
DCTN1 Binds to TDP-43 and Regulates TDP-43 Aggregation

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
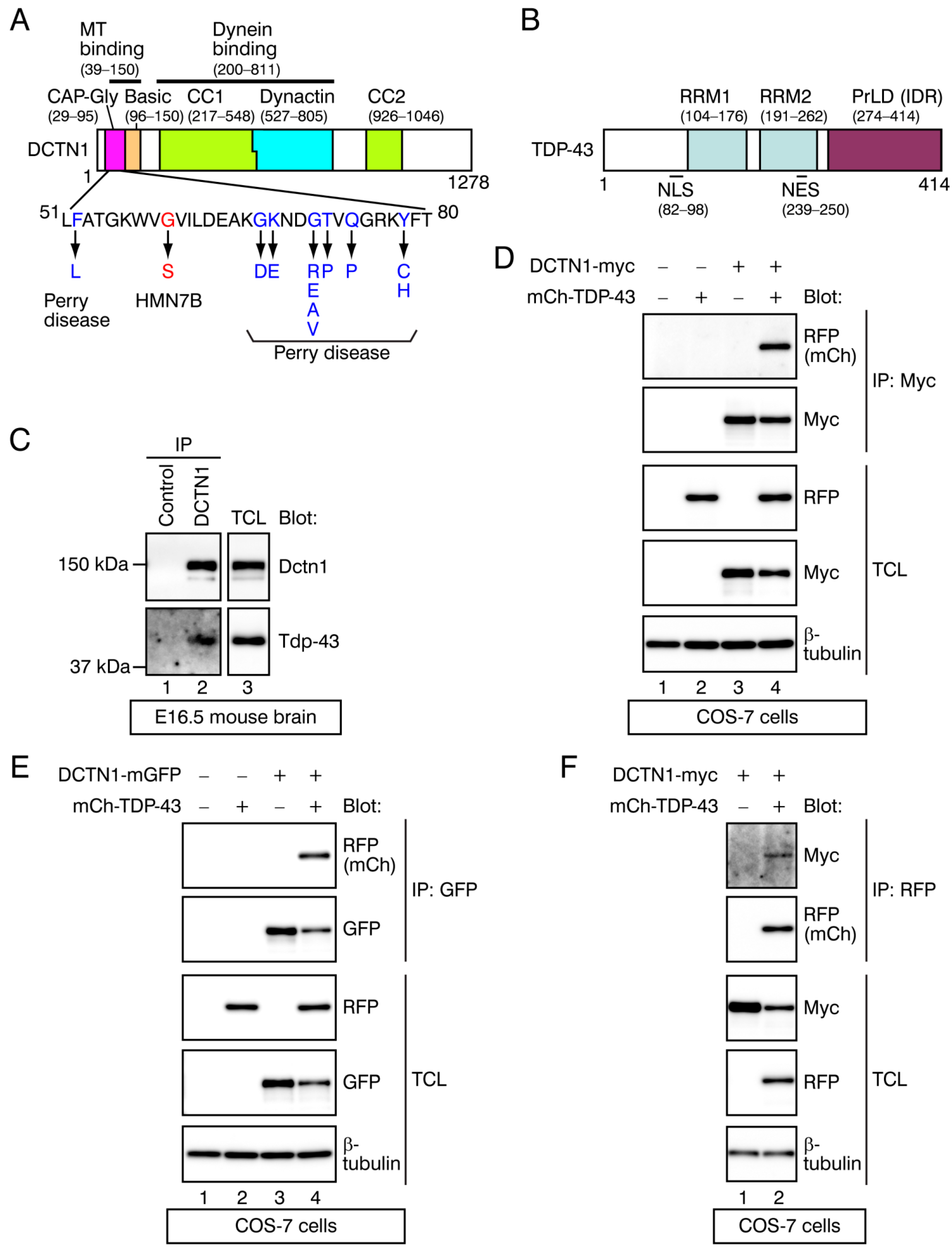
2.1. Identification of TDP-43 as a DCTN1-Interacting Protein
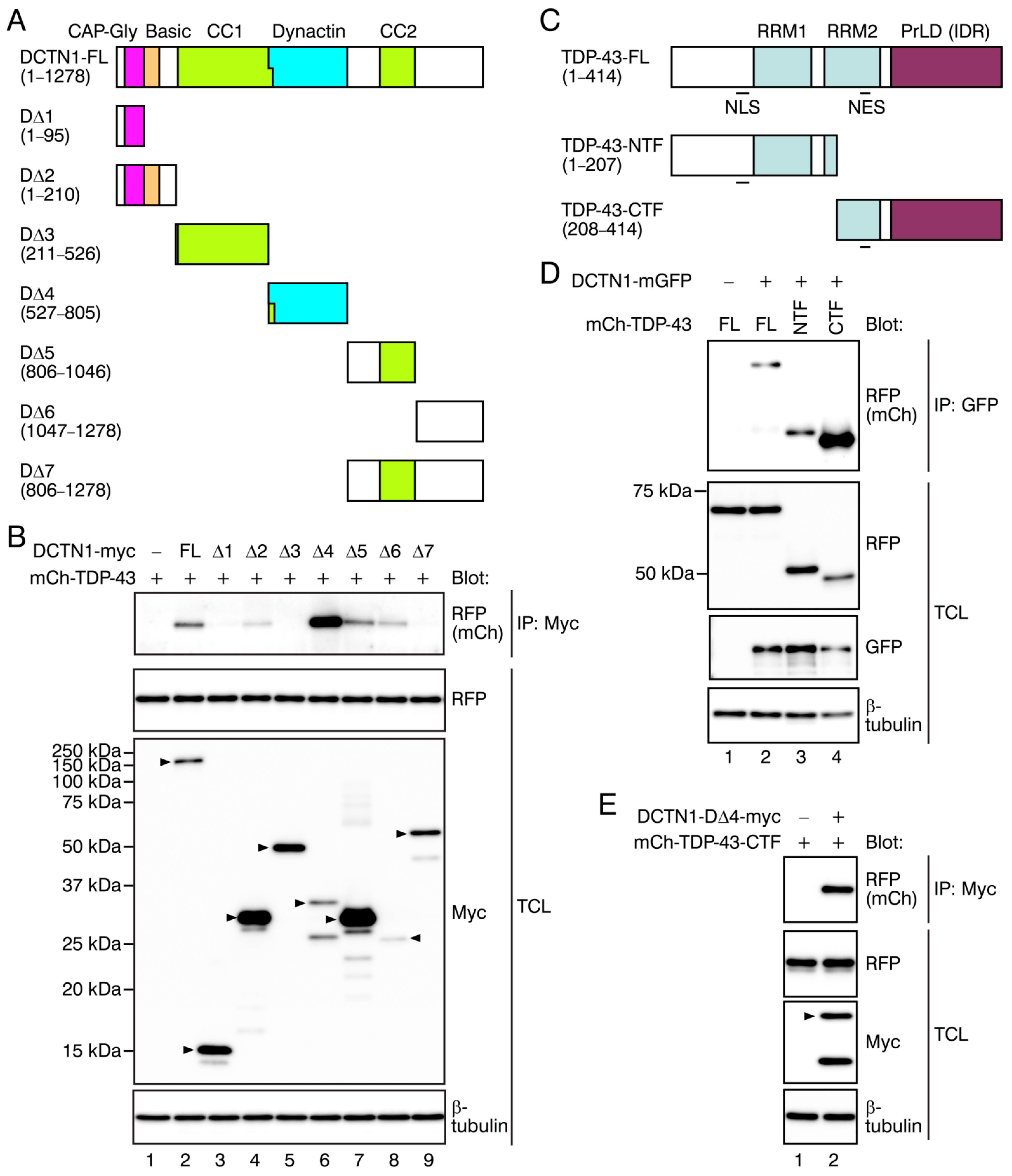
2.2. Determination of the Interacting Regions within DCTN1 and TDP-43
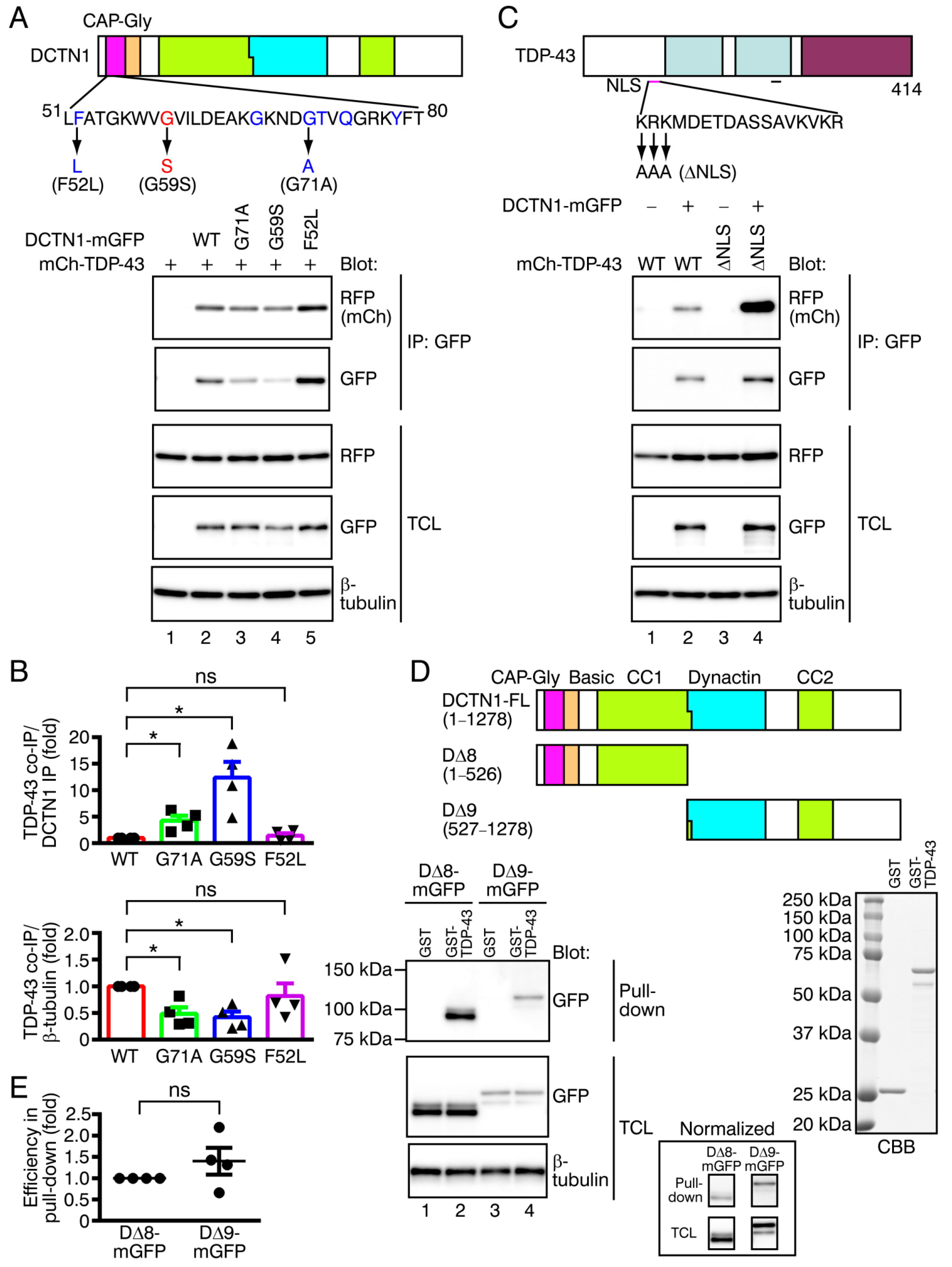
2.3. Effects of Point Mutations in DCTN1 and TDP-43 on Their Interactions
2.4. In Vitro Binding between DCTN1 and TDP-43
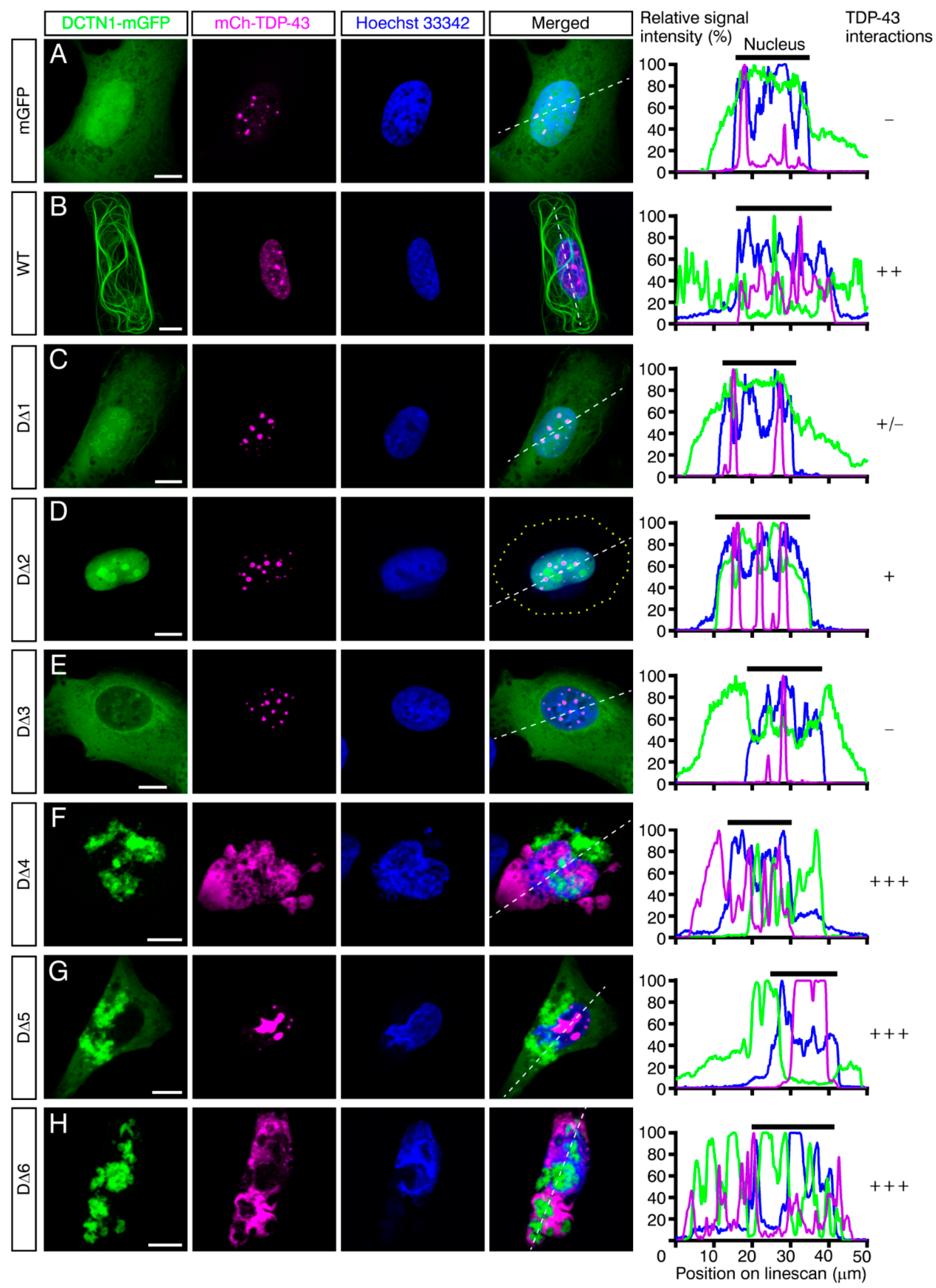
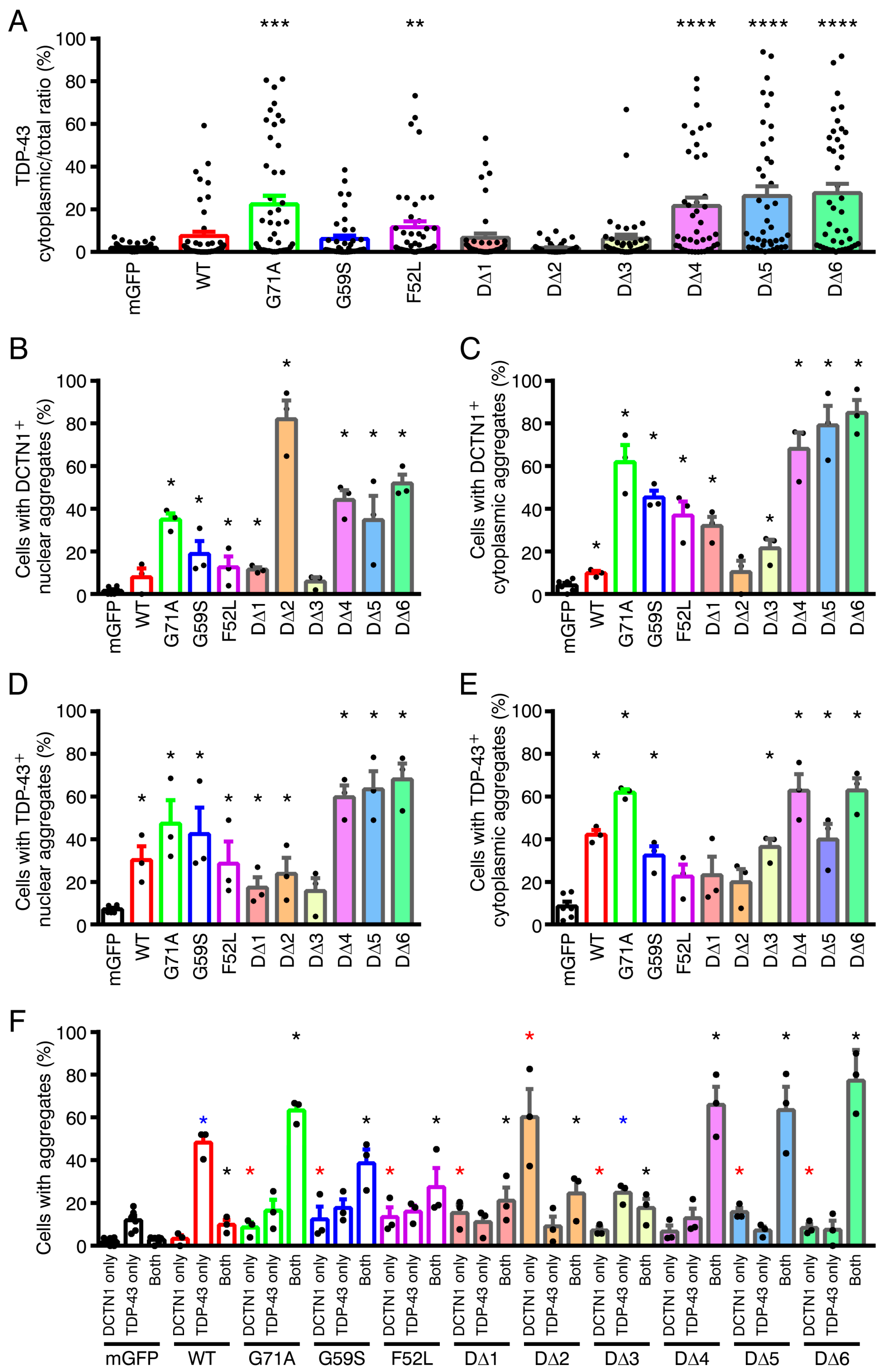
2.5. Effects of Disease-Linked Point Mutants and Truncated Mutants of DCTN1 on the Subcellular Localization and Aggregation of TDP-43
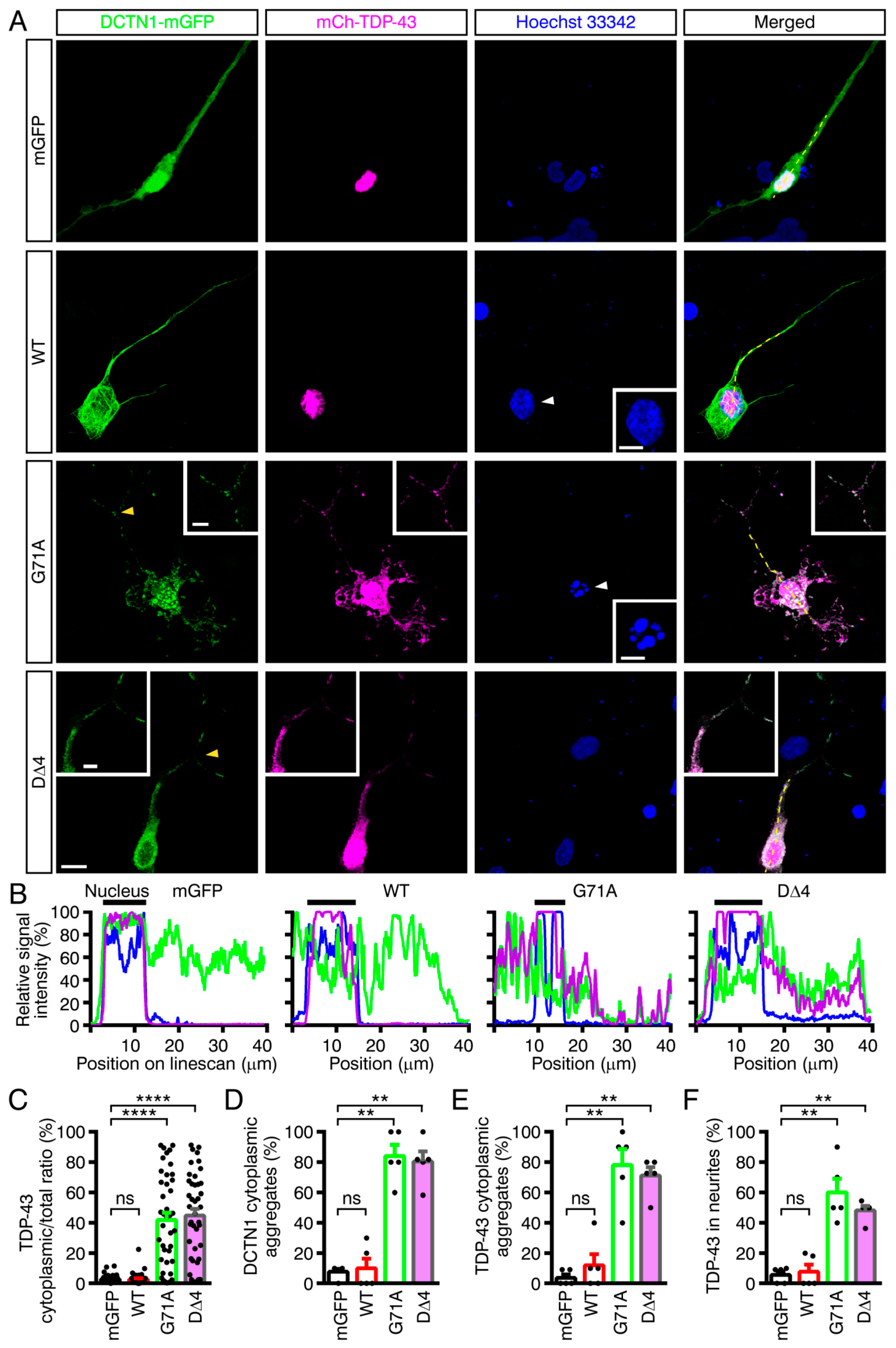
2.6. Modeling Mutant DCTN1-Induced TDP-43 Aggregation in Human iPSC-Derived Neurons
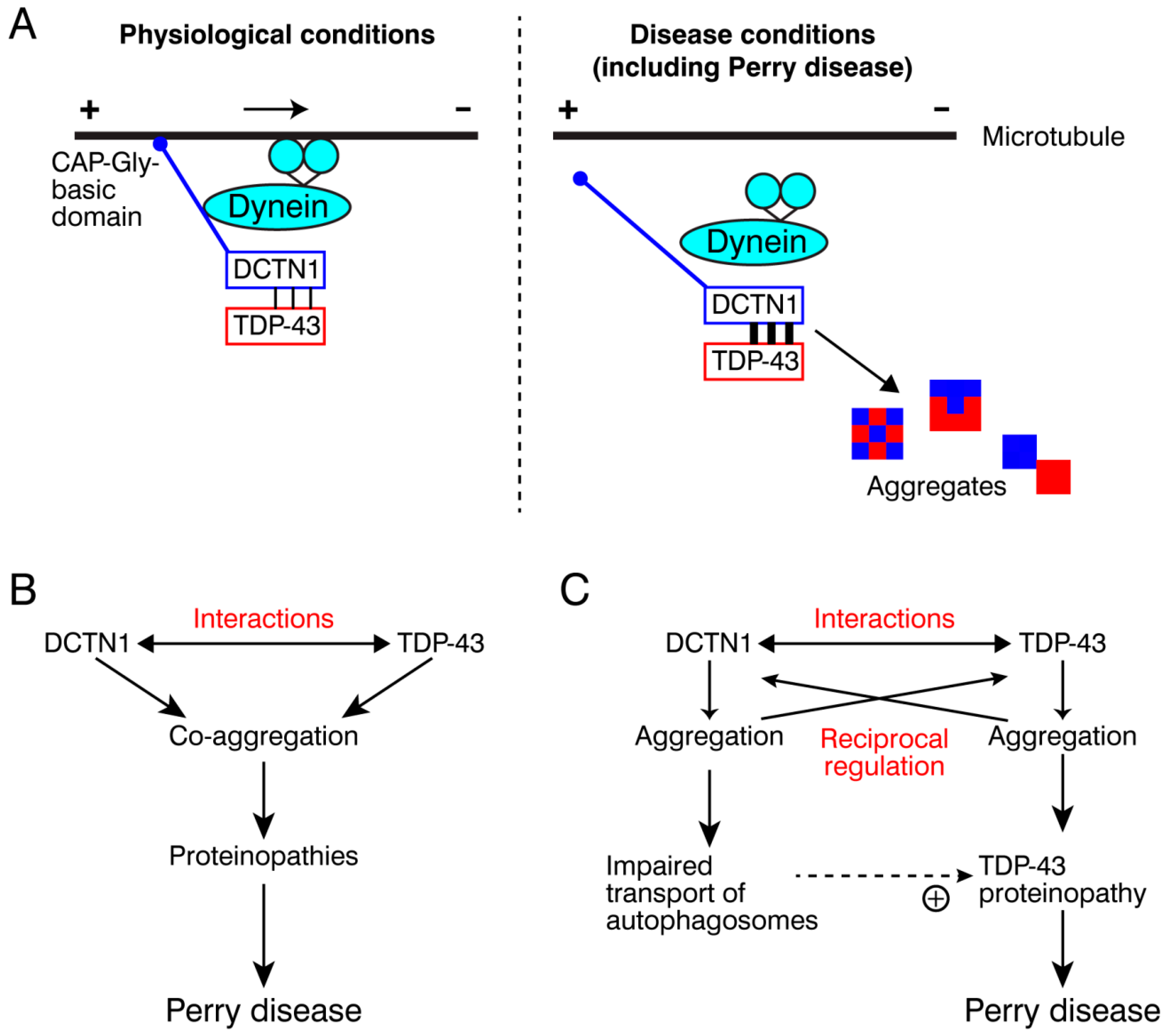
3. Discussion
3.1. DCTN1 Regulates the Cytoplasmic–Nuclear Transport of TDP-43
3.2. How Does Dynein Interact with the DCTN1-TDP-43 Complex?
3.3. Multivalent Interactions between DCTN1 and TDP-43
3.4. How Does DCTN1 Dysregulation Lead to TDP-43 Aggregation?
3.5. Different Effects of Perry Disease- and HMN7B-Linked Mutations on DCTN1 Function
4. Materials and Methods
4.1. Protocol Approval in Biological and Animal Experiments
4.2. Expression Plasmids
4.3. Antibodies
4.4. Cell Culture and Transfection
4.5. Coimmunoprecipitation, GST Pull-Down, and Western Blotting
4.6. Microscopy and Quantification
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ALS | Amyotrophic lateral sclerosis |
| CAP-Gly | Cytoskeleton-associated protein glycine-rich |
| CC | Coiled-coil |
| DCTN1 | Dynactin subunit 1 |
| FTD | Frontotemporal dementia |
| GFP | Green fluorescent protein |
| GST | Glutathione-S-transferase |
| HMN7B | Distal hereditary motor neuropathy 7B |
| IDR | Intrinsically disordered region |
| IP | Immunoprecipitation |
| iPSC | Induced pluripotent stem cell |
| LLPS | Liquid-liquid phase separation |
| MT | Microtubule |
| NCPR | Net charge per residue |
| NES | Nuclear export signal |
| NLS | Nuclear localization signal |
| PCR | Polymerase chain reaction |
| PONDR | Predictor of naturally disordered regions |
| PrLD | Prion-like domain |
| RFP | Red fluorescent protein |
| RNP | Ribonucleoprotein |
| TCL | Total cell lysates |
| TDP-43 | TAR DNA-binding protein 43 |
References
- Schroer, T.A. Dynactin. Annu. Rev. Cell Dev. Biol. 2004, 20, 759–779. [Google Scholar] [CrossRef] [PubMed]
- Olenick, M.A.; Holzbaur, E.L.F. Dynein activators and adaptors at a glance. J. Cell Sci. 2019, 132, jcs227132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holzbaur, E.L.F.; Hammarback, J.A.; Paschal, B.M.; Kravit, N.G.; Pfister, K.K.; Vallee, R.B. Homology of a 150 K cytoplasmic dynein-associated polypeptide with the Drosophila gene Glued. Nat. Cell Biol. 1991, 351, 579–583. [Google Scholar] [CrossRef]
- Gill, S.R.; Schroer, T.A.; Szilak, I.; Steuer, E.R.; Sheetz, M.P.; Cleveland, D.W. Dynactin, a conserved, ubiquitously expressed component of an activator of vesicle motility mediated by cytoplasmic dynein. J. Cell Biol. 1991, 115, 1639–1650. [Google Scholar] [CrossRef]
- Karki, S.; Holzbaur, E.L.F. Affinity Chromatography Demonstrates a Direct Binding between Cytoplasmic Dynein and the Dynactin Complex. J. Biol. Chem. 1995, 270, 28806–28811. [Google Scholar] [CrossRef] [Green Version]
- Vaughan, K.T.; Vallee, R.B. Cytoplasmic dynein binds dynactin through a direct interaction between the intermediate chains and p150Glued. J. Cell Biol. 1995, 131, 1507–1516. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Foster, H.E.; Rondelet, A.; Lacey, S.E.; Bahi-Buisson, N.; Bird, A.W.; Carter, A.P. Cryo-EM Reveals How Human Cytoplasmic Dynein Is Auto-inhibited and Activated. Cell 2017, 169, 1303–1314.e18. [Google Scholar] [CrossRef] [Green Version]
- Urnavicius, L.; Lau, C.K.; Elshenawy, M.M.; Morales-Rios, E.; Motz, C.; Yildiz, A.; Carter, A.P. Cryo-EM shows how dynactin recruits two dyneins for faster movement. Nat. Cell Biol. 2018, 554, 202–206. [Google Scholar] [CrossRef]
- Reck-Peterson, S.L.; Redwine, W.B.; Vale, R.D.; Carter, A.P. The cytoplasmic dynein transport machinery and its many cargoes. Nat. Rev. Mol. Cell Biol. 2018, 19, 382–398. [Google Scholar] [CrossRef]
- Canty, J.T.; Yildiz, A. Activation and Regulation of Cytoplasmic Dynein. Trends Biochem. Sci. 2020, 45, 440–453. [Google Scholar] [CrossRef]
- Waterman-Storer, C.M.; Karki, S.; Holzbaur, E.L. The p150Glued component of the dynactin complex binds to both microtubules and the actin-related protein centractin (Arp-1). Proc. Natl. Acad. Sci. USA 1995, 92, 1634–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaughan, K.T.; Tynan, S.H.; Faulkner, N.E.; Echeverri, C.J.; Vallee, R.B. Colocalization of cytoplasmic dynein with dynactin and CLIP-170 at microtubule distal ends. J. Cell Sci. 1999, 112, 1437–1447. [Google Scholar] [PubMed]
- Moughamian, A.J.; Holzbaur, E.L. Dynactin Is Required for Transport Initiation from the Distal Axon. Neuron 2012, 74, 331–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloyd, T.E.; Machamer, J.; O’Hara, K.; Kim, J.H.; Collins, S.E.; Wong, M.Y.; Sahin, B.; Imlach, W.; Yang, Y.; Levitan, E.S.; et al. The p150Glued CAP-Gly Domain Regulates Initiation of Retrograde Transport at Synaptic Termini. Neuron 2012, 74, 344–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Culver–Hanlon, T.L.; Lex, S.A.; Stephens, A.D.; Quintyne, N.J.; King, S.J. A microtubule-binding domain in dynactin increases dynein processivity by skating along microtubules. Nat. Cell Biol. 2006, 8, 264–270. [Google Scholar] [CrossRef]
- Lazarus, J.E.; Moughamian, A.J.; Tokito, M.K.; Holzbaur, E.L.F. Dynactin Subunit p150Glued Is a Neuron-Specific Anti-Catastrophe Factor. PLoS Biol. 2013, 11, e1001611. [Google Scholar] [CrossRef] [Green Version]
- Goodson, H.V.; Skube, S.B.; Stalder, R.; Valetti, C.; Kreis, T.E.; Morrison, E.E.; Schroer, T.A. CLIP-170 interacts with dynactin complex and the APC-binding protein EB1 by different mechanisms. Cell Motil. Cytoskelet. 2003, 55, 156–173. [Google Scholar] [CrossRef]
- Lomakin, A.J.; Semenova, I.; Zaliapin, I.; Kraikivski, P.; Nadezhdina, E.; Slepchenko, B.M.; Akhmanova, A.; Rodionov, V. CLIP-170-Dependent Capture of Membrane Organelles by Microtubules Initiates Minus-End Directed Transport. Dev. Cell 2009, 17, 323–333. [Google Scholar] [CrossRef] [Green Version]
- Akhmanova, A.; Steinmetz, M.O. Tracking the ends: A dynamic protein network controls the fate of microtubule tips. Nat. Rev. Mol. Cell Biol. 2008, 9, 309–322. [Google Scholar] [CrossRef]
- Winton, M.J.; Igaz, L.M.; Wong, M.M.; Kwong, L.K.; Trojanowski, J.Q.; Lee, V.M.-Y. Disturbance of Nuclear and Cytoplasmic TAR DNA-binding Protein (TDP-43) Induces Disease-like Redistribution, Sequestration, and Aggregate Formation. J. Biol. Chem. 2008, 283, 13302–13309. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Kim, H.J.; Wang, H.; Monaghan, J.; Freyermuth, F.; Sung, J.C.; O’Donovan, K.; Fare, C.M.; Diaz, Z.; Singh, N.; et al. Nuclear-Import Receptors Reverse Aberrant Phase Transitions of RNA-Binding Proteins with Prion-like Domains. Cell 2018, 173, 677–692.e20. [Google Scholar] [CrossRef] [Green Version]
- Engelender, S.; Sharp, A.H.; Colomer, V.; Tokito, M.K.; Lanahan, A.; Worley, P.F.; Holzbaur, E.L.; Ross, C.A. Huntingtin-associated protein 1 (HAP1) interacts with the p150Glued subunit of dynactin. Hum. Mol. Genet. 1997, 6, 2205–2212. [Google Scholar] [CrossRef] [Green Version]
- Li, S.-H.; Gutekunst, C.-A.; Hersch, S.M.; Li, X.-J. Interaction of Huntingtin-Associated Protein with Dynactin P150Glued. J. Neurosci. 1998, 18, 1261–1269. [Google Scholar] [CrossRef] [Green Version]
- Caviston, J.P.; Ross, J.L.; Antony, S.M.; Tokito, M.; Holzbaur, E.L.F. Huntingtin facilitates dynein/dynactin-mediated vesicle transport. Proc. Natl. Acad. Sci. USA 2007, 104, 10045–10050. [Google Scholar] [CrossRef] [Green Version]
- Caviston, J.P.; Holzbaur, E.L.F. Huntingtin as an essential integrator of intracellular vesicular trafficking. Trends Cell Biol. 2009, 19, 147–155. [Google Scholar] [CrossRef] [Green Version]
- Wong, Y.C.; Holzbaur, E.L.F. The Regulation of Autophagosome Dynamics by Huntingtin and HAP1 Is Disrupted by Expression of Mutant Huntingtin, Leading to Defective Cargo Degradation. J. Neurosci. 2014, 34, 1293–1305. [Google Scholar] [CrossRef] [Green Version]
- Ikenaka, K.; Kawai, K.; Katsuno, M.; Huang, Z.; Jiang, Y.-M.; Iguchi, Y.; Kobayashi, K.; Kimata, T.; Waza, M.; Tanaka, F.; et al. dnc-1/dynactin 1 Knockdown Disrupts Transport of Autophagosomes and Induces Motor Neuron Degeneration. PLoS ONE 2013, 8, e54511. [Google Scholar] [CrossRef] [Green Version]
- Ratti, A.; Buratti, E. Physiological functions and pathobiology of TDP-43 and FUS/TLS proteins. J. Neurochem. 2016, 138, 95–111. [Google Scholar] [CrossRef]
- Birsa, N.; Bentham, M.P.; Fratta, P. Cytoplasmic functions of TDP-43 and FUS and their role in ALS. Semin. Cell Dev. Biol. 2020, 99, 193–201. [Google Scholar] [CrossRef]
- Ayala, Y.M.; Zago, P.; D’Ambrogio, A.; Xu, Y.-F.; Petrucelli, L.; Buratti, E.; Baralle, F.E. Structural determinants of the cellular localization and shuttling of TDP-43. J. Cell Sci. 2008, 121, 3778–3785. [Google Scholar] [CrossRef] [Green Version]
- Johnson, B.S.; Snead, D.; Lee, J.J.; McCaffery, J.M.; Shorter, J.; Gitler, A.D. TDP-43 Is Intrinsically Aggregation-prone, and Amyotrophic Lateral Sclerosis-linked Mutations Accelerate Aggregation and Increase Toxicity. J. Biol. Chem. 2009, 284, 20329–20339. [Google Scholar] [CrossRef] [Green Version]
- Boeynaems, S.; Alberti, S.; Fawzi, N.L.; Mittag, T.; Polymenidou, M.; Rousseau, F.; Schymkowitz, J.; Shorter, J.; Wolozin, B.; Bosch, L.V.D.; et al. Protein Phase Separation: A New Phase in Cell Biology. Trends Cell Biol. 2018, 28, 420–435. [Google Scholar] [CrossRef] [Green Version]
- Uyar, B.; Weatheritt, R.J.; Dinkel, H.; Davey, N.E.; Gibson, T.J. Proteome-wide analysis of human disease mutations in short linear motifs: Neglected players in cancer? Mol. BioSyst. 2014, 10, 2626–2642. [Google Scholar] [CrossRef] [Green Version]
- Tziortzouda, P.; Bosch, L.V.D.; Hirth, F. Triad of TDP43 control in neurodegeneration: Autoregulation, localization and aggregation. Nat. Rev. Neurosci. 2021, 22, 197–208. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef]
- Kwong, L.K.; Neumann, M.; Sampathu, D.M.; Lee, V.M.-Y.; Trojanowski, J.Q. TDP-43 proteinopathy: The neuropathology underlying major forms of sporadic and familial frontotemporal lobar degeneration and motor neuron disease. Acta Neuropathol. 2007, 114, 63–70. [Google Scholar] [CrossRef]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 Mutations in Familial and Sporadic Amyotrophic Lateral Sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef]
- Kabashi, E.; Valdmanis, P.N.; Dion, P.A.; Spiegelman, D.; McConkey, B.J.; Velde, C.V.; Bouchard, J.-P.; Lacomblez, L.; Pochigaeva, K.; Salachas, F.; et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 2008, 40, 572–574. [Google Scholar] [CrossRef]
- Ling, S.-C.; Polymenidou, M.; Cleveland, D.W. Converging Mechanisms in ALS and FTD: Disrupted RNA and Protein Homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef] [Green Version]
- Dugger, B.N.; Dickson, D.W. Pathology of Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035. [Google Scholar] [CrossRef]
- Schiavo, G.; Greensmith, L.; Hafezparast, M.; Fisher, E.M. Cytoplasmic dynein heavy chain: The servant of many masters. Trends Neurosci. 2013, 36, 641–651. [Google Scholar] [CrossRef]
- Moughamian, A.J.; Holzbaur, E.L. Cytoplasmic dynein dysfunction and neurodegenerative disease. Dyneins 2018, 2, 286–315. [Google Scholar] [CrossRef]
- Puls, I.; Jonnakuty, C.; LaMonte, B.H.; Holtzbaur, E.L.F.; Tokito, M.; Mann, E.; Floeter, M.K.; Bidus, K.; Drayna, D.; Oh, S.J.; et al. Mutant dynactin in motor neurons disease. Nat. Genet. 2003, 33, 455–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, T.L.; Bratty, P.J.A.; Hansen, S.; Kennedy, J.; Urquhart, N.; Dolman, C.L. Hereditary Mental Depression and Parkinsonism With Taurine Deficiency. Arch. Neurol. 1975, 32, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Tsuboi, Y.; Wszolek, Z.K.; Kusuhara, T.; Doh-Ura, K.; Yamada, T. Japanese family with parkinsonism, depression, weight loss, and central hypoventilation. Neurology 2002, 58, 1025–1030. [Google Scholar] [CrossRef] [PubMed]
- Farrer, M.J.; Hulihan, M.M.; Kachergus, J.M.; Dächsel, J.C.; Stoessl, A.J.; Grantier, L.L.; Calne, S.; Calne, D.B.; Lechevalier, B.; Chapon, F.; et al. DCTN1 mutations in Perry syndrome. Nat. Genet. 2009, 41, 163–165. [Google Scholar] [CrossRef]
- Wider, C.; Dickson, D.W.; Stoessl, A.J.; Tsuboi, Y.; Chapon, F.; Gutmann, L.; Lechevalier, B.; Calne, D.B.; Personett, D.A.; Hulihan, M.; et al. Pallidonigral TDP-43 pathology in Perry syndrome. Park. Relat. Disord. 2009, 15, 281–286. [Google Scholar] [CrossRef] [Green Version]
- Mishima, T.; Fujioka, S.; Tomiyama, H.; Yabe, I.; Kurisaki, R.; Fujii, N.; Neshige, R.; Ross, O.A.; Farrer, M.J.; Dickson, D.W.; et al. Establishing diagnostic criteria for Perry syndrome. J. Neurol. Neurosurg. Psychiatry 2017, 89, 482–487. [Google Scholar] [CrossRef] [Green Version]
- Mishima, T.; Koga, S.; Lin, W.-L.; Kasanuki, K.; Castanedes-Casey, M.; Wszolek, Z.K.; Oh, S.J.; Tsuboi, Y.; Dickson, D.W. Perry Syndrome: A Distinctive Type of TDP-43 Proteinopathy. J. Neuropathol. Exp. Neurol. 2017, 76, 676–682. [Google Scholar] [CrossRef]
- Čierny, M.; Hooshmand, S.I.; Fee, D.; Tripathi, S.; Dsouza, N.R.; Kirschner, A.L.P.; Zimmermann, M.T.; Brennan, R. Novel destabilizing Dynactin variant (DCTN1 p.Tyr78His) in patient with Perry syndrome. Park. Relat. Disord. 2020, 77, 110–113. [Google Scholar] [CrossRef]
- Hosaka, Y.; Inoshita, T.; Shiba-Fukushima, K.; Cui, C.; Arano, T.; Imai, Y.; Hattori, N. Reduced TDP-43 Expression Improves Neuronal Activities in a Drosophila Model of Perry Syndrome. EBioMedicine 2017, 21, 218–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, J.; Milpetz, F.; Bork, P.; Ponting, C.P. SMART, a simple modular architecture research tool: Identification of signaling domains. Proc. Natl. Acad. Sci. USA 1998, 95, 5857–5864. [Google Scholar] [CrossRef] [Green Version]
- Trigg, J.; Gutwin, K.; Keating, A.E.; Berger, B. Multicoil2: Predicting Coiled Coils and Their Oligomerization States from Sequence in the Twilight Zone. PLoS ONE 2011, 6, e23519. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Romero, P.; Rani, M.; Dunker, A.K.; Obradovic, Z. Predicting Protein Disorder for N-, C-, and Internal Regions. Genome Inform. 1999, 10, 30–40. [Google Scholar]
- Holehouse, A.S.; Das, R.K.; Ahad, J.N.; Richardson, M.O.; Pappu, R.V. CIDER: Resources to Analyze Sequence-Ensemble Relationships of Intrinsically Disordered Proteins. Biophys. J. 2017, 112, 16–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anurag, M.; Singh, G.P.; Dash, D. Location of disorder in coiled coilproteins is influenced by its biological role and subcellular localization: A GO-based study on human proteome. Mol. BioSyst. 2011, 8, 346–352. [Google Scholar] [CrossRef]
- King, S.J.; Brown, C.L.; Maier, K.C.; Quintyne, N.J.; Schroer, T.A. Analysis of the Dynein-Dynactin Interaction In Vitro and In Vivo. Mol. Biol. Cell 2003, 14, 5089–5097. [Google Scholar] [CrossRef]
- Igaz, L.M.; Kwong, L.K.; Chen-Plotkin, A.; Winton, M.J.; Unger, T.L.; Xu, Y.; Neumann, M.; Trojanowski, J.Q.; Lee, V.M.-Y. Expression of TDP-43 C-terminal Fragments in Vitro Recapitulates Pathological Features of TDP-43 Proteinopathies. J. Biol. Chem. 2009, 284, 8516–8524. [Google Scholar] [CrossRef] [Green Version]
- Chou, C.-C.; Zhang, Y.; Umoh, M.E.; Vaughan, S.W.; Lorenzini, I.; Liu, F.; Sayegh, M.; Donlin-Asp, P.G.; Chen, Y.H.; Duong, D.M.; et al. TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat. Neurosci. 2018, 21, 228–239. [Google Scholar] [CrossRef] [Green Version]
- Lai, C.; Lin, X.; Chandran, J.; Shim, H.; Yang, W.-J.; Cai, H. The G59S Mutation in p150glued Causes Dysfunction of Dynactin in Mice. J. Neurosci. 2007, 27, 13982–13990. [Google Scholar] [CrossRef] [PubMed]
- Araki, E.; Tsuboi, Y.; Daechsel, J.; Milnerwood, A.; Vilariño-Güell, C.; Fujii, N.; Mishima, T.; Oka, T.; Hara, H.; Fukae, J.; et al. A NovelDCTN1mutation with late-onset parkinsonism and frontotemporal atrophy. Mov. Disord. 2014, 29, 1201–1204. [Google Scholar] [CrossRef] [PubMed]
- Gasset-Rosa, F.; Lu, S.; Yu, H.; Chen, C.; Melamed, Z.; Guo, L.; Shorter, J.; Da Cruz, S.; Cleveland, D.W. Cytoplasmic TDP-43 De-mixing Independent of Stress Granules Drives Inhibition of Nuclear Import, Loss of Nuclear TDP-43, and Cell Death. Neuron 2019, 102, 339–357.e7. [Google Scholar] [CrossRef] [Green Version]
- Mishima, T.; Ishikawa, T.; Imamura, K.; Kondo, T.; Koshiba, Y.; Takahashi, R.; Takahashi, J.; Watanabe, A.; Fujii, N.; Tsuboi, Y.; et al. Cytoplasmic aggregates of dynactin in iPSC-derived tyrosine hydroxylase-positive neurons from a patient with Perry syndrome. Park. Relat. Disord. 2016, 30, 67–72. [Google Scholar] [CrossRef]
- Mishima, T.; Fujioka, S.; Fukae, J.; Yuasa-Kawada, J.; Tsuboi, Y. Modeling Parkinson’s Disease and Atypical Parkinsonian Syndromes Using Induced Pluripotent Stem Cells. Int. J. Mol. Sci. 2018, 19, 3870. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Pak, C.; Han, Y.; Ahlenius, H.; Zhang, Z.; Chanda, S.; Marro, S.; Patzke, C.; Acuna, C.; Covy, J.; et al. Rapid Single-Step Induction of Functional Neurons from Human Pluripotent Stem Cells. Neuron 2013, 78, 785–798. [Google Scholar] [CrossRef] [Green Version]
- Matsushita, M.; Nakatake, Y.; Arai, I.; Ibata, K.; Kohda, K.; Goparaju, S.K.; Murakami, M.; Sakota, M.; Chikazawa-Nohtomi, N.; Ko, S.B.; et al. Neural differentiation of human embryonic stem cells induced by the transgene-mediated overexpression of single transcription factors. Biochem. Biophys. Res. Commun. 2017, 490, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, F.K.; Okamoto, S.; Mitsui, J.; Sone, T.; Ishikawa, M.; Yamamoto, Y.; Kanegae, Y.; Nakatake, Y.; Imaizumi, K.; Ishiura, H.; et al. The pathogenesis linked to coenzyme Q10 insufficiency in iPSC-derived neurons from patients with multiple-system atrophy. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Ishikawa, M.; Fujimori, K.; Maeda, T.; Kushima, I.; Arioka, Y.; Mori, D.; Nakatake, Y.; Yamagata, B.; Nio, S.; et al. In Vitro Modeling of the Bipolar Disorder and Schizophrenia Using Patient-Derived Induced Pluripotent Stem Cells with Copy Number Variations of PCDH15 and RELN. eNeuro 2019, 6, 0403–0418.2019. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, K.-I.; Saiki, S.; Furuya, N.; Yamada, D.; Imamichi, Y.; Li, Y.; Kawajiri, S.; Sasaki, H.; Koike, M.; Tsuboi, Y.; et al. p150glued-Associated Disorders Are Caused by Activation of Intrinsic Apoptotic Pathway. PLoS ONE 2014, 9, e94645. [Google Scholar] [CrossRef]
- Watanabe, S.; Kaneko, K.; Yamanaka, K. Accelerated Disease Onset with Stabilized Familial Amyotrophic Lateral Sclerosis (ALS)-linked Mutant TDP-43 Proteins. J. Biol. Chem. 2013, 288, 3641–3654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, J.P.; Brown, R.H.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nat. Cell Biol. 2016, 539, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Münch, C.; Sedlmeier, R.; Meyer, T.; Homberg, V.; Sperfeld, A.D.; Kurt, A.; Prudlo, J.; Peraus, G.; Hanemann, C.O.; Stumm, G.; et al. Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology 2004, 63, 724–726. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Seki, N.; Ishiura, H.; Mitsui, J.; Matsukawa, T.; Kishino, A.; Onodera, O.; Aoki, M.; Shimozawa, N.; Murayama, S.; et al. Development of a High-Throughput Microarray-Based Resequencing System for Neurological Disorders and Its Application to Molecular Genetics of Amyotrophic Lateral Sclerosis. Arch. Neurol. 2008, 65, 1326–1332. [Google Scholar] [CrossRef] [PubMed]
- Borg, R.; Wismayer, M.F.; Bonavia, K.; Wismayer, A.F.; Vella, M.; Van Vugt, J.J.F.A.; Kenna, B.J.; Kenna, K.P.; Vassallo, N.; Veldink, J.H.; et al. Genetic analysis of ALS cases in the isolated island population of Malta. Eur. J. Hum. Genet. 2021, 29, 604–614. [Google Scholar] [CrossRef] [PubMed]
- Giannakakou, P.; Sackett, D.L.; Ward, Y.; Webster, K.R.; Blagosklonny, M.V.; Fojo, T. p53 is associated with cellular microtubules and is transported to the nucleus by dynein. Nat. Cell Biol. 2000, 2, 709–717. [Google Scholar] [CrossRef]
- Hanz, S.; Perlson, E.; Willis, D.; Zheng, J.-Q.; Massarwa, R.; Huerta, J.J.; Koltzenburg, M.; Kohler, M.; Van-Minnen, J.; Twiss, J.L.; et al. Axoplasmic Importins Enable Retrograde Injury Signaling in Lesioned Nerve. Neuron 2003, 40, 1095–1104. [Google Scholar] [CrossRef] [Green Version]
- Mikenberg, I.; Widera, D.; Kaus, A.; Kaltschmidt, B.; Kaltschmidt, C. Transcription Factor NF-κB Is Transported to the Nucleus via Cytoplasmic Dynein/Dynactin Motor Complex in Hippocampal Neurons. PLoS ONE 2007, 2, e589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldberg, M.W. Nuclear pore complex tethers to the cytoskeleton. Semin. Cell Dev. Biol. 2017, 68, 52–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, G.D.; Coyaud, É.; Gonçalves, J.; Mojarad, B.A.; Liu, Y.; Wu, Q.; Gheiratmand, L.; Comartin, D.; Tkach, J.M.; Cheung, S.W.; et al. A Dynamic Protein Interaction Landscape of the Human Centrosome-Cilium Interface. Cell 2015, 163, 1484–1499. [Google Scholar] [CrossRef] [Green Version]
- Yoshizawa, T.; Ali, R.; Jiou, J.; Fung, H.Y.J.; Burke, K.A.; Kim, S.J.; Lin, Y.; Peeples, W.B.; Saltzberg, D.; Soniat, M.; et al. Nuclear Import Receptor Inhibits Phase Separation of FUS through Binding to Multiple Sites. Cell 2018, 173, 693–705.e22. [Google Scholar] [CrossRef] [Green Version]
- Hofweber, M.; Hutten, S.; Bourgeois, B.; Spreitzer, E.; Niedner-Boblenz, A.; Schifferer, M.; Ruepp, M.-D.; Simons, M.; Niessing, D.; Madl, T.; et al. Phase Separation of FUS Is Suppressed by Its Nuclear Import Receptor and Arginine Methylation. Cell 2018, 173, 706–719.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qamar, S.; Wang, G.; Randle, S.J.; Ruggeri, F.S.; Varela, J.A.; Lin, J.Q.; Phillips, E.C.; Miyashita, A.; Williams, D.; Ströhl, F.; et al. FUS Phase Separation Is Modulated by a Molecular Chaperone and Methylation of Arginine Cation-π Interactions. Cell 2018, 173, 720–734.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, I.D.; Buchanan, C.N.; Zdradzinski, M.D.; Sahoo, P.K.; Smith, T.P.; Thames, E.; Kar, A.N.; Twiss, J.L. The functional organization of axonal mRNA transport and translation. Nat. Rev. Neurosci. 2021, 22, 77–91. [Google Scholar] [CrossRef]
- Fernandopulle, M.S.; Lippincott-Schwartz, J.; Ward, M.E. RNA transport and local translation in neurodevelopmental and neurodegenerative disease. Nat. Neurosci. 2021, 1–11, in press. [Google Scholar] [CrossRef]
- Alami, N.H.; Smith, R.B.; Carrasco, M.A.; Williams, L.A.; Winborn, C.S.; Han, S.S.; Kiskinis, E.; Winborn, B.; Freibaum, B.D.; Kanagaraj, A.; et al. Axonal Transport of TDP-43 mRNA Granules Is Impaired by ALS-Causing Mutations. Neuron 2014, 81, 536–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polymenidou, M.; Lagier-Tourenne, C.; Hutt, K.R.; Huelga, S.C.; Moran, J.; Liang, T.Y.; Ling, S.-C.; Sun, E.; Wancewicz, E.; Mazur, C.; et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 2011, 14, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Dienstbier, M.; Boehl, F.; Li, X.; Bullock, S.L. Egalitarian is a selective RNA-binding protein linking mRNA localization signals to the dynein motor. Genes Dev. 2009, 23, 1546–1558. [Google Scholar] [CrossRef] [Green Version]
- Deacon, S.W.; Serpinskaya, A.S.; Vaughan, P.S.; Fanarraga, M.L.; Vernos, I.; Vaughan, K.T.; Gelfand, V.I. Dynactin is required for bidirectional organelle transport. J. Cell Biol. 2003, 160, 297–301. [Google Scholar] [CrossRef]
- Urnavicius, L.; Zhang, K.; Diamant, A.G.; Motz, C.; Schlager, M.A.; Yu, M.; Patel, N.A.; Robinson, C.V.; Carter, A.P. The structure of the dynactin complex and its interaction with dynein. Science 2015, 347, 1441–1446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.J.; Taylor, J.P. Lost in Transportation: Nucleocytoplasmic Transport Defects in ALS and Other Neurodegenerative Diseases. Neuron 2017, 96, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita-Kawada, M.; Hasegawa, H.; Hongu, T.; Yanagi, S.; Kanaho, Y.; Masai, I.; Mishima, T.; Chen, X.; Tsuboi, Y.; Rao, Y.; et al. A crucial role for Arf6 in the response of commissural axons to Slit. Development 2019, 146, dev172106. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deshimaru, M.; Kinoshita-Kawada, M.; Kubota, K.; Watanabe, T.; Tanaka, Y.; Hirano, S.; Ishidate, F.; Hiramoto, M.; Ishikawa, M.; Uehara, Y.; et al. DCTN1 Binds to TDP-43 and Regulates TDP-43 Aggregation. Int. J. Mol. Sci. 2021, 22, 3985. https://doi.org/10.3390/ijms22083985
Deshimaru M, Kinoshita-Kawada M, Kubota K, Watanabe T, Tanaka Y, Hirano S, Ishidate F, Hiramoto M, Ishikawa M, Uehara Y, et al. DCTN1 Binds to TDP-43 and Regulates TDP-43 Aggregation. International Journal of Molecular Sciences. 2021; 22(8):3985. https://doi.org/10.3390/ijms22083985
Chicago/Turabian StyleDeshimaru, Manami, Mariko Kinoshita-Kawada, Kaori Kubota, Takuya Watanabe, Yasuyoshi Tanaka, Saito Hirano, Fumiyoshi Ishidate, Masaki Hiramoto, Mitsuru Ishikawa, Yoshinari Uehara, and et al. 2021. "DCTN1 Binds to TDP-43 and Regulates TDP-43 Aggregation" International Journal of Molecular Sciences 22, no. 8: 3985. https://doi.org/10.3390/ijms22083985