
Where and Why Modeling Amyotrophic Lateral Sclerosis



1
Preclinical Neuroscience, IRCCS Santa Lucia Foundation, 00143 Rome, Italy
2
Institute for Systems Analysis and Computer Science “A. Ruberti”, National Research Council (IASI—CNR), 00185 Rome, Italy
*
Author to whom correspondence should be addressed.
†
Equally contributing authors.
Int. J. Mol. Sci. 2021, 22(8), 3977; https://doi.org/10.3390/ijms22083977
Submission received: 18 March 2021
/
Revised: 8 April 2021
/
Accepted: 9 April 2021
/
Published: 12 April 2021
(This article belongs to the Special Issue Modeling Neurological Disorders in Experimental Animals: New Insights and Emerging Roles)
Abstract
:Over the years, researchers have leveraged a host of different in vivo models in order to dissect amyotrophic lateral sclerosis (ALS), a neurodegenerative/neuroinflammatory disease that is heterogeneous in its clinical presentation and is multigenic, multifactorial and non-cell autonomous. These models include both vertebrates and invertebrates such as yeast, worms, flies, zebrafish, mice, rats, guinea pigs, dogs and, more recently, non-human primates. Despite their obvious differences and peculiarities, only the concurrent and comparative analysis of these various systems will allow the untangling of the causes and mechanisms of ALS for finally obtaining new efficacious therapeutics. However, harnessing these powerful organisms poses numerous challenges. In this context, we present here an updated and comprehensive review of how eukaryotic unicellular and multicellular organisms that reproduce a few of the main clinical features of the disease have helped in ALS research to dissect the pathological pathways of the disease insurgence and progression. We describe common features as well as discrepancies among these models, highlighting new insights and emerging roles for experimental organisms in ALS.
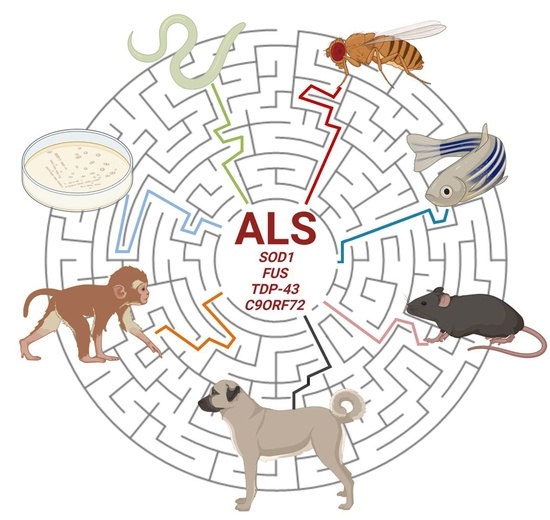
1. Introduction
Amyotrophic lateral sclerosis (ALS) was identified more than two hundred years ago and incredible scientific progress has been made during the past decades although there is still a shortage of information and many hypotheses have still to be confirmed about its complex pathogenesis [1,2]. Very recently, a high-throughput-omics analysis has contributed to the identification of candidate causal and risk ALS genes, molecular alterations and derangements of pathways along with their impact on the outcome of the disease [3,4]. Most research has been carried out and achievements also obtained thanks to the exploitation of several different in vitro and in vivo experimental models. These provide essential clues for: (a) understanding the genesis of motor neuron death as a hallmark of the disease; (b) distinguishing the overall contribution of genes, protein targets and mechanisms involved in the insurgence and progression of ALS; (c) validating new biomarkers for diagnostic purposes; (d) discovering and repurposing new treatments comprising drugs, antibodies, stem cells and gene therapies [5,6,7,8,9]. However, the use of a single model cannot per se discern and redirect the pathological process of the disease. Only the integrated and comparative scrutiny of different ALS organisms will be conceivably able to provide the right answers to the several still open questions about ALS. The present work will provide an updated review of the experimental ALS models generated in both invertebrate and vertebrate organisms that have helped to promote fruitful research in the ALS field.
2. ALS: An Old But Unbeaten Disease
2.1. ALS Genes
A major part of ALS cases is classified as “sporadic” (sALS) while up to 10% are defined as “familial” (fALS) in origin. Variations in a number of genes and loci are associated with ALS susceptibility. More than 50 genes have been associated with fALS [10] and in the present review we will focus on the most commonly mutated fALS genes (SOD1, FUS, TDP-43 and C9ORF72), which are, overall, involved in more than half of fALS cases and for which many models have been developed so far.
SOD1 was the first gene to be recognized as ALS-linked [11,12]; it encodes for a ubiquitous Cu/Zn superoxide dismutase that catalyzes the dismutation of superoxide radicals to hydrogen peroxide and dioxygen. Over the years, more than 150 mutations of the SOD1 gene have been related to ALS [13] but the mechanism by which mutant SOD1 triggers motor neuron damage remains still unclear. To date, overwhelming evidence supports the combination of both loss- and gain-of-toxic-functions linked to SOD1 mutations [14,15,16] and leading to the perturbation of mitochondrial dynamics, protein folding, axonal transport and cellular metabolism [17,18]. While mutant SOD1 is recognized as a pathological cause of about 20–25% of fALS cases, the wild type or misfolded SOD1 is implicated also in a significant fraction of sALS cases [19]. Mutant SOD1 protein expression in non-neuronal cells is also involved in ALS pathogenesis [20,21].
Another ALS-related gene is FUS, coding for the multirole protein Fused-in-Sarcoma/Translocated in Liposarcoma (FUS/TLS) present in RNA-containing stress granules [22,23,24] and involved in RNA metabolism [25,26,27] and gene expression [28]. The protein belongs to the FET (FUS-EWS-TAF15) RNA-binding protein family and the gene was recently described as bicistronic, encoding also for an additional peptide (altFUS) [29]. Since 2009, several mutations in the FUS gene have been linked to ALS (with a frequency of 2.8% for fALS cases and 0.3% for sALS cases) [30,31,32,33,34,35], many of which affect FUS nuclear localization, impairing its role as a regulator of transcription and RNA maturation and causing toxic FUS aggregates in the cytoplasm [36,37,38,39,40,41]. Mutant FUS also confers loss- and gain-of-functions in the nucleus by inducing the abnormal formation of dysfunctional subnuclear bodies known as paraspeckles of which FUS is an essential component [42].
Found as the primary component of neuronal inclusions in ALS and fronto-temporal dementia (FTD) patients [43,44], TAR-binding protein 43 (TDP-43) is involved in several functions relating to RNA metabolism such as mRNA stability [45], miRNA processing [25] and splicing regulation [46]. More than 50 different point mutations in the TARDBP gene (encoding for TDP-43) have been identified as ALS-causing (frequency: 4.2% in fALS; 0.8% in sALS) [34,35], altering its shuttle role from the nucleus to the cytoplasm and therefore modulating its RNA-related functions and determining the formation of aggregates [47]. In spinal motor neurons, TDP-43 binds to low molecular weight neurofilament mRNA, thus suggesting roles also in regulating axonal stability or damage. Moreover, TDP-43 participates in the non-homologous end joining enzymatic pathway and repair of double-strand DNA breaks in stem cell-derived motor neurons while its absence is correlated with increased DNA breaks [48]. Finally, a hyperphosphorylated, ubiquitinated and cleaved form of TDP-43 is considered to be a disease-relevant protein in ALS [49].
The Chromosome 9 Open Reading Frame 72 (C9ORF72) gene contains 12 exons and encodes for a little characterized protein that has a pivotal role in autophagy modulation, recently described as part of a guanine nucleotide exchange factor complex [50,51]. In a healthy condition, the hexanucleotide GGGGCC (G4C2) within the first intron may be repeated from 2 to 23 times but its aberrant expansion reaching hundreds or thousands of repeats has been found in ALS and FTD patients and is considered the most common genetic cause of fALS. It is also detected in occasional patients with sALS with a frequency of 39% and 7%, respectively [52,53,54,55,56]. Although still debated, mounting evidence suggests that C9ORF72-ALS pathogenesis is the result of a cascade of multiple cellular mechanisms that include: (i) RNA toxicity depending on G4C2 hexanucleotide repeat expansions (HRE) [57,58]; (ii) aggregation of toxic dipeptide repeat proteins (DPR) produced from the HRE region through Repeat-Associated Non-ATG (RAN) translation [59,60,61]; (iii) reduced levels of C9ORF72 protein causing disease by loss-of-function mechanisms [62]. Although not mutually exclusive, all of these mechanisms may account for the disease-causing effects of the hexanucleotide C9ORF72 expansions.
2.2. ALS Diagnostic Criteria
ALS is defined as a specific subtype of progressively degenerative motor neuron diseases that comprise primary lateral sclerosis, progressive muscular atrophy and progressive bulbar palsy and that are more generally classified as anterior horn cell diseases. These also include spinal muscular atrophy types I, II and III in children, type IV in adults and Kennedy’s disease, one of the most frequent disorders to be misdiagnosed as ALS [63,64]. However, ALS is not a disease that only harms motor neurons and several patients are currently reported to display extra motor deficits such as cognitive-behavioral disturbances, sensory nerve conduction failures and extrapyramidal alterations [65], thus questioning the classification of ALS as a mere motor neuron disease subtype.
For several years, the only accepted criterion for diagnosing ALS was the electrodiagnostic examination, the chief laboratory methodology for testing the neuromuscular system, originally formulated by Lambert [66]. This diagnostic approach was then improved, included and revised in the newest ALS diagnostic tool for correct ALS diagnosis, i.e., the revised El Escorial Criteria [67,68].
Based on these criteria, ALS abnormalities must be present in multiple muscles with peripheral innervation from different nerve roots and in multiple limbs. In the revised El Escorial Criteria, the electrodiagnostic analysis is corroborated by accurate anamnesis and physical examination also via neuroimaging and laboratory testing, aimed at excluding other diseases that might mimic a generalized pathology of motor neurons. For instance, a reformulation of the electromyography and new diagnostic methods including transcranial magnetic stimulation, magnetic resonance imaging voxel-based morphometry and diffuse tensor imaging have also been introduced with the aim of further improving the diagnosis of ALS [69]. More recently, novel neuroradiological evidence sustained a procedure not only for diagnosing but also, most importantly, for staging sALS based on transactivation response TDP-43 pathology. According to this evidence, a corticofugal axonal model for the progression of ALS can be hypothesized whereby the pathology originates in the primary motor cortex and spreads via axonal projections to subcortical structures and to lower motor neurons [70]. Parallel experimental studies have also indicated the cell-to-cell propagation of aggregated or truncated TDP-43 protein suggesting a direct transmission of TDP-43 inclusions to contiguous cells [71]. An additional hypothesis in line with the anatomical spreading of TDP-43 aggregates now suggests a trans-synaptic transmission of TDP-43 [72]. In any case, the perturbation of cellular clearance seems to be accepted as a likely factor for modifying TDP-43 aggregation and disease spreading.
Despite this increasingly accumulated evidence [73], an accurate and early diagnosis of ALS is still impossible without reliable biomarkers. Although the explanation of some genetic and molecular bases of ALS have produced further advancement, early diagnostic detection remains challenging.
3. Modeling ALS in Different Systems
3.1. ALS In Vitro Models
An experimental tool that has greatly helped in understanding cellular and molecular mechanisms of ALS is represented by different neuronal cell lines expressing ALS genes. Since the first use of NSC-34 cells (a mouse neural hybrid cell line produced by a fusion of motor neuron-enriched embryonic mouse spinal cord neurons with mouse neuroblastoma cells [74]) stably transfected with a vector expressing a human wild type or G93A mutant SOD1 [75,76,77,78,79], several works have continued to prove the resemblance of NSC-34 with motor neurons and their reliability as a cellular model of ALS. Similarly, the human neuroblastoma SH-SY5Y cell line has improved our understanding of not only SOD1 but also TDP-43-ALS pathology [80,81,82].
A great improvement in ALS cellular research was then provided by the exploitation of human-induced pluripotent stem cells (iPSCs) [83] that introduced the advantage of generating cells directly from individual patients with familial or sporadic ALS mutations [84,85]. In addition to motor neurons, iPSCs can also mimic other cell types associated with ALS such as astrocytes, microglia and oligodendrocytes, well known to be directly involved in the disease. Although iPSC technology eliminates the species-specificity problem inherent with the use of animal models, it remains an in vitro system with several limitations; for instance, a lack of tissue connectivity and functional complexity.
3.2. Use of Unicellular Eukaryotes for Modeling ALS
Despite unicellular models not being able to recapitulate complex disease phenotypes for certain, the completely sequenced genome [86] and the presence of an ortholog for many human disease genes [87] render the eukaryote budding yeast Saccharomyces cerevisiae a good candidate for where to model different pathologies [88,89,90]. As such, this model organism has been used to set up pioneering experiments identifying several different ALS genetic modifiers. For instance, a yeast ALS model led to the discovery that ATAXIN-2 modulates TDP-43 toxicity. Moreover, in yeast, Ataxin-2 carrying a mild expansion of a CAG trinucleotide was identified as an ALS disease gene [91]. Likewise, the first evidence that stress granule components act as TDP-43 modifiers [92] and that nucleocytoplasmic transport is linked to C9ORF72-ALS [93] was carried out through unbiased yeast genetic screening.
Human SOD1 expression in yeast is reported to assume a gain-of-toxic-function due to the loss of protein stability also perturbing metabolic regulation and enhancing the senescence process [94]. Interestingly, the heteromeric interaction between the wild type and mutant SOD1-A4V exacerbates the toxic effect and impairs the antioxidant response in SOD1-deficient yeast cells [95]. Yeast SOD1-deficiency is fully complemented by the expression of the human wild type SOD1, given the high identity between the two isoforms [96].
FUS ectopic overexpression in yeast inhibits the ubiquitin-proteasome system [97] and determines the formation of toxic cytoplasmic aggregates that co-localize with P-bodies and stress granules [24,87,98,99]. Removing the RNA recognition motif of FUS does not act on aggregates production but rescues FUS-induced toxicity, thus highlighting that the interaction between FUS and RNA is essential for FUS toxic action [98]. Consistently, a recent genetic screening in yeast identified several human genes acting as FUS toxicity suppressors, many of which were reported as RNA-binding proteins [100].
When expressed in yeast at high levels, TDP-43 forms subcellular aggregates that inhibit cell growth and exert a cytotoxic effect [101,102]. Interestingly, a yeast genetic screening revealed that the depletion of the Dbr1 debranching enzyme, whose role is to eliminate intronic lariats during the splicing process, becomes a strong suppressor of TDP-43-induced toxicity. Non-removed intronic lariats act as traps for TDP-43, thus impeding its interference with cellular RNAs and RNA-binding proteins [103,104]. Moreover, a recent report showed that TDP-43-induced cell death is considerably reduced in yeast deleted for CNC1 (Cyclin C), DNM1 (Dynamin-related) or YBH3 (Bax inhibitor) genes, all involved in the oxidative stress response pathway at different levels [105].
Although Saccharomyces cerevisiae cannot offer insights concerning many aspects of the ALS pathomechanism as it is a unicellular organism, studies on yeasts have uncovered several key features of cytotoxicity and proteinopathy. Clearly, to gain a more comprehensive view of ALS pathology, it is mandatory to utilize higher complexity invertebrate and vertebrate models.
3.3. ALS Disease in Caenorhabditis elegans
A model system that is increasingly accepted and exploited in ALS is the nematode Caenorhabditis elegans, which is among the most intensely studied organisms in modern experimental biology. The simple and compact nervous system formed by 302 neurons and equipped with multiple synapses is well studied and amenable to neural circuit identification and analysis [106,107]. As such, Caenorhabditis elegans has been used to identify many genes affecting the nervous system function, development and disease states, given the fact that about 30% of human genes have a functional ortholog in the C. elegans genome [107]. Moreover, several mechanisms underlying the processing of sensory information, the generation of specific motor outputs and locomotion circuits or the control of behavioral states can be easily analyzed in the nematode. As many cellular stress and survival pathways are conserved in worms, transgenic Caenorhabditis elegans has been extensively used to model ALS as well [108].
In particular, the ubiquitous expression of mutant SOD1 prevents the natural biological response to oxidative stress and determines protein aggregation [109] while mutant SOD1 expression throughout the worm’s entire nervous system results in locomotion defects and an impaired neuronal transmission [110]. Interestingly, the generation of single-copy/knock-in mutant SOD1 models (A4V, H71Y, L84V, G93A and G85R) allowed for the dissection of the specific impact of different mutations on cholinergic versus glutamatergic motor neuron degeneration, showing that ALS pathogenesis is neuronal subtype-specific and characterized by both gain-of-toxic-function and loss-of-function [111].
Regarding FUS, Murakami and colleagues expressed several FUS mutations and two truncated FUS proteins throughout the worm’s nervous system. Interestingly, only the mutations causing aggregation produced aberrant motor phenotypes that could not be rescued by the expression of a wild type FUS, thus suggesting a gain-of-function mechanism caused by these mutations [112]. Electron microscopy and electrophysiology assays moreover showed that the transgenic expression of a wild type or mutant FUS in Caenorhabditis elegans caused a decreased transmission from motor neurons to muscles and impaired synaptic vesicle docking at neuromuscular junctions [113].
The first TDP-43 overexpression model in the nematode was developed by Ash and co-authors. The pan-neuronal expression of human TDP-43 or Caenorhabditis elegans tdp-1 generates worms with uncoordinated, slow movements and the fasciculation of the GABAergic motor neurons [114,115]. Moreover, targeting the expression of mutant TDP-43 in worm GABAergic motor neurons causes age-dependent progressive paralysis, GABAergic neurodegeneration and impairment of synapses [116]. Moreover, mutant exogenous TDP-43 induces oxidative stress and the abnormal expression of endogenous tdp-1 with a decline in neuronal functions and lifespan; coherently, tdp-1 deletion determines the rescue of these phenotypes [117]. Locomotor defects in TDP-43 mutant worms were also rescued by α-metil-α-phenylsuccinimide treatment as shown by a disease modifier screening [118]. Recently, it has been established that hyperphosphorylation of TDP-43 interferes with protein homeostasis, resulting in neurotoxicity. Among the kinases involved, cell division cycle kinase 7 directly participates in TDP-43 phosphorylation because its inhibition reduces the phosphorylation of TDP-43 in Caenorhabditis elegans [119].
The nematode ortholog of C9ORF72 is named alfa-1 (ALS/FTD associated gene homolog). The decreased expression of alfa-1 or loss-of-function of the mutant alfa-1 causes motility defects, neurodegeneration (specifically of motor neurons) and sensitivity to osmotic stress [120]. Although further characterization remains to be done, it is interesting that the loss of alfa-1 is linked to defective neuronal integrity specifically of GABAergic motor neurons in worms. Transgenic worms expressing a (G4C2)29 construct showed paralysis and lethality [121].
Although C. elegans modeling has allowed the recapitulation of a few aspects of ALS-related neurodegeneration and toxicity, it should be clearly considered that the nematode is a quite simple organism, lacking defined tissues and organs including a brain and blood. However, large screening using Caenorhabditis elegans for investigating genetic interactions among disease genes and targeting specific aspects of neurodegeneration seems promising and likely to show relevance also in higher organisms.
3.4. ALS Research by the Drosophila melanogaster Model
For more than a century Drosophila melanogaster has represented a valuable choice of where to perform studies from genetic to physiological fields of biology. Together with a copious progeny production at each generation, the availability of specific transgenesis programs allowing the fine-tuned control of transgene expression [122,123,124,125] has rendered Drosophila a very good platform for ALS modeling and therapeutic compound screening. This adds to the fact that transgenic flies expressing disease genes are able to reliably reproduce a few ALS symptoms (locomotor disabilities, cellular inclusions, mitochondrial dysfunction and early death) and that disease progression can be monitored through different kinds of tests [126]. For all of these reasons, most of the mutated genes known to be involved in ALS have been modeled in flies to assess their potential contribution to pathological phenotypes [127,128].
Transgenic flies expressing the human SOD1 gene carrying point mutations (G85R, A4V, G37R, G41D) exhibit motor neuron dysfunction, climbing impairment, focal SOD1 aggregates, damaged mitochondria and oxidative stress [129,130,131]. Interestingly, different antioxidant compounds have been reported to be neuroprotective in SOD1-ALS Drosophila models, ameliorating motor performances, extending lifespan and lowering SOD1 cytoplasmic inclusions [132,133,134,135,136].
Flies expressing mutant human or Drosophila FUS (cabeza) showed progressive neurodegeneration, damaged photoreceptors and neuronal complications [137,138,139,140]. Interestingly, all defects were rescued by introducing a wild type human FUS transgene in a mutant cabeza background [141]. Studies on FUS transgenic flies have demonstrated that human FUS-induced toxicity is reduced by inhibiting nuclear export, thus confirming the involvement of nucleocytoplasmic transport in ALS pathogenesis [142,143]. Of note, a recent ALS modifier screening in two different Drosophila models carrying either a mutant FUS or mutant TARDBP human transgenes identified a complex array of enhancers and suppressors, many of which were able to rescue the ALS phenotype in both pathogenic conditions [144]. The identification of disease modifiers effective in different ALS models is surely key evidence of shared cellular and molecular pathways involved in many forms of ALS regardless of the causative genes. For instance, the overexpression of ter94, the fly ortholog of fALS-related gene valosin-containing protein (VCP) [145], was reported to be protective in a Drosophila model of cabeza-knockdown [146]. Additional studies on Drosophila allowed the correlation of FUS-induced neurodegeneration with many other cellular processes such as transcription and translation regulation [147,148], piRNA biogenesis [149], stress granule assembly [22,23,24,150,151] and Hippo-signaling pathways [152,153], shedding further light on the complex pathogenesis of ALS.
The overexpression of mutant or a wild type human TDP-43 as well as the overexpression of the TBPH (TARDBP fly ortholog) gene [139,154,155,156,157,158,159,160] in Drosophila impacts on lifespan, motor abilities, axonal transport and eclosion from the pupal case. TBPH depletion also causes locomotor disturbances and reduces the lifespan [159,161]. Of note, different potential therapeutic approaches have been found through Drosophila TDP-43 transgenic models. In particular, the modulation of autophagy [162,163,164], mitophagy [165], mitochondrial dynamics [166], glucose and lipid metabolism [167,168,169] and stress granule dynamics [92,170,171,172,173] have been reported to exert a positive effect on fly motor defects and lifespan.
The ectopic expression of an expanded GGGCC hexanucleotide or toxic dipeptide repeats in fly tissues causes ommatidia disorganization, motor defects and neuromuscular junction anomalies [174,175,176,177,178]. Recent studies on these gain-of-function Drosophila models revealed that different cellular processes impact on C9ORF72-ALS pathogenesis such as transcription [177,178,179,180], translation [181], nucleocytoplasmic transport [174,182,183,184] and protein degradation [185]. Despite the many advantages presented by the Drosophila model, it is clear that the highest limitation of this organism is the anatomical difference between the human and the fly brain together with the impossibility of conducting behavioral assays for which vertebrate studies are surely more informative.
3.5. ALS Pathogenesis in Danio rerio
Among the common model systems of ALS, a newcomer in the field is Danio rerio (zebrafish) that is becoming a widespread organism for investigating the disease owning to a lot of benefits in comparison with other vertebrate models such as a short life cycle, very high fertility and, most of all, larval transparency. Furthermore, zebrafish have a nervous system and main neurotransmitters remarkably similar to humans. Beside the high similarity with the human genome, genes strictly implicated in neurodegenerative diseases are also highly conserved between humans and zebrafish. Another advantage of using zebrafish is that the genome is easily modifiable [186,187,188].
In particular, transgenic Danio rerio overexpressing mutant SOD1 exhibited the main features of ALS (the loss of motor neurons, muscle degeneration, damage to neuromuscular junctions and impaired motor performance) culminating in decreased endurance in a swim test and the reduction of survival [189]. Moreover, in zebrafish expressing mutant SOD1, motor impairment, protein misfolding, ER-Golgi transport dysfunction and cytoplasmic mislocalization of TDP-43 were rescued by the redox function of the protein disulfide isomerase, suggesting that redox regulation is critical in maintaining cellular homeostasis [190].
The knockdown of endogenous fus or the overexpression of some human defective fus alleles in zebrafish generates a pathologic motor phenotype that is rescued by co-expressing the wild type human fus but not ALS-related fus mutations. The wild type human FUS also rescues the tardbp knockdown phenotype, suggesting a common pathogenic pathway. However, the co-expression of mutant sod1 and mutant fus aggravates motor function more than the single overexpression of mutant sod1 [186,191].
The expression of mutant tardbp (the gene encoding TDP-43) in zebrafish embryos induces motor neuron axonopathy, which was rescued by co-expressing the wild type but not mutant human TARDBP genes [192]. Recently, Campanari and collaborators confirmed in zebrafish the “distal axonopathy” hypothesis of ALS, according to which the pathological alterations take place at the neuromuscular junction during the presymptomatic phases of the disease. Zebrafish TDP-43 knockdown indeed resulted in early deficiencies of motor functions and neuromuscular junction disassembly. Moreover, the authors established that a partially depleted TDP-43 affected acetylcholinesterase expression, thus identifying this enzyme as a limiting factor regulating the connection between muscle and motor neurons [193].
Knockdown of the C9ORF72 zebrafish ortholog induces axonopathy of motor neurons and locomotor deficits that, of note, are rescued by overexpressing human C9ORF72 mRNA transcripts [194,195]. Conversely, G4C2 HRE results in RNA foci and dipeptide repeat formation leading to a significant increase of apoptotic cells, toxicity and motor axon abnormalities [196,197,198].
3.6. Modeling ALS Phenotypes in Mouse, Rat and Guinea Pig Models
As the mouse (Mus musculus) has a development and a genome similar to humans, it is the most used system to reproduce neurodegenerative diseases including ALS [199]. Although much less used than mice, rats (Rattus norvegicus) and guinea pigs (Cavia porcellus) also have physiological characteristics like humans and, in some ways, are better models than mice because of their relatively larger brain and body size and higher stress resistance to experimental manipulation.
After discovering several mutations in the gene encoding the SOD1 enzyme as major causes of ALS [11], the most studied animal model has been the SOD1-G93A mouse [200]. However, because additional mutations have been discovered through the years, other mouse models have been developed such as the SOD1-D83G, SOD1-D90A and SOD1-G37R mice and are the most studied [201]. A common feature of these genetic variants with different copper-binding abilities and causing different phenotypes is to mimic the main features of ALS and to recapitulate the human disease [6,202]. For instance, the SOD1-G93A mouse displays abundant cytoplasmic inclusions and aggregates in degenerating motor neurons of ventral horns of the spinal cord together with reactive gliosis. Furthermore, impaired neuromuscular junction, extensive inflammation in the spinal cord and disruption of endothelial barrier integrity have been observed [9,203,204,205,206,207,208,209]. These features are also typical of the human pathology [210]. A major disadvantage of the SOD1-G93A mouse model is the absence of motor neuron degeneration in the cerebral cortex, one of the main features of the human disease [211]. However, further studies have shown a progressive decrease in dendritic length and spine density in the pyramidal neurons of the motor cortex, functional and structural neuronal alterations associated with abnormal glutamate activity in the motor cortex of presymptomatic G93A mice [212,213]. Interestingly, the transgenic SOD1-D83G mouse model showed a degeneration of motor neurons not only in the spinal cord but also in the cerebral cortex and particularly during early life but without showing any sign of paralysis in adult life, which was different from the SOD1-G93A mouse model [214]. Although the SOD1-D90A mutation appears to be less toxic than others, homozygous mice develop a fatal motor neuron disease with a slower progression and bladder disturbances similar to those observed in human ALS patients are homozygous for the D90A mutation. These mice accumulate SOD1 aggregates, inclusions and vacuoles in the ventral horns of the spinal cord [215]. Finally, SOD1-G37R mice also develop a progressive motor neuron disease. In particular, low levels of mutant SOD1 accumulation affect only the lower motor neurons while higher levels determine a more severe phenotype and also hit other neuronal populations. The main pathologic alteration of SOD1-G37R mice is the presence of vacuoles originating from mitochondrial debris in axons and dendrites [216].
Recently, Magota and co-workers demonstrated in the SOD1-G93A rat model that blood-spinal cord barrier destruction can directly contribute to motor neuron degeneration. The authors observed that the intravenous infusion of healthy mesenchymal stem cells to these rats delayed disease progression, preserved barrier function, increased the expression of the neurotrophic factor Neurturin and preserved motor neurons [217].
Fus mutations can now also be exogenously expressed in both rats and mice to mimic ALS [218]. In the rat model, motor dysfunction and paralysis, neuronal loss, gliosis and protein aggregation are observed in the brain and spinal cord. In the mouse model, in addition to protein aggregation and the consequent neurodegeneration of motor neurons, inhibition of the protein transport between the Golgi complex and endoplasmic reticulum and damage to the neuromuscular junction have also been described [219,220]. Moreover, Ishigaki and co-workers reported that Fus knockdown in the mouse hippocampus leads to Tau hyperphosphorylation, neuronal death and reduced adult neurogenesis. Furthermore, these animals exhibit phenotypic behavior typical of FTD such as limitations in socialization, hyperactivity and disinhibition [221,222].
In transgenic TDP-43 mice, damage to cortical and spinal motor neurons, axonal degeneration, mitochondrial dysfunction, gliosis and loss of neuromuscular junctions have been observed [44,223,224]. Recently, transgenic TDP-43 mice with a pathological accumulation of cytoplasmic TDP-43 were used to study DNA damage, an additional pathological feature of ALS. It was shown that the lack of DNA repair activity by TDP-43 mislocalization caused DNA damage [225].
Mice with C9ORF72 mutations exhibit movement defects and cognitive deficits in addition to neuronal death, gliosis, weight loss, neuromuscular junction disruption, myopathy and behavioral changes such as hyperactivity and anxiety [53,54,210,219,226,227,228,229,230]. On the contrary, mice knockouts for C9ORF72 do not show neurodegeneration or motor alterations [231].
On the other hand, the loss of C9ORF72 protein in rats requires the synergistic action of excitotoxicity to cause neuronal injury and disease progression. The ablation of C9ORF72 protein in rats and treatment with glutamate analogue kainic acid indeed stimulates the release of excitatory neurotransmitters resulting in an increased susceptibility of motor neurons to excitotoxicity, motor neuron degeneration and motor deficits [232].
Guinea pigs were also initially used to model ALS by adopting a diet deficient in ascorbic acid, which caused muscle atrophy, motor neuron degeneration and demyelination of the pyramidal tract [233]. However, guinea pigs were mainly used in pioneering studies to highlight an autoimmune etiology for ALS. In particular, defects of motor neurons have been experimentally induced by immunizing guinea pigs with purified bovine spinal motor neurons or homogenates from the ventral horns of healthy bovine spinal cords [234]. The serum from either immunized guinea pigs or from ALS patients was next used to infect mice through intraperitoneal inoculations. These mice demonstrated an increased acetylcholine release from axon terminals [235], IgG accumulation, increased intracellular calcium and morphological and functional defects of spinal motor neurons in part resembling ALS disease features [236,237].
Despite the many advantages and knowledge acquired by the use of these ALS mice, rats and guinea pigs, rodent modeling requires a high maintenance cost and a time-consuming investigation.
3.7. Canine ALS Modeling
Due to clinical and molecular similarities, canine degenerative myelopathy (CDM) is the only spontaneously occurring pathology that represents a model for human SOD1-related ALS [238]. Precisely, CDM is characterized by the progressive degeneration of axons, muscle atrophy, hyporeflexia, astrocytosis, peripheral demyelination and SOD1 inclusions [238,239,240]. Interestingly, T18S and E40K missense mutations in the canine SOD1 gene are the only mutations reported as CDM causative [239,241]. Different from ALS, whose SOD1 causative mutations are inherited as dominant, CDM shows recessive inheritance with a reduced penetrance [239]. Both T18S and E40K mutations do not cause disruption in the dismutase domain and impact only on protein hydrophobicity but not stability although promote the formation of aggregates and thus suggesting a gain-of-toxic-function [242,243].
Recent insights about the glia contribution to CDM describe a demyelination of oligodendrocytes [240], an increase of arginase 1-expressing microglia in the proximity of motor neurons [244] and the upregulation of CB2 receptors in activated astrocytes as the main cellular responses and biological markers of the disease [245]. Of note, these pathological features confirm those previously described in ALS rodent models and in patients [206,246,247].
Taking into account the high pathogenic similarity between spontaneous CDM and SOD1-ALS and the analogous complexity of human and canine nervous system, studies on dog tissues may help to better dissect the still unknown pathomechanisms of ALS and thus support the progress for therapeutic developments. However, it should be considered that dogs with CDM are often euthanized at early phases of the disease so the tissues potentially available for investigative studies can provide information only about early disease stages.
3.8. Non-Human Primate Models of ALS
Non-human primates (NHPs) are the evolutionarily closest species to humans and share many structural, cognitive and functional features. In particular, they have the capability to exert high brain functions such as thinking, reasoning, decision making and judging because of the development of a complex network of brain connections involving the neocortex and prefrontal cortex [248,249]. These features, together with neuroanatomical and genetic similarities, make NHPs a very desirable ALS model.
Callithrix jacchus, the marmoset, is one of the most commonly used NHPs to validate potential therapeutic strategies and to generate neurodegenerative diseases models thanks to its small size, easy handling and rapidity to obtain transgenic forms [250,251,252]. For instance, a SOD1-ALS marmoset model was obtained through the intrathecal delivery of an adeno-associated virus encoding an artificial SOD1-specific microRNA, determining the reduction of SOD1 levels both in motor neurons and spinal cord sections [253]. By the same technique, a Macaca fascicularis SOD1-ALS model was recently obtained [254]. Likewise, a stereotaxic injection in Callithrix jacchus of a FUS-targeting shRNA was used to obtain a FUS-ALS marmoset model [255]. A TDP-43 overexpressing model was realized in Macaca fascicularis through the cervical injection of an adeno-associated virus containing a TDP-43 coding sequence. All of these monkeys exhibited muscle atrophy, progressive motor weakness, TDP-43 mislocalization and Cystatin C aggregates reminiscent of ALS patients [256], thus confirming the faithful correspondence between patient symptoms and what can be reproduced in monkeys.
Despite the clear advantage of these models, gene therapy approaches in NHPs need without a doubt further development but because of the ethical issue complexity, they are only a promising route to treat ALS.
4. Concluding Remarks
In this review we have described how several unicellular and multicellular organisms including vertebrate and invertebrate eukaryotes such as yeast, worms, flies, zebrafish, mice, rats, guinea pigs, dogs and, more recently, non-human primates, can serve as model systems to efficiently untangle the complicated ALS pathogenesis (Table 1). Indeed, a thorough molecular analysis demonstrated that each of these organisms has greatly contributed to highlighting specific features of the disease among which are excitotoxicity, oxidative stress, endoplasmic reticulum stress, mitophagy, autophagy, proteinopathy, gliosis and inflammation. However, by analyzing all of the major available information, we have come to the conclusion that understanding the causes and mechanisms of ALS would better require the concurrent and comparative analysis of these animal models although harnessing these powerful organisms still poses numerous challenges about their translational power. Indeed, comparative research deals with models based on interspecies and intraspecies variations can sometimes even complicate the task of explaining the disease itself.
If we then ask why these models have become so widespread in preclinical analyses, we have to recognize their high flexibility in reproducing a few of the main clinical features of the disease, at least in part and in their own ways. By translationally investigating lifespan, motor capabilities (through swimming, climbing and running tests), morphological/histological modifications, motor neuron excitability and signal transduction mechanisms at cellular and molecular levels, these organisms have indeed proven to be instrumental in identifying new efficacious genetic modifiers and therapeutic agents against the insurgence and progression of the disease. Nevertheless, we are aware that they will never become surrogates for clinical testing.
Overall, this review has described common features but also discrepancies among experimental organisms in ALS, underlining new insights and emerging roles. In other words, we have illustrated the promises and problems of extrapolating ALS findings from animal models.
Author Contributions
Conceptualization, C.V.; writing—original draft preparation, review and editing, F.L., S.A., C.V.; graphical representation, F.L., S.A., C.V. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Italian Ministry of Health through Fondazione Santa Lucia IRCCS Ricerca Corrente.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Graphical abstract was created with BioRender.com.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Yerbury, J.J.; Farrawell, N.E.; McAlary, L. Proteome Homeostasis Dysfunction: A Unifying Principle in ALS Pathogenesis. Trends Neurosci. 2020, 43, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Yousefian-Jazi, A.; Seol, Y.; Kim, J.; Ryu, H.L.; Lee, J.; Ryu, H. Pathogenic Genome Signatures That Damage Motor Neurons in Amyotrophic Lateral Sclerosis. Cells 2020, 9, 2687. [Google Scholar] [CrossRef] [PubMed]
- Volonté, C.; Morello, G.; Spampinato, A.G.; Amadio, S.; Apolloni, S.; D’Agata, V.; Cavallaro, S. Omics-based exploration and functional validation of neurotrophic factors and histamine as therapeutic targets in ALS. Ageing Res. Rev. 2020, 62, 101121. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Akiyama, T.; Warita, H.; Aoki, M. Omics Approach to Axonal Dysfunction of Motor Neurons in Amyotrophic Lateral Sclerosis (ALS). Front. Neurosci. 2020, 14, 194. [Google Scholar] [CrossRef] [PubMed]
- Gittings, L.M.; Sattler, R. Recent advances in understanding amyotrophic lateral sclerosis and emerging therapies. Fac. Rev. 2020, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Gautier, O.; Tassoni-Tsuchida, E.; Ma, X.R.; Gitler, A.D. ALS Genetics: Gains, Losses, and Implications for Future Therapies. Neuron 2020, 108, 822–842. [Google Scholar] [CrossRef] [PubMed]
- Sivandzade, F.; Cucullo, L. Regenerative Stem Cell Therapy for Neurodegenerative Diseases: An Overview. Int. J. Mol. Sci. 2021, 22, 2153. [Google Scholar] [CrossRef]
- Scaricamazza, S.; Salvatori, I.; Ferri, A.; Valle, C. Skeletal Muscle in ALS: An Unappreciated Therapeutic Opportunity? Cells 2021, 10, 525. [Google Scholar] [CrossRef]
- Volonté, C.; Apolloni, S.; Sabatelli, M. Histamine beyond its effects on allergy: Potential therapeutic benefits for the treatment of Amyotrophic Lateral Sclerosis (ALS). Pharmacol. Ther. 2019, 202, 120–131. [Google Scholar] [CrossRef]
- Shatunov, A.; Al-Chalabi, A. The genetic architecture of ALS. Neurobiol. Dis. 2021, 147, 105156. [Google Scholar] [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.C.; Hentati, A.; Donaldson, D.H.; Goto, J.; O’Regan, J.P.; Deng, H.-X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Deng, H.X.; Hentati, A.; Tainer, J.A.; Iqbal, Z.; Cayabyab, A.; Hung, W.Y.; Getzoff, E.D.; Hu, P.; Herzfeldt, B.; Roos, R.P.; et al. Amyotrophic lateral sclerosis and structural defects in Cu,Zn superoxide dismutase. Science 1993, 261, 1047–1051. [Google Scholar] [CrossRef]
- Chattopadhyay, M.; Valentine, J.S. Aggregation of Copper–Zinc Superoxide Dismutase in Familial and Sporadic ALS. Antioxid. Redox Signal. 2009, 11, 1603–1614. [Google Scholar] [CrossRef] [Green Version]
- Şahin, A.; Held, A.; Bredvik, K.; Major, P.; Achilli, T.-M.; Kerson, A.G.; Wharton, K.; Stilwell, G.; Reenan, R. Human SOD1 ALS Mutations in a Drosophila Knock-In Model Cause Severe Phenotypes and Reveal Dosage-Sensitive Gain- and Loss-of-Function Components. Genetics 2017, 205, 707–723. [Google Scholar] [CrossRef]
- Bruijn, L.I.; Houseweart, M.K.; Kato, S.; Anderson, K.L.; Anderson, S.D.; Ohama, E.; Reaume, A.G.; Scott, R.W.; Cleveland, D.W. Aggregation and Motor Neuron Toxicity of an ALS-Linked SOD1 Mutant Independent from Wild-Type SOD1. Science 1998, 281, 1851–1854. [Google Scholar] [CrossRef] [Green Version]
- Yim, M.B.; Kang, J.H.; Yim, H.S.; Kwak, H.S.; Chock, P.B.; Stadtman, E.R. A gain-of-function of an amyotrophic lateral sclerosis-associated Cu,Zn-superoxide dismutase mutant: An enhancement of free radical formation due to a decrease in Km for hydrogen peroxide. Proc. Natl. Acad. Sci. USA 1996, 93, 5709–5714. [Google Scholar] [CrossRef] [Green Version]
- Ilieva, H.; Polymenidou, M.; Cleveland, D.W. Non–cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J. Cell Biol. 2009, 187, 761–772. [Google Scholar] [CrossRef] [Green Version]
- Calió, M.L.; Henriques, E.; Siena, A.; Bertoncini, C.R.A.; Gil-Mohapel, J.; Rosenstock, T.R. Mitochondrial Dysfunction, Neurogenesis, and Epigenetics: Putative Implications for Amyotrophic Lateral Sclerosis Neurodegeneration and Treatment. Front. Neurosci. 2020, 14, 679. [Google Scholar] [CrossRef]
- Paré, B.; Lehmann, M.; Beaudin, M.; Nordström, U.; Saikali, S.; Julien, J.-P.; Gilthorpe, J.D.; Marklund, S.L.; Cashman, N.R.; Andersen, P.M.; et al. Misfolded SOD1 pathology in sporadic Amyotrophic Lateral Sclerosis. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Boillée, S.; Velde, C.V.; Cleveland, D.W. ALS: A Disease of Motor Neurons and Their Nonneuronal Neighbors. Neuron 2006, 52, 39–59. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, K.; Boillee, S.; Roberts, E.A.; Garcia, M.L.; McAlonis-Downes, M.; Mikse, O.R.; Cleveland, D.W.; Goldstein, L.S.B. Mutant SOD1 in cell types other than motor neurons and oligodendrocytes accelerates onset of disease in ALS mice. Proc. Natl. Acad. Sci. USA 2008, 105, 7594–7599. [Google Scholar] [CrossRef] [Green Version]
- Bosco, D.A.; Lemay, N.; Ko, H.K.; Zhou, H.; Burke, C.; Kwiatkowski, T.J.; Sapp, P.; McKenna-Yasek, D.; Brown, R.H.; Hayward, L.J. Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum. Mol. Genet. 2010, 19, 4160–4175. [Google Scholar] [CrossRef] [Green Version]
- Daigle, J.G.; Krishnamurthy, K.; Ramesh, N.; Casci, I.; Monaghan, J.; McAvoy, K.; Godfrey, E.W.; Daniel, D.C.; Johnson, E.M.; Monahan, Z.; et al. Pur-alpha regulates cytoplasmic stress granule dynamics and ameliorates FUS toxicity. Acta Neuropathol. 2016, 131, 605–620. [Google Scholar] [CrossRef] [Green Version]
- Gal, J.; Zhang, J.; Kwinter, D.M.; Zhai, J.; Jia, H.; Jia, J.; Zhu, H. Nuclear localization sequence of FUS and induction of stress granules by ALS mutants. Neurobiol. Aging 2011, 32, 2323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregory, R.I.; Yan, K.-P.; Amuthan, G.; Chendrimada, T.; Doratotaj, B.; Cooch, N.; Shiekhattar, R. The Microprocessor complex mediates the genesis of microRNAs. Nat. Cell Biol. 2004, 432, 235–240. [Google Scholar] [CrossRef]
- Tan, A.Y.; Manley, J.L. TLS Inhibits RNA Polymerase III Transcription. Mol. Cell. Biol. 2009, 30, 186–196. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Embree, L.J.; Tsai, S.; Hickstein, D.D. Oncoprotein TLS Interacts with Serine-Arginine Proteins Involved in RNA Splicing. J. Biol. Chem. 1998, 273, 27761–27764. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Arai, S.; Song, X.; Reichart, D.; Du, K.; Pascual, G.; Tempst, P.; Rosenfeld, M.G.; Glass, C.K.; Kurokawa, R. Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nat. Cell Biol. 2008, 454, 126–130. [Google Scholar] [CrossRef]
- Brunet, M.A.; Jacques, J.; Nassari, S.; Tyzack, G.E.; McGoldrick, P.; Zinman, L.; Jean, S.; Robertson, J.; Patani, R.; Roucou, X. The FUS gene is dual-coding with both proteins contributing to FUS -mediated toxicity. EMBO Rep. 2021, 22, e50640. [Google Scholar] [CrossRef]
- Kwiatkowski, T.J., Jr.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS Gene on Chromosome 16 Cause Familial Amyotrophic Lateral Sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef] [Green Version]
- Vance, C.; Rogelj, B.; Hortobágyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA Processing Protein, Cause Familial Amyotrophic Lateral Sclerosis Type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modigliani, S.D.; Morlando, M.; Errichelli, L.; Sabatelli, M.; Bozzoni, I. An ALS-associated mutation in the FUS 3′-UTR disrupts a microRNA–FUS regulatory circuitry. Nat. Commun. 2014, 5, 4335. [Google Scholar] [CrossRef] [PubMed]
- Sabatelli, M.; Moncada, A.; Conte, A.; Lattante, S.; Marangi, G.; Luigetti, M.; Lucchini, M.; Mirabella, M.; Romano, A.; Del Grande, A.; et al. Mutations in the 3′ untranslated region of FUS causing FUS overexpression are associated with amyotrophic lateral sclerosis. Hum. Mol. Genet. 2013, 22, 4748–4755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapeli, K.; Martinez, F.J.; Yeo, G.W. Genetic mutations in RNA-binding proteins and their roles in ALS. Qual. Life Res. 2017, 136, 1193–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, Z.-Y.; Zhou, Z.-R.; Che, C.-H.; Liu, C.-Y.; He, R.-L.; Huang, H.-P. Genetic epidemiology of amyotrophic lateral sclerosis: A systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2017, 88, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Lyashchenko, A.K.; Lu, L.; Nasrabady, S.E.; Elmaleh, M.; Mendelsohn, M.; Nemes, A.; Tapia, J.C.; Mentis, G.Z.; Shneider, N.A. ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat. Commun. 2016, 7, 10465. [Google Scholar] [CrossRef] [PubMed]
- Naumann, M.; Pal, A.; Goswami, A.; Lojewski, X.; Japtok, J.; Vehlow, A.; Naujock, M.; Günther, R.; Jin, M.; Stanslowsky, N.; et al. Impaired DNA damage response signaling by FUS-NLS mutations leads to neurodegeneration and FUS aggregate formation. Nat. Commun. 2018, 9, 1–17. [Google Scholar] [CrossRef]
- Lagier-Tourenne, C.; Polymenidou, M.; Hutt, K.R.; Vu, A.Q.; Baughn, M.; Huelga, S.C.; Clutario, K.M.; Ling, S.-C.; Liang, T.Y.; Mazur, C.; et al. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat. Neurosci. 2012, 15, 1488–1497. [Google Scholar] [CrossRef]
- Zhou, Y.; Liu, S.; Liu, G.; Öztürk, A.; Hicks, G.G. ALS-Associated FUS Mutations Result in Compromised FUS Alternative Splicing and Autoregulation. PLoS Genet. 2013, 9, e1003895. [Google Scholar] [CrossRef] [Green Version]
- Murakami, T.; Qamar, S.; Lin, J.Q.; Schierle, G.S.K.; Rees, E.; Miyashita, A.; Costa, A.R.; Dodd, R.B.; Chan, F.T.; Michel, C.H.; et al. ALS/FTD Mutation-Induced Phase Transition of FUS Liquid Droplets and Reversible Hydrogels into Irreversible Hydrogels Impairs RNP Granule Function. Neuron 2015, 88, 678–690. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, J.; Birsa, N.; Milioto, C.; McLaughlin, M.; Ule, A.M.; Robaldo, D.; Eberle, A.B.; Kräuchi, R.; Bentham, M.; Brown, A.-L.; et al. FUS ALS-causative mutations impair FUS autoregulation and splicing factor networks through intron retention. Nucleic Acids Res. 2020, 48, 6889–6905. [Google Scholar] [CrossRef]
- An, H.; Skelt, L.; Notaro, A.; Highley, J.R.; Fox, A.H.; La Bella, V.; Buchman, V.L.; Shelkovnikova, T.A. ALS-linked FUS mutations confer loss and gain of function in the nucleus by promoting excessive formation of dysfunctional paraspeckles. Acta Neuropathol. Commun. 2019, 7, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [Green Version]
- Volkening, K.; Leystra-Lantz, C.; Yang, W.; Jaffee, H.; Strong, M.J. Tar DNA binding protein of 43 kDa (TDP-43), 14-3-3 proteins and copper/zinc superoxide dismutase (SOD1) interact to modulate NFL mRNA stability. Implications for altered RNA processing in amyotrophic lateral sclerosis (ALS). Brain Res. 2009, 1305, 168–182. [Google Scholar] [CrossRef]
- Buratti, E.; Baralle, F.E. Characterization and Functional Implications of the RNA Binding Properties of Nuclear Factor TDP-43, a Novel Splicing Regulator ofCFTR Exon 9. J. Biol. Chem. 2001, 276, 36337–36343. [Google Scholar] [CrossRef] [Green Version]
- Suk, T.R.; Rousseaux, M.W.C. The role of TDP-43 mislocalization in amyotrophic lateral sclerosis. Mol. Neurodegener. 2020, 15, 1–16. [Google Scholar] [CrossRef]
- Mitra, J.; Guerrero, E.N.; Hegde, P.M.; Liachko, N.F.; Wang, H.; Vasquez, V.; Gao, J.; Pandey, A.; Taylor, J.P.; Kraemer, B.C.; et al. Motor neuron disease-associated loss of nuclear TDP-43 is linked to DNA double-strand break repair defects. Proc. Natl. Acad. Sci. USA 2019, 116, 4696–4705. [Google Scholar] [CrossRef] [Green Version]
- Brauer, S.; Zimyanin, V.; Hermann, A. Prion-like properties of disease-relevant proteins in amyotrophic lateral sclerosis. J. Neural Transm. 2018, 125, 591–613. [Google Scholar] [CrossRef]
- Pang, W.; Hu, F. Cellular and physiological functions of C9ORF72 and implications for ALS/FTD. J. Neurochem. 2020. [Google Scholar] [CrossRef]
- Sellier, C.; Campanari, M.; Corbier, C.J.; Gaucherot, A.; Kolb-Cheynel, I.; Oulad-Abdelghani, M.; Ruffenach, F.; Page, A.; Ciura, S.; Kabashi, E.; et al. Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J. 2016, 35, 1276–1297. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.; Poulter, M.; Hensman, D.; Rohrer, J.D.; Mahoney, C.J.; Adamson, G.; Campbell, T.; Uphill, J.; Borg, A.; Fratta, P.; et al. Large C9ORF72 Hexanucleotide Repeat Expansions Are Seen in Multiple Neurodegenerative Syndromes and Are More Frequent Than Expected in the UK Population. Am. J. Hum. Genet. 2013, 92, 345–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [Green Version]
- Smith, B.N.; Newhouse, S.; Shatunov, A.; Vance, C.; Topp, S.; Johnson, L.; Miller, J.W.; Lee, Y.; Troakes, C.; Scott, K.M.; et al. The C9ORF72 expansion mutation is a common cause of ALS+/−FTD in Europe and has a single founder. Eur. J. Hum. Genet. 2012, 21, 102–108. [Google Scholar] [CrossRef]
- Shepheard, S.R.; Parker, M.D.; Cooper-Knock, J.; Verber, N.S.; Tuddenham, L.; Heath, P.; Beauchamp, N.; Place, E.; Sollars, E.S.A.; Turner, M.R.; et al. Value of systematic genetic screening of patients with amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2021. [Google Scholar] [CrossRef]
- Gendron, T.F.; Bieniek, K.F.; Zhang, Y.-J.; Jansen-West, K.; Ash, P.E.A.; Caulfield, T.; Daughrity, L.; Dunmore, J.H.; Castanedes-Casey, M.; Chew, J.; et al. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. 2013, 126, 829–844. [Google Scholar] [CrossRef] [Green Version]
- Gendron, T.F.; Belzil, V.V.; Zhang, Y.-J.; Petrucelli, L. Mechanisms of toxicity in C9FTLD/ALS. Acta Neuropathol. 2014, 127, 359–376. [Google Scholar] [CrossRef] [Green Version]
- Ash, P.E.; Bieniek, K.F.; Gendron, T.F.; Caulfield, T.; Lin, W.-L.; DeJesus-Hernandez, M.; van Blitterswijk, M.M.; Jansen-West, K.; Paul, J.W.; Rademakers, R.; et al. Unconventional Translation of C9ORF72 GGGGCC Expansion Generates Insoluble Polypeptides Specific to c9FTD/ALS. Neuron 2013, 77, 639–646. [Google Scholar] [CrossRef] [Green Version]
- Zu, T.; Gibbens, B.; Doty, N.S.; Gomes-Pereira, M.; Huguet, A.; Stone, M.D.; Margolis, J.; Peterson, M.; Markowski, T.W.; Ingram, M.A.C.; et al. Non-ATG–initiated translation directed by microsatellite expansions. Proc. Natl. Acad. Sci. USA 2010, 108, 260–265. [Google Scholar] [CrossRef] [Green Version]
- Cleary, J.D.; Ranum, L.P. New developments in RAN translation: Insights from multiple diseases. Curr. Opin. Genet. Dev. 2017, 44, 125–134. [Google Scholar] [CrossRef]
- Waite, A.J.; Bäumer, D.; East, S.; Neal, J.; Morris, H.R.; Ansorge, O.; Blake, D.J. Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiol. Aging 2014, 35, 1779. [Google Scholar] [CrossRef] [Green Version]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef] [Green Version]
- Oskarsson, B.; Gendron, T.F.; Staff, N.P. Amyotrophic Lateral Sclerosis: An Update for 2018. Mayo Clin. Proc. 2018, 93, 1617–1628. [Google Scholar] [CrossRef] [Green Version]
- Silani, V.; Ludolph, A.; Fornai, F. The emerging picture of ALS: A multisystem, not only a “motor neuron disease”. Arch. Ital. Biol. 2018, 155, 153–158. [Google Scholar] [CrossRef]
- Lambert, E.H.; Mulder, D.W. Electromyographic studies in amyotrophic lateral sclerosis. Proc. Staff. Meet. Mayo Clin. 1957, 32, 441–446. [Google Scholar]
- Wilbourn, A.J. Clinical neurophysiology in the diagnosis of amyotrophic lateral sclerosis: The Lambert and the El Escorial criteria. J. Neurol. Sci. 1998, 160, S25–S29. [Google Scholar] [CrossRef]
- Ludolph, A.C.; Drory, V.E.; Hardiman, O.; Nakano, I.; Ravits, J.; Robberecht, W.; Shefner, J.M.; WFN Research Group on ALS/MND. A revision of the El Escorial criteria—2015. Amyotroph. Lateral Scler. Front. Degener. 2015, 16, 291–292. [Google Scholar] [CrossRef]
- Li, D.-W.; Liu, M.; Cui, B.; Fang, J.; Guan, Y.-Z.; Ding, Q.; Li, X.; Cui, L. The Awaji criteria increases the diagnostic sensitivity of the revised El Escorial criteria for amyotrophic lateral sclerosis diagnosis in a Chinese population. PLoS ONE 2017, 12, e0171522. [Google Scholar] [CrossRef]
- Verde, F.; Del Tredici, K.; Braak, H. The multisystem degeneration amyotrophic lateral sclerosis—Neuropathological staging and clinical translation. Arch. Ital. Biol. 2018, 155, 210–227. [Google Scholar] [CrossRef]
- Jo, M.; Lee, S.; Jeon, Y.-M.; Kim, S.; Kwon, Y.; Kim, H.-J. The role of TDP-43 propagation in neurodegenerative diseases: Integrating insights from clinical and experimental studies. Exp. Mol. Med. 2020, 52, 1652–1662. [Google Scholar] [CrossRef] [PubMed]
- Riku, Y. Reappraisal of the anatomical spreading and propagation hypothesis about TDP-43 aggregation in amyotrophic lateral sclerosis and frontotemporal lobar degeneration. Neuropathology 2020, 40, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Štětkářová, I.; Ehler, E. Diagnostics of Amyotrophic Lateral Sclerosis: Up to Date. Diagnostics 2021, 11, 231. [Google Scholar] [CrossRef] [PubMed]
- Cashman, N.R.; Durham, H.D.; Blusztajn, J.K.; Oda, K.; Tabira, T.; Shaw, I.T.; Dahrouge, S.; Antel, J.P. Neuroblastoma × spinal cord (NSC) hybrid cell lines resemble developing motor neurons. Dev. Dyn. 1992, 194, 209–221. [Google Scholar] [CrossRef]
- Cookson, M.R.; Menzies, F.M.; Manning, P.; Eggett, C.J.; Figlewicz, D.A.; McNeil, C.J.; Shaw, P.J. Cu/Zn superoxide dismutase (SOD1) mutations associated with familial amyotrophic lateral sclerosis (ALS) affect cellular free radical release in the presence of oxidative stress. Amyotroph. Lateral Scler. 2002, 3, 75–85. [Google Scholar] [CrossRef]
- Allen, S.; Heath, P.R.; Kirby, J.; Wharton, S.B.; Cookson, M.R.; Menzies, F.M.; Banks, R.E.; Shaw, P.J. Analysis of the Cytosolic Proteome in a Cell Culture Model of Familial Amyotrophic Lateral Sclerosis Reveals Alterations to the Proteasome, Antioxidant Defenses, and Nitric Oxide Synthetic Pathways. J. Biol. Chem. 2003, 278, 6371–6383. [Google Scholar] [CrossRef] [Green Version]
- Rizzardini, M.; Mangolini, A.; Lupi, M.; Ubezio, P.; Bendotti, C.; Cantoni, L. Low levels of ALS-linked Cu/Zn superoxide dismutase increase the production of reactive oxygen species and cause mitochondrial damage and death in motor neuron-like cells. J. Neurol. Sci. 2005, 232, 95–103. [Google Scholar] [CrossRef]
- Ferri, A.; Cozzolino, M.; Crosio, C.; Nencini, M.; Casciati, A.; Gralla, E.B.; Rotilio, G.; Valentine, J.S.; Carrì, M.T. Familial ALS-superoxide dismutases associate with mitochondria and shift their redox potentials. Proc. Natl. Acad. Sci. USA 2006, 103, 13860–13865. [Google Scholar] [CrossRef] [Green Version]
- Gomes, C.; Palma, A.S.; Almeida, R.; Regalla, M.; McCluskey, L.F.; Trojanowski, J.Q.; Costa, J. Establishment of a cell model of ALS disease: Golgi apparatus disruption occurs independently from apoptosis. Biotechnol. Lett. 2007, 30, 603–610. [Google Scholar] [CrossRef]
- Sala, G.; Beretta, S.; Ceresa, C.; Mattavelli, L.; Zoia, C.; Tremolizzo, L.; Ferri, A.; Carrì, M.T.; Ferrarese, C. Impairment of glutamate transport and increased vulnerability to oxidative stress in neuroblastoma SH-SY5Y cells expressing a Cu,Zn superoxide dismutase typical of familial amyotrophic lateral sclerosis. Neurochem. Int. 2005, 46, 227–234. [Google Scholar] [CrossRef]
- Hu, W.; Liu, X.; Wang, S.; Sun, G.; Zhao, R.; Lu, H. SecinH3 Attenuates TDP-43 p.Q331K-Induced Neuronal Toxicity by Suppressing Endoplasmic Reticulum Stress and Enhancing Autophagic Flux. IUBMB Life 2019, 71, 192–199. [Google Scholar] [CrossRef] [Green Version]
- Giannini, M.; Bayona-Feliu, A.; Sproviero, D.; Barroso, S.I.; Cereda, C.; Aguilera, A. TDP-43 mutations link Amyotrophic Lateral Sclerosis with R-loop homeostasis and R loop-mediated DNA damage. PLoS Genet. 2020, 16, e1009260. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-Y.; Ting, H.-C.; Liu, C.-A.; Su, H.-L.; Chiou, T.-W.; Lin, S.-Z.; Harn, H.-J.; Ho, T.-J. Induced Pluripotent Stem Cell (iPSC)-Based Neurodegenerative Disease Models for Phenotype Recapitulation and Drug Screening. Molecules 2020, 25, 2000. [Google Scholar] [CrossRef]
- Karagiannis, P.; Inoue, H. ALS, a cellular whodunit on motor neuron degeneration. Mol. Cell. Neurosci. 2020, 107, 103524. [Google Scholar] [CrossRef]
- Goffeau, A.; Barrell, B.G.; Bussey, H.; Davis, R.W.; Dujon, B.; Feldmann, H.; Galibert, F.; Hoheisel, J.D.; Jacq, C.; Johnston, M.; et al. Life with 6000 Genes. Science 1996, 274, 546–567. [Google Scholar] [CrossRef] [Green Version]
- Kryndushkin, D.; Shewmaker, F. Modeling ALS and FTLD proteinopathies in yeast: An efficient approach for studying protein aggregation and toxicity. Prion 2011, 5, 250–257. [Google Scholar] [CrossRef] [Green Version]
- Chernoff, Y.O.; Grizel, A.V.; Rubel, A.A.; Zelinsky, A.A.; Chandramowlishwaran, P.; Chernova, T.A. Application of yeast to studying amyloid and prion diseases. Adv. Genet. 2020, 105, 293–380. [Google Scholar] [CrossRef]
- Rencus-Lazar, S.; DeRowe, Y.; Adsi, H.; Gazit, E.; Laor, D. Yeast Models for the Study of Amyloid-Associated Disorders and Development of Future Therapy. Front. Mol. Biosci. 2019, 6, 15. [Google Scholar] [CrossRef] [Green Version]
- Di Gregorio, S.E.; Duennwald, M.L. ALS Yeast Models—Past Success Stories and New Opportunities. Front. Mol. Neurosci. 2018, 11, 394. [Google Scholar] [CrossRef]
- Elden, A.C.; Kim, H.-J.; Hart, M.P.; Chen-Plotkin, A.S.; Johnson, B.S.; Fang, X.; Armakola, M.; Geser, F.; Greene, R.; Lu, M.M.; et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nat. Cell Biol. 2010, 466, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-J.; Raphael, A.R.; LaDow, E.S.; McGurk, L.; Weber, R.A.; Trojanowski, J.Q.; Lee, V.M.-Y.; Finkbeiner, S.; Gitler, A.D.; Bonini, N.M. Therapeutic modulation of eIF2α phosphorylation rescues TDP-43 toxicity in amyotrophic lateral sclerosis disease models. Nat. Genet. 2014, 46, 152–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jovičić, A.; Mertens, J.; Boeynaems, S.; Bogaert, E.; Chai, N.; Yamada, S.B.; Paul, J.W.; Sun, S.; Herdy, J.R.; Bieri, G.; et al. Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat. Neurosci. 2015, 18, 1226–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastow, E.L.; Peswani, A.R.; Tarrant, D.S.J.; Pentland, D.R.; Chen, X.; Morgan, A.; Staniforth, G.L.; Tullet, J.M.; Rowe, M.L.; Howard, M.J.; et al. New links between SOD1 and metabolic dysfunction from a yeast model of Amyotrophic Lateral Sclerosis (ALS). J. Cell Sci. 2016, 129, 4118–4129. [Google Scholar] [CrossRef] [Green Version]
- Brasil, A.D.A.; De Carvalho, M.D.C.; Gerhardt, E.; Queiroz, D.D.; Pereira, M.D.; Outeiro, T.F.; Eleutherio, E.C.A. Characterization of the activity, aggregation, and toxicity of heterodimers of WT and ALS-associated mutant Sod1. Proc. Natl. Acad. Sci. USA 2019, 116, 25991–26000. [Google Scholar] [CrossRef] [Green Version]
- Rabizadeh, S.; Gralla, E.B.; Borchelt, D.R.; Gwinn, R.; Valentine, J.S.; Sisodia, S.; Wong, P.; Lee, M.; Hahn, H.; Bredesen, D.E. Mutations associated with amyotrophic lateral sclerosis convert superoxide dismutase from an antiapoptotic gene to a proapoptotic gene: Studies in yeast and neural cells. Proc. Natl. Acad. Sci. USA 1995, 92, 3024–3028. [Google Scholar] [CrossRef] [Green Version]
- Park, S.-K.; Arslan, F.; Kanneganti, V.; Barmada, S.J.; Purushothaman, P.; Verma, S.C.; Liebman, S.W. Overexpression of a conserved HSP40 chaperone reduces toxicity of several neurodegenerative disease proteins. Prion 2018, 12, 16–22. [Google Scholar] [CrossRef]
- Sun, Z.; Diaz, Z.; Fang, X.; Hart, M.P.; Chesi, A.; Shorter, J.; Gitler, A.D. Molecular Determinants and Genetic Modifiers of Aggregation and Toxicity for the ALS Disease Protein FUS/TLS. PLoS Biol. 2011, 9, e1000614. [Google Scholar] [CrossRef]
- Ju, S.; Tardiff, D.F.; Han, H.; Divya, K.; Zhong, Q.; Maquat, L.E.; Bosco, D.A.; Hayward, L.J.; Brown, R.H., Jr.; Lindquist, S.; et al. A Yeast Model of FUS/TLS-Dependent Cytotoxicity. PLoS Biol. 2011, 9, e1001052. [Google Scholar] [CrossRef] [Green Version]
- Hayden, E.; Chen, S.; Chumley, A.; Xia, C.; Zhong, Q.; Ju, S. A Genetic Screen for Human Genes Suppressing FUS Induced Toxicity in Yeast. G3 Genes Genomes Genet. 2020, 10, 1843–1852. [Google Scholar] [CrossRef] [Green Version]
- Johnson, B.S.; Snead, D.; Lee, J.J.; McCaffery, J.M.; Shorter, J.; Gitler, A.D. TDP-43 Is Intrinsically Aggregation-prone, and Amyotrophic Lateral Sclerosis-linked Mutations Accelerate Aggregation and Increase Toxicity. J. Biol. Chem. 2009, 284, 20329–20339. [Google Scholar] [CrossRef] [Green Version]
- Johnson, B.S.; McCaffery, J.M.; Lindquist, S.; Gitler, A.D. A yeast TDP-43 proteinopathy model: Exploring the molecular determinants of TDP-43 aggregation and cellular toxicity. Proc. Natl. Acad. Sci. USA 2008, 105, 6439–6444. [Google Scholar] [CrossRef] [Green Version]
- Armakola, M.; Higgins, M.J.; Figley, M.D.; Barmada, S.J.; Scarborough, E.A.; Diaz, Z.; Fang, X.; Shorter, J.; Krogan, N.J.; Finkbeiner, S.; et al. Inhibition of RNA lariat debranching enzyme suppresses TDP-43 toxicity in ALS disease models. Nat. Genet. 2012, 44, 1302–1309. [Google Scholar] [CrossRef] [Green Version]
- Figley, M.D.; Gitler, A.D. Yeast genetic screen reveals novel therapeutic strategy for ALS. Rare Dis. 2013, 1, e24420. [Google Scholar] [CrossRef] [Green Version]
- Bharathi, V.; Girdhar, A.; Patel, B.K. Role of CNC1 gene in TDP-43 aggregation-induced oxidative stress-mediated cell death in S. cerevisiae model of ALS. BBA Mol. Cell Res. 2021, 1868, 118993. [Google Scholar] [CrossRef]
- Yemini, E.; Jucikas, T.; Grundy, L.J.; Brown, A.E.; Schafer, W.R. A database of Caenorhabditis elegans behavioral phenotypes. Nat. Methods 2013, 10, 877–879. [Google Scholar] [CrossRef] [Green Version]
- Therrien, M.; Parker, J.A. Worming forward: Amyotrophic lateral sclerosis toxicity mechanisms and genetic interactions in Caenorhabditis elegans. Front. Genet. 2014, 5, 85. [Google Scholar] [CrossRef] [Green Version]
- Caldwell, K.A.; Willicott, C.W.; Caldwell, G.A. Modeling neurodegeneration in Caenorhabditis elegans. Dis. Model. Mech. 2020, 13, dmm046110. [Google Scholar] [CrossRef]
- Oeda, T.; Shimohama, S.; Kitagawa, N.; Kohno, R.; Imura, T.; Shibasaki, H.; Ishii, N. Oxidative stress causes abnormal accumulation of familial amyotrophic lateral sclerosis-related mutant SOD1 in transgenic Caenorhabditis elegans. Hum. Mol. Genet. 2001, 10, 2013–2023. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Farr, G.W.; Hall, D.H.; Li, F.; Furtak, K.; Dreier, L.; Horwich, A.L. An ALS-Linked Mutant SOD1 Produces a Locomotor Defect Associated with Aggregation and Synaptic Dysfunction When Expressed in Neurons of Caenorhabditis elegans. PLoS Genet. 2009, 5, e1000350. [Google Scholar] [CrossRef] [Green Version]
- Baskoylu, S.N.; Yersak, J.; O’Hern, P.; Grosser, S.; Simon, J.; Kim, S.; Schuch, K.; Dimitriadi, M.; Yanagi, K.S.; Lins, J.; et al. Single copy/knock-in models of ALS SOD1 in C. elegans suggest loss and gain of function have different contributions to cholinergic and glutamatergic neurodegeneration. PLoS Genet. 2018, 14, e1007682. [Google Scholar] [CrossRef] [Green Version]
- Murakami, T.; Yang, S.-P.; Xie, L.; Kawano, T.; Fu, D.; Mukai, A.; Bohm, C.; Chen, F.; Robertson, J.; Suzuki, H.; et al. ALS mutations in FUS cause neuronal dysfunction and death in Caenorhabditis elegans by a dominant gain-of-function mechanism. Hum. Mol. Genet. 2011, 21, 1–9. [Google Scholar] [CrossRef]
- Markert, S.M.; Skoruppa, M.; Yu, B.; Mulcahy, B.; Zhen, M.; Gao, S.; Sendtner, M.; Stigloher, C. Overexpression of an ALS-associated FUS mutation in C. elegans disrupts NMJ morphology and leads to defective neuromuscular transmission. Biol. Open 2020, 9, bio055129. [Google Scholar] [CrossRef]
- Ash, P.E.; Zhang, Y.-J.; Roberts, C.M.; Saldi, T.; Hutter, H.; Buratti, E.; Petrucelli, L.; Link, C.D. Neurotoxic effects of TDP-43 overexpression in C. elegans. Hum. Mol. Genet. 2010, 19, 3206–3218. [Google Scholar] [CrossRef]
- Liachko, N.F.; Guthrie, C.R.; Kraemer, B.C. Phosphorylation Promotes Neurotoxicity in a Caenorhabditis elegans Model of TDP-43 Proteinopathy. J. Neurosci. 2010, 30, 16208–16219. [Google Scholar] [CrossRef]
- Vaccaro, A.; Tauffenberger, A.; Aggad, D.; Rouleau, G.; Drapeau, P.; Parker, J.A. Mutant TDP-43 and FUS Cause Age-Dependent Paralysis and Neurodegeneration in C. elegans. PLoS ONE 2012, 7, e31321. [Google Scholar] [CrossRef]
- Vaccaro, A.; Tauffenberger, A.; Ash, P.E.A.; Carlomagno, Y.; Petrucelli, L.; Parker, J.A. TDP-1/TDP-43 Regulates Stress Signaling and Age-Dependent Proteotoxicity in Caenorhabditis elegans. PLoS Genet. 2012, 8, e1002806. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.Q.; Pontifex, M.G.; Phelan, M.M.; Pidathala, C.; Kraemer, B.C.; Barclay, J.W.; Berry, N.G.; O’Neill, P.M.; Burgoyne, R.D.; Morgan, A. α-Methyl-α-phenylsuccinimide ameliorates neurodegeneration in a C. elegans model of TDP-43 proteinopathy. Neurobiol. Dis. 2018, 118, 40–54. [Google Scholar] [CrossRef]
- Rojas-Prats, E.; Martinez-Gonzalez, L.; Gonzalo-Consuegra, C.; Liachko, N.F.; Perez, C.; Ramírez, D.; Kraemer, B.C.; Martin-Requero, Á.; Perez, D.I.; Gil, C.; et al. Targeting nuclear protein TDP-43 by cell division cycle kinase 7 inhibitors: A new therapeutic approach for amyotrophic lateral sclerosis. Eur. J. Med. Chem. 2021, 210, 112968. [Google Scholar] [CrossRef]
- Therrien, M.; Rouleau, G.A.; Dion, P.A.; Parker, J.A. Deletion of C9ORF72 Results in Motor Neuron Degeneration and Stress Sensitivity in C. elegans. PLoS ONE 2013, 8, e83450. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Hao, L.; Saur, T.; Joyal, K.; Zhao, Y.; Zhai, D.; Li, J.; Pribadi, M.; Coppola, G.; Cohen, B.M.; et al. Forward Genetic Screen in Caenorhabditis elegans Suggests F57A10.2 and acp-4 as Suppressors of C9ORF72 Related Phenotypes. Front. Mol. Neurosci. 2016, 9, 113. [Google Scholar] [CrossRef] [PubMed]
- Brand, A.H.; Perrimon, N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 1993, 118, 401–415. [Google Scholar] [PubMed]
- Caygill, E.E.; Brand, A.H. The GAL4 System: A Versatile System for the Manipulation and Analysis of Gene Expression. Methods Mol. Biol. 2016, 1478, 33–52. [Google Scholar] [CrossRef] [PubMed]
- Potter, C.J.; Tasic, B.; Russler, E.V.; Liang, L.; Luo, L. The Q System: A Repressible Binary System for Transgene Expression, Lineage Tracing, and Mosaic Analysis. Cell 2010, 141, 536–548. [Google Scholar] [CrossRef] [Green Version]
- Lai, S.-L.; Lee, T. Genetic mosaic with dual binary transcriptional systems in Drosophila. Nat. Neurosci. 2006, 9, 703–709. [Google Scholar] [CrossRef]
- Azuma, Y.; Mizuta, I.; Tokuda, T.; Mizuno, T. Amyotrophic Lateral Sclerosis Model. Adv. Exp. Med. Biol. 2018, 1076, 79–95. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Omori, K.; Asada, S.; Yoshida, H. Epigenetic Regulation of ALS and CMT: A Lesson from Drosophila Models. Int. J. Mol. Sci. 2021, 22, 491. [Google Scholar] [CrossRef]
- Layalle, S.; They, L.; Ourghani, S.; Raoul, C.; Soustelle, L. Amyotrophic Lateral Sclerosis Genes in Drosophila melanogaster. Int. J. Mol. Sci. 2021, 22, 904. [Google Scholar] [CrossRef]
- Watson, M.R.; Lagow, R.D.; Xu, K.; Zhang, B.; Bonini, N.M. A Drosophila Model for Amyotrophic Lateral Sclerosis Reveals Motor Neuron Damage by Human SOD1. J. Biol. Chem. 2008, 283, 24972–24981. [Google Scholar] [CrossRef] [Green Version]
- Elia, A.J.; Parkes, T.L.; Kirby, K.; George-Hyslop, P.S.; Boulianne, G.-H.L.; Phillips, J.P.; Hilliker, A.J.; St, P. Expression of human FALS SOD in motorneurons of Drosophila. Free. Radic. Biol. Med. 1999, 26, 1332–1338. [Google Scholar] [CrossRef]
- Bahadorani, S.; Mukai, S.T.; Rabie, J.; Beckman, J.S.; Phillips, J.P.; Hilliker, A.J. Expression of zinc-deficient human superoxide dismutase in Drosophila neurons produces a locomotor defect linked to mitochondrial dysfunction. Neurobiol. Aging 2013, 34, 2322–2330. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Liang, W.; Wang, H.; Yang, Y.; Wang, T.; Wang, S.; Wang, X.; Wang, Y.; Feng, H. γ-Oryzanol mitigates oxidative stress and prevents mutant SOD1-Related neurotoxicity in Drosophila and cell models of amyotrophic lateral sclerosis. Neuropharmacology 2019, 160, 107777. [Google Scholar] [CrossRef]
- Zhang, C.; Yang, Y.; Liang, W.; Wang, T.; Wang, S.; Wang, X.; Wang, Y.; Jiang, H.; Feng, H. Neuroprotection by urate on the mutant hSOD1-related cellular and Drosophila models of amyotrophic lateral sclerosis: Implication for GSH synthesis via activating Akt/GSK3β/Nrf2/GCLC pathways. Brain Res. Bull. 2019, 146, 287–301. [Google Scholar] [CrossRef]
- Wang, T.; Cheng, J.; Wang, S.; Wang, X.; Jiang, H.; Yang, Y.; Wang, Y.; Zhang, C.; Liang, W.; Feng, H. α-Lipoic acid attenuates oxidative stress and neurotoxicity via the ERK/Akt-dependent pathway in the mutant hSOD1 related Drosophila model and the NSC34 cell line of amyotrophic lateral sclerosis. Brain Res. Bull. 2018, 140, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wang, S.; Wang, X.; Jiang, H.; Yang, Y.; Wang, Y.; Cheng, J.; Zhang, C.; Liang, W.; Feng, H. Fisetin Exerts Antioxidant and Neuroprotective Effects in Multiple Mutant hSOD1 Models of Amyotrophic Lateral Sclerosis by Activating ERK. Neuroscience 2018, 379, 152–166. [Google Scholar] [CrossRef]
- De Rose, F.; Marotta, R.; Talani, G.; Catelani, T.; Solari, P.; Poddighe, S.; Borghero, G.; Marrosu, F.; Sanna, E.; Kasture, S.; et al. Differential effects of phytotherapic preparations in the hSOD1 Drosophila melanogaster model of ALS. Sci. Rep. 2017, 7, 41059. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Yang, M.; Deng, J.; Chen, X.; Ye, Y.; Zhu, L.; Liu, J.; Ye, H.; Shen, Y.; Li, Y.; et al. Expression of human FUS protein in Drosophila leads to progressive neurodegeneration. Protein Cell 2011, 2, 477–486. [Google Scholar] [CrossRef] [Green Version]
- Sasayama, H.; Shimamura, M.; Tokuda, T.; Azuma, Y.; Yoshida, T.; Mizuno, T.; Nakagawa, M.; Fujikake, N.; Nagai, Y.; Yamaguchi, M. Knockdown of the Drosophila Fused in Sarcoma (FUS) Homologue Causes Deficient Locomotive Behavior and Shortening of Motoneuron Terminal Branches. PLoS ONE 2012, 7, e39483. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, K.R.; Godena, V.K.; Hewitt, V.L.; Whitworth, A.J. Axonal transport defects are a common phenotype in Drosophila models of ALS. Hum. Mol. Genet. 2016, 25, 2378–2392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanson, N.A.; Maltare, A.; King, H.; Smith, R.; Kim, J.H.; Taylor, J.P.; Lloyd, T.E.; Pandey, U.B. A Drosophila model of FUS-related neurodegeneration reveals genetic interaction between FUS and TDP-43. Hum. Mol. Genet. 2011, 20, 2510–2523. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.-W.; Brent, J.R.; Tomlinson, A.; Shneider, N.A.; McCabe, B.D. The ALS-associated proteins FUS and TDP-43 function together to affect Drosophila locomotion and life span. J. Clin. Investig. 2011, 121, 4118–4126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steyaert, J.; Scheveneels, W.; Vanneste, J.; Van Damme, P.; Robberecht, W.; Callaerts, P.; Bogaert, E.; Bosch, L.V.D. FUS-induced neurotoxicity in Drosophila is prevented by downregulating nucleocytoplasmic transport proteins. Hum. Mol. Genet. 2018, 27, 4103–4116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fallini, C.; Khalil, B.; Smith, C.L.; Rossoll, W. Traffic jam at the nuclear pore: All roads lead to nucleocytoplasmic transport defects in ALS/FTD. Neurobiol. Dis. 2020, 140, 104835. [Google Scholar] [CrossRef]
- Kankel, M.W.; Sen, A.; Lu, L.; Theodorou, M.; Dimlich, D.N.; McCampbell, A.; Henderson, C.E.; Shneider, N.A.; Artavanis-Tsakonas, S. Amyotrophic Lateral Sclerosis Modifiers in Drosophila Reveal the Phospholipase D Pathway as a Potential Therapeutic Target. Genetics 2020, 215, 747–766. [Google Scholar] [CrossRef]
- Johnson, J.O.; Mandrioli, J.; Benatar, M.; Abramzon, Y.; Van Deerlin, V.M.; Trojanowski, J.Q.; Gibbs, J.R.; Brunetti, M.; Gronka, S.; Wuu, J.; et al. Exome Sequencing Reveals VCP Mutations as a Cause of Familial ALS. Neuron 2010, 68, 857–864. [Google Scholar] [CrossRef] [Green Version]
- Azuma, Y.; Tokuda, T.; Shimamura, M.; Kyotani, A.; Sasayama, H.; Yoshida, T.; Mizuta, I.; Mizuno, T.; Nakagawa, M.; Fujikake, N.; et al. Identification of ter94, Drosophila VCP, as a strong modulator of motor neuron degeneration induced by knockdown of Caz, Drosophila FUS. Hum. Mol. Genet. 2014, 23, 3467–3480. [Google Scholar] [CrossRef] [Green Version]
- Mallik, M.; Catinozzi, M.; Hug, C.B.; Zhang, L.; Wagner, M.; Bussmann, J.; Bittern, J.; Mersmann, S.; Klämbt, C.; Drexler, H.C.; et al. Xrp1 genetically interacts with the ALS-associated FUS orthologue caz and mediates its toxicity. J. Cell Biol. 2018, 217, 3947–3964. [Google Scholar] [CrossRef] [Green Version]
- Di Salvio, M.; Piccinni, V.; Gerbino, V.; Mantoni, F.; Camerini, S.; Lenzi, J.; Rosa, A.L.; Chellini, L.; Loreni, F.; Carri, M.T.; et al. Pur-alpha functionally interacts with FUS carrying ALS-associated mutations. Cell Death Dis. 2015, 6, e1943. [Google Scholar] [CrossRef] [Green Version]
- Wakisaka, K.T.; Tanaka, R.; Hirashima, T.; Muraoka, Y.; Azuma, Y.; Yoshida, H.; Tokuda, T.; Asada, S.; Suda, K.; Ichiyanagi, K.; et al. Novel roles of Drosophila FUS and Aub responsible for piRNA biogenesis in neuronal disorders. Brain Res. 2019, 1708, 207–219. [Google Scholar] [CrossRef]
- Casci, I.; Krishnamurthy, K.; Kour, S.; Tripathy, V.; Ramesh, N.; Anderson, E.N.; Marrone, L.; Grant, R.A.; Oliver, S.; Gochenaur, L.; et al. Muscleblind acts as a modifier of FUS toxicity by modulating stress granule dynamics and SMN localization. Nat. Commun. 2019, 10, 1–20. [Google Scholar] [CrossRef]
- Vance, C.; Scotter, E.L.; Nishimura, A.L.; Troakes, C.; Mitchell, J.C.; Kathe, C.; Urwin, H.; Manser, C.; Miller, C.C.; Hortobágyi, T.; et al. ALS mutant FUS disrupts nuclear localization and sequesters wild-type FUS within cytoplasmic stress granules. Hum. Mol. Genet. 2013, 22, 2676–2688. [Google Scholar] [CrossRef]
- Azuma, Y.; Tokuda, T.; Kushimura, Y.; Yamamoto, I.; Mizuta, I.; Mizuno, T.; Nakagawa, M.; Ueyama, M.; Nagai, Y.; Iwasaki, Y.; et al. Hippo, Drosophila MST, is a novel modifier of motor neuron degeneration induced by knockdown of Caz, Drosophila FUS. Exp. Cell Res. 2018, 371, 311–321. [Google Scholar] [CrossRef]
- Gogia, N.; Sarkar, A.; Mehta, A.S.; Ramesh, N.; Deshpande, P.; Kango-Singh, M.; Pandey, U.B.; Singh, A. Inactivation of Hippo and cJun-N-terminal Kinase (JNK) signaling mitigate FUS mediated neurodegeneration in vivo. Neurobiol. Dis. 2020, 140, 104837. [Google Scholar] [CrossRef]
- Estes, P.S.; Daniel, S.G.; McCallum, A.P.; Boehringer, A.V.; Sukhina, A.S.; Zwick, R.A.; Zarnescu, D.C. Motor neurons and glia exhibit specific individualized responses to TDP-43 expression in a Drosophila model of amyotrophic lateral sclerosis. Dis. Model. Mech. 2013, 6, 721–733. [Google Scholar] [CrossRef] [Green Version]
- Estes, P.S.; Boehringer, A.; Zwick, R.; Tang, J.E.; Grigsby, B.; Zarnescu, D.C. Wild-type and A315T mutant TDP-43 exert differential neurotoxicity in a Drosophila model of ALS. Hum. Mol. Genet. 2011, 20, 2308–2321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanson, K.A.; Kim, S.H.; Wassarman, D.A.; Tibbetts, R.S. Ubiquilin Modifies TDP-43 Toxicity in a Drosophila Model of Amyotrophic Lateral Sclerosis (ALS). J. Biol. Chem. 2010, 285, 11068–11072. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Ray, P.; Rao, E.J.; Shi, C.; Guo, W.; Chen, X.; Woodruff, E.A.; Fushimi, K.; Wu, J.Y. A Drosophila model for TDP-43 proteinopathy. Proc. Natl. Acad. Sci. USA 2010, 107, 3169–3174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaper, D.C.; Adachi, Y.; Lazarou, L.; Greenstein, M.; Simoes, F.A.; Di Domenico, A.; Solomon, D.A.; Lowe, S.; Alsubaie, R.; Cheng, D.; et al. Drosophila TDP-43 dysfunction in glia and muscle cells cause cytological and behavioural phenotypes that characterize ALS and FTLD. Hum. Mol. Genet. 2013, 22, 3883–3893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, M.-J.; Cheng, C.-W.; Shen, C.-K.J. Neuronal Function and Dysfunction of Drosophila dTDP. PLoS ONE 2011, 6, e20371. [Google Scholar] [CrossRef] [Green Version]
- Langellotti, S.; Romano, V.; Romano, G.; Klima, R.; Feiguin, F.; Cragnaz, L.; Romano, M.; Baralle, F.E. A novel Drosophila model of TDP-43 proteinopathies: N-terminal sequences combined with the Q/N domain induce protein functional loss and locomotion defects. Dis. Model. Mech. 2016, 9, 659–669. [Google Scholar] [CrossRef] [Green Version]
- Feiguin, F.; Godena, V.K.; Romano, G.; D’Ambrogio, A.; Klima, R.; Baralle, F.E. Depletion of TDP-43 affects Drosophila motoneurons terminal synapsis and locomotive behavior. FEBS Lett. 2009, 583, 1586–1592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Füllgrabe, J.; Jackson, A.; Sanchez, M.J.; Karabiyik, C.; et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, C.-W.; Lin, M.-J.; Shen, C.-K.J. Rapamycin alleviates pathogenesis of a new Drosophila model of ALS-TDP. J. Neurogenetics 2015, 29, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Donde, A.; Sun, M.; Jeong, Y.H.; Wen, X.; Ling, J.; Lin, S.; Braunstein, K.; Nie, S.; Wang, S.; Chen, L.; et al. Upregulation of ATG7 attenuates motor neuron dysfunction associated with depletion of TARDBP/TDP-43. Autophagy 2020, 16, 672–682. [Google Scholar] [CrossRef]
- Sun, X.; Duan, Y.; Qin, C.; Li, J.-C.; Duan, G.; Deng, X.; Ni, J.; Cao, X.; Xiang, K.; Tian, K.; et al. Distinct multilevel misregulations of Parkin and PINK1 revealed in cell and animal models of TDP-43 proteinopathy. Cell Death Dis. 2018, 9, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Khalil, B.; Cabirol-Pol, M.-J.; Miguel, L.; Whitworth, A.J.; Lecourtois, M.; Liévens, J.-C. Enhancing Mitofusin/Marf ameliorates neuromuscular dysfunction in Drosophila models of TDP-43 proteinopathies. Neurobiol. Aging 2017, 54, 71–83. [Google Scholar] [CrossRef]
- Manzo, E.; Lorenzini, I.; Barrameda, D.; O’Conner, A.G.; Barrows, J.M.; Starr, A.; Kovalik, T.; Rabichow, B.E.; Lehmkuhl, E.M.; Shreiner, D.D.; et al. Glycolysis upregulation is neuroprotective as a compensatory mechanism in ALS. eLife 2019, 8. [Google Scholar] [CrossRef]
- Manzo, E.; O’Conner, A.G.; Barrows, J.M.; Shreiner, D.D.; Birchak, G.J.; Zarnescu, D.C. Medium-Chain Fatty Acids, Beta-Hydroxybutyric Acid and Genetic Modulation of the Carnitine Shuttle Are Protective in a Drosophila Model of ALS Based on TDP-43. Front. Mol. Neurosci. 2018, 11, 182. [Google Scholar] [CrossRef]
- Joardar, A.; Manzo, E.; Zarnescu, D.C. Metabolic Dysregulation in Amyotrophic Lateral Sclerosis: Challenges and Opportunities. Curr. Genet. Med. Rep. 2017, 5, 108–114. [Google Scholar] [CrossRef]
- Coyne, A.N.; Siddegowda, B.B.; Estes, P.S.; Johannesmeyer, J.; Kovalik, T.; Daniel, S.G.; Pearson, A.; Bowser, R.; Zarnescu, D.C. Futsch/MAP1B mRNA Is a Translational Target of TDP-43 and Is Neuroprotective in a Drosophila Model of Amyotrophic Lateral Sclerosis. J. Neurosci. 2014, 34, 15962–15974. [Google Scholar] [CrossRef] [Green Version]
- Duan, Y.; Du, A.; Gu, J.; Duan, G.; Wang, C.; Gui, X.; Ma, Z.; Qian, B.; Deng, X.; Zhang, K.; et al. PARylation regulates stress granule dynamics, phase separation, and neurotoxicity of disease-related RNA-binding proteins. Cell Res. 2019, 29, 233–247. [Google Scholar] [CrossRef] [Green Version]
- McGurk, L.; Gomes, E.; Guo, L.; Mojsilovic-Petrovic, J.; Tran, V.; Kalb, R.G.; Shorter, J.; Bonini, N.M. Poly(ADP-Ribose) Prevents Pathological Phase Separation of TDP-43 by Promoting Liquid Demixing and Stress Granule Localization. Mol. Cell 2018, 71, 703–717. [Google Scholar] [CrossRef] [Green Version]
- Matsukawa, K.; Kukharsky, M.S.; Park, S.-K.; Park, S.; Watanabe, N.; Iwatsubo, T.; Hashimoto, T.; Liebman, S.W.; Shelkovnikova, T.A. Long non-coding RNA NEAT1_1 ameliorates TDP-43 toxicity in in vivo models of TDP-43 proteinopathy. RNA Biol. 2021, 1–9. [Google Scholar] [CrossRef]
- Freibaum, B.D.; Lu, Y.; Lopez-Gonzalez, R.; Kim, N.C.; Almeida, S.; Lee, K.-H.; Badders, N.; Valentine, M.; Miller, B.L.; Wong, P.C.; et al. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nat. Cell Biol. 2015, 525, 129–133. [Google Scholar] [CrossRef]
- Mizielinska, S.; Grönke, S.; Niccoli, T.; Ridler, C.E.; Clayton, E.L.; Devoy, A.; Moens, T.; Norona, F.E.; Woollacott, I.O.C.; Pietrzyk, J.; et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science 2014, 345, 1192–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.; Poidevin, M.; Li, X.; Li, Y.; Shu, L.; Nelson, D.L.; Li, H.; Hales, C.M.; Gearing, M.; Wingo, T.S.; et al. Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 7778–7783. [Google Scholar] [CrossRef] [Green Version]
- Goodman, L.D.; Prudencio, M.; Kramer, N.J.; Martinez-Ramirez, L.F.; Srinivasan, A.R.; Lan, M.; Parisi, M.J.; Zhu, Y.; Chew, J.; Cook, C.N.; et al. Toxic expanded GGGGCC repeat transcription is mediated by the PAF1 complex in C9orf72-associated FTD. Nat. Neurosci. 2019, 22, 863–874. [Google Scholar] [CrossRef]
- Kramer, N.J.; Carlomagno, Y.; Zhang, Y.-J.; Almeida, S.; Cook, C.N.; Gendron, T.F.; Prudencio, M.; Van Blitterswijk, M.; Belzil, V.; Couthouis, J.; et al. Spt4 selectively regulates the expression of C9orf72 sense and antisense mutant transcripts. Science 2016, 353, 708–712. [Google Scholar] [CrossRef] [Green Version]
- Goodman, L.D.; Bonini, N.M. New Roles for Canonical Transcription Factors in Repeat Expansion Diseases. Trends Genet. 2020, 36, 81–92. [Google Scholar] [CrossRef]
- Hirtreiter, A.; Damsma, G.E.; Cheung, A.C.M.; Klose, D.; Grohmann, D.; Vojnic, E.; Martin, A.C.R.; Cramer, P.; Werner, F. Spt4/5 stimulates transcription elongation through the RNA polymerase clamp coiled-coil motif. Nucleic Acids Res. 2010, 38, 4040–4051. [Google Scholar] [CrossRef]
- Goodman, L.D.; Prudencio, M.; Srinivasan, A.R.; Rifai, O.M.; Lee, V.M.-Y.; Petrucelli, L.; Bonini, N.M. eIF4B and eIF4H mediate GR production from expanded G4C2 in a Drosophila model for C9orf72-associated ALS. Acta Neuropathol. Commun. 2019, 7, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Donnelly, C.J.; Haeusler, A.R.; Grima, J.C.; Machamer, J.B.; Steinwald, P.; Daley, E.L.; Miller, S.J.; Cunningham, K.M.; Vidensky, S.; et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nat. Cell Biol. 2015, 525, 56–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunningham, K.M.; Maulding, K.; Ruan, K.; Senturk, M.; Grima, J.C.; Sung, H.; Zuo, Z.; Song, H.; Gao, J.; Dubey, S.; et al. TFEB/Mitf links impaired nuclear import to autophagolysosomal dysfunction in C9-ALS. eLife 2020, 9. [Google Scholar] [CrossRef]
- Boeynaems, S.; Bogaert, E.; Michiels, E.; Gijselinck, I.; Sieben, A.; Jovičić, A.; De Baets, G.; Scheveneels, W.; Steyaert, J.; Cuijt, I.; et al. Drosophila screen connects nuclear transport genes to DPR pathology in c9ALS/FTD. Sci. Rep. 2016, 6, 20877. [Google Scholar] [CrossRef]
- Azoulay-Ginsburg, S.; Di Salvio, M.; Weitman, M.; Afri, M.; Ribeiro, S.; Ebbinghaus, S.; Cestra, G.; Gruzman, A. Chemical chaperones targeted to the endoplasmic reticulum (ER) and lysosome prevented neurodegeneration in a C9orf72 repeat expansion drosophila amyotrophic lateral sclerosis (ALS) model. Pharmacol. Rep. 2021, 73, 536–550. [Google Scholar] [CrossRef]
- Babin, P.J.; Goizet, C.; Raldúa, D. Zebrafish models of human motor neuron diseases: Advantages and limitations. Prog. Neurobiol. 2014, 118, 36–58. [Google Scholar] [CrossRef]
- Abugable, A.A.; Morris, J.L.; Palminha, N.M.; Zaksauskaite, R.; Ray, S.; El-Khamisy, S.F. DNA repair and neurological disease: From molecular understanding to the development of diagnostics and model organisms. DNA Repair 2019, 81, 102669. [Google Scholar] [CrossRef]
- Shaw, C.A.; Morrice, J.R.; Gregory-Evans, C.Y. Animal models of amyotrophic lateral sclerosis: A comparison of model validity. Neural Regen. Res. 2018, 13, 2050–2054. [Google Scholar] [CrossRef]
- Ramesh, T.; Lyon, A.N.; Pineda, R.H.; Wang, C.; Janssen, P.M.L.; Canan, B.D.; Burghes, A.H.M.; Beattie, C.E. A genetic model of amyotrophic lateral sclerosis in zebrafish displays phenotypic hallmarks of motoneuron disease. Dis. Model. Mech. 2010, 3, 652–662. [Google Scholar] [CrossRef] [Green Version]
- Parakh, S.; Shadfar, S.; Perri, E.R.; Ragagnin, A.M.; Piattoni, C.V.; Fogolín, M.B.; Yuan, K.C.; Shahheydari, H.; Don, E.K.; Thomas, C.J.; et al. The Redox Activity of Protein Disulfide Isomerase Inhibits ALS Phenotypes in Cellular and Zebrafish Models. iScience 2020, 23, 101097. [Google Scholar] [CrossRef]
- Kabashi, E.; Bercier, V.; Lissouba, A.; Liao, M.; Brustein, E.; Rouleau, G.A.; Drapeau, P. FUS and TARDBP but Not SOD1 Interact in Genetic Models of Amyotrophic Lateral Sclerosis. PLoS Genet. 2011, 7, e1002214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabashi, E.; Lin, L.; Tradewell, M.L.; Dion, P.A.; Bercier, V.; Bourgouin, P.; Rochefort, D.; Hadj, S.B.; Durham, H.D.; Velde, C.V.; et al. Gain and loss of function of ALS-related mutations of TARDBP (TDP-43) cause motor deficits in vivo. Hum. Mol. Genet. 2009, 19, 671–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campanari, M.-L.; Marian, A.; Ciura, S.; Kabashi, E. TDP-43 Regulation of AChE Expression Can Mediate ALS-Like Phenotype in Zebrafish. Cells 2021, 10, 221. [Google Scholar] [CrossRef] [PubMed]
- Ciura, S.; Lattante, S.; Le Ber, I.; Latouche, M.; Tostivint, H.; Brice, A.; Kabashi, E. Loss of function of C9orf72 causes motor deficits in a zebrafish model of Amyotrophic Lateral Sclerosis. Ann. Neurol. 2013, 74, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Fortier, G.; Butti, Z.; Patten, S.A. Modelling C9orf72-Related Amyotrophic Lateral Sclerosis in Zebrafish. Biomedicines. 2020, 8, 440. [Google Scholar] [CrossRef]
- Lee, Y.-B.; Chen, H.-J.; Peres, J.N.; Gomez-Deza, J.; Attig, J.; Štalekar, M.; Troakes, C.; Nishimura, A.L.; Scotter, E.L.; Vance, C.; et al. Hexanucleotide Repeats in ALS/FTD Form Length-Dependent RNA Foci, Sequester RNA Binding Proteins, and Are Neurotoxic. Cell Rep. 2013, 5, 1178–1186. [Google Scholar] [CrossRef] [Green Version]
- Swinnen, B.; Bento-Abreu, A.; Gendron, T.F.; Boeynaems, S.; Bogaert, E.; Nuyts, R.; Timmers, M.; Scheveneels, W.; Hersmus, N.; Wang, J.; et al. A zebrafish model for C9orf72 ALS reveals RNA toxicity as a pathogenic mechanism. Acta Neuropathol. 2018, 135, 427–443. [Google Scholar] [CrossRef] [Green Version]
- Shaw, M.P.; Higginbottom, A.; McGown, A.; Castelli, L.M.; James, E.; Hautbergue, G.M.; Shaw, P.J.; Ramesh, T.M. Stable transgenic C9orf72 zebrafish model key aspects of the ALS/FTD phenotype and reveal novel pathological features. Acta Neuropathol. Commun. 2018, 6, 125. [Google Scholar] [CrossRef] [Green Version]
- De Giorgio, F.; Maduro, C.; Fisher, E.M.C.; Acevedo-Arozena, A. Transgenic and physiological mouse models give insights into different aspects of amyotrophic lateral sclerosis. Dis. Model. Mech. 2019, 12, dmm037424. [Google Scholar] [CrossRef] [Green Version]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef]
- Sosa, M.A.G.; De Gasperi, R.; Elder, G.A. Modeling human neurodegenerative diseases in transgenic systems. Qual. Life Res. 2011, 131, 535–563. [Google Scholar] [CrossRef]
- Tokuda, E.; Okawa, E.; Watanabe, S.; Ono, S.-I.; Marklund, S.L. Dysregulation of intracellular copper homeostasis is common to transgenic mice expressing human mutant superoxide dismutase-1s regardless of their copper-binding abilities. Neurobiol. Dis. 2013, 54, 308–319. [Google Scholar] [CrossRef]
- Apolloni, S.; Caputi, F.; Pignataro, A.; Amadio, S.; Fabbrizio, P.; Ammassari-Teule, M.; Volonté, C. Histamine Is an Inducer of the Heat Shock Response in SOD1-G93A Models of ALS. Int. J. Mol. Sci. 2019, 20, 3793. [Google Scholar] [CrossRef] [Green Version]
- Apolloni, S.; Amadio, S.; Fabbrizio, P.; Morello, G.; Spampinato, A.G.; Latagliata, E.C.; Salvatori, I.; Proietti, D.; Ferri, A.; Madaro, L.; et al. Histaminergic transmission slows progression of amyotrophic lateral sclerosis. J. Cachex Sarcopenia Muscle 2019, 10, 872–893. [Google Scholar] [CrossRef] [Green Version]
- Fabbrizio, P.; Apolloni, S.; Bianchi, A.; Salvatori, I.; Valle, C.; Lanzuolo, C.; Bendotti, C.; Nardo, G.; Volonté, C. P2X7 activation enhances skeletal muscle metabolism and regeneration in SOD1G93A mouse model of amyotrophic lateral sclerosis. Brain Pathol. 2019, 30, 272–282. [Google Scholar] [CrossRef] [Green Version]
- Volonté, C.; Amadio, S.; Fabbrizio, P.; Apolloni, S. Functional microglia neurotransmitters in amyotrophic lateral sclerosis. Semin. Cell Dev. Biol. 2019, 94, 121–128. [Google Scholar] [CrossRef]
- Proietti, D.; Giordani, L.; De Bardi, M.; D’Ercole, C.; Lozanoska-Ochser, B.; Amadio, S.; Volontè, C.; Marinelli, S.; Muchir, A.; Bouchè, M.; et al. Activation of skeletal muscle-resident glial cells upon nerve injury. JCI Insight 2021. [Google Scholar] [CrossRef]
- Lepore, E.; Casola, I.; Dobrowolny, G.; Musarò, A. Neuromuscular Junction as an Entity of Nerve-Muscle Communication. Cells 2019, 8, 906. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Kang, Y.; Zhou, Y.; Li, X.; Lan, J.; Wu, L.; Feng, X.; Peng, Y. ALS-causing SOD1 mutants regulate occludin phosphorylation/ubiquitination and endocytic trafficking via the ITCH/Eps15/Rab5 axis. Neurobiol. Dis. 2021, 153, 105315. [Google Scholar] [CrossRef]
- Alrafiah, A. From Mouse Models to Human Disease: An Approach for Amyotrophic Lateral Sclerosis. Vivo 2018, 32, 983–998. [Google Scholar] [CrossRef] [Green Version]
- Nijssen, J.; Comley, L.H.; Hedlund, E. Motor neuron vulnerability and resistance in amyotrophic lateral sclerosis. Acta Neuropathol. 2017, 133, 863–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fogarty, M.J.; Mu, E.W.H.; Noakes, P.G.; Lavidis, N.A.; Bellingham, M.C. Marked changes in dendritic structure and spine density precede significant neuronal death in vulnerable cortical pyramidal neuron populations in the SOD1G93A mouse model of amyotrophic lateral sclerosis. Acta Neuropathol. Commun. 2016, 4, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saba, L.; Viscomi, M.T.; Caioli, S.; Pignataro, A.; Bisicchia, E.; Pieri, M.; Molinari, M.; Ammassari-Teule, M.; Zona, C. Altered Functionality, Morphology, and Vesicular Glutamate Transporter Expression of Cortical Motor Neurons from a Presymptomatic Mouse Model of Amyotrophic Lateral Sclerosis. Cereb. Cortex 2016, 26, 1512–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joyce, P.I.; McGoldrick, P.; Saccon, R.A.; Weber, W.; Fratta, P.; West, S.J.; Zhu, N.; Carter, S.; Phatak, V.; Stewart, M.; et al. A novel SOD1-ALS mutation separates central and peripheral effects of mutant SOD1 toxicity. Hum. Mol. Genet. 2015, 24, 1883–1897. [Google Scholar] [CrossRef] [Green Version]
- Jonsson, P.A.; Graffmo, K.S.; Brännström, T.; Nilsson, M.P.; Andersen, P.M.; Marklund, S.L. Motor Neuron Disease in Mice Expressing the Wild Type-Like D90A Mutant Superoxide Dismutase-1. J. Neuropathol. Exp. Neurol. 2006, 65, 1126–1136. [Google Scholar] [CrossRef] [Green Version]
- Wong, P.C.; Pardo, C.A.; Borchelt, D.R.; Lee, M.K.; Copeland, N.G.; Jenkins, N.A.; Sisodia, S.S.; Cleveland, D.W.; Price, D.L. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 1995, 14, 1105–1116. [Google Scholar] [CrossRef] [Green Version]
- Magota, H.; Sasaki, M.; Kataoka-Sasaki, Y.; Oka, S.; Ukai, R.; Kiyose, R.; Onodera, R.; Kocsis, J.D.; Honmou, O. Intravenous infusion of mesenchymal stem cells delays disease progression in the SOD1G93A transgenic amyotrophic lateral sclerosis rat model. Brain Res. 2021, 1757, 147296. [Google Scholar] [CrossRef]
- Blair, I.P.; Williams, K.L.; Warraich, S.T.; Durnall, J.C.; Thoeng, A.D.; Manavis, J.; Blumbergs, P.C.; Vucic, S.; Kiernan, M.C.; Nicholson, G.A. FUS mutations in amyotrophic lateral sclerosis: Clinical, pathological, neurophysiological and genetic analysis. J. Neurol. Neurosurg. Psychiatry 2009, 81, 639–645. [Google Scholar] [CrossRef] [Green Version]
- Gois, A.M.; Mendonça, D.M.; Freire, M.A.M.; Santos, J.R. In Vitro And In Vivo Models of Amyotrophic Lateral Sclerosis: An Updated Overview. Brain Res. Bull. 2020, 159, 32–43. [Google Scholar] [CrossRef]
- Ling, S.-C.; Dastidar, S.G.; Tokunaga, S.; Ho, W.Y.; Lim, K.; Ilieva, H.; Parone, P.A.; Tyan, S.-H.; Tse, T.M.; Chang, J.-C.; et al. Overriding FUS autoregulation in mice triggers gain-of-toxic dysfunctions in RNA metabolism and autophagy-lysosome axis. eLife 2019, 8. [Google Scholar] [CrossRef]
- Ishigaki, S.; Fujioka, Y.; Okada, Y.; Riku, Y.; Udagawa, T.; Honda, D.; Yokoi, S.; Endo, K.; Ikenaka, K.; Takagi, S.; et al. Altered Tau Isoform Ratio Caused by Loss of FUS and SFPQ Function Leads to FTLD-like Phenotypes. Cell Rep. 2017, 18, 1118–1131. [Google Scholar] [CrossRef]
- Udagawa, T.; Fujioka, Y.; Tanaka, M.; Honda, D.; Yokoi, S.; Riku, Y.; Ibi, D.; Nagai, T.; Yamada, K.; Watanabe, H.; et al. FUS regulates AMPA receptor function and FTLD/ALS-associated behaviour via GluA1 mRNA stabilization. Nat. Commun. 2015, 6, 7098. [Google Scholar] [CrossRef]
- Van Deerlin, V.M.; Leverenz, J.B.; Bekris, L.M.; Bird, T.D.; Yuan, W.; Elman, L.B.; Clay, D.; Wood, E.M.; Chen-Plotkin, A.S.; Martinez-Lage, M.; et al. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: A genetic and histopathological analysis. Lancet Neurol. 2008, 7, 409–416. [Google Scholar] [CrossRef] [Green Version]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 Mutations in Familial and Sporadic Amyotrophic Lateral Sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef]
- Konopka, A.; Whelan, D.R.; Jamali, S.; Perri, E.; Shahheydari, H.; Toth, R.P.; Parakh, S.; Robinson, T.; Cheong, A.; Mehta, P.; et al. Impaired NHEJ repair in amyotrophic lateral sclerosis is associated with TDP-43 mutations. Mol. Neurodegener. 2020, 15, 1–28. [Google Scholar] [CrossRef]
- Riemslagh, F.W.; van der Toorn, E.C.; Verhagen, R.F.M.; Maas, A.; Bosman, L.W.J.; Hukema, R.K.; Willemsen, R. Inducible expression of human C9ORF72 36 × G4C2 hexanucleotide repeats is sufficient to cause RAN translation and rapid muscular atrophy in mice. Dis. Model. Mech. 2021, 14, dmm044842. [Google Scholar] [CrossRef]
- Herranz-Martin, S.; Chandran, J.; Lewis, K.; Mulcahy, P.; Higginbottom, A.; Walker, C.; Valenzuela, I.M.-P.Y.; Jones, R.A.; Coldicott, I.; Iannitti, T.; et al. Viral delivery of C9orf72 hexanucleotide repeat expansions in mice leads to repeat-length-dependent neuropathology and behavioural deficits. Dis. Model. Mech. 2017, 10, 859–868. [Google Scholar] [CrossRef] [Green Version]
- Chew, J.; Gendron, T.F.; Prudencio, M.; Sasaguri, H.; Zhang, Y.-J.; Castanedes-Casey, M.; Lee, C.W.; Jansen-West, K.; Kurti, A.; Murray, M.E.; et al. C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science 2015, 348, 1151–1154. [Google Scholar] [CrossRef] [Green Version]
- Schludi, M.H.; Becker, L.; Garrett, L.; Gendron, T.F.; Zhou, Q.; Schreiber, F.; Popper, B.; Dimou, L.; Strom, T.M.; Winkelmann, J.; et al. Spinal poly-GA inclusions in a C9orf72 mouse model trigger motor deficits and inflammation without neuron loss. Acta Neuropathol. 2017, 134, 241–254. [Google Scholar] [CrossRef]
- Liu, Y.; Pattamatta, A.; Zu, T.; Reid, T.; Bardhi, O.; Borchelt, D.R.; Yachnis, A.T.; Ranum, L.P. C9orf72 BAC Mouse Model with Motor Deficits and Neurodegenerative Features of ALS/FTD. Neuron 2016, 90, 521–534. [Google Scholar] [CrossRef] [Green Version]
- Koppers, M.; Blokhuis, A.M.; Westeneng, H.; Terpstra, M.L.; Zundel, C.A.C.; De Sá, R.V.; Schellevis, R.D.; Waite, A.J.; Blake, D.J.; Veldink, J.H.; et al. C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann. Neurol. 2015, 78, 426–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, W.; Zhang, L.; Sun, C.; Gao, X.; Guan, F.; Li, J.; Chen, W.; Ma, Y.; Zhang, L. Knock in of a hexanucleotide repeat expansion in the C9orf72 gene induces ALS in rats. Anim. Model. Exp. Med. 2020, 3, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Jager, W.D.H. Experimental amyotrophic lateral sclerosis in the Guinea-Pig. J. Neurol. Sci. 1985, 67, 133–142. [Google Scholar] [CrossRef]
- Smith, R.; Engelhardt, J.I.; Tajti, J.; Appel, S.H. Experimental immune-mediated motor neuron diseases: Models for human ALS. Brain Res. Bull. 1993, 30, 373–380. [Google Scholar] [CrossRef]
- Appel, S.H.; Engelhardt, J.I.; Garcia, J.; Stefani, E. Immunoglobulins from animal models of motor neuron disease and from human amyotrophic lateral sclerosis patients passively transfer physiological abnormalities to the neuromuscular junction. Proc. Natl. Acad. Sci. USA 1991, 88, 647–651. [Google Scholar] [CrossRef] [Green Version]
- Engelhardt, J.I.; Siklós, L.; Appel, S.H. Altered Calcium Homeostasis and Ultrastructure in Motoneurons of Mice Caused by Passively Transferred Anti-motoneuronal IgG. J. Neuropathol. Exp. Neurol. 1997, 56, 21–39. [Google Scholar] [CrossRef] [Green Version]
- Obál, I.; Nógrádi, B.; Meszlényi, V.; Patai, R.; Ricken, G.; Kovacs, G.G.; Tripolszki, K.; Széll, M.; Siklós, L.; Engelhardt, J.I. Experimental Motor Neuron Disease Induced in Mice with Long-Term Repeated Intraperitoneal Injections of Serum from ALS Patients. Int. J. Mol. Sci. 2019, 20, 2573. [Google Scholar] [CrossRef] [Green Version]
- Nardone, R.; Höller, Y.; Taylor, A.C.; Lochner, P.; Tezzon, F.; Golaszewski, S.; Brigo, F.; Trinka, E. Canine degenerative myelopathy: A model of human amyotrophic lateral sclerosis. Zoology 2016, 119, 64–73. [Google Scholar] [CrossRef]
- Awano, T.; Johnson, G.S.; Wade, C.M.; Katz, M.L.; Johnson, G.C.; Taylor, J.F.; Perloski, M.; Biagi, T.; Baranowska, I.; Long, S.; et al. Genome-wide association analysis reveals aSOD1mutation in canine degenerative myelopathy that resembles amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2009, 106, 2794–2799. [Google Scholar] [CrossRef] [Green Version]
- Golubczyk, D.; Malysz-Cymborska, I.; Kalkowski, L.; Janowski, M.; Coates, J.R.; Wojtkiewicz, J.; Maksymowicz, W.; Walczak, P. The Role of Glia in Canine Degenerative Myelopathy: Relevance to Human Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2019, 56, 5740–5748. [Google Scholar] [CrossRef] [Green Version]
- Wininger, F.; Zeng, R.; Johnson, G.; Katz, M.; Johnson, G.; Bush, W.; Jarboe, J.; Coates, J. Degenerative Myelopathy in a Bernese Mountain Dog with a Novel SOD1 Missense Mutation. J. Veter. Intern. Med. 2011, 25, 1166–1170. [Google Scholar] [CrossRef]
- Crisp, M.J.; Beckett, J.; Coates, J.R.; Miller, T.M. Canine degenerative myelopathy: Biochemical characterization of superoxide dismutase 1 in the first naturally occurring non-human amyotrophic lateral sclerosis model. Exp. Neurol. 2013, 248, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, S.; Kamatari, Y.O.; Kuwahara, Y.; Hara, H.; Yamato, O.; Maeda, S.; Kamishina, H.; Honda, R. Canine SOD1 harboring E40K or T18S mutations promotes protein aggregation without reducing the global structural stability. PeerJ 2020, 8, e9512. [Google Scholar] [CrossRef]
- Toedebusch, C.M.; Snyder, J.C.; Jones, M.R.; Garcia, V.B.; Johnson, G.C.; Villalón, E.L.; Coates, J.R.; Garcia, M.L. Arginase-1 expressing microglia in close proximity to motor neurons were increased early in disease progression in canine degenerative myelopathy, a model of amyotrophic lateral sclerosis. Mol. Cell. Neurosci. 2018, 88, 148–157. [Google Scholar] [CrossRef]
- Fernández-Trapero, M.; Espejo-Porras, F.; Rodríguez-Cueto, C.; Coates, J.R.; Pérez-Díaz, C.; De Lago, E.; Fernández-Ruiz, J. Upregulation of CB2 receptors in reactive astrocytes in canine degenerative myelopathy, a disease model of amyotrophic lateral sclerosis. Dis. Model. Mech. 2017, 10, 551–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, K.E.; Rasmussen, A.L.; Bennett, W.; King, A.; West, A.K.; Chung, R.S.; Chuah, M.I. Microglia and motor neurons during disease progression in the SOD1G93A mouse model of amyotrophic lateral sclerosis: Changes in arginase1 and inducible nitric oxide synthase. J. Neuroinflammation 2014, 11, 55. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Morrison, B.M.; Li, Y.; Lengacher, S.; Farah, M.H.; Hoffman, P.N.; Liu, Y.; Tsingalia, A.; Jin, L.; Zhang, P.-W.; et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nat. Cell Biol. 2012, 487, 443–448. [Google Scholar] [CrossRef]
- Verdier, J.-M.; Acquatella, I.; Lautier, C.; Devau, G.; Trouche, S.; Lasbleiz, C.; Mestre-Francés, N. Lessons from the analysis of nonhuman primates for understanding human aging and neurodegenerative diseases. Front. Neurosci. 2015, 9, 64. [Google Scholar] [CrossRef] [Green Version]
- Herculano-Houzel, S.; Mota, B.; Wong, P.; Kaas, J.H. Connectivity-driven white matter scaling and folding in primate cerebral cortex. Proc. Natl. Acad. Sci. USA 2010, 107, 19008–19013. [Google Scholar] [CrossRef] [Green Version]
- Mansfield, K. Marmoset models commonly used in biomedical research. Comp. Med. 2003, 53, 383–392. [Google Scholar] [PubMed]
- Sasaki, E.; Suemizu, H.; Shimada, A.; Hanazawa, K.; Oiwa, R.; Kamioka, M.; Tomioka, I.; Sotomaru, Y.; Hirakawa, R.; Eto, T.; et al. Generation of transgenic non-human primates with germline transmission. Nature 2009, 459, 523–527. [Google Scholar] [CrossRef]
- Miller, C.T.; Freiwald, W.A.; Leopold, D.A.; Mitchell, J.F.; Silva, A.C.; Wang, X. Marmosets: A Neuroscientific Model of Human Social Behavior. Neuron 2016, 90, 219–233. [Google Scholar] [CrossRef]
- Borel, F.; Gernoux, G.; Cardozo, B.; Metterville, J.P.; Cabrera, G.T.; Song, L.; Su, Q.; Gao, G.P.; Elmallah, M.K.; Brown, R.H.; et al. Therapeutic rAAVrh10 Mediated SOD1 Silencing in Adult SOD1G93A Mice and Nonhuman Primates. Hum. Gene Ther. 2016, 27, 19–31. [Google Scholar] [CrossRef] [Green Version]
- Borel, F.; Gernoux, G.; Sun, H.; Stock, R.; Blackwood, M.; Brown, R.H., Jr.; Mueller, C. Safe and effective superoxide dismutase 1 silencing using artificial microRNA in macaques. Sci. Transl. Med. 2018, 10, eaau6414. [Google Scholar] [CrossRef] [Green Version]
- Endo, K.; Ishigaki, S.; Masamizu, Y.; Fujioka, Y.; Watakabe, A.; Yamamori, T.; Hatanaka, N.; Nambu, A.; Okado, H.; Katsuno, M.; et al. Silencing of FUS in the common marmoset (Callithrix jacchus) brain via stereotaxic injection of an adeno-associated virus encoding shRNA. Neurosci. Res. 2018, 130, 56–64. [Google Scholar] [CrossRef]
- Uchida, A.; Sasaguri, H.; Kimura, N.; Tajiri, M.; Ohkubo, T.; Ono, F.; Sakaue, F.; Kanai, K.; Hirai, T.; Sano, T.; et al. Non-human primate model of amyotrophic lateral sclerosis with cytoplasmic mislocalization of TDP-43. Brain 2012, 135, 833–846. [Google Scholar] [CrossRef]

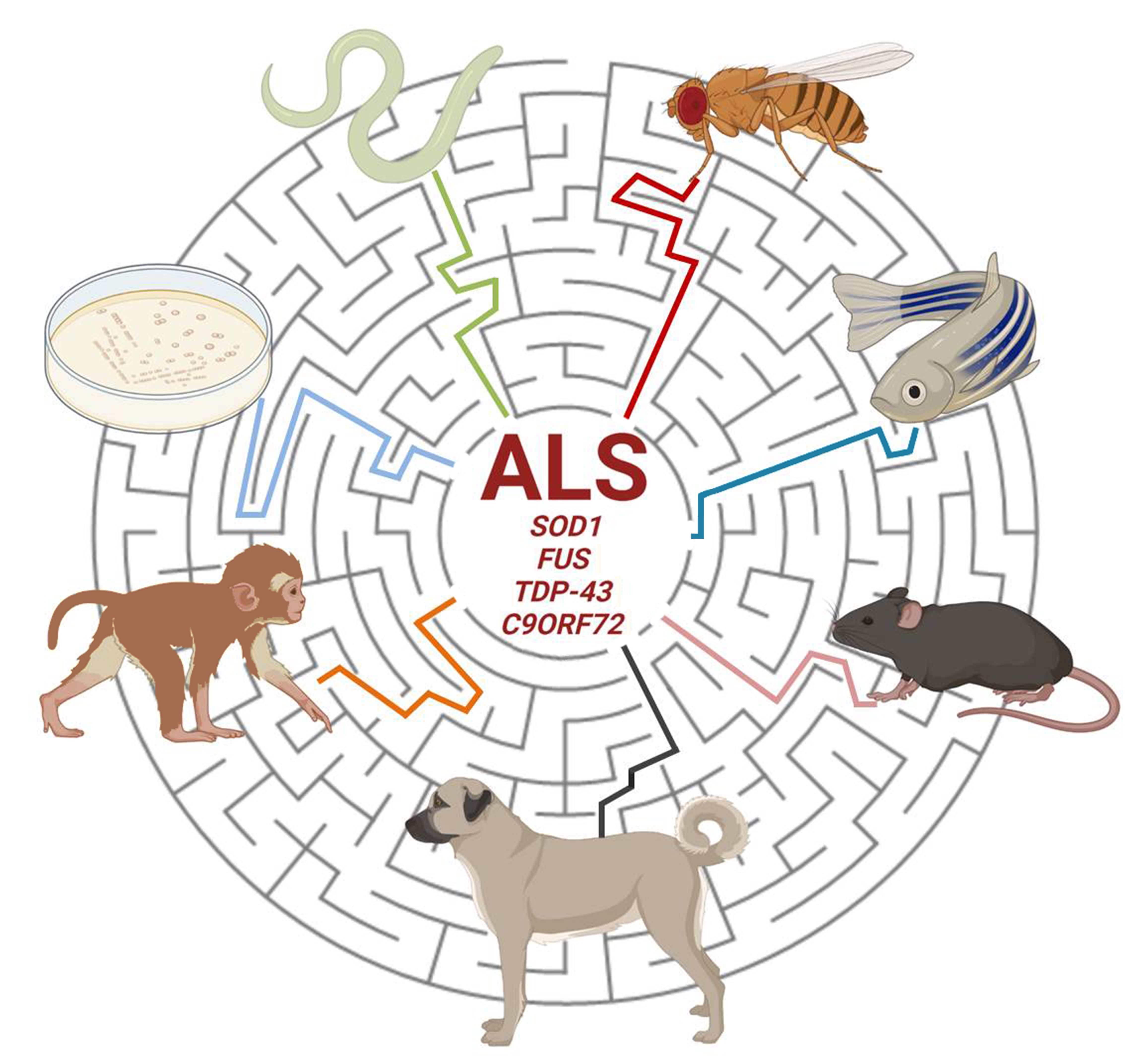{kind=link}
Table 1.
Where and why modeling amyotrophic lateral sclerosis (ALS).
| Where | Why | Why Not |
|---|---|---|
| Cell Lines | Neuron-like phenotype | Lack of system complexity |
| iPSCs | fALS and sALS modeling | Lack of system complexity |
| Yeast | Very suitable for investigating cytotoxicity and proteinopathy aspects | Unicellular organism |
| Worms | Easy and precise investigation of neuronal circuits | Brain and blood absence |
| Flies | Fast and reliable reproduction of ALS symptoms | Anatomical difference with the human nervous system |
| Zebrafish | Nervous system and neurotransmitters similar to humans | Lack of upper motor neurons |
| Rodents | Nervous system and neurotransmitters very similar to humans | High maintenance costs and time-consuming investigation |
| Canines | Spontaneously occurring disease, nervous system as complex as humans | Only early-stage disease tissues available |
| Non-Human Primates | Neuroanatomical, cognitive and functional similarities with humans | Ethical issues |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Liguori, F.; Amadio, S.; Volonté, C. Where and Why Modeling Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2021, 22, 3977. https://doi.org/10.3390/ijms22083977
AMA Style
Liguori F, Amadio S, Volonté C. Where and Why Modeling Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences. 2021; 22(8):3977. https://doi.org/10.3390/ijms22083977
Chicago/Turabian StyleLiguori, Francesco, Susanna Amadio, and Cinzia Volonté. 2021. "Where and Why Modeling Amyotrophic Lateral Sclerosis" International Journal of Molecular Sciences 22, no. 8: 3977. https://doi.org/10.3390/ijms22083977
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.