
Rapidly Growing Protein-Centric Technologies to Extensively Identify Protein–RNA Interactions: Application to the Analysis of Co-Transcriptional RNA Processing


Abstract
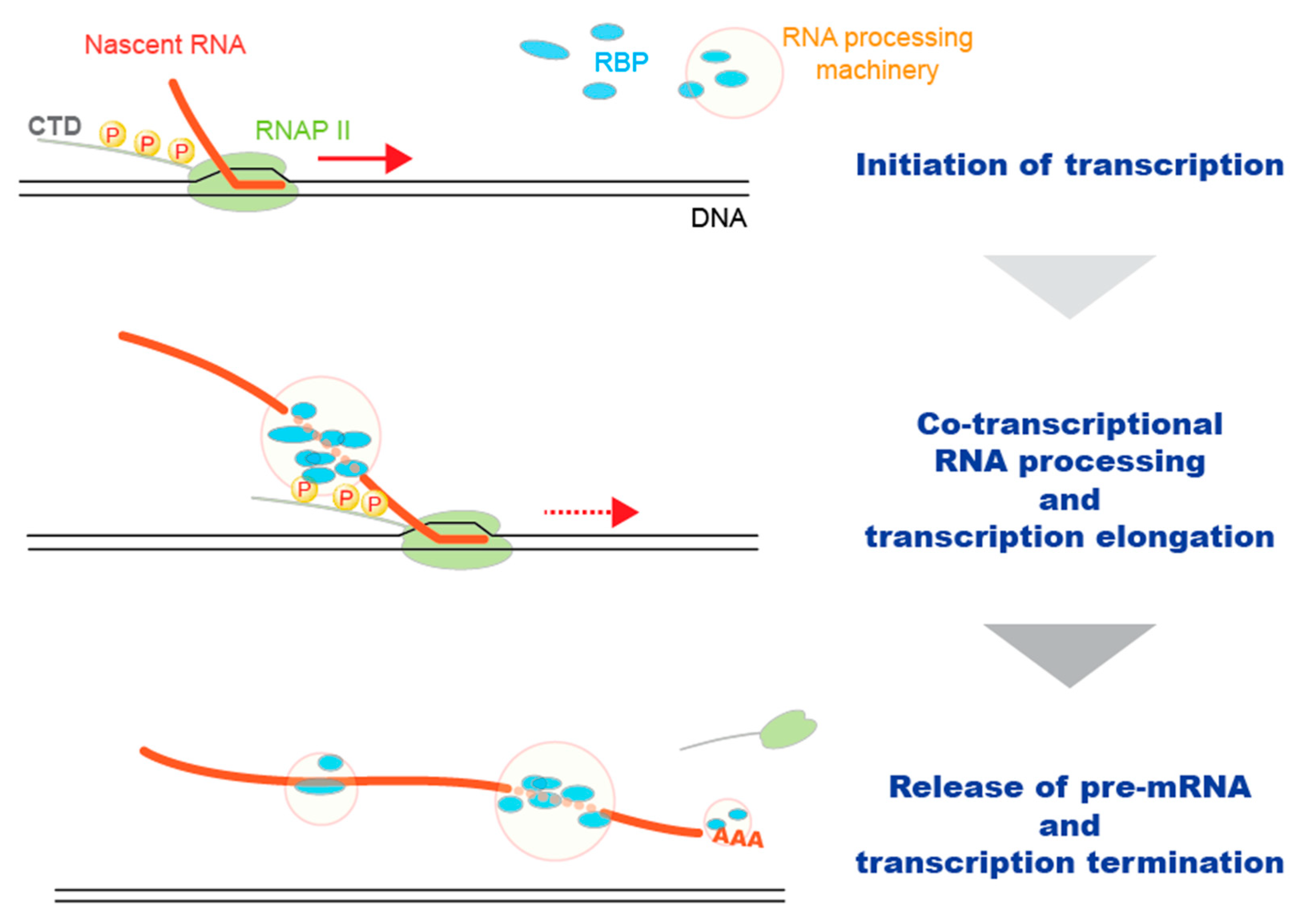
:1. Introduction
2. General Methods to Identify Protein–RNA Interaction Sites in RNA
2.1. Development of Methods to Detect Protein–RNA Interaction Sites
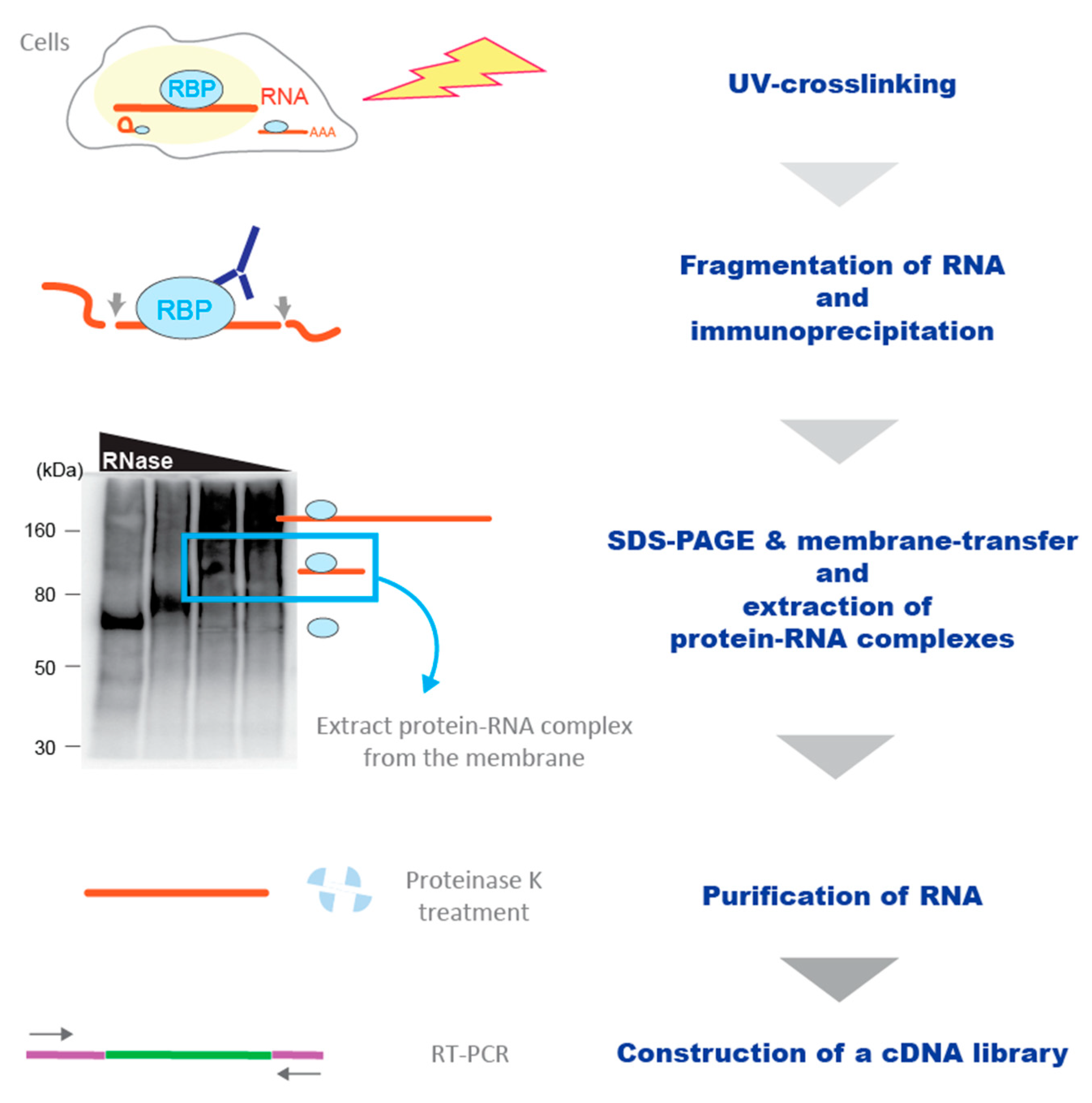
2.2. Crosslinking and Immunoprecipitation (CLIP)
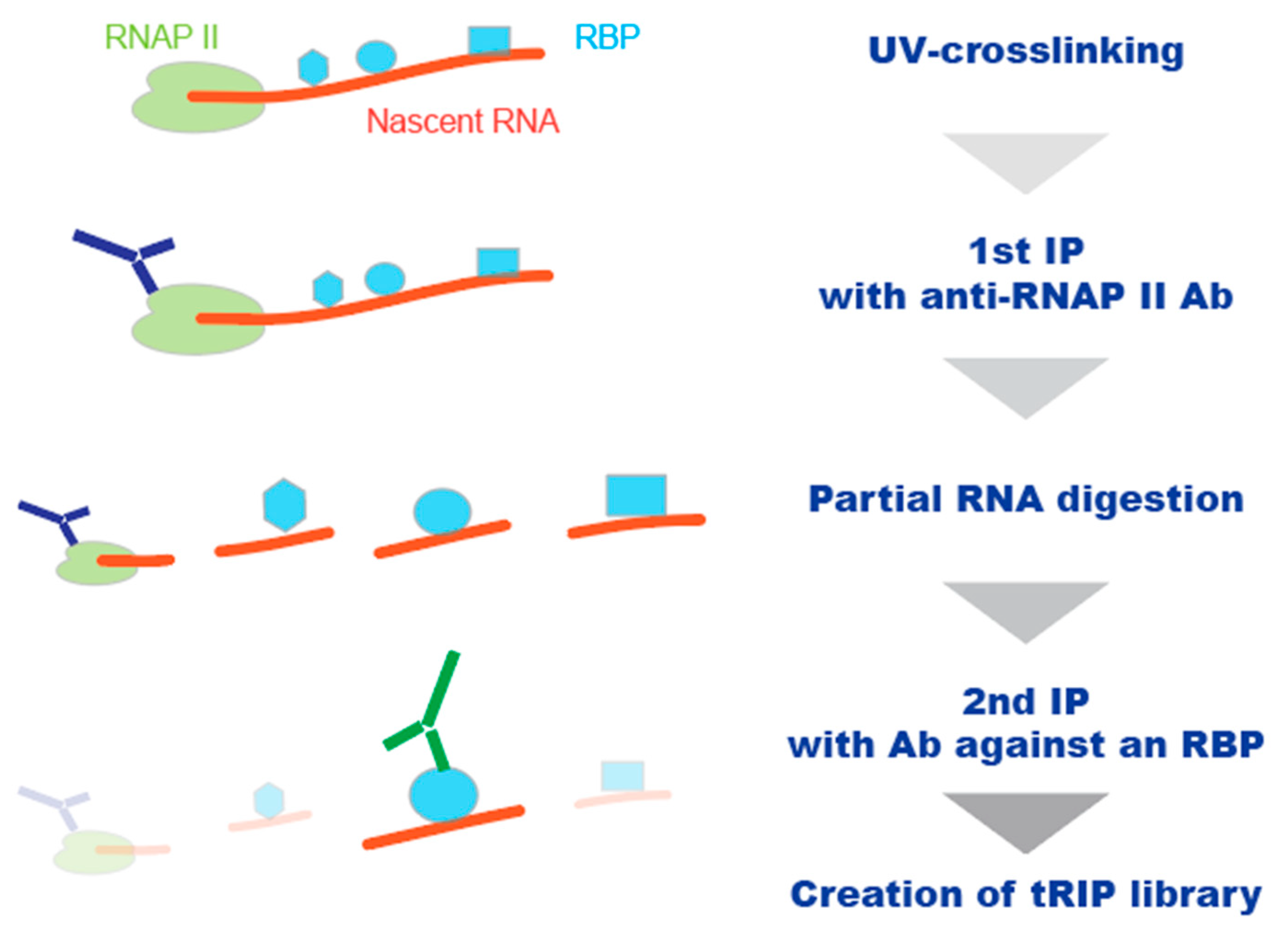
2.3. Enhancement of the Sensitivity of the CLIP Methodology
3. Other Strategies to Detect Protein–RNA Interactions in Living Cells
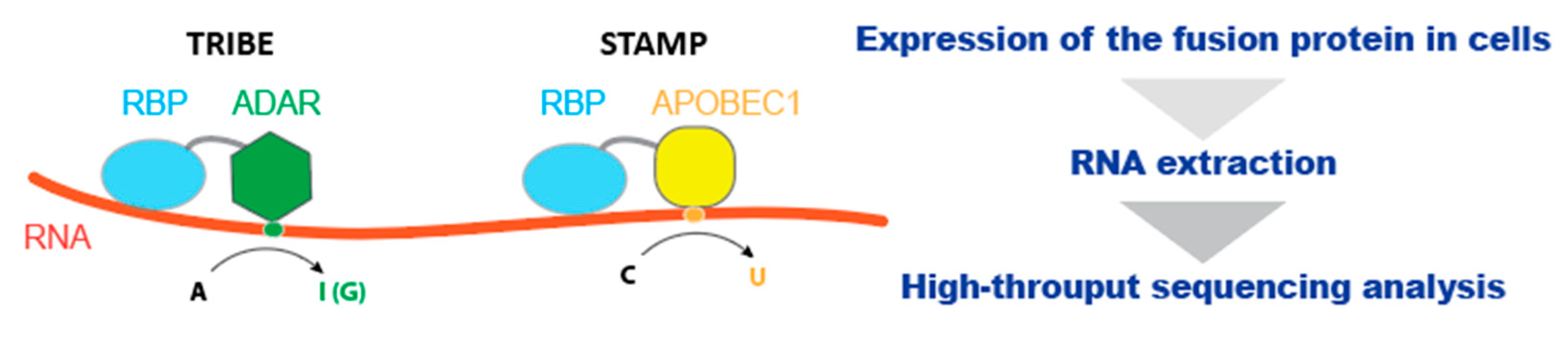
3.1. The Utilization of RNA-Editing Mechanism
3.2. The Utilization of the Proximity Labeling System
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviation
| 5′ capping | a 7-methyl guanosine cap at the 5′ end |
| ALS | Amyotrophic lateral sclerosis |
| APA | Alternative polyadenylation |
| APEX-RIP | The combination of proximity ligation with crosslinking of protein-RNA interactions by formaldehyde |
| CAP-seq | Chromophore-assisted proximity labeling and sequencing |
| CIMS | Crosslink-induced mutation sites |
| CITS | Crosslink-induced truncation sites |
| CLIP | Crosslinking and immunoprecipitation |
| CLIP-seq | High-throughput sequencing of the cDNA library generated by CLIP |
| CTD | C-terminal domain of RNAP II |
| eCLIP | Enhanced CLIP |
| ENCODE | The Encyclopedia of DNA Elements |
| FA-crosslinking | Formaldehyde-crosslinking |
| hnRNPs | Heterogeneous ribonuclear proteins |
| IP | Immunoprecipitation |
| irCLIP | Infrared-CLIP |
| KIN-CLIP | Kinetic crosslinking and immunoprecipitation |
| m6A | Methyl adenosine 6 |
| MW | Molecular weight |
| NET-seq | Native elongating transcript sequencing |
| PL | Proximity labeling |
| PrismNet | Protein-RNA Interaction by Structure-informed Modeling using deep neural NETwork |
| Proximity-CLIP | The combination of proximity ligation with crosslinking of protein–RNA interactions by UV |
| RBP | RNA-binding proteins |
| RIP | RNA immunoprecipitation |
| RNAP II | RNA polymerase II |
| scRNA-seq | Single-cell RNA-sequencing |
| SDS-PAGE | SDS-polyacrylamide gel |
| SRSFs | Serine/arginine-rich splicing factors |
| STAMP | Surveying Targets by APOBEC Mediated Profiling |
| TEX | Terminator 5′-phosphate-dependent exonuclease |
| TGRIT | Thermostable group II intron reverse transcriptase |
| TRIBE | Targets of RNA-binding proteins identified by editing |
| tRIP | Target RNA immunoprecipitation |
References
- Bentley, D.L. Coupling mRNA processing with transcription in time and space. Nat. Rev. Genet. 2014, 15, 163–175. [Google Scholar] [CrossRef] [Green Version]
- Herzel, L.; Ottoz, D.S.M.; Alpert, T.; Neugebauer, K.M. Splicing and transcription touch base: Co-transcriptional spliceosome assembly and function. Nat. Rev. Mol. Cell Biol. 2017, 18, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Kaida, D.; Berg, M.G.; Younis, I.; Kasim, M.; Singh, L.N.; Wan, L.; Dreyfuss, G. U1 snRNP protects pre-mRNAs from premature cleavage and polyadenylation. Nature 2010, 468, 664–668. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Zhou, Y.; Pandit, S.; Huang, J.; Li, H.; Lin, C.Y.; Xiao, R.; Burge, C.B.; Fu, X.D. SR Proteins Collaborate with 7SK and Promoter-Associated Nascent RNA to Release Paused Polymerase. Cell 2013, 153, 855–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuda, A.; Takeda, J.; Okuno, T.; Okamoto, T.; Ohkawara, B.; Ito, M.; Ishigaki, S.; Sobue, G.; Ohno, K. Position-specific binding of FUS to nascent RNA regulates mRNA length. Genes Dev. 2015, 29, 1045–1057. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, A.; Iida, K.; Tsubota, T.; Hosokawa, M.; Denawa, M.; Brown, J.B.; Ninomiya, K.; Ito, M.; Kimura, H.; Abe, T.; et al. Loss of Sfpq Causes Long-Gene Transcriptopathy in the Brain. Cell Rep. 2018, 23, 1326–1341. [Google Scholar] [CrossRef] [PubMed]
- Harlen, K.M.; Churchman, L.S. The code and beyond: Transcription regulation by the RNA polymerase II carboxy-terminal domain. Nat. Rev. Mol. Cell Biol. 2017, 18, 263–273. [Google Scholar] [CrossRef]
- Nojima, T.; Rebelo, K.; Gomes, T.; Grosso, A.R.; Proudfoot, N.J.; Carmo-Fonseca, M. RNA Polymerase II Phosphorylated on CTD Serine 5 Interacts with the Spliceosome during Co-transcriptional Splicing. Mol. Cell 2018, 72, 369–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trendel, J.; Schwarzl, T.; Horos, R.; Prakash, A.; Bateman, A.; Hentze, M.W.; Krijgsveld, J. The Human RNA-Binding Proteome and Its Dynamics during Translational Arrest. Cell 2019, 176, 391–403. [Google Scholar] [CrossRef] [Green Version]
- Wahl, M.C.; Will, C.L.; Luhrmann, R. The spliceosome: Design principles of a dynamic RNP machine. Cell 2009, 136, 701–718. [Google Scholar] [CrossRef] [Green Version]
- So, B.R.; Di, C.; Cai, Z.; Venters, C.C.; Guo, J.; Oh, J.M.; Arai, C.; Dreyfuss, G. A Complex of U1 snRNP with Cleavage and Polyadenylation Factors Controls Telescripting, Regulating mRNA Transcription in Human Cells. Mol. Cell 2019. [Google Scholar] [CrossRef] [PubMed]
- Gerstberger, S.; Hafner, M.; Tuschl, T. A census of human RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 829–845. [Google Scholar] [CrossRef] [PubMed]
- Popova, V.V.; Kurshakova, M.M.; Kopytova, D.V. [Methods to study the RNA-protein interactions]. Mol. Biol. 2015, 49, 472–481. [Google Scholar] [CrossRef]
- Ramanathan, M.; Porter, D.F.; Khavari, P.A. Methods to study RNA-protein interactions. Nat. Methods 2019, 16, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Hafner, M.; Katsantoni, M.; Köster, T.; Marks, J.; Mukherjee, J.; Staiger, D.; Ule, J.; Zavolan, M.J.N.R.M.P. CLIP and complementary methods. Nat. Rev. Methods Primers 2021, 1, 1–23. [Google Scholar] [CrossRef]
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef]
- Ray, D.; Kazan, H.; Cook, K.B.; Weirauch, M.T.; Najafabadi, H.S.; Li, X.; Gueroussov, S.; Albu, M.; Zheng, H.; Yang, A.; et al. A compendium of RNA-binding motifs for decoding gene regulation. Nature 2013, 499, 172–177. [Google Scholar] [CrossRef] [Green Version]
- Lambert, N.; Robertson, A.; Jangi, M.; McGeary, S.; Sharp, P.A.; Burge, C.B. RNA Bind-n-Seq: Quantitative assessment of the sequence and structural binding specificity of RNA binding proteins. Mol. Cell 2014, 54, 887–900. [Google Scholar] [CrossRef] [Green Version]
- Cartegni, L.; Wang, J.; Zhu, Z.; Zhang, M.Q.; Krainer, A.R. ESEfinder: A web resource to identify exonic splicing enhancers. Nucleic Acids Res. 2003, 31, 3568–3571. [Google Scholar] [CrossRef] [Green Version]
- Piva, F.; Giulietti, M.; Burini, A.B.; Principato, G. SpliceAid 2: A database of human splicing factors expression data and RNA target motifs. Hum. Mutat. 2012, 33, 81–85. [Google Scholar] [CrossRef]
- Buckanovich, R.J.; Darnell, R.B. The neuronal RNA binding protein Nova-1 recognizes specific RNA targets in vitro and in vivo. Mol. Cell. Biol. 1997, 17, 3194–3201. [Google Scholar] [CrossRef] [Green Version]
- Peritz, T.; Zeng, F.; Kannanayakal, T.J.; Kilk, K.; Eiriksdottir, E.; Langel, U.; Eberwine, J. Immunoprecipitation of mRNA-protein complexes. Nat. Protoc. 2006, 1, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Tenenbaum, S.A.; Lager, P.J.; Carson, C.C.; Keene, J.D. Ribonomics: Identifying mRNA subsets in mRNP complexes using antibodies to RNA-binding proteins and genomic arrays. Methods 2002, 26, 191–198. [Google Scholar] [CrossRef]
- Keene, J.D.; Komisarow, J.M.; Friedersdorf, M.B. RIP-Chip: The isolation and identification of mRNAs, microRNAs and protein components of ribonucleoprotein complexes from cell extracts. Nat. Protoc. 2006, 1, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Sephton, C.F.; Cenik, C.; Kucukural, A.; Dammer, E.B.; Cenik, B.; Han, Y.; Dewey, C.M.; Roth, F.P.; Herz, J.; Peng, J.; et al. Identification of neuronal RNA targets of TDP-43-containing ribonucleoprotein complexes. J. Biol. Chem. 2011, 286, 1204–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Ohsumi, T.K.; Kung, J.T.; Ogawa, Y.; Grau, D.J.; Sarma, K.; Song, J.J.; Kingston, R.E.; Borowsky, M.; Lee, J.T. Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol. Cell 2010, 40, 939–953. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, C.O.; Friedersdorf, M.; Keene, J.D. Quantifying RNA binding sites transcriptome-wide using DO-RIP-seq. RNA 2017, 23, 32–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagenmakers, A.J.; Reinders, R.J.; van Venrooij, W.J. Cross-linking of mRNA to proteins by irradiation of intact cells with ultraviolet light. Eur. J. Biochem. 1980, 112, 323–330. [Google Scholar] [CrossRef]
- Niranjanakumari, S.; Lasda, E.; Brazas, R.; Garcia-Blanco, M.A. Reversible cross-linking combined with immunoprecipitation to study RNA-protein interactions in vivo. Methods 2002, 26, 182–190. [Google Scholar] [CrossRef]
- Wang, Z.; Rana, T.M. Probing RNA-protein interactions by psoralen photocrosslinking. Methods Mol. Biol. 1999, 118, 49–62. [Google Scholar] [CrossRef]
- Zook, D.E.; Fahnestock, S.R. Covalent cross-linking of ribosomal RNA and proteins by methylene blue-sensitized photooxidation. Biochim. Biophys. Acta 1978, 517, 400–406. [Google Scholar] [CrossRef]
- Lee, F.C.Y.; Ule, J. Advances in CLIP Technologies for Studies of Protein-RNA Interactions. Mol. Cell 2018, 69, 354–369. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, J.R. Ultraviolet light-induced crosslinking of mRNA to proteins. Nucleic Acids Res. 1979, 6, 715–732. [Google Scholar] [CrossRef]
- Darnell, R.B. HITS-CLIP: Panoramic views of protein–RNA regulation in living cells. Wiley Interdiscip. Rev. RNA 2010, 1, 266–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ule, J.; Jensen, K.B.; Ruggiu, M.; Mele, A.; Ule, A.; Darnell, R.B. CLIP identifies Nova-regulated RNA networks in the brain. Science 2003, 302, 1212–1215. [Google Scholar] [CrossRef] [PubMed]
- Licatalosi, D.D.; Mele, A.; Fak, J.J.; Ule, J.; Kayikci, M.; Chi, S.W.; Clark, T.A.; Schweitzer, A.C.; Blume, J.E.; Wang, X.; et al. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature 2008, 456, 464–469. [Google Scholar] [CrossRef] [Green Version]
- Hafner, M.; Landthaler, M.; Burger, L.; Khorshid, M.; Hausser, J.; Berninger, P.; Rothballer, A.; Ascano, M., Jr.; Jungkamp, A.C.; Munschauer, M.; et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 2010, 141, 129–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Darnell, R.B. Mapping in vivo protein-RNA interactions at single-nucleotide resolution from HITS-CLIP data. Nat. Biotechnol. 2011, 29, 607–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishore, S.; Jaskiewicz, L.; Burger, L.; Hausser, J.; Khorshid, M.; Zavolan, M. A quantitative analysis of CLIP methods for identifying binding sites of RNA-binding proteins. Nat. Methods 2011. [Google Scholar] [CrossRef] [PubMed]
- Konig, J.; Zarnack, K.; Rot, G.; Curk, T.; Kayikci, M.; Zupan, B.; Turner, D.J.; Luscombe, N.M.; Ule, J. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat. Struct. Mol. Biol. 2010, 17, 909–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Nostrand, E.L.; Freese, P.; Pratt, G.A.; Wang, X.; Wei, X.; Xiao, R.; Blue, S.M.; Chen, J.Y.; Cody, N.A.L.; Dominguez, D.; et al. A large-scale binding and functional map of human RNA-binding proteins. Nature 2020, 583, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Rot, G.; Wang, Z.; Huppertz, I.; Modic, M.; Lence, T.; Hallegger, M.; Haberman, N.; Curk, T.; von Mering, C.; Ule, J. High-Resolution RNA Maps Suggest Common Principles of Splicing and Polyadenylation Regulation by TDP-43. Cell Rep. 2017, 19, 1056–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller-McNicoll, M.; Botti, V.; de Jesus Domingues, A.M.; Brandl, H.; Schwich, O.D.; Steiner, M.C.; Curk, T.; Poser, I.; Zarnack, K.; Neugebauer, K.M. SR proteins are NXF1 adaptors that link alternative RNA processing to mRNA export. Genes Dev. 2016, 30, 553–566. [Google Scholar] [CrossRef] [Green Version]
- Katz, Y.; Wang, E.T.; Airoldi, E.M.; Burge, C.B. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat. Methods 2010, 7, 1009–1015. [Google Scholar] [CrossRef]
- Takeda, J.I.; Masuda, A.; Ohno, K. Six GU-rich (6GUR) FUS-binding motifs detected by normalization of CLIP-seq by Nascent-seq. Gene 2017, 618, 57–64. [Google Scholar] [CrossRef]
- Martin, G.; Gruber, A.R.; Keller, W.; Zavolan, M. Genome-wide analysis of pre-mRNA 3′ end processing reveals a decisive role of human cleavage factor I in the regulation of 3′ UTR length. Cell Rep. 2012, 1, 753–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Xu, K.; Huang, W.; Yang, Y.T.; Li, P.; Tang, L.; Xiong, T.; Zhang, Q.C. Predicting dynamic cellular protein-RNA interactions by deep learning using in vivo RNA structures. Cell Res. 2021, 31, 495–516. [Google Scholar] [CrossRef]
- Linder, B.; Grozhik, A.V.; Olarerin-George, A.O.; Meydan, C.; Mason, C.E.; Jaffrey, S.R. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Methods 2015, 12, 767–772. [Google Scholar] [CrossRef]
- Ke, S.; Alemu, E.A.; Mertens, C.; Gantman, E.C.; Fak, J.J.; Mele, A.; Haripal, B.; Zucker-Scharff, I.; Moore, M.J.; Park, C.Y.; et al. A majority of m6A residues are in the last exons, allowing the potential for 3′ UTR regulation. Genes Dev. 2015, 29, 2037–2053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugimoto, Y.; Vigilante, A.; Darbo, E.; Zirra, A.; Militti, C.; D’Ambrogio, A.; Luscombe, N.M.; Ule, J. hiCLIP reveals the in vivo atlas of mRNA secondary structures recognized by Staufen 1. Nature 2015, 519, 491–494. [Google Scholar] [CrossRef] [Green Version]
- Kramer, K.; Sachsenberg, T.; Beckmann, B.M.; Qamar, S.; Boon, K.L.; Hentze, M.W.; Kohlbacher, O.; Urlaub, H. Photo-cross-linking and high-resolution mass spectrometry for assignment of RNA-binding sites in RNA-binding proteins. Nat. Methods 2014, 11, 1064–1070. [Google Scholar] [CrossRef]
- Leitner, A.; Dorn, G.; Allain, F.H. Combining Mass Spectrometry (MS) and Nuclear Magnetic Resonance (NMR) Spectroscopy for Integrative Structural Biology of Protein-RNA Complexes. Cold Spring Harb Perspect Biol. 2019, 11. [Google Scholar] [CrossRef] [Green Version]
- Jutzi, D.; Campagne, S.; Schmidt, R.; Reber, S.; Mechtersheimer, J.; Gypas, F.; Schweingruber, C.; Colombo, M.; von Schroetter, C.; Loughlin, F.E.; et al. Aberrant interaction of FUS with the U1 snRNA provides a molecular mechanism of FUS induced amyotrophic lateral sclerosis. Nat. Commun. 2020, 11, 6341. [Google Scholar] [CrossRef] [PubMed]
- Zarnegar, B.J.; Flynn, R.A.; Shen, Y.; Do, B.T.; Chang, H.Y.; Khavari, P.A. irCLIP platform for efficient characterization of protein-RNA interactions. Nat. Methods 2016, 13, 489–492. [Google Scholar] [CrossRef] [PubMed]
- Van Nostrand, E.L.; Pratt, G.A.; Shishkin, A.A.; Gelboin-Burkhart, C.; Fang, M.Y.; Sundararaman, B.; Blue, S.M.; Nguyen, T.B.; Surka, C.; Elkins, K.; et al. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat. Methods 2016, 13, 508–514. [Google Scholar] [CrossRef]
- Masuda, A.; Kawachi, T.; Takeda, J.I.; Ohkawara, B.; Ito, M.; Ohno, K. tRIP-seq reveals repression of premature polyadenylation by co-transcriptional FUS-U1 snRNP assembly. EMBO Rep. 2020, 21, e49890. [Google Scholar] [CrossRef] [PubMed]
- Sasagawa, Y.; Nikaido, I.; Hayashi, T.; Danno, H.; Uno, K.D.; Imai, T.; Ueda, H.R. Quartz-Seq: A highly reproducible and sensitive single-cell RNA sequencing method, reveals non-genetic gene-expression heterogeneity. Genome Biol. 2013, 14, R31. [Google Scholar] [CrossRef] [Green Version]
- Masuda, A.; Takeda, J.; Ohno, K. FUS-mediated regulation of alternative RNA processing in neurons: Insights from global transcriptome analysis. Wiley Interdiscip. Rev. RNA 2016, 7, 330–340. [Google Scholar] [CrossRef]
- Sharma, D.; Zagore, L.L.; Brister, M.M.; Ye, X.; Crespo-Hernandez, C.E.; Licatalosi, D.D.; Jankowsky, E. The kinetic landscape of an RNA-binding protein in cells. Nature 2021, 591, 152–156. [Google Scholar] [CrossRef]
- McMahon, A.C.; Rahman, R.; Jin, H.; Shen, J.L.; Fieldsend, A.; Luo, W.; Rosbash, M. TRIBE: Hijacking an RNA-Editing Enzyme to Identify Cell-Specific Targets of RNA-Binding Proteins. Cell 2016, 165, 742–753. [Google Scholar] [CrossRef] [Green Version]
- Zinshteyn, B.; Nishikura, K. Adenosine-to-inosine RNA editing. Wiley Interdiscip. Rev. Syst. Biol. Med. 2009, 1, 202–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.; Rahman, R.; Rosbash, M. Mechanistic implications of enhanced editing by a HyperTRIBE RNA-binding protein. RNA 2018, 24, 173–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brannan, K.W.; Chaim, I.A.; Marina, R.J.; Yee, B.A.; Kofman, E.R.; Lorenz, D.A.; Jagannatha, P.; Dong, K.D.; Madrigal, A.A.; Underwood, J.G.; et al. Robust single-cell discovery of RNA targets of RNA-binding proteins and ribosomes. Nat. Methods 2021, 18, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Mayer, A.; di Iulio, J.; Maleri, S.; Eser, U.; Vierstra, J.; Reynolds, A.; Sandstrom, R.; Stamatoyannopoulos, J.A.; Churchman, L.S. Native elongating transcript sequencing reveals human transcriptional activity at nucleotide resolution. Cell 2015, 161, 541–554. [Google Scholar] [CrossRef] [Green Version]
- Nojima, T.; Gomes, T.; Grosso, A.R.; Kimura, H.; Dye, M.J.; Dhir, S.; Carmo-Fonseca, M.; Proudfoot, N.J. Mammalian NET-Seq Reveals Genome-wide Nascent Transcription Coupled to RNA Processing. Cell 2015, 161, 526–540. [Google Scholar] [CrossRef] [Green Version]
- Martell, J.D.; Deerinck, T.J.; Sancak, Y.; Poulos, T.L.; Mootha, V.K.; Sosinsky, G.E.; Ellisman, M.H.; Ting, A.Y. Engineered ascorbate peroxidase as a genetically encoded reporter for electron microscopy. Nat. Biotechnol. 2012, 30, 1143–1148. [Google Scholar] [CrossRef] [Green Version]
- Roux, K.J.; Kim, D.I.; Raida, M.; Burke, B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 2012, 196, 801–810. [Google Scholar] [CrossRef] [Green Version]
- Branon, T.C.; Bosch, J.A.; Sanchez, A.D.; Udeshi, N.D.; Svinkina, T.; Carr, S.A.; Feldman, J.L.; Perrimon, N.; Ting, A.Y. Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol. 2018, 36, 880–887. [Google Scholar] [CrossRef]
- Benhalevy, D.; Anastasakis, D.G.; Hafner, M. Proximity-CLIP provides a snapshot of protein-occupied RNA elements in subcellular compartments. Nat. Methods 2018, 15, 1074–1082. [Google Scholar] [CrossRef]
- Kaewsapsak, P.; Shechner, D.M.; Mallard, W.; Rinn, J.L.; Ting, A.Y. Live-cell mapping of organelle-associated RNAs via proximity biotinylation combined with protein-RNA crosslinking. eLife 2017, 6. [Google Scholar] [CrossRef]
- Fazal, F.M.; Han, S.; Parker, K.R.; Kaewsapsak, P.; Xu, J.; Boettiger, A.N.; Chang, H.Y.; Ting, A.Y. Atlas of Subcellular RNA Localization Revealed by APEX-Seq. Cell 2019, 178, 473–490. [Google Scholar] [CrossRef] [PubMed]
- Lam, S.S.; Martell, J.D.; Kamer, K.J.; Deerinck, T.J.; Ellisman, M.H.; Mootha, V.K.; Ting, A.Y. Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat. Methods 2015, 12, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Tang, W.; Li, Z.; Zou, Z.; Zhou, Y.; Li, R.; Xiong, T.; Wang, J.; Zou, P. Mapping spatial transcriptome with light-activated proximity-dependent RNA labeling. Nat. Chem. Biol. 2019, 15, 1110–1119. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CLIP | irCLIP | eCLIP | tRIP | |
|---|---|---|---|---|
| UV crosslinking | √ | √ | √ | √ |
| Cell lysis and IP | √ | √ | √ | √ |
| RNase treatment | Total cell lysates | on beads | Total cell lysates | on beads |
| Dephosphorylation of RNA | √ | √ | √ | – |
| 5′ end labeling | √ | Replaced with infrared dye-labeling of 3′ linker | √ | – |
| 3′ linker ligation | √ | √ | √ | √ |
| SDS-PAGE | √ | √ | √ | Replaced with deadenylase- and TEX-treatments |
| Transfer to membranes | √ | √ | √ | |
| Cut membrane | √ | √ | √ | |
| Proteinase K treatment | √ | √ | √ | √ |
| Purification of RNA | Phenol/chloroform | Phenol/chloroform | Phenol/chloroform | Column purification |
| ETOH precipitation | ETOH precipitation | Column purification | – | |
| Reverse transcription | √ | √ | √ | √ |
| Purification of cDNA/RNA | – | Pull-down with streptavidin beads | Silane-beads purification | – |
| Modification of 5′ end | 5′ linker ligation | Circularization of cDNA | 5′ linker ligation | PolyA tailing |
| Purification of cDNA/RNA | Phenol/chloroform ETOH precipitation | Silane-beads purification | Silane-beads purification | – |
| PCR amplification | √ | √ | √ | √ |
| Hands-on time | 4 days | 3 days | 4 days | 2 days |
| Method | Advantages | Disadvantages | Analysis of the Interactions Specific to the RNAP II Machinery |
|---|---|---|---|
| RIP |
|
|
|
| CLIP irCLIP [54] eCLIP [55] tRIP [56] |
|
|
|
| KIN-CLIP [59] |
|
|
|
| RNA editing TRIBE [60] STAMP [63] |
|
|
|
| Proximity labeling Proximity-CLIP [69] APEX-RIP [70] CAP-seq [73] |
|
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Masuda, A.; Kawachi, T.; Ohno, K. Rapidly Growing Protein-Centric Technologies to Extensively Identify Protein–RNA Interactions: Application to the Analysis of Co-Transcriptional RNA Processing. Int. J. Mol. Sci. 2021, 22, 5312. https://doi.org/10.3390/ijms22105312
Masuda A, Kawachi T, Ohno K. Rapidly Growing Protein-Centric Technologies to Extensively Identify Protein–RNA Interactions: Application to the Analysis of Co-Transcriptional RNA Processing. International Journal of Molecular Sciences. 2021; 22(10):5312. https://doi.org/10.3390/ijms22105312
Chicago/Turabian StyleMasuda, Akio, Toshihiko Kawachi, and Kinji Ohno. 2021. "Rapidly Growing Protein-Centric Technologies to Extensively Identify Protein–RNA Interactions: Application to the Analysis of Co-Transcriptional RNA Processing" International Journal of Molecular Sciences 22, no. 10: 5312. https://doi.org/10.3390/ijms22105312