
Dysregulation of Muscle-Specific MicroRNAs as Common Pathogenic Feature Associated with Muscle Atrophy in ALS, SMA and SBMA: Evidence from Animal Models and Human Patients

 , , , , ,
, , , , ,  , , , , , ,
, , , , , ,  , and
, and
Abstract
:1. Introduction
2. Results
2.1. Dysregulated Expression of MyomiRs in G93A-SOD1, Δ7SMA, and AR113Q Mouse Muscle Tissue in Relation to Disease Progression
2.2. Altered Expression of MyomiR Targets in G93A-SOD1, Δ7SMA, and AR113Q Muscle Tissue in Relation to Disease Progression
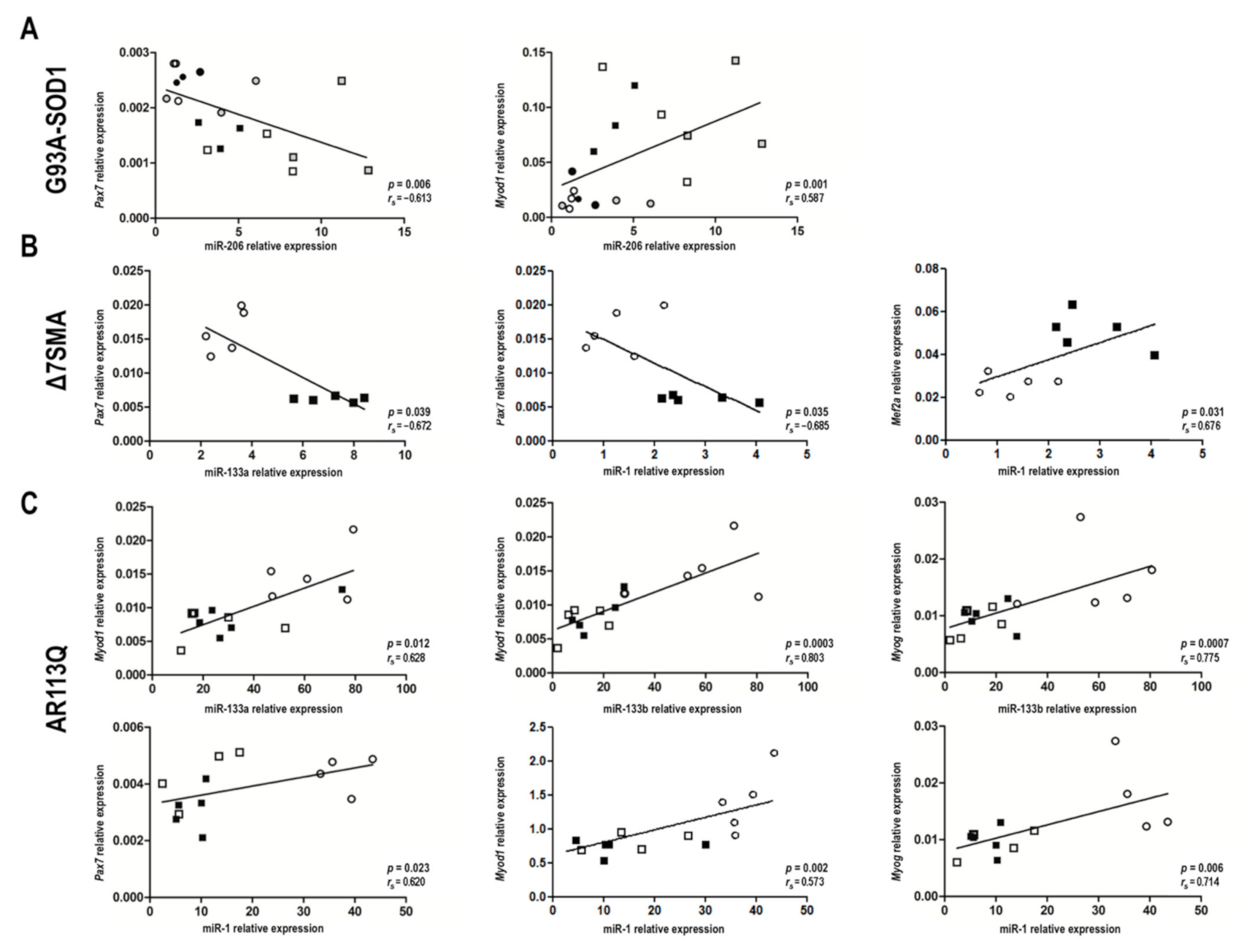
2.3. MyomiR-mRNA Target Correlations in Muscle Tissue of G93A-SOD1, Δ7SMA, and AR113Q Mice
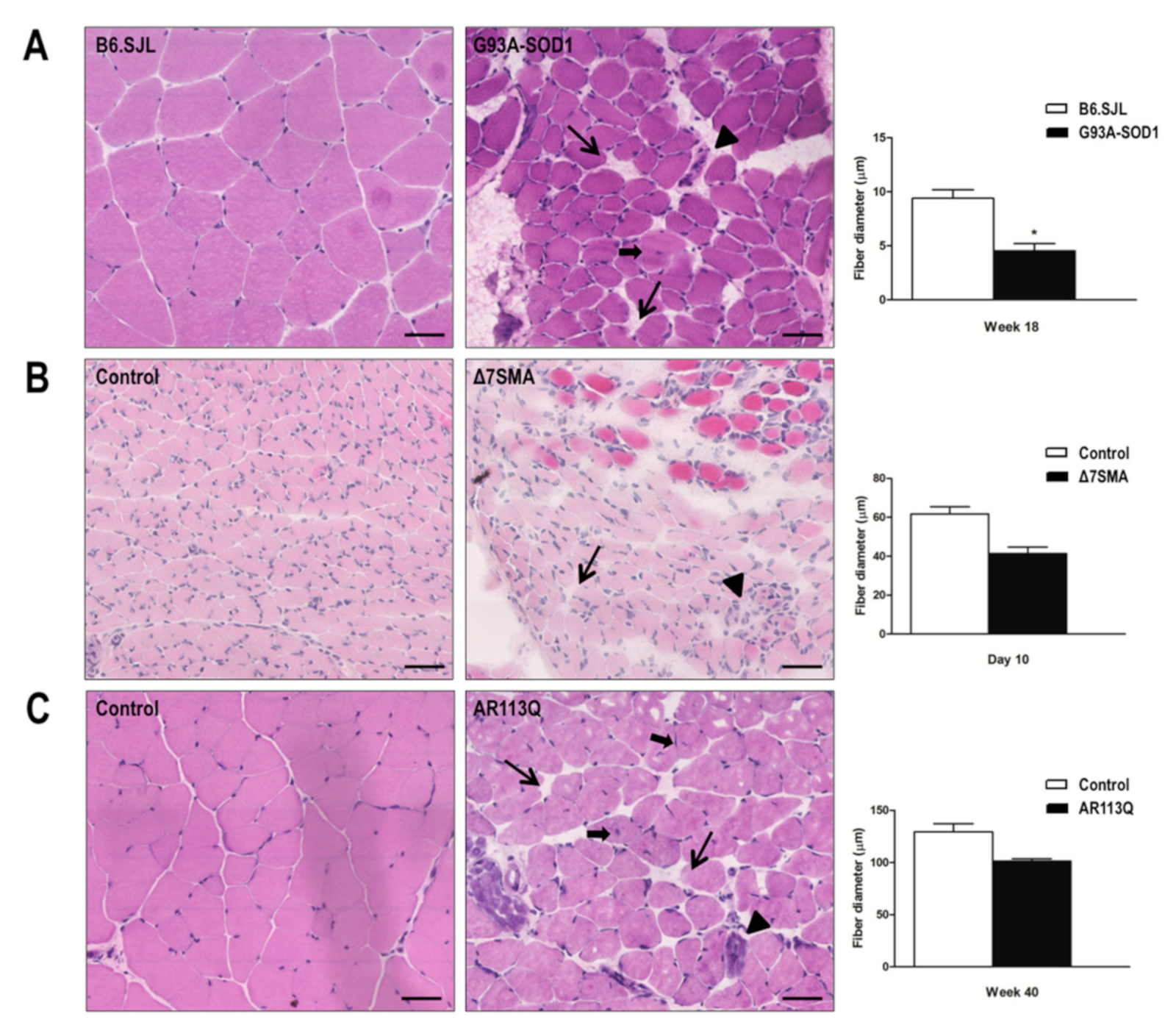
2.4. Alteration of G93A-SOD1, Δ7SMA, and AR113Q Muscle Architecture at Late Disease Stage
2.5. Altered MyomiR Expression in Serum Samples of ALS, SMA, and SBMA Patients
3. Discussion
4. Materials and Methods
4.1. Animal Models
4.2. Quantitative Real-Time PCR to Assess MyomiR Expression in Mouse Muscle Tissues
4.3. MyomiR Target Gene Prediction and Analysis in Mouse Muscle Tissues
4.4. Western Blotting Assay
4.5. Histological Analysis
4.6. Patients and Biological Samples
4.7. Quantitative Real-Time PCR to Assess MyomiRs in Patient Serum Samples
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Al-Chalabi, A.; Visscher, P.M. Motor neuron disease: Common genetic variants and the heritability of ALS. Nat. Rev. Neurol. 2014, 10, 549–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Therrien, M.; Dion, P.A.; Rouleau, G.A. ALS: Recent Developments from Genetics Studies. Curr. Neurol. Neurosci. Rep. 2016, 16, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wijesekera, L.C.; Leigh, P.N. Amyotrophic lateral sclerosis. Orphanet. J. Rare. Dis. 2009, 4, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, P.J. Molecular and cellular pathways of neurodegeneration in motor neurone disease. J. Neurol. Neurosurg. Psychiatry 2005, 76, 1046–1057. [Google Scholar] [CrossRef] [Green Version]
- Cleveland, D.W.; Rothstein, J.D. From charcot to lou gehrig: Deciphering selective motor neuron death in als. Nat. Rev. Neurosci. 2001, 2, 806–819. [Google Scholar] [CrossRef]
- Barber, S.C.; Shaw, P.J. Oxidative stress in ALS: Key role in motor neuron injury and therapeutic target. Free. Radic. Biol. Med. 2010, 48, 629–641. [Google Scholar] [CrossRef]
- Kolb, S.J.; Kissel, J.T. Spinal Muscular Atrophy. Neurol. Clin. 2015, 33, 831–846. [Google Scholar] [CrossRef] [Green Version]
- Gubitz, A.K.; Feng, W.; Dreyfuss, G. The SMN complex. Exp. Cell Res. 2004, 296, 51–56. [Google Scholar] [CrossRef]
- Bowerman, M.; Murray, L.M.; Scamps, F.; Schneider, B.L.; Kothary, R.; Raoul, C. Pathogenic commonalities between spinal muscular atrophy and amyotrophic lateral sclerosis: Converging roads to therapeutic development. Eur. J. Med. Genet. 2018, 61, 685–698. [Google Scholar] [CrossRef]
- Tosolini, A.P.; Sleigh, J.N. Motor Neuron Gene Therapy: Lessons from Spinal Muscular Atrophy for Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2017, 10, 405. [Google Scholar] [CrossRef] [Green Version]
- La Spada, A.R.; Wilson, E.M.; Lubahn, D.B.; Harding, A.E.; Fischbeck, K.H. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nat. Cell Biol. 1991, 352, 77–79. [Google Scholar] [CrossRef]
- Cortes, C.J.; Ling, S.-C.; Guo, L.T.; Hung, G.; Tsunemi, T.; Ly, L.; Tokunaga, S.; Lopez, E.; Sopher, B.L.; Bennett, C.F.; et al. Muscle Expression of Mutant Androgen Receptor Accounts for Systemic and Motor Neuron Disease Phenotypes in Spinal and Bulbar Muscular Atrophy. Neuron 2014, 82, 295–307. [Google Scholar] [CrossRef] [Green Version]
- Rusmini, P.; Bolzoni, E.; Crippa, V.; Onesto, E.; Sau, D.; Galbiati, M.; Piccolella, M.; Poletti, A. Proteasomal and autophagic degradative activities in spinal and bulbar muscular atrophy. Neurobiol. Dis. 2010, 40, 361–369. [Google Scholar] [CrossRef]
- Rusmini, P.; Polanco, M.J.; Cristofani, R.; Cicardi, M.E.; Meroni, M.; Galbiati, M.; Piccolella, M.; Messi, E.; Giorgetti, E.; Lieberman, A.P.; et al. Aberrant Autophagic Response in The Muscle of a Knock-in Mouse Model of Spinal and Bulbar Muscular Atrophy. Sci. Rep. 2015, 5, 15174. [Google Scholar] [CrossRef] [Green Version]
- Galbiati, M.; Crippa, V.; Rusmini, P.; Cristofani, R.; Cicardi, M.E.; Giorgetti, E.; Onesto, E.; Messi, E.; Poletti, A. ALS-related misfolded protein management in motor neurons and muscle cells. Neurochem. Int. 2014, 79, 70–78. [Google Scholar] [CrossRef]
- Marcuzzo, S.; Zucca, I.; Mastropietro, A.; de Rosbo, N.K.; Cavalcante, P.; Tartari, S.; Bonanno, S.; Preite, L.; Mantegazza, R.; Bernasconi, P. Hind limb muscle atrophy precedes cerebral neuronal degeneration in G93A-SOD1 mouse model of amyo-trophic lateral sclerosis: A longitudinal MRI study. Exp. Neurol. 2014, 231, 30–37. [Google Scholar] [CrossRef]
- Marcuzzo, S.; Bonanno, S.; Figini, M.; Scotti, A.; Zucca, I.; Minati, L.; Riva, N.; Domi, T.; Fossaghi, A.; Quattrini, A.; et al. A longitudinal DTI and histological study of the spinal cord reveals early pathological alterations in G93A-SOD1 mouse model of amyotrophic lateral sclerosis. Exp. Neurol. 2017, 293, 43–52. [Google Scholar] [CrossRef]
- Valsecchi, V.; Boido, M.; De Amicis, E.; Piras, A.; Vercelli, A. Expression of Muscle-Specific MiRNA 206 in the Progression of Disease in a Murine SMA Model. PLoS ONE 2015, 10, e0128560. [Google Scholar] [CrossRef]
- Brooks, B.R.; Thisted, R.A.; Appel, S.H.; Bradley, W.G.; Olney, R.K.; Berg, J.E.; Pope, L.E.; Smith, R.A.; AVP-923 ALS Study Group. Treatment of pseudobulbar affect in ALS with dextromethorphan/quinidine: A randomized trial. Neurology 2004, 63, 1364–1370. [Google Scholar] [CrossRef]
- Chiu, A.Y.; Zhai, P.; Canto, M.C.D.; Peters, T.M.; Kwon, Y.W.; Prattis, S.M.; Gurney, M.E. Age-Dependent Penetrance of Disease in a Transgenic Mouse Model of Familial Amyotrophic Lateral Sclerosis. Mol. Cell. Neurosci. 1995, 6, 349–362. [Google Scholar] [CrossRef]
- Dupuis, L.; Echaniz-Laguna, A. Skeletal Muscle in Motor Neuron Diseases: Therapeutic Target and Delivery Route for Potential Treatments. Curr. Drug Targets 2010, 11, 1250–1261. [Google Scholar] [CrossRef] [PubMed]
- Frey, D.; Schneider, C.; Xu, L.; Borg, J.; Spooren, W.; Caroni, P. Early and Selective Loss of Neuromuscular Synapse Subtypes with Low Sprouting Competence in Motoneuron Diseases. J. Neurosci. 2000, 20, 2534–2542. [Google Scholar] [CrossRef] [PubMed]
- Dobrowolny, G.; Aucello, M.; Rizzuto, E.; Beccafico, S.; Mammucari, C.; Bonconpagni, S.; Belia, S.; Wannenes, F.; Nicoletti, C.; Del Prete, Z.; et al. Skeletal Muscle Is a Primary Target of SOD1G93A-Mediated Toxicity. Cell Metab. 2008, 8, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.; Martin, L.J. Skeletal muscle-restricted expression of human SOD1 causes motor neuron degeneration in transgenic mice. Hum. Mol. Genet. 2010, 19, 2284–2302. [Google Scholar] [CrossRef] [Green Version]
- Loeffler, J.-P.; Picchiarelli, G.; Dupuis, L.; De Aguilar, J.-L.G. The Role of Skeletal Muscle in Amyotrophic Lateral Sclerosis. Brain Pathol. 2016, 26, 227–236. [Google Scholar] [CrossRef]
- Zhang, Z.; Lotti, F.; Dittmar, K.; Younis, I.; Wan, L.; Kasim, M.; Dreyfuss, G. SMN Deficiency Causes Tissue-Specific Perturbations in the Repertoire of snRNAs and Widespread Defects in Splicing. Cell 2008, 133, 585–600. [Google Scholar] [CrossRef] [Green Version]
- Giavazzi, A.; Setola, V.; Simonati, A.; Battaglia, G. Neuronal-specific roles of the survival motor neuron protein: Evidence from survival motor neuron expression patterns in the developing human central nervous system. J. Neuropathol. Exp. Neurol. 2006, 65, 267–277. [Google Scholar] [CrossRef] [Green Version]
- Setola, V.; Terao, M.; Locatelli, D.; Bassanini, S.; Garattini, E.; Battaglia, G. Axonal-SMN (a-SMN), a protein isoform of the survival motor neuron gene, is specifically involved in axonogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 1959–1964. [Google Scholar] [CrossRef] [Green Version]
- Fidziańska, A.; Goebel, H.H.; Warlo, I. Acute infantile spinal muscular atrophy. Muscle apoptosis as a proposed pathogenetic mechanism. Brain 1990, 113, 433–445. [Google Scholar] [CrossRef]
- Stevens, L.; Bastide, B.; Maurage, C.A.; Dupont, E.; Montel, V.; Cieniewski-Bernard, C.; Cuisset, J.M.; Vallee, L.; Mounier, Y. Childhood spinal muscular atrophy induces alterations in contractile and regulatory protein isoform expressions. Neuropathol. Appl. Neurobiol. 2008, 34, 659–670. [Google Scholar] [CrossRef]
- D’Errico, P.; Boido, M.; Piras, A.; Valsecchi, V.; De Amicis, E.; Locatelli, D.; Capra, S.; Vagni, F.; Vercelli, A.; Battaglia, G. Selective vulnerability of spinal and cortical motor neuron subpopulations in delta7 SMA mice. PLoS ONE 2013, 8, e82654. [Google Scholar] [CrossRef]
- Nicole, S.; Desforges, B.; Millet, G.; Lesbordes, J.; Diaz, C.C.; Vertes, D.; Cao, M.L.; De Backer, F.; Languille, L.; Roblot, N.; et al. Intact satellite cells lead to remarkable protection against Smn gene defect in differentiated skeletal muscle. J. Cell Biol. 2003, 161, 571–582. [Google Scholar] [CrossRef]
- Shafey, D.; Côté, P.D.; Kothary, R. Hypomorphic Smn knockdown C2C12 myoblasts reveal intrinsic defects in myoblast fusion and myotube morphology. Exp. Cell Res. 2005, 311, 49–61. [Google Scholar] [CrossRef]
- Park, G.-H.; Kariya, S.; Monani, U.R. Spinal Muscular Atrophy: New and Emerging Insights from Model Mice. Curr. Neurol. Neurosci. Rep. 2010, 10, 108–117. [Google Scholar] [CrossRef] [Green Version]
- Fayzullina, S.; Martin, L.J. Skeletal Muscle DNA Damage Precedes Spinal Motor Neuron DNA Damage in a Mouse Model of Spinal Muscular Atrophy (SMA). PLoS ONE 2014, 9, e93329. [Google Scholar] [CrossRef]
- Mo, K.; Razak, Z.; Rao, P.; Yu, Z.; Adachi, H.; Katsuno, M.; Sobue, G.; Lieberman, A.P.; Westwood, J.T.; Monks, D.A. Microarray Analysis of Gene Expression by Skeletal Muscle of Three Mouse Models of Kennedy Disease/Spinal Bulbar Muscular Atrophy. PLoS ONE 2010, 5, e12922. [Google Scholar] [CrossRef]
- Malena, A.; Pennuto, M.; Tezze, C.; Querin, G.; D’Ascenzo, C.; Silani, V.; Cenacchi, G.; Scaramozza, A.; Romito, S.; Morandi, L.; et al. Androgen-dependent impairment of myogenesis in spinal and bulbar muscular atrophy. Acta Neuropathol. 2013, 126, 109–121. [Google Scholar] [CrossRef]
- Ramzan, F.; McPhail, M.; Rao, P.; Mo, K.; Halievski, K.; Swift-Gallant, A.; Mendoza-Viveros, L.; Cheng, H.Y.; Monks, D.A. Distinct Etiological Roles for Myocytes and Motor Neurons in a Mouse Model of Kennedy’s Disease/Spinobulbar Muscular Atrophy. J. Neurosci. 2015, 35, 6444–6451. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Dadgar, N.; Allbertelli, M.; Gruis, K.; Jordan, C.; Robins, D.M.; Lieberman, A.P. Androgen dependent pathology demonstrates myopathic contribution to the Kennedy disease phenotype in a mouse knock-in model. J. Clin. Investig. 2006, 116, 2663–2672. [Google Scholar] [CrossRef]
- Chivet, M.; Marchioretti, C.; Pirazzini, M.; Piol, D.; Scaramuzzino, C.; Polanco, M.J.; Romanello, V.; Zuccaro, E.; Parodi, S.; D’Antonio, M.; et al. Polyglutamine-Expanded Androgen Receptor Alteration of Skeletal Muscle Homeostasis and Myonuclear Aggregation Are Affected by Sex, Age and Muscle Metabolism. Cells 2020, 9, 325. [Google Scholar] [CrossRef] [Green Version]
- Lieberman, A.P.; Yu, Z.; Murray, S.; Peralta, R.; Low, A.; Guo, S.; Yu, X.X.; Cortes, C.J.; Bennett, C.F.; Monia, B.P.; et al. Peripheral Androgen Receptor Gene Suppression Rescues Disease in Mouse Models of Spinal and Bulbar Muscular Atrophy. Cell Rep. 2014, 7, 774–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haramati, S.; Chapnik, E.; Sztainberg, Y.; Eilam, R.; Zwang, R.; Gershoni, N.; McGlinn, E.; Heiser, P.W.; Wills, A.-M.; Wirguin, I.; et al. miRNA malfunction causes spinal motor neuron disease. Proc. Natl. Acad. Sci. USA 2010, 107, 13111–13116. [Google Scholar] [CrossRef] [Green Version]
- Kye, M.J.; Gonçalves, I.C. The role of miRNA in motor neuron disease. Front. Cell Neurosci. 2014, 8, 15. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.F.; Mandel, E.M.; Thomson, J.M.; Wu, Q.; Callis, T.E.; Hammond, S.M.; Conlon, F.L.; Wang, D.Z. The Role of MicroRNA-1 and MicroRNA-133 in Skeletal Muscle Proliferation and Differentiation. Nat. Genet. 2005, 38, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-F.; Callis, T.E.; Wang, D.-Z. microRNAs and muscle disorders. J. Cell Sci. 2009, 122, 13–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horak, M.; Novakab, J.; Bienertova-Vaskuac, J. Muscle-specific microRNAs in skeletal muscle development. Dev. Biol. 2016, 410, 1–13. [Google Scholar] [CrossRef]
- Ge, Y.; Chen, J. MicroRNAs in skeletal myogenesis. Cell Cycle 2011, 10, 441–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.-F.; Tao, Y.; Li, J.; Deng, Z.; Yan, Z.; Xiao, X.; Wang, D. microRNA-1 and microRNA-206 regulate skeletal muscle satellite cell proliferation and differentiation by repressing Pax7. J. Cell Biol. 2010, 190, 867–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, F.; Zhang, W.; Geyra, A.; Kang, J.-S.; Krauss, R.S. Positive Regulation of Myogenic bHLH Factors and Skeletal Muscle Development by the Cell Surface Receptor CDO. Dev. Cell 2004, 7, 843–854. [Google Scholar] [CrossRef] [Green Version]
- Ma, G.; Wang, Y.; Li, Y.; Cui, L.; Zhao, Y.; Zhao, B.; Li, K. MiR-206, a Key Modulator of Skeletal Muscle Development and Disease. Int. J. Biol. Sci. 2015, 11, 345–352. [Google Scholar] [CrossRef]
- Liu, N.; Nelson, B.R.; Bezprozvannaya, S.; Shelton, J.M.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Requirement of MEF2A, C, and D for skeletal muscle regeneration. Proc. Natl. Acad. Sci. USA 2014, 111, 4109–4114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, A.H.; Valdez, G.; Moresi, V.; Qi, X.; McAnally, J.; Elliott, J.L.; Bassel-Duby, R.; Sanes, J.R.; Olson, E.N. MicroRNA-206 Delays ALS Progression and Promotes Regeneration of Neuromuscular Synapses in Mice. Science 2009, 326, 1549–1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovanda, A.; Leonardis, L.; Zidar, J.; Koritnik, B.; Dolenc-Groselj, L.; Kovacic, S.R.; Curk, T.; Rogelj, B. Differential expression of microRNAs and other small RNAs in muscle tissue of patients with ALS and healthy age-matched controls. Sci. Rep. 2018, 8, 5609. [Google Scholar] [CrossRef] [PubMed]
- Tasca, E.; Pegoraro, V.; Merico, A.; Angelini, C. Circulating microRNAs as biomarkers of muscle differentiation and atrophy in ALS. Clin. Neuropathol. 2016, 35, 22–30. [Google Scholar] [CrossRef]
- Pegoraro, V.; Marozzo, R.; Angelini, C. MicroRNAs and HDAC4 protein expression in the skeletal muscle of ALS patients. Clin. Neuropathol. 2020, 39, 105–114. [Google Scholar] [CrossRef]
- Bruneteau, G.; Simonet, T.; Bauché, S.; Mandjee, N.; Malfatti, E.; Girard, E.; Tanguy, M.-L.; Béhin, A.; Khiami, F.; Sariali, E.; et al. Muscle histone deacetylase 4 upregulation in amyotrophic lateral sclerosis: Potential role in reinnervation ability and disease progression. Brain 2013, 136, 2359–2368. [Google Scholar] [CrossRef]
- Si, Y.; Cui, X.; Crossman, D.K.; Hao, J.; Kazamel, M.; Kwon, Y.; King, P.H. Muscle microRNA signatures as biomarkers of disease progression in amyotrophic lateral sclerosis. Neurobiol. Dis. 2018, 114, 85–94. [Google Scholar] [CrossRef]
- Bonanno, S.; Marcuzzo, S.; Malacarne, C.; Giagnorio, E.; Masson, R.; Zanin, R.; Arnoldi, M.T.; Andreetta, F.; Simoncini, O.; Venerando, A.; et al. Circulating MyomiRs as potential biomarkers to monitor response to Nusinersen in pediatric SMA patients. Biomedicines 2020, 8, 21. [Google Scholar] [CrossRef] [Green Version]
- Toivonen, J.M.; Manzano, R.; Oliván, S.; Zaragoza, P.; García-Redondo, A.; Osta, R. MicroRNA-206: A Potential Circulating Biomarker Candidate for Amyotrophic Lateral Sclerosis. PLoS ONE 2014, 9, e89065. [Google Scholar] [CrossRef] [Green Version]
- Jensen, L.; Jørgensen, L.H.; Bech, R.D.; Frandsen, U.; Schrøder, H.D. Skeletal Muscle Remodelling as a Function of Disease Progression in Amyotrophic Lateral Sclerosis. BioMed Res. Int. 2016, 2016, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Pegoraro, V.; Merico, A.; Angelini, C. Micro-RNAs in ALS muscle: Differences in gender, age at onset and disease duration. J. Neurol. Sci. 2017, 380, 58–63. [Google Scholar] [CrossRef] [Green Version]
- Dweep, H.; Sticht, C.; Pandey, P.; Gretz, N. miRWalk-database: Prediction of possible miRNA binding sites by “walking” the genes of three genomes. J. Biomed. Inform. 2011, 44, 839–847. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Kumar, R.M.; Penn, B.H.; Berkes, C.A.; Kooperberg, C.; Boyer, L.A.; Young, R.A.; Tapscott, S.J. Global and gene-specific analyses show distinct roles for Myod and Myog at a common set of promoters. EMBO J. 2006, 25, 502–511. [Google Scholar] [CrossRef] [Green Version]
- Du, C.; Jin, Y.Q.; Qi, J.J.; Ji, Z.X.; Li, S.Y.; An, G.S.; Jia, H.T.; Ni, J.H. Effects of myogenin on expression of late muscle genes through MyoD-dependent chromatin remodeling ability of myogenin. Mol. Cells 2012, 34, 133–142. [Google Scholar] [CrossRef] [Green Version]
- Meroni, M.; Crippa, V.; Cristofani, R.; Rusmini, P.; Cicardi, M.E.; Messi, E.; Piccolella, M.; Tedesco, B.; Ferrari, V.; Sorarù, G.; et al. Transforming growth factor beta 1 signaling is altered in the spinal cord and muscle of amyotrophic lateral sclerosis mice and patients. Neurobiol. Aging 2019, 82, 48–59. [Google Scholar] [CrossRef]
- McCarthy, J.J. The MyomiR Network in Skeletal Muscle Plasticity. Exerc. Sport Sci. Rev. 2011, 39, 150–154. [Google Scholar] [CrossRef]
- Di Pietro, L.; Baranzini, M.; Berardinelli, M.G.; Lattanzi, W.; Monforte, M.; Tasca, G.; Conte, A.; Logroscino, G.; Michetti, F.; Ricci, E.; et al. Potential therapeutic targets for ALS: MIR206, MIR208b and MIR499 are modulated during disease progression in the skeletal muscle of patients. Sci. Rep. 2017, 7, 9538. [Google Scholar] [CrossRef]
- Rinchetti, P.; Rizzuti, M.; Faravelli, I.; Corti, S. MicroRNA Metabolism and Dysregulation in Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2017, 55, 2617–2630. [Google Scholar] [CrossRef] [Green Version]
- Catalanotto, C.; Cogoni, C.; Zardo, G. MicroRNA in Control of Gene Expression: An Overview of Nuclear Functions. Int. J. Mol. Sci. 2016, 17, 1712. [Google Scholar] [CrossRef] [Green Version]
- Diaz, G.; Zamboni, F.; Tice, A.; Farci, P. Integrated ordination of miRNA and mRNA expression profiles. BMC Genom. 2015, 16, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Rao, P.K.; Kumar, R.M.; Farkhondeh, M.; Baskerville, S.; Lodish, H.F. Myogenic factors that regulate expression of muscle-specific microRNAs. Proc. Natl. Acad. Sci. USA 2006, 103, 8721–8726. [Google Scholar] [CrossRef] [Green Version]
- Calvo, A.C.; Manzano, R.; Atencia-Cibreiro, G.; Oliván, S.; Muñoz, M.J.; Zaragoza, P.; Cordero-Vázquez, P.; Esteban-Pérez, J.; García-Redondo, A.; Osta, R. Genetic Biomarkers for ALS Disease in Transgenic SOD1G93A Mice. PLoS ONE 2012, 7, e32632. [Google Scholar] [CrossRef] [Green Version]
- Catapano, F.; Zaharieva, I.; Scoto, M.; Marrosu, E.; Morgan, J.; Muntoni, F.; Zhou, H. Altered Levels of MicroRNA-9, -206, and -132 in Spinal Muscular Atrophy and Their Response to Antisense Oligonucleotide Therapy. Mol. Ther. Nucleic Acids 2016, 5, e331. [Google Scholar] [CrossRef] [Green Version]
- Borisov, A.B.; Carlson, B.M. Cell death in denervated skeletal muscle is distinct from classical apoptosis. Anat. Rec. 2000, 258, 305–318. [Google Scholar] [CrossRef]
- Cacchiarelli, D.; Legnini, I.; Martone, J.; Cazzella, V.; D’Amico, A.; Bertini, E.; Bozzoni, I. miRNAs as serum biomarkers for Duchenne muscular dystrophy. EMBO Mol. Med. 2011, 3, 258–265. [Google Scholar] [CrossRef]
- Pegoraro, V.; Merico, A.; Angelini, C. MyomiRNAs Dysregulation in ALS Rehabilitation. Brain Sci. 2019, 9, 8. [Google Scholar] [CrossRef] [Green Version]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef]
- Le, T.T.; Pham, L.T.; Butchbach, M.E.; Zhang, H.L.; Monani, U.R.; Coovert, D.D.; Gavrilina, T.O.; Xing, L.; Bassell, G.J.; Burghes, A.H. SMNΔ7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum. Mol. Genet. 2005, 14, 845–857. [Google Scholar] [CrossRef]
- Song, J.; Bai, Z.; Han, W.; Zhang, J.; Meng, H.; Bi, J.; Ma, X.; Han, S.; Zhang, Z. Identification of Suitable Reference Genes for qPCR Analysis of Serum microRNA in Gastric Cancer Patients. Dig. Dis. Sci. 2011, 57, 897–904. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SOD1-Mutated ALS (n = 14) | SBMA (n = 10) | Healthy Controls (n = 17) | SMA (n = 23) | Anti-NMDA Receptor Encephalitis (n = 19) | |
|---|---|---|---|---|---|
| Sex (F/M) | 7/7 | 0/10 | 8/9 | 13/10 | 12/7 |
| Age at serum collection (Years, mean ± SD) | 50 ± 12.2 | 56.3 ± 5.92 | 48.64 ± 11.33 | 6.86 ± 3.33 | 13.13 ± 5.62 |
| Disease duration (Years, mean ± SD) | 2.73 ± 3 | 15.9 ± 9.48 | - | 5.15 ± 2.95 | - |
| Disease-related information | Sporadic/familial SOD1-ALS 8/6 | CAG expansion (38 to 62 repeats) | - | SMN1 deletion Type II/III 17/6 | Autoimmune encephalitis |
| Muscle function at time of serum collection | 14 pts with limb weakness (walkers), of which 4 with dysphagia | 4 pts with fatigability; 1 pt with muscle cramps; 1 pt with limb weakness (walker); 4 pts with dysphagia and limb weakness (3 walkers, 1 sitter) | - | 18 sitters; 5 walkers | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malacarne, C.; Galbiati, M.; Giagnorio, E.; Cavalcante, P.; Salerno, F.; Andreetta, F.; Cagnoli, C.; Taiana, M.; Nizzardo, M.; Corti, S.; et al. Dysregulation of Muscle-Specific MicroRNAs as Common Pathogenic Feature Associated with Muscle Atrophy in ALS, SMA and SBMA: Evidence from Animal Models and Human Patients. Int. J. Mol. Sci. 2021, 22, 5673. https://doi.org/10.3390/ijms22115673
Malacarne C, Galbiati M, Giagnorio E, Cavalcante P, Salerno F, Andreetta F, Cagnoli C, Taiana M, Nizzardo M, Corti S, et al. Dysregulation of Muscle-Specific MicroRNAs as Common Pathogenic Feature Associated with Muscle Atrophy in ALS, SMA and SBMA: Evidence from Animal Models and Human Patients. International Journal of Molecular Sciences. 2021; 22(11):5673. https://doi.org/10.3390/ijms22115673
Chicago/Turabian StyleMalacarne, Claudia, Mariarita Galbiati, Eleonora Giagnorio, Paola Cavalcante, Franco Salerno, Francesca Andreetta, Cinza Cagnoli, Michela Taiana, Monica Nizzardo, Stefania Corti, and et al. 2021. "Dysregulation of Muscle-Specific MicroRNAs as Common Pathogenic Feature Associated with Muscle Atrophy in ALS, SMA and SBMA: Evidence from Animal Models and Human Patients" International Journal of Molecular Sciences 22, no. 11: 5673. https://doi.org/10.3390/ijms22115673