
Inflammatory Microenvironment and Specific T Cells in Myeloproliferative Neoplasms: Immunopathogenesis and Novel Immunotherapies

 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. Hit the Road JAK: JAK-STAT Signaling at the Dangerous Crossroads between Inflammation and Clonal Myeloproliferation
3. Tainted Neighborhood: The Emerging Role of the Bone Marrow Niche
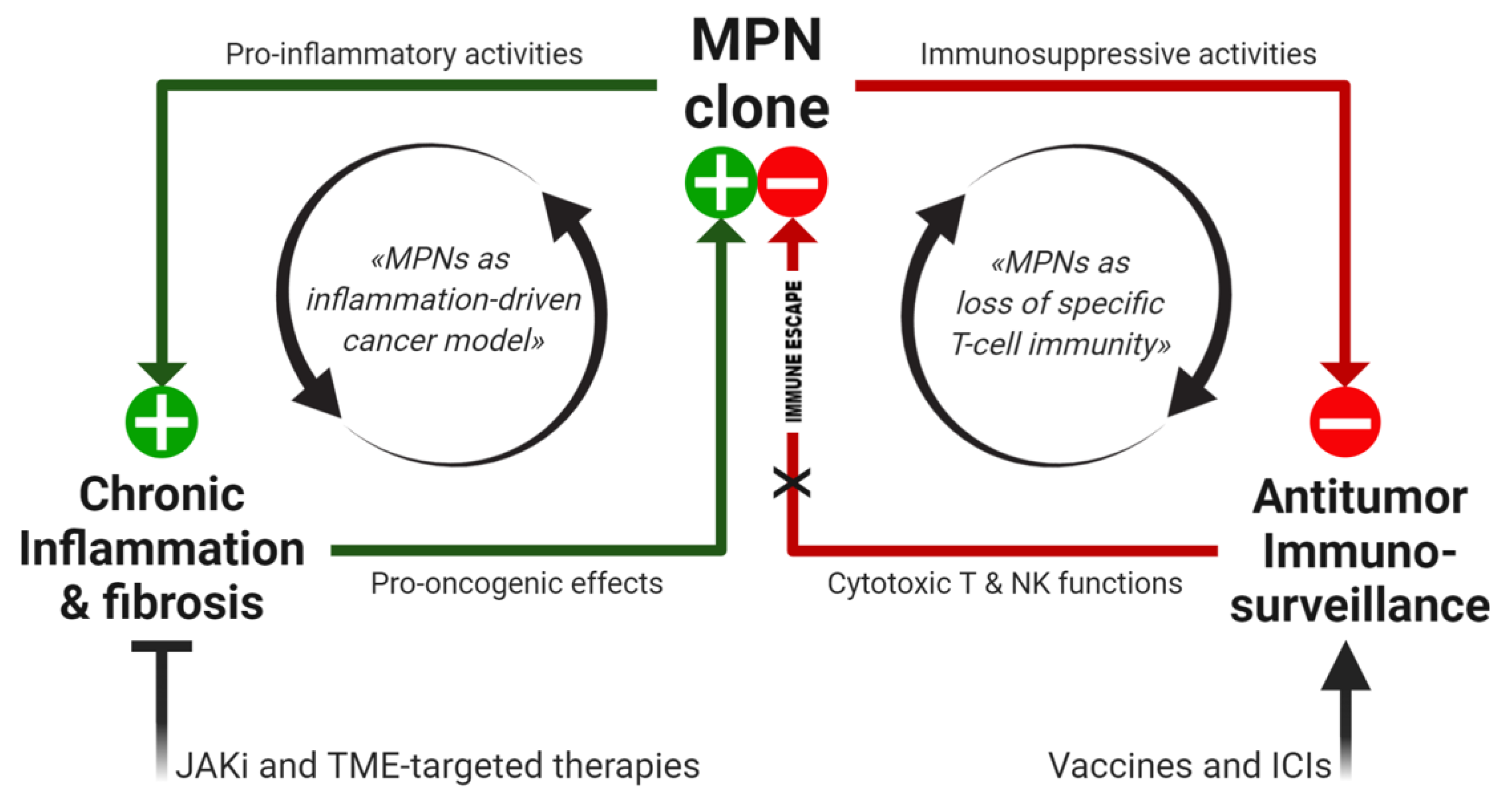
4. The Perfect Storm: Combining Inflammation and Specific Mechanisms of Tumor Immune Escape
5. Novel Mutant Hunters: The Emergence of JAK2/CALR Mutation-Targeted T Cell Immunity
6. Looking for the Achilles’ Heel in MPNs: Innovative Targeted Therapies
6.1. Targeting the Microenvironment
6.2. Targeting CD123
6.3. Checkpoint Inhibitors
6.4. Vaccination
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Spivak, J.L. Myeloproliferative Neoplasms. N. Engl. J. Med. 2017, 376, 2168–2181. [Google Scholar] [CrossRef] [Green Version]
- Shallis, R.M.; Wang, R.; Davidoff, A.; Ma, X.; Podoltsev, N.A.; Zeidan, A.M. Epidemiology of the Classical Myeloproliferative Neoplasms: The Four Corners of an Expansive and Complex Map. Blood Rev. 2020, 42, 100706. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Guglielmelli, P.; Larson, D.R.; Finke, C.; Wassie, E.A.; Pieri, L.; Gangat, N.; Fjerza, R.; Belachew, A.A.; Lasho, T.L.; et al. Long-Term Survival and Blast Transformation in Molecularly Annotated Essential Thrombocythemia, Polycythemia Vera, and Myelofibrosis. Blood 2014, 124, 2507–2513. [Google Scholar] [CrossRef] [PubMed]
- Moliterno, A.R.; Ginzburg, Y.Z.; Hoffman, R. Clinical Insights into the Origins of Thrombosis in Myeloproliferative Neoplasms. Blood 2020. [Google Scholar] [CrossRef]
- Harrison, C.N.; Koschmieder, S.; Foltz, L.; Guglielmelli, P.; Flindt, T.; Koehler, M.; Mathias, J.; Komatsu, N.; Boothroyd, R.N.; Spierer, A.; et al. The Impact of Myeloproliferative Neoplasms (MPNs) on Patient Quality of Life and Productivity: Results from the International MPN Landmark Survey. Ann. Hematol. 2017, 96, 1653–1665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luque Paz, D.; Jouanneau-Courville, R.; Riou, J.; Ianotto, J.-C.; Boyer, F.; Chauveau, A.; Renard, M.; Chomel, J.-C.; Cayssials, E.; Gallego-Hernanz, M.-P.; et al. Leukemic Evolution of Polycythemia Vera and Essential Thrombocythemia: Genomic Profiles Predict Time to Transformation. Blood Adv. 2020, 4, 4887–4897. [Google Scholar] [CrossRef] [PubMed]
- Zoi, K.; Cross, N.C.P. Genomics of Myeloproliferative Neoplasms. J. Clin. Oncol. 2017, 35, 947–954. [Google Scholar] [CrossRef]
- Vainchenker, W.; Constantinescu, S.N. JAK/STAT Signaling in Hematological Malignancies. Oncogene 2013, 32, 2601–2613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, L.M.; Tong, W.; Levine, R.L.; Scott, M.A.; Beer, P.A.; Stratton, M.R.; Futreal, P.A.; Erber, W.N.; McMullin, M.F.; Harrison, C.N.; et al. JAK2 Exon 12 Mutations in Polycythemia Vera and Idiopathic Erythrocytosis. N. Engl. J. Med. 2007, 356, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Pardanani, A.D.; Levine, R.L.; Lasho, T.; Pikman, Y.; Mesa, R.A.; Wadleigh, M.; Steensma, D.P.; Elliott, M.A.; Wolanskyj, A.P.; Hogan, W.J.; et al. MPL515 Mutations in Myeloproliferative and Other Myeloid Disorders: A Study of 1182 Patients. Blood 2006, 108, 3472–3476. [Google Scholar] [CrossRef] [Green Version]
- Klampfl, T.; Gisslinger, H.; Harutyunyan, A.S.; Nivarthi, H.; Rumi, E.; Milosevic, J.D.; Them, N.C.C.; Berg, T.; Gisslinger, B.; Pietra, D.; et al. Somatic Mutations of Calreticulin in Myeloproliferative Neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390. [Google Scholar] [CrossRef] [Green Version]
- Nangalia, J.; Massie, C.E.; Baxter, E.J.; Nice, F.L.; Gundem, G.; Wedge, D.C.; Avezov, E.; Li, J.; Kollmann, K.; Kent, D.G.; et al. Somatic CALR Mutations in Myeloproliferative Neoplasms with Nonmutated JAK2. N. Engl. J. Med. 2013, 369, 2391–2405. [Google Scholar] [CrossRef] [Green Version]
- Vainchenker, W.; Plo, I.; Marty, C.; Varghese, L.N.; Constantinescu, S.N. The Role of the Thrombopoietin Receptor MPL in Myeloproliferative Neoplasms: Recent Findings and Potential Therapeutic Applications. Expert Rev. Hematol. 2019, 12, 437–448. [Google Scholar] [CrossRef]
- Elf, S.; Abdelfattah, N.S.; Chen, E.; Perales-Patón, J.; Rosen, E.A.; Ko, A.; Peisker, F.; Florescu, N.; Giannini, S.; Wolach, O.; et al. Mutant Calreticulin Requires Both Its Mutant C-Terminus and the Thrombopoietin Receptor for Oncogenic Transformation. Cancer Discov. 2016, 6, 368–381. [Google Scholar] [CrossRef] [Green Version]
- Pecquet, C.; Balligand, T.; Chachoua, I.; Roy, A.; Vertenoeil, G.; Colau, D.; Fertig, E.; Marty, C.; Nivarthi, H.; Defour, J.-P.; et al. Secreted Mutant Calreticulins As Rogue Cytokines Trigger Thrombopoietin Receptor Activation Specifically in CALR Mutated Cells: Perspectives for MPN Therapy. Blood 2018, 132, 4. [Google Scholar] [CrossRef]
- Prins, D.; González Arias, C.; Klampfl, T.; Grinfeld, J.; Green, A.R. Mutant Calreticulin in the Myeloproliferative Neoplasms. HemaSphere 2020, 4, e333. [Google Scholar] [CrossRef]
- Barbui, T.; Thiele, J.; Gisslinger, H.; Kvasnicka, H.M.; Vannucchi, A.M.; Guglielmelli, P.; Orazi, A.; Tefferi, A. The 2016 WHO Classification and Diagnostic Criteria for Myeloproliferative Neoplasms: Document Summary and in-Depth Discussion. Blood Cancer J. 2018, 8, 15. [Google Scholar] [CrossRef]
- Lundberg, P.; Karow, A.; Nienhold, R.; Looser, R.; Hao-Shen, H.; Nissen, I.; Girsberger, S.; Lehmann, T.; Passweg, J.; Stern, M.; et al. Clonal Evolution and Clinical Correlates of Somatic Mutations in Myeloproliferative Neoplasms. Blood 2014, 123, 2220–2228. [Google Scholar] [CrossRef] [Green Version]
- Grabek, J.; Straube, J.; Bywater, M.; Lane, S.W. MPN: The Molecular Drivers of Disease Initiation, Progression and Transformation and Their Effect on Treatment. Cells 2020, 9, 1901. [Google Scholar] [CrossRef] [PubMed]
- McPherson, S.; McMullin, M.F.; Mills, K. Epigenetics in Myeloproliferative Neoplasms. J. Cell. Mol. Med. 2017, 21, 1660–1667. [Google Scholar] [CrossRef] [Green Version]
- Homaei Hadad, E.; Pezeshki, S.M.S.; Shahrabi, S.; Saki Malehi, A.; Saki, N. Co-Existence of Mutations in Myeloproliferative Neoplasms and Their Clinical Significance: A Prognostic Approach. Expert Rev. Hematol. 2020, 13, 1289–1301. [Google Scholar] [CrossRef] [PubMed]
- Rumi, E.; Trotti, C.; Vanni, D.; Casetti, I.C.; Pietra, D.; Sant’Antonio, E. The Genetic Basis of Primary Myelofibrosis and Its Clinical Relevance. Int. J. Mol. Sci. 2020, 21, 8885. [Google Scholar] [CrossRef]
- Loberg, M.A.; Bell, R.K.; Goodwin, L.O.; Eudy, E.; Miles, L.A.; SanMiguel, J.M.; Young, K.; Bergstrom, D.E.; Levine, R.L.; Schneider, R.K.; et al. Sequentially Inducible Mouse Models Reveal That Npm1 Mutation Causes Malignant Transformation of Dnmt3a-Mutant Clonal Hematopoiesis. Leukemia 2019, 33, 1635–1649. [Google Scholar] [CrossRef]
- Forghieri, F.; Paolini, A.; Morselli, M.; Bigliardi, S.; Bonacorsi, G.; Leonardi, G.; Coluccio, V.; Maccaferri, M.; Fantuzzi, V.; Faglioni, L.; et al. NPM1 Mutations May Reveal Acute Myeloid Leukemia in Cases Otherwise Morphologically Diagnosed as Myelodysplastic Syndromes or Myelodysplastic/Myeloproliferative Neoplasms. Leuk. Lymphoma 2015, 56, 3222–3226. [Google Scholar] [CrossRef]
- Patel, S.S.; Ho, C.; Ptashkin, R.N.; Sadigh, S.; Bagg, A.; Geyer, J.T.; Xu, M.L.; Prebet, T.; Mason, E.F.; Seegmiller, A.C.; et al. Clinicopathologic and Genetic Characterization of Nonacute NPM1-Mutated Myeloid Neoplasms. Blood Adv. 2019, 3, 1540–1545. [Google Scholar] [CrossRef] [Green Version]
- 26. Forghieri, F.; Nasillo, V.; Paolini, A.; Bettelli, F.; Pioli, V.; Giusti, D.; Gilioli, A.; Colasante, C.; Acquaviva, G.; Riva, G.; et al. NPM1-Mutated Myeloid Neoplasms with <20% Blasts: A Really Distinct Clinico-Pathologic Entity? Int. J. Mol. Sci. 2020, 21, 8975. [Google Scholar] [CrossRef]
- Perner, E. Heidel Roles of JAK2 in Aging, Inflammation, Hematopoiesis and Malignant Transformation. Cells 2019, 8, 854. [Google Scholar] [CrossRef] [Green Version]
- Hammarén, H.M.; Virtanen, A.T.; Raivola, J.; Silvennoinen, O. The Regulation of JAKs in Cytokine Signaling and Its Breakdown in Disease. Cytokine 2019, 118, 48–63. [Google Scholar] [CrossRef]
- Barosi, G. An Immune Dysregulation in MPN. Curr. Hematol. Malig. Rep. 2014, 9, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Koschmieder, S.; Mughal, T.I.; Hasselbalch, H.C.; Barosi, G.; Valent, P.; Kiladjian, J.-J.; Jeryczynski, G.; Gisslinger, H.; Jutzi, J.S.; Pahl, H.L.; et al. Myeloproliferative Neoplasms and Inflammation: Whether to Target the Malignant Clone or the Inflammatory Process or Both. Leukemia 2016, 30, 1018–1024. [Google Scholar] [CrossRef]
- Mendez Luque, L.F.; Blackmon, A.L.; Ramanathan, G.; Fleischman, A.G. Key Role of Inflammation in Myeloproliferative Neoplasms: Instigator of Disease Initiation, Progression and Symptoms. Curr. Hematol. Malig. Rep. 2019, 14, 145–153. [Google Scholar] [CrossRef]
- Koschmieder, S.; Chatain, N. Role of Inflammation in the Biology of Myeloproliferative Neoplasms. Blood Rev. 2020, 42, 100711. [Google Scholar] [CrossRef] [PubMed]
- Hasselbalch, H.C. Perspectives on Chronic Inflammation in Essential Thrombocythemia, Polycythemia Vera, and Myelofibrosis: Is Chronic Inflammation a Trigger and Driver of Clonal Evolution and Development of Accelerated Atherosclerosis and Second Cancer? Blood 2012, 119, 3219–3225. [Google Scholar] [CrossRef] [Green Version]
- Lussana, F.; Rambaldi, A. Inflammation and Myeloproliferative Neoplasms. J. Autoimmun. 2017, 85, 58–63. [Google Scholar] [CrossRef]
- Landtblom, A.R.; Bower, H.; Andersson, T.M.-L.; Dickman, P.W.; Samuelsson, J.; Björkholm, M.; Kristinsson, S.Y.; Hultcrantz, M. Second Malignancies in Patients with Myeloproliferative Neoplasms: A Population-Based Cohort Study of 9379 Patients. Leukemia 2018, 32, 2203–2210. [Google Scholar] [CrossRef] [PubMed]
- Brabrand, M.; Frederiksen, H. Risks of Solid and Lymphoid Malignancies in Patients with Myeloproliferative Neoplasms: Clinical Implications. Cancers 2020, 12, 61. [Google Scholar] [CrossRef]
- Hasselbalch, H.C.; Skov, V.; Kjær, L.; Sørensen, T.L.; Ellervik, C.; Wienecke, T. Myeloproliferative Blood Cancers as a Human Neuroinflammation Model for Development of Alzheimer’s Disease: Evidences and Perspectives. J. Neuroinflammation 2020, 17, 248. [Google Scholar] [CrossRef] [PubMed]
- Bak, M.; Jess, T.; Flachs, E.M.; Zwisler, A.-D.; Juel, K.; Frederiksen, H. Risk of Inflammatory Bowel Disease in Patients with Chronic Myeloproliferative Neoplasms: A Danish Nationwide Cohort Study. Cancers 2020, 12, 2700. [Google Scholar] [CrossRef]
- Hasselbalch, H.C. Chronic Inflammation as a Promotor of Mutagenesis in Essential Thrombocythemia, Polycythemia Vera and Myelofibrosis. A Human Inflammation Model for Cancer Development? Leuk. Res. 2013, 37, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.; Sajid, Z.; Pedersen, R.K.; Gudmand-Hoeyer, J.; Ellervik, C.; Skov, V.; Kjær, L.; Pallisgaard, N.; Kruse, T.A.; Thomassen, M.; et al. Mathematical Modelling as a Proof of Concept for MPNs as a Human Inflammation Model for Cancer Development. PLoS ONE 2017, 12, e0183620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjørn, M.E.; Hasselbalch, H.C. The Role of Reactive Oxygen Species in Myelofibrosis and Related Neoplasms. Mediat. Inflamm. 2015, 2015, 648090. [Google Scholar] [CrossRef] [Green Version]
- Longhitano, L.; Li Volti, G.; Giallongo, C.; Spampinato, M.; Barbagallo, I.; Di Rosa, M.; Romano, A.; Avola, R.; Tibullo, D.; Palumbo, G.A. The Role of Inflammation and Inflammasome in Myeloproliferative Disease. J. Clin. Med. 2020, 9, 2334. [Google Scholar] [CrossRef] [PubMed]
- Marty, C.; Lacout, C.; Droin, N.; Le Couédic, J.-P.; Ribrag, V.; Solary, E.; Vainchenker, W.; Villeval, J.-L.; Plo, I. A Role for Reactive Oxygen Species in JAK2V617F Myeloproliferative Neoplasm Progression. Leukemia 2013, 27, 2187–2195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemmati, S.; Haque, T.; Gritsman, K. Inflammatory Signaling Pathways in Preleukemic and Leukemic Stem Cells. Front. Oncol. 2017, 7, 265. [Google Scholar] [CrossRef] [Green Version]
- Allegra, A.; Pioggia, G.; Tonacci, A.; Casciaro, M.; Musolino, C.; Gangemi, S. Synergic Crosstalk between Inflammation, Oxidative Stress, and Genomic Alterations in BCR-ABL-Negative Myeloproliferative Neoplasm. Antioxidants 2020, 9, 1037. [Google Scholar] [CrossRef]
- Hermouet, S.; Godard, A.; Pineau, D.; Corre, I.; Raher, S.; Lippert, E.; Jacques, Y. Abnormal Production of Interleukin (IL)-11 and IL-8 in Polycythaemia Vera. Cytokine 2002, 20, 178–183. [Google Scholar] [CrossRef]
- Panteli, K.E.; Hatzimichael, E.C.; Bouranta, P.K.; Katsaraki, A.; Seferiadis, K.; Stebbing, J.; Bourantas, K.L. Serum Interleukin (IL)-1, IL-2, SIL-2Ra, IL-6 and Thrombopoietin Levels in Patients with Chronic Myeloproliferative Diseases. Br. J. Haematol. 2005, 130, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Alonci, A.; Allegra, A.; Bellomo, G.; Penna, G.; D’Angelo, A.; Quartarone, E.; Musolino, C. Evaluation of Circulating Endothelial Cells, VEGF and VEGFR2 Serum Levels in Patients with Chronic Myeloproliferative Diseases. Hematol. Oncol. 2008, 26, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Allegra, A.; Alonci, A.; Bellomo, G.; D’Angelo, A.; Granata, A.; Russo, S.; Quartarone, E.; Musolino, C. Evaluation of Interleukin-17 Serum Levels in Patients with Chronic Myeloproliferative Diseases. Tumori 2009, 95, 404–405. [Google Scholar] [CrossRef] [PubMed]
- Pourcelot, E.; Trocme, C.; Mondet, J.; Bailly, S.; Toussaint, B.; Mossuz, P. Cytokine Profiles in Polycythemia Vera and Essential Thrombocythemia Patients: Clinical Implications. Exp. Hematol. 2014, 42, 360–368. [Google Scholar] [CrossRef]
- Boissinot, M.; Cleyrat, C.; Vilaine, M.; Jacques, Y.; Corre, I.; Hermouet, S. Anti-Inflammatory Cytokines Hepatocyte Growth Factor and Interleukin-11 Are over-Expressed in Polycythemia Vera and Contribute to the Growth of Clonal Erythroblasts Independently of JAK2V617F. Oncogene 2011, 30, 990–1001. [Google Scholar] [CrossRef] [Green Version]
- Jutzi, J.S.; Pahl, H.L. The Hen or the Egg: Inflammatory Aspects of Murine MPN Models. Mediat. Inflamm. 2015, 2015, 101987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefferi, A.; Vaidya, R.; Caramazza, D.; Finke, C.; Lasho, T.; Pardanani, A. Circulating Interleukin (IL)-8, IL-2R, IL-12, and IL-15 Levels Are Independently Prognostic in Primary Myelofibrosis: A Comprehensive Cytokine Profiling Study. J. Clin. Oncol. 2011, 29, 1356–1363. [Google Scholar] [CrossRef] [PubMed]
- Masselli, E.; Pozzi, G.; Gobbi, G.; Merighi, S.; Gessi, S.; Vitale, M.; Carubbi, C. Cytokine Profiling in Myeloproliferative Neoplasms: Overview on Phenotype Correlation, Outcome Prediction, and Role of Genetic Variants. Cells 2020, 9, 2136. [Google Scholar] [CrossRef]
- Agarwal, A.; Morrone, K.; Bartenstein, M.; Zhao, Z.J.; Verma, A.; Goel, S. Bone Marrow Fibrosis in Primary Myelofibrosis: Pathogenic Mechanisms and the Role of TGF-β. Stem Cell Investig. 2016, 3, 5. [Google Scholar] [CrossRef]
- Wehrle, J.; Seeger, T.S.; Schwemmers, S.; Pfeifer, D.; Bulashevska, A.; Pahl, H.L. Transcription Factor Nuclear Factor Erythroid-2 Mediates Expression of the Cytokine Interleukin 8, a Known Predictor of Inferior Outcome in Patients with Myeloproliferative Neoplasms. Haematologica 2013, 98, 1073–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaidya, R.; Gangat, N.; Jimma, T.; Finke, C.M.; Lasho, T.L.; Pardanani, A.; Tefferi, A. Plasma Cytokines in Polycythemia Vera: Phenotypic Correlates, Prognostic Relevance, and Comparison with Myelofibrosis. Am. J. Hematol. 2012, 87, 1003–1005. [Google Scholar] [CrossRef] [PubMed]
- Flamant, L.; Toffoli, S.; Raes, M.; Michiels, C. Hypoxia Regulates Inflammatory Gene Expression in Endothelial Cells. Exp. Cell Res. 2009, 315, 733–747. [Google Scholar] [CrossRef] [PubMed]
- Kleppe, M.; Kwak, M.; Koppikar, P.; Riester, M.; Keller, M.; Bastian, L.; Hricik, T.; Bhagwat, N.; McKenney, A.S.; Papalexi, E.; et al. JAK-STAT Pathway Activation in Malignant and Nonmalignant Cells Contributes to MPN Pathogenesis and Therapeutic Response. Cancer Discov. 2015, 5, 316–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elliott, M.A.; Verstovsek, S.; Dingli, D.; Schwager, S.M.; Mesa, R.A.; Li, C.Y.; Tefferi, A. Monocytosis Is an Adverse Prognostic Factor for Survival in Younger Patients with Primary Myelofibrosis. Leuk. Res. 2007, 31, 1503–1509. [Google Scholar] [CrossRef]
- Barraco, D.; Cerquozzi, S.; Gangat, N.; Patnaik, M.M.; Lasho, T.; Finke, C.; Hanson, C.A.; Ketterling, R.P.; Pardanani, A.; Tefferi, A. Monocytosis in Polycythemia Vera: Clinical and Molecular Correlates. Am. J. Hematol. 2017, 92, 640–645. [Google Scholar] [CrossRef] [Green Version]
- Fisher, D.A.C.; Miner, C.A.; Engle, E.K.; Hu, H.; Collins, T.B.; Zhou, A.; Allen, M.J.; Malkova, O.N.; Oh, S.T. Cytokine Production in Myelofibrosis Exhibits Differential Responsiveness to JAK-STAT, MAP Kinase, and NFκB Signaling. Leukemia 2019, 33, 1978–1995. [Google Scholar] [CrossRef] [PubMed]
- Lai, H.Y.; Brooks, S.A.; Craver, B.M.; Morse, S.J.; Nguyen, T.K.; Haghighi, N.; Garbati, M.R.; Fleischman, A.G. Defective Negative Regulation of Toll-like Receptor Signaling Leads to Excessive TNF-α in Myeloproliferative Neoplasm. Blood Adv. 2019, 3, 122–131. [Google Scholar] [CrossRef] [Green Version]
- Spanoudakis, E.; Papoutselis, M.; Bazdiara, I.; Lamprianidi, E.; Kordella, X.; Tilkeridis, C.; Tsatalas, C.; Kotsianidis, I. The JAK2V617F Point Mutation Increases the Osteoclast Forming Ability of Monocytes in Patients with Chronic Myeloproliferative Neoplasms and Makes Their Osteoclasts More Susceptible to JAK2 Inhibition. Mediterr. J. Hematol. Infect. Dis. 2018, 10, e2018058. [Google Scholar] [CrossRef] [PubMed]
- Verstovsek, S.; Manshouri, T.; Pilling, D.; Bueso-Ramos, C.E.; Newberry, K.J.; Prijic, S.; Knez, L.; Bozinovic, K.; Harris, D.M.; Spaeth, E.L.; et al. Role of Neoplastic Monocyte-Derived Fibrocytes in Primary Myelofibrosis. J. Exp. Med. 2016, 213, 1723–1740. [Google Scholar] [CrossRef] [PubMed]
- Øbro, N.F.; Grinfeld, J.; Belmonte, M.; Irvine, M.; Shepherd, M.S.; Rao, T.N.; Karow, A.; Riedel, L.M.; Harris, O.B.; Baxter, E.J.; et al. Longitudinal Cytokine Profiling Identifies GRO-α and EGF as Potential Biomarkers of Disease Progression in Essential Thrombocythemia. HemaSphere 2020, 4, e371. [Google Scholar] [CrossRef]
- Kleppe, M.; Koche, R.; Zou, L.; van Galen, P.; Hill, C.E.; Dong, L.; De Groote, S.; Papalexi, E.; Hanasoge Somasundara, A.V.; Cordner, K.; et al. Dual Targeting of Oncogenic Activation and Inflammatory Signaling Increases Therapeutic Efficacy in Myeloproliferative Neoplasms. Cancer Cell 2018, 33, 29–43.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, E.K.; Luo, M.; Rauh, M.J. Clonal Hematopoiesis and Inflammation: Partners in Leukemogenesis and Comorbidity. Exp. Hematol. 2020, 83, 85–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Zhao, K.; Shen, Q.; Han, Y.; Gu, Y.; Li, X.; Zhao, D.; Liu, Y.; Wang, C.; Zhang, X.; et al. Tet2 Is Required to Resolve Inflammation by Recruiting Hdac2 to Specifically Repress IL-6. Nature 2015, 525, 389–393. [Google Scholar] [CrossRef] [Green Version]
- Cull, A.H.; Snetsinger, B.; Buckstein, R.; Wells, R.A.; Rauh, M.J. Tet2 Restrains Inflammatory Gene Expression in Macrophages. Exp. Hematol. 2017, 55, 56–70.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Z.; Kotzin, J.J.; Ramdas, B.; Chen, S.; Nelanuthala, S.; Palam, L.R.; Pandey, R.; Mali, R.S.; Liu, Y.; Kelley, M.R.; et al. Inhibition of Inflammatory Signaling in Tet2 Mutant Preleukemic Cells Mitigates Stress-Induced Abnormalities and Clonal Hematopoiesis. Cell Stem Cell 2018, 23, 833–849.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, S.J.; Scadden, D.T. The Bone Marrow Niche for Haematopoietic Stem Cells. Nature 2014, 505, 327–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitroulis, I.; Kalafati, L.; Bornhäuser, M.; Hajishengallis, G.; Chavakis, T. Regulation of the Bone Marrow Niche by Inflammation. Front. Immunol. 2020, 11, 1540. [Google Scholar] [CrossRef]
- Korn, C.; Méndez-Ferrer, S. Myeloid Malignancies and the Microenvironment. Blood 2017, 129, 811–822. [Google Scholar] [CrossRef] [Green Version]
- Mullally, A.; Poveromo, L.; Schneider, R.K.; Al-Shahrour, F.; Lane, S.W.; Ebert, B.L. Distinct Roles for Long-Term Hematopoietic Stem Cells and Erythroid Precursor Cells in a Murine Model of Jak2V617F-Mediated Polycythemia Vera. Blood 2012, 120, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, P.; Takizawa, H.; Kubovcakova, L.; Guo, G.; Hao-Shen, H.; Dirnhofer, S.; Orkin, S.H.; Manz, M.G.; Skoda, R.C. Myeloproliferative Neoplasms Can Be Initiated from a Single Hematopoietic Stem Cell Expressing JAK2-V617F. J. Exp. Med. 2014, 211, 2213–2230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, E.; Schneider, R.K.; Breyfogle, L.J.; Rosen, E.A.; Poveromo, L.; Elf, S.; Ko, A.; Brumme, K.; Levine, R.; Ebert, B.L.; et al. Distinct Effects of Concomitant Jak2V617F Expression and Tet2 Loss in Mice Promote Disease Progression in Myeloproliferative Neoplasms. Blood 2015, 125, 327–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mead, A.J.; Mullally, A. Myeloproliferative Neoplasm Stem Cells. Blood 2017, 129, 1607–1616. [Google Scholar] [CrossRef] [Green Version]
- Schepers, K.; Pietras, E.M.; Reynaud, D.; Flach, J.; Binnewies, M.; Garg, T.; Wagers, A.J.; Hsiao, E.C.; Passegué, E. Myeloproliferative Neoplasia Remodels the Endosteal Bone Marrow Niche into a Self-Reinforcing Leukemic Niche. Cell Stem Cell 2013, 13, 285–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Raimondo, F.; Palumbo, G.A.; Molica, S.; Giustolisi, R. Angiogenesis in Chronic Myeloproliferative Diseases. Acta Haematol. 2001, 106, 177–183. [Google Scholar] [CrossRef]
- Dragoni, S.; Reforgiato, M.; Zuccolo, E.; Poletto, V.; Lodola, F.; Ruffinatti, F.A.; Bonetti, E.; Guerra, G.; Barosi, G.; Rosti, V.; et al. Dysregulation of VEGF-Induced Proangiogenic Ca2+ Oscillations in Primary Myelofibrosis-Derived Endothelial Colony-Forming Cells. Exp. Hematol. 2015, 43, 1019–1030.e3. [Google Scholar] [CrossRef] [PubMed]
- Hoermann, G.; Greiner, G.; Valent, P. Cytokine Regulation of Microenvironmental Cells in Myeloproliferative Neoplasms. Mediat. Inflamm. 2015, 2015, 869242. [Google Scholar] [CrossRef] [Green Version]
- Zahr, A.A.; Salama, M.E.; Carreau, N.; Tremblay, D.; Verstovsek, S.; Mesa, R.; Hoffman, R.; Mascarenhas, J. Bone Marrow Fibrosis in Myelofibrosis: Pathogenesis, Prognosis and Targeted Strategies. Haematologica 2016, 101, 660–671. [Google Scholar] [CrossRef] [Green Version]
- Vannucchi, A.M.; Bianchi, L.; Paoletti, F.; Pancrazzi, A.; Torre, E.; Nishikawa, M.; Zingariello, M.; Di Baldassarre, A.; Rana, R.A.; Lorenzini, R.; et al. A Pathobiologic Pathway Linking Thrombopoietin, GATA-1, and TGF-Beta1 in the Development of Myelofibrosis. Blood 2005, 105, 3493–3501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teodorescu, P.; Pasca, S.; Jurj, A.; Gafencu, G.; Joelsson, J.-P.; Selicean, S.; Moldovan, C.; Munteanu, R.; Onaciu, A.; Tigu, A.-B.; et al. Transforming Growth Factor β-Mediated Micromechanics Modulates Disease Progression in Primary Myelofibrosis. J. Cell. Mol. Med. 2020, 24, 11100–11110. [Google Scholar] [CrossRef]
- Gleitz, H.F.E.; Dugourd, A.J.F.; Leimkühler, N.B.; Snoeren, I.A.M.; Fuchs, S.N.R.; Menzel, S.; Ziegler, S.; Kröger, N.; Triviai, I.; Büsche, G.; et al. Increased CXCL4 Expression in Hematopoietic Cells Links Inflammation and Progression of Bone Marrow Fibrosis in MPN. Blood 2020, 136, 2051–2064. [Google Scholar] [CrossRef]
- Ramos, T.L.; Sánchez-Abarca, L.I.; Rosón-Burgo, B.; Redondo, A.; Rico, A.; Preciado, S.; Ortega, R.; Rodríguez, C.; Muntión, S.; Hernández-Hernández, Á.; et al. Mesenchymal Stromal Cells (MSC) from JAK2+ Myeloproliferative Neoplasms Differ from Normal MSC and Contribute to the Maintenance of Neoplastic Hematopoiesis. PLoS ONE 2017, 12, e0182470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, R.K.; Mullally, A.; Dugourd, A.; Peisker, F.; Hoogenboezem, R.; Van Strien, P.M.H.; Bindels, E.M.; Heckl, D.; Büsche, G.; Fleck, D.; et al. Gli1+ Mesenchymal Stromal Cells Are a Key Driver of Bone Marrow Fibrosis and an Important Cellular Therapeutic Target. Cell Stem Cell 2017, 20, 785–800.e8. [Google Scholar] [CrossRef] [Green Version]
- Decker, M.; Martinez-Morentin, L.; Wang, G.; Lee, Y.; Liu, Q.; Leslie, J.; Ding, L. Leptin-Receptor-Expressing Bone Marrow Stromal Cells Are Myofibroblasts in Primary Myelofibrosis. Nat. Cell Biol. 2017, 19, 677–688. [Google Scholar] [CrossRef]
- Pilling, D.; Fan, T.; Huang, D.; Kaul, B.; Gomer, R.H. Identification of Markers That Distinguish Monocyte-Derived Fibrocytes from Monocytes, Macrophages, and Fibroblasts. PLoS ONE 2009, 4, e7475. [Google Scholar] [CrossRef]
- Manshouri, T.; Verstovsek, S.; Harris, D.M.; Veletic, I.; Zhang, X.; Post, S.M.; Bueso-Ramos, C.E.; Estrov, Z. Primary Myelofibrosis Marrow-Derived CD14+/CD34- Monocytes Induce Myelofibrosis-like Phenotype in Immunodeficient Mice and Give Rise to Megakaryocytes. PLoS ONE 2019, 14, e0222912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eyden, B. The Myofibroblast: A Study of Normal, Reactive and Neoplastic Tissues, with an Emphasis on Ultrastructure. J. Submicrosc. Cytol. Pathol. 2007, 7–166, 231–296. [Google Scholar]
- Eyden, B. The Myofibroblast: Phenotypic Characterization as a Prerequisite to Understanding Its Functions in Translational Medicine. J. Cell. Mol. Med. 2008, 12, 22–37. [Google Scholar] [CrossRef] [Green Version]
- Desterke, C.; Martinaud, C.; Ruzehaji, N.; Le Bousse-Kerdilès, M.-C. Inflammation as a Keystone of Bone Marrow Stroma Alterations in Primary Myelofibrosis. Mediat. Inflamm. 2015, 2015, 415024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arranz, L.; Sánchez-Aguilera, A.; Martín-Pérez, D.; Isern, J.; Langa, X.; Tzankov, A.; Lundberg, P.; Muntión, S.; Tzeng, Y.-S.; Lai, D.-M.; et al. Neuropathy of Haematopoietic Stem Cell Niche Is Essential for Myeloproliferative Neoplasms. Nature 2014, 512, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C.; Novetsky, A.; Chen, C.; Novetsky, A.D. Plasma Matrix Metalloproteinase and Tissue Inhibitor of Metalloproteinase in Patients with Agnogenic Myeloid Metaplasia or Idiopathic Primary Myelofibrosis. Br. J. Haematol. 2002, 119, 709–712. [Google Scholar] [CrossRef]
- Liu, G.M.; Zhang, L.J.; Fu, J.Z.; Liang, W.T.; Cheng, Z.Y.; Bai, P.; Bian, Y.S.; Wan, J.S. [Regulation of Ruxolitinib on matrix metalloproteinase in JAK2V617F positive myeloroliferative neoplasms cells]. Zhonghua Xue Ye Xue Za Zhi 2017, 38, 140–145. [Google Scholar] [CrossRef]
- Lucero, H.A.; Kagan, H.M. Lysyl Oxidase: An Oxidative Enzyme and Effector of Cell Function. Cell. Mol. Life Sci. 2006, 63, 2304–2316. [Google Scholar] [CrossRef]
- Eliades, A.; Papadantonakis, N.; Bhupatiraju, A.; Burridge, K.A.; Johnston-Cox, H.A.; Migliaccio, A.R.; Crispino, J.D.; Lucero, H.A.; Trackman, P.C.; Ravid, K. Control of Megakaryocyte Expansion and Bone Marrow Fibrosis by Lysyl Oxidase. J. Biol. Chem. 2011, 286, 27630–27638. [Google Scholar] [CrossRef] [Green Version]
- Abbonante, V.; Chitalia, V.; Rosti, V.; Leiva, O.; Matsuura, S.; Balduini, A.; Ravid, K. Upregulation of Lysyl Oxidase and Adhesion to Collagen of Human Megakaryocytes and Platelets in Primary Myelofibrosis. Blood 2017, 130, 829–831. [Google Scholar] [CrossRef] [Green Version]
- Tadmor, T.; Bejar, J.; Attias, D.; Mischenko, E.; Sabo, E.; Neufeld, G.; Vadasz, Z. The Expression of Lysyl-Oxidase Gene Family Members in Myeloproliferative Neoplasms. Am. J. Hematol. 2013, 88, 355–358. [Google Scholar] [CrossRef]
- Skov, V.; Larsen, T.S.; Thomassen, M.; Riley, C.H.; Jensen, M.K.; Bjerrum, O.W.; Kruse, T.A.; Hasselbalch, H.C. Molecular Profiling of Peripheral Blood Cells from Patients with Polycythemia Vera and Related Neoplasms: Identification of Deregulated Genes of Significance for Inflammation and Immune Surveillance. Leuk. Res. 2012, 36, 1387–1392. [Google Scholar] [CrossRef]
- Skov, V.; Thomassen, M.; Riley, C.H.; Jensen, M.K.; Bjerrum, O.W.; Kruse, T.A.; Hasselbalch, H.C.; Larsen, T.S. Gene Expression Profiling with Principal Component Analysis Depicts the Biological Continuum from Essential Thrombocythemia over Polycythemia Vera to Myelofibrosis. Exp. Hematol. 2012, 40, 771–780.e19. [Google Scholar] [CrossRef]
- Skov, V.; Riley, C.H.; Thomassen, M.; Larsen, T.S.; Jensen, M.K.; Bjerrum, O.W.; Kruse, T.A.; Hasselbalch, H.C. Whole Blood Transcriptional Profiling Reveals Significant Down-Regulation of Human Leukocyte Antigen Class I and II Genes in Essential Thrombocythemia, Polycythemia Vera and Myelofibrosis. Leuk. Lymphoma 2013, 54, 2269–2273. [Google Scholar] [CrossRef]
- Riley, C.H.; Hansen, M.; Brimnes, M.K.; Hasselbalch, H.C.; Bjerrum, O.W.; Straten, P.T.; Svane, I.M.; Jensen, M.K. Expansion of Circulating CD56 bright Natural Killer Cells in Patients with JAK2-Positive Chronic Myeloproliferative Neoplasms during Treatment with Interferon-α. Eur. J. Haematol. 2015, 94, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Romano, M.; Sollazzo, D.; Trabanelli, S.; Barone, M.; Polverelli, N.; Perricone, M.; Forte, D.; Luatti, S.; Cavo, M.; Vianelli, N.; et al. Mutations in JAK2 and Calreticulin Genes Are Associated with Specific Alterations of the Immune System in Myelofibrosis. OncoImmunology 2017, 6, e1345402. [Google Scholar] [CrossRef] [Green Version]
- Humblet-Baron, S.; Barber, J.S.; Roca, C.P.; Lenaerts, A.; Koni, P.A.; Liston, A. Murine Myeloproliferative Disorder as a Consequence of Impaired Collaboration between Dendritic Cells and CD4 T Cells. Blood 2019, 133, 319–330. [Google Scholar] [CrossRef]
- Riley, C.H.; Jensen, M.K.; Brimnes, M.K.; Hasselbalch, H.C.; Bjerrum, O.W.; Straten, P.T.; Svane, I.M. Increase in Circulating CD4+CD25+Foxp3+ T Cells in Patients with Philadelphia-Negative Chronic Myeloproliferative Neoplasms during Treatment with IFN-α. Blood 2011, 118, 2170–2173. [Google Scholar] [CrossRef] [Green Version]
- Keohane, C.; Kordasti, S.; Seidl, T.; Perez Abellan, P.; Thomas, N.S.B.; Harrison, C.N.; McLornan, D.P.; Mufti, G.J. JAK Inhibition Induces Silencing of T Helper Cytokine Secretion and a Profound Reduction in T Regulatory Cells. Br. J. Haematol. 2015, 171, 60–73. [Google Scholar] [CrossRef] [Green Version]
- Bizymi, N.; Bjelica, S.; Kittang, A.O.; Mojsilovic, S.; Velegraki, M.; Pontikoglou, C.; Roussel, M.; Ersvær, E.; Santibañez, J.F.; Lipoldová, M.; et al. Myeloid-Derived Suppressor Cells in Hematologic Diseases: Promising Biomarkers and Treatment Targets. HemaSphere 2019, 3, e168. [Google Scholar] [CrossRef]
- Wang, J.C.; Kundra, A.; Andrei, M.; Baptiste, S.; Chen, C.; Wong, C.; Sindhu, H. Myeloid-Derived Suppressor Cells in Patients with Myeloproliferative Neoplasm. Leuk. Res. 2016, 43, 39–43. [Google Scholar] [CrossRef]
- Rachidi, S.; Metelli, A.; Riesenberg, B.; Wu, B.X.; Nelson, M.H.; Wallace, C.; Paulos, C.M.; Rubinstein, M.P.; Garrett-Mayer, E.; Hennig, M.; et al. Platelets Subvert T Cell Immunity against Cancer via GARP-TGFβ Axis. Sci. Immunol. 2017, 2. [Google Scholar] [CrossRef] [Green Version]
- Prestipino, A.; Emhardt, A.J.; Aumann, K.; O’Sullivan, D.; Gorantla, S.P.; Duquesne, S.; Melchinger, W.; Braun, L.; Vuckovic, S.; Boerries, M.; et al. Oncogenic JAK2 V617F Causes PD-L1 Expression, Mediating Immune Escape in Myeloproliferative Neoplasms. Sci. Transl. Med. 2018, 10, eaam7729. [Google Scholar] [CrossRef] [Green Version]
- Kjaer, L.; Holmström, M.O.; Cordua, S.; Andersen, M.H.; Svane, I.M.; Thomassen, M.; Kruse, T.A.; Pallisgaard, N.; Skov, V.; Hasselbalch, H.C. Sorted Peripheral Blood Cells Identify CALR Mutations in B- and T-Lymphocytes. Leuk. Lymphoma 2018, 59, 973–977. [Google Scholar] [CrossRef]
- Liu, P.; Zhao, L.; Loos, F.; Marty, C.; Xie, W.; Martins, I.; Lachkar, S.; Qu, B.; Waeckel-Énée, E.; Plo, I.; et al. Immunosuppression by Mutated Calreticulin Released from Malignant Cells. Mol. Cell 2020, 77, 748–760.e9. [Google Scholar] [CrossRef] [PubMed]
- Robert, C. A Decade of Immune-Checkpoint Inhibitors in Cancer Therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef]
- Elsallab, M.; Levine, B.L.; Wayne, A.S.; Abou-El-Enein, M. CAR T-Cell Product Performance in Haematological Malignancies before and after Marketing Authorisation. Lancet Oncol. 2020, 21, e104–e116. [Google Scholar] [CrossRef]
- Goebeler, M.-E.; Bargou, R.C. T Cell-Engaging Therapies—BiTEs and Beyond. Nat. Rev. Clin. Oncol. 2020, 17, 418–434. [Google Scholar] [CrossRef]
- Biernacki, M.A.; Bleakley, M. Neoantigens in Hematologic Malignancies. Front. Immunol. 2020, 11, 121. [Google Scholar] [CrossRef]
- Chen, C.I.-U.; Maecker, H.T.; Lee, P.P. Development and Dynamics of Robust T-Cell Responses to CML under Imatinib Treatment. Blood 2008, 111, 5342–5349. [Google Scholar] [CrossRef]
- Riva, G.; Luppi, M.; Barozzi, P.; Quadrelli, C.; Basso, S.; Vallerini, D.; Zanetti, E.; Morselli, M.; Forghieri, F.; Maccaferri, M.; et al. Emergence of BCR-ABL-Specific Cytotoxic T Cells in the Bone Marrow of Patients with Ph+ Acute Lymphoblastic Leukemia during Long-Term Imatinib Mesylate Treatment. Blood 2010, 115, 1512–1518. [Google Scholar] [CrossRef] [PubMed]
- Riva, G.; Luppi, M.; Quadrelli, C.; Barozzi, P.; Basso, S.; Vallerini, D.; Zanetti, E.; Morselli, M.; Forghieri, F.; Maccaferri, M.; et al. BCR-ABL-Specific Cytotoxic T Cells in the Bone Marrow of Patients with Ph(+) Acute Lymphoblastic Leukemia during Second-Generation Tyrosine-Kinase Inhibitor Therapy. Blood Cancer J. 2011, 1, e30. [Google Scholar] [CrossRef] [PubMed]
- Greiner, J.; Ono, Y.; Hofmann, S.; Schmitt, A.; Mehring, E.; Götz, M.; Guillaume, P.; Döhner, K.; Mytilineos, J.; Döhner, H.; et al. Mutated Regions of Nucleophosmin 1 Elicit Both CD4(+) and CD8(+) T-Cell Responses in Patients with Acute Myeloid Leukemia. Blood 2012, 120, 1282–1289. [Google Scholar] [CrossRef] [PubMed]
- Forghieri, F.; Riva, G.; Lagreca, I.; Barozzi, P.; Vallerini, D.; Morselli, M.; Paolini, A.; Bresciani, P.; Colaci, E.; Maccaferri, M.; et al. Characterization and Dynamics of Specific T Cells against Nucleophosmin-1 (NPM1)-Mutated Peptides in Patients with NPM1-Mutated Acute Myeloid Leukemia. Oncotarget 2019, 10, 869–882. [Google Scholar] [CrossRef] [Green Version]
- Goodyear, O.C.; Pratt, G.; McLarnon, A.; Cook, M.; Piper, K.; Moss, P. Differential Pattern of CD4+ and CD8+ T-Cell Immunity to MAGE-A1/A2/A3 in Patients with Monoclonal Gammopathy of Undetermined Significance (MGUS) and Multiple Myeloma. Blood 2008, 112, 3362–3372. [Google Scholar] [CrossRef]
- Comoli, P.; Basso, S.; Riva, G.; Barozzi, P.; Guido, I.; Gurrado, A.; Quartuccio, G.; Rubert, L.; Lagreca, I.; Vallerini, D.; et al. BCR-ABL-Specific T-Cell Therapy in Ph+ ALL Patients on Tyrosine-Kinase Inhibitors. Blood 2017, 129, 582–586. [Google Scholar] [CrossRef] [Green Version]
- Lulla, P.; Heslop, H.E. Fall of the Mutants: T Cells Targeting BCR-ABL. Blood 2017, 129, 539–540. [Google Scholar] [CrossRef] [Green Version]
- Holmström, M.O.; Hjortsø, M.D.; Ahmad, S.M.; Met, Ö.; Martinenaite, E.; Riley, C.; Straten, P.; Svane, I.M.; Hasselbalch, H.C.; Andersen, M.H. The JAK2V617F Mutation Is a Target for Specific T Cells in the JAK2V617F-Positive Myeloproliferative Neoplasms. Leukemia 2017, 31, 495–498. [Google Scholar] [CrossRef] [PubMed]
- Holmström, M.O.; Riley, C.H.; Svane, I.M.; Hasselbalch, H.C.; Andersen, M.H. The CALR Exon 9 Mutations Are Shared Neoantigens in Patients with CALR Mutant Chronic Myeloproliferative Neoplasms. Leukemia 2016, 30, 2413–2416. [Google Scholar] [CrossRef]
- Holmström, M.O.; Martinenaite, E.; Ahmad, S.M.; Met, Ö.; Friese, C.; Kjær, L.; Riley, C.H.; Thor Straten, P.; Svane, I.M.; Hasselbalch, H.C.; et al. The Calreticulin (CALR) Exon 9 Mutations Are Promising Targets for Cancer Immune Therapy. Leukemia 2018, 32, 429–437. [Google Scholar] [CrossRef]
- Cimen Bozkus, C.; Roudko, V.; Finnigan, J.P.; Mascarenhas, J.; Hoffman, R.; Iancu-Rubin, C.; Bhardwaj, N. Immune Checkpoint Blockade Enhances Shared Neoantigen-Induced T-Cell Immunity Directed against Mutated Calreticulin in Myeloproliferative Neoplasms. Cancer Discov. 2019, 9, 1192–1207. [Google Scholar] [CrossRef] [PubMed]
- Holmström, M.O.; Ahmad, S.M.; Klausen, U.; Bendtsen, S.K.; Martinenaite, E.; Riley, C.H.; Svane, I.M.; Kjær, L.; Skov, V.; Ellervik, C.; et al. High Frequencies of Circulating Memory T Cells Specific for Calreticulin Exon 9 Mutations in Healthy Individuals. Blood Cancer J. 2019, 9, 8. [Google Scholar] [CrossRef] [PubMed]
- Holmström, M.O.; Riley, C.H.; Skov, V.; Svane, I.M.; Hasselbalch, H.C.; Andersen, M.H. Spontaneous T-Cell Responses against the Immune Check Point Programmed-Death-Ligand 1 (PD-L1) in Patients with Chronic Myeloproliferative Neoplasms Correlate with Disease Stage and Clinical Response. OncoImmunology 2018, 7, e1433521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jørgensen, M.A.; Holmström, M.O.; Martinenaite, E.; Riley, C.H.; Hasselbalch, H.C.; Andersen, M.H. Spontaneous T-Cell Responses against Arginase-1 in the Chronic Myeloproliferative Neoplasms Relative to Disease Stage and Type of Driver Mutation. OncoImmunology 2018, 7, e1468957. [Google Scholar] [CrossRef] [Green Version]
- Schischlik, F.; Jäger, R.; Rosebrock, F.; Hug, E.; Schuster, M.; Holly, R.; Fuchs, E.; Milosevic Feenstra, J.D.; Bogner, E.; Gisslinger, B.; et al. Mutational Landscape of the Transcriptome Offers Putative Targets for Immunotherapy of Myeloproliferative Neoplasms. Blood 2019, 134, 199–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLornan, D.P.; Yakoub-Agha, I.; Robin, M.; Chalandon, Y.; Harrison, C.N.; Kroger, N. State-of-the-Art Review: Allogeneic Stem Cell Transplantation for Myelofibrosis in 2019. Haematologica 2019, 104, 659–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuykendall, A.T.; Talati, C.; Al Ali, N.; Sweet, K.; Padron, E.; Sallman, D.A.; Lancet, J.E.; List, A.F.; Zuckerman, K.S.; Komrokji, R.S. The Treatment Landscape of Myelofibrosis before and After Ruxolitinib Approval. Clin. Lymphoma Myeloma Leuk. 2017, 17, e45–e53. [Google Scholar] [CrossRef] [PubMed]
- Cervantes, F.; Pereira, A. Does Ruxolitinib Prolong the Survival of Patients with Myelofibrosis? Blood 2017, 129, 832–837. [Google Scholar] [CrossRef]
- Bose, P.; Verstovsek, S. JAK Inhibition for the Treatment of Myelofibrosis: Limitations and Future Perspectives. HemaSphere 2020, 4, e424. [Google Scholar] [CrossRef]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.W.N.; Miller, C.B.; Silver, R.T.; Talpaz, M.; et al. Long-Term Treatment with Ruxolitinib for Patients with Myelofibrosis: 5-Year Update from the Randomized, Double-Blind, Placebo-Controlled, Phase 3 COMFORT-I Trial. J. Hematol. Oncol. J. Hematol. Oncol. 2017, 10, 55. [Google Scholar] [CrossRef] [Green Version]
- Palandri, F.; Breccia, M.; Bonifacio, M.; Polverelli, N.; Elli, E.M.; Benevolo, G.; Tiribelli, M.; Abruzzese, E.; Iurlo, A.; Heidel, F.H.; et al. Life after Ruxolitinib: Reasons for Discontinuation, Impact of Disease Phase, and Outcomes in 218 Patients with Myelofibrosis. Cancer 2020, 126, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.A.; Odenike, O. The Next Generation of JAK Inhibitors: An Update on Fedratinib, Momelotonib, and Pacritinib. Curr. Hematol. Malig. Rep. 2020, 15, 409–418. [Google Scholar] [CrossRef]
- Harrison, C.N.; Schaap, N.; Mesa, R.A. Management of Myelofibrosis after Ruxolitinib Failure. Ann. Hematol. 2020, 99, 1177–1191. [Google Scholar] [CrossRef] [Green Version]
- Newberry, K.J.; Patel, K.; Masarova, L.; Luthra, R.; Manshouri, T.; Jabbour, E.; Bose, P.; Daver, N.; Cortes, J.; Kantarjian, H.; et al. Clonal Evolution and Outcomes in Myelofibrosis after Ruxolitinib Discontinuation. Blood 2017, 130, 1125–1131. [Google Scholar] [CrossRef] [Green Version]
- Ciurea, S.O.; Merchant, D.; Mahmud, N.; Ishii, T.; Zhao, Y.; Hu, W.; Bruno, E.; Barosi, G.; Xu, M.; Hoffman, R. Pivotal Contributions of Megakaryocytes to the Biology of Idiopathic Myelofibrosis. Blood 2007, 110, 986–993. [Google Scholar] [CrossRef]
- Malara, A.; Abbonante, V.; Zingariello, M.; Migliaccio, A.; Balduini, A. Megakaryocyte Contribution to Bone Marrow Fibrosis: Many Arrows in the Quiver. Mediterr. J. Hematol. Infect. Dis. 2018, 10, e2018068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Psaila, B.; Wang, G.; Rodriguez-Meira, A.; Li, R.; Heuston, E.F.; Murphy, L.; Yee, D.; Hitchcock, I.S.; Sousos, N.; O’Sullivan, J.; et al. Single-Cell Analyses Reveal Megakaryocyte-Biased Hematopoiesis in Myelofibrosis and Identify Mutant Clone-Specific Targets. Mol. Cell 2020, 78, 477–492.e8. [Google Scholar] [CrossRef]
- Gisslinger, H.; Gotic, M.; Holowiecki, J.; Penka, M.; Thiele, J.; Kvasnicka, H.-M.; Kralovics, R.; Petrides, P.E.; ANAHYDRET Study Group. Anagrelide Compared with Hydroxyurea in WHO-Classified Essential Thrombocythemia: The ANAHYDRET Study, a Randomized Controlled Trial. Blood 2013, 121, 1720–1728. [Google Scholar] [CrossRef] [Green Version]
- Espasandin, Y.R.; Glembotsky, A.C.; Grodzielski, M.; Lev, P.R.; Goette, N.P.; Molinas, F.C.; Marta, R.F.; Heller, P.G. Anagrelide Platelet-Lowering Effect Is Due to Inhibition of Both Megakaryocyte Maturation and Proplatelet Formation: Insight into Potential Mechanisms. J. Thromb. Haemost. 2015, 13, 631–642. [Google Scholar] [CrossRef]
- Magnaghi-Jaulin, L.; Eot-Houllier, G.; Gallaud, E.; Giet, R. Aurora A Protein Kinase: To the Centrosome and Beyond. Biomolecules 2019, 9, 28. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Nagata, Y.; Yu, G.; Nguyen, H.G.; Jones, M.R.; Toselli, P.; Jackson, C.W.; Tatsuka, M.; Todokoro, K.; Ravid, K. Aberrant Quantity and Localization of Aurora-B/AIM-1 and Survivin during Megakaryocyte Polyploidization and the Consequences of Aurora-B/AIM-1-Deregulated Expression. Blood 2004, 103, 3717–3726. [Google Scholar] [CrossRef]
- Wen, Q.; Goldenson, B.; Silver, S.J.; Schenone, M.; Dancik, V.; Huang, Z.; Wang, L.-Z.; Lewis, T.A.; An, W.F.; Li, X.; et al. Identification of Regulators of Polyploidization Presents Therapeutic Targets for Treatment of AMKL. Cell 2012, 150, 575–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gangat, N.; Marinaccio, C.; Swords, R.; Watts, J.M.; Gurbuxani, S.; Rademaker, A.; Fought, A.J.; Frankfurt, O.; Altman, J.K.; Wen, Q.J.; et al. Aurora Kinase A Inhibition Provides Clinical Benefit, Normalizes Megakaryocytes, and Reduces Bone Marrow Fibrosis in Patients with Myelofibrosis: A Phase I Trial. Clin. Cancer Res. 2019, 25, 4898–4906. [Google Scholar] [CrossRef] [Green Version]
- Sprüssel, A.; Schulte, J.H.; Weber, S.; Necke, M.; Händschke, K.; Thor, T.; Pajtler, K.W.; Schramm, A.; König, K.; Diehl, L.; et al. Lysine-Specific Demethylase 1 Restricts Hematopoietic Progenitor Proliferation and Is Essential for Terminal Differentiation. Leukemia 2012, 26, 2039–2051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jutzi, J.S.; Kleppe, M.; Dias, J.; Staehle, H.F.; Shank, K.; Teruya-Feldstein, J.; Gambheer, S.M.M.; Dierks, C.; Rienhoff, H.Y.; Levine, R.L.; et al. LSD1 Inhibition Prolongs Survival in Mouse Models of MPN by Selectively Targeting the Disease Clone. HemaSphere 2018, 2, e54. [Google Scholar] [CrossRef] [PubMed]
- Pettit, K.; Gerds, A.T.; Yacoub, A.; Watts, J.M.; Tartaczuch, M.; Bradley, T.J.; Shortt, J.; Stevenson, W.S.; Curtin, N.J.; Rossetti, J.M.; et al. A Phase 2a Study of the LSD1 Inhibitor Img-7289 (Bomedemstat) for the Treatment of Myelofibrosis. Blood 2019, 134, 556. [Google Scholar] [CrossRef]
- Verstovsek, S.; Talpaz, M.; Wadleigh, M.; Palmer, J.; Isidori, A.; te Boekhorst, P.; Savona, M.; Gotlib, J.; Hasserjian, R.; Pozdnyakova, O.; et al. S828 A Randomized, Double Blind Phase 2 Study of 3 Different Doses of PRM-151 in Patients with Myelofibrosis who Were Previously Treated with or Ineligible for Ruxolitinib. HemaSphere 2019, 3, 367. [Google Scholar] [CrossRef]
- Rossi, C.; Zini, R.; Rontauroli, S.; Ruberti, S.; Prudente, Z.; Barbieri, G.; Bianchi, E.; Salati, S.; Genovese, E.; Bartalucci, N.; et al. Role of TGF-Β1/MiR-382-5p/SOD2 Axis in the Induction of Oxidative Stress in CD34+ Cells from Primary Myelofibrosis. Mol. Oncol. 2018, 12, 2102–2123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iancu-Rubin, C.; Mosoyan, G.; Wang, J.; Kraus, T.; Sung, V.; Hoffman, R. Stromal Cell-Mediated Inhibition of Erythropoiesis Can Be Attenuated by Sotatercept (ACE-011), an Activin Receptor Type II Ligand Trap. Exp. Hematol. 2013, 41, 155–166.e17. [Google Scholar] [CrossRef]
- Carrancio, S.; Markovics, J.; Wong, P.; Leisten, J.; Castiglioni, P.; Groza, M.C.; Raymon, H.K.; Heise, C.; Daniel, T.; Chopra, R.; et al. An Activin Receptor IIA Ligand Trap Promotes Erythropoiesis Resulting in a Rapid Induction of Red Blood Cells and Haemoglobin. Br. J. Haematol. 2014, 165, 870–882. [Google Scholar] [CrossRef] [Green Version]
- Naymagon, L.; Mascarenhas, J. Myelofibrosis-Related Anemia: Current and Emerging Therapeutic Strategies. HemaSphere 2017, 1, e1. [Google Scholar] [CrossRef]
- Fenaux, P.; Kiladjian, J.J.; Platzbecker, U. Luspatercept for the Treatment of Anemia in Myelodysplastic Syndromes and Primary Myelofibrosis. Blood 2019, 133, 790–794. [Google Scholar] [CrossRef] [Green Version]
- Fenaux, P.; Platzbecker, U.; Mufti, G.J.; Garcia-Manero, G.; Buckstein, R.; Santini, V.; Díez-Campelo, M.; Finelli, C.; Cazzola, M.; Ilhan, O.; et al. Luspatercept in Patients with Lower-Risk Myelodysplastic Syndromes. N. Engl. J. Med. 2020, 382, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Bose, P.; Daver, N.; Jabbour, E.J.; Pike, A.; Newberry, K.J.; Zhou, L.; Pierce, S.; Wang, X.; Kantarjian, H.M.; Verstovsek, S. Phase-2 Study of Sotatercept (ACE-011) in Myeloproliferative Neoplasm-Associated Myelofibrosis and Anemia. Blood 2016, 128, 478. [Google Scholar] [CrossRef]
- Gerds, A.T.; Vannucchi, A.M.; Passamonti, F.; Kremyanskaya, M.; Gotlib, J.R.; Palmer, J.M.; McCaul, K.; Ribrag, V.; Mead, A.J.; Harrison, C.N.; et al. A Phase 2 Study of Luspatercept in Patients with Myelofibrosis-Associated Anemia. Blood 2019, 134, 557. [Google Scholar] [CrossRef]
- Varricchio, L.; Mascarenhas, J.; Migliaccio, A.R.; O’Connor-McCourt, M.; Tremblay, G.; Denis, J.-F.; Iancu-Rubin, C.; Hoffman, R. AVID200, a Potent Trap for TGF-β Ligands Inhibits TGF-Β1 Signaling in Human Myelofibrosis. Blood 2018, 132, 1791. [Google Scholar] [CrossRef]
- Mascarenhas, J.; Li, T.; Sandy, L.; Newsom, C.; Petersen, B.; Godbold, J.; Hoffman, R. Anti-Transforming Growth Factor-β Therapy in Patients with Myelofibrosis. Leuk. Lymphoma 2014, 55, 450–452. [Google Scholar] [CrossRef] [PubMed]
- Yue, L.; Bartenstein, M.; Zhao, W.; Ho, W.T.; Han, Y.; Murdun, C.; Mailloux, A.W.; Zhang, L.; Wang, X.; Budhathoki, A.; et al. Efficacy of ALK5 Inhibition in Myelofibrosis. JCI Insight 2017, 2, e90932. [Google Scholar] [CrossRef] [Green Version]
- Herlihy, N.; Harrison, C.N.; McLornan, D.P. Exploitation of the Neural-Hematopoietic Stem Cell Niche Axis to Treat Myeloproliferative Neoplasms. Haematologica 2019, 104, 639–641. [Google Scholar] [CrossRef]
- Drexler, B.; Passweg, J.R.; Tzankov, A.; Bigler, M.; Theocharides, A.P.; Cantoni, N.; Keller, P.; Stussi, G.; Ruefer, A.; Benz, R.; et al. The Sympathomimetic Agonist Mirabegron Did Not Lower JAK2-V617F Allele Burden, but Restored Nestin-Positive Cells and Reduced Reticulin Fibrosis in Patients with Myeloproliferative Neoplasms: Results of Phase II Study SAKK 33/14. Haematologica 2019, 104, 710–716. [Google Scholar] [CrossRef] [Green Version]
- Leiva, O.; Ng, S.K.; Chitalia, S.; Balduini, A.; Matsuura, S.; Ravid, K. The Role of the Extracellular Matrix in Primary Myelofibrosis. Blood Cancer J. 2017, 7, e525. [Google Scholar] [CrossRef] [PubMed]
- Schilter, H.; Findlay, A.D.; Perryman, L.; Yow, T.T.; Moses, J.; Zahoor, A.; Turner, C.I.; Deodhar, M.; Foot, J.S.; Zhou, W.; et al. The Lysyl Oxidase like 2/3 Enzymatic Inhibitor, PXS-5153A, Reduces Crosslinks and Ameliorates Fibrosis. J. Cell. Mol. Med. 2019, 23, 1759–1770. [Google Scholar] [CrossRef] [Green Version]
- Leiva, O.; Ng, S.K.; Matsuura, S.; Chitalia, V.; Lucero, H.; Findlay, A.; Turner, C.; Jarolimek, W.; Ravid, K. Novel Lysyl Oxidase Inhibitors Attenuate Hallmarks of Primary Myelofibrosis in Mice. Int. J. Hematol. 2019, 110, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Verstovsek, S.; Savona, M.R.; Mesa, R.A.; Dong, H.; Maltzman, J.D.; Sharma, S.; Silverman, J.; Oh, S.T.; Gotlib, J. A Phase 2 Study of Simtuzumab in Patients with Primary, Post-Polycythaemia Vera or Post-Essential Thrombocythaemia Myelofibrosis. Br. J. Haematol. 2017, 176, 939–949. [Google Scholar] [CrossRef] [Green Version]
- Guglielmelli, P.; Rotunno, G.; Pacilli, A.; Rumi, E.; Rosti, V.; Delaini, F.; Maffioli, M.; Fanelli, T.; Pancrazzi, A.; Pieri, L.; et al. Prognostic Impact of Bone Marrow Fibrosis in Primary Myelofibrosis. A Study of the AGIMM Group on 490 Patients. Am. J. Hematol. 2016, 91, 918–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagley, C.J.; Woodcock, J.M.; Stomski, F.C.; Lopez, A.F. The Structural and Functional Basis of Cytokine Receptor Activation: Lessons from the Common Beta Subunit of the Granulocyte-Macrophage Colony-Stimulating Factor, Interleukin-3 (IL-3), and IL-5 Receptors. Blood 1997, 89, 1471–1482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broughton, S.E.; Dhagat, U.; Hercus, T.R.; Nero, T.L.; Grimbaldeston, M.A.; Bonder, C.S.; Lopez, A.F.; Parker, M.W. The GM-CSF/IL-3/IL-5 Cytokine Receptor Family: From Ligand Recognition to Initiation of Signaling. Immunol. Rev. 2012, 250, 277–302. [Google Scholar] [CrossRef] [PubMed]
- El Achi, H.; Dupont, E.; Paul, S.; Khoury, J.D. CD123 as a Biomarker in Hematolymphoid Malignancies: Principles of Detection and Targeted Therapies. Cancers 2020, 12, 87. [Google Scholar] [CrossRef]
- Testa, U.; Pelosi, E.; Frankel, A. CD 123 Is a Membrane Biomarker and a Therapeutic Target in Hematologic Malignancies. Biomark. Res. 2014, 2, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardanani, A.; Lasho, T.; Chen, D.; Kimlinger, T.K.; Finke, C.; Zblewski, D.; Patnaik, M.M.; Reichard, K.K.; Rowinsky, E.; Hanson, C.A.; et al. Aberrant Expression of CD123 (Interleukin-3 Receptor-α) on Neoplastic Mast Cells. Leukemia 2015, 29, 1605–1608. [Google Scholar] [CrossRef]
- Jordan, C.T.; Upchurch, D.; Szilvassy, S.J.; Guzman, M.L.; Howard, D.S.; Pettigrew, A.L.; Meyerrose, T.; Rossi, R.; Grimes, B.; Rizzieri, D.A.; et al. The Interleukin-3 Receptor Alpha Chain Is a Unique Marker for Human Acute Myelogenous Leukemia Stem Cells. Leukemia 2000, 14, 1777–1784. [Google Scholar] [CrossRef] [Green Version]
- Testa, U.; Riccioni, R.; Militi, S.; Coccia, E.; Stellacci, E.; Samoggia, P.; Latagliata, R.; Mariani, G.; Rossini, A.; Battistini, A.; et al. Elevated Expression of IL-3Rα in Acute Myelogenous Leukemia Is Associated with Enhanced Blast Proliferation, Increased Cellularity, and Poor Prognosis. Blood 2002, 100, 2980–2988. [Google Scholar] [CrossRef] [PubMed]
- Lasho, T.; Finke, C.; Kimlinger, T.K.; Zblewski, D.; Chen, D.; Patnaik, M.M.; Hanson, C.A.; Brooks, C.; Tefferi, A.; Pardanani, A. Expression of CD123 (IL-3R-Alpha), a Therapeutic Target of SL-401, on Myeloproliferative Neoplasms. Blood 2014, 124, 5577. [Google Scholar] [CrossRef]
- Testa, U.; Pelosi, E.; Castelli, G. CD123 as a Therapeutic Target in the Treatment of Hematological Malignancies. Cancers 2019, 11, 1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pemmaraju, N.; Lane, A.A.; Sweet, K.L.; Stein, A.S.; Vasu, S.; Blum, W.; Rizzieri, D.A.; Wang, E.S.; Duvic, M.; Sloan, J.M.; et al. Tagraxofusp in Blastic Plasmacytoid Dendritic-Cell Neoplasm. N. Engl. J. Med. 2019, 380, 1628–1637. [Google Scholar] [CrossRef] [PubMed]
- Mani, R.; Goswami, S.; Gopalakrishnan, B.; Ramaswamy, R.; Wasmuth, R.; Tran, M.; Mo, X.; Gordon, A.; Bucci, D.; Lucas, D.M.; et al. The Interleukin-3 Receptor CD123 Targeted SL-401 Mediates Potent Cytotoxic Activity against CD34+CD123+ Cells from Acute Myeloid Leukemia/Myelodysplastic Syndrome Patients and Healthy Donors. Haematologica 2018, 103, 1288–1297. [Google Scholar] [CrossRef] [Green Version]
- Lane, A.A. Targeting CD123 in AML. Clin. Lymphoma Myeloma Leuk. 2020, 20 (Suppl. 1), S67–S68. [Google Scholar] [CrossRef]
- Pemmaraju, N.; Gupta, V.; Ali, H.; Yacoub, A.; Wang, E.S.; Lee, S.; Schiller, G.J.; Sardone, M.; Wysowskyj, H.; Chen, J.; et al. Results from a Phase 1/2 Clinical Trial of Tagraxofusp (SL-401) in Patients with Intermediate, or High Risk, Relapsed/Refractory Myelofibrosis. Blood 2019, 134, 558. [Google Scholar] [CrossRef]
- Igarashi, Y.; Sasada, T. Cancer Vaccines: Toward the Next Breakthrough in Cancer Immunotherapy. J. Immunol. Res. 2020, 2020, 5825401. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Drug | Target (Mechanism) | Condition(s) | Trial (Phase) | Reference | Results/Comments |
|---|---|---|---|---|---|
| PRM-151 | Recombinant human pentraxin-2 | MF | NCT01981850 (Phase II) | Verstovsek et al., 2019 [157] | ↓fibrosis, ↓transfusions, only modest SI |
| Fresolimumab (GC1008) | TGF-β (MoAb) | MF | NCT01291784 (Phase I) | Mascarenhas et al., 2014 [167] | Only 3 patients enrolled; early termination |
| Sotatercept (ACE-011) | TGF-β (ligand trap) | MF with anemia | NCT01712308 (Phase II) | Bose et al., 2016 [164] | ±ruxolitinibEndpoint: ↑Hb ORR (monotherapy) = 35% |
| Luspatercept (ACE-536) | TGF-β (ligand trap) | MF with anemia | NCT03194542 (Phase II) | Gerds et al., 2019 [165] | ±ruxolitinibEndpoint: ↑Hb ORR (monotherapy) = 20% |
| AVID200 | TGF-β1/β3 (ligand trap) | MF | NCT03895112 (Phase I/Ib) | Still recruiting; no results published yet | |
| Galunisertib (LY2157299) | ALK5 (kinase inhibitor) | MF | Preclinical | Yue et al., 2017 [168] | Murine models only: ↓fibrosis |
| Alisertib(MLN8237) | AURKA (kinase inhibitor) | MF (or AMKL) | NCT02530619 (Phase I) | Gangat et al., 2019 [153] | SVR = 29%, TI = 8%, SI = 23%, ↓fibrosis |
| Bomedemstat (IMG-7289) | LSD1 (small molecule inhibitor) | MF | NCT03136185 (Phase II) | Pettit et al., 2019 [156] | Still recruiting; interim results: SI, slight SVR in a subset of patients |
| Mirabegron | β-3 adrenergic agonist | JAK2V617F+ MPNs | NCT02311569 (Phase II) | Drexler et al., 2019 [170] | ↑nestin + MSCs, mild ↓fibrosis, ↓MK clusters, ↔ JAK2V617F allele burden |
| Simtuzumab (GS-6624) | LOXL2 (MoAb) | MF | NCT01369498 (Phase II) | Verstovsek et al., 2017 [174] | ±ruxolitinib ↓Fibrosis in 36.7%; limited overall efficacy |
| GANT61 | Gli1/Hedgehog (small molecule inhibitor) | MF | Preclinical | Schneider et al., 2017 [88] | Murine and human in vitro models: ↓fibrosis, ↓myofibroblastic phenotype |
| Drug | Target (Mechanism) | Condition(s) | Trial (Phase) | Reference | Results/Comments |
|---|---|---|---|---|---|
| Tagraxofusp (SL-401) | CD123 (MoAb) | MF (or CMML) | NCT02268253 (Phase I/II) | Pemmaraju et al., 2019 [188] | Still recruiting; SVR = 53%, SI = 45% |
| Nivolumab | PD-1 (MoAb) | MF | NCT02421354 (Phase II) | - | Terminated early for lack of efficacy |
| Pembrolizumab | PD-1 (MoAb) | PV, MF | NCT03065400 (Phase II) | Cimen Bozkus et al., 2019 [131] | Ongoing; ↑reactivity to CALR mutant epitopes in vivo and in vitro |
| Durvalumab | PD-L1 (MoAb) | MF | NCT02871323 (Phase I) | Withdrawn before patients’ enrolment | |
| Ipilimumab * | CTLA-4 (MoAb) | MPNs (among other conditions) | NCT01822509 (Phase I/Ib) | Ongoing; aims: assessment of AEs and best dose of ipilimumab or nivolumab | |
| CALRLong36 peptide (exon 9 mut) vaccine | Mutated CALR (vaccine) | CALR+ PMF and ET | NCT03566446 (Phase I) | Aims: assessment of AEs and T cell cytokine release against the target antigen | |
| PD-L1Long (19–27) ArgLong2 (169–206) vaccine | PD-L1 and ARG1 (vaccine) | ET, PV | NCT04051307 (Phase I/II) | Expected effects: ↑specific T cell responses, ↑killing of mutant cells, ↓ARG1, ↓PD-L1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nasillo, V.; Riva, G.; Paolini, A.; Forghieri, F.; Roncati, L.; Lusenti, B.; Maccaferri, M.; Messerotti, A.; Pioli, V.; Gilioli, A.; et al. Inflammatory Microenvironment and Specific T Cells in Myeloproliferative Neoplasms: Immunopathogenesis and Novel Immunotherapies. Int. J. Mol. Sci. 2021, 22, 1906. https://doi.org/10.3390/ijms22041906
Nasillo V, Riva G, Paolini A, Forghieri F, Roncati L, Lusenti B, Maccaferri M, Messerotti A, Pioli V, Gilioli A, et al. Inflammatory Microenvironment and Specific T Cells in Myeloproliferative Neoplasms: Immunopathogenesis and Novel Immunotherapies. International Journal of Molecular Sciences. 2021; 22(4):1906. https://doi.org/10.3390/ijms22041906
Chicago/Turabian StyleNasillo, Vincenzo, Giovanni Riva, Ambra Paolini, Fabio Forghieri, Luca Roncati, Beatrice Lusenti, Monica Maccaferri, Andrea Messerotti, Valeria Pioli, Andrea Gilioli, and et al. 2021. "Inflammatory Microenvironment and Specific T Cells in Myeloproliferative Neoplasms: Immunopathogenesis and Novel Immunotherapies" International Journal of Molecular Sciences 22, no. 4: 1906. https://doi.org/10.3390/ijms22041906