
Mitochondrial Lipid Homeostasis at the Crossroads of Liver and Heart Diseases

 ,
,
Abstract
:1. Introduction
Association between Liver and Heart Disease
2. Liver as a Central Organ for Lipid Metabolism
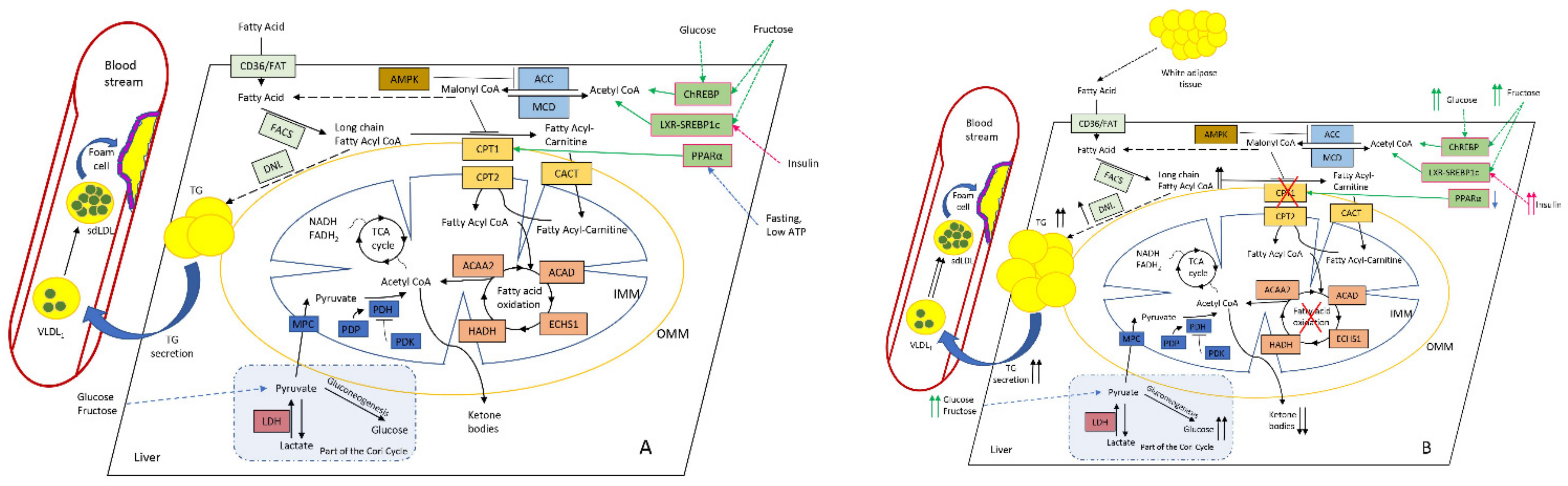
2.1. Lipids Homeostasis in the Liver Mitochondria: Fatty-Acid β-Oxidation
2.2. Interplay and Co-Regulation with Glucose Metabolism
2.3. Role of Perilipin 5 in NAFLD and Atherosclerosis
2.4. Role of the Liver Mitochondria in the Development of CVD-Promoting Dyslipidemia
3. Cardioprotection
4. Pharmaceutical Strategies to Treat NAFLD and Reduce CVD Risk
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACAA2 | acetyl-CoA acyltransferase 2 |
| ACAD | acyl-CoA dehydrogenase |
| ACC | acetyl-CoA carboxylase |
| AITC | allyl isothiocyanate |
| AMPK | AMP-activated protein kinase |
| ANT2 | ADP/ATP translocase 2 |
| APOE | apolipoprotein E |
| ATF4 | activating transcription factor 4 |
| BAT | brown adipose tissue |
| CACT | acylcarnitine translocase |
| CASP1 | caspase-1 |
| CAT | catalase |
| CCl4 | carbon tetrachloride |
| CCN1 | cellular communication network factor 1 |
| CES1 | carboxylesterase 1 |
| ChO | chitosan oligosaccharide |
| CHOP | C/EBP homologous protein |
| ChREBP | carbohydrate-responsive element-binding protein |
| ClC-2 | chloride voltage-gated channel 2 |
| CLOCK | circadian locomotor output cycles kaput |
| CPT | carnitine palmitoyltransferase |
| CREBH | CAMP-responsive element-binding protein, hepatic-specific |
| CRLS1 | cardiolipin synthase 1 |
| CVD | Cardiovascular diseases |
| DAGs | diacylglycerols |
| DNL | de novo lipogenesis |
| DPP4 | dipeptidyl peptidase-4 |
| ECHS1 | enoyl-CoA hydratase, short chain 1 |
| ELAVL1 | RNA-binding protein HuR |
| ERK1/2 | extracellular signal-regulated kinases 1 and 2 |
| ETC | electron transport chain |
| FA | fatty acid |
| FABP1 | fatty-acid-binding protein 1 |
| FACS | fatty-acid synthase |
| FAT | fatty-acid translocase |
| FGF21 | fibroblast growth factor 21 |
| FGFR1 | FGF receptor 1 |
| FNDC5 | fibronectin type III domain-containing protein 5 |
| FOH | farnesol |
| FRS2α | fibroblast growth factor receptor substrate 2 alpha |
| GCN2 | general control nonderepressible 2 |
| GDF15 | growth/differentiation factor 15 |
| GGPPS | geranylgeranyl pyrophosphate synthase |
| GNMT | glycine N-methyltransferase |
| GRK2 | G protein-coupled receptor kinase 2 |
| HADH | hydroxyacyl-CoA dehydrogenase |
| HC | high cholesterol |
| HCC | hepatocellular carcinoma |
| HDL-C | high-density lipoprotein cholesterol |
| IL-6 | Interleukin 6 |
| IMP2 | insulin-like growth factor 2 mRNA binding protein 2 |
| IGFBP1 | Insulin-like growth factor-binding protein 1 |
| IR | insulin resistance |
| IRS1 | insulin receptor substrate 1 |
| JNK | c-Jun N-terminal kinase |
| KLB | β-Klotho |
| LAMP2A | lysosome-associated membrane protein 2A |
| LAP1 | lamina-associated polypeptide 1 |
| LCAD | long-chain acyl-CoA dehydrogenase |
| LCFAs | long-chain FAs |
| LCHAD | long-chain 3-hydoxyacyl-CoA dehydrogenase |
| LDH | lactate dehydrogenase |
| LDL | low-density lipoprotein |
| LKB1 | liver kinase B1 |
| LPGAT1 | lysophosphatidylglycerol acyltransferase 1 |
| LRP1 | LDL receptor-related protein-1 |
| LXR | liver X receptor |
| MCD | malonyl-CoA decarboxylase |
| MCJ | methylation-controlled J protein |
| MFN2 | mitofusin 2 |
| MetS | metabolic syndrome |
| MPC | mitochondrial pyruvate carrier 1 |
| MUPs | major urinary proteins |
| NAFLD | non-alcoholic fatty liver disease |
| NASH | non-alcoholic steatohepatitis |
| NF-kB | nuclear factor kappa B |
| NLRP3 | NLR family pyrin domain-containing 3 |
| OS | oxidative stress |
| PA | palmitate |
| Plin5 | perilipin 5 |
| PCs | phosphatidylcholines |
| PDH | pyruvate dehydrogenase |
| PDK | pyruvate dehydrogenase kinase |
| PDP | pyruvate dehydrogenase phosphatase |
| PRX5 | peroxiredoxin |
| PTP1B | protein tyrosine phosphatase non-receptor type 1 |
| ROCK1 | rho-kinase 1 |
| RON | macrophage-stimulating 1 receptor |
| S100A11 | S100 calcium-binding protein A11 |
| SFA | saturated fatty acids |
| SIRT1 | sirtuin 1 |
| SLUG | snail family transcriptional repressor 2 |
| SMOC2 | secreted modular calcium-binding protein 2 |
| SOD1 | Cu/Zn-superoxide dismutase |
| SREBP1c | sterol regulatory element-binding protein-1c |
| STK25 | serine/threonine kinase 25 |
| T2DM | type 2 diabetes mellitus |
| TBK1 | TANK-binding kinase 1 |
| TC | total cholesterol |
| TCA cycle | tricarboxylic acid cycle |
| TGF-β | transforming growth factor beta |
| TFF3 | trefoil factor 3 |
| TM6SF2 | transmembrane 6 superfamily member 2 |
| TNF-α | tumor necrosis factor alpha |
| UCP2 | uncoupling protein 2 |
| VSIG4 | V-set and immunoglobulin domain-containing protein-4 |
| XBP1 | Xbp1-X-box binding protein 1 |
References
- Kasper, P.; Martin, A.; Lang, S.; Kütting, F.; Goeser, T.; Demir, M.; Steffen, H.-M. NAFLD and cardiovascular diseases: A clinical review. Clin. Res. Cardiol. 2020. [Google Scholar] [CrossRef]
- Baratta, F.; Pastori, D.; Angelico, F.; Balla, A.; Paganini, A.M.; Cocomello, N.; Ferro, D.; Violi, F.; Sanyal, A.J.; Del Ben, M. Nonalcoholic Fatty Liver Disease and Fibrosis Associated With Increased Risk of Cardiovascular Events in a Prospective Study. Clin. Gastroenterol. Hepatol. 2020, 18, 2324–2331.e4. [Google Scholar] [CrossRef]
- Liu, Y.; Zhong, G.-C.; Tan, H.-Y.; Hao, F.-B.; Hu, J.-J. Nonalcoholic fatty liver disease and mortality from all causes, cardiovascular disease, and cancer: A meta-analysis. Sci. Rep. 2019, 9, 11124. [Google Scholar] [CrossRef] [Green Version]
- Przybyszewski, E.M.; Targher, G.; Roden, M.; Corey, K.E. Nonalcoholic Fatty Liver Disease and Cardiovascular Disease. Clin. Liver Dis. 2021, 17, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Stols-Gonçalves, D.; Hovingh, G.K.; Nieuwdorp, M.; Holleboom, A.G. NAFLD and Atherosclerosis: Two Sides of the Same Dysmetabolic Coin? Trends Endocrinol. Metab. 2019, 30, 891–902. [Google Scholar] [CrossRef] [PubMed]
- Meex, R.C.R.; Watt, M.J. Hepatokines: Linking nonalcoholic fatty liver disease and insulin resistance. Nat. Rev. Endocrinol. 2017, 13, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Chiriac, S.; Stanciu, C.; Girleanu, I.; Cojocariu, C.; Sfarti, C.; Singeap, A.-M.; Cuciureanu, T.; Huiban, L.; Muzica, C.M.; Zenovia, S.; et al. Nonalcoholic Fatty Liver Disease and Cardiovascular Diseases: The Heart of the Matter. Can. J. Gastroenterol. Hepatol. 2021, 2021, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. The changing landscape of atherosclerosis. Nature 2021, 592, 524–533. [Google Scholar] [CrossRef]
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgözoğlu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Primer 2019, 5, 56. [Google Scholar] [CrossRef]
- Niederseer, D.; Wernly, B.; Aigner, E.; Stickel, F.; Datz, C. NAFLD and Cardiovascular Diseases: Epidemiological, Mechanistic and Therapeutic Considerations. J. Clin. Med. 2021, 10, 467. [Google Scholar] [CrossRef]
- Oni, E.; Budoff, M.J.; Zeb, I.; Li, D.; Veledar, E.; Polak, J.F.; Blankstein, R.; Wong, N.D.; Blaha, M.J.; Agatston, A.; et al. Nonalcoholic Fatty Liver Disease Is Associated With Arterial Distensibility and Carotid Intima-Media Thickness: (from the Multi-Ethnic Study of Atherosclerosis). Am. J. Cardiol. 2019, 124, 534–538. [Google Scholar] [CrossRef]
- Xin, Z.; Zhu, Y.; Wang, S.; Liu, S.; Xu, M.; Wang, T.; Lu, J.; Chen, Y.; Zhao, Z.; Wang, W.; et al. Associations of subclinical atherosclerosis with nonalcoholic fatty liver disease and fibrosis assessed by non-invasive score. Liver Int. 2020, 40, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Koo, B.K.; Allison, M.A.; Criqui, M.H.; Denenberg, J.O.; Wright, C.M. The association between liver fat and systemic calcified atherosclerosis. J. Vasc. Surg. 2020, 71, 204–211.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandrasekharan, K.; Alazawi, W. Genetics of Non-Alcoholic Fatty Liver and Cardiovascular Disease: Implications for Therapy? Front. Pharmacol. 2020, 10, 1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sazonova, M.A.; Ryzhkova, A.I.; Sinyov, V.V.; Galitsyna, E.V.; Melnichenko, A.A.; Demakova, N.A.; Sobenin, I.A.; Shkurat, T.P.; Orekhov, A.N. Mitochondrial Genome Mutations Associated with Myocardial Infarction. Dis. Markers 2018, 2018, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Mitrofanov, K.Y.; Zhelankin, A.V.; Shiganova, G.M.; Sazonova, M.A.; Bobryshev, Y.V.; Postnov, A.Y.; Sobenin, I.A.; Orekhov, A.N. Analysis of mitochondrial DNA heteroplasmic mutations A1555G, C3256T, T3336C, С5178А, G12315A, G13513A, G14459A, G14846A and G15059A in CHD patients with the history of myocardial infarction. Exp. Mol. Pathol. 2016, 100, 87–91. [Google Scholar] [CrossRef]
- Dabravolski, S.A.; Bezsonov, E.E.; Baig, M.S.; Popkova, T.V.; Nedosugova, L.V.; Starodubova, A.V.; Orekhov, A.N. Mitochondrial Mutations and Genetic Factors Determining NAFLD Risk. Int. J. Mol. Sci. 2021, 22, 4459. [Google Scholar] [CrossRef]
- Sliz, E.; Sebert, S.; Würtz, P.; Kangas, A.J.; Soininen, P.; Lehtimäki, T.; Kähönen, M.; Viikari, J.; Männikkö, M.; Ala-Korpela, M.; et al. NAFLD risk alleles in PNPLA3, TM6SF2, GCKR and LYPLAL1 show divergent metabolic effects. Hum. Mol. Genet. 2018, 27, 2214–2223. [Google Scholar] [CrossRef] [Green Version]
- Simons, N.; Isaacs, A.; Koek, G.H.; Kuč, S.; Schaper, N.C.; Brouwers, M.C.G.J. PNPLA3, TM6SF2, and MBOAT7 Genotypes and Coronary Artery Disease. Gastroenterology 2017, 152, 912–913. [Google Scholar] [CrossRef] [Green Version]
- Brouwers, M.C.G.J.; Simons, N.; Stehouwer, C.D.A.; Isaacs, A. Non-alcoholic fatty liver disease and cardiovascular disease: Assessing the evidence for causality. Diabetologia 2020, 63, 253–260. [Google Scholar] [CrossRef] [Green Version]
- Käräjämäki, A.J.; Hukkanen, J.; Kauma, H.; Kesäniemi, Y.A.; Ukkola, O. Metabolic syndrome but not genetic polymorphisms known to induce NAFLD predicts increased total mortality in subjects with NAFLD (OPERA study). Scand. J. Clin. Lab. Investig. 2020, 80, 106–113. [Google Scholar] [CrossRef]
- Grimaudo, S.; Pipitone, R.M.; Pennisi, G.; Celsa, C.; Cammà, C.; Di Marco, V.; Barcellona, M.R.; Boemi, R.; Enea, M.; Giannetti, A.; et al. Association Between PNPLA3 rs738409 C>G Variant and Liver-Related Outcomes in Patients With Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2020, 18, 935–944.e3. [Google Scholar] [CrossRef] [PubMed]
- Haas, J.T.; Vonghia, L.; Mogilenko, D.A.; Verrijken, A.; Molendi-Coste, O.; Fleury, S.; Deprince, A.; Nikitin, A.; Woitrain, E.; Ducrocq-Geoffroy, L.; et al. Transcriptional network analysis implicates altered hepatic immune function in NASH development and resolution. Nat. Metab. 2019, 1, 604–614. [Google Scholar] [CrossRef] [PubMed]
- Gehrke, N.; Schattenberg, J.M. Metabolic Inflammation—A Role for Hepatic Inflammatory Pathways as Drivers of Comorbidities in Nonalcoholic Fatty Liver Disease? Gastroenterology 2020, 158, 1929–1947.e6. [Google Scholar] [CrossRef]
- Chávez-Talavera, O.; Tailleux, A.; Lefebvre, P.; Staels, B. Bile Acid Control of Metabolism and Inflammation in Obesity, Type 2 Diabetes, Dyslipidemia, and Nonalcoholic Fatty Liver Disease. Gastroenterology 2017, 152, 1679–1694.e3. [Google Scholar] [CrossRef]
- Vos, D.Y.; van de Sluis, B. Function of the endolysosomal network in cholesterol homeostasis and metabolic-associated fatty liver disease (MAFLD). Mol. Metab. 2021, 101146. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Lee, H.S.; Cho, A.-R.; Lee, Y.-J.; Kwon, Y.-J. Non-Alcoholic Fatty Liver Disease Is an Independent Risk Factor for LDL Cholesterol Target Level. Int. J. Environ. Res. Public. Health 2021, 18, 3442. [Google Scholar] [CrossRef]
- Armandi, A.; Rosso, C.; Caviglia, G.P.; Bugianesi, E. Insulin Resistance across the Spectrum of Nonalcoholic Fatty Liver Disease. Metabolites 2021, 11, 155. [Google Scholar] [CrossRef]
- Fujii, H.; Kawada, N.; Japan Study Group of NAFLD (JSG-NAFLD). The Role of Insulin Resistance and Diabetes in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2020, 21, 3863. [Google Scholar] [CrossRef] [PubMed]
- Ryan, J.D.; Armitage, A.E.; Cobbold, J.F.; Banerjee, R.; Borsani, O.; Dongiovanni, P.; Neubauer, S.; Morovat, R.; Wang, L.M.; Pasricha, S.-R.; et al. Hepatic iron is the major determinant of serum ferritin in NAFLD patients. Liver Int. 2018, 38, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.A.; Yang, Y.; Zhang, L.; Sun, Z.; Jia, G.; Parrish, A.R.; Sowers, J.R. Insulin resistance, cardiovascular stiffening and cardiovascular disease. Metabolism 2021, 119, 154766. [Google Scholar] [CrossRef] [PubMed]
- Poznyak, A.; Grechko, A.V.; Poggio, P.; Myasoedova, V.A.; Alfieri, V.; Orekhov, A.N. The Diabetes Mellitus–Atherosclerosis Connection: The Role of Lipid and Glucose Metabolism and Chronic Inflammation. Int. J. Mol. Sci. 2020, 21, 1835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, X.; Kaminga, A.C.; Chen, J.; Luo, M.; Luo, J. Fetuin-A and Fetuin-B in Non-Alcoholic Fatty Liver Disease: A Meta-Analysis and Meta-Regression. Int. J. Environ. Res. Public. Health 2020, 17, 2735. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Xiao, J.; Zhao, Z.; Wang, M.; Wang, Y.; Xin, Y. Systematic Review and Meta-analysis of Circulating Fetuin-A Levels in Nonalcoholic Fatty Liver Disease. J. Clin. Transl. Hepatol. 2020, 9, 3. [Google Scholar] [CrossRef]
- Jiménez, M.C.; Sun, Q.; Schürks, M.; Hu, F.B.; Manson, J.E.; Rexrode, K.M. Circulating Fetuin-A and Risk of Ischemic Stroke in Women. Clin. Chem. 2014, 60, 165–173. [Google Scholar] [CrossRef] [Green Version]
- Lichtenauer, M.; Wernly, B.; Paar, V.; Rohm, I.; Jung, C.; Yilmaz, A.; Hoppe, U.C.; Schulze, P.C.; Kretzschmar, D.; Pistulli, R. Specifics of fetuin-A levels in distinct types of chronic heart failure. J. Clin. Lab. Anal. 2018, 32, e22179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catanzaro, R.; Selvaggio, F.; Sciuto, M.; Zanoli, L.; Yazdani, A.; He, F.; Marotta, F. Triglycerides to high-density lipoprotein cholesterol ratio for diagnosing nonalcoholic fatty liver disease. Minerva Gastroenterol. 2021. [Google Scholar] [CrossRef]
- Tutunchi, H.; Naeini, F.; Ebrahimi-mameghani, M.; Mobasseri, M.; Naghshi, S.; Ostadrahimi, A. The association of the steatosis severity, NAFLD fibrosis score and FIB-4 index with atherogenic dyslipidaemia in adult patients with NAFLD: A cross-sectional study. Int. J. Clin. Pract. 2021, 75, e14131. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Leon, E.; Connelly, M.A.; Konomi, J.V.; Caltharp, S.; Cleeton, R.; Vos, M.B. Increased atherogenic lipoprotein profile in children with NON-ALCOHOLIC steatohepatitis. Pediatr. Obes. 2020, 15, e12648. [Google Scholar] [CrossRef]
- Di Costanzo, A.; Ronca, A.; D’Erasmo, L.; Manfredini, M.; Baratta, F.; Pastori, D.; Di Martino, M.; Ceci, F.; Angelico, F.; Del Ben, M.; et al. HDL-Mediated Cholesterol Efflux and Plasma Loading Capacities Are Altered in Subjects with Metabolically- but Not Genetically Driven Non-Alcoholic Fatty Liver Disease (NAFLD). Biomedicines 2020, 8, 625. [Google Scholar] [CrossRef]
- Adorni, M.P.; Ronda, N.; Bernini, F.; Zimetti, F. High Density Lipoprotein Cholesterol Efflux Capacity and Atherosclerosis in Cardiovascular Disease: Pathophysiological Aspects and Pharmacological Perspectives. Cells 2021, 10, 574. [Google Scholar] [CrossRef]
- Gordon, S.M.; Amar, M.J.; Jeiran, K.; Stagliano, M.; Staller, E.; Playford, M.P.; Mehta, N.N.; Vaisar, T.; Remaley, A.T. Effect of niacin monotherapy on high density lipoprotein composition and function. Lipids Health Dis. 2020, 19, 190. [Google Scholar] [CrossRef] [PubMed]
- Aissa, A.F.; Tryndyak, V.; de Conti, A.; Melnyk, S.; Gomes, T.D.U.H.; Bianchi, M.L.P.; James, S.J.; Beland, F.A.; Antunes, L.M.G.; Pogribny, I.P. Effect of methionine-deficient and methionine-supplemented diets on the hepatic one-carbon and lipid metabolism in mice. Mol. Nutr. Food Res. 2014, 58, 1502–1512. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Guan, Y.; Yang, X.; Xia, Z.; Wu, J. Association of Serum Homocysteine Levels with Histological Severity of NAFLD. J. Gastrointestin. Liver Dis. 2020, 29, 51–58. [Google Scholar] [CrossRef]
- Yuan, S.; Mason, A.M.; Carter, P.; Burgess, S.; Larsson, S.C. Homocysteine, B vitamins, and cardiovascular disease: A Mendelian randomization study. BMC Med. 2021, 19, 97. [Google Scholar] [CrossRef] [PubMed]
- Djuric, D.; Jakovljevic, V.; Zivkovic, V.; Srejovic, I. Homocysteine and homocysteine-related compounds: An overview of the roles in the pathology of the cardiovascular and nervous systems. Can. J. Physiol. Pharmacol. 2018, 96, 991–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fricker, Z.P.; Pedley, A.; Massaro, J.M.; Vasan, R.S.; Hoffmann, U.; Benjamin, E.J.; Long, M.T. Liver Fat Is Associated With Markers of Inflammation and Oxidative Stress in Analysis of Data From the Framingham Heart Study. Clin. Gastroenterol. Hepatol. 2019, 17, 1157–1164.e4. [Google Scholar] [CrossRef]
- Katsarou, A.; Moustakas, I.I.; Pyrina, I.; Lembessis, P.; Koutsilieris, M.; Chatzigeorgiou, A. Metabolic inflammation as an instigator of fibrosis during non-alcoholic fatty liver disease. World J. Gastroenterol. 2020, 26, 1993–2011. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, K.J.; eun Yoo, M.; Kim, G.; Yoon, H.; Jo, K.; Youn, J.-C.; Yun, M.; Park, J.Y.; Shim, C.Y.; et al. Association of non-alcoholic steatohepatitis with subclinical myocardial dysfunction in non-cirrhotic patients. J. Hepatol. 2018, 68, 764–772. [Google Scholar] [CrossRef]
- Chiu, L.S.; Pedley, A.; Massaro, J.M.; Benjamin, E.J.; Mitchell, G.F.; McManus, D.D.; Aragam, J.; Vasan, R.S.; Cheng, S.; Long, M.T. The association of non-alcoholic fatty liver disease and cardiac structure and function—Framingham Heart Study. Liver Int. 2020, 40, 2445–2454. [Google Scholar] [CrossRef]
- Hodson, L. Hepatic fatty acid synthesis and partitioning: The effect of metabolic and nutritional state. Proc. Nutr. Soc. 2019, 78, 126–134. [Google Scholar] [CrossRef] [Green Version]
- Roumans, K.H.M.; Basset Sagarminaga, J.; Peters, H.P.F.; Schrauwen, P.; Schrauwen-Hinderling, V.B. Liver fat storage pathways: Methodologies and dietary effects. Curr. Opin. Lipidol. 2021, 32, 9–15. [Google Scholar] [CrossRef]
- Smith, G.I.; Shankaran, M.; Yoshino, M.; Schweitzer, G.G.; Chondronikola, M.; Beals, J.W.; Okunade, A.L.; Patterson, B.W.; Nyangau, E.; Field, T.; et al. Insulin resistance drives hepatic de novo lipogenesis in nonalcoholic fatty liver disease. J. Clin. Investig. 2020, 130, 1453–1460. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Tian, R.; She, Z.; Cai, J.; Li, H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2020, 152, 116–141. [Google Scholar] [CrossRef]
- Itoh, M.; Ogawa, Y.; Suganami, T. Chronic inflammation as a molecular basis of nonalcoholic steatohepatitis: Role of macrophages and fibroblasts in the liver. Nagoya J. Med. Sci. 2020, 82, 391–397. [Google Scholar] [PubMed]
- Chornyi, S.; IJlst, L.; van Roermund, C.W.T.; Wanders, R.J.A.; Waterham, H.R. Peroxisomal Metabolite and Cofactor Transport in Humans. Front. Cell Dev. Biol. 2021, 8, 613892. [Google Scholar] [CrossRef] [PubMed]
- Miotto, P.M.; Petrick, H.L.; Holloway, G.P. Acute insulin deprivation results in altered mitochondrial substrate sensitivity conducive to greater fatty acid transport. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E345–E353. [Google Scholar] [CrossRef] [PubMed]
- Brown, Z.J.; Fu, Q.; Ma, C.; Kruhlak, M.; Zhang, H.; Luo, J.; Heinrich, B.; Yu, S.J.; Zhang, Q.; Wilson, A.; et al. Carnitine palmitoyltransferase gene upregulation by linoleic acid induces CD4+ T cell apoptosis promoting HCC development. Cell Death Dis. 2018, 9, 620. [Google Scholar] [CrossRef]
- Fernandes, G.W.; Bocco, B.M.L.C. Hepatic mediators of lipid metabolism and ketogenesis: Focus on fatty liver and diabetes. Curr. Diabetes Rev. 2020, 16. [Google Scholar] [CrossRef] [PubMed]
- Adeva-Andany, M.M.; Carneiro-Freire, N.; Seco-Filgueira, M.; Fernández-Fernández, C.; Mouriño-Bayolo, D. Mitochondrial β-oxidation of saturated fatty acids in humans. Mitochondrion 2019, 46, 73–90. [Google Scholar] [CrossRef]
- Colbert, C.L.; Kim, C.-W.; Moon, Y.-A.; Henry, L.; Palnitkar, M.; McKean, W.B.; Fitzgerald, K.; Deisenhofer, J.; Horton, J.D.; Kwon, H.J. Crystal structure of Spot 14, a modulator of fatty acid synthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 18820–18825. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.H.; Kim, N.H.; Yun, J.S.; Cho, E.S.; Cha, Y.H.; Cho, S.B.; Lee, S.-H.; Cha, S.Y.; Kim, S.-Y.; Choi, J.; et al. Snail augments fatty acid oxidation by suppression of mitochondrial ACC2 during cancer progression. Life Sci. Alliance 2020, 3, e202000683. [Google Scholar] [CrossRef]
- Harriman, G.; Greenwood, J.; Bhat, S.; Huang, X.; Wang, R.; Paul, D.; Tong, L.; Saha, A.K.; Westlin, W.F.; Kapeller, R.; et al. Acetyl-CoA carboxylase inhibition by ND-630 reduces hepatic steatosis, improves insulin sensitivity, and modulates dyslipidemia in rats. Proc. Natl. Acad. Sci. USA 2016, 113, E1796–E1805. [Google Scholar] [CrossRef] [Green Version]
- Goedeke, L.; Bates, J.; Vatner, D.F.; Perry, R.J.; Wang, T.; Ramirez, R.; Li, L.; Ellis, M.W.; Zhang, D.; Wong, K.E.; et al. Acetyl-CoA Carboxylase Inhibition Reverses NAFLD and Hepatic Insulin Resistance but Promotes Hypertriglyceridemia in Rodents: Hepatology. Hepatology 2018, 68, 2197–2211. [Google Scholar] [CrossRef] [Green Version]
- Boudaba, N.; Marion, A.; Huet, C.; Pierre, R.; Viollet, B.; Foretz, M. AMPK Re-Activation Suppresses Hepatic Steatosis but its Downregulation Does Not Promote Fatty Liver Development. EBioMedicine 2018, 28, 194–209. [Google Scholar] [CrossRef] [Green Version]
- Ming, Y.; Yin, Y.; Sun, Z. Interaction of Nuclear Receptor Subfamily 4 Group A Member 1 (Nr4a1) and Liver Linase B1 (LKB1) Mitigates Type 2 Diabetes Mellitus by Activating Monophosphate-Activated Protein Kinase (AMPK)/Sirtuin 1 (SIRT1) Axis and Inhibiting Nuclear Factor-kappa B (NF-κB) Activation. Med. Sci. Monit. 2020, 26, e920278-1. [Google Scholar] [PubMed]
- Martínez-Jiménez, V.; Cortez-Espinosa, N.; Rodríguez-Varela, E.; Vega-Cárdenas, M.; Briones-Espinoza, M.; Ruíz-Rodríguez, V.M.; López-López, N.; Briseño-Medina, A.; Turiján-Espinoza, E.; Portales-Pérez, D.P. Altered levels of sirtuin genes (SIRT1, SIRT2, SIRT3 and SIRT6) and their target genes in adipose tissue from individual with obesity. Diabetes Metab. Syndr. Clin. Res. Rev. 2019, 13, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Kathirvel, E.; Morgan, K.; French, S.W.; Morgan, T.R. Acetyl-l-carnitine and lipoic acid improve mitochondrial abnormalities and serum levels of liver enzymes in a mouse model of nonalcoholic fatty liver disease. Nutr. Res. 2013, 33, 932–941. [Google Scholar] [CrossRef] [PubMed]
- Jun, D.W.; Cho, W.K.; Jun, J.H.; Kwon, H.J.; Jang, K.-S.; Kim, H.-J.; Jeon, H.J.; Lee, K.N.; Lee, H.L.; Lee, O.Y.; et al. Prevention of free fatty acid-induced hepatic lipotoxicity by carnitine via reversal of mitochondrial dysfunction. Liver Int. 2011, 31, 1315–1324. [Google Scholar] [CrossRef] [PubMed]
- Frigini, E.N.; Barrera, E.E.; Pantano, S.; Porasso, R.D. Role of membrane curvature on the activation/deactivation of Carnitine Palmitoyltransferase 1A: A coarse grain molecular dynamic study. Biochim. Biophys. Acta BBA Biomembr. 2020, 1862, 183094. [Google Scholar] [CrossRef]
- Auguet, T.; Berlanga, A.; Guiu-Jurado, E.; Martinez, S.; Porras, J.; Aragonès, G.; Sabench, F.; Hernandez, M.; Aguilar, C.; Sirvent, J.; et al. Altered Fatty Acid Metabolism-Related Gene Expression in Liver from Morbidly Obese Women with Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2014, 15, 22173–22187. [Google Scholar] [CrossRef] [Green Version]
- Naguib, G.; Morris, N.; Yang, S.; Fryzek, N.; Haynes-Williams, V.; Huang, W.A.; Norman-Wheeler, J.; Rotman, Y. Dietary fatty acid oxidation is decreased in non-alcoholic fatty liver disease: A palmitate breath test study. Liver Int. 2020, 40, 590–597. [Google Scholar] [CrossRef]
- Lee, J.; Choi, J.; Selen Alpergin, E.S.; Zhao, L.; Hartung, T.; Scafidi, S.; Riddle, R.C.; Wolfgang, M.J. Loss of Hepatic Mitochondrial Long-Chain Fatty Acid Oxidation Confers Resistance to Diet-Induced Obesity and Glucose Intolerance. Cell Rep. 2017, 20, 655–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Cristofano, M.; Ferramosca, A.; Di Giacomo, M.; Fusco, C.; Boscaino, F.; Luongo, D.; Vera Rotondi, A.; Maurano, F.; Cocca, E.; Mazzarella, G.; et al. Mechanisms underlying the hormetic effect of conjugated linoleic acid: Focus on Nrf2, mitochondria and NADPH oxidases. Free Radic. Biol. Med. 2021, 167, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Selen, E.S.; Choi, J.; Wolfgang, M.J. Discordant hepatic fatty acid oxidation and triglyceride hydrolysis leads to liver disease. JCI Insight 2021, 6, e135626. [Google Scholar] [CrossRef] [PubMed]
- Luukkonen, P.K.; Dufour, S.; Lyu, K.; Zhang, X.-M.; Hakkarainen, A.; Lehtimäki, T.E.; Cline, G.W.; Petersen, K.F.; Shulman, G.I.; Yki-Järvinen, H. Effect of a ketogenic diet on hepatic steatosis and hepatic mitochondrial metabolism in nonalcoholic fatty liver disease. Proc. Natl. Acad. Sci. USA 2020, 117, 7347–7354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fletcher, J.A.; Deja, S.; Satapati, S.; Fu, X.; Burgess, S.C.; Browning, J.D. Impaired ketogenesis and increased acetyl-CoA oxidation promote hyperglycemia in human fatty liver. JCI Insight 2019, 4, e127737. [Google Scholar] [CrossRef] [PubMed]
- Bjørndal, B.; Alterås, E.K.; Lindquist, C.; Svardal, A.; Skorve, J.; Berge, R.K. Associations between fatty acid oxidation, hepatic mitochondrial function, and plasma acylcarnitine levels in mice. Nutr. Metab. 2018, 15, 10. [Google Scholar] [CrossRef]
- Yin, J.; Zhu, Y.; Malik, V.; Li, X.; Peng, X.; Zhang, F.F.; Shan, Z.; Liu, L. Intake of Sugar-Sweetened and Low-Calorie Sweetened Beverages and Risk of Cardiovascular Disease: A Meta-Analysis and Systematic Review. Adv. Nutr. 2021, 12, 89–101. [Google Scholar] [CrossRef]
- Haslam, D.E.; Peloso, G.M.; Herman, M.A.; Dupuis, J.; Lichtenstein, A.H.; Smith, C.E.; McKeown, N.M. Beverage Consumption and Longitudinal Changes in Lipoprotein Concentrations and Incident Dyslipidemia in US Adults: The Framingham Heart Study. J. Am. Heart Assoc. 2020, 9, e014083. [Google Scholar] [CrossRef]
- Mamounis, K.J.; Yasrebi, A.; Roepke, T.A. Linoleic acid causes greater weight gain than saturated fat without hypothalamic inflammation in the male mouse. J. Nutr. Biochem. 2017, 40, 122–131. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; O’Connell, J.F.; Carlson, O.D.; González-Mariscal, I.; Kim, Y.; Moaddel, R.; Ghosh, P.; Egan, J.M. Linoleic acid in diets of mice increases total endocannabinoid levels in bowel and liver: Modification by dietary glucose. Obes. Sci. Pract. 2019, 5, 383–394. [Google Scholar] [CrossRef] [Green Version]
- Chandra, A.; Røsjø, H.; Svensson, M.; Vigen, T.; Ihle-Hansen, H.; Orstad, E.B.; Rønning, O.M.; Lyngbakken, M.N.; Nygård, S.; Berge, T.; et al. Plasma linoleic acid levels and cardiovascular risk factors: Results from the Norwegian ACE 1950 Study. Eur. J. Clin. Nutr. 2020, 74, 1707–1717. [Google Scholar] [CrossRef]
- Softic, S.; Gupta, M.K.; Wang, G.-X.; Fujisaka, S.; O’Neill, B.T.; Rao, T.N.; Willoughby, J.; Harbison, C.; Fitzgerald, K.; Ilkayeva, O.; et al. Divergent effects of glucose and fructose on hepatic lipogenesis and insulin signaling. J. Clin. Investig. 2017, 127, 4059–4074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiStefano, J.K. Fructose-mediated effects on gene expression and epigenetic mechanisms associated with NAFLD pathogenesis. Cell. Mol. Life Sci. 2020, 77, 2079–2090. [Google Scholar] [CrossRef] [PubMed]
- Lanaspa, M.A.; Sanchez-Lozada, L.G.; Choi, Y.-J.; Cicerchi, C.; Kanbay, M.; Roncal-Jimenez, C.A.; Ishimoto, T.; Li, N.; Marek, G.; Duranay, M.; et al. Uric Acid Induces Hepatic Steatosis by Generation of Mitochondrial Oxidative Stress. J. Biol. Chem. 2012, 287, 40732–40744. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.-J.; Shin, H.-S.; Choi, H.S.; Park, J.-W.; Jo, I.; Oh, E.-S.; Lee, K.-Y.; Lee, B.-H.; Johnson, R.J.; Kang, D.-H. Uric acid induces fat accumulation via generation of endoplasmic reticulum stress and SREBP-1c activation in hepatocytes. Lab. Investig. 2014, 94, 1114–1125. [Google Scholar] [CrossRef] [Green Version]
- Vavrova, E.; Lenoir, V.; Alves-Guerra, M.-C.; Denis, R.G.; Castel, J.; Esnous, C.; Dyck, J.R.B.; Luquet, S.; Metzger, D.; Bouillaud, F.; et al. Muscle expression of a malonyl-CoA-insensitive carnitine palmitoyltransferase-1 protects mice against high-fat/high-sucrose diet-induced insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E649–E660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Weeghel, M.; Abdurrachim, D.; Nederlof, R.; Argmann, C.A.; Houtkooper, R.H.; Hagen, J.; Nabben, M.; Denis, S.; Ciapaite, J.; Kolwicz, S.C.; et al. Increased cardiac fatty acid oxidation in a mouse model with decreased malonyl-CoA sensitivity of CPT1B. Cardiovasc. Res. 2018, 114, 1324–1334. [Google Scholar] [CrossRef] [PubMed]
- Softic, S.; Meyer, J.G.; Wang, G.-X.; Gupta, M.K.; Batista, T.M.; Lauritzen, H.P.M.M.; Fujisaka, S.; Serra, D.; Herrero, L.; Willoughby, J.; et al. Dietary Sugars Alter Hepatic Fatty Acid Oxidation via Transcriptional and Post-translational Modifications of Mitochondrial Proteins. Cell Metab. 2019, 30, 735–753.e4. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, Y.-L.; Qiu, G.; Pian, L.; Guo, L.; Cao, H.; Liu, J.; Zhao, Y.; Li, X.; Xu, Z.; et al. Hepatic neddylation targets and stabilizes electron transfer flavoproteins to facilitate fatty acid β-oxidation. Proc. Natl. Acad. Sci. USA 2020, 117, 2473–2483. [Google Scholar] [CrossRef]
- Montagna, C.; Cirotti, C.; Rizza, S.; Filomeni, G. When S -Nitrosylation Gets to Mitochondria: From Signaling to Age-Related Diseases. Antioxid. Redox Signal. 2020, 32, 884–905. [Google Scholar] [CrossRef]
- Wang, C.; Zhao, Y.; Gao, X.; Li, L.; Yuan, Y.; Liu, F.; Zhang, L.; Wu, J.; Hu, P.; Zhang, X.; et al. Perilipin 5 improves hepatic lipotoxicity by inhibiting lipolysis. Hepatology 2015, 61, 870–882. [Google Scholar] [CrossRef]
- Jin, Y.; Tan, Y.; Chen, L.; Liu, Y.; Ren, Z. Reactive Oxygen Species Induces Lipid Droplet Accumulation in HepG2 Cells by Increasing Perilipin 2 Expression. Int. J. Mol. Sci. 2018, 19, 3445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trevino, M.B.; Mazur-Hart, D.; Machida, Y.; King, T.; Nadler, J.; Galkina, E.V.; Poddar, A.; Dutta, S.; Imai, Y. Liver Perilipin 5 Expression Worsens Hepatosteatosis But Not Insulin Resistance in High Fat-Fed Mice. Mol. Endocrinol. 2015, 29, 1414–1425. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Zheng, S.; Attie, A.D.; Keller, M.P.; Bernlohr, D.A.; Blaner, W.S.; Newberry, E.P.; Davidson, N.O.; Chen, A. Perilipin 5 and liver fatty acid binding protein function to restore quiescence in mouse hepatic stellate cells. J. Lipid Res. 2018, 59, 416–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.; Chen, A. Perilipin 5 restores the formation of lipid droplets in activated hepatic stellate cells and inhibits their activation. Lab. Investig. 2016, 96, 791–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keenan, S.N.; Meex, R.C.; Lo, J.C.Y.; Ryan, A.; Nie, S.; Montgomery, M.K.; Watt, M.J. Perilipin 5 Deletion in Hepatocytes Remodels Lipid Metabolism and Causes Hepatic Insulin Resistance in Mice. Diabetes 2019, 68, 543–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Sreenivasan, U.; Hu, H.; Saladino, A.; Polster, B.M.; Lund, L.M.; Gong, D.; Stanley, W.C.; Sztalryd, C. Perilipin 5, a lipid droplet-associated protein, provides physical and metabolic linkage to mitochondria. J. Lipid Res. 2011, 52, 2159–2168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Loh, K.; Song, Z.; Yang, H.; Zhang, Y.; Lin, S. Update on glycerol-3-phosphate acyltransferases: The roles in the development of insulin resistance. Nutr. Diabetes 2018, 8, 34. [Google Scholar] [CrossRef] [PubMed]
- Benador, I.Y.; Veliova, M.; Mahdaviani, K.; Petcherski, A.; Wikstrom, J.D.; Assali, E.A.; Acín-Pérez, R.; Shum, M.; Oliveira, M.F.; Cinti, S.; et al. Mitochondria Bound to Lipid Droplets Have Unique Bioenergetics, Composition, and Dynamics that Support Lipid Droplet Expansion. Cell Metab. 2018, 27, 869–885.e6. [Google Scholar] [CrossRef] [Green Version]
- Tan, Y.; Jin, Y.; Wang, Q.; Huang, J.; Wu, X.; Ren, Z. Ren Perilipin 5 Protects against Cellular Oxidative Stress by Enhancing Mitochondrial Function in HepG2 Cells. Cells 2019, 8, 1241. [Google Scholar] [CrossRef] [Green Version]
- Asimakopoulou, A.; Engel, K.M.; Gassler, N.; Bracht, T.; Sitek, B.; Buhl, E.M.; Kalampoka, S.; Pinoé-Schmidt, M.; van Helden, J.; Schiller, J.; et al. Deletion of Perilipin 5 Protects against Hepatic Injury in Nonalcoholic Fatty Liver Disease via Missing Inflammasome Activation. Cells 2020, 9, 1346. [Google Scholar] [CrossRef]
- Asimakopoulou, A.; Vucur, M.; Luedde, T.; Schneiders, S.; Kalampoka, S.; Weiss, T.; Weiskirchen, R. Perilipin 5 and Lipocalin 2 Expression in Hepatocellular Carcinoma. Cancers 2019, 11, 385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, P.; Li, M.; Han, X.; Bi, Y.; Zhang, W.; Wu, Z.; Wu, G. Perilipin 5 deficiency promotes atherosclerosis progression through accelerating inflammation, apoptosis, and oxidative stress. J. Cell. Biochem. 2019, 120, 19107–19123. [Google Scholar] [CrossRef]
- Liu, Q.; Zhou, Z.; Liu, P.; Zhang, S. Comparative proteomic study of liver lipid droplets and mitochondria in mice housed at different temperatures. FEBS Lett. 2019, 593, 2118–2138. [Google Scholar] [CrossRef] [PubMed]
- Badenes, M.; Amin, A.; González-García, I.; Félix, I.; Burbridge, E.; Cavadas, M.; Ortega, F.J.; de Carvalho, É.; Faísca, P.; Carobbio, S.; et al. Deletion of iRhom2 protects against diet-induced obesity by increasing thermogenesis. Mol. Metab. 2020, 31, 67–84. [Google Scholar] [CrossRef]
- Giles, D.A.; Moreno-Fernandez, M.E.; Stankiewicz, T.E.; Graspeuntner, S.; Cappelletti, M.; Wu, D.; Mukherjee, R.; Chan, C.C.; Lawson, M.J.; Klarquist, J.; et al. Thermoneutral housing exacerbates nonalcoholic fatty liver disease in mice and allows for sex-independent disease modeling. Nat. Med. 2017, 23, 829–838. [Google Scholar] [CrossRef]
- Mass Sanchez, P.B.; Krizanac, M.; Weiskirchen, R.; Asimakopoulos, A. Understanding the Role of Perilipin 5 in Non-Alcoholic Fatty Liver Disease and Its Role in Hepatocellular Carcinoma: A Review of Novel Insights. Int. J. Mol. Sci. 2021, 22, 5284. [Google Scholar] [CrossRef] [PubMed]
- Herker, E.; Vieyres, G.; Beller, M.; Krahmer, N.; Bohnert, M. Lipid Droplet Contact Sites in Health and Disease. Trends Cell Biol. 2021, 31, 345–358. [Google Scholar] [CrossRef]
- Chen, Z.; Qin, H.; Qiu, S.; Chen, G.; Chen, Y. Correlation of triglyceride to high-density lipoprotein cholesterol ratio with nonalcoholic fatty liver disease among the non-obese Chinese population with normal blood lipid levels: A retrospective cohort research. Lipids Health Dis. 2019, 18, 162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, X.Y.; Shi, D.; Ding, J.; Cheng, Z.Y.; Li, H.Y.; Li, J.S.; Pu, H.Q.; Yang, A.M.; He, C.L.; Zhang, J.P.; et al. Total cholesterol to high-density lipoprotein cholesterol ratio is a significant predictor of nonalcoholic fatty liver: Jinchang cohort study. Lipids Health Dis. 2019, 18, 47. [Google Scholar] [CrossRef] [PubMed]
- Kunutsor, S.K.; Zaccardi, F.; Karppi, J.; Kurl, S.; Laukkanen, J.A. Is High Serum LDL/HDL Cholesterol Ratio an Emerging Risk Factor for Sudden Cardiac Death? Findings from the KIHD Study. J. Atheroscler. Thromb. 2017, 24, 600–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Aicha, S.; Badimon, L.; Vilahur, G. Advances in HDL: Much More than Lipid Transporters. Int. J. Mol. Sci. 2020, 21, 732. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.; Liu, B.; Huang, K.; Huang, K. Enoyl coenzyme A hydratase 1 protects against high-fat-diet-induced hepatic steatosis and insulin resistance. Biochem. Biophys. Res. Commun. 2018, 499, 403–409. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, J.; Sun, H.; Liu, X.; Zheng, Y.; Xu, D.; Wang, J.; Jia, D.; Han, X.; Liu, F.; et al. Defective Phosphatidylglycerol Remodeling Causes Hepatopathy, Linking Mitochondrial Dysfunction to Hepatosteatosis. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 763–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, C.; Xiong, H.; Hu, Y.; Wang, W.; Mei, G.; Wang, H.; Li, Y.; Zhou, Z.; Meng, F.; Zhang, P.; et al. Cardiolipin Synthase 1 Ameliorates NASH Through Activating Transcription Factor 3 Transcriptional Inactivation. Hepatology 2020, 72, 1949–1967. [Google Scholar] [CrossRef]
- Hernández-Alvarez, M.I.; Sebastián, D.; Vives, S.; Ivanova, S.; Bartoccioni, P.; Kakimoto, P.; Plana, N.; Veiga, S.R.; Hernández, V.; Vasconcelos, N.; et al. Deficient Endoplasmic Reticulum-Mitochondrial Phosphatidylserine Transfer Causes Liver Disease. Cell 2019, 177, 881–895.e17. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Tussy, P.; Fernández-Ramos, D.; Lopitz-Otsoa, F.; Simón, J.; Barbier-Torres, L.; Gomez-Santos, B.; Nuñez-Garcia, M.; Azkargorta, M.; Gutiérrez-de Juan, V.; Serrano-Macia, M.; et al. miR-873-5p targets mitochondrial GNMT-Complex II interface contributing to non-alcoholic fatty liver disease. Mol. Metab. 2019, 29, 40–54. [Google Scholar] [CrossRef]
- Liu, R.; Chen, L.; Wang, Y.; Zhang, G.; Cheng, Y.; Feng, Z.; Bai, X.; Liu, J. High ratio of ω-3/ω-6 polyunsaturated fatty acids targets mTORC1 to prevent high-fat diet-induced metabolic syndrome and mitochondrial dysfunction in mice. J. Nutr. Biochem. 2020, 79, 108330. [Google Scholar] [CrossRef]
- Kang, S.G.; Choi, M.J.; Jung, S.-B.; Chung, H.K.; Chang, J.Y.; Kim, J.T.; Kang, Y.E.; Lee, J.H.; Hong, H.J.; Jun, S.M.; et al. Differential roles of GDF15 and FGF21 in systemic metabolic adaptation to the mitochondrial integrated stress response. iScience 2021, 24, 102181. [Google Scholar] [CrossRef] [PubMed]
- Seitz, S.; Kwon, Y.; Hartleben, G.; Jülg, J.; Sekar, R.; Krahmer, N.; Najafi, B.; Loft, A.; Gancheva, S.; Stemmer, K.; et al. Hepatic Rab24 controls blood glucose homeostasis via improving mitochondrial plasticity. Nat. Metab. 2019, 1, 1009–1026. [Google Scholar] [CrossRef]
- Barbier-Torres, L.; Fortner, K.A.; Iruzubieta, P.; Delgado, T.C.; Giddings, E.; Chen, Y.; Champagne, D.; Fernández-Ramos, D.; Mestre, D.; Gomez-Santos, B.; et al. Silencing hepatic MCJ attenuates non-alcoholic fatty liver disease (NAFLD) by increasing mitochondrial fatty acid oxidation. Nat. Commun. 2020, 11, 3360. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Zhang, Y.; Park, S.-Y.; Joseph, A.-M.; Han, C.; Park, H.-J.; Kalavalapalli, S.; Chun, S.-K.; Morgan, D.; Kim, J.-S.; et al. Mitochondrial ATP transporter depletion protects mice against liver steatosis and insulin resistance. Nat. Commun. 2017, 8, 14477. [Google Scholar] [CrossRef]
- Cruces-Sande, M.; Vila-Bedmar, R.; Arcones, A.C.; González-Rodríguez, Á.; Rada, P.; Gutiérrez-de-Juan, V.; Vargas-Castrillón, J.; Iruzubieta, P.; Sánchez-González, C.; Formentini, L.; et al. Involvement of G protein-coupled receptor kinase 2 (GRK2) in the development of non-alcoholic steatosis and steatohepatitis in mice and humans. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2018, 1864, 3655–3667. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.-W.; Addy, C.; Kusunoki, J.; Anderson, N.N.; Deja, S.; Fu, X.; Burgess, S.C.; Li, C.; Ruddy, M.; Chakravarthy, M.; et al. Acetyl CoA Carboxylase Inhibition Reduces Hepatic Steatosis but Elevates Plasma Triglycerides in Mice and Humans: A Bedside to Bench Investigation. Cell Metab. 2017, 26, 394–406.e6. [Google Scholar] [CrossRef] [PubMed]
- Takagi, H.; Ikehara, T.; Kashiwagi, Y.; Hashimoto, K.; Nanchi, I.; Shimazaki, A.; Nambu, H.; Yukioka, H. ACC2 Deletion Enhances IMCL Reduction Along With Acetyl-CoA Metabolism and Improves Insulin Sensitivity in Male Mice. Endocrinology 2018, 159, 3007–3019. [Google Scholar] [CrossRef]
- Huang, H.; Lee, S.-H.; Sousa-Lima, I.; Kim, S.S.; Hwang, W.M.; Dagon, Y.; Yang, W.-M.; Cho, S.; Kang, M.-C.; Seo, J.A.; et al. Rho-kinase/AMPK axis regulates hepatic lipogenesis during overnutrition. J. Clin. Investig. 2018, 128, 5335–5350. [Google Scholar] [CrossRef] [Green Version]
- Li, C.-X.; Gao, J.-G.; Wan, X.-Y.; Chen, Y.; Xu, C.-F.; Feng, Z.-M.; Zeng, H.; Lin, Y.-M.; Ma, H.; Xu, P.; et al. Allyl isothiocyanate ameliorates lipid accumulation and inflammation in nonalcoholic fatty liver disease via the Sirt1/AMPK and NF-κB signaling pathways. World J. Gastroenterol. 2019, 25, 5120–5133. [Google Scholar] [CrossRef]
- Goetzman, E.S.; Bharathi, S.S.; Zhang, Y.; Zhao, X.-J.; Dobrowolski, S.F.; Peasley, K.; Sims-Lucas, S.; Monga, S.P. Impaired mitochondrial medium-chain fatty acid oxidation drives periportal macrovesicular steatosis in sirtuin-5 knockout mice. Sci. Rep. 2020, 10, 18367. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zhao, Q.; Song, N.; Yan, Z.; Lin, R.; Wu, S.; Jiang, L.; Hong, S.; Xie, J.; Zhou, H.; et al. AdipoR1/AdipoR2 dual agonist recovers nonalcoholic steatohepatitis and related fibrosis via endoplasmic reticulum-mitochondria axis. Nat. Commun. 2020, 11, 5807. [Google Scholar] [CrossRef]
- Liu, J.; Jiang, S.; Zhao, Y.; Sun, Q.; Zhang, J.; Shen, D.; Wu, J.; Shen, N.; Fu, X.; Sun, X.; et al. Geranylgeranyl diphosphate synthase (GGPPS) regulates non-alcoholic fatty liver disease (NAFLD)-fibrosis progression by determining hepatic glucose/fatty acid preference under high-fat diet conditions: GGPPS regulates NAFLD-fibrosis by glycolysis. J. Pathol. 2018, 246, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Wang, Y.; Zhang, J.; Liu, B.; Deng, X.; Xin, S.; Xu, K. N-glycosylation of CREBH improves lipid metabolism and attenuates lipotoxicity in NAFLD by modulating PPARα and SCD-1. FASEB J. 2020, 34, 15338–15363. [Google Scholar] [CrossRef]
- Regué, L.; Minichiello, L.; Avruch, J.; Dai, N. Liver-specific deletion of IGF2 mRNA binding protein-2/IMP2 reduces hepatic fatty acid oxidation and increases hepatic triglyceride accumulation. J. Biol. Chem. 2019, 294, 11944–11951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pant, A.; Rondini, E.A.; Kocarek, T.A. Farnesol induces fatty acid oxidation and decreases triglyceride accumulation in steatotic HepaRG cells. Toxicol. Appl. Pharmacol. 2019, 365, 61–70. [Google Scholar] [CrossRef]
- Wu, X.; Zheng, H.; Yang, R.; Luan, X.; Zhang, L.; Jin, Q.; Jin, Y.; Xue, J. Mouse trefoil factor 3 ameliorated high-fat-diet-induced hepatic steatosis via increasing peroxisome proliferator-activated receptor-α-mediated fatty acid oxidation. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E436–E445. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Mera, P.; Casas, J.; Salvador, J.; Rodríguez, A.; Alonso, S.; Sebastián, D.; Soler-Vázquez, M.C.; Montironi, C.; Recalde, S.; et al. Liver CPT1A gene therapy reduces diet-induced hepatic steatosis in mice and highlights potential lipid biomarkers for human NAFLD. FASEB J. 2020, 34, 11816–11837. [Google Scholar] [CrossRef]
- Mohammed, S.; Nicklas, E.H.; Thadathil, N.; Selvarani, R.; Royce, G.H.; Kinter, M.; Richardson, A.; Deepa, S.S. Role of necroptosis in chronic hepatic inflammation and fibrosis in a mouse model of increased oxidative stress. Free Radic. Biol. Med. 2021, 164, 315–328. [Google Scholar] [CrossRef]
- Kim, M.H.; Seong, J.B.; Huh, J.-W.; Bae, Y.C.; Lee, H.-S.; Lee, D.-S. Peroxiredoxin 5 ameliorates obesity-induced non-alcoholic fatty liver disease through the regulation of oxidative stress and AMP-activated protein kinase signaling. Redox Biol. 2020, 28, 101315. [Google Scholar] [CrossRef]
- Shin, S.-K.; Cho, H.-W.; Song, S.-E.; Bae, J.-H.; Im, S.-S.; Hwang, I.; Ha, H.; Song, D.-K. Ablation of catalase promotes non-alcoholic fatty liver via oxidative stress and mitochondrial dysfunction in diet-induced obese mice. Pflüg. Arch. Eur. J. Physiol. 2019, 471, 829–843. [Google Scholar] [CrossRef]
- Liu, S.; Yuan, J.; Yue, W.; Bi, Y.; Shen, X.; Gao, J.; Xu, X.; Lu, Z. GCN2 deficiency protects against high fat diet induced hepatic steatosis and insulin resistance in mice. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2018, 1864, 3257–3267. [Google Scholar] [CrossRef]
- Luo, X.; Shi, X.; Sun, Z.; Xiao, J.; Song, H.; Lu, G.; Xu, X. GCN2 Deficiency Enhances Protective Effects of Exercise on Hepatic Steatosis. BioMed Res. Int. 2020, 2020, 1–10. [Google Scholar]
- Feng, W.; Lei, T.; Wang, Y.; Feng, R.; Yuan, J.; Shen, X.; Wu, Y.; Gao, J.; Ding, W.; Lu, Z. GCN2 deficiency ameliorates cardiac dysfunction in diabetic mice by reducing lipotoxicity and oxidative stress. Free Radic. Biol. Med. 2019, 130, 128–139. [Google Scholar] [CrossRef]
- Wang, Y.; Lei, T.; Yuan, J.; Wu, Y.; Shen, X.; Gao, J.; Feng, W.; Lu, Z. GCN2 deficiency ameliorates doxorubicin-induced cardiotoxicity by decreasing cardiomyocyte apoptosis and myocardial oxidative stress. Redox Biol. 2018, 17, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Pi, H.; Liu, M.; Xi, Y.; Chen, M.; Tian, L.; Xie, J.; Chen, M.; Wang, Z.; Yang, M.; Yu, Z.; et al. Long-term exercise prevents hepatic steatosis: A novel role of FABP1 in regulation of autophagy-lysosomal machinery. FASEB J. 2019, 33, 11870–11883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, J.N.; Dey, A.; Cai, J.; Zhang, J.; Tian, Y.; Kennett, M.; Ma, Y.; Liang, T.J.; Patterson, A.D.; Hankey-Giblin, P.A. Metabolic Profiling Reveals Aggravated Non-Alcoholic Steatohepatitis in High-Fat High-Cholesterol Diet-Fed Apolipoprotein E-Deficient Mice Lacking Ron Receptor Signaling. Metabolites 2020, 10, 326. [Google Scholar] [CrossRef]
- Yu, Y.; Liu, Y.; An, W.; Song, J.; Zhang, Y.; Zhao, X. STING-mediated inflammation in Kupffer cells contributes to progression of nonalcoholic steatohepatitis. J. Clin. Investig. 2018, 129, 546–555. [Google Scholar] [CrossRef]
- Nardo, A.D.; Grün, N.G.; Zeyda, M.; Dumanic, M.; Oberhuber, G.; Rivelles, E.; Helbich, T.H.; Markgraf, D.F.; Roden, M.; Claudel, T.; et al. Impact of osteopontin on the development of non-alcoholic liver disease and related hepatocellular carcinoma. Liver Int. 2020, 40, 1620–1633. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, A.; Haller, A.; Cash, J.G.; Nam, C.; Igel, E.; Roebroek, A.J.M.; Hui, D.Y. Mutation in the distal NPxY motif of LRP1 alleviates dietary cholesterol-induced dyslipidemia and tissue inflammation. J. Lipid Res. 2021, 62, 100012. [Google Scholar] [CrossRef]
- Li, B.-T.; Sun, M.; Li, Y.-F.; Wang, J.-Q.; Zhou, Z.-M.; Song, B.-L.; Luo, J. Disruption of the ERLIN–TM6SF2–APOB complex destabilizes APOB and contributes to non-alcoholic fatty liver disease. PLoS Genet. 2020, 16, e1008955. [Google Scholar] [CrossRef]
- Ma, S.Y.; Sun, K.S.; Zhang, M.; Zhou, X.; Zheng, X.H.; Tian, S.Y.; Liu, Y.S.; Chen, L.; Gao, X.; Ye, J.; et al. Disruption of Plin5 degradation by CMA causes lipid homeostasis imbalance in NAFLD. Liver Int. 2020, 40, 2427–2438. [Google Scholar] [CrossRef] [PubMed]
- Khare, T.; Khare, S.; Angdisen, J.J.; Zhang, Q.; Stuckel, A.; Mooney, B.P.; Ridenhour, S.E.; Gitan, R.S.; Hammoud, G.M.; Ibdah, J.A. Defects in long-chain 3-hydroxy acyl-CoA dehydrogenase lead to hepatocellular carcinoma: A novel etiology of hepatocellular carcinoma. Int. J. Cancer 2020, 147, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Queiroz, J.; Hussain, M.M. Nonalcoholic fatty liver disease in CLOCK mutant mice. J. Clin. Investig. 2020. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lin, H.; Jiang, L.; Shang, Q.; Yin, L.; Lin, J.D.; Wu, W.-S.; Rui, L. Hepatic Slug epigenetically promotes liver lipogenesis, fatty liver disease, and type 2 diabetes. J. Clin. Investig. 2020, 130, 2992–3004. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.-Y.; Hernandez-Ono, A.; Fedotova, T.; Östlund, C.; Lee, M.J.; Gibeley, S.B.; Liang, C.-C.; Dauer, W.T.; Ginsberg, H.N.; Worman, H.J. Nuclear envelope–localized torsinA-LAP1 complex regulates hepatic VLDL secretion and steatosis. J. Clin. Investig. 2019, 129, 4885–4900. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Gonzales, N.M.; Lonard, D.M.; Putluri, N.; Zhu, B.; Dacso, C.C.; York, B.; O’Malley, B.W. XBP1 links the 12-hour clock to NAFLD and regulation of membrane fluidity and lipid homeostasis. Nat. Commun. 2020, 11, 6215. [Google Scholar] [CrossRef]
- Huh, J.Y.; Reilly, S.M.; Abu-Odeh, M.; Murphy, A.N.; Mahata, S.K.; Zhang, J.; Cho, Y.; Seo, J.B.; Hung, C.-W.; Green, C.R.; et al. TANK-Binding Kinase 1 Regulates the Localization of Acyl-CoA Synthetase ACSL1 to Control Hepatic Fatty Acid Oxidation. Cell Metab. 2020, 32, 1012–1027.e7. [Google Scholar] [CrossRef]
- Liou, C.-J.; Wei, C.-H.; Chen, Y.-L.; Cheng, C.-Y.; Wang, C.-L.; Huang, W.-C. Fisetin Protects Against Hepatic Steatosis Through Regulation of the Sirt1/AMPK and Fatty Acid β-Oxidation Signaling Pathway in High-Fat Diet-Induced Obese Mice. Cell. Physiol. Biochem. 2018, 49, 1870–1884. [Google Scholar] [CrossRef]
- Liou, C.-J.; Wu, S.-J.; Shen, S.-C.; Chen, L.-C.; Chen, Y.-L.; Huang, W.-C. Phloretin ameliorates hepatic steatosis through regulation of lipogenesis and Sirt1/AMPK signaling in obese mice. Cell Biosci. 2020, 10, 114. [Google Scholar] [CrossRef]
- Tao, W.; Sun, W.; Liu, L.; Wang, G.; Xiao, Z.; Pei, X.; Wang, M. Chitosan Oligosaccharide Attenuates Nonalcoholic Fatty Liver Disease Induced by High Fat Diet through Reducing Lipid Accumulation, Inflammation and Oxidative Stress in C57BL/6 Mice. Mar. Drugs 2019, 17, 645. [Google Scholar] [CrossRef] [Green Version]
- Domínguez-Pérez, M.; Simoni-Nieves, A.; Rosales, P.; Nuño-Lámbarri, N.; Rosas-Lemus, M.; Souza, V.; Miranda, R.U.; Bucio, L.; Uribe Carvajal, S.; Marquardt, J.U.; et al. Cholesterol burden in the liver induces mitochondrial dynamic changes and resistance to apoptosi. J. Cell. Physiol. 2019, 234, 7213–7223. [Google Scholar] [CrossRef] [PubMed]
- Lindquist, C.; Bjørndal, B.; Bakke, H.G.; Slettom, G.; Karoliussen, M.; Rustan, A.C.; Thoresen, G.H.; Skorve, J.; Nygård, O.K.; Berge, R.K. A mitochondria-targeted fatty acid analogue influences hepatic glucose metabolism and reduces the plasma insulin/glucose ratio in male Wistar rats. PLoS ONE 2019, 14, e0222558. [Google Scholar] [CrossRef] [PubMed]
- Muyyarikkandy, M.S.; McLeod, M.; Maguire, M.; Mahar, R.; Kattapuram, N.; Zhang, C.; Surugihalli, C.; Muralidaran, V.; Vavilikolanu, K.; Mathews, C.E.; et al. Branched chain amino acids and carbohydrate restriction exacerbate ketogenesis and hepatic mitochondrial oxidative dysfunction during NAFLD. FASEB J. 2020, 34, 14832–14849. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-C.; Hsieh, Y.-C.; Chan, C.-C.; Sun, H.-J.; Huang, Y.-H.; Hou, M.-C.; Lin, H.-C. Human relaxin-2 attenuates hepatic steatosis and fibrosis in mice with non-alcoholic fatty liver disease. Lab. Investig. 2019, 99, 1203–1216. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Xie, B.; Fan, M.; Candas-Green, D.; Jiang, J.X.; Wei, R.; Wang, Y.; Chen, H.-W.; Hu, Y.; Li, J.J. Low-Level Saturated Fatty Acid Palmitate Benefits Liver Cells by Boosting Mitochondrial Metabolism via CDK1-SIRT3-CPT2 Cascade. Dev. Cell 2020, 52, 196–209.e9. [Google Scholar] [CrossRef] [Green Version]
- Lian, J.; Watts, R.; Quiroga, A.D.; Beggs, M.R.; Alexander, R.T.; Lehner, R. Ces1d deficiency protects against high-sucrose diet-induced hepatic triacylglycerol accumulation. J. Lipid Res. 2019, 60, 880–891. [Google Scholar] [CrossRef]
- Li, Y.; Sun, J.-P.; Wang, J.; Lu, W.-H.; Xie, L.-Y.; Lv, J.; Li, H.-X.; Yang, S.-F. Expression of Vsig4 attenuates macrophage-mediated hepatic inflammation and fibrosis in high fat diet (HFD)-induced mice. Biochem. Biophys. Res. Commun. 2019, 516, 858–865. [Google Scholar] [CrossRef]
- Ju, L.; Sun, Y.; Xue, H.; Chen, L.; Gu, C.; Shao, J.; Lu, R.; Luo, X.; Wei, J.; Ma, X.; et al. CCN1 promotes hepatic steatosis and inflammation in non-alcoholic steatohepatitis. Sci. Rep. 2020, 10, 3201. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.; Shin, E.; Bae, J.; Cho, Y.; Lee, J.-Y.; Lee, Y.; Lee, B.-W.; Kang, E.S.; Cha, B.-S. Dipeptidyl peptidase-4 inhibitor protects against non-alcoholic steatohepatitis in mice by targeting TRAIL receptor-mediated lipoapoptosis via modulating hepatic dipeptidyl peptidase-4 expression. Sci. Rep. 2020, 10, 19429. [Google Scholar] [CrossRef]
- Cansby, E.; Nuñez-Durán, E.; Magnusson, E.; Amrutkar, M.; Booten, S.L.; Kulkarni, N.M.; Svensson, L.T.; Borén, J.; Marschall, H.-U.; Aghajan, M.; et al. Targeted Delivery of Stk25 Antisense Oligonucleotides to Hepatocytes Protects Mice Against Nonalcoholic Fatty Liver Disease. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 597–618. [Google Scholar] [CrossRef] [Green Version]
- Yuting, Y.; Lifeng, F.; Qiwei, H. Secreted modular calcium-binding protein 2 promotes high fat diet (HFD)-induced hepatic steatosis through enhancing lipid deposition, fibrosis and inflammation via targeting TGF-β1. Biochem. Biophys. Res. Commun. 2019, 509, 48–55. [Google Scholar] [CrossRef]
- Zhang, Z.; Zong, C.; Jiang, M.; Hu, H.; Cheng, X.; Ni, J.; Yi, X.; Jiang, B.; Tian, F.; Chang, M.-W.; et al. Hepatic HuR modulates lipid homeostasis in response to high-fat diet. Nat. Commun. 2020, 11, 3067. [Google Scholar] [CrossRef] [PubMed]
- Canivet, C.M.; Bonnafous, S.; Rousseau, D.; Leclere, P.S.; Lacas-Gervais, S.; Patouraux, S.; Sans, A.; Luci, C.; Bailly-Maitre, B.; Iannelli, A.; et al. Hepatic FNDC5 is a potential local protective factor against Non-Alcoholic Fatty Liver. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2020, 1866, 165705. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, Z.; Li, C.; Zhu, T.; Gao, J.; Zhou, H.; Zheng, Y.; Chang, Q.; Wang, M.; Wu, J.; et al. S100A11 Promotes Liver Steatosis via FOXO1-Mediated Autophagy and Lipogenesis. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 697–724. [Google Scholar] [CrossRef] [PubMed]
- Mirea, A.-M.; Stienstra, R.; Kanneganti, T.-D.; Tack, C.J.; Chavakis, T.; Toonen, E.J.M.; Joosten, L.A.B. Mice Deficient in the IL-1β Activation Genes Prtn3, Elane, and Casp1 Are Protected Against the Development of Obesity-Induced NAFLD. Inflammation 2020, 43, 1054–1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, D.; Cui, H.; Zhang, Y. Lack of ClC-2 Alleviates High Fat Diet-Induced Insulin Resistance and Non-Alcoholic Fatty Liver Disease. Cell. Physiol. Biochem. 2018, 45, 2187–2198. [Google Scholar] [CrossRef]
- Shum, M.; Shintre, C.A.; Althoff, T.; Gutierrez, V.; Segawa, M.; Saxberg, A.D.; Martinez, M.; Adamson, R.; Young, M.R.; Faust, B.; et al. ABCB10 exports mitochondrial biliverdin, driving metabolic maladaptation in obesity. Sci. Transl. Med. 2021, 13, eabd1869. [Google Scholar] [CrossRef]
- McGlaughon, J.L.; Pasquali, M.; Wallace, K.; Ross, J.; Senol-Cosar, O.; Shen, W.; Weaver, M.A.; Feigenbaum, A.; Lyon, E.; Enns, G.M.; et al. Assessing the strength of evidence for genes implicated in fatty acid oxidation disorders using the ClinGen clinical validity framework. Mol. Genet. Metab. 2019, 128, 122–128. [Google Scholar] [CrossRef]
- Li, Q.; Tomcik, K.; Zhang, S.; Puchowicz, M.A.; Zhang, G.-F. Dietary regulation of catabolic disposal of 4-hydroxynonenal analogs in rat liver. Free Radic. Biol. Med. 2012, 52, 1043–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wanders, R.J.A.; Vaz, F.M.; Waterham, H.R.; Ferdinandusse, S. Fatty Acid Oxidation in Peroxisomes: Enzymology, Metabolic Crosstalk with Other Organelles and Peroxisomal Disorders. In Peroxisome Biology: Experimental Models, Peroxisomal Disorders and Neurological Diseases; Lizard, G., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2020; Volume 1299, pp. 55–70. ISBN 978-3-030-60203-1. [Google Scholar]
- Dahan, N.; Francisco, T.; Falter, C.; Rodrigues, T.; Kalel, V.; Kunze, M.; Hansen, T.; Schliebs, W.; Erdmann, R. Current advances in the function and biogenesis of peroxisomes and their roles in health and disease. Histochem. Cell Biol. 2021, 155, 513–524. [Google Scholar] [CrossRef]
- Bharathi, S.S.; Zhang, Y.; Gong, Z.; Muzumdar, R.; Goetzman, E.S. Role of mitochondrial acyl-CoA dehydrogenases in the metabolism of dicarboxylic fatty acids. Biochem. Biophys. Res. Commun. 2020, 527, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Grünig, D.; Duthaler, U.; Krähenbühl, S. Effect of Toxicants on Fatty Acid Metabolism in HepG2 Cells. Front. Pharmacol. 2018, 9, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Choi, J.; Aja, S.; Scafidi, S.; Wolfgang, M.J. Loss of Adipose Fatty Acid Oxidation Does Not Potentiate Obesity at Thermoneutrality. Cell Rep. 2016, 14, 1308–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuevas-Ramos, D.; Mehta, R.; Aguilar-Salinas, C.A. Fibroblast Growth Factor 21 and Browning of White Adipose Tissue. Front. Physiol. 2019, 10, 37. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, K.D.; Qiu, Y.; Cui, X.; Goh, Y.P.S.; Mwangi, J.; David, T.; Mukundan, L.; Brombacher, F.; Locksley, R.M.; Chawla, A. Alternatively activated macrophages produce catecholamines to sustain adaptive thermogenesis. Nature 2011, 480, 104–108. [Google Scholar] [CrossRef] [Green Version]
- Ménégaut, L.; Thomas, C.; Lagrost, L.; Masson, D. Fatty acid metabolism in macrophages: A target in cardio-metabolic diseases. Curr. Opin. Lipidol. 2016, 1. [Google Scholar] [CrossRef]
- Nomura, M.; Liu, J.; Yu, Z.-X.; Yamazaki, T.; Yan, Y.; Kawagishi, H.; Rovira, I.I.; Liu, C.; Wolfgang, M.J.; Mukouyama, Y.; et al. Macrophage fatty acid oxidation inhibits atherosclerosis progression. J. Mol. Cell. Cardiol. 2019, 127, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Wek, R.C. Role of eIF2α Kinases in Translational Control and Adaptation to Cellular Stress. Cold Spring Harb. Perspect. Biol. 2018, 10, a032870. [Google Scholar] [CrossRef]
- Masson, G.R. Towards a model of GCN2 activation. Biochem. Soc. Trans. 2019, 47, 1481–1488. [Google Scholar] [CrossRef] [Green Version]
- Brouwers, M.C.G.J.; Simons, N.; Stehouwer, C.D.A.; Koek, G.H.; Schaper, N.C.; Isaacs, A. Relationship Between Nonalcoholic Fatty Liver Disease Susceptibility Genes and Coronary Artery Disease: Hepatology Communications. Hepatol. Commun. 2019, 3, 587–596. [Google Scholar] [CrossRef]
- Krawczyk, M.; Portincasa, P.; Lammert, F. PNPLA3-associated steatohepatitis: Toward a gene-based classification of fatty liver disease. Semin. Liver Dis. 2013, 33, 369–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smagris, E.; BasuRay, S.; Li, J.; Huang, Y.; Lai, K.V.; Gromada, J.; Cohen, J.C.; Hobbs, H.H. Pnpla3I148M knockin mice accumulate PNPLA3 on lipid droplets and develop hepatic steatosis. Hepatology 2015, 61, 108–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- BasuRay, S.; Smagris, E.; Cohen, J.C.; Hobbs, H.H. The PNPLA3 variant associated with fatty liver disease (I148M) accumulates on lipid droplets by evading ubiquitylation. Hepatology 2017, 66, 1111–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luukkonen, P.K.; Nick, A.; Hölttä-Vuori, M.; Thiele, C.; Isokuortti, E.; Lallukka-Brück, S.; Zhou, Y.; Hakkarainen, A.; Lundbom, N.; Peltonen, M.; et al. Human PNPLA3-I148M variant increases hepatic retention of polyunsaturated fatty acids. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Saki, S.; Saki, N.; Poustchi, H.; Malekzadeh, R. Assessment of Genetic Aspects of Non-alcoholic Fatty Liver and Premature Cardiovascular Events. Middle East J. Dig. Dis. 2020, 12, 65–88. [Google Scholar] [CrossRef]
- Fan, Y.; Lu, H.; Guo, Y.; Zhu, T.; Garcia-Barrio, M.T.; Jiang, Z.; Willer, C.J.; Zhang, J.; Chen, Y.E. Hepatic Transmembrane 6 Superfamily Member 2 Regulates Cholesterol Metabolism in Mice. Gastroenterology 2016, 150, 1208–1218. [Google Scholar] [CrossRef] [Green Version]
- Wright, F.A.; Bonzerato, C.G.; Sliter, D.A.; Wojcikiewicz, R.J.H. The erlin2 T65I mutation inhibits erlin1/2 complex–mediated inositol 1,4,5-trisphosphate receptor ubiquitination and phosphatidylinositol 3-phosphate binding. J. Biol. Chem. 2018, 293, 15706–15714. [Google Scholar] [CrossRef] [Green Version]
- Saeed, N.; Nadeau, B.; Shannon, C.; Tincopa, M. Evaluation of Dietary Approaches for the Treatment of Non-Alcoholic Fatty Liver Disease: A Systematic Review. Nutrients 2019, 11, 3064. [Google Scholar] [CrossRef] [Green Version]
- Shojaee-Moradie, F.; Cuthbertson, D.J.; Barrett, M.; Jackson, N.C.; Herring, R.; Thomas, E.L.; Bell, J.; Kemp, G.J.; Wright, J.; Umpleby, A.M. Exercise Training Reduces Liver Fat and Increases Rates of VLDL Clearance But Not VLDL Production in NAFLD. J. Clin. Endocrinol. Metab. 2016, 101, 4219–4228. [Google Scholar] [CrossRef] [Green Version]
- Lassailly, G.; Caiazzo, R.; Ntandja-Wandji, L.-C.; Gnemmi, V.; Baud, G.; Verkindt, H.; Ningarhari, M.; Louvet, A.; Leteurtre, E.; Raverdy, V.; et al. Bariatric Surgery Provides Long-term Resolution of Nonalcoholic Steatohepatitis and Regression of Fibrosis. Gastroenterology 2020, 159, 1290–1301.e5. [Google Scholar] [CrossRef]
- Aminian, A.; Zajichek, A.; Arterburn, D.E.; Wolski, K.E.; Brethauer, S.A.; Schauer, P.R.; Kattan, M.W.; Nissen, S.E. Association of Metabolic Surgery With Major Adverse Cardiovascular Outcomes in Patients With Type 2 Diabetes and Obesity. JAMA 2019, 322, 1271. [Google Scholar] [CrossRef] [PubMed]
- Deprince, A.; Haas, J.T.; Staels, B. Dysregulated lipid metabolism links NAFLD to cardiovascular disease. Mol. Metab. 2020, 42, 101092. [Google Scholar] [CrossRef] [PubMed]
- Jensen-Cody, S.O.; Potthoff, M.J. Hepatokines and metabolism: Deciphering communication from the liver. Mol. Metab. 2021, 44, 101138. [Google Scholar] [CrossRef]
- Smati, S.; Régnier, M.; Fougeray, T.; Polizzi, A.; Fougerat, A.; Lasserre, F.; Lukowicz, C.; Tramunt, B.; Guillaume, M.; Burnol, A.-F.; et al. Regulation of hepatokine gene expression in response to fasting and feeding: Influence of PPAR-α and insulin-dependent signalling in hepatocytes. Diabetes Metab. 2020, 46, 129–136. [Google Scholar] [CrossRef]
- Uebanso, T.; Taketani, Y.; Yamamoto, H.; Amo, K.; Tanaka, S.; Arai, H.; Takei, Y.; Masuda, M.; Yamanaka-Okumura, H.; Takeda, E. Liver X receptor negatively regulates fibroblast growth factor 21 in the fatty liver induced by cholesterol-enriched diet. J. Nutr. Biochem. 2012, 23, 785–790. [Google Scholar] [CrossRef] [PubMed]
- Fisher, F.M.; Maratos-Flier, E. Understanding the Physiology of FGF21. Annu. Rev. Physiol. 2016, 78, 223–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barb, D.; Bril, F.; Kalavalapalli, S.; Cusi, K. Plasma Fibroblast Growth Factor 21 Is Associated With Severity of Nonalcoholic Steatohepatitis in Patients With Obesity and Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2019, 104, 3327–3336. [Google Scholar] [CrossRef]
- Pereira, R.O.; Marti, A.; Olvera, A.C.; Tadinada, S.M.; Bjorkman, S.H.; Weatherford, E.T.; Morgan, D.A.; Westphal, M.; Patel, P.H.; Kirby, A.K.; et al. OPA1 deletion in brown adipose tissue improves thermoregulation and systemic metabolism via FGF21. eLife 2021, 10, e66519. [Google Scholar] [CrossRef]
- Kim, A.M.; Somayaji, V.R.; Dong, J.Q.; Rolph, T.P.; Weng, Y.; Chabot, J.R.; Gropp, K.E.; Talukdar, S.; Calle, R.A. Once-weekly administration of a long-acting fibroblast growth factor 21 analogue modulates lipids, bone turnover markers, blood pressure and body weight differently in obese people with hypertriglyceridaemia and in non-human primates. Diabetes Obes. Metab. 2017, 19, 1762–1772. [Google Scholar] [CrossRef]
- Sanyal, A.; Charles, E.D.; Neuschwander-Tetri, B.A.; Loomba, R.; Harrison, S.A.; Abdelmalek, M.F.; Lawitz, E.J.; Halegoua-DeMarzio, D.; Kundu, S.; Noviello, S.; et al. Pegbelfermin (BMS-986036), a PEGylated fibroblast growth factor 21 analogue, in patients with non-alcoholic steatohepatitis: A randomised, double-blind, placebo-controlled, phase 2a trial. Lancet 2018, 392, 2705–2717. [Google Scholar] [CrossRef]
- Kaufman, A.; Abuqayyas, L.; Denney, W.S.; Tillman, E.J.; Rolph, T. AKR-001, an Fc-FGF21 Analog, Showed Sustained Pharmacodynamic Effects on Insulin Sensitivity and Lipid Metabolism in Type 2 Diabetes Patients. Cell Rep. Med. 2020, 1, 100057. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.; Hanouneh, I.A.; Noureddin, M.; Rolph, T.; Alkhouri, N. Fibroblast growth factor (FGF)-21 based therapies: A magic bullet for nonalcoholic fatty liver disease (NAFLD)? Expert Opin. Investig. Drugs 2020, 29, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.M.; Yang, E.H.; Quan, W.; Nam, E.H.; Cheon, H.G. Discovery of a novel fibroblast activation protein (FAP) inhibitor, BR103354, with anti-diabetic and anti-steatotic effects. Sci. Rep. 2020, 10, 21280. [Google Scholar] [CrossRef] [PubMed]
- Kokkinos, J.; Tang, S.; Rye, K.-A.; Ong, K.L. The role of fibroblast growth factor 21 in atherosclerosis. Atherosclerosis 2017, 257, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Tabari, F.S.; Karimian, A.; Parsian, H.; Rameshknia, V.; Mahmoodpour, A.; Majidinia, M.; Maniati, M.; Yousefi, B. The roles of FGF21 in atherosclerosis pathogenesis. Rev. Endocr. Metab. Disord. 2019. [Google Scholar] [CrossRef]
- Wei, W.; Dutchak, P.A.; Wang, X.; Ding, X.; Wang, X.; Bookout, A.L.; Goetz, R.; Mohammadi, M.; Gerard, R.D.; Dechow, P.C.; et al. Fibroblast growth factor 21 promotes bone loss by potentiating the effects of peroxisome proliferator-activated receptor. Proc. Natl. Acad. Sci. USA 2012, 109, 3143–3148. [Google Scholar] [CrossRef] [Green Version]
- Charles, E.D.; Neuschwander-Tetri, B.A.; Pablo Frias, J.; Kundu, S.; Luo, Y.; Tirucherai, G.S.; Christian, R. Pegbelfermin (BMS-986036), PEGylated FGF21, in Patients with Obesity and Type 2 Diabetes: Results from a Randomized Phase 2 Study. Obesity 2019, 27, 41–49. [Google Scholar] [CrossRef]
- Kenneally, S.; Sier, J.H.; Moore, J.B. Efficacy of dietary and physical activity intervention in non-alcoholic fatty liver disease: A systematic review. BMJ Open Gastroenterol. 2017, 4, e000139. [Google Scholar] [CrossRef]


{kind=link}
| Gene/Target | Mutant/Line | Results | Reference |
|---|---|---|---|
| Mitochondrial Functions | |||
| Ech1 (Enoyl coenzyme A hydratase 1) | Ech1 OE; C57BL/6 | Ech1 OE ameliorates lipid accumulation, liver injury, dyslipidemia, and IR. | [115] |
| LPGAT1 (Lysophosphatidylglycerol Acyltransferase 1) | LPGAT1−/−; C57BL/6 | LPGAT1 deficiency protected mice from diet-induced obesity, but led to hepatopathy, insulin resistance, and NAFLD as a consequence of OS, mitochondrial DNA depletion, and mitochondrial dysfunction. | [116] |
| CRLS1 (Cardiolipin synthase 1) | CRLS1−/−; C57BL/6 | Crls deficiency resulted in a prominently aggravated lipid metabolism disorder, inflammation, and fibrosis; CRLS1 suppressed ATF3 expression and inhibits its activity in palmitic-acid-stimulated hepatocytes | [117] |
| MFN2 (Mitofusin 2) | Liver-specific Mfn2 KO mice; C57BL/6J | Mouse NASH models present lower levels of Mfn2 in the liver, and the re-expression of Mfn2 in the liver ameliorates the NASH phenotype. Hepatic Mfn2 ablation causes a NASH-like phenotype that progresses to liver cancer with age. Mfn2 binds to and participates in the transfer of PS. Hepatic Mfn2 deficiency causes a reduced transfer of PS from ER to mitochondria, which leads to reduced PS synthesis and ER stress, in turn causing inflammation, fibrosis, and liver cancer. | [118] |
| Mitochondrial GNMT (glycine N-methyltransferase)-Complex II | Liver-specific repression of the GNMT by miR-873-5p; C57BL/6 | GNMT expression is controlled by miR-873-5p in the hepatocytes, leading to disruptions in mitochondrial functionality. NASH therapies based on anti-miR-873-5p resolve lipid accumulation, inflammation, and fibrosis by enhancing fatty-acid β-oxidation in the mitochondria. | [119] |
| mTORC1 | Diet with high/low ratio of ω-3/ω-6 polyunsaturated fatty acids; C57BL/6 | Body weight, atherosclerosis marker, insulin signal index, and level of lipid accumulation in the liver were significantly lowered in the high group. Expressions of p-mTOR and raptor were inhibited by high ω-3 PUFAs. High ω-3 PUFAs depressed p-mTOR and raptor expressions, regulated ETC and TCA cycle pathway, and increased activities of mitochondrial complexes I, II, III, IV, and V. | [120] |
| GADD45GIP1 (CRIF1); GDF15 and FGF21 | Liver-specific Crif1-deleted mice; GDF15 and FGF21 null mice; C57BL/6J | Crif1 KO mice showed lower hepatic lipid accumulation, which was associated with lower hepatic expression of Srebp1, Srebp1c, and Cd36; Crif1 KO mice were resistant to diet-induced obesity and protected against hepatic steatosis and insulin resistance when fed an HFD. | [121] |
| RAB24 | Human-delivered samples; Hepa1-6 cells; FGF21 and RAB24 KO mice; C57BL/6N | Rab24 directly interacts with FIS1, thus regulating mitochondrial turnover. Reduction of Rab24 causes reduced mitochondrial fission resulting in enhanced energy usage. Rab24 KO reassembles the fasting state, whereby mitochondria are metabolically reprogrammed towards higher respiration through enhanced connectivity and bioenergetic efficiency. | [122] |
| MCJ (Methylation-controlled J protein) | Leptin receptor mutant (Leprdb/J); C57BL/6J | The therapeutic inhibition of MCJ expression in vivo enhances FAO in the liver in a NASH model. The enhanced FAO resulting from inhibiting MCJ is due to enhanced Complex I activity. In vivo treatment of siMCJ of mice with NASH increases β-oxidation and decreases lipid accumulation in the liver, but does not increase ROS production. | [123] |
| ANT2 (ADP/ATP translocase 2) | Liver-specific Ant2 cKO mice; ANT2 inhibition by carboxyatractyloside (CATR) | Targeted disruption of Ant2 in mouse liver enhances uncoupled respiration without damaging mitochondrial integrity and liver functions. Liver-specific Ant2 KO mice are leaner and resistant to hepatic steatosis, obesity and insulin resistance under a lipogenic diet. | [124] |
| GRK2 (G protein-coupled receptor kinase 2) | GRK2 hemizygous mice | GRK2± mice were protected from HFD-induced NAFLD. GRK2± mice preserved hepatic protective mechanisms as enhanced autophagy and mitochondrial fusion and biogenesis, together with reduced endoplasmic reticulum stress. Enhanced GRK2 expression potentiated palmitic-acid-triggered lipid accumulation in human hepatocytes directly relating GRK2 levels to steatosis. | [125] |
| AMPK-CPT Signalling Pathway | |||
| ACC1; ACC2 (Acetyl-CoA carboxylase) | KKAy or C57BL/6J mice; ACC1 and ACC2 liver-specific KO | Deletion of ACCs decreased PUFA concentrations in the liver due to reduced malonyl-CoA. PUFA deficiency induced SREBP-1c, which increased GPAT1 expression and VLDL secretion. Thus, inhibiting lipogenesis in humans reduced hepatic steatosis, but inhibiting ACC resulted in hypertriglyceridemia due to activation of SREBP-1c and increased VLDL secretion. | [126] |
| ACC2 | ACC2 KO mice; C57BL/6J | The global deletion of ACC2 enhances lipid disposal without competing with glucose metabolism at the whole-body and skeletal-muscle levels. This successful lipid reduction is characterized by a decreased acetyl-CoA pool in skeletal muscle, which is accounted for by enhanced TCA cycle activity and acetyl-CoA conversion into acetylcarnitine. | [127] |
| ROCK1 (Rho-kinase 1) | Liver-specific ROCK1 deletion; C57BL/6J | Mice lacking ROCK1 in the liver were resistant to diet-induced obesity owing to increased energy expenditure and thermogenic gene expression. Treatment with metformin reduced hepatic lipid accumulation by inactivating ROCK1, resulting in activation of AMPK downstream signalling. | [128] |
| SIRT1 (Sirtuin 1), AMPKα (AMP-activated protein kinase); AITC (Allyl isothiocyanate) treatment | Sirt1 and AMPKα; AML-12 cells; C57BL/6 | AITC attenuates inflammation by inhibiting the NF-κB signalling pathway in vitro and de novo lipogenesis, and promotes FAO by activating the Sirt1/AMPK signalling pathway in vitro. | [129] |
| SIRT5; LCAD (Long-chain acyl-CoA dehydrogenase) | SIRT5 and LCAD knockout C57BL/6 | Medium-chain triglycerides (MCT), containing C8–C12 FA degradation, was significantly reduced in the Sirt5KO liver. This decrease was localized to the mitochondrial β-oxidation pathway, as Sirt5KO mice exhibited no change in peroxisomal C12 β-oxidation. ER ω-oxidation was increased in Sirt5KO liver. LCAD KO mice developed periportal macrovesicular steatosis when fed coconut oil. | [130] |
| Adiponectin-based agonist JT003 | HepG2 and human hepatic activated stellate cell line LX2; C57BL/6J | AdipoRs dual agonist JT003 with a longer half-life could ameliorate NASH and related liver fibrosis via AMPK, PPARα, and PI3K-Akt signal pathways. JT003 treatment significantly improves the function of the ER–mitochondrial axis, which contributes to the reduced HSC activation. | [131] |
| GGPPS (Geranylgeranyl pyrophosphate synthase) | Liver-specific GGPPS deletion; C57BL/6J | Long-term HFD decreases GGPPS expression, which shifts the fuel preference from FAs toward glucose. Liver-specific Ggpps deficiency drives the Warburg effect by impairing mitochondria function, and induces hepatic inflammation. Ggpps deficiency enhances the hyper-farnesylation of liver kinase B1 and promotes metabolic reprogramming by regulating AMPK activity. | [132] |
| Mouse CREBH (CAMP-responsive element-binding protein, hepatic-specific) site-directed mutagenesis, transfection (OE) | Mouse AML-12 cells, human hepatocyte: HepG2 and HEK293T cells; C57BL/6J | N-glycosylation of CREBH modulated the production of PPARα and activation of SCD-1 by interfering with the recognition of CRE in their promoters, inducing CREBH/PPARα and CREBH/SCD-1 interaction. This subsequently improved the synthesis of hepatic lipids and sterols and relieved inflammation, lipotoxicity, and lipid peroxidation. | [133] |
| IMP2 (Insulin-like growth factor 2 MRNA binding Protein 2) | Hepatocyte-specific IMP2 knockout; C57BL/6 | IMP2 binds and stabilizes the mRNAs encoding the critical regulators of hepatic fatty-acid oxidation, PPARα and CPT-1A; loss of IMP2 diminishes the abundance of those mRNAs, resulting in reduced mitochondrial fatty-acid oxidation. Mice with hepatic IMP2 deficiency fed an HFD show a modest, progressive accumulation of hepatic triglycerides beyond that of HFD-fed controls, ultimately reflected in elevated circulating triglycerides and mildly elevated blood glucose. | [134] |
| FOH (Farnesol) | Steatotic HepaRG cells | FOH treatment increases FAO and decreases TG accumulation in steatotic HepaRG cells, which is likely attributable to PPARα-mediated induction of mitochondrial FAO. | [135] |
| TFF3 (Trefoil factor 3) | TFF3 KO; C57BL/6 | Tff3 binds the promoter of PPAR and up-regulates hepatic FAO. | [136] |
| CPT1A (Human carnitine palmitoyltransferase 1A) | C57BL/6 | Expression of hCPT1AM (a mutated isoform that is insensitive to malonyl-CoA) enhanced hepatic FAO and autophagy, reduced liver steatosis, and improved glucose homeostasis. | [137] |
| CPT2 (Carnitine palmitoyltransferase 2) | Liver-specific deficiency of CPT2; C57BL/6 | Cpt2L−/− mice were resistant to HFD-induced obesity and glucose intolerance with an absence of liver damage, although they exhibited serum dyslipidemia, hepatic oxidative stress, and systemic carnitine deficiency. Feeding an HFD induced hepatokines in mice, with a loss of hepatic fatty-acid oxidation that enhanced systemic energy expenditure and suppressed adiposity. | [73] |
| Antioxidant | |||
| SOD1 (Cu/Zn-superoxide dismutase) | Sod1−/−; C57BL/6 | Excess fat accumulation in the livers of Sod1KO mice due to impaired VLDL secretion leads to NAFLD, and the high OS triggers necroptosis in the liver, leading to the generation of DAMPs. The DAMPs activate macrophages and the inflammasome leading to the production of pro-inflammatory cytokines, resulting in non-resolving chronic inflammation. | [138] |
| PRX5 (Peroxiredoxin) | PRX5 KO; C57BL/6J; HepG2 cells | Prx5 ameliorated FFA-induced ROS overproduction and lipid accumulation in HepG2 cells. Prx5 overexpression ameliorated hepatic steatosis by regulating lipogenesis and hepatic inflammation. Additionally, upon NAFLD induction, the expression of lipogenesis-related proteins increased more among Prx5 KO mice than among WT mice. | [139] |
| CAT (catalase) | CAT knockout C57BL/6; HepG2 cells; | The fat accumulation, lipid peroxidation, and H2O2 release were significantly elevated in HFD CAT KO mice. The liver mitochondria tended to be more severely damaged, and mitochondrial DNA copy number and cellular ATP concentrations were significantly lower in CAT KO mice. In CAT KO HepG2 cells, fatty-acid treatment causes accelerated cellular lipid accumulation and depressed mitochondrial biogenesis. | [140] |
| General Lipid Metabolism | |||
| GCN2 (General control nonderepressible 2) | Gcn2−/−; H9C2 cells; C57BL/6 | Gcn2−/− significantly attenuated HFD-induced liver dysfunction, hepatic steatosis, and insulin resistance; Exercised GCN2-deficient mice have enhanced efficacy in improving hepatic steatosis and liver lipid metabolism, at least partially, via the AMPK/SIRT1/PPARα pathway. GCN2 deficiency protects cardiac function by reducing lipid accumulation, OS, and cell death by inhibiting eIF2α -ATF4-CHOP signalling. | [141,142,143,144] |
| FABP1 (Fatty-acid-binding protein 1) | FABP1 OE; C57BL/6 | Exercise down-regulated the FABP1 signalling pathway, which was most closely associated with lipid metabolism. Liver-specific overexpression of FABP1 abolished the protective effect of exercise in NAFLD mice. Exercise significantly increased autophagic flux via restoring lysosomal function, including lysosomal proteolysis and lysosomal acidification maintenance, contributing to enhancement in autophagic clearance and subsequent alleviation of hepatic steatosis. | [145] |
| APOE (Apolipoprotein E) and RON (Macrophage stimulating 1 receptor) | ApoE−/−/Ron−/−; C57BL/6 | Double KO mice had features of steatosis, inflammation, OS, and hepatocyte damage, as well as increased accumulation of FAs in the liver and decreased levels of bile acids. | [146] |
| STING (Tmem173) | STING−/−; C57BL/6 | STING deficiency attenuated steatosis, fibrosis, and inflammation; increased fasting glucose levels in mice independently of insulin; reduced levels of cholesterol, triglycerides, and LDL in serum; enhanced levels of HDL; reduced levels of mtDNA in hepatocytes, TNF-α, and IL-6 expression in cultured Kupffer cells; and reduced mRNA levels of Col1A1 and α -SMA in livers. | [147] |
| SPP1 (Osteopontin) | Spp1−/−; C57BL/6 | Spp1−/− mice had increased lipid accumulation, high levels of ALT, fatty-acid translocase (CD36/FAT), pro-fibrogenic markers (Col1a1, Col 4a1, Timp1), and insulin secretion; while hepatic FOXO1 was downregulated. | [148] |
| LRP1 (LDL receptor-related protein-1) | LRP1 with distal NPxY motif mutation; C57BL/6 | Dysfunction of LRP1 is protective against HFHC diet-induced dyslipidemia, fatty liver disease, and neuroinflammation. | [149] |
| TM6SF2 (Transmembrane 6 superfamily member 2) | Tm6sf2−/−; C57BL/6 | APOB and ER lipid raft protein (ERLIN) 1 and 2 were TM6SF2-interacting proteins. ERLINs and TM6SF2 mutually bound and stabilized each other. TM6SF2 bound and stabilized APOB via two luminal loops. ERLINs did not interact with APOB directly, but still increased APOB stability through stabilizing TM6SF2. Defective APOB stabilization, as a result of ERLIN or TM6SF2 deficiency or E167K mutation, is a key factor contributing to NAFLD. | [150] |
| LAMP2A (Lysosome-associated membrane protein 2A) | LAMP2Afl/fl Cre+; C57BL/6 | LAMP2A reduction resulted in decreased levels of (chaperone-mediated autophagy) CMA-positive regulators. Deleting LAMP2A hindered lipid droplet (LD) breakdown, but not LD formation. The disruption of CMA-induced perilipin 5 (Plin5) degradation was an obstacle to LD breakdown, explaining the lipid homeostasis imbalance in NAFLD. | [151] |
| LCHAD (Long-chain 3-hydoxyacyl-CoA dehydrogenase) | LCHAD heterozygous mice | LCHAD mice developed significant hepatic steatosis starting at a young age (3 months old) and HCC at an older age (>13 months old) without any evidence of fibrosis or cirrhosis. LCHAD defects predispose to HCC, and mitochondrial dysfunction plays an important role in HCC pathogenesis. | [152] |
| CLOCK (Circadian locomotor output cycles kaput) and APOE (Apolipoprotein E) | ClkΔ19/Δ19, Apoe−/−; C57BL/6 | CLOCK regulates HIF1α protein levels by binding to the E-boxes in the promoters and modulating the expression of PHD proteins that regulate HIF1α protein stability. HIF1α binds to the Cd36 promoter to increase the expression of CD36 and uptake of fatty acids by the liver. Thus, a regulatory mechanism involving circadian CLOCK, hypoxia signalling, and lipid metabolism protects against NAFLD. | [153] |
| SLUG (Snail family transcriptional repressor 2) | SlugΔhep; C57BL/6 | Slug is a new lipogenic TF that promotes de novo lipogenesis by an epigenetic mechanism. Hepatocyte-specific deletion of Slug inhibited the hepatic lipogenic program and protected against obesity-associated NAFLD, IR, and glucose intolerance; Slug-associated Lsd1 mediates lipogenesis by demethylating H3K9 on the Fasn promoter, suggesting a new demethylation lipogenic insulin/Slug/Lsd1/H3K9 pathway that promotes NAFLD and T2DM. | [154] |
| LAP1 (Lamina-associated polypeptide 1) and TOR1A (TorsinA), an AAA+ ATPase | Lap1fl/fl and Tor1afl/fl; C57BL/6 | The torsinA/LAP1 pathway regulates VLDL secretion and liver fat accumulation. Conditional deletion of either LAP1 or torsinA from hepatocytes caused profound steatosis. | [155] |
| XBP1 (Xbp1-X-box binding protein 1) | AlbCre;Xbp1flx/flx; C57BL/6 | XBP1 is a 12 h clock manner that regulates gene expression, cellular membrane fluidity, and mitochondrial utilization of fatty-acid and glucose substrates; XBP1 provides temporal transcriptional regulation of the key metabolic enzymes such as SCD1, LPCAT3, and LCAT. | [156] |
| TBK1 (TANK-binding kinase 1) | Liver-specific TBK1 knockout; C57BL/6J | TBK1 impacts lipid metabolism via binding to the key rate-limiting enzyme ACSL1. In the fasted state, TBK1 expression is induced, but remains inactive, and can serve as a molecular scaffold to localize ACSL1 to the mitochondrial outer membrane, thus facilitating fatty-acid β-oxidation. In the absence of TBK1, fasting-stimulated ACSL1 localization to mitochondria is blunted, driving the localization of the enzyme to the ER for fatty-acid re-esterification. | [157] |
| Supplementation | |||
| Fisetin injection | FL83B cells; C57BL/6 | Fisetin treatment had decreased body weight and epididymal adipose tissue weight; reduced liver LD and hepatocyte steatosis, and alleviated serum FFA and leptin concentrations; significantly decreased FAS; and significantly increased phosphorylation of AMPKα and the production of sirt-1 and CPT1 in the liver tissue. In vitro, fisetin decreased lipid accumulation and increased lipolysis and β-oxidation in hepatocytes. | [158] |
| Phloretin supplementation/treatment | HepG2 cells; C57BL/6 | Phloretin significantly reduced excessive lipid accumulation and decreased SREBP-1c, blocking the expression of FAS in oleic acid-induced HepG2 cells. Phloretin increased Sirt1 and phosphorylation of AMPK to suppress ACC expression, reducing FA synthesis in hepatocytes. | [159] |
| Chitosan oligosaccharide (ChO) treatment | Inflammation and OS; C57BL/6 | ChO treatment decreases the serum levels of AST and ALT, Il-6, Il-1β, Tnf-α, and lowers lipid accumulation; and induces higher expression of fatty β-oxidation-related genes PPARα and CPT1, and OS-related genes (NQO1, HO-1, GSTA1. | [160] |
| HC (High cholesterol) diet | Mitochondrial parameters; C57BL/6 | HC-diet-induced liver damage and dysfunction, associated with a decrease in mitochondrial membrane potential and ATP production, increased cholesterol levels in the organelle. Mitochondria adapt to high levels of cholesterol content, increasing fission and decreasing apoptosis, while damaged hepatocytes do not enter apoptosis and proliferate, perpetuating liver damage. | [161] |
| A mitochondria-targeted fatty acid analogue 1-triple TTA | Hepatic glucose homeostasis; Wistar rats | The mitochondrially targeted fatty-acid analogue 1-triple TTA seemed to lower hepatic glucose and glycogen levels by inhibition of gluconeogenesis. This was also linked to a reduction in glucose oxidation accompanied by reduced pyruvate dehydrogenase activity and stimulation of ME1 and G6PD, favouring a shift from glucose to FAO. | [162] |
| Glucose and fructose supplementation | Hepatic glucose homeostasis; C57BL/6 | Glucose and fructose increased ChREBP-β levels, and fructose supplementation uniquely increased SREBP1c and downstream fatty-acid-synthesis genes, resulting in reduced liver insulin signalling. In contrast, glucose enhanced total ChREBP expression and triglyceride synthesis, but was associated with improved hepatic insulin signalling. | [84] |
| Feeding with 1.5X branched-chain amino acids (BCAAs) | Ketogenesis and hepatic mitochondrial oxidation; C57BL/6 | Long-term exposure of the mice to the BCAA-modified diet resulted in a chronic ketogenic environment. Metabolic profiling demonstrated that chronic induction of the hepatic mitochondrial oxidative networks (β-oxidation, ketogenesis, TCA cycle) occurred together with lower rates of lipogenesis in the liver of the ketogenic mice. | [163] |
| Recombinant human relaxin-2 supplementation | C57BL/6 | Human relaxin-2 attenuated steatosis and increased phosphorylation of IRS1, Akt eNOS, and activated genes that regulate fatty-acid oxidation and suppressed ACC. | [164] |
| Fructose supplementation | Hepatic FAO; C57BL/6 | Fructose supplementation increased fatty-acid synthesis mediated via upregulation of SREBP1c and ChREBP-β, in which glucose supplementation increased TG synthesis associated with upregulation of ChREBP. Fructose increased hepatic malonyl-CoA levels and increased acetylation of ACADL and CPT1a, while glucose supplementation resulted in increased acetylation of HADA/B. | [90] |
| Supplementation with SFA (saturated fatty acids) | SFA-induced lipotoxicity; C57BL/6 | High-level palmitate (HPA) induces lipotoxic effects in liver cells, while low-level PA (LPA) increases mitochondrial functions and alleviates the injuries induced by HPA or by hepatotoxic agent CCl4 (carbon tetrachloride). LPA-mediated mitochondrial homeostasis is regulated by CDK1-mediated SIRT3 phosphorylation, which in turn deacetylates and dimerizes CPT2 to enhance FAO. | [165] |
| Other | |||
| CES1 (Carboxylesterase 1) | Ces1−/−; C57BL/6 | Ces1d-deficient mice were protected from HSD-induced hepatic lipid accumulation. Ces1d deficiency leads to activation of AMPK and inhibitory phosphorylation of ACC. | [166] |
| VSIG4 (V-set and immunoglobulin domain-containing protein-4) | Vsig4−/−; C57BL/6 | Loss of Vsig4 accelerated the severity of lipid deposition, fibrosis, and the inflammatory response via the NF-kB and TGFβ 1 signalling pathways. | [167] |
| CCN1 (Cellular communication network factor 1) | C57BL/6 | CCN1 OE up-regulates the expression of fatty-acid metabolism-associated genes; it increased the expression of cleaved caspase 3 and the pro-apoptotic protein Bax. | [168] |
| DPP4 (Dipeptidyl peptidase-4) | DPP4 inhibitor (DPP4i); C57BL/6J; HepG2 cell line | DPP4i administration reduced serum liver enzyme and hepatic triglyceride levels and markedly improved hepatic steatosis and fibrosis in the AMLN-diet-induced NASH model. DPP4i may efficiently attenuate the pathogenesis of AMLN diet-induced NASH in mice by suppressing lipotoxicity-induced apoptosis. | [169] |
| STK25 (Serine/threonine kinase 25) | Liver-specific triantennary GalNAc-conjugated ASO targeting STK 25; C57BL/6J | Hepatocyte-targeting GalNAc-Stk25 ASO in obese mice effectively ameliorated steatosis, inflammatory infiltration, hepatic stellate-cell activation, nutritional fibrosis, and hepatocellular damage in the liver, without any systemic toxicity or local tolerability concerns. Also, treated mice were protected against HF-diet-induced hepatic oxidative stress and had improved mitochondrial function. | [170] |
| SMOC2 (Secreted modular calcium-binding protein 2) | SMOC2 KO; C57BL/6 | SMOC2 expression promoted hepatic steatosis by interacting with TGF-β 1 to regulate lipid metabolism, fibrosis, and inflammation. | [171] |
| ELAVL1 (RNA-binding protein HuR) | Hepatocyte-specific HuR knockout; C57BL/6 | HuR controls the production of CYCS, NDUFB6, UQCRB, and APOB; preserves the ability of mitochondria to produce energy; and maintains lipid homeostasis. | [172] |
| FNDC5 (Fibronectin type III domain-containing protein 5) | C57BL/6; mouse primary hepatocytes and HepG2 cells | FNDC5 regulates liver steatosis, and insulin resistance and injury, thus limiting the development and progression of NAFLD; irisin promotes mitochondrial biogenesis and reduces OS. | [173] |
| S100A11 (S100 calcium-binding protein A11) | Ghr-floxed x(B6; FVB-Tg (adipoq-Cre)1Evdr/J) | A high-fat diet promotes liver S100A11 expression, which may interact with HDAC6 to block its binding to FOXO1, releasing or increasing the acetylation of FOXO1, thus activating autophagy and lipogenesis, and accelerating lipid accumulation and liver steatosis. | [174] |
| CASP1 (Caspase-1), CASP11, PR3 (Prtn3), and NE (Elane) | Casp1−/−/Casp11−/−/NE−/−/PR3−/−; C57BL/6 | Mutants were protected from developing diet-induced weight gain, liver steatosis, and adipose tissue inflammation | [175] |
| ClC-2 (Chloride voltage-gated channel 2) | Chloride channel 2 KO mice; C57BL/6; HepG2 cells | Knockdown of ClC-2 in liver-attenuated HFD-induced weight gain, obesity, hepatocellular ballooning, and liver lipid accumulation and fibrosis, accompanied by reduced plasma FFA, TG, TC, ALT, AST, glucose, and insulin levels. HFD-fed ClC-2 KO mice showed inhibited hepatic lipid accumulation via regulating lipid metabolism through decreasing SREBP-1c expression and its downstream targets (such as FAS, HMGCR, and ACCα). | [176] |
| ABCB10 (ATP-binding cassette sub-family B member 10, mitochondrial) | Liver-specific deficiency of ABCB10; C57BL/6 | ABCB10 was identified as a mitochondrial biliverdin exporter. Diet-induced obese mice with liver-specific ABCB10 deletion were protected from steatosis and hyperglycemia, and had improved insulin-mediated suppression of glucose production and decreased expression of lipogenic SREBP-1c. Protection was concurrent with enhanced mitochondrial function and increased inactivation of PTP1B (protein tyrosine phosphatase non-receptor type 1), a phosphatase that disrupts insulin signalling and elevates SREBP1c expression. Thus, as a lipophilic hydrogen peroxide scavenger, bilirubin was the maladaptive effector linked to ABCB10 function. | [177] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dabravolski, S.A.; Bezsonov, E.E.; Baig, M.S.; Popkova, T.V.; Orekhov, A.N. Mitochondrial Lipid Homeostasis at the Crossroads of Liver and Heart Diseases. Int. J. Mol. Sci. 2021, 22, 6949. https://doi.org/10.3390/ijms22136949
Dabravolski SA, Bezsonov EE, Baig MS, Popkova TV, Orekhov AN. Mitochondrial Lipid Homeostasis at the Crossroads of Liver and Heart Diseases. International Journal of Molecular Sciences. 2021; 22(13):6949. https://doi.org/10.3390/ijms22136949
Chicago/Turabian StyleDabravolski, Siarhei A., Evgeny E. Bezsonov, Mirza S. Baig, Tatyana V. Popkova, and Alexander N. Orekhov. 2021. "Mitochondrial Lipid Homeostasis at the Crossroads of Liver and Heart Diseases" International Journal of Molecular Sciences 22, no. 13: 6949. https://doi.org/10.3390/ijms22136949