Copper Toxicity Links to Pathogenesis of Alzheimer’s Disease and Therapeutics Approaches



1
CAS Center for Excellence in Biotic Interactions, College of Life Science, University of Chinese Academy of Sciences, Yuquan Road 19, Beijing 100049, China
2
School of Medical and Health Sciences, Edith Cowan University, Perth WA6027, Australia
3
College of Life Science, Agricultural University of Hebei, Baoding 071000, China
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(20), 7660; https://doi.org/10.3390/ijms21207660
Submission received: 25 August 2020
/
Revised: 24 September 2020
/
Accepted: 29 September 2020
/
Published: 16 October 2020
(This article belongs to the Special Issue Functional Modification of Neuronal Networks)
Abstract
:Alzheimer’s disease (AD) is an irreversible, age-related progressive neurological disorder, and the most common type of dementia in aged people. Neuropathological lesions of AD are neurofibrillary tangles (NFTs), and senile plaques comprise the accumulated amyloid-beta (Aβ), loaded with metal ions including Cu, Fe, or Zn. Some reports have identified metal dyshomeostasis as a neurotoxic factor of AD, among which Cu ions seem to be a central cationic metal in the formation of plaque and soluble oligomers, and have an essential role in the AD pathology. Cu-Aβ complex catalyzes the generation of reactive oxygen species (ROS) and results in oxidative damage. Several studies have indicated that oxidative stress plays a crucial role in the pathogenesis of AD. The connection of copper levels in AD is still ambiguous, as some researches indicate a Cu deficiency, while others show its higher content in AD, and therefore there is a need to increase and decrease its levels in animal models, respectively, to study which one is the cause. For more than twenty years, many in vitro studies have been devoted to identifying metals’ roles in Aβ accumulation, oxidative damage, and neurotoxicity. Towards the end, a short review of the modern therapeutic approach in chelation therapy, with the main focus on Cu ions, is discussed. Despite the lack of strong proofs of clinical advantage so far, the conjecture that using a therapeutic metal chelator is an effective strategy for AD remains popular. However, some recent reports of genetic-regulating copper transporters in AD models have shed light on treating this refractory disease. This review aims to succinctly present a better understanding of Cu ions’ current status in several AD features, and some conflicting reports are present herein.
1. Introduction
Alzheimer’s disease (AD) is a multifactorial, complex brain disease defined by progressive cognitive decline, heterogeneity of behavioral presentations, and dementia in older people [1,2,3]. In 1907, Alois Alzheimer was the first to identify a mental decline with amyloid plaques and neurofibrillary tangles found in most dementia symptoms [4,5]. This disorder’s main risk factor is old age, because the elderly are more prone to diseases, affecting 10% of people aged 65, and this proportion rises by about three times for people aged 85 and older [6,7]. AD typically destroys neurons, and their connection with the brain regions such as the entorhinal cortex and hippocampus area, the parts of the brain essential in forming memories [8]. This disorder disrupts processes necessary for healthy neurons, such as communication, metabolism, and repair [9,10]. Ultimately, the disease is fatal. It is one of the leading causes of death [11] that we are currently unable to stop or cure because the underlying etiology is poorly understood at present [11,12].
Unfortunately, the treatment of AD has often been delayed in general because it is diagnosed only after prominent signs of cognitive deterioration [13], and this is all due to the lack of awareness of cognitive problems on the part of patients and patients’ families [14]. Clinical detection of this disorder is only possible when the symptoms are advanced enough to show visible behavior or cognitive changes [15]. There could be enough time to halt or slow this disorder’s development with early AD identification before complete onset [15]. Indeed, currently, there is no such treatment for AD [16], and approved drugs that have insignificant effects at altering the pathophysiological course of this disorder [17,18], due to the disease developing from a combination of lifestyle, environment, and genetic risk factors that affect the brain over time [19,20].
One of the most common neuropathological hallmarks of AD is the misfolding and aggregation of amyloid plaques-extracellular insoluble deposits of the β-amyloid peptides [21], and the intracellular formed NFTs (neurofibrillary tangles) [22], leading to the loss of communication between nerve cells, causes brain damage and shrinkage [23]. Posterior cingulated cortex (PCC) [24], entorhinal cortex (EC) [25], hippocampus (HIP) [26] (the first part to be affected by AD), middle temporal gyrus (MTG) (role in cognitive functions such as language processing), and superior frontal gyrus (SFG) (helps in memory) [27,28] are the regions affected in this multifactorial neurological disorder. Some studies have identified the impaired function of the middle temporal gyrus [29] and superior frontal gyrus in AD [30].
Extracellular deposits of Aβ peptides in Alzheimer’s are the main pathological events in AD [31,32,33,34]. Senile plaques or amyloid plaques mainly consist of small amyloid beta-peptides (Aβ) (up to 42 or 43 amino acids long) [35]. These are β amyloid precursor protein (APP) metabolites, derived by proteolytic sequential cleavage, first through β-secretase and then with γ-secretase, in the amyloidogenic pathway of producing peptides (Aβ), which contain 39 to 43 amino acids [36]. The APP (main isoforms, APP(695), APP(751), and APP(770)) is a type 1 transmembrane glycoprotein, which is essential for neurogenesis, neurite outgrowth, neuronal guidance, synapse formation, and repair [37,38,39]. The reason for neuritic plaques (senile plaques) forming in AD is due to irregularity between the production and removal of the beta-amyloid protein that accumulates [7]. Hence, the amyloid cascade hypothesis postulates that aggregation and accumulation of Aβ is the first pathological event in AD onset and initiates a cycle of adverse physiological changes that lead to neurodegeneration.
Another study has investigated Aβ aggregations in the senile plaques and co-localization of adenosine receptors in the AD [40]. Recently, some investigations have been done on adenosine, a purine ribonucleoside, because of its neuromodulator and neuroprotection function in neurological disorders [41,42]. It is present in all cells containing glia and neurons, initiates its biological process by four G-protein coupled receptors (GPCRs), namely, the A1, A2A,…A2BAR [43,44]. It has a role in regulating and integrating neuronal excitability, affecting many essential brain activities like sleep, memory, and neural plasticity [45,46,47]. Much research has analyzed adenosine effects via its receptors A1 and A2A in AD [48]. Nonselective blockage or modulation of these two receptors could protect cognitive impairment, making them innovative feasible therapeutic agents for AD [49]. Hippocampus, a brain region important for memory, learning, and neurogenesis [50,51,52], is one of the earliest affected brain regions that tends to exhibit the most rapid volume loss in the disease progression, and its pathology was found to be central to AD [50,53,54].
The hippocampus is a sensitive part of the brain to the dysfunctional homeostasis of transition metals, more so than any other brain region. Much research has also identified another brain part, the cortex, which is damaged by AD [55,56,57], linked with motor function, planning, organization, argumentation, feeling, and language processing [58]. NFTs are mainly composed of the microtubule-associated protein tau, predominantly expressed in the neurons under physiological conditions. This protein is mis-sorted into the somatodendritic compartment due to the tau sorting process’s failure, which is another essential factor that aggregates in AD [59]. Microtubules are essential components of a neuron’s cytoskeletal system, required for several fundamental cellular and dendritic processes, such as neuronal migration, polarity, axonal production, and differentiation [60,61]. Abnormal Aβ production might lead to the activation of tau mis-sorting, inducing tau pathology [62,63].
Multivalent metal ions such as copper (Cu) [64,65,66], zinc (Zn) [67,68], and iron (Fe) [69,70] are reported to be at higher levels in Alzheimer’s senile plaques [71,72]; while the connection of these metal ions with Aβ aggregation is still not well known. Indeed, some evidence from transgenic animal studies shows that Cu accumulates in senile plaques in the brains of 5 × FAD and Tg-SwDI/NOS2−/− mice models with neurodegeneration, as compared to PSAPP, where no Cu deposition has been seen among the mice with less neurodegeneration [73]. Much research has accumulated on Zn and Cu ions’ altered homeostasis as the central pathological hallmark [74,75,76] and shows the link of proteins related to Cu metabolism with this multifactorial AD [77].
Considerable research has suggested that Cu dyshomeostasis contributes to the onset of the most common neurodegenerative disorders besides AD, including Parkinson’s disease, prion-mediated encephalopathies, Huntington’s disease (HD), and amyotrophic lateral sclerosis (ALS) [78,79,80,81]. Hence, circumstances leading to a higher or lower copper concentration can be hazardous to health, such as Menkes diseases, a genetic disorder of Cu deficiency [82,83]. Furthermore, an autosomal recessive disorder, Wilson disease (WD), caused by defects of the ATP7B gene with excessive copper deposition in the body and patients’ brain examinations have shown copper concentration eight times greater than the controls [84].
Contradicting reports about the Cu concentration in AD has been reported. Some researches indicate a copper deficiency [85,86,87,88], while the majority show its higher level in AD, and therefore, reducing its level is required [89,90,91,92,93,94,95]. Investigations have grown exponentially in the neurodegenerative disorder fields over the past two decades. However, AD’s exact etiology is still not well understood, and as such, there is no successful therapeutic option available for this disorder to date [96,97].This literature review aims to present current knowledge regarding Cu’s role in AD. Towards the end, a short review of feasible therapeutics/strategies recommended for solving the problems associated with the metal’s implication in AD has also been discussed.
2. Copper Ion Implication in AD
Like other body parts, the brain contains many necessary transition metal ions, such as cobalt, copper, chromium, iron, zinc, and non-essential metals. Generally, the brain is the part of the body that contains the highest amount of transition metal ions content per weight. In comparison, the content of the copper ion in the brain is 0.004 g per kg [98]. It is an important chemical component of cell biology because it can receive and donate electrons. Once delivered and spread in a body, the cycle of Cu ions as the cupric ion (Cu2+) in its higher oxidation state and cuprous (reduced) form (Cu+), often joined to cuproenzymes with a small proportion as labile Cu, which was named as free or unbound Cu [99]. As a redox catalyst, Cu is necessary for many enzymes’ catalytic activity, regulating various cellular, biochemical, and regulatory processes. This metal plays an essential role in the catalytic centers of metalloproteins, electron transfer (ET) sites, and structural components.
Many studies have provided information concerning high serum levels of non-Cp–Cu, which results in reduced cognitive function, and the rate of mild cognitive impairment (MCI) to AD increased [90,100,101]. Postmortem biochemical analyses of the AD brain have revealed the reduced total soluble Cu levels, while its content within insoluble neuritic plaques is raised [102,103,104]. However, despite the decreased total Cu level in the central nervous system (CNS), elevated levels of redox-active exchangeable Cu are found in the Brodmann (BA46) and the temporal lobe (BA22) areas. AD cortical tissue has an increased propensity to bind exchangeable Cu2+ with increasing oxidative damage and neuropathological alterations, which have been seen in Alzheimer’s cases [105].
2.1. Copper and Amyloid-Beta Precursor Protein
Many authors have cited several models on copper implication in AD. Indeed, the most approved have put forward the “gain-of-function” of Aβ after binding Cu2+ ions [106]. Alternatively, current hypotheses suggesting “loss-of-function” of Aβ as the pathology of this disorder [94,107]. However, APP exports metal from neurons and a lower level of the soluble, functional Aβ monomer may lead to copper accumulation in the cell [107].
APP can bind to Cu2+ and reduce it to Cu1+ through its copper-binding domain (CuBD). APP can strongly bind Cu2+ to the N-terminal resulting in the decrement of copper ions [108]. Genetic studies of animal models have suggested that the APP-induced conversion of Cu2+ to Cu+ increases copper ion removal from the brain; this process could justify the point of why Alzheimer cases show lower brain level and higher Cu content in their serum-plasma [100,101,103,109,110]. However, Cu-binding with the N-terminal domain of APP may manage other functions of this protein, including synaptogenic function, stability, and metabolism [111,112,113,114]. Interestingly, the lower the copper content of the brain, the higher the ratio of endocytosed APP, and the generation of Aβ maybe works as a defense mechanism to stop the unnecessary loss of Cu [114,115]. Though newly produced intracellular Aβ can remove Cu, it probably leads to dyshomeostasis of copper ions and Aβ peptide deposition into plaques. This process results in neuritic plaques formation because Cu ions increase Aβ accumulation and cell damage due to the production of reactive oxygen species in AD [116,117,118].
Amyloid plaques or senile plaques mostly consist of the Aβ peptides, the essential peptide whose presence at a nanomolar concentration is shown by numerous studies in the cerebrospinal fluid (CSF) as well as in serum [119,120]. However, the TASTPM animal model study pointed out that the concentration of Cu in the Alzheimer’s brain does not link to plaques deposition [121]. The affinity of Cu2+ ions for Aβ peptides is very high [122,123], and it also increases the portion of beta-sheet and alpha-helix in Aβ proteins, which may be the cause of its aggregation [124]. So, β-amyloid deposition is the reason behind pathological alterations in AD, and its clearance when patients are immunized does not stop this disorder [125,126,127,128,129]. However, some scientific studies reported the presence of neuritic plaques in the brains of cognitively healthy elderly [130,131,132,133].
The soluble oligomers obtained from the culturing of cells possess high chemical resistance and protect against its conversion into monomers via several degrading factors and maintain the presence of covalent cross-links in them [134,135]. Binding of Cu2+ ions increases dityrosine-linked β-amyloid dimers as observed in vitro studies of this neurological disorder [136,137,138,139]. This dimer structure switches from parallel to anti-parallel in the presence of Cu2+ and this process is regulated with the occupied binding sites of Cu [140]. Moreover, the same scholars later demonstrated that the nanomolar content of Cu2+ has no impact on peptide–peptide bonds of dityrosine-linked β-amyloid dimers [141]. Another study has shown that Cu2+ ions binding results in structural variations in the β-amyloid dimers, causing oligomer-defining interactions, including N-terminal interaction in them [142]. The mutant dimer does not make dityrosine cross-links because of tyrosine10 (Y10) mutation to alanine on Aβ, and it is not linked to neurotoxicity (Figure 1) [143].
A meta-analysis [144] and the following investigations [105,145] revealed a lower total Cu in AD, while the level of labile Cu is higher in most of the brain areas affected by this disease [105]. Alzheimer’s brain tissues and the cortexes of transgenic animals with severe brain damage showed the high Cu2+ ions-binding capacity [105,146]. Additionally, the APPsw/0 mouse model study reported parenchymal Aβ plaques, but no damage to neurons has been observed (Table 1) [147,148].
In relation to neuroinflammation, copper performs essential roles in the activation of microglia. However, there is insufficient data present about this. Scientific studies have suggested that Cu increases Aβ toxicity, and the microglia with fibrillar Aβ results in phenotypic activation, and the activated microglia is neurotoxic and causes neurodegeneration [160]. Cu-Aβ complex causes activation of microglia and the release of tumor necrosis factor-α (TNF-α) and nitric oxide (NO) in an NF-kappa B dependent pathway [89,161]. Recently, the study by Kitazawa (2016) [162] indicated that copper-Aβ complex attenuated microglial phagocytosis of BV2 and improved the release of TNF-α and interleukin-1 beta (IL-1β), which results in reducing expression of lipoprotein receptor-related protein-1 (LRP-1). Reduction in the level of LRP-1 leads to further impairment in the transcytotic Aβ clearance and increased neuroinflammation [163]. Indeed, a study showed that a trace level of Cu increases the Aβ induced neurotoxicity in the cholesterol-fed mouse through the inflammatory pathway; but, no effects of inflammation have been seen when treated with copper or cholesterol only [164]. Activated microglia expresses the ATP7A, also known as the Menkes protein (MNK), which is indicated to be gathered around the plaques by histological investigations.
Interestingly, the expression of ATP7A has been determined to be increased by interferon-gamma (IFN-γ), which is a pro-inflammatory cytokine but not by TNF-alpha or IL-1beta [165]. The inflammatory process linked with AD has been shown accompanied by the altered microglial copper homeostasis in the disease. Remarkably, the copper-deficient diet-fed mice showed symptoms of activated microglia and astrocytes, proposing Cu homeostasis is required under physiological conditions to stop neuroinflammation [166]. Moreover, some studies have suggested that copper homeostasis controls pro-inflammatory and anti-inflammatory phenotypes shift in microglia cell, by the nitric oxide regulation and disruption of S-nitrosothiol signaling [167,168]. However, further study is needed to understand the underlying etiology of how Cu controls the CNS immune responses, especially its function in the clearance of pathological hallmarks of AD, such as β-amyloid and tau, which may provide a new drug target for AD.
Cu2+ induced fibril formation at physiological pH because it is a highly pH-dependent process. However, amorphous aggregation occurs under the acidic environment [142,169]. Hence, the misfolding of Aβ40 and Aβ42 in the brain are neuropathological hallmarks of AD. Moreover, the molecular mechanism of its aggregation in vivo is still unclear. However, metal ions affect their deposition in vitro. Aβ42 aggregates much faster than the most common form Aβ40, and more toxic to neurons than Aβ40, even though Aβ42 differs from Aβ40 by only two (IA) amino acid residues at the C-terminal end. As Aβ40 contains more than one binding site of Cu, the second Cu2+-binding site interferes with the aggregation of Aβ40 to the amyloid fibrillar state in a proton-rich environment [152].
2.2. Copper and Tau Protein
Autophagic-lysosomal flux is a lysosome-dependent cellular degradation program that plays an essential role in the clearance process of abnormally modified cellular proteins. Much data have suggested that endo-lysosomal/autophagic dysfunction is responsible for soluble oligomeric forms and insoluble forms of tau aggregation (Figure 2) [170,171].
The tau protein has also been investigated for its Cu binding, which plays an essential role in NFTs production [157,172,173]. Tau protein shows redox activity when it binds to copper, causing oxidative damage to the brain tissues [174]. However, despite tau as the main pathological hallmark of AD, only a small proportion of research has been done to check its link with Cu’s dyshomeostasis. Further studies are needed to evaluate Cu function in tau kinases and phosphatases and their role in cognitive impairment. These works will also increase our knowledge of the neuropathology of AD (Table 1).
2.3. Copper and ROS Production
Reactive oxygen species (ROS) generation is associated with a redox-active copper ion complex with aggregated Aβ, which has been identified to contribute to oxidative stress and damage to neuronal cells in AD [175].Copper-Aβ fibrils complex produces hydrogen peroxide (H2O2) in the presence of ascorbic acid, a biological reductant [175,176]. Increment in the ratio of (Cu-Aβ) leads to the production of H2O2, hydroxyl radicals (OH•), and misfolding of proteins (aberrant aggregates) shifts from amyloid fibrils to amorphous aggregates [116]. While initial studies have shown ROS being dangerous to causing neurodegeneration, the recently gathered data suggest some ROS action is necessary for cognition function and memory development [177,178,179,180,181]. According to some results, it has been suggested that the Cu-Aβ complex generates less ROS than unbound Cu ions [182]. Cu ions interaction with Aβ results in its accumulation under a slightly acidic environment and promotes ROS production in Alzheimer’s patients [183]. The production of ROS due to metal ions such as Cu leads to oxidative damages to Aβ peptide. This oxidized β-amyloid has been seen in senile or amyloid plaques during in vivo studies [184]. Some in vitro data imply that oligomeric and fibrillar forms of Aβ prevent hydrogen peroxide production at high concentrations of Cu2+. Additionally, amyloid fibrils produce less hydrogen peroxide than that in the oligomeric state [185].
However, the pro-oxidant function of the Cu-Aβ complex is not confirmed yet, because this complex is more effective in ROS generation than several tested biological relevant Cu-peptides and Cu-binding proteins [186] but less efficient than loosely-bound Cu [182,187,188,189]. It is usually stated that hydrogen peroxide production is a two-electron oxidation process; however, current research has indicated the production of superoxide (O2−) as an intermediate in hydrogen peroxide formation via Cu-Aβ complex and oxygen [190]. Cu is redox-active and, when bound to Aβ, catalytically cycles between the Cu+1 and Cu+2 oxidative states to generate ROS such as O•−2, OH•, and H2O2. Thus, the coordination of amyloid-β with Cu ions plays a significant role since ROS generation is a metal-catalyzed process, termed as a catalytic in-between state [191]. Therefore, computational studies have also examined Cu’s role in that state and its reactivity to the substrates such as oxygen or hydrogen peroxide [192,193,194].
Toxicity due to Cu ions in AD has been linked with the oxidant form of Cu ions such as Cu2+ [65,195]. Considerable studies found that the elimination of Cu+ from Aβ inhibits the production of Aβ oligomers and oxidative damage [196], and Cu1+ has a stronger affinity to monomeric Aβ peptide than Cu2+, which leads us to propose that Cu1+ cation is principal in the oxidation state in vivo [197].
Contrarily, the same metal ions also exist as catalytic metal ions, like Cu in SOD1, where they stop producing the H2O2. This also proves the value of coordination compounds. Copper can be in both pro-oxidants and antioxidants, which depends on its coordination position in compounds. However, in AD, higher production of ROS or less activity of the enzymes which degrade ROS results in an imbalance of pro-oxidants and antioxidants form, which cause oxidative damage on biomolecules [176]. Therefore, it is now clear how important it is to regulate copper ions’ metabolism in terms of content, transportation, storage, and association with active sites.
Abnormal Cu homeostasis increases the levels of free or loosely bound copper, which often produces ROS [98]. These ions can also attach to off-target biomolecules and disrupt their functional roles, leading to higher chances of oxidative damage.
Perhaps the Cu-Aβ complex is directly linked with ROS generation, so most of the studies have been associated with Cu-Aβ complex, suggesting a direct link between AD and oxidative damage [182]. Remarkably, all of the above data highlights the point that Cu is highly toxic in excess amounts and responsible for its participation in a redox-cycling reaction, which produces ROS that results in much damage to biomolecules such as carbohydrates, nucleic acids, lipids, and proteins. So, a higher level of free Cu ions causes more toxicity to the cells and eventually leads to cell death. Therefore, cellular Cu should be tightly controlled (Figure 3) [99].
2.4. Copper Deficiency and Cholesterol Rich in AD
A lower level of net copper was observed in the TgCRND8 mice model, with parenchymal amyloid aggregation but no loss of neurons [148,198]. The reason for early-onset familial AD is mutations in genes of proteins essential for mammalian systems in copper ion uptake [199]. As discussed before, a meta-analysis indicates a copper deficiency in the brain of Alzheimer’s cases [144]. Another study also showed a copper deficiency in the deceased Alzheimer’s brain’s defective regions with dementia symptoms [18]. Cu concentration of the elderly has a direct relation with Aβ aggregation [86]. While proteolytic cleavage of APP is a two-step pathway; non-amyloidogenic APP processing pathway and amyloidogenic pathway, in the presence and deficiency of Cu, respectively. Some results suggest the interaction of copper ions with a γ-secretase complex can inhibit amyloid production [200].
Based on the results of comparing blood copper levels of AD patients with healthy controls, which shows a significant reduction in copper ion, it has been hypothesized that Cu deficiency can lead to pathological hallmarks of AD [201,202]. An alternative study, which describes meta-analyses results of the copper quantification in serum-plasma and the brain, suggested Cu deficiency in the brain is a symptom of Cu dyshomeostasis, which relates to Wilson’s disease [203]. While, the dietary copper addition leads to an increase of intracellular copper concentration in APP/PS1 AD mice [155], which has been shown in parenchymal Aβ plaques, a decrease of AD pathology, but no loss of neurons seen [147,148]. Interestingly, some results showed that in AD patients, Cu deficiency does not have any link to their diet [201].
There is substantial proof about the amyloidogenic pathway’s connection with lipid raft, which is a particular cholesterol-rich microdomain. Although the deposition of Cu ions is associated with their cellular deficiency, the Cu level in lipid rafts has been inversely associated with the cellular Cu level, the simultaneous enrichment of Aβ and Cu within lipid rafts leads to higher redox-active Cu-Aβ complex formation in the absence of Cu conditions of AD [114,204]. Seemingly, a high-cholesterol rich diet plays an essential role in the AD pathology. Indeed, many investigations determined that lipids are a necessary part of this disorder [205,206,207]. Higher Cu2+ and lipid content in the neurodegenerative diseases have also been described by some recent scientific studies [208,209]. When the transgenic AD mice treated with Cu and cholesterol-fed diet, the ratio of Aβ42/Aβ40 increased, and a significant difference in the visuospatial memory was identified [208]; furthermore, in the rabbit brain, Aβ accumulation increases with the feeding of cholesterol food and Cu containing water (Cu ion in the form of Cu sulfate) [210].
Cholesterol-rich regions have also been detected for the enzyme activity of the cleavage of APP to amyloid proteins in AD brains [114,211]. Amyloid proteins attached to the plasma membrane surface and the Ca2+ ions help to penetrate the phospholipid bilayer [212]. The formation of Aβ22-35 channels is a cholesterol-dependent process and regulated with small cholesterol (~30 mol%) in phospholipid membranes. However, these channels cause an imbalance in Ca2+ homeostasis in neuronal cells and result in the bring-up of the Ca hypothesis of Alzheimer’s disease (Figure 4) [213,214]. Contrarily, Cu ions do not cause neurotoxicity in the absence of amyloid peptides [153]. Earlier unsuccessful therapeutic efforts and recent results about the aggregation of Aβ peptides in the cholesterol-rich regions (lipid rafts) lead to a different hypothesis that soluble Aβ oligomers (AβOs) associated with the cell membrane are responsible for neurotoxicity in AD [215,216,217].
3. Contradictory Results about Copper Level in AD
In the body cells, Cu is absorbed through a high-affinity copper transporter Ctr1, incorporating cuprous (Cu+) ions from the intestinal microvilli’s surface. Little is known about Cu2+ absorption, which is probably absorbed by divalent metal transporter 1 (DMT1) or other shared metal transporters [218]. Ctr1is responsible for the majority (~70%) of Cu import into mammalian cells, from which Cu is passed to glutathione, which carries Cu through the cytoplasm [219]. The absorbed copper ions will be targeted to Cu-binding chaperones and enzymes in different cell compartments such as cytosolic, mitochondrial, and Golgi. In the cytosol, Cu chaperone for superoxide dismutase 1 (SOD1), CCS, mediates Cu+ loading. A recent study suggested that the direct transfer of copper from Ctr1 to chaperones and then passing it to SOD1 is via forming a Ctr1-CCS-SOD1 complex [218]. Besides CCS, soluble copper chaperones such as Atox1 and Cox17 can also escort Cu+ from Ctr1 in the cytosolic pool to facilitate copper supply to their specific target compartments [220].
Consequently, in the absence of Ctr1, other pathways to absorbed Cu ions are unavailable to the organism because of the sequestration of copper in the sub-apical vesicles. This has been confirmed by making the intestinal epithelial cell-specific knockout of the Ctr1 (Ctr1int/int) mice, which manifested severe Cu deficiency, and the majority died within three weeks of post-birth [221]. A considerable portion of ingested cuprous ions are passed into circulation in enterocytes to reach different tissues by Atox1/ATPase routes. The mouse model with inactivated ATOX1/ATP7A routes showed defects in Cu distribution, which leads to pathological variations in many organs, especially the brain [222].
In the CNS, Cu deficiency has been found in the hippocampus and amygdala regions of Alzheimer’s patients, which causes severe histopathologic alterations in AD. Additionally, scientific research has put forward that the frontal cortex tissue of Alzheimer’s patients had an increased susceptibility for exchangeable copper (CuEXC), which is associated with the overproduction of free radicals (ROS) in AD [223].
In the CSF of the AD patients, there is no significant change in Cu concentration as compared to that of the healthy cases (HC) [224]. Furthermore, within peripheral fluids, abnormal homeostasis of copper ions has been intensively investigated. The relevant data point to increased [224,225], decreased [88,226], or unchanged [227] serum or plasma Cu in Alzheimer’s patients. Many other scientific analyses have also reported excessive free or diffusible copper in serum [224,226,228]. However, Rembach (2013) has suggested the possibility of decreased non-CP copper levels-copper that is not bound to ceruloplasmin in mild cognitive impairment (MCI) and AD, which leads to a decline of copper-dependent biochemical activities in AD [229], such as reducing SOD1 activity of erythrocytes [88].
Cu association for AD is ambiguous as some substantial researches showed Cu deficiency in AD and, hence, it is required to increase Cu levels [86,87,88]. In contrast, many different scientific pieces of evidence demonstrated Cu overload, and thus it is necessary to reduce it [90,91,92,93,94,95]. The main updated explanation so far is that the abnormal Cu homeostasis is due to an increment in the labile Cu ions and a reduced attachment to proteins [107,174].
Until 2012, the published contradictory scientific researches fueled the debate of copper concentrations in AD. So far, to check Cu levels in various biological specimens of AD patients, such as serum, plasma, and CSF, six meta-analyses have been done during the past six years. Studies published from 1984 to 2017 have been included in these meta-analyses [100,101,224,230,231,232], which give unambiguous results: overall and unbound Cu both are present in higher concentrations in the serum-plasma samples of the AD patients compared to that in the healthy cases [230]. According to the very recent meta-analysis, which includes a total of 35 pieces of research: eighteen report an increase, fourteen show no change, and one reports a decrease in Cu level in the serum-plasma of this disorder [232]. Subsequently, three more studies have been published, stating increased Cu2+ ions level in Alzheimer’s compared to that in the healthy controls [233,234,235].
These recent researches have contributed considerably to the explanation of the previous controversy. In blood, a higher level of free plasma Cu, which has been identified in 50–60% of Alzheimer’s patients, can explain the higher level of serum Cu in AD [145,174,233,236]. Another earlier research also observed an increased concentration of serum copper ions in a special kind of AD (Alzheimer’s disease epsilon four apolipoprotein E allele carriers) [237]. According to some scientific investigation, a genetic basis may be the reason for this particular type of AD [237,238,239,240].
4. Therapeutics to Tackle Copper Ions in AD
Despite the exponential growth of scientific literature published in the neurodegenerative disorders area, especially for AD, the exact etiology of AD is still not well understood. To date, there is no successful therapeutic option available for this disorder [96,241]. While there is no cure, there are five FDA-approved medications to cope with the symptoms of AD, which may prevent this disease from getting worse over time [242].
In vitro, removal of Cu2+ from Aβ prevents its accumulation [243,244,245], leads to its degradation, stops hydroxyl radical (•OH) production and oxidative damage, and finally reduces cell death [245]. For the effects as mentioned above, researches have suggested potential metal chelation therapy for AD [246,247,248,249,250,251,252]. Nevertheless, the challenge is to build selective and specific metal chelators, as metal ions play crucial roles in Alzheimer’s brains. The first metal chelator made for arsenic toxicity in the 1940s was 2,3-Dimercaptopropanol (BAL) [253,254]. Much later, followed by the same approach, the first-generation of metal chelator, a lipophilic small molecule clioquinol (5-chloro-7-iodo-8HQ or CQ) was introduced at the end of the 1990s [244,255]. Transgenic mouse models treated with CQ showed promise by reducing Aβ accumulation by 50%. CQ reduced Aβ aggregation during Phase II trials and improved cognitive behavior, but failed to provide sufficient evidence of a positive clinical benefit in a larger clinical trial [256,257]. Furthermore, patients exhibited some severe side effects, including neurotoxicity and mutagenicity; therefore, further clinical trials of CQ were stopped.
The most progressive chelator so far is PBT2 (5,7-dichloro-2-((dimethylamino)methyl)) [258], a second-generation of scaffold-based chelator, which has been inspired by CQ and also showed excellent antioxidant properties [259,260,261]. It is a more effective Zn/Cu ionophore than CQ, which could decrease H2O2 formation, have greater BBB (blood-brain barrier) permeability, higher solubility, and could also inhibit Cu and Zn induced Aβ accumulation in vitro [261,262]. PBT2 treatment targets metal-induced damage [263], and most importantly, it prevents the loss of necessary metal ions from the body such as the kidney, liver, lungs, and brain [241,264]. It also shifts the Alzheimer’s phenotype within days by reducing insoluble Aβ levels by ~30% [261] and alters tau and synaptophysin protein levels’ phosphorylation. Interestingly, a lowered level of insoluble total and elevated levels of the soluble total tau has been shown in the treatment with PBT2 [259]. Despite the effects mentioned above, the results of human clinical trials are not up to the mark according to some studies [241,265]. However, some scholars have denied this idea [266]. Results from the phase IIb, the randomized clinical trial, were not as promising even though phase Ib/IIa preclinical trials demonstrated significant reductions in Aβ levels and improvement in various aspects of cognitive functioning.
The research in the Tg 2576 transgenic mice model has shown parenchymal plaque [147,148], indicating that metal chelators help slow disease progression and remove Cu ions only helpful in the initial stages of the AD [267]. While PS1 and PS2 play roles in Cu2+ uptake, tissue-specific knocking down of the single presenilins ortholog (PSN) in Drosophila reduces Cu2+ levels and increases its susceptibility to oxidative insult [199]. It was observed that the silencing of PSN in flies had less sensitivity to excess dietary Cu due to the reduced copper uptake. BLOC-1 physically interacts with ATP7A, and disruption of the Drosophila’s dysbindin/BLOC-1 complex affects copper homeostasis in both mammalian cells and Drosophila [268].
Different approaches have been used to treat the pathological hallmarks of the multifactorial AD due to Cu dyshomeostasis, including the metal chelation therapy [261,269,270,271]. Restoring the intracellular copper decreases β-amyloid production, which was found through a mechanism that depends on the activation of phosphatidylinositol 3-kinase (PI3K)/PI3K-Akt pathway, and JNK (Jun N-terminal kinase) [200,272]. Moreover, lately, studies have observed increased intracellular Cu inhibited AD-causing Aβ peptide by direct targeting of presenilin (PS1 or PS2) subunits and nicastrin (NCT) in the γ-secretase complex [64,200,273]. Hence, higher intracellular Cu levels can improve cognitive function as well, by preventing β-amyloid aggregation and tau phosphorylation [155,274,275]. There is proof of the bis(thiosemicarbazone) copper(II) complex having the immunomodulatory potential [276,277], and greater BBB permeability. It inhibits microglial as well as astrocytic inflammatory responses and also has a role in the decrement of bacterial lipopolysaccharide (LPS) induced inflammation [278]. Some researchers have suggested that excess dietary Cu intake increases AD risks, and diets with measured copper should be supported. Additionally, a study conduct with a small amount of Cu in drinking water results in rising levels of amyloid peptides in the brain [162,207], a process that appears to be linked with dysfunction of LRP1-mediated efflux of Aβ from the brain [279] in vascular smooth muscle cells [280,281]. The median intake of copper from food among children and adolescents aged 2–19 years, the recommended daily allowance (RDA) ranges from 800 to 1000 mcg/day. In adults aged 20 and older, 1400–1700 mcg/day is recommended. Despite the difficulties, balancing Cu homeostasis has numerous advantages, and can be a potential drug target for this progressive, neurological disorder [89,156,282,283,284].
For other neurological disorders, such as Wilson’s disease (WD), a different chelating agent, tetrathiomolybdate (TTM, an ammonium salt), appears to be a promising alternative, which can act by inhibiting copper uptake. TTM had the advantages of being fast-acting and did not lead to neurological deterioration in WD patients. It can restore normal copper balance without increasing serum “free copper” within several weeks compared to other copper chelators or zinc salts requiring several months [285]. However, the ammonium formulation has been proven too unstable for routine use, so clinical experience with them remains limited. Of note, bis-choline salt of TTM, WTX101, has recently become available on a named patient basis in the USA and Europe. This complex is more stable than TTM and phase III FOCUS study compared to standard of care (SoC) in WD patients was started in 2018, with results expected in 2020 [286].
5. Multifunctional Chelators (MFCs) to Control Metal Mediated Abnormalities
Because multiple pathological variables are involved in the pathology of AD, therapeutics or drugs target a single mechanism that is not enough to treat this disorder. New therapeutics or medicines that can target multiple factors at the same time can be beneficial for patients suffering from neurodegenerative disorders. It is now undeniable that the next generation of therapies should have the ability to target the different causes of disease progression at the same time [287]. Neurodegenerative disorder drugs must have more than one of the following properties to be useful for these diseases such as control of the production of ROS, the ability of metal chelating, and greater BBB permeability, a decrease of β-Amyloid peptide deposition (Figure 5), and last but not least, of regulating enzymes associated with the mechanism of the disease, for example, acetylcholinesterase [288,289].
Bifunctional metal chelators (BFCs) were suggested to treat multifactorial AD because they have the ability of both metals chelating and binding with amyloids. Substantial research has been made in this field in the last decade [259,263,290,291]. The fluorescent dye thioflavin T (ThT), also known as Basic Yellow 1 or CI 49005, has been widely used to detect amyloid fibrils [292] in both in vivo and in vitro studies. The first bifunctional chelator designed was XH1 (Figure 6), connecting various molecular fragments of different specificities to make a hybrid molecule [293]. Its structure is composed of two-terminal thioflavin-T-derived moieties, which are attached by a DTPA (diethylene triamine penta-acetic acid) binding unit.
Besides metal ions’ chelation role in Alzheimer’s, new chelating molecules such as phenyl benzotriazole followed by the same design principle of thioflavin-T(ThT), correlating them with dipicolylamine or pyrinophane type metal chelators have been reported by the studies [263,287,291,294]. Several studies have analyzed the deposition of Aβ1-42 in the deficiencies and presence of essential metal ions [291,295]. Franz and co-workers have used persuasive strategies to design prochelators in 2006. These chelators only work in the presence of oxidative stress and inhibit essential ion loss like Cu and Zn of metalloproteins. Followed by the same approach, boronic ester (BSIH), an excellent first-generation prochelator metal affinity group, was composed. It works as an iron chelation with salicylaldehyde isonicotinoyl hydrazone (SIH), in the presence of H2O2 [296].
Various analogs of boronic ester, such as boronic esters (BSIH, BSBH) and acids (BASIH), have been investigated to check their performance as metal chelators [297]. Currently, many promising molecular scaffolds using the same strategy are exploring their effect on the multifactorial AD [298,299,300,301]. Choi (2011) and Hindo (2009) [302,303] reported the derived compounds and analyzed their impact on metal-binding properties, β-amyloid deposition both in deficiency and the presence of metal ions. Currently, small novel compounds such as 2,2-bipyridine (bpy) derivatives (1–4) and other N,N-dimethylaniline including novel N-bidentate ligands, have been described to show good results for the treatment of multifactorial AD [304,305]. The effects of several flavonoids such as myricetin and EGCG (epigallocatechin gallate) have also been tested for this disorder [306,307,308].
While Orvig described salen-type Schiff-bases in addition to other chelating agents for the first time, it has also been connected with carbohydrate moieties [259,309,310,311,312]. In vivo investigation of the diacetylbis(N(4)-methylthiosemicarbazonato) copper(II) (CuII(ATSM)) (Figure 6) compound has seen its protection against nitrosative damage of peroxynitrite and an increase of survival in the ALS mouse models [313]. Unfortunately, it was not studied much for metal chelation and protection against oxidative damage in AD. Many multifunctional molecules have been rationally designed to simultaneously resist and target various pathological hallmarks of the brain disorders, and their effects have been checked and reviewed by many studies [290,314]. Finally, despite all tremendous efforts that have been made for the success of multifunctional molecules as a potential therapy, only a handful of them show promising results in human clinical trials. Indeed the usage of these compounds to target the multi-mechanisms of the neurodegenerative diseases, its potential causes of failure also need to be understood, which has been reviewed elsewhere [122,265]. Overall, it is an effective strategy and will hopefully provide a better therapeutic option for Alzheimer’s cases when the compounds made become more tissue targeting specific.
6. Efficacy of Therapeutic Chelation
There are a large number of metal chelators that have been developed to cure AD. Indeed, only a few of them made their way to clinical trials [315,316]. To discriminate the bulk of chelation therapies, which mostly link with the release of heavy metal poisoning, these used therapeutic chelators have been named ionophores, metallochaperones, and MPACs (metal-protein attenuating compounds). CQ (PBT1) and PBT2 are the most popular MPACs for AD, both were designed based on old chemistry with different applications, and the term MPACs was popularly used because it was a belief that PBT1 and PBT2 cause deaccumulation of β-amyloid plaques loaded with Cu and Zn ions [317,318]. Terdendate ligands (L), such as PBT2, make bonds in a 1:1 ratio (Cullin 1), and a distortion occurs at 5-coordinate 1:2 ratio (Cullin 2) form, while the copper(II)-bound form of this class terdentate 8HQ, comprising peptides and side chains of proteins is predicting a ternary metal ion complex.
The word “ionophore” was used for a large number of cellular metal uptake experiments in vitro [261,319,320], while the name “metallochaperone” has now been suggested most of the times. Ionophores constitute a distinct subset of metal-binding drugs capable of moving multiple members of a given ion across cell membranes. The main difference lies between chelator and ionophores is in the functional result of the metal complex. Traditional medicinal chelating agents result in excretion of the absorbed metal from its receptor site into the system where it cannot exert toxicity and can make them bio-unavailable. In contrast, ionophores generally form lipophilic metal complexes that make the membrane permeable to specific ions, creating a more or less selective channel to particular ions. Hence, there is a possibility that 8HQs as carrier ionophores can work in the hydrophobic environment of several plasma membranes (PMs), and the Cu, which is not removed from 8HQs ligands, causes localization to phospholipid bilayer, and this results in 8HQs interference with bonds of heavy metals of essential regulatory enzymes [321,322,323] due to the formation of the ternary complex. Interestingly, the generation of ROS can be detected by adding such ligands to the culture of neural stem cells [324], in contrast to the founding principle of therapeutic chelation therapy [325].
Some therapeutic benefits of 8HQ therapeutic chelators have been suggested by transgenic animal studies of AD [261,319]. Indeed, chelation therapy in human clinical tests has not provided any satisfactory results so far. Chelation therapy using D-penicillamine has also not presented any proof of improving disease pathology and has to stop earlier in initial phases due to its side effects, causing some to question the usage of 8HQs [326]. Independent evaluation of the human clinical trials from 2006 to 2014 using 8HQs, frequently reported no advantage to AD patients [327,328,329]. Despite all these discouraging signs, the hypothesis is that 8HQs was successful in two human clinical trials. Post-hoc study of the Phase 2A trials claimed that PBT2 improves cognition in AD [330], though, the results of this clinical trial are in question. Another study by Ayton (2013) [331] also claimed the positive outcome of clinical trials. Therefore, some researchers were still showing promise for these compounds [325,332].
In short, according to Drew (2017) [265], there is a preference in describing results of clinical trials of Cu chelation as positive and helpful for Alzheimer’s cases, which results in continued checking of different chelators for the well-defined targets to treat the dyshomeostasis of metals in AD.
7. Conclusions
Collectively, studies strongly advocate that dyshomeostasis of Cu, leads to the onset and progression of AD. Earlier researches have recognized amyloid plaques as toxic factors in AD. Though 20–40% of healthy cases have amyloid plaques, as illustrated by some studies [333]. Furthermore, cell death often leads to amyloid plaque formation in the brain. While mounting evidence implicates ROS in the AD etiology, loosely bound copper ions are very efficient catalysts for ROS generation by a copper-amyloid complex [105,334].
Some studies indicate an increased liable pool of Cu in the brain [105,230] responsible for Cu deficiency. The reason of Cu deficiency seems to be an essential factor in AD. Copper deficiency leads to Cu enrichment in lipid rafts, so maybe an elevation in lipid raft domains could be the reason for Cu deficiency in the brain; thus, lipid raft domains could be an efficient drug target. Studies indicated that the disrupted lipid rafts (by omega-3 fatty acids) slowed the progression of AD [215,335]. Another direction for research depends on the feasibility of developing novel therapeutic approaches to work against this disease.
The proposal for direct chelation therapy of Cu ions to work in this disorder is still in discussion [265]. Support for the lowering cellular Cu levels comes from the Drosophila model of AD, where although copper chelation or genetic knockdown of copper transporters (Ctr1C) decreased the expression of Aβ degrading proteases but rescued the toxic phenotype [336]. Similar results were also observed by silencing the expression of Ctr1B, or when copper exporter DmATP7 [336] and dMTF-1 or MtnA [94] were overexpressed in the nervous system of the Aβ transgenic flies. These flies exhibited improved neurodegeneration, locomotion, longevity, and a reduction in Cu-Aβ complex-induced oxidative stress.
Furthermore, in parallel, antibody-based treatment for Aβ aggregation is now developing and providing safe results as well [337]. Research organizations should come to the same standpoint regarding the experimental requirements and procedures to be used, to avoid different and ambiguous results for such serious matters. In this context, all the struggles for a better understanding of AD pathology’s molecular mechanisms and developing innovative therapeutic approaches should be appreciated.
Funding
This work was supported by the National Natural Science Foundation of China (31571042), Beijing Municipal Natural Science Foundation (7202129), the Special Fund for University Teachers, the Institute of Scientific Research Cooperation at the Chinese Academy of Sciences (Y55201BY00, KJRH2015-011), the Key Basic Research Project of Applied Basic Research Program of Hebei Province (18966315D), and One Hundred Outstanding Creative Talents Support Program of Hebei (BR2-218).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Villemagne, V.L.; Doré, V.; Burnham, S.C.; Masters, C.L.; Rowe, C.C. Imaging tau and amyloid-β proteinopathies in Alzheimer disease and other conditions. Nat. Rev. Neurol. 2018, 14, 225–236. [Google Scholar] [CrossRef]
- Mullane, K.; Williams, M. Alzheimer’s disease beyond amyloid: Can the repetitive failures of amyloid-targeted therapeutics inform future approaches to dementia drug discovery? Biochem. Pharmacol. 2020, 177, 113945. [Google Scholar] [CrossRef]
- Testai, F.D.; Gorelick, P.B. Definition and Concept of Vascular Cognitive Impairment. In Stroke Revisited: Vascular Cognitive Impairment; Springer: Berlin/Heidelberg, Germany, 2020; pp. 1–14. [Google Scholar]
- Barber, R.C. The Genetics of Alzheimer’s Disease. Scientifica 2012, 2012, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.K.; Srivastav, S.; Yadav, A.K.; Srikrishna, S.; Perry, G. Overview of Alzheimer’s Disease and Some Therapeutic Approaches Targeting Aβ by Using Several Synthetic and Herbal Compounds. Oxid. Med. Cell. Longev. 2016, 2016, 7361613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerreiro, R.; Bras, J. The age factor in Alzheimer’s disease. Genome Med. 2015, 7, 106. [Google Scholar] [CrossRef] [Green Version]
- Wan, L.; Nie, G.; Zhang, J.; Luo, Y.; Zhang, P.; Zhang, Z.; Zhao, B. β-Amyloid peptide increases levels of iron content and oxidative stress in human cell and Caenorhabditis elegans models of Alzheimer disease. Free Radic. Biol. Med. 2011, 50, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Michaelis, M.L.; Michaelis, E.K. Functional Genomics of Brain Aging and Alzheimers Disease: Focus on Selective Neuronal Vulnerability. Curr. Genom. 2010, 11, 618–633. [Google Scholar] [CrossRef] [Green Version]
- Cummings, J.L.; Vinters, H.V.; Cole, G.M.; Khachaturian, Z.S. Alzheimer’s disease: Etiologies, pathophysiology, cognitive reserve, and treatment opportunities. Neurology 1998, 51 (Suppl. 1), S2–S17. [Google Scholar] [CrossRef]
- Sheng, M.; Sabatini, B.L.; Sudhof, T.C. Synapses and Alzheimer’s Disease. Cold Spring Harb. Perspect. Biol. 2012, 4, a005777. [Google Scholar] [CrossRef] [Green Version]
- Santos, C.Y.; Snyder, P.J.; Wu, W.-C.; Zhang, M.; Echeverria, A.; Alber, J. Pathophysiologic relationship between Alzheimer’s disease, cerebrovascular disease, and cardiovascular risk: A review and synthesis. Alzheimers Dement. Diagn. Assess. Dis. Monit. 2017, 7, 69–87. [Google Scholar] [CrossRef] [Green Version]
- van Norden, A.G.W.; van Dijk, E.J.; de Laat, K.F.; Scheltens, P.; OldeRikkert, M.G.M.; de Leeuw, F.E. Dementia: Alzheimer pathology and vascular factors: From mutually exclusive to interaction. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2012, 1822, 340–349. [Google Scholar] [CrossRef] [Green Version]
- Judge, D.; Roberts, J.; Khandker, R.; Ambegaonkar, B.; Black, C.M. Physician perceptions about the barriers to prompt diagnosis of mild cognitive impairment and Alzheimer’s disease. Int. J. Alzheimers Dis. 2019, 2019. [Google Scholar] [CrossRef] [Green Version]
- Leskovjan, A.C.; Kretlow, A.; Lanzirotti, A.; Barrea, R.; Vogt, S.; Miller, L.M. Increased brain iron coincides with early plaque formation in a mouse model of Alzheimer’s disease. NeuroImage 2011, 55, 32–38. [Google Scholar] [CrossRef] [Green Version]
- Torosyan, N.; Silverman, D.H.S. Neuronuclear Imaging in the Evaluation of Dementia and Mild Decline in Cognition. Semin. Nucl. Med. 2012, 42, 415–422. [Google Scholar] [CrossRef] [Green Version]
- Akhondzadeh, S. New Hopes for Treatment of Alzheimer’s Disease. Avicenna J. Med. Biotechnol. 2017, 10, 1. [Google Scholar]
- Conte-Daban, A.; Day, A.; Faller, P.; Hureau, C. How Zn can impede Cu detoxification by chelating agents in Alzheimer’s disease: A proof-of-concept study. Dalton Trans. 2016, 45, 15671–15678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickart, L.; Vasquez-Soltero, J.; Margolina, A. The Effect of the Human Peptide GHK on Gene Expression Relevant to Nervous System Function and Cognitive Decline. Brain Sci. 2017, 7, 20. [Google Scholar] [CrossRef] [Green Version]
- Mielke, M.M.; Leoutsakos, J.-M.; Corcoran, C.D.; Green, R.C.; Norton, M.C.; Welsh-Bohmer, K.A.; Tschanz, J.T.; Lyketsos, C.G. Effects of Food and Drug Administration-approved medications for Alzheimer’s disease on clinical progression. Alzheimers Dement. 2012, 8, 180–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nabeshima, T.; Kim, H.-C. Involvement of Genetic and Environmental Factors in the Onset of Depression. Exp. Neurobiol. 2013, 22, 235. [Google Scholar] [CrossRef] [Green Version]
- Ashraf, G.M.; Greig, N.H.; Khan, T.A.; Hassan, I.; Tabrez, S.; Shakil, S.; Sheikh, I.A.; Zaidi, S.K.; Akram, M.; Jabir, N.R.; et al. Protein misfolding and aggregation in Alzheimer’s disease and type 2 diabetes mellitus. CNS Neurol. Disord. Drug Targets 2014, 13, 1280–1293. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological Alterations in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef]
- Furcila, D.; Domínguez-Álvaro, M.; DeFelipe, J.; Alonso-Nanclares, L.J.F.I.N. Subregional Density of Neurons, Neurofibrillary Tangles and Amyloid Plaques in the Hippocampus of Patients With Alzheimer’s Disease. Front. Neuroanat. 2019, 13. [Google Scholar] [CrossRef] [PubMed]
- Leech, R.; Sharp, D.J. The role of the posterior cingulate cortex in cognition and disease. Brain A J. Neurol. 2014, 137 (Pt1), 12–32. [Google Scholar] [CrossRef] [Green Version]
- Maass, A.; Schütze, H.; Speck, O.; Yonelinas, A.; Tempelmann, C.; Heinze, H.-J.; Berron, D.; Cardenas-Blanco, A.; Brodersen, K.H.; Stephan, K.E.; et al. Laminar activity in the hippocampus and entorhinal cortex related to novelty and episodic encoding. Nat. Commun. 2014, 5, 5547. [Google Scholar] [CrossRef] [Green Version]
- Mu, Y.; Gage, F.H. Adult hippocampal neurogenesis and its role in Alzheimer’s disease. Mol. Neurodegener. 2011, 6, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bubbico, G.; Chiacchiaretta, P.; Parenti, M.; Di Marco, M.; Panara, V.; Sepede, G.; Ferretti, A.; Perrucci, M.G. Effects of second language learning on the plastic aging brain: Functional connectivity, cognitive decline, and reorganization. Front. Neurosci. 2019, 13, 423. [Google Scholar] [CrossRef]
- Jablonska, K.; Piotrowska, M.; Bednarek, H.; Szymaszek, A.; Marchewka, A.; Wypych, M.; Szelag, E. Maintenance vs. Manipulation in Auditory Verbal Working Memory in the Elderly: New Insights Based on Temporal Dynamics of Information Processing in the Millisecond Time Range. Front. Aging Neurosci. 2020, 12, 194. [Google Scholar] [CrossRef]
- Liu, X.; Chen, W.; Tu, Y.; Hou, H.; Huang, X.; Chen, X.; Guo, Z.; Bai, G. The abnormal functional connectivity between the hypothalamus and the temporal gyrus underlying depression in Alzheimer’s disease patients. Front. Aging Neurosci. 2018, 10, 37. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, L.G.; Jackowski, A.P.; Oliveira, M.O.; Ribeiro Flor, Y.M.; Souza, A.A.; Bueno, O.F.; Brucki, S.M. The thickness of posterior cortical areas is related to executive dysfunction in Alzheimer’s disease. Clinics 2014, 69, 28–37. [Google Scholar] [CrossRef]
- Kreutzer, A.G.; Yoo, S.; Spencer, R.K.; Nowick, J.S. Stabilization, Assembly, and Toxicity of Trimers Derived from Aβ. J. Am. Chem. Soc. 2017, 139, 966–975. [Google Scholar] [CrossRef]
- Sepulcre, J.; Schultz, A.P.; Sabuncu, M.; Gomez-Isla, T.; Chhatwal, J.; Becker, A.; Sperling, R.; Johnson, K.A. In Vivo Tau, Amyloid, and Gray Matter Profiles in the Aging Brain. J. Neurosci. 2016, 36, 7364–7374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buendia, I.; Parada, E.; Navarro, E.; León, R.; Negredo, P.; Egea, J.; López, M.G. Subthreshold Concentrations of Melatonin and Galantamine Improves Pathological AD-Hallmarks in Hippocampal Organotypic Cultures. Mol. Neurobiol. 2016, 53, 3338–3348. [Google Scholar] [CrossRef] [PubMed]
- Reitz, C. Alzheimer’s Disease and the Amyloid Cascade Hypothesis: A Critical Review. Int. J. Alzheimers Dis. 2012, 2012, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.P.; LeVine, H. Alzheimer’s Disease and the Amyloid-β Peptide. J. Alzheimers Dis. 2010, 19, 311–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Multhaup, G.; Huber, O.; Buée, L.; Galas, M.-C. Amyloid Precursor Protein (APP) Metabolites APP Intracellular Fragment (AICD), Aβ42, and Tau in Nuclear Roles. J. Biol. Chem. 2015, 290, 23515–23522. [Google Scholar] [CrossRef] [Green Version]
- Nicolas, M.; Hassan, B.A. Amyloid precursor protein and neural development. Development 2014, 141, 2543–2548. [Google Scholar] [CrossRef] [Green Version]
- Nalivaeva, N.N.; Turner, A.J. The amyloid precursor protein: A biochemical enigma in brain development, function and disease. FEBS Lett. 2013, 587, 2046–2054. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.-D.; Chan, C.H.-S.; Ma, Q.-H.; Xu, X.-H.; Xiao, Z.-C.; Tan, E.-K. The roles of amyloid precursor protein (APP) in neurogenesis. Cell Adhes. Migr. 2011, 5, 280–292. [Google Scholar] [CrossRef] [Green Version]
- Franco, R.; Rivas-Santisteban, R.; Casanovas, M.; Lillo, A.; Saura, C.A.; Navarro, G.J.C. Adenosine A2A Receptor Antagonists Affects NMDA Glutamate Receptor Function. Potential to Address Neurodegeneration in Alzheimer’s Disease. Cells 2020, 9, 1075. [Google Scholar] [CrossRef]
- Cortés, A.; Gracia, E.; Moreno, E.; Mallol, J.; Lluís, C.; Canela, E.I.; Casadó, V. Moonlighting Adenosine Deaminase: A Target Protein for Drug Development. Med. Res. Rev. 2015, 35, 85–125. [Google Scholar] [CrossRef]
- Maiuolo, J.; Oppedisano, F.; Gratteri, S.; Muscoli, C.; Mollace, V. Regulation of uric acid metabolism and excretion. Int. J. Cardiol. 2016, 213, 8–14. [Google Scholar] [CrossRef] [Green Version]
- Cunha, R.A. Adenosine as a neuromodulator and as a homeostatic regulator in the nervous system: Different roles, different sources and different receptors. Neurochem. Int. 2001, 38, 107–125. [Google Scholar] [CrossRef]
- Sheth, S.; Brito, R.; Mukherjea, D.; Rybak, L.; Ramkumar, V. Adenosine Receptors: Expression, Function and Regulation. Int. J. Mol. Sci. 2014, 15, 2024–2052. [Google Scholar] [CrossRef] [Green Version]
- Rau, A.R.; Ariwodola, O.J.; Weiner, J.L. Postsynaptic Adenosine A2A Receptors Modulate Intrinsic Excitability of Pyramidal Cells in the Rat Basolateral Amygdala. Int. J. Neuropsychopharmacol. 2015, 18, 1075. [Google Scholar] [CrossRef] [PubMed]
- Gorgoni, M.; D’Atri, A.; Lauri, G.; Rossini, P.M.; Ferlazzo, F.; De Gennaro, L. Is Sleep Essential for Neural Plasticity in Humans, and How Does It Affect Motor and Cognitive Recovery? Neural Plast. 2013, 2013, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Liu, N.; Jacobson, K.A.; Gavrilova, O.; Reitman, M.L. Physiology and effects of nucleosides in mice lacking all four adenosine receptors. PLoS Biol. 2019, 17, e3000161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angulo, E.; Casadó, V.; Mallol, J.; Canela, E.I.; Viñals, F.; Ferrer, I.; Lluis, C.; Franco, R. A1 adenosine receptors accumulate in neurodegenerative structures in Alzheimer disease and mediate both amyloid precursor protein processing and tau phosphorylation and translocation. Brain Pathol. 2003, 13, 440–451. [Google Scholar] [CrossRef]
- Liu, Y.J.; Chen, J.; Li, X.; Zhou, X.; Hu, Y.M.; Chu, S.F.; Peng, Y.; Chen, N.H. Research progress on adenosine in central nervous system diseases. CNS Neurosci. Ther. 2019, 25, 899–910. [Google Scholar] [CrossRef]
- Lazarov, O.; Hollands, C. Hippocampal neurogenesis: Learning to remember. Prog. Neurobiol. 2016, 138–140, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Bengtsson, S.K.S.; Johansson, M.; Bäckström, T. Long-term continuous allopregnanolone elevation causes memory decline and hippocampus shrinkage, in female wild-type B6 mice. Horm. Behav. 2016, 78, 160–167. [Google Scholar] [CrossRef]
- Ben Ahmed, O.; Benois-Pineau, J.; Allard, M.; Ben Amar, C.; Catheline, G. Classification of Alzheimer’s disease subjects from MRI using hippocampal visual features. Multimed. Tools Appl. 2015, 74, 1249–1266. [Google Scholar] [CrossRef] [Green Version]
- La Joie, R.; Perrotin, A.; de La Sayette, V.; Egret, S.; Doeuvre, L.; Belliard, S.; Eustache, F.; Desgranges, B.; Chételat, G. Hippocampal subfield volumetry in mild cognitive impairment, Alzheimer’s disease and semantic dementia. NeuroImage Clin. 2013, 3, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, O.; Walterfang, M.; Looi, J.C.L.; Malykhin, N.; Östberg, P.; Zandbelt, B.; Styner, M.; Paniagua, B.; Velakoulis, D.; Örndahl, E.; et al. Hippocampal Shape Analysis in Alzheimer’s Disease and Frontotemporal Lobar Degeneration Subtypes. J. Alzheimers Dis. 2012, 30, 355–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, Y.; Lee, K.H.; Choi, K.Y.; Lee, J.J.; Kim, B.C.; Kwon, G.-R. Alzheimer’s Disease Diagnosis Based on Cortical and Subcortical Features. J. Healthc. Eng. 2019, 2019, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Castillo, E.; Leon, J.; Mazzei, G.; Abolhassani, N.; Haruyama, N.; Saito, T.; Saido, T.; Hokama, M.; Iwaki, T.; Ohara, T.; et al. Comparative profiling of cortical gene expression in Alzheimer’s disease patients and mouse models demonstrates a link between amyloidosis and neuroinflammation. Sci. Rep. 2017, 7, 17762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, M.E.; Lu, A.T.; Bennett, D.A.; Horvath, S. Epigenetic age of the pre-frontal cortex is associated with neuritic plaques, amyloid load, and Alzheimer’s disease related cognitive functioning. Aging 2015, 7, 1198–1211. [Google Scholar] [CrossRef]
- Leisman, G.; Moustafa, A.A.; Shafir, T. Thinking, Walking, Talking: Integratory Motor and Cognitive Brain Function. Front. Public Health 2016, 4, 94. [Google Scholar] [CrossRef] [Green Version]
- Götz, J.; Xia, D.; Leinenga, G.; Chew, Y.L.; Nicholas, H. What Renders TAU Toxic. Front. Neurol. 2013, 4, 72. [Google Scholar] [CrossRef] [Green Version]
- Lasser, M.; Tiber, J.; Lowery, L.A. The Role of the Microtubule Cytoskeleton in Neurodevelopmental Disorders. Front. Cell. Neurosci. 2018, 12, 165. [Google Scholar] [CrossRef] [Green Version]
- Kapitein, L.C.; Hoogenraad, C.C. Building the Neuronal Microtubule Cytoskeleton. Neuron 2015, 87, 492–506. [Google Scholar] [CrossRef] [Green Version]
- Morris, M.; Knudsen, G.M.; Maeda, S.; Trinidad, J.C.; Ioanoviciu, A.; Burlingame, A.L.; Mucke, L. Tau post-translational modifications in wild-type and human amyloid precursor protein transgenic mice. Nat. Neurosci. 2015, 18, 1183–1189. [Google Scholar] [CrossRef]
- Zempel, H.; Mandelkow, E. Lost after translation: Missorting of Tau protein and consequences for Alzheimer disease. Trends Neurosci. 2014, 37, 721–732. [Google Scholar] [CrossRef]
- Bagheri, S.; Squitti, R.; Haertlé, T.; Siotto, M.; Saboury, A.A. Role of Copper in the Onset of Alzheimer’s Disease Compared to Other Metals. Front. Aging Neurosci. 2018, 9, 446. [Google Scholar] [CrossRef] [PubMed]
- Greenough, M.A.; Ramírez Munoz, A.; Bush, A.I.; Opazo, C.M. Metallo-pathways to Alzheimer’s disease: Lessons from genetic disorders of copper trafficking. Metallomics 2016, 8, 831–839. [Google Scholar] [CrossRef]
- Xu, J.; Begley, P.; Church, S.J.; Patassini, S.; McHarg, S.; Kureishy, N.; Hollywood, K.A.; Waldvogel, H.J.; Liu, H.; Zhang, S.; et al. Elevation of brain glucose and polyol-pathway intermediates with accompanying brain-copper deficiency in patients with Alzheimer’s disease: Metabolic basis for dementia. Sci. Rep. 2016, 6, 27524. [Google Scholar] [CrossRef] [PubMed]
- Mezentsev, Y.V.; Medvedev, A.E.; Kechko, O.I.; Makarov, A.A.; Ivanov, A.S.; Mantsyzov, A.B.; Kozin, S.A. Zinc-induced heterodimer formation between metal-binding domains of intact and naturally modified amyloid-beta species: Implication to amyloid seeding in Alzheimer’s disease? J. Biomol. Struct. Dyn. 2016, 34, 2317–2326. [Google Scholar] [CrossRef] [PubMed]
- Kulikova, A.A.; Makarov, A.A.; Kozin, S.A. Roles of zinc ions and structural polymorphism of β-amyloid in the development of Alzheimer’s disease. Mol. Biol. 2015, 49, 217–230. [Google Scholar] [CrossRef]
- Sands, S.A.; Leung-Toung, R.; Wang, Y.; Connelly, J.; LeVine, S.M. Enhanced Histochemical Detection of Iron in Paraffin Sections of Mouse Central Nervous System Tissue. ASN Neuro 2016, 8, 175909141667097. [Google Scholar] [CrossRef] [Green Version]
- James, S.A.; Churches, Q.I.; de Jonge, M.D.; Birchall, I.E.; Streltsov, V.; McColl, G.; Adlard, P.A.; Hare, D.J. Iron, Copper, and Zinc Concentration in Aβ Plaques in the APP/PS1 Mouse Model of Alzheimer’s Disease Correlates with Metal Levels in the Surrounding Neuropil. ACS Chem. Neurosci. 2017, 8, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Summers, K.L.; Schilling, K.M.; Roseman, G.; Markham, K.A.; Dolgova, N.V.; Kroll, T.; Sokaras, D.; Millhauser, G.L.; Pickering, I.J.; George, G.N. X-ray absorption spectroscopy investigations of copper (II) coordination in the human amyloid β peptide. Inorg. Chem. 2019, 58, 6294–6311. [Google Scholar] [CrossRef]
- Ji, M.; Arbel, M.; Zhang, L.; Freudiger, C.W.; Hou, S.S.; Lin, D.; Yang, X.; Bacskai, B.J.; Xie, X.S. Label-free imaging of amyloid plaques in Alzheimer’s disease with stimulated Raman scattering microscopy. Sci. Adv. 2018, 4, eaat7715. [Google Scholar] [CrossRef] [Green Version]
- Bourassa, M.W.; Leskovjan, A.C.; Tappero, R.V.; Farquhar, E.R.; Colton, C.A.; Van Nostrand, W.E.; Miller, L.M. Elevated copper in the amyloid plaques and iron in the cortex are observed in mouse models of Alzheimer’s disease that exhibit neurodegeneration. Biomed. Spectrosc. Imaging 2013, 2, 129–139. [Google Scholar] [CrossRef] [Green Version]
- Arnal, N.; Castillo, O.; de Alaniz, M.J.T.; Marra, C.A. Effects of Copper and/or Cholesterol Overload on Mitochondrial Function in a Rat Model of Incipient Neurodegeneration. Int. J. Alzheimers Dis. 2013, 2013, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.; Ye, J.; Zhou, S.; Pi, R.; Dou, J.; Zang, L.; Chen, X.; Chao, X.; Li, W.; Liu, M.; et al. The effects of chronic copper exposure on the amyloid protein metabolisim associated genes’ expression in chronic cerebral hypoperfused rats. Neurosci. Lett. 2012, 518, 14–18. [Google Scholar] [CrossRef]
- Hozumi, I.; Hasegawa, T.; Honda, A.; Ozawa, K.; Hayashi, Y.; Hashimoto, K.; Yamada, M.; Koumura, A.; Sakurai, T.; Kimura, A.; et al. Patterns of levels of biological metals in CSF differ among neurodegenerative diseases. J. Neurol. Sci. 2011, 303, 95–99. [Google Scholar] [CrossRef]
- Pal, A.; Kumar, A.; Prasad, R. Predictive association of copper metabolism proteins with Alzheimer’s disease and Parkinson’s disease: A preliminary perspective. Biometals 2014, 27, 25–31. [Google Scholar] [CrossRef]
- Xiao, G.; Fan, Q.; Wang, X.; Zhou, B. Huntington disease arises from a combinatory toxicity of polyglutamine and copper binding. Proc. Natl. Acad. Sci. USA 2013, 110, 14995–15000. [Google Scholar] [CrossRef] [Green Version]
- Siggs, O.M.; Cruite, J.T.; Du, X.; Rutschmann, S.; Masliah, E.; Beutler, B.; Oldstone, M.B.A. Disruption of copper homeostasis due to a mutation of Atp7a delays the onset of prion disease. Proc. Natl. Acad. Sci. USA 2012, 109, 13733–13738. [Google Scholar] [CrossRef] [Green Version]
- Rose, F.; Hodak, M.; Bernholc, J. Mechanism of copper(II)-induced misfolding of Parkinson’s disease protein. Sci. Rep. 2011, 1, 11. [Google Scholar] [CrossRef] [Green Version]
- Souza, P.C.; Thallmair, S.; Marrink, S.J.; Mera-Adasme, R. An Allosteric Pathway in Copper, Zinc Superoxide Dismutase Unravels the Molecular Mechanism of the G93A Amyotrophic Lateral Sclerosis-Linked Mutation. J. Phys. Chem. lett. 2019, 10, 7740–7744. [Google Scholar] [CrossRef]
- Delangle, P.; Mintz, E. Chelation therapy in Wilson’s disease: From d-penicillamine to the design of selective bioinspired intracellular Cu(i) chelators. Dalton Trans. 2012, 41, 6359–6370. [Google Scholar] [CrossRef] [PubMed]
- Lutsenko, S.; Bhattacharjee, A.; Hubbard, A.L. Copper handling machinery of the brain. Metallomics 2010, 2, 596–608. [Google Scholar] [CrossRef] [PubMed]
- Squitti, R.; Ghidoni, R.; Simonelli, I.; Ivanova, I.D.; Colabufo, N.A.; Zuin, M.; Benussi, L.; Binetti, G.; Cassetta, E.; Rongioletti, M. Copper dyshomeostasis in Wilson disease and Alzheimer’s disease as shown by serum and urine copper indicators. J. Trace Elem. Med. Biol. 2018, 45, 181–188. [Google Scholar] [CrossRef]
- Xu, J.; Church, S.J.; Patassini, S.; Begley, P.; Waldvogel, H.J.; Curtis, M.A.; Faull, R.L.M.; Unwin, R.D.; Cooper, G.J.S. Evidence for widespread, severe brain copper deficiency in Alzheimer’s dementia. Metallomics 2017, 9, 1106–1119. [Google Scholar] [CrossRef] [Green Version]
- Exley, C.; House, E.; Polwart, A.; Esiri, M.M. Brain Burdens of Aluminum, Iron, and Copper and their Relationships with Amyloid-β Pathology in 60 Human Brains. J. Alzheimers Dis. 2012, 31, 725–730. [Google Scholar] [CrossRef]
- Kaden, D.; Bush, A.I.; Danzeisen, R.; Bayer, T.A.; Multhaup, G. Disturbed Copper Bioavailability in Alzheimer’s Disease. Int. J. Alzheimers Dis. 2011, 2011, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Vural, H.; Demirin, H.; Kara, Y.; Eren, I.; Delibas, N. Alterations of plasma magnesium, copper, zinc, iron and selenium concentrations and some related erythrocyte antioxidant enzyme activities in patients with Alzheimer’s disease. J. Trace Elem. Med. Biol. 2010, 24, 169–173. [Google Scholar] [CrossRef]
- Yu, J.; Luo, X.; Xu, H.; Ma, Q.; Yuan, J.; Li, X.; Chang, R.C.-C.; Qu, Z.; Huang, X.; Zhuang, Z.; et al. Identification of the Key Molecules Involved in Chronic Copper Exposure-Aggravated Memory Impairment in Transgenic Mice of Alzheimer’s Disease Using Proteomic Analysis. J. Alzheimers Dis. 2015, 44, 455–469. [Google Scholar] [CrossRef]
- Squitti, R.; Siotto, M.; Polimanti, R. Low-copper diet as a preventive strategy for Alzheimer’s disease. Neurobiol. Aging 2014, 35, S40–S50. [Google Scholar] [CrossRef] [Green Version]
- Brewer, G.J. Alzheimer’s disease causation by copper toxicity and treatment with zinc. Front. Aging Neurosci. 2014, 6, 92. [Google Scholar] [CrossRef]
- Ceccom, J.; Coslédan, F.; Halley, H.; Francès, B.; Lassalle, J.M.; Meunier, B. Copper Chelator Induced Efficient Episodic Memory Recovery in a Non-Transgenic Alzheimer’s Mouse Model. PLoS ONE 2012, 7, e43105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eskici, G.; Axelsen, P.H. Copper and Oxidative Stress in the Pathogenesis of Alzheimer’s Disease. Biochemistry 2012, 51, 6289–6311. [Google Scholar] [CrossRef] [PubMed]
- Hua, H.; Münter, L.; Harmeier, A.; Georgiev, O.; Multhaup, G.; Schaffner, W. Toxicity of Alzheimer’s disease-associated Aβ peptide is ameliorated in a Drosophila model by tight control of zinc and copper availability. Biol. Chem. 2011, 392, 919–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.; Zhang, J.; Liu, N.; Luo, Y.; Zhao, B. Copper ions influence the toxicity of β-amyloid(1–42) in a concentration-dependent manner in a Caenorhabditis elegans model of Alzheimer’s disease. Sci. China Life Sci. 2011, 54, 527–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and future treatments for Alzheimer’s disease. Ther. Adv. Neurol. Disord. 2013, 6, 19–33. [Google Scholar] [CrossRef] [Green Version]
- Karthivashan, G.; Ganesan, P.; Park, S.-Y.; Kim, J.-S.; Choi, D.-K. Therapeutic strategies and nano-drug delivery applications in management of ageing Alzheimer’s disease. Drug Deliv. 2018, 25, 307–320. [Google Scholar] [CrossRef] [Green Version]
- Faller, P.; Hureau, C. A Bioinorganic View of Alzheimer’s Disease: When Misplaced Metal Ions (Re)direct the Electrons to the Wrong Target. Chem. A Eur. J. 2012, 18, 15910–15920. [Google Scholar] [CrossRef]
- Chen, J.; Jiang, Y.; Shi, H.; Peng, Y.; Fan, X.; Li, C.J.P.A.E. The molecular mechanisms of copper metabolism and its roles in human diseases. Pflüg. Arch. Eur. J. Physiol. 2020, 472, 1415–1429. [Google Scholar] [CrossRef]
- Wang, Z.-X.; Tan, L.; Wang, H.-F.; Ma, J.; Liu, J.; Tan, M.-S.; Sun, J.-H.; Zhu, X.-C.; Jiang, T.; Yu, J.-T. Serum Iron, Zinc, and Copper Levels in Patients with Alzheimer’s Disease: A Replication Study and Meta-Analyses. J. Alzheimers Dis. 2015, 47, 565–581. [Google Scholar] [CrossRef]
- Ventriglia, M.; Bucossi, S.; Panetta, V.; Squitti, R. Copper in Alzheimer’s Disease: A Meta-Analysis of Serum, Plasma, and Cerebrospinal Fluid Studies. J. Alzheimers Dis. 2012, 30, 981–984. [Google Scholar] [CrossRef] [Green Version]
- Miller, L.M.; Wang, Q.; Telivala, T.P.; Smith, R.J.; Lanzirotti, A.; Miklossy, J. Synchrotron-based infrared and X-ray imaging shows focalized accumulation of Cu and Zn co-localized with β-amyloid deposits in Alzheimer’s disease. J. Struct. Biol. 2006, 155, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Rembach, A.; Hare, D.J.; Lind, M.; Fowler, C.J.; Cherny, R.A.; McLean, C.; Bush, A.I.; Masters, C.L.; Roberts, B.R. Decreased Copper in Alzheimer’s Disease Brain Is Predominantly in the Soluble Extractable Fraction. Int. J. Alzheimers Dis. 2013, 2013, 1–7. [Google Scholar] [CrossRef]
- Lovell, M.A.; Robertson, J.D.; Teesdale, W.J.; Campbell, J.L.; Markesbery, W.R. Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 1998, 158, 47–52. [Google Scholar] [CrossRef]
- James, S.A.; Volitakis, I.; Adlard, P.A.; Duce, J.A.; Masters, C.L.; Cherny, R.A.; Bush, A.I. Elevated labile Cu is associated with oxidative pathology in Alzheimer disease. Free Radic. Biol. Med. 2012, 52, 298–302. [Google Scholar] [CrossRef]
- Castellani, R.J.; Plascencia-Villa, G.; Perry, G. The amyloid cascade and Alzheimer’s disease therapeutics: Theory versus observation. Lab. Investing. 2019, 99, 958–970. [Google Scholar] [CrossRef]
- Kepp, K.P. Alzheimer’s disease due to loss of function: A new synthesis of the available data. Prog. Neurobiol. 2016, 143, 36–60. [Google Scholar] [CrossRef] [Green Version]
- Multhaup, G.; Schlicksupp, A.; Hesse, L.; Beher, D.; Ruppert, T.; Masters, C.L.; Beyreuther, K. The Amyloid Precursor Protein of Alzheimer’s Disease in the Reduction of Copper(II) to Copper(I). Science 1996, 271, 1406–1409. [Google Scholar] [CrossRef]
- Maynard, C.J.; Cappai, R.; Volitakis, I.; Cherny, R.A.; White, A.R.; Beyreuther, K.; Masters, C.L.; Bush, A.I.; Li, Q.-X. Overexpression of Alzheimer’s Disease Amyloid-β Opposes the Age-dependent Elevations of Brain Copper and Iron. J. Biol. Chem. 2002, 277, 44670–44676. [Google Scholar] [CrossRef] [Green Version]
- Squitti, R.; Ghidoni, R.; Siotto, M.; Ventriglia, M.; Benussi, L.; Paterlini, A.; Magri, M.; Binetti, G.; Cassetta, E.; Caprara, D.; et al. Value of serum nonceruloplasmin copper for prediction of mild cognitive impairment conversion to Alzheimer disease. Ann. Neurol. 2014, 75, 574–580. [Google Scholar] [CrossRef]
- Baumkotter, F.; Schmidt, N.; Vargas, C.; Schilling, S.; Weber, R.; Wagner, K.; Fiedler, S.; Klug, W.; Radzimanowski, J.; Nickolaus, S.; et al. Amyloid Precursor Protein Dimerization and Synaptogenic Function Depend on Copper Binding to the Growth Factor-Like Domain. J. Neurosci. 2014, 34, 11159–11172. [Google Scholar] [CrossRef] [Green Version]
- Noda, Y.; Asada, M.; Kubota, M.; Maesako, M.; Watanabe, K.; Uemura, M.; Kihara, T.; Shimohama, S.; Takahashi, R.; Kinoshita, A.; et al. Copper enhances APP dimerization and promotes Aβ production. Neurosci. Lett. 2013, 547, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Spoerri, L.; Vella, L.J.; Pham, C.L.L.; Barnham, K.J.; Cappai, R. The Amyloid Precursor Protein Copper Binding Domain Histidine Residues 149 and 151 Mediate APP Stability and Metabolism. J. Biol. Chem. 2012, 287, 26840–26853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, Y.H.; Robb, E.L.; Volitakis, I.; Ho, M.; Evin, G.; Li, Q.-X.; Culvenor, J.G.; Masters, C.L.; Cherny, R.A.; Bush, A.I. Paradoxical Condensation of Copper with Elevated β-Amyloid in Lipid Rafts under Cellular Copper Deficiency Conditions. J. Biol. Chem. 2009, 284, 21899–21907. [Google Scholar] [CrossRef] [Green Version]
- Acevedo, K.M.; Hung, Y.H.; Dalziel, A.H.; Li, Q.-X.; Laughton, K.; Wikhe, K.; Rembach, A.; Roberts, B.; Masters, C.L.; Bush, A.I.; et al. Copper Promotes the Trafficking of the Amyloid Precursor Protein. J. Biol. Chem. 2011, 286, 8252–8262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayes, J.; Tinker-Mill, C.; Kolosov, O.; Zhang, H.; Tabner, B.J.; Allsop, D. β-Amyloid Fibrils in Alzheimer Disease Are Not Inert When Bound to Copper Ions but Can Degrade Hydrogen Peroxide and Generate Reactive Oxygen Species. J. Biol. Chem. 2014, 289, 12052–12062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atwood, C.S.; Scarpa, R.C.; Huang, X.; Moir, R.D.; Jones, W.D.; Fairlie, D.P.; Tanzi, R.E.; Bush, A.I. Characterization of copper interactions with alzheimer amyloid beta peptides: Identification of an attomolar-affinity copper binding site on amyloid beta1-42. J. Neurochem. 2000, 75, 1219–1233. [Google Scholar] [CrossRef]
- White, A.R.; Reyes, R.; Mercer, J.F.B.; Camakaris, J.; Zheng, H.; Bush, A.I.; Multhaup, G.; Beyreuther, K.; Masters, C.L.; Cappai, R. Copper levels are increased in the cerebral cortex and liver of APP and APLP2 knockout mice. Brain Res. 1999, 842, 439–444. [Google Scholar] [CrossRef]
- Pate, K.M.; Murphy, R.M. Cerebrospinal Fluid Proteins as Regulators of Beta-amyloid Aggregation and Toxicity. Israel J. Chem. 2017, 57, 602–612. [Google Scholar] [CrossRef]
- Vigo-Pelfrey, C.; Lee, D.; Keim, P.; Lieberburg, I.; Schenk, D.B. Rapid Communication: Characterization of ?-Amyloid Peptide from Human Cerebrospinal Fluid. J. Neurochem. 1993, 61, 1965–1968. [Google Scholar] [CrossRef]
- Torres, J.B.; Andreozzi, E.M.; Dunn, J.T.; Siddique, M.; Szanda, I.; Howlett, D.R.; Sunassee, K.; Blower, P.J. PET Imaging of Copper Trafficking in a Mouse Model of Alzheimer Disease. J. Nucl. Med. 2016, 57, 109–114. [Google Scholar] [CrossRef] [Green Version]
- Doig, A.J.; del Castillo-Frias, M.P.; Berthoumieu, O.; Tarus, B.; Nasica-Labouze, J.; Sterpone, F.; Nguyen, P.H.; Hooper, N.M.; Faller, P.; Derreumaux, P. Why Is Research on Amyloid-β Failing to Give New Drugs for Alzheimer’s Disease? ACS Chem. Neurosci. 2017, 8, 1435–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mital, M.; Wezynfeld, N.E.; Frączyk, T.; Wiloch, M.Z.; Wawrzyniak, U.E.; Bonna, A.; Tumpach, C.; Barnham, K.J.; Haigh, C.L.; Bal, W.; et al. A Functional Role for Aβ in Metal Homeostasis? N-Truncation and High-Affinity Copper Binding. Angew. Chem. Int. Ed. 2015, 54, 10460–10464. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.-L.; Sun, Y.-X.; Jiang, Z.-F. Cu(II) Potentiation of Alzheimer A?1-40 Cytotoxicity and Transition on its Secondary Structure. Acta Biochim. Biophys. Sin. 2006, 38, 765–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, G.P.; Clark, I.A.; Vissel, B. Inconsistencies and Controversies Surrounding the Amyloid Hypothesis of Alzheimer’s Disease. Acta Neuropathol. Commun. 2014, 2, 135. [Google Scholar] [CrossRef] [Green Version]
- Wright, A.L.; Zinn, R.; Hohensinn, B.; Konen, L.M.; Beynon, S.B.; Tan, R.P.; Clark, I.A.; Abdipranoto, A.; Vissel, B. Neuroinflammation and Neuronal Loss Precede Aβ Plaque Deposition in the hAPP-J20 Mouse Model of Alzheimer’s Disease. PLoS ONE 2013, 8, e59586. [Google Scholar] [CrossRef] [Green Version]
- Xie, H.; Hou, S.; Jiang, J.; Sekutowicz, M.; Kelly, J.; Bacskai, B.J. Rapid cell death is preceded by amyloid plaque-mediated oxidative stress. Proc. Natl. Acad. Sci. USA 2013, 110, 7904–7909. [Google Scholar] [CrossRef] [Green Version]
- Bittner, T.; Burgold, S.; Dorostkar, M.M.; Fuhrmann, M.; Wegenast-Braun, B.M.; Schmidt, B.; Kretzschmar, H.; Herms, J. Amyloid plaque formation precedes dendritic spine loss. Acta Neuropathol. 2012, 124, 797–807. [Google Scholar] [CrossRef] [Green Version]
- Holmes, C.; Boche, D.; Wilkinson, D.; Yadegarfar, G.; Hopkins, V.; Bayer, A.; Jones, R.W.; Bullock, R.; Love, S.; Neal, J.W.; et al. Long-term effects of Aβ42 immunisation in Alzheimer’s disease: Follow-up of a randomised, placebo-controlled phase I trial. Lancet 2008, 372, 216–223. [Google Scholar] [CrossRef] [Green Version]
- Ricciarelli, R.; Fedele, E. The Amyloid Cascade Hypothesis in Alzheimer’s Disease: It’s Time to Change Our Mind. Curr. Neuropharmacol. 2017, 15, 926–935. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, R.H. Brain aging, Alzheimer’s disease, and mitochondria. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2011, 1812, 1630–1639. [Google Scholar] [CrossRef] [Green Version]
- Esparza, T.J.; Zhao, H.; Cirrito, J.R.; Cairns, N.J.; Bateman, R.J.; Holtzman, D.M.; Brody, D.L. Amyloid-beta oligomerization in Alzheimer dementia versus high-pathology controls. Ann. Neurol. 2013, 73, 104–119. [Google Scholar] [CrossRef] [PubMed]
- Sperling, R.A.; Aisen, P.S.; Beckett, L.A.; Bennett, D.A.; Craft, S.; Fagan, A.M.; Iwatsubo, T.; Jack, C.R.; Kaye, J.; Montine, T.J.; et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 280–292. [Google Scholar] [CrossRef] [Green Version]
- Naylor, R.; Hill, A.; Barnham, K. Is Covalently Crosslinked Aβ Responsible for Synaptotoxicity in Alzheimers Disease? Curr. Alzheimer Res. 2008, 5, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Lesné, S.; Koh, M.T.; Kotilinek, L.; Kayed, R.; Glabe, C.G.; Yang, A.; Gallagher, M.; Ashe, K.H. A specific amyloid-β protein assembly in the brain impairs memory. Nature 2006, 440, 352–357. [Google Scholar] [CrossRef]
- Al-Hilaly, Y.K.; Williams, T.L.; Stewart-Parker, M.; Ford, L.; Skaria, E.; Cole, M.; Bucher, W.; Morris, K.L.; Sada, A.; Thorpe, J.R.; et al. A central role for dityrosine crosslinking of Amyloid-β in Alzheimer’s disease. Acta Neuropathol. Commun. 2013, 1, 83. [Google Scholar] [CrossRef] [Green Version]
- Kok, W.M.; Cottam, J.M.; Ciccotosto, G.D.; Miles, L.A.; Karas, J.A.; Scanlon, D.B.; Roberts, B.R.; Parker, M.W.; Cappai, R.; Barnham, K.J.; et al. Synthetic dityrosine-linked β-amyloid dimers form stable, soluble, neurotoxic oligomers. Chem. Sci. 2013, 4, 4449–4454. [Google Scholar] [CrossRef]
- Atwood, C.S.; Perry, G.; Zeng, H.; Kato, Y.; Jones, W.D.; Ling, K.-Q.; Huang, X.; Moir, R.D.; Wang, D.; Sayre, L.M.; et al. Copper Mediates Dityrosine Cross-Linking of Alzheimer’s Amyloid-β. Biochemistry 2004, 43, 560–568. [Google Scholar] [CrossRef]
- Streltsov, V.A.; Titmuss, S.J.; Epa, V.C.; Barnham, K.J.; Masters, C.L.; Varghese, J.N. The Structure of the Amyloid-β Peptide High-Affinity Copper II Binding Site in Alzheimer Disease. Biophys. J. 2008, 95, 3447–3456. [Google Scholar] [CrossRef] [Green Version]
- Hane, F.; Tran, G.; Attwood, S.J.; Leonenko, Z. Cu(2+) affects amyloid-β (1-42) aggregation by increasing peptide-peptide binding forces. PLoS ONE 2013, 8, e59005. [Google Scholar] [CrossRef] [Green Version]
- Hane, F.T.; Hayes, R.; Lee, B.Y.; Leonenko, Z. Effect of Copper and Zinc on the Single Molecule Self-Affinity of Alzheimer’s Amyloid-β Peptides. PLoS ONE 2016, 11, e0147488. [Google Scholar] [CrossRef]
- Lv, Z.; Condron, M.M.; Teplow, D.B.; Lyubchenko, Y.L. Nanoprobing of the Effect of Cu2+ Cations on Misfolding, Interaction and Aggregation of Amyloid β Peptide. J. Neuroimmune Pharmacol. 2013, 8, 262–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnham, K.J.; Haeffner, F.; Ciccotosto, G.D.; Curtain, C.C.; Tew, D.; Mavros, C.; Beyreuther, K.; Carrington, D.; Masters, C.L.; Cherny, R.A.; et al. Tyrosine gated electron transfer is key to the toxic mechanism of Alzheimer’s disease β-amyloid. FASEB J. 2004, 18, 1427–1429. [Google Scholar] [CrossRef] [PubMed]
- Schrag, M.; Mueller, C.; Oyoyo, U.; Smith, M.A.; Kirsch, W.M. Iron, zinc and copper in the Alzheimer’s disease brain: A quantitative meta-analysis. Some insight on the influence of citation bias on scientific opinion. Prog. Neurobiol. 2011, 94, 296–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, S.T.; Harry, G.J.; Hayden, K.M.; Szabo, D.T.; Birnbaum, L. Comparison of Metal Levels between Postmortem Brain and Ventricular Fluid in Alzheimer’s Disease and Nondemented Elderly Controls. Toxicol. Sci. 2016, 150, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.; Muzik, O.; Gatson, J.; Kernie, S.G.; Diaz-Arrastia, R. Assessment of Traumatic Brain Injury by Increased 64Cu Uptake on 64CuCl2 PET/CT. J. Nucl. Med. 2015, 56, 1252–1257. [Google Scholar] [CrossRef] [Green Version]
- Elder, G.A.; Gama Sosa, M.A.; De Gasperi, R. Transgenic Mouse Models of Alzheimer’s Disease. Mt. Sinai J. Med. A J. Transl. Personal Med. 2010, 77, 69–81. [Google Scholar] [CrossRef] [Green Version]
- Sasaguri, H.; Nilsson, P.; Hashimoto, S.; Nagata, K.; Saito, T.; De Strooper, B.; Hardy, J.; Vassar, R.; Winblad, B.; Saido, T.C. APP mouse models for Alzheimer’s disease preclinical studies. EMBO J. 2017, 36, 2473–2487. [Google Scholar] [CrossRef]
- Chen, W.-T.; Liao, Y.-H.; Yu, H.-M.; Cheng, I.H.; Chen, Y.-R. Distinct Effects of Zn 2+, Cu 2+, Fe 3+, and Al 3+ on Amyloid-β Stability, Oligomerization, and Aggregation. J. Biol. Chem. 2011, 286, 9646–9656. [Google Scholar] [CrossRef] [Green Version]
- Ha, C.; Ryu, J.; Park, C.B. Metal Ions Differentially Influence the Aggregation and Deposition of Alzheimer’s β-Amyloid on a Solid Template. Biochemistry 2007, 46, 6118–6125. [Google Scholar] [CrossRef] [PubMed]
- Karr, J.W.; Szalai, V.A. Cu(II) Binding to Monomeric, Oligomeric, and Fibrillar Forms of the Alzheimer’s Disease Amyloid-β Peptide. Biochemistry 2008, 47, 5006–5016. [Google Scholar] [CrossRef]
- Mold, M.; Ouro-Gnao, L.; Wieckowski, B.M.; Exley, C. Copper prevents amyloid-β1–42 from forming amyloid fibrils under near-physiological conditions in vitro. Sci. Rep. 2013, 3, 1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarell, C.J.; Wilkinson, S.R.; Viles, J.H. Substoichiometric Levels of Cu 2+ Ions Accelerate the Kinetics of Fiber Formation and Promote Cell Toxicity of Amyloid-β from Alzheimer Disease. J. Biol. Chem. 2010, 285, 41533–41540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tõugu, V.; Karafin, A.; Zovo, K.; Chung, R.S.; Howells, C.; West, A.K.; Palumaa, P. Zn(II)- and Cu(II)-induced non-fibrillar aggregates of amyloid-β (1-42) peptide are transformed to amyloid fibrils, both spontaneously and under the influence of metal chelators. J. Neurochem. 2009, 110, 1784–1795. [Google Scholar] [CrossRef]
- Crouch, P.J.; Hung, L.W.; Adlard, P.A.; Cortes, M.; Lal, V.; Filiz, G.; Perez, K.A.; Nurjono, M.; Caragounis, A.; Du, T.; et al. Increasing Cu bioavailability inhibits A oligomers and tau phosphorylation. Proc. Natl. Acad. Sci. USA 2009, 106, 381–386. [Google Scholar] [CrossRef] [Green Version]
- Kitazawa, M.; Cheng, D.; LaFerla, F.M. Chronic copper exposure exacerbates both amyloid and tau pathology and selectively dysregulates cdk5 in a mouse model of AD. J. Neurochem. 2009, 108, 1550–1560. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.-X.; Du, J.-T.; Zeng, Z.-Y.; Wu, W.-H.; Zhao, Y.-F.; Kanazawa, K.; Ishizuka, Y.; Nemoto, T.; Nakanishi, H.; Li, Y.-M. Copper (II) modulates in vitro aggregation of a tau peptide. Peptides 2007, 28, 2229–2234. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.; Liu, G.; Zhao, Y.; Shi, Z.; Zheng, Q.; Bu, G.; Xu, H.; Zhang, Y.-W. The role of copper and the copper-related protein CUTA in mediating APP processing and Aβ generation. Neurobiol. Aging 2015, 36, 1310–1315. [Google Scholar] [CrossRef] [PubMed]
- White, A.R.; Multhaup, G.; Galatis, D.; McKinstry, W.J.; Parker, M.W.; Pipkorn, R.; Beyreuther, K.; Masters, C.L.; Cappai, R. Contrasting, species-dependent modulation of copper-mediated neurotoxicity by the Alzheimer’s disease amyloid precursor protein. J. Neurosci. Off. J. Soc. Neurosci. 2002, 22, 365–376. [Google Scholar] [CrossRef] [Green Version]
- Mandrekar-Colucci, S.; Landreth, G.E. Microglia and inflammation in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2010, 9, 156–167. [Google Scholar] [CrossRef]
- Lopes da Silva, S.; Vellas, B.; Elemans, S.; Luchsinger, J.; Kamphuis, P.; Yaffe, K.; Sijben, J.; Groenendijk, M.; Stijnen, T. Plasma nutrient status of patients with Alzheimer’s disease: Systematic review and meta-analysis. Alzheimers Dement. 2014, 10, 485–502. [Google Scholar] [CrossRef] [Green Version]
- Kitazawa, M.; Hsu, H.-W.; Medeiros, R. Copper Exposure Perturbs Brain Inflammatory Responses and Impairs Clearance of Amyloid-Beta. Toxicol. Sci. 2016, 152, 194–204. [Google Scholar] [CrossRef] [Green Version]
- Newcombe, E.A.; Camats-Perna, J.; Silva, M.L.; Valmas, N.; Huat, T.J.; Medeiros, R. Inflammation: The link between comorbidities, genetics, and Alzheimer’s disease. J. Neuroinflamm. 2018, 15, 276. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Wu, D.-m.; Zheng, Y.-l.; Sun, D.-x.; Hu, B.; Shan, Q.; Zhang, Z.-f.; Fan, S.-h. Trace amounts of copper exacerbate beta amyloid-induced neurotoxicity in the cholesterol-fed mice through TNF-mediated inflammatory pathway. Brain. Behav. Immun. 2009, 23, 193–203. [Google Scholar] [CrossRef]
- Zheng, Z.; White, C.; Lee, J.; Peterson, T.S.; Bush, A.I.; Sun, G.Y.; Weisman, G.A.; Petris, M.J. Altered microglial copper homeostasis in a mouse model of Alzheimer’s disease. J. Neurochem. 2010, 114, 1630–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zucconi, G.G.; Cipriani, S.; Scattoni, R.; Balgkouranidou, I.; Hawkins, D.P.; Ragnarsdottir, K.V. Copper deficiency elicits glial and neuronal response typical of neurodegenerative disorders. Neuropathol. Appl. Neurobiol. 2007, 33, 212–225. [Google Scholar] [CrossRef] [PubMed]
- Rossi-George, A.; Guo, C.-J.; Oakes, B.L.; Gow, A.J. Copper modulates the phenotypic response of activated BV2 microglia through the release of nitric oxide. Nitric Oxide 2012, 27, 201–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi-George, A.; Guo, C.-J. Copper disrupts S-nitrosothiol signaling in activated BV2 microglia. Neurochem. Int. 2016, 99, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Bin, Y.; Li, X.; He, Y.; Chen, S.; Xiang, J. Amyloid-β peptide (1–42) aggregation induced by copper ions under acidic conditions. Acta Biochim. Biophys. Sin. 2013, 45, 570–577. [Google Scholar] [CrossRef] [Green Version]
- Hamano, T.; Gendron, T.F.; Causevic, E.; Yen, S.-H.; Lin, W.-L.; Isidoro, C.; DeTure, M.; Ko, L.-W. Autophagic-lysosomal perturbation enhances tau aggregation in transfectants with induced wild-type tau expression. Eur. J. Neurosci. 2008, 27, 1119–1130. [Google Scholar] [CrossRef]
- Wang, Y.; Martinez-Vicente, M.; Krüger, U.; Kaushik, S.; Wong, E.; Mandelkow, E.-M.; Cuervo, A.M.; Mandelkow, E. Tau fragmentation, aggregation and clearance: The dual role of lysosomal processing. Hum. Mol. Genet. 2009, 18, 4153–4170. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q.-F.; Li, Y.-M.; Du, J.-T.; Kanazawa, K.; Nemoto, T.; Nakanishi, H.; Zhao, Y.-F. Binding of copper (II) ion to an Alzheimer’s tau peptide as revealed by MALDI-TOF MS, CD, and NMR. Biopolymers 2005, 79, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Li, Y.; Du, J.; Liu, H.; Kanazawa, K.; Nemoto, T.; Nakanishi, H.; Zhao, Y. Copper binding properties of a tau peptide associated with Alzheimer’s disease studied by CD, NMR, and MALDI-TOF MS. Peptides 2006, 27, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Squitti, R.; Siotto, M.; Arciello, M.; Rossi, L. Non-ceruloplasmin bound copper and ATP7B gene variants in Alzheimer’s disease. Metallomics 2016, 8, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathy, S.; Yoo, B.; McElheny, D.; Tay, W.; Ishii, Y. Capturing a Reactive State of Amyloid Aggregates. J. Biol. Chem. 2014, 289, 9998–10010. [Google Scholar] [CrossRef] [Green Version]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Hu, D. Aging-Dependent Alterations in Synaptic Plasticity and Memory in Mice That Overexpress Extracellular Superoxide Dismutase. J. Neurosci. 2006, 26, 3933–3941. [Google Scholar] [CrossRef]
- Kamsler, A.; Segal, M. Hydrogen peroxide modulation of synaptic plasticity. J. Neurosci. Off. J. Soc. Neurosci. 2003, 23, 269–276. [Google Scholar] [CrossRef]
- Kamsler, A.; Segal, M. Hydrogen Peroxide As a Diffusible Signal Molecule in Synaptic Plasticity. Mol. Neurobiol. 2004, 29, 167–178. [Google Scholar] [CrossRef]
- Kishida, K.T.; Klann, E. Sources and Targets of Reactive Oxygen Species in Synaptic Plasticity and Memory. Antioxid. Redox Signal 2007, 9, 233–244. [Google Scholar] [CrossRef] [Green Version]
- Knapp, L.T.; Klann, E. Role of reactive oxygen species in hippocampal long-term potentiation: Contributory or inhibitory? J. Neurosci. Res. 2002, 70, 1–7. [Google Scholar] [CrossRef]
- Nakamura, M.; Shishido, N.; Nunomura, A.; Smith, M.A.; Perry, G.; Hayashi, Y.; Nakayama, K.; Hayashi, T. Three Histidine Residues of Amyloid-β Peptide Control the Redox Activity of Copper and Iron. Biochemistry 2007, 46, 12737–12743. [Google Scholar] [CrossRef] [PubMed]
- Cheignon, C.; Faller, P.; Testemale, D.; Hureau, C.; Collin, F. Metal-catalyzed oxidation of Aβ and the resulting reorganization of Cu binding sites promote ROS production. Metallomics 2016, 8, 1081–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naslund, J.; Schierhorn, A.; Hellman, U.; Lannfelt, L.; Roses, A.D.; Tjernberg, L.O.; Silberring, J.; Gandy, S.E.; Winblad, B.; Greengard, P. Relative abundance of Alzheimer A beta amyloid peptide variants in Alzheimer disease and normal aging. Proc. Natl. Acad. Sci. USA 1994, 91, 8378–8382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, C.-L.; Wu, W.-H.; Liu, Q.; Sun, X.; Ma, Y.; Zhao, Y.-F.; Li, Y.-M. Dual functions of β-amyloid oligomer and fibril in Cu(II)-induced H2O2 production. Regul. Pept. 2010, 163, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Guilloreau, L.; Combalbert, S.; Sournia-Saquet, A.; Mazarguil, H.; Faller, P. Redox Chemistry of Copper–Amyloid-β: The Generation of Hydroxyl Radical in the Presence of Ascorbate is Linked to Redox-Potentials and Aggregation State. ChemBioChem 2007, 8, 1317–1325. [Google Scholar] [CrossRef] [PubMed]
- Chassaing, S.; Collin, F.; Dorlet, P.; Gout, J.; Hureau, C.; Faller, P. Copper and heme-mediated Abeta toxicity: Redox chemistry, Abeta oxidations and anti-ROS compounds. Curr. Top. Med. Chem. 2012, 12, 2573–2595. [Google Scholar] [CrossRef] [PubMed]
- Baruch-Suchodolsky, R.; Fischer, B. Soluble Amyloid β 1−28 −Copper(I)/Copper(II)/Iron(II) Complexes Are Potent Antioxidants in Cell-Free Systems. Biochemistry 2008, 47, 7796–7806. [Google Scholar] [CrossRef]
- Nadal, R.C.; Rigby, S.E.J.; Viles, J.H. Amyloid β−Cu 2+ Complexes in both Monomeric and Fibrillar Forms Do Not Generate H 2 O 2 Catalytically but Quench Hydroxyl Radicals. Biochemistry 2008, 47, 11653–11664. [Google Scholar] [CrossRef]
- Reybier, K.; Ayala, S.; Alies, B.; Rodrigues, J.V.; Bustos Rodriguez, S.; La Penna, G.; Collin, F.; Gomes, C.M.; Hureau, C.; Faller, P. Free Superoxide is an Intermediate in the Production of H 2 O 2 by Copper(I)-Aβ Peptide and O 2. Angew. Chem. Int. Ed. 2016, 55, 1085–1089. [Google Scholar] [CrossRef]
- Cheignon, C.; Jones, M.; Atrián-Blasco, E.; Kieffer, I.; Faller, P.; Collin, F.; Hureau, C. Identification of key structural features of the elusive Cu–Aβ complex that generates ROS in Alzheimer’s disease. Chem. Sci. 2017, 8, 5107–5118. [Google Scholar] [CrossRef] [Green Version]
- Prosdocimi, T.; De Gioia, L.; Zampella, G.; Bertini, L. On the generation of OH· radical species from H2O2 by Cu(I) amyloid beta peptide model complexes: A DFT investigation. JBIC J. Biol. Inorg. Chem. 2016, 21, 197–212. [Google Scholar] [CrossRef] [PubMed]
- Mirats, A.; Alí-Torres, J.; Rodríguez-Santiago, L.; Sodupe, M.; La Penna, G. Dioxygen activation in the Cu–amyloid β complex. Phys. Chem. Chem. Phys. 2015, 17, 27270–27274. [Google Scholar] [CrossRef] [Green Version]
- La Penna, G.; Hureau, C.; Andreussi, O.; Faller, P. Identifying, by first-principles simulations, Cu[amyloid-β] species making Fenton-type reactions in Alzheimer’s disease. J. Phy. Chem. B 2013, 117, 16455–16467. [Google Scholar] [CrossRef]
- Brewer, G.J. Divalent Copper as a Major Triggering Agent in Alzheimer’s Disease. J. Alzheimers Dis. 2015, 46, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Atrián-Blasco, E.; Cerrada, E.; Conte-Daban, A.; Testemale, D.; Faller, P.; Laguna, M.; Hureau, C. Copper (I) targeting in the Alzheimer’s disease context: A first example using the biocompatible PTA ligand. Metallomics 2015, 7, 1229–1232. [Google Scholar] [CrossRef] [PubMed]
- Feaga, H.A.; Maduka, R.C.; Foster, M.N.; Szalai, V.A. Affinity of Cu + for the Copper-Binding Domain of the Amyloid-β Peptide of Alzheimer’s Disease. Inorg. Chem. 2011, 50, 1614–1618. [Google Scholar] [CrossRef] [PubMed]
- Phinney, A.L.; Drisaldi, B.; Schmidt, S.D.; Lugowski, S.; Coronado, V.; Liang, Y.; Horne, P.; Yang, J.; Sekoulidis, J.; Coomaraswamy, J.; et al. In vivo reduction of amyloid- by a mutant copper transporter. Proc. Natl. Acad. Sci. USA 2003, 100, 14193–14198. [Google Scholar] [CrossRef] [Green Version]
- Southon, A.; Greenough, M.A.; Ganio, G.; Bush, A.I.; Burke, R.; Camakaris, J. Presenilin Promotes Dietary Copper Uptake. PLoS ONE 2013, 8, e62811. [Google Scholar] [CrossRef] [Green Version]
- Gerber, H.; Wu, F.; Dimitrov, M.; Garcia Osuna, G.M.; Fraering, P.C. Zinc and Copper Differentially Modulate Amyloid Precursor Protein Processing by γ-Secretase and Amyloid-β Peptide Production. J. Biol. Chem. 2017, 292, 3751–3767. [Google Scholar] [CrossRef] [Green Version]
- Giacoppo, S.; Galuppo, M.; Calabrò, R.S.; D’Aleo, G.; Marra, A.; Sessa, E.; Bua, D.G.; Potortì, A.G.; Dugo, G.; Bramanti, P.; et al. Heavy Metals and Neurodegenerative Diseases: An Observational Study. Biol. Trace Elem. Res. 2014, 161, 151–160. [Google Scholar] [CrossRef]
- Klevay, L.M. Alzheimer’s disease as copper deficiency. Med. Hypotheses 2008, 70, 802–807. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, N.; Iso, H.; Kitanaka, N.; Kitanaka, J.; Eguchi, H.; Ookawara, T.; Ozawa, K.; Shimoda, S.; Yoshihara, D.; Takemura, M.; et al. Effects of copper metabolism on neurological functions in Wistar and Wilson’s disease model rats. Biochem. Biophys. Res. Commun. 2006, 349, 1079–1086. [Google Scholar] [CrossRef] [PubMed]
- Cater, M.A.; McInnes, K.T.; Li, Q.-X.; Volitakis, I.; La Fontaine, S.; Mercer, J.F.B.; Bush, A.I. Intracellular copper deficiency increases amyloid-β secretion by diverse mechanisms. Biochem. J. 2008, 412, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Brewer, G.J. Copper excess, zinc deficiency, and cognition loss in Alzheimer’s disease. BioFactors 2012, 38, 107–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, M.C.; Evans, D.A.; Tangney, C.C.; Bienias, J.L.; Schneider, J.A.; Wilson, R.S.; Scherr, P.A. Dietary Copper and High Saturated and trans Fat Intakes Associated With Cognitive Decline. Arch. Neurol. 2006, 63, 1085–1088. [Google Scholar] [CrossRef] [Green Version]
- Sparks, D.L.; Schreurs, B.G. Trace amounts of copper in water induce -amyloid plaques and learning deficits in a rabbit model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2003, 100, 11065–11069. [Google Scholar] [CrossRef] [Green Version]
- Arnal, N.; Morel, G.R.; de Alaniz, M.J.T.; Castillo, O.; Marra, C.A. Role of Copper and Cholesterol Association in the Neurodegenerative Process. Int. J. Alzheimers Dis. 2013, 2013, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Wong, B.X.; Hung, Y.H.; Bush, A.I.; Duce, J.A. Metals and cholesterol: Two sides of the same coin in Alzheimer’s disease pathology. Front. Aging Neurosci. 2014, 6, 91. [Google Scholar] [CrossRef] [Green Version]
- Larry Sparks, D. Cholesterol, Copper, and Accumulation of Thioflavine S-Reactive Alzheimer’s-Like Amyloid β in Rabbit Brain. J. Mol. Neurosci. 2004, 24, 097–104. [Google Scholar] [CrossRef]
- Riddell, D.R.; Christie, G.; Hussain, I.; Dingwall, C. Compartmentalization of β-secretase (Asp2) into low-buoyant density, noncaveolar lipid rafts. Curr. Biol. 2001, 11, 1288–1293. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Zheng, J. Cholesterol Promotes the Interaction of Alzheimer β-Amyloid Monomer with Lipid Bilayer. J. Mol. Biol. 2012, 421, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Di Scala, C.; Yahi, N.; Boutemeur, S.; Flores, A.; Rodriguez, L.; Chahinian, H.; Fantini, J. Common molecular mechanism of amyloid pore formation by Alzheimer’s β-amyloid peptide and α-synuclein. Sci. Rep. 2016, 6, 28781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Scala, C.; Troadec, J.-D.; Lelièvre, C.; Garmy, N.; Fantini, J.; Chahinian, H. Mechanism of cholesterol-assisted oligomeric channel formation by a short Alzheimer β-amyloid peptide. J. Neurochem. 2014, 128, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Arbor, S.C.; LaFontaine, M.; Cumbay, M. Amyloid-beta Alzheimer targets - protein processing, lipid rafts, and amyloid-beta pores. Yale J. Biol. Med. 2016, 89, 5–21. [Google Scholar] [PubMed]
- Drolle, E.; Hane, F.; Lee, B.; Leonenko, Z. Atomic force microscopy to study molecular mechanisms of amyloid fibril formation and toxicity in Alzheimer’s disease. Drug Metab. Rev. 2014, 46, 207–223. [Google Scholar] [CrossRef]
- Kotler, S.A.; Walsh, P.; Brender, J.R.; Ramamoorthy, A. Differences between amyloid-β aggregation in solution and on the membrane: Insights into elucidation of the mechanistic details of Alzheimer’s disease. Chem. Soc. Rev. 2014, 43, 6692–6700. [Google Scholar] [CrossRef] [Green Version]
- Skopp, A.; Boyd, S.D.; Ullrich, M.S.; Liu, L.; Winkler, D.D. Copper–zinc superoxide dismutase (Sod1) activation terminates interaction between its copper chaperone (Ccs) and the cytosolic metal-binding domain of the copper importer Ctr1. Biometals 2019, 32, 695–705. [Google Scholar] [CrossRef] [Green Version]
- Ackerman, C.M.; Chang, C.J. Copper signaling in the brain and beyond. J. Biol. Chem. 2018, 293, 4628–4635. [Google Scholar] [CrossRef] [Green Version]
- Witt, B.; Schaumlöffel, D.; Schwerdtle, T. Subcellular Localization of Copper—Cellular Bioimaging with Focus on Neurological Disorders. Int. J. Mol. Sci. 2020, 21, 2341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nose, Y.; Wood, L.K.; Kim, B.-E.; Prohaska, J.R.; Fry, R.S.; Spears, J.W.; Thiele, D.J. Ctr1 is an apical copper transporter in mammalian intestinal epithelial cells in vivo that is controlled at the level of protein stability. J. Biol. Chem. 2010, 285, 32385–32392. [Google Scholar] [CrossRef] [Green Version]
- Hodgkinson, V.L.; Dale, J.M.; Garcia, M.L.; Weisman, G.A.; Lee, J.; Gitlin, J.D.; Petris, M.J. X-linked spinal muscular atrophy in mice caused by autonomous loss of ATP7A in the motor neuron. J. Pathol. 2015, 236, 241–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deibel, M.A.; Ehmann, W.D.; Markesbery, W.R. Copper, iron, and zinc imbalances in severely degenerated brain regions in Alzheimer’s disease: Possible relation to oxidative stress. J. Neurol. Sci. 1996, 143, 137–142. [Google Scholar] [CrossRef]
- Bucossi, S.; Ventriglia, M.; Panetta, V.; Salustri, C.; Pasqualetti, P.; Mariani, S.; Siotto, M.; Rossini, P.M.; Squitti, R. Copper in Alzheimer’s Disease: A Meta-Analysis of Serum, Plasma, and Cerebrospinal Fluid Studies. J. Alzheimers Dis. 2011, 24, 175–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Squitti, R.; Lupoi, D.; Pasqualetti, P.; Dal Forno, G.; Vernieri, F.; Chiovenda, P.; Rossi, L.; Cortesi, M.; Cassetta, E.; Rossini, P.M. Elevation of serum copper levels in Alzheimer’s disease. Neurology 2002, 59, 1153–1161. [Google Scholar] [CrossRef]
- Brewer, G.J.; Kanzer, S.H.; Zimmerman, E.A.; Celmins, D.F.; Heckman, S.M.; Dick, R. Copper and Ceruloplasmin Abnormalities in Alzheimer’s Disease. Am. J. Alzheimers Dis. Other Dement. 2010, 25, 490–497. [Google Scholar] [CrossRef]
- Ozcankaya, R.; Delibas, N. Malondialdehyde, superoxide dismutase, melatonin, iron, copper, and zinc blood concentrations in patients with Alzheimer disease: Cross-sectional study. Croat. Med. J. 2002, 43, 28–32. [Google Scholar]
- Squitti, R.; Ghidoni, R.; Scrascia, F.; Benussi, L.; Panetta, V.; Pasqualetti, P.; Moffa, F.; Bernardini, S.; Ventriglia, M.; Binetti, G.; et al. Free Copper Distinguishes Mild Cognitive Impairment Subjects from Healthy Elderly Individuals. J. Alzheimers Dis. 2011, 23, 239–248. [Google Scholar] [CrossRef]
- Rembach, A.; Doecke, J.D.; Roberts, B.R.; Watt, A.D.; Faux, N.G.; Volitakis, I.; Pertile, K.K.; Rumble, R.L.; Trounson, B.O.; Fowler, C.J.; et al. Longitudinal Analysis of Serum Copper and Ceruloplasmin in Alzheimer’s Disease. J. Alzheimers Dis. 2013, 34, 171–182. [Google Scholar] [CrossRef] [Green Version]
- Squitti, R.; Simonelli, I.; Ventriglia, M.; Siotto, M.; Pasqualetti, P.; Rembach, A.; Doecke, J.; Bush, A.I. Meta-Analysis of Serum Non-Ceruloplasmin Copper in Alzheimer’s Disease. J. Alzheimers Dis. 2013, 38, 809–822. [Google Scholar] [CrossRef]
- Schrag, M.; Mueller, C.; Zabel, M.; Crofton, A.; Kirsch, W.M.; Ghribi, O.; Squitti, R.; Perry, G. Oxidative stress in blood in Alzheimer’s disease and mild cognitive impairment: A meta-analysis. Neurobiol. Dis. 2013, 59, 100–110. [Google Scholar] [CrossRef]
- Li, D.-D.; Zhang, W.; Wang, Z.-Y.; Zhao, P. Serum Copper, Zinc, and Iron Levels in Patients with Alzheimer’s Disease: A Meta-Analysis of Case-Control Studies. Front. Aging Neurosci. 2017, 9, 300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talwar, P.; Grover, S.; Sinha, J.; Chandna, P.; Agarwal, R.; Kushwaha, S.; Kukreti, R. Multifactorial Analysis of a Biomarker Pool for Alzheimer Disease Risk in a North Indian Population. Dement. Geriatr. Cogn. Disord. 2017, 44, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Pu, Z.; Xu, W.; Lin, Y.; He, J.; Huang, M. Oxidative Stress Markers and Metal Ions are Correlated With Cognitive Function in Alzheimer’s Disease. Am. J. Alzheimers Dis. Other Dement. 2017, 32, 353–359. [Google Scholar] [CrossRef]
- Guan, C.; Dang, R.; Cui, Y.; Liu, L.; Chen, X.; Wang, X.; Zhu, J.; Li, D.; Li, J.; Wang, D. Characterization of plasma metal profiles in Alzheimer’s disease using multivariate statistical analysis. PLoS ONE 2017, 12, e0178271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tecchio, F.; Vecchio, F.; Ventriglia, M.; Porcaro, C.; Miraglia, F.; Siotto, M.; Rossini, P.M.; Rongioletti, M.; Squitti, R. Non-Ceruloplasmin Copper Distinguishes A Distinct Subtype of Alzheimer’s Disease: A Study of EEG-Derived Brain Activity. Curr. Alzheimer Res. 2016, 13, 1374–1384. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, C.; Martin, T.; Cacho, J.; Brenas, M.T.; Arroyo, T.; Garcia, B.; Navajo, J.A.; González-Buitrago, J.M. Serum zinc, copper, insulin and lipids in Alzheimer’s disease epsilon 4 apolipoprotein E allele carriers. Eur. J. Clin. Investing. 1999, 29, 637–642. [Google Scholar] [CrossRef] [PubMed]
- Squitti, R.; Ventriglia, M.; Gennarelli, M.; Colabufo, N.A.; El Idrissi, I.G.; Bucossi, S.; Mariani, S.; Rongioletti, M.; Zanetti, O.; Congiu, C.; et al. Non-Ceruloplasmin Copper Distincts Subtypes in Alzheimer’s Disease: A Genetic Study of ATP7B Frequency. Mol. Neurobiol. 2017, 54, 671–681. [Google Scholar] [CrossRef]
- Mercer, S.W.; Wang, J.; Burke, R. In Vivo Modeling of the Pathogenic Effect of Copper Transporter Mutations That Cause Menkes and Wilson Diseases, Motor Neuropathy, and Susceptibility to Alzheimer’s Disease. J. Biol. Chem. 2017, 292, 4113–4122. [Google Scholar] [CrossRef] [Green Version]
- Squitti, R.; Polimanti, R.; Siotto, M.; Bucossi, S.; Ventriglia, M.; Mariani, S.; Vernieri, F.; Scrascia, F.; Trotta, L.; Rossini, P.M. ATP7B Variants as Modulators of Copper Dyshomeostasis in Alzheimer’s Disease. Neuro Mol. Med. 2013, 15, 515–522. [Google Scholar] [CrossRef]
- Hegde, M.L.; Bharathi, P.; Suram, A.; Venugopal, C.; Jagannathan, R.; Poddar, P.; Srinivas, P.; Sambamurti, K.; Rao, K.J.; Scancar, J.; et al. Challenges Associated with Metal Chelation Therapy in Alzheimer’s Disease. J. Alzheimers Dis. 2009, 17, 457–468. [Google Scholar] [CrossRef] [Green Version]
- Y.Y. Szeto, J.; J.G. Lewis, S. Current Treatment Options for Alzheimer’s Disease and Parkinson’s Disease Dementia. Curr. Neuropharmacol. 2016, 14, 326–338. [Google Scholar] [CrossRef] [PubMed]
- Behbehani, G.R.; Barzegar, L.; Mohebbian, M.; Saboury, A.A. A Comparative Interaction between Copper Ions with Alzheimer’s β Amyloid Peptide and Human Serum Albumin. Bioinorg. Chem. Appl. 2012, 2012, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Cherny, R.A.; Atwood, C.S.; Xilinas, M.E.; Gray, D.N.; Jones, W.D.; McLean, C.A.; Barnham, K.J.; Volitakis, I.; Fraser, F.W.; Kim, Y.-S.; et al. Treatment with a Copper-Zinc Chelator Markedly and Rapidly Inhibits β-Amyloid Accumulation in Alzheimer’s Disease Transgenic Mice. Neuron 2001, 30, 665–676. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.-h.; Lei, P.; Liu, Q.; Hu, J.; Gunn, A.P.; Chen, M.-s.; Rui, Y.-f.; Su, X.-y.; Xie, Z.-p.; Zhao, Y.-F.; et al. Sequestration of Copper from β-Amyloid Promotes Selective Lysis by Cyclen-Hybrid Cleavage Agents. J. Biol. Chem. 2008, 283, 31657–31664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, T.; Yang, L.; Zhang, C.; Wang, Y.; Ma, X.; Wang, K.; Luo, J.; Yao, C.; Wang, X.; Wang, X. A copper–amyloid-β targeted fluorescent chelator as a potential theranostic agent for Alzheimer’s disease. Inorg. Chem. Front. 2016, 3, 1572–1581. [Google Scholar] [CrossRef]
- Geng, J.; Li, M.; Wu, L.; Ren, J.; Qu, X. Liberation of Copper from Amyloid Plaques: Making a Risk Factor Useful for Alzheimer’s Disease Treatment. J. Med. Chem. 2012, 55, 9146–9155. [Google Scholar] [CrossRef] [PubMed]
- Hauser-Davis, R.A.; de Freitas, L.V.; Cukierman, D.S.; Cruz, W.S.; Miotto, M.C.; Landeira-Fernandez, J.; Valiente-Gabioud, A.A.; Fernández, C.O.; Rey, N.A. Disruption of zinc and copper interactions with Aβ(1–40) by a non-toxic, isoniazid-derived, hydrazone: A novel biometal homeostasis restoring agent in Alzheimer’s disease therapy? Metallomics 2015, 7, 743–747. [Google Scholar] [CrossRef]
- Hung, V.W.S.; Bressan, L.P.; Seo, K.; Kerman, K. Electroanalysis of Natural Compounds as Copper Chelating Agents for Alzheimer’s Disease Therapy. Electroanalysis 2015, 27, 2670–2678. [Google Scholar] [CrossRef]
- Liu, G.; Garrett, M.R.; Men, P.; Zhu, X.; Perry, G.; Smith, M.A. Nanoparticle and other metal chelation therapeutics in Alzheimer disease. Bioch. Biophys. Acta (BBA) Mol. Basis Dis. 2005, 1741, 246–252. [Google Scholar] [CrossRef] [Green Version]
- Moret, V.; Laras, Y.; Pietrancosta, N.; Garino, C.; Quéléver, G.; Rolland, A.; Mallet, B.; Norreel, J.-C.; Kraus, J.-L. 1,1′-Xylyl bis-1,4,8,11-tetraaza cyclotetradecane: A new potential copper chelator agent for neuroprotection in Alzheimer’s disease. Its comparative effects with clioquinol on rat brain copper distribution. Bioorg. Med. Chem. Lett. 2006, 16, 3298–3301. [Google Scholar] [CrossRef]
- Nguyen, M.; Robert, A.; Sournia-Saquet, A.; Vendier, L.; Meunier, B. Characterization of New Specific Copper Chelators as Potential Drugs for the Treatment of Alzheimer’s Disease. Chem. A Eur. J. 2014, 20, 6771–6785. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.E.; Orvig, C. Medicinal Inorganic Chemistry Approaches to Passivation and Removal of Aberrant Metal Ions in Disease. Chem. Rev. 2009, 109, 4885–4910. [Google Scholar] [CrossRef] [PubMed]
- Kosnett, M.J. The Role of Chelation in the Treatment of Arsenic and Mercury Poisoning. J. Med. Toxicol. 2013, 9, 347–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cahoon, L. The curious case of clioquinol. Nat. Med. 2009, 15, 356–359. [Google Scholar] [CrossRef] [PubMed]
- Barnham, K.J.; Bush, A.I. Metals in Alzheimer’s and Parkinson’s Diseases. Curr. Opin. Chem. Biol. 2008, 12, 222–228. [Google Scholar] [CrossRef]
- Bush, A.I. Drug development based on the metals hypothesis of Alzheimer’s disease. J. Alzheimers Dis. 2008, 15, 223–240. [Google Scholar] [CrossRef] [Green Version]
- Liang, S.H.; Southon, A.G.; Fraser, B.H.; Krause-Heuer, A.M.; Zhang, B.; Shoup, T.M.; Lewis, R.; Volitakis, I.; Han, Y.; Greguric, I.; et al. Novel Fluorinated 8-Hydroxyquinoline Based Metal Ionophores for Exploring the Metal Hypothesis of Alzheimer’s Disease. ACS Med. Chem. Lett. 2015, 6, 1025–1029. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Rodríguez, C.; Telpoukhovskaia, M.; Orvig, C. The art of building multifunctional metal-binding agents from basic molecular scaffolds for the potential application in neurodegenerative diseases. Coord. Chem. Rev. 2012, 256, 2308–2332. [Google Scholar] [CrossRef]
- Prachayasittikul, V.; Prachayasittikul, V.; Prachayasittikul, S.; Ruchirawat, S. 8-Hydroxyquinolines: A review of their metal chelating properties and medicinal applications. Drug Des. Devel. Ther. 2013, 7, 1157. [Google Scholar] [CrossRef] [Green Version]
- Adlard, P.A.; Cherny, R.A.; Finkelstein, D.I.; Gautier, E.; Robb, E.; Cortes, M.; Volitakis, I.; Liu, X.; Smith, J.P.; Perez, K.; et al. Rapid Restoration of Cognition in Alzheimer’s Transgenic Mice with 8-Hydroxy Quinoline Analogs Is Associated with Decreased Interstitial Aβ. Neuron 2008, 59, 43–55. [Google Scholar] [CrossRef] [Green Version]
- Rana, M.; Sharma, A.K. Cu and Zn interactions with Aβ peptides: Consequence of coordination on aggregation and formation of neurotoxic soluble Aβ oligomers. Metallomics 2019, 11, 64–84. [Google Scholar] [CrossRef] [PubMed]
- Rana, M.; Cho, H.-J.; Roy, T.K.; Mirica, L.M.; Sharma, A.K. Azo-dyes based small bifunctional molecules for metal chelation and controlling amyloid formation. Inorg. Chim. Acta 2018, 471, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Crouch, P.J.; Savva, M.S.; Hung, L.W.; Donnelly, P.S.; Mot, A.I.; Parker, S.J.; Greenough, M.A.; Volitakis, I.; Adlard, P.A.; Cherny, R.A.; et al. The Alzheimer’s therapeutic PBT2 promotes amyloid-β degradation and GSK3 phosphorylation via a metal chaperone activity. J. Neurochem. 2011, 119, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Drew, S.C. The Case for Abandoning Therapeutic Chelation of Copper Ions in Alzheimer’s Disease. Front. Neurosci. 2017, 11, 317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Squitti, R.; Salustri, C.; Rongioletti, M.; Siotto, M. Commentary: The Case for Abandoning Therapeutic Chelation of Copper Ions in Alzheimer’s Disease. Front. Neurol. 2017, 8, 503. [Google Scholar] [CrossRef] [Green Version]
- Quinn, J.F.; Harris, C.J.; Cobb, K.E.; Domes, C.; Ralle, M.; Brewer, G.; Wadsworth, T.L. A Copper-Lowering Strategy Attenuates Amyloid Pathology in a Transgenic Mouse Model of Alzheimer’s Disease. J. Alzheimers Dis. 2010, 21, 903–914. [Google Scholar] [CrossRef] [Green Version]
- Gokhale, A.; Vrailas-Mortimer, A.; Larimore, J.; Comstra, H.S.; Zlatic, S.A.; Werner, E.; Manvich, D.F.; Iuvone, P.M.; Weinshenker, D.; Faundez, V. Neuronal copper homeostasis susceptibility by genetic defects in dysbindin, a schizophrenia susceptibility factor. Hum. Mol. Genet. 2015, 24, 5512–5523. [Google Scholar] [CrossRef] [Green Version]
- Grossi, C.; Francese, S.; Casini, A.; Rosi, M.C.; Luccarini, I.; Fiorentini, A.; Gabbiani, C.; Messori, L.; Moneti, G.; Casamenti, F. Clioquinol Decreases Amyloid-β Burden and Reduces Working Memory Impairment in a Transgenic Mouse Model of Alzheimer’s Disease. J. Alzheimers Dis. 2009, 17, 423–440. [Google Scholar] [CrossRef]
- Matlack, K.E.S.; Tardiff, D.F.; Narayan, P.; Hamamichi, S.; Caldwell, K.A.; Caldwell, G.A.; Lindquist, S. Clioquinol promotes the degradation of metal-dependent amyloid-β (Aβ) oligomers to restore endocytosis and ameliorate A toxicity. Proc. Natl. Acad. Sci. USA 2014, 111, 4013–4018. [Google Scholar] [CrossRef] [Green Version]
- Segal-Gavish, H.; Danino, O.; Barhum, Y.; Ben-Zur, T.; Shai, E.; Varon, D.; Offen, D.; Fischer, B. A Multifunctional Biocompatible Drug Candidate is Highly Effective in Delaying Pathological Signs of Alzheimer’s Disease in 5XFAD Mice. J. Alzheimers Dis. 2017, 58, 389–400. [Google Scholar] [CrossRef]
- Donnelly, P.S.; Caragounis, A.; Du, T.; Laughton, K.M.; Volitakis, I.; Cherny, R.A.; Sharples, R.A.; Hill, A.F.; Li, Q.-X.; Masters, C.L.; et al. Selective Intracellular Release of Copper and Zinc Ions from Bis(thiosemicarbazonato) Complexes Reduces Levels of Alzheimer Disease Amyloid-β Peptide. J. Biol. Chem. 2008, 283, 4568–4577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venugopal, C.; Demos, C.M.; Rao, K.S.J.; Pappolla, M.A.; Sambamurti, K. Beta-secretase: Structure, function, and evolution. CNS Neurol. Disord. Drug Targets 2008, 7, 278–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenche, V.B.; Barnham, K.J. Alzheimer’s disease & metals: Therapeutic opportunities. Br. J. Pharmacol. 2011, 163, 211–219. [Google Scholar] [PubMed] [Green Version]
- Ribarič, S. Peptides as Potential Therapeutics for Alzheimer’s Disease. Molecules 2018, 23, 283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenzie-Nickson, S.; Bush, A.I.; Barnham, K.J.J. Bis(thiosemicarbazone) Metal Complexes as Therapeutics for Neurodegenerative Diseases. Curr. Top. Med. Chem. 2016, 16, 3058–3068. [Google Scholar] [CrossRef]
- Paterson, B.M.; Cullinane, C.; Crouch, P.J.; White, A.R.; Barnham, K.J.; Roselt, P.D.; Noonan, W.; Binns, D.; Hicks, R.J.; Donnelly, P.S. Modification of Biodistribution and Brain Uptake of Copper Bis(thiosemicarbazonato) Complexes by the Incorporation of Amine and Polyamine Functional Groups. Inorg. Chem. 2019, 58, 4540–4552. [Google Scholar] [CrossRef]
- Choo, X.Y.; Liddell, J.R.; Huuskonen, M.T.; Grubman, A.; Moujalled, D.; Roberts, J.; Kysenius, K.; Patten, L.; Quek, H.; Oikari, L.E.; et al. CuII(atsm) Attenuates Neuroinflammation. Front. Neurosci. 2018, 12, 668. [Google Scholar] [CrossRef]
- Ramanathan, A.; Nelson, A.R.; Sagare, A.P.; Zlokovic, B.V. Impaired vascular-mediated clearance of brain amyloid beta in Alzheimer’s disease: The role, regulation and restoration of LRP1. Front. Aging Neurosci. 2015, 7, 136. [Google Scholar] [CrossRef] [Green Version]
- Kanekiyo, T.; Liu, C.-C.; Shinohara, M.; Li, J.; Bu, G. LRP1 in brain vascular smooth muscle cells mediates local clearance of Alzheimer’s amyloid-β. J. Neurosci Off. J. Soc. Neurosci. 2012, 32, 16458–16465. [Google Scholar] [CrossRef] [Green Version]
- Singh, I.; Sagare, A.P.; Coma, M.; Perlmutter, D.; Gelein, R.; Bell, R.D.; Deane, R.J.R.; Zhong, E.; Parisi, M.; Ciszewski, J.; et al. Low levels of copper disrupt brain amyloid-β homeostasis by altering its production and clearance. Proc. Natl. Acad. Sci. USA 2013, 110, 14771–14776. [Google Scholar] [CrossRef] [Green Version]
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Sengupta, U.; Clos, A.L.; Jackson, G.R.; Kayed, R. Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol. Neurodegener. 2011, 6, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tai, H.-C.; Serrano-Pozo, A.; Hashimoto, T.; Frosch, M.P.; Spires-Jones, T.L.; Hyman, B.T. The Synaptic Accumulation of Hyperphosphorylated Tau Oligomers in Alzheimer Disease Is Associated With Dysfunction of the Ubiquitin-Proteasome System. Am. J. Pathol. 2012, 181, 1426–1435. [Google Scholar] [CrossRef] [Green Version]
- Ayton, S.; Lei, P.; Bush, A.I. Biometals and their therapeutic implications in Alzheimer’s disease. Neurotherapeutics 2015, 12, 109–120. [Google Scholar] [CrossRef] [Green Version]
- Stremmel, W. Bis-choline Tetrathiomolybdate as Old Drug in a New Design for Wilson’s Disease: Good for Brain and Liver? Hepatology 2019, 69, 901. [Google Scholar] [CrossRef] [PubMed]
- Weiss, K.H.; Askari, F.K.; Czlonkowska, A.; Ferenci, P.; Bronstein, J.M.; Bega, D.; Ala, A.; Nicholl, D.; Flint, S.; Olsson, L.; et al. Bis-choline tetrathiomolybdate in patients with Wilson’s disease: An open-label, multicentre, phase 2 study. Lancet Gastroenterol. Hepatol. 2017, 2, 869–876. [Google Scholar] [CrossRef]
- Sharma, A.K.; Kim, J.J.; Prior, J.T.; Hawco, N.J.; Rath, N.P.; Kim, J.J.; Mirica, L.M. Small Bifunctional Chelators That Do Not Disaggregate Amyloid β Fibrils Exhibit Reduced Cellular Toxicity. Inorg. Chem. 2014, 53, 11376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartholomä, M.D. Recent developments in the design of bifunctional chelators for metal-based radiopharmaceuticals used in Positron Emission Tomography. Inorg. Chim. Acta 2012, 389, 36–51. [Google Scholar] [CrossRef]
- Rajasekhar, K.; Madhu, C.; Govindaraju, T. Natural Tripeptide-Based Inhibitor of Multifaceted Amyloid β Toxicity. ACS Chem. Neurosci. 2016, 7, 1300–1310. [Google Scholar] [CrossRef]
- Savelieff, M.G.; DeToma, A.S.; Derrick, J.S.; Lim, M.H. The Ongoing Search for Small Molecules to Study Metal-Associated Amyloid-β Species in Alzheimer’s Disease. Acc. Chem. Res. 2014, 47, 2475–2482. [Google Scholar] [CrossRef]
- Sharma, A.K.; Pavlova, S.T.; Kim, J.J.; Finkelstein, D.; Hawco, N.J.; Rath, N.P.; Kim, J.J.; Mirica, L.M. Bifunctional Compounds for Controlling Metal-Mediated Aggregation of the Aβ 42 Peptide. J. Am. Chem. Soc. 2012, 134, 6625–6636. [Google Scholar] [CrossRef] [Green Version]
- LeVine, H. Quantification of beta-sheet amyloid fibril structures with thioflavin T. Methods Enzymol. 1999, 309, 274–284. [Google Scholar]
- Dedeoglu, A.; Cormier, K.; Payton, S.; Tseitlin, K.A.; Kremsky, J.N.; Lai, L.; Li, X.; Moir, R.D.; Tanzi, R.E.; Bush, A.I.; et al. Preliminary studies of a novel bifunctional metal chelator targeting Alzheimer’s amyloidogenesis. Exp. Gerontol. 2004, 39, 1641–1649. [Google Scholar] [CrossRef] [PubMed]
- Bandara, N.; Sharma, A.K.; Krieger, S.; Schultz, J.W.; Han, B.H.; Rogers, B.E.; Mirica, L.M. Evaluation of 64 Cu-Based Radiopharmaceuticals that Target Aβ Peptide Aggregates as Diagnostic Tools for Alzheimer’s Disease. J. Am. Chem. Soc. 2017, 139, 12550–12558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.; Kim, J.I.; Na, S.; Eom, K. Metal ions affect the formation and stability of amyloid β aggregates at multiple length scales. Phys. Chem. Chem. Phys. 2018, 20, 8951–8961. [Google Scholar] [CrossRef] [PubMed]
- Charkoudian, L.K.; Pham, D.M.; Franz, K.J. A Pro-Chelator Triggered by Hydrogen Peroxide Inhibits Iron-Promoted Hydroxyl Radical Formation. J. Am. Chem. Soc. 2006, 128, 12424–12425. [Google Scholar] [CrossRef]
- Charkoudian, L.K.; Pham, D.M.; Kwon, A.M.; Vangeloff, A.D.; Franz, K.J. Modifications of boronic ester pro-chelators triggered by hydrogen peroxide tune reactivity to inhibit metal-promoted oxidative stress. Dalton Trans. 2007, 43, 5031–5042. [Google Scholar] [CrossRef]
- Dickens, M.G.; Franz, K.J. A prochelator activated by hydrogen peroxide prevents metal-induced amyloid Beta aggregation. Chembiochem A Eur. J. Chem. Biol. 2010, 11, 59–62. [Google Scholar] [CrossRef] [Green Version]
- Folk, D.S.; Franz, K.J. A Prochelator Activated by β-Secretase Inhibits Aβ Aggregation and Suppresses Copper-Induced Reactive Oxygen Species Formation. J. Am. Chem. Soc. 2010, 132, 4994–4995. [Google Scholar] [CrossRef] [Green Version]
- Hyman, L.M.; Franz, K.J. A cell-permeable fluorescent prochelator responds to hydrogen peroxide and metal ions by decreasing fluorescence. Inorg. Chim. Acta 2012, 380, 125–134. [Google Scholar] [CrossRef] [Green Version]
- Perez, L.R.; Franz, K.J. Minding metals: Tailoring multifunctional chelating agents for neurodegenerative disease. Dalton Trans. 2010, 39, 2177–2187. [Google Scholar] [CrossRef]
- Choi, J.-S.; Braymer, J.J.; Park, S.K.; Mustafa, S.; Chae, J.; Lim, M.H. Synthesis and characterization of IMPY derivatives that regulate metal-induced amyloid-β aggregation. Metallomics 2011, 3, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Hindo, S.S.; Mancino, A.M.; Braymer, J.J.; Liu, Y.; Vivekanandan, S.; Ramamoorthy, A.; Lim, M.H. Small Molecule Modulators of Copper-Induced Aβ Aggregation. J. Am. Chem. Soc. 2009, 131, 16663–16665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, Y.; Lee, H.J.; Kim, M.; Nam, G.; Lee, S.J.C.; Cho, J.; Park, C.-M.; Lim, M.H. Strategic Design of 2,2′-Bipyridine Derivatives to Modulate Metal–Amyloid-β Aggregation. Inorg. Chem. 2017, 56, 6695–6705. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Lee, H.J.; Kim, K.Y.; Lee, S.J.C.; Suh, J.-M.; Cho, J.; Chae, J.; Lim, M.H. Tuning Structures and Properties for Developing Novel Chemical Tools toward Distinct Pathogenic Elements in Alzheimer’s Disease. ACS Chem. Neurosci. 2018, 9, 800–808. [Google Scholar] [CrossRef]
- Hyung, S.J.; DeToma, A.S.; Brender, J.R.; Lee, S.; Vivekanandan, S.; Kochi, A.; Choi, J.S.; Ramamoorthy, A.; Ruotolo, B.T.; Lim, M.H. Insights into antiamyloidogenic properties of the green tea extract (-)-epigallocatechin-3-gallate toward metal-associated amyloid- species. Proc. Natl. Acad. Sci. USA 2013, 110, 3743–3748. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Park, H.M.; Hyung, S.-J.; DeToma, A.S.; Kim, C.; Ruotolo, B.T.; Lim, M.H. Exploring the reactivity of flavonoid compounds with metal-associated amyloid-β species. Dalton Trans. 2012, 41, 6558–6566. [Google Scholar] [CrossRef] [Green Version]
- DeToma, A.S.; Choi, J.-S.; Braymer, J.J.; Lim, M.H. Myricetin: A Naturally Occurring Regulator of Metal-Induced Amyloid-β Aggregation and Neurotoxicity. Chembiochem 2011, 12, 1198–1201. [Google Scholar] [CrossRef]
- Scott, L.E.; Telpoukhovskaia, M.; Rodríguez-Rodríguez, C.; Merkel, M.; Bowen, M.L.; Page, B.D.G.; Green, D.E.; Storr, T.; Thomas, F.; Allen, D.D.; et al. N-Aryl-substituted 3-(β-D-glucopyranosyloxy)-2-methyl-4(1H)-pyridinones as agents for Alzheimer’s therapy. Chem. Sci. 2011, 2, 642–648. [Google Scholar] [CrossRef]
- Scott, L.E.; Page, B.D.G.; Patrick, B.O.; Orvig, C. Altering pyridinone N-substituents to optimise activity as potential prodrugs for Alzheimer’s disease. Dalton Trans. 2008, 6364–6367. [Google Scholar] [CrossRef]
- Storr, T.; Scott, L.E.; Bowen, M.L.; Green, D.E.; Thompson, K.H.; Schugar, H.J.; Orvig, C. Glycosylated tetrahydrosalens as multifunctional molecules for Alzheimer’s therapy. Dalton Trans. 2009, 16, 3034–3043. [Google Scholar] [CrossRef]
- Storr, T.; Merkel, M.; Song-Zhao, G.X.; Scott, L.E.; Green, D.E.; Bowen, M.L.; Thompson, K.H.; Patrick, B.O.; Schugar, H.J.; Orvig, C. Synthesis, Characterization, and Metal Coordinating Ability of Multifunctional Carbohydrate-Containing Compounds for Alzheimer’s Therapy. J. Am. Chem. Soc. 2007, 129, 7453–7463. [Google Scholar] [CrossRef] [PubMed]
- Soon, C.P.W.; Donnelly, P.S.; Turner, B.J.; Hung, L.W.; Crouch, P.J.; Sherratt, N.A.; Tan, J.-L.; Lim, N.K.H.; Lam, L.; Bica, L.; et al. Diacetylbis( N (4)-methylthiosemicarbazonato) Copper(II) (Cu II (atsm)) Protects against Peroxynitrite-induced Nitrosative Damage and Prolongs Survival in Amyotrophic Lateral Sclerosis Mouse Model. J. Biol. Chem. 2011, 286, 44035–44044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savelieff, M.G.; Nam, G.; Kang, J.; Lee, H.J.; Lee, M.; Lim, M.H. Development of Multifunctional Molecules as Potential Therapeutic Candidates for Alzheimer’s Disease, Parkinson’s Disease, and Amyotrophic Lateral Sclerosis in the Last Decade. Chem. Rev. 2019, 119, 1221–1322. [Google Scholar] [CrossRef] [PubMed]
- Robert, A.; Liu, Y.; Nguyen, M.; Meunier, B. Regulation of Copper and Iron Homeostasis by Metal Chelators: A Possible Chemotherapy for Alzheimer’s Disease. Acc. Chem. Res. 2015, 48, 1332–1339. [Google Scholar] [CrossRef] [PubMed]
- Telpoukhovskaia, M.A.; Orvig, C. Werner coordination chemistry and neurodegeneration. Chem. Soc. Rev. 2013, 42, 1836–1846. [Google Scholar] [CrossRef]
- Cherny, R.A.; Legg, J.T.; McLean, C.A.; Fairlie, D.P.; Huang, X.; Atwood, C.S.; Beyreuther, K.; Tanzi, R.E.; Masters, C.L.; Bush, A.I. Aqueous Dissolution of Alzheimer’s Disease Aβ Amyloid Deposits by Biometal Depletion. J. Biol. Chem. 1999, 274, 23223–23228. [Google Scholar] [CrossRef] [Green Version]
- Lannfelt, L.; Blennow, K.; Zetterberg, H.; Batsman, S.; Ames, D.; Harrison, J.; Masters, C.L.; Targum, S.; Bush, A.I.; Murdoch, R.; et al. Safety, efficacy, and biomarker findings of PBT2 in targeting Aβ as a modifying therapy for Alzheimer’s disease: A phase IIa, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2008, 7, 779–786. [Google Scholar] [CrossRef]
- Adlard, P.A.; Bica, L.; White, A.R.; Nurjono, M.; Filiz, G.; Crouch, P.J.; Donnelly, P.S.; Cappai, R.; Finkelstein, D.I.; Bush, A.I. Metal Ionophore Treatment Restores Dendritic Spine Density and Synaptic Protein Levels in a Mouse Model of Alzheimer’s Disease. PLoS ONE 2011, 6, e17669. [Google Scholar] [CrossRef] [Green Version]
- White, A.R.; Du, T.; Laughton, K.M.; Volitakis, I.; Sharples, R.A.; Xilinas, M.E.; Hoke, D.E.; Holsinger, R.M.D.; Evin, G.; Cherny, R.A.; et al. Degradation of the Alzheimer Disease Amyloid β-Peptide by Metal-dependent Up-regulation of Metalloprotease Activity. J. Biol. Chem. 2006, 281, 17670–17680. [Google Scholar] [CrossRef] [Green Version]
- Kawamura, K.; Kuroda, Y.; Sogo, M.; Fujimoto, M.; Inui, T.; Mitsui, T. Superoxide dismutase as a target of clioquinol-induced neurotoxicity. Biochem. Biophys. Res. Commun. 2014, 452, 181–185. [Google Scholar] [CrossRef]
- King, O.N.F.; Li, X.S.; Sakurai, M.; Kawamura, A.; Rose, N.R.; Ng, S.S.; Quinn, A.M.; Rai, G.; Mott, B.T.; Beswick, P.; et al. Quantitative High-Throughput Screening Identifies 8-Hydroxyquinolines as Cell-Active Histone Demethylase Inhibitors. PLoS ONE 2010, 5, e15535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martirosyan, A. Actions of a Histone Deacetylase Inhibitor NSC3852 (5-Nitroso-8-quinolinol) Link Reactive Oxygen Species to Cell Differentiation and Apoptosis in MCF-7 Human Mammary Tumor Cells. J. Pharmacol. Exp. Ther. 2006, 317, 546–552. [Google Scholar] [CrossRef] [Green Version]
- Haigh, C.L.; Tumpach, C.; Collins, S.J.; Drew, S.C. A 2-Substituted 8-Hydroxyquinoline Stimulates Neural Stem Cell Proliferation by Modulating ROS Signalling. Cell Biochem. Biophys. 2016, 74, 297–306. [Google Scholar] [CrossRef]
- Barnham, K.J.; Bush, A.I. Biological metals and metal-targeting compounds in major neurodegenerative diseases. Chem. Soc. Rev. 2014, 43, 6727–6749. [Google Scholar] [CrossRef] [Green Version]
- Squitti, R.; Rossini, P.M.; Cassetta, E.; Moffa, F.; Pasqualetti, P.; Cortesi, M.; Colloca, A.; Rossi, L.; Finazzi-Agro, A. d-penicillamine reduces serum oxidative stress in Alzheimer’s disease patients. Eur. J. Clin. Investing. 2002, 32, 51–59. [Google Scholar] [CrossRef]
- Jenagaratnam, L.; McShane, R. Clioquinol for the treatment of Alzheimer’s Disease; Jenagaratnam, L., Ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2006. [Google Scholar]
- Sampson, E.; Jenagaratnam, L.; McShane, R. Metal protein attenuating compounds for the treatment of Alzheimer’s disease. Cochrane Database Syst. Rev. 2008, CD005380. [Google Scholar]
- Sampson, E.L.; Jenagaratnam, L.; McShane, R. Metal Protein Attenuating Compounds for the Treatment of Alzheimer’s Dementia; Sampson, E.L., Ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2014. [Google Scholar]
- Faux, N.G.; Ritchie, C.W.; Gunn, A.; Rembach, A.; Tsatsanis, A.; Bedo, J.; Harrison, J.; Lannfelt, L.; Blennow, K.; Zetterberg, H.; et al. PBT2 Rapidly Improves Cognition in Alzheimer’s Disease: Additional Phase II Analyses. J. Alzheimers Dis. 2010, 20, 509–516. [Google Scholar] [CrossRef] [Green Version]
- Ayton, S.; Lei, P.; Bush, A.I. Metallostasis in Alzheimer’s disease. Free Radic. Biol. Med. 2013, 62, 76–89. [Google Scholar] [CrossRef]
- Ryan, T.M.; Roberts, B.R.; McColl, G.; Hare, D.J.; Doble, P.A.; Li, Q.X.; Lind, M.; Roberts, A.M.; Mertens, H.D.T.; Kirby, N.; et al. Stabilization of Nontoxic Aβ-Oligomers: Insights into the Mechanism of Action of Hydroxyquinolines in Alzheimer’s Disease. J. Neurosci. 2015, 35, 2871–2884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jack, C.R.; Knopman, D.S.; Jagust, W.J.; Shaw, L.M.; Aisen, P.S.; Weiner, M.W.; Petersen, R.C.; Trojanowski, J.Q. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010, 9, 119–128. [Google Scholar] [CrossRef] [Green Version]
- Arce-Varas, N.; Abate, G.; Prandelli, C.; Martínez, C.; Cuetos, F.; Menéndez, M.; Marziano, M.; Cabrera-García, D.; Fernández-Sánchez, M.T.; Novelli, A.; et al. Comparison of Extracellular and Intracellular Blood Compartments Highlights Redox Alterations in Alzheimer’s and Mild Cognitive Impairment Patients. Curr. Alzheimer Res. 2017, 14, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dannenberger, D.; Nuernberg, G.; Renne, U.; Nuernberg, K.; Langhammer, M.; Huber, K.; Breier, B. High-fat diets rich in ω-3 or ω-6 polyunsaturated fatty acids have distinct effects on lipid profiles and lipid peroxidation in mice selected for either high body weight or leanness. Nutrition 2013, 29, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Lang, M.; Fan, Q.; Wang, L.; Zheng, Y.; Xiao, G.; Wang, X.; Wang, W.; Zhong, Y.; Zhou, B. Inhibition of human high-affinity copper importer Ctr1 orthologous in the nervous system of Drosophila ameliorates Aβ42-induced Alzheimer’s disease–like symptoms. Neurobiol. Aging 2013, 34, 2604–2612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
Figure 1.
Copper’s role in the aggregation of Aβ peptides in the neuritic plaques of AD. (A) Cu2+ complexes with beta-amyloid peptides lead to dityrosine-linked β-amyloid dimer formation, which is neurotoxic and resists degradation into monomers. (B) Cu binding with the Y10A mutant peptide causes no neurotoxicity, dityrosine cross-linking, and degrades to monomers via degrading agents
Figure 1.
Copper’s role in the aggregation of Aβ peptides in the neuritic plaques of AD. (A) Cu2+ complexes with beta-amyloid peptides lead to dityrosine-linked β-amyloid dimer formation, which is neurotoxic and resists degradation into monomers. (B) Cu binding with the Y10A mutant peptide causes no neurotoxicity, dityrosine cross-linking, and degrades to monomers via degrading agents

Figure 2.
Dysfunction of autophagy and tau protein neurofibrillary tangles (NFTs) in the hippocampus of AD. Oligomeric Aβ-induced ROS production results in oxidative damage and mitochondrial dysfunction, in which hyperphosphorylated tau protein and NFTs produce through an imbalance of various protein kinases and phosphatases. These events lead to autophagic dysfunction and aggregated tau protein to neuronal loss in Alzheimer’s disease.
Figure 2.
Dysfunction of autophagy and tau protein neurofibrillary tangles (NFTs) in the hippocampus of AD. Oligomeric Aβ-induced ROS production results in oxidative damage and mitochondrial dysfunction, in which hyperphosphorylated tau protein and NFTs produce through an imbalance of various protein kinases and phosphatases. These events lead to autophagic dysfunction and aggregated tau protein to neuronal loss in Alzheimer’s disease.

Figure 3.
Redox cycling of Cu2+/Cu+ with Aβ peptides leads to the production of hydrogen peroxide. Unstable reactive oxygen species (ROS) production from H2O2 results in oxidative stress, leading to mitochondrial dysfunction, oxidative cellular damage, and neuronal loss. Cytotoxic end-products of lipid peroxidation malondialdehyde (MDA) and 4-hydroxy-2-nonenal (HNE) promote cell death.
Figure 3.
Redox cycling of Cu2+/Cu+ with Aβ peptides leads to the production of hydrogen peroxide. Unstable reactive oxygen species (ROS) production from H2O2 results in oxidative stress, leading to mitochondrial dysfunction, oxidative cellular damage, and neuronal loss. Cytotoxic end-products of lipid peroxidation malondialdehyde (MDA) and 4-hydroxy-2-nonenal (HNE) promote cell death.

Figure 4.
Schematic representation of the effects and correlation between Cu and cholesterol-rich lipid rafts in Alzheimer’s disease. (A) The enzymes that are present in lipid rafts are responsible for the cleavage of APP to Aβ peptide. (B) Cu deficient AD brains lead to copper accumulation in lipid rafts, and rising concentrations of Cu results in higher Aβ production due to an increase in β-secretase activity. (C) Calcium-permeable pores formed by small oligomers of Aβ peptides. These pores are calcium channels and disrupt cellular Ca2+ homeostasis, eventually leading to neuronal death.
Figure 4.
Schematic representation of the effects and correlation between Cu and cholesterol-rich lipid rafts in Alzheimer’s disease. (A) The enzymes that are present in lipid rafts are responsible for the cleavage of APP to Aβ peptide. (B) Cu deficient AD brains lead to copper accumulation in lipid rafts, and rising concentrations of Cu results in higher Aβ production due to an increase in β-secretase activity. (C) Calcium-permeable pores formed by small oligomers of Aβ peptides. These pores are calcium channels and disrupt cellular Ca2+ homeostasis, eventually leading to neuronal death.

Figure 5.
Multifunctional compounds (MFCs) target for Alzheimer’s Disease.

Figure 6.
Structure of bifunctional chelating agents from various research groups’ reports as discussed in the text.
Figure 6.
Structure of bifunctional chelating agents from various research groups’ reports as discussed in the text.


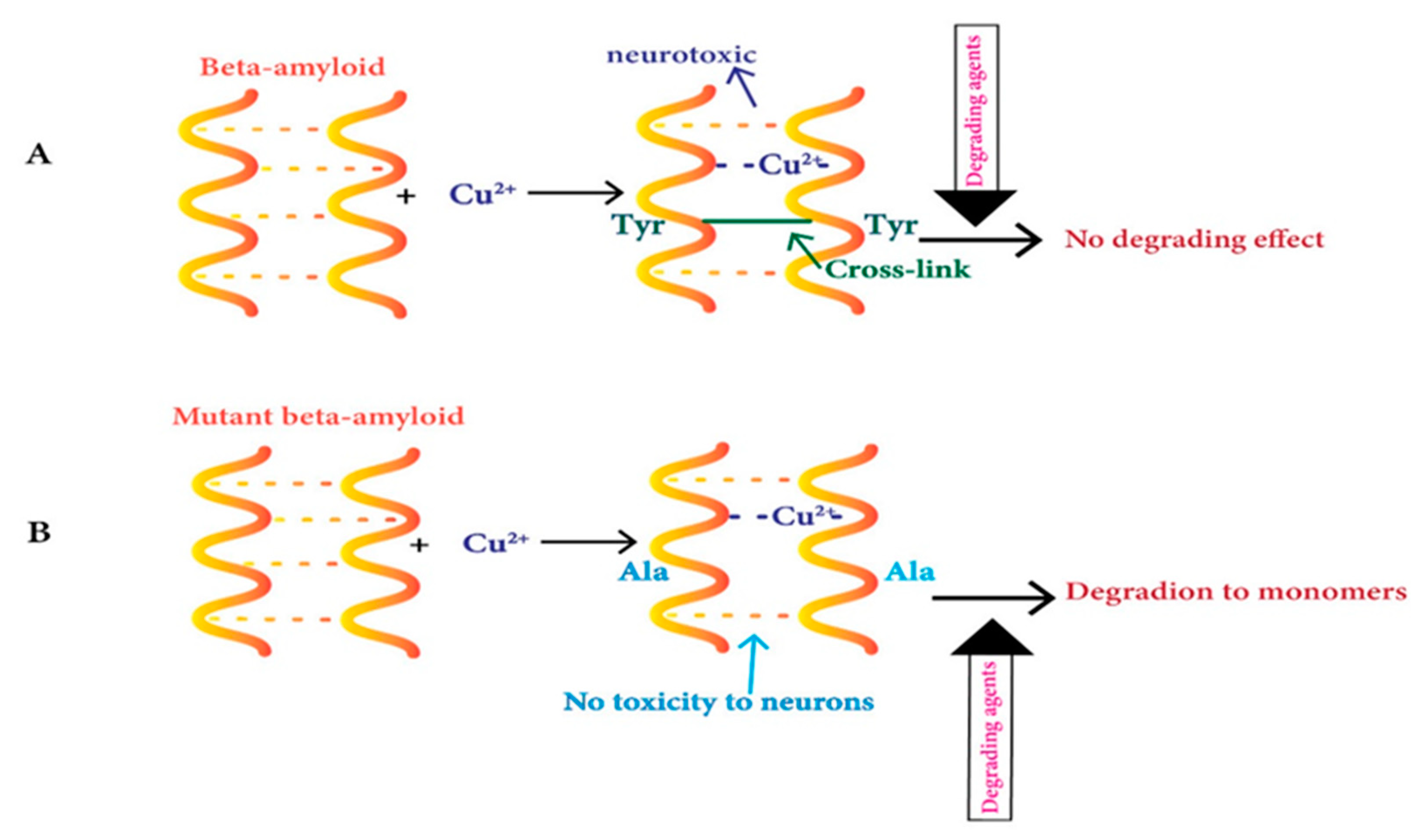{kind=link}
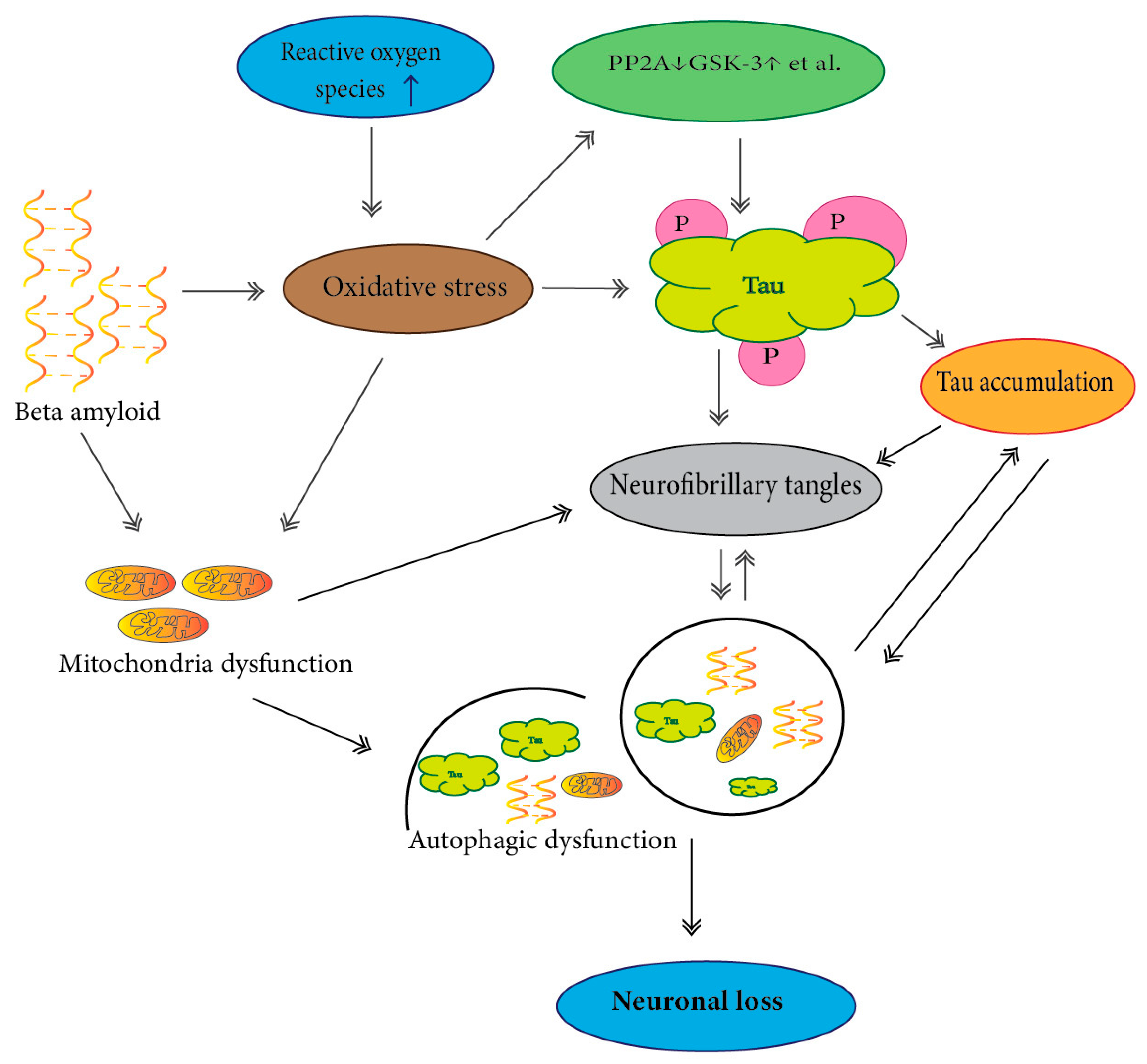{kind=link}
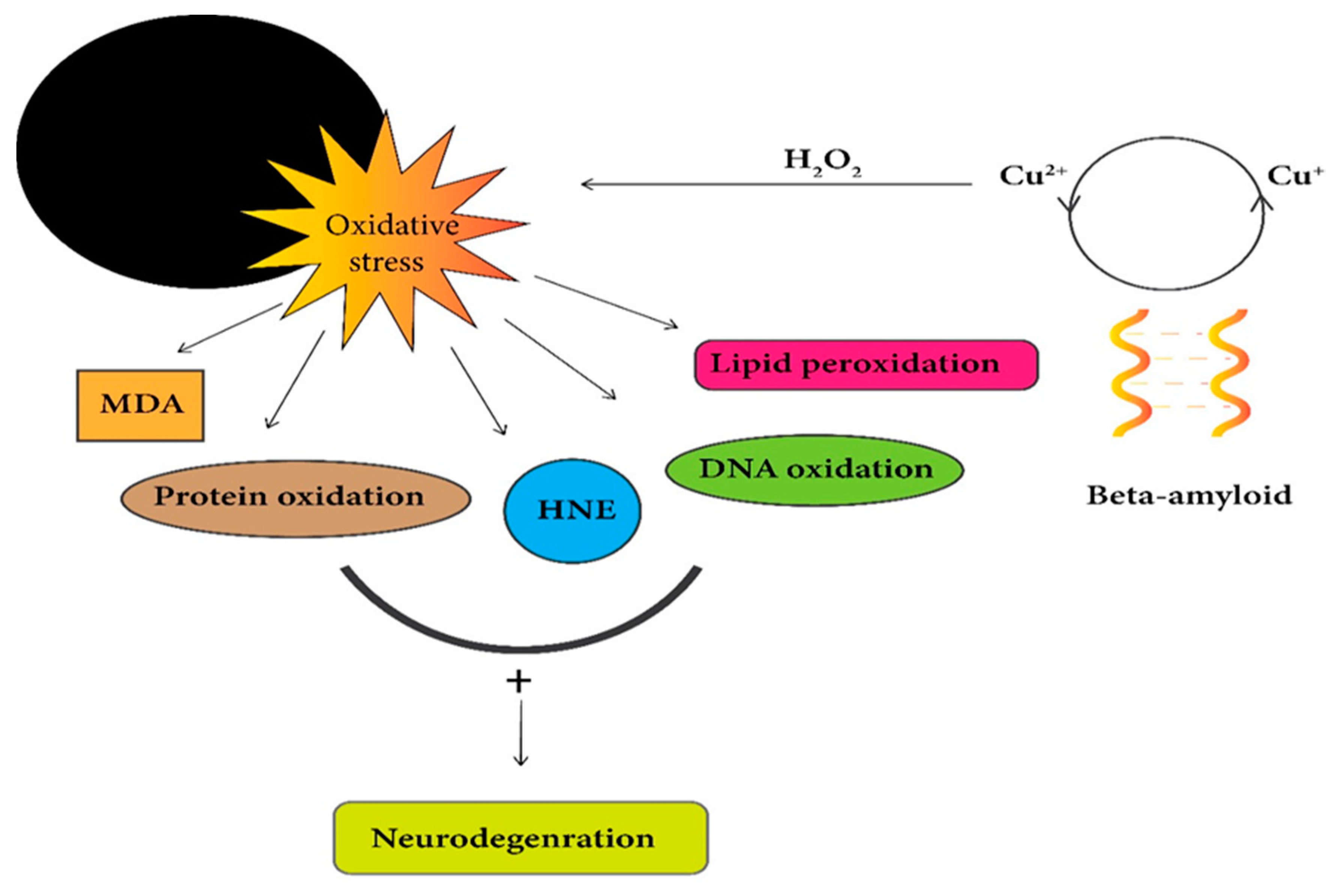{kind=link}
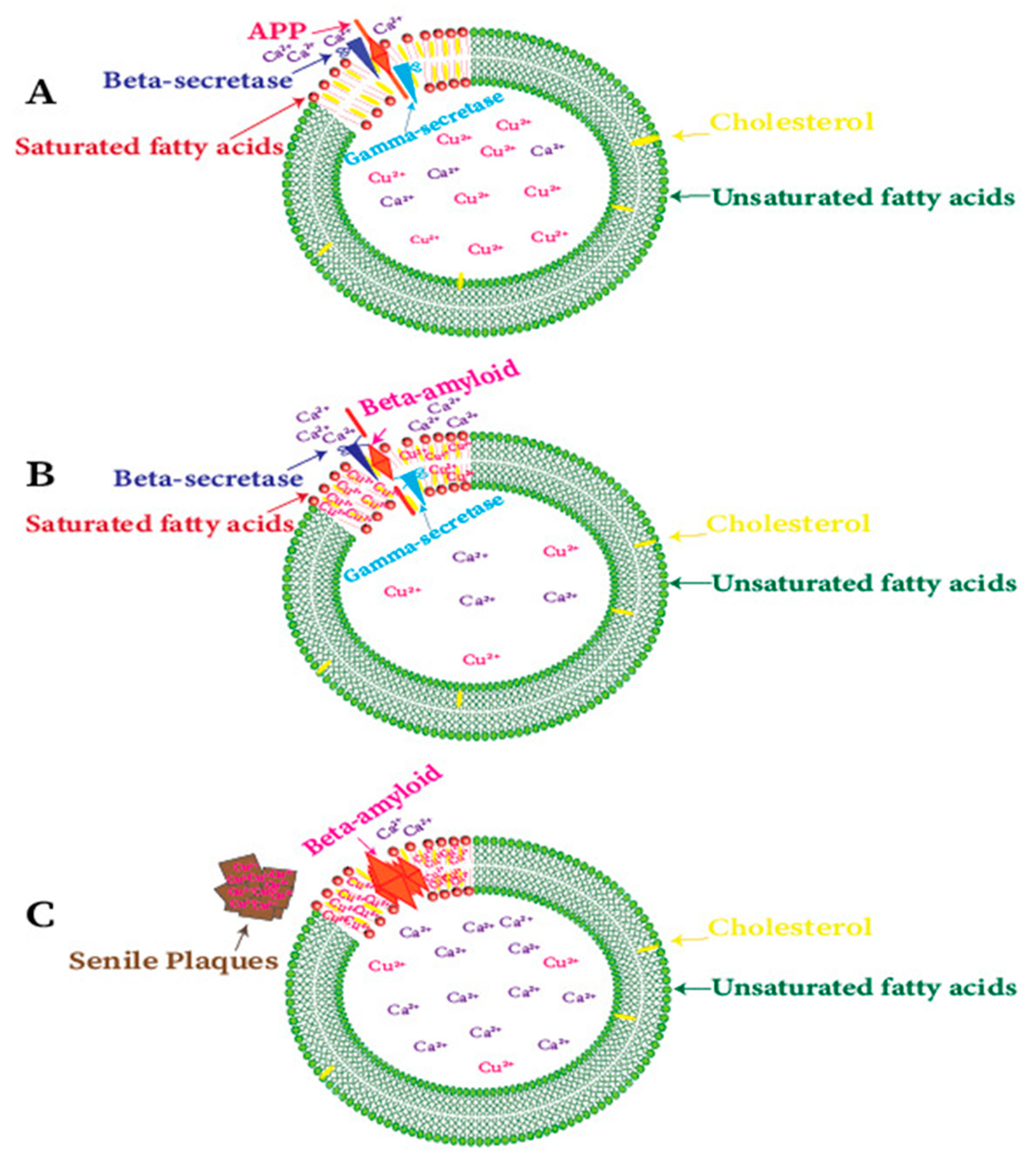{kind=link}
{kind=link}
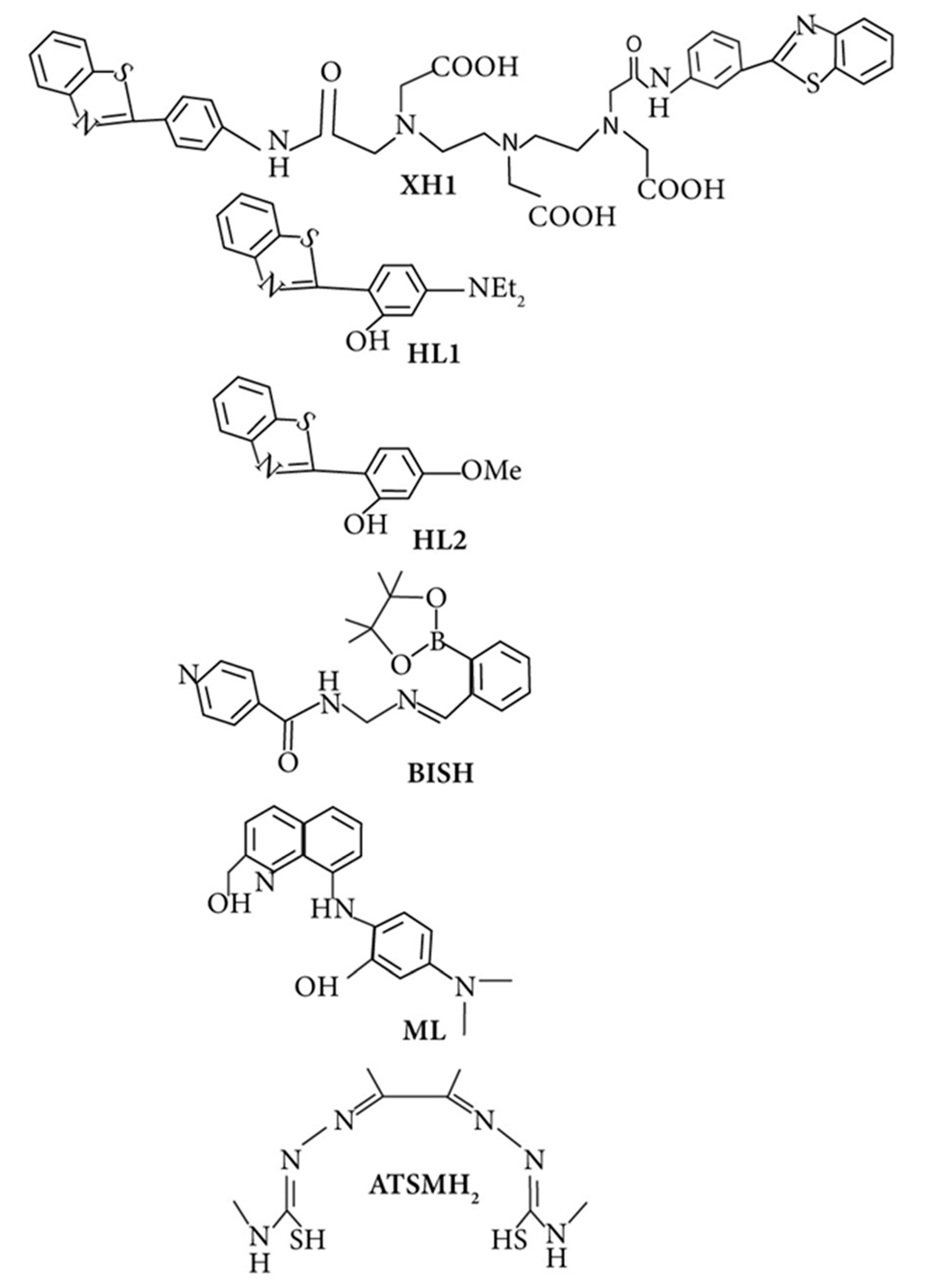{kind=link}
Table 1.
Copper ions’ effects on selected metal-binding proteins implicated in Alzheimer’s disease.
| Protein. | Effect | Animal and Cell Model |
|---|---|---|
| Amyloid-β | Cu plays a role in modulating the aggregation of amyloid-β and decreases toxicity; nevertheless, the presence of copper insoluble amyloid-β accelerate apoptotic cell death. Sub-stoichiometric levels of copper(II are rendered Aβ aggregation and cause more neurotoxicity. | A synthetic peptide (Aβ2535), HEK293 cell,PC-12,and primary hippocampal cells [149,150,151,152,153,154]. |
| Tau | Plays in modulating phosphorylation. Plays in modulating Aβ aggregation. | Triple-transgenic mice model of AD (3xTg-AD), SHSY5Y human neuroblastoma cells, and Alzheimer’s disease transgenic mouse model [155,156]. A peptide from tau possesses a repeat microtubule-binding domain [157]. |
| Amyloid precursor protein | Increase expression levels and distribution of APP and amyloid-β, respectively. Copper has promoted traffic and redistribution of APP. Increases Cu2+ mediated oxidative stress as well as APP ectodomain neuronal cell death. | APP/PS1transgenic mice, N2a cells, primary cortical neurons, MDCK-APP-cherry cells, polarized epithelial cells, SH-SY5Y cells [112,115,156,158]. Recombination of amyloid precursor protein (APP), APP mutant cells, and primary neuronal cell lines [159]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ejaz, H.W.; Wang, W.; Lang, M. Copper Toxicity Links to Pathogenesis of Alzheimer’s Disease and Therapeutics Approaches. Int. J. Mol. Sci. 2020, 21, 7660. https://doi.org/10.3390/ijms21207660
AMA Style
Ejaz HW, Wang W, Lang M. Copper Toxicity Links to Pathogenesis of Alzheimer’s Disease and Therapeutics Approaches. International Journal of Molecular Sciences. 2020; 21(20):7660. https://doi.org/10.3390/ijms21207660
Chicago/Turabian StyleEjaz, Hafza Wajeeha, Wei Wang, and Minglin Lang. 2020. "Copper Toxicity Links to Pathogenesis of Alzheimer’s Disease and Therapeutics Approaches" International Journal of Molecular Sciences 21, no. 20: 7660. https://doi.org/10.3390/ijms21207660
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.