.; Shao, H.; Du, Z.; Ng, J.C.; Peng, C. The cotransplantation of olfactory ensheathing cells with bone marrow mesenchymal stem cells exerts antiapoptotic effects in adult rats after spinal cord injury. Stem Cells Int. 2015, 2015, 516215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gazdic, M.; Volarevic, V.; Harrell, C.R.; Fellabaum, C.; Jovicic, N.; Arsenijevic, N.; Stojkovic, M. Stem cells therapy for spinal cord injury. Int. J. Mol. Sci. 2018, 19, 1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Chen, H.; Duan, Z.; Chen, K.; Liu, Z.; Zhang, L.; Yao, D.; Li, B. The effects of co-transplantation of olfactory ensheathing cells and schwann cells on local inflammation environment in the contused spinal cord of rats. Mol. Neurobiol. 2017, 54, 943–953. [Google Scholar] [CrossRef]
- Lin, L.; Lin, H.; Bai, S.; Zheng, L.; Zhang, X. Bone marrow mesenchymal stem cells (BMSCs) improved functional recovery of spinal cord injury partly by promoting axonal regeneration. Neurochem. Int. 2018, 115, 80–84. [Google Scholar] [CrossRef]
- Kigerl, K.A.; Gensel, J.C.; Ankeny, D.P.; Alexander, J.K.; Donnelly, D.J.; Popovich, P.G. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J. Neurosci. 2009, 29, 13435–13444. [Google Scholar] [CrossRef] [Green Version]
- Yao, R.; Murtaza, M.; Velasquez, J.T.; Todorovic, M.; Rayfield, A.; Ekberg, J.; Barton, M.; St John, J. Olfactory ensheathing cells for spinal cord injury: Sniffing out the issues. Cell Transplant. 2018, 27, 879–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashammakhi, N.; Kim, H.J.; Ehsanipour, A.; Bierman, R.D.; Kaarela, O.; Xue, C.; Khademhosseini, A.; Seidlits, S.K. Regenerative therapies for spinal cord injury. Tissue Eng. Part B Rev. 2019, 25, 471–491. [Google Scholar] [CrossRef] [PubMed]
- Tsintou, M.; Dalamagkas, K.; Seifalian, A.M. Advances in regenerative therapies for spinal cord injury: A biomaterials approach. Neural Regen. Res. 2015, 10, 726. [Google Scholar]
- Colombo, E.; Farina, C. Astrocytes: Key regulators of neuroinflammation. Trends Immunol. 2016, 37, 608–620. [Google Scholar] [CrossRef]
- Cekanaviciute, E.; Buckwalter, M.S. Astrocytes: Integrative regulators of neuroinflammation in stroke and other neurological diseases. Neurotherapeutics 2016, 13, 685–701. [Google Scholar] [CrossRef]
- Pineau, I.; Sun, L.; Bastien, D.; Lacroix, S. Astrocytes initiate inflammation in the injured mouse spinal cord by promoting the entry of neutrophils and inflammatory monocytes in an IL-1 receptor/MyD88-dependent fashion. Brain Behav. Immunity 2010, 24, 540–553. [Google Scholar] [CrossRef] [PubMed]
- Haroon, F.; Drögemüller, K.; Händel, U.; Brunn, A.; Reinhold, D.; Nishanth, G.; Mueller, W.; Trautwein, C.; Ernst, M.; Deckert, M.; et al. Gp130-dependent astrocytic survival is critical for the control of autoimmune central nervous system inflammation. J. Immunol. 2011, 186, 6521–6531. [Google Scholar] [CrossRef] [PubMed]
- Neirinckx, V.; Coste, C.; Franzen, R.; Gothot, A.; Rogister, B.; Wislet, S. Neutrophil contribution to spinal cord injury and repair. J. Neuroinflamm. 2014, 11, 150. [Google Scholar] [CrossRef] [Green Version]
- Stirling, D.P.; Liu, S.; Kubes, P.; Yong, V.W. Depletion of Ly6G/Gr-1 leukocytes after spinal cord injury in mice alters wound healing and worsens neurological outcome. J. Neurosci. 2009, 29, 753–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, V.H.; Teeling, J. Microglia and macrophages of the central nervous system: The contribution of microglia priming and systemic inflammation to chronic neurodegeneration. Semin. Immunopathol. 2013, 35, 601–612. [Google Scholar] [CrossRef] [Green Version]
- Herz, J.; Filiano, A.J.; Smith, A.; Yogev, N.; Kipnis, J. Myeloid cells in the central nervous system. Immunity 2017, 46, 943–956. [Google Scholar] [CrossRef] [Green Version]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef]
- Kroner, A.; Greenhalgh, A.D.; Zarruk, J.G.; dos Santos, R.P.; Gaestel, M.; David, S. TNF and increased intracellular iron alter macrophage polarization to a detrimental M1 phenotype in the injured spinal cord. Neuron 2014, 83, 1098–1116. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef]
- Francos-Quijorna, I.; Amo-Aparicio, J.; Martinez-Muriana, A.; López-Vales, R. IL-4 drives microglia and macrophages toward a phenotype conducive for tissue repair and functional recovery after spinal cord injury. Glia 2016, 64, 2079–2092. [Google Scholar] [CrossRef] [Green Version]
- Walsh, J.T.; Zheng, J.; Smirnov, I.; Lorenz, U.; Tung, K.; Kipnis, J. Regulatory T cells in central nervous system injury: A double-edged sword. J. Immunol. 2014, 193, 5013–5022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zajarias-Fainsod, D.; Carrillo-Ruiz, J.; Mestre, H.; Grijalva, I.; Madrazo, I.; Ibarra, A. Autoreactivity against myelin basic protein in patients with chronic paraplegia. Eur. Spine J. 2012, 21, 964–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshizaki, A.; Miyagaki, T.; DiLillo, D.J.; Matsushita, T.; Horikawa, M.; Kountikov, E.I.; Spolski, R.; Poe, J.C.; Leonard, W.J.; Tedder, T.F. Regulatory B cells control T-cell autoimmunity through IL-21-dependent cognate interactions. Nature 2012, 491, 264–268. [Google Scholar] [CrossRef]
- Popovich, P.G.; Tovar, C.A.; Wei, P.; Fisher, L.; Jakeman, L.B.; Basso, D.M. A reassessment of a classic neuroprotective combination therapy for spinal cord injured rats: LPS/pregnenolone/indomethacin. Exp. Neurol. 2012, 233, 677–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hakan, T.; Toklu, H.Z.; Biber, N.; Celik, H.; Erzik, C.; Oğünç, A.V.; Spolski, R.; Poe, J.C.; Leonard, W.J.; Şener, G. Meloxicam exerts neuroprotection on spinal cord trauma in rats. Int. J. Neurosci. 2011, 121, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.R.; Ma, Y.; Guo, H.H.; Lu, Z.D.; Jin, Q.H. Therapeutic efficacy of cyclosporin A against spinal cord injury in rats with hyperglycemia. Mol. Med. Rep. 2018, 17, 4369–4375. [Google Scholar] [CrossRef] [Green Version]
- Saganová, K.; Gálik, J.; Blaško, J.; Korimová, A.; Račeková, E.; Vanický, I. Immunosuppressant FK506: Focusing on neuroprotective effects following brain and spinal cord injury. Life Sci. 2012, 91, 77–82. [Google Scholar] [CrossRef]
- Martiñón, S.; García, E.; Gutierrez-Ospina, G.; Mestre, H.; Ibarra, A. Development of protective autoimmunity by immunization with a neural-derived peptide is ineffective in severe spinal cord injury. PLoS ONE 2012, 7, e32027. [Google Scholar] [CrossRef] [Green Version]
- Afshari, K.; Dehdashtian, A.; Haddadi, N.S.; Haj-Mirzaian, A.; Iranmehr, A.; Ebrahimi, M.A.; Tavangar, S.M.; Faghir-Ghanesefat, H.; Mohammadi, F.; Rahimi, N.; et al. Anti-inflammatory effects of Metformin improve the neuropathic pain and locomotor activity in spinal cord injured rats: Introduction of an alternative therapy. Spinal Cord 2018, 56, 1032–1041. [Google Scholar] [CrossRef]
- Nourbakhsh, B.; Waubant, E. Neurodegeneration and Remyelination in Multiple Sclerosis. In Multiple Sclerosis: A Mechanistic View; Academic Press: Cambridge, MA, USA, 2016; pp. 311–337. [Google Scholar]
- Wu, X.; Xu, X.M. RhoA/Rho kinase in spinal cord injury. Neural Regen. Res. 2016, 11, 23–27. [Google Scholar]
- Chelyshev, Y.A.; Ismagilov, M.F.; Mukhamedshina, Y.O.; Povysheva, T.V.; Boychuk, N.V. Rho/ROCK signaling pathway after spinal cord injury. Neurol. Bull. 2017, 49, 71–77. [Google Scholar]
- Tuladhar, A.; Mitrousis, N.; Führmann, T.; Shoichet, M.S. Central Nervous System. In Translational Regenerative Medicine; Academic Press: Cambridge, MA, USA, 2015; pp. 415–435. [Google Scholar]
- Duncan, I.D.; Brower, A.; Kondo, Y.; Curlee, J.F.; Schultz, R.D. Extensive remyelination of the CNS leads to functional recovery. Proc. Natl. Acad. Sci. USA 2009, 106, 6832–6836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, S.K.; Michaels, N.J.; Ilyntskyy, S.; Keough, M.B.; Kovalchuk, O.; Yong, V.W. Multimodal enhancement of remyelination by exercise with a pivotal role for oligodendroglial PGC1α. Cell Rep. 2018, 24, 3167–3179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badner, A.; Siddiqui, A.M.; Fehlings, M.G. Spinal cord injuries: How could cell therapy help? Expert Opin. Biol. Ther. 2017, 17, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Ha, K.Y.; Molon, J.N.; Kim, Y.H. Bone marrow–derived mesenchymal stem cell transplantation for chronic spinal cord injury in rats: Comparative study between intralesional and intravenous transplantation. Spine 2013, 38, E1065–E1074. [Google Scholar] [CrossRef]
- Fry, E.J.; Chagnon, M.J.; López-Vales, R.; Tremblay, M.L.; David, S. Corticospinal tract regeneration after spinal cord injury in receptor protein tyrosine phosphatase sigma deficient mice. Glia 2010, 58, 423–433. [Google Scholar] [CrossRef]
- Kim, M.; Park, S.R.; Choi, B.H. Biomaterial scaffolds used for the regeneration of spinal cord injury (SCI). Histol. Histopathol. 2011, 29. [Google Scholar] [CrossRef]
- Fu, P.C.; Tang, R.H.; Yu, Z.Y.; Xie, M.J.; Wang, W.; Luo, X. The Rho-associated kinase inhibitors Y27632 and fasudil promote microglial migration in the spinal cord via the ERK signaling pathway. Neural Regen. Res. 2018, 13, 677. [Google Scholar]
- Kabu, S.; Gao, Y.; Kwon, B.K.; Labhasetwar, V. Drug delivery, cell-based therapies, and tissue engineering approaches for spinal cord injury. J. Control. Release 2015, 219, 141–154. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phases | Signaling Molecules | Source | Class | Function | Ref. |
|---|---|---|---|---|---|
| Phase I | Thrombin | Serum | Protease | Clot formation &astrocyte proliferation | [50] |
| ATP | Neuron, oligodendrocytes & astrocytes | Neurotransmitters | Microglia chemotaxis & reactive astrogliosis | [41,42,43] | |
| Glutamate | Neuron, oligodendrocytes & astrocytes | Neurotransmitters | Microglia chemotaxis & reactive astrogliosis | [52] | |
| Phase I & II | Alarmins (HMGB1) | Damaged cells | DAMPs | Pro-inflammatory signals & increase phagocytosis | [53] |
| S100s | [53] | ||||
| DNA | [53] | ||||
| PAMPs (LPS) | Microbes | [54] | |||
| IL-1b, TNFa, INFg | Astrocyte, microglia & Leukocytes | Cytokines Chemokines | Pro-inflammatory regulation | [53] | |
| IL-6, CCL2 | Leukocyte instruction & astrocyte scar formation | [52] | |||
| CD200, CD47 | Neurons | NIRegs | Protection of healthy self | [53] | |
| Phase III | Neurotrophins & BDNF | Neurons & Astrocytes | Neural Remodeling | Synapse remodeling | [54] |
| Thmbs, C1q | Astrocytes & Microglia | Synapse formation & pruning | [55] | ||
| Perineuronal net | Astrocytes & O progenitor cell | Restrict terminal sprouting | [55] | ||
| Phase I & III | MMP-9 | Serum & Microglia | OPC proliferation, remyelination & neovascular remodeling | [55] | |
| Kallikreins | Astrocytes, Microglia, Neurons & Serum | Proteases | Proinflammatory & demyelination | [56] | |
| Serpins | Astrocytes, Microglia & O progenitor cells | Inhibit deleterious protease | [55] | ||
| FGF | Astrocytes, Neuron & Endothelia | Growth Factors & Morphogens | Fibrotic scar, ECM & neovascular remodeling | [41,43] | |
| VEGF | Endothelia, Fibroblast & Astrocytes | Neovascular remodeling & remyelination | [55] | ||
| PDGF-B | Endothelia & Astrocytes | ||||
| PDGF-A | |||||
| Phase II & III | Endothelin, EGF, BMP | Neuron, Astrocytes & O progenitor cells | Growth Factors, Morphogens | Astrocyte proliferation & glial scar formation | [57,58] |
| Sr. No. | Compound | Class | Receptor | Mechanism of Action | Reference |
|---|---|---|---|---|---|
| 1 | Gacyclidine (GK-11) | Tenocyclidine, closely related to phencyclidine | Noncompetitive NMDA receptor | Inhibits formation of ischemic SCI lesion. | [62] |
| 2 | NBQX | 2, 3-Dihydroxy-6-nitro-7-sulfamoylbenzoquinoxaline | AMPA/kainate receptor antagonist | Enhances mitochondrial functions and retard ROS and RNS formation. | [63] |
| 3 | Topirmate | 2,3:4,5-Bis-O-(1-methylethylidene)-beta-D-fructopyranose sulfamate | AMPA receptor antagonist | Promotes neuroprotective activity, improves tissue recovery, oligodendrocytes and motor function. NBQX and topiramate both showed powerful neuroprotective activity in female rat model. | [63,64] |
| 4 | APV | 2- Amino-5-phosphovaleric acid | NMDA receptor antagonist | Block glutamate activation and transport. | [64] |
| 5 | Magnesium | element | Non-competitive NMDA receptor antagonist | Reduces excitotoxicity and inflammation. | [9] |
| 6 | AP4A (Diadenosine tetraposphate) | Putative alarmone | Pirinergic receptor partial agonists and even act as antagonists in presence of the full agonist of P2 receptors (P2 are ATP receptors) | Reduces ATP-dependent excitotoxicity related death by both lowering the intracellular calcium response and decreasing the expression of P2 receptors. | [65] |
| Sr. No | Compound | Class | Group | Mechanism of Action | Ref. |
|---|---|---|---|---|---|
| 1. | Tetrodotoxin (TTX) | Guanidine | Na+ channel blocker | TTX block cellular Na+/Ca+2 exchange, membrane depolarization, and glutamate release and block neuronal cell death. TTX also improve motor function. | [66] |
| 2. | Riluzole | Benzothiazole | Voltage-gated Na+ channel blocker | Inhibit glutamate transmission and reduces glutamate associated excitotoxicity in neuronal tissue. Stop Na+ efflux and H+ influx within neurons and prevent neuronal acidosis. | [68,69,70] |
| 3. | Mexiletine | local anesthetic, antiarrhythmic agent, similar to lidocaine | Na+ channel blocker | Stop demyelination of neuronal tissues and evoke motor function recovery. Decreases lipid peroxidation, evokes motor function and promote neuroprotection. | [70] |
| 4. | Phenytoin | Hydantoin derivative | Na+ channel blocker | Block cellular Na+/Ca+2 exchange and promote neuroprotection | [70] |
| 5. | Nimodipine | Dihydropridinic | L-type VGCCs | Reduces malondialdehyde (MDA) levels, macrophages marker ED1activation and activation of myeloperoxidases (MPo). Prevent oxidative damage by reduction of macrophages infiltration to injured tissues. | [71] |
| 6. | Mibefradil | Posicor | T-type VGCCs | Selective blockade of transient, low-voltage-activated (T-type) calcium channels | [72] |
| 7. | Trimethadione | oxazolidinedione | T-type VGCCs | Selective blockade of transient, low-voltage-activated (T-type) calcium channels | [72] |
| Sr. No. | Compound | Class | MOA | Ref. |
|---|---|---|---|---|
| 1. | Bilobalide | Terpenoids from Ginkogo biloba leaves extract | Showed neuroprotective action on neurons and schwann cells by inhibiting ROS formation and apoptosis, It also modifies cytochrome-C oxidase subunit I level and regulates mitochondrial functions | [100] |
| 2. | Centella asiatica (L.) Urban (CA) | (pegaga) malay & Chinese traditional medicine | It acts as a brain tonic, which improve memory, it was also found to improve spinal cord recovery in organotypic rat model | [101] |
| 3. | MLC601 & MLC901 | Neuroaid | It is a combination of natural products, that has shown to be safe and to aid neurological recovery after brain & spinal injuries and have a potential role in improving recovery after SCI | [103] |
| 4. | Kaitocephalin | Eupenicillium shearii extract | Potent glutamate receptors (AMPA & NMDA) antagonist and inhibit glutamate excitotoxicity | [104] |
| 5. | Myricetin | Flavonoid | Inhibits glutamate excitotoxicity by stopping NMDAR receptor phosphorylation and reducing Ca+2 overloads | [105] |
| 6. | Curcumin | Curcuminoids of turmeric (Curcuma longa) | Exert neuroprotective activity by restoration of glutathione S transferase (GST), glutathione peroxidases (GPx) and MnSOD (manganese superoxide dismutase) activity | [102] |
| Sr. No. | Compound | Class | MOA | Ref. |
|---|---|---|---|---|
| 1. | Indomethacin | Non-steroidal anti-inflammatory drug (NSAID) is a nonselective cyclooxygenase inhibitor (COX) | It inhibits prostaglandin production and prevents tissue necrosis. Indomethacin prevents RhoA synthesis (RhoA prevents axonal growth), prevent oligodendrocytes loss and axonal myelination. | [140,142] |
| 2. | Meloxicam | COX2 inhibitor | It inhibits prostaglandin synthesis, reduces oxidative stress and provides neuroprotection by inhibiting the production of ROS, LPO, GSH and DNA fragmentation. | [143] |
| 3. | Cyclosporine A | Immunosuppressant | It inhibits helper T lymphocytes, cytotoxic and inflammatory responses in macrophages, expression of nitric oxide synthase, production of tumor necrosis factor (TNF-α) and reduce expression of IL-1, IL-2, and IL-6 | [139] |
| 4. | Tacrolimus (FK506) | Immunosuppressant (isolated from Streptomyces tsukubanensis) | It possesses neuroprotective effect on T cells and modulates inflammation. It also inhibits caspase-3, NF-kB and promotes oligodendroglial survival. | [140] |
| 5. | A91 (87-99 immunogenic sequence) | Neural peptide INDP | It promotes neuroprotection by activating T-lymphocytes, Th2 anti-inflammatory activity and promote brain-derived neurotropic factor (BDNF). INDP inhibits iNOS expression, ON production and LPO generation after SCI prevents apoptosis. | [141,142] |
| 6. | Metformin | Hypoglycemic drug, AMP-protein kinase (AMPK), an agonist. | It inhibits apoptosis by inhibiting mTOR and p70S6K pathways, promote autophagy and inhibit NF-kB inflammation. It also regulate TNFα and IL-1β inflammatory cytokines | [142] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anjum, A.; Yazid, M.D.; Fauzi Daud, M.; Idris, J.; Ng, A.M.H.; Selvi Naicker, A.; Ismail, O.H.R.; Athi Kumar, R.K.; Lokanathan, Y. Spinal Cord Injury: Pathophysiology, Multimolecular Interactions, and Underlying Recovery Mechanisms. Int. J. Mol. Sci. 2020, 21, 7533. https://doi.org/10.3390/ijms21207533
Anjum A, Yazid MD, Fauzi Daud M, Idris J, Ng AMH, Selvi Naicker A, Ismail OHR, Athi Kumar RK, Lokanathan Y. Spinal Cord Injury: Pathophysiology, Multimolecular Interactions, and Underlying Recovery Mechanisms. International Journal of Molecular Sciences. 2020; 21(20):7533. https://doi.org/10.3390/ijms21207533
Chicago/Turabian StyleAnjum, Anam, Muhammad Da’in Yazid, Muhammad Fauzi Daud, Jalilah Idris, Angela Min Hwei Ng, Amaramalar Selvi Naicker, Ohnmar Htwe@ Rashidah Ismail, Ramesh Kumar Athi Kumar, and Yogeswaran Lokanathan. 2020. "Spinal Cord Injury: Pathophysiology, Multimolecular Interactions, and Underlying Recovery Mechanisms" International Journal of Molecular Sciences 21, no. 20: 7533. https://doi.org/10.3390/ijms21207533