
Problems of Pathogenesis and Pathogenetic Therapy of COVID-19 from the Perspective of the General Theory of Pathological Systems (General Pathological Processes)


Abstract
:1. Introduction
2. Systemic Reactions in COVID-19
2.1. Signs of Acute Phase Response and Hypercytokinemia in COVID-19
- (a)
- (b)
- (c)
- It is necessary to account for the instability of individual SIR indicator concentrations in the blood (especially cytokines) and use for this integral SIR criteria with the preliminary establishment of specific levels of pathobiological and diagnostic significance for each SIR indicator, followed by their integration into a single scale [4,5,57].
2.2. The Main Factors of Systemic Alteration in COVID-19
- (a)
- Systemic hypoxia (an obvious phenomenon in ARDS);
- (b)
- Entry of tissue factor (TF) into the bloodstream or increased expression of its membrane form (CD142) on the endothelium’s surface, entry of other hemostasis and complement activators into the blood; accumulation of microbial-pathogen-associated molecular patterns (PAMP) and endogenous-damage-associated molecular patterns (DAMP) in blood, acting on cells through pattern-recognition receptors (PRR) [5];
- (c)
- Systemic activation relationship of leukocytes and macrophages despite the loss of their antiviral efficacy [58];
- (d)
- The entry into the bloodstream of a significant number of inflammatory mediators with the development of the phenomenon of excitotoxicity.
2.2.1. Hypoxia
2.2.2. Cell Activation by Pattern-Recognition Receptors
2.2.3. Metabolic Causes of Tissue Damage, the Possible Role of Chronic Low-Grade Inflammation in the Pathogenesis of COVID-19
2.2.4. Tissue Damage by Activated T Cells and Macrophages; Assessment Issues in COVID-19 Cytokine Release Syndrome and Cytokine Storm Syndrome
2.3. Endothelial Dysfunction and Microcirculatory Disorders in COVID-19
2.4. Dysfunction of the Hypothalamic–Pituitary–Adrenal System (HPA)
3. COVID-19 and Typical Pathological Processes
3.1. General Characteristics of Canonical Inflammation
3.2. Cellular Pro-Inflammatory Stress as an Elementary, Integral Functional System of Pathological Processes
- Oxidative stress.
- Cell response to the DNA damage.
- Mitochondrial stress, including mitochondrial unfolded protein response (UPRmt).
- Stress of the endoplasmic reticulum (ER), including calcium-dependent mechanisms and UPRER.
- Response of inducible heat shock proteins (HSPs), including their participation in the UPR.
- Inhibition (during cell growth) or intensification of autophagy processes (utilization of altered organelles and macromolecules) and other manifestations of lysosomal stress.
- Education by inflammasomes.
- Formation of stress, non-coding microRNAs (miRNAs).
- Formation of an intracellular network of signaling pathways of cellular stress.
- Formation of pro-inflammatory receptor and secretory cell phenotype.
3.3. Tissue Stress as a Common Basis of Pathology and Extreme Human Physiology; Non-Canonical Variants of Inflammation
3.4. Possible Connection of COVID-19 with the Mechanisms of Cellular Stress and General Pathological Processes
- (1)
- ChSLGI as the basis for comorbid pathologies that are risk factors for critical complications. These pathologies include morbid obesity, metabolic syndrome, and type 2 diabetes, which are known risk factors for the development of critical conditions in ChSLGI [18,22,177,222] and should be considered in Cov infections. The accumulation of aberrant metabolites and their transport forms (including modified lipoproteins) in the blood, latent microcirculation disorders, an increase in the pro-inflammatory phenotype of many cell types, and the dysfunction of neuroendocrine and paracrine regulation of organs and systems are all driving factors in these pathologies.
- (2)
- Inflammation of the classical type, mainly on the territory of the respiratory and intestinal organs, which forms a functional barrier that limits the spread of the virus in the body. The insufficient efficiency of canonical inflammation determines the possibility of generalization of viruses in the body (an additional factor of systemic alteration), excessive production of cytokines (as factors of excitotoxicity), and the development of noncardiogenic pulmonary edema (ARDS).
- (3)
- Systemic inflammation, which is a typical critical complication of COVID-19, the clinical manifestations of SI are complexes of resuscitation syndromes. For the SI development, the integral factors of systemic alteration must reach a level sufficient for the development of the “systemic inflammatory microcirculation” phenomenon.
- (4)
- The possible influence of transferred SI on the course of chronic comorbid diseases requires additional prospective studies.

4. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Malik, Y.S.; Sircar, S.; Bhat, S.; Sharun, K.; Dhama, K.; Dadar, M.; Tiwari, R.; Chaicumpa, W. Emerging Novel Coronavirus (2019-NCoV)-Current Scenario, Evolutionary Perspective Based on Genome Analysis and Recent Developments. Vet. Q. 2020, 40, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Bellocchio, L.; Bordea, I.R.; Ballini, A.; Lorusso, F.; Hazballa, D.; Isacco, C.G.; Malcangi, G.; Inchingolo, A.D.; Dipalma, G.; Inchingolo, F.; et al. Environmental Issues and Neurological Manifestations Associated with COVID-19 Pandemic: New Aspects of the Disease? Int. J. Environ. Res. Public Health 2020, 17, 8049. [Google Scholar] [CrossRef]
- Dhama, K.; Khan, S.; Tiwari, R.; Sircar, S.; Bhat, S.; Malik, Y.S.; Singh, K.P.; Chaicumpa, W.; Bonilla-Aldana, D.K.; Rodriguez-Morales, A.J. Coronavirus Disease 2019-COVID-19. Clin. Microbiol Rev. 2020, 33, e00028. [Google Scholar] [CrossRef]
- Zotova, N.V.; Chereshnev, V.A.; Gusev, E.Y. Systemic Inflammation: Methodological Approaches to Identification of the Common Pathological Process. PLoS ONE 2016, 11, e0155138. [Google Scholar] [CrossRef] [PubMed]
- Gusev, E.Y.; Zotova, N.V. Cellular Stress and General Pathological Processes. Curr. Pharm. Des. 2019, 25, 251–297. [Google Scholar] [CrossRef]
- Harrison, A.G.; Lin, T.; Wang, P. Mechanisms of SARS-CoV-2 Transmission and Pathogenesis. Trends Immunol. 2020, 41, 1100–1115. [Google Scholar] [CrossRef]
- Verkhivker, G.M. Molecular Simulations and Network Modeling Reveal an Allosteric Signaling in the SARS-CoV-2 Spike Proteins. J. Proteome Res. 2020, 19, 4587–4608. [Google Scholar] [CrossRef] [PubMed]
- Vickers, C.; Hales, P.; Kaushik, V.; Dick, L.; Gavin, J.; Tang, J.; Godbout, K.; Parsons, T.; Baronas, E.; Hsieh, F.; et al. Hydrolysis of Biological Peptides by Human Angiotensin-Converting Enzyme-Related Carboxypeptidase. J. Biol. Chem. 2002, 277, 14838–14843. [Google Scholar] [CrossRef] [Green Version]
- Simões e Silva, A.C.; Silveira, K.D.; Ferreira, A.J.; Teixeira, M.M. ACE2, Angiotensin-(1-7) and Mas Receptor Axis in Inflammation and Fibrosis. Br. J. Pharmacol. 2013, 169, 477–492. [Google Scholar] [CrossRef] [Green Version]
- Hasan, S.S.; Capstick, T.; Ahmed, R.; Kow, C.S.; Mazhar, F.; Merchant, H.A.; Zaidi, S.T.R. Mortality in COVID-19 Patients with Acute Respiratory Distress Syndrome and Corticosteroids Use: A Systematic Review and Meta-Analysis. Expert Rev. Respir. Med. 2020, 14, 1149–1163. [Google Scholar] [CrossRef]
- Fan, E.; Beitler, J.R.; Brochard, L.; Calfee, C.S.; Ferguson, N.D.; Slutsky, A.S.; Brodie, D. COVID-19-Associated Acute Respiratory Distress Syndrome: Is a Different Approach to Management Warranted? Lancet Respir. Med. 2020, 8, 816–821. [Google Scholar] [CrossRef]
- Grasselli, G.; Tonetti, T.; Protti, A.; Langer, T.; Girardis, M.; Bellani, G.; Laffey, J.; Carrafiello, G.; Carsana, L.; Rizzuto, C.; et al. Pathophysiology of COVID-19-Associated Acute Respiratory Distress Syndrome: A Multicentre Prospective Observational Study. Lancet Respir. Med. 2020, 8, 1201–1208. [Google Scholar] [CrossRef]
- Camporota, L.; Chiumello, D.; Busana, M.; Gattinoni, L.; Marini, J.J. Pathophysiology of COVID-19-Associated Acute Respiratory Distress Syndrome. Lancet Respir. Med. 2021, 9, e1. [Google Scholar] [CrossRef]
- Asakura, H.; Ogawa, H. COVID-19-Associated Coagulopathy and Disseminated Intravascular Coagulation. Int. J. Hematol. 2021, 113, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Peiris, J.S.M.; Chu, C.M.; Cheng, V.C.C.; Chan, K.S.; Hung, I.F.N.; Poon, L.L.M.; Law, K.I.; Tang, B.S.F.; Hon, T.Y.W.; Chan, C.S.; et al. Clinical Progression and Viral Load in a Community Outbreak of Coronavirus-Associated SARS Pneumonia: A Prospective Study. Lancet 2003, 361, 1767–1772. [Google Scholar] [CrossRef] [Green Version]
- Devaux, C.A.; Rolain, J.-M.; Raoult, D. ACE2 Receptor Polymorphism: Susceptibility to SARS-CoV-2, Hypertension, Multi-Organ Failure, and COVID-19 Disease Outcome. J. Microbiol. Immunol. Infect. 2020, 53, 425–435. [Google Scholar] [CrossRef]
- Madabhavi, I.; Sarkar, M.; Kadakol, N. COVID-19: A Review. Monaldi Arch. Chest Dis. 2020, 90. [Google Scholar] [CrossRef]
- Motaib, I.; Zbiri, S.; Elamari, S.; Dini, N.; Chadli, A.; El Kettani, C. Obesity and Disease Severity Among Patients With COVID-19. Cureus 2021, 13, e13165. [Google Scholar] [CrossRef]
- Hilser, J.R.; Han, Y.; Biswas, S.; Gukasyan, J.; Cai, Z.; Zhu, R.; Tang, W.H.W.; Deb, A.; Lusis, A.J.; Hartiala, J.A.; et al. Association of Serum HDL-Cholesterol and Apolipoprotein A1 Levels with Risk of Severe SARS-CoV-2 Infection. J. Lipid Res. 2021, 62, 100061. [Google Scholar] [CrossRef]
- O’Hearn, M.; Liu, J.; Cudhea, F.; Micha, R.; Mozaffarian, D. Coronavirus Disease 2019 Hospitalizations Attributable to Cardiometabolic Conditions in the United States: A Comparative Risk Assessment Analysis. J. Am. Heart Assoc. 2021, 10, e019259. [Google Scholar] [CrossRef]
- Stefan, N.; Birkenfeld, A.L.; Schulze, M.B. Global Pandemics Interconnected—Obesity, Impaired Metabolic Health and COVID-19. Nat. Rev. Endocrinol. 2021, 17, 135–149. [Google Scholar] [CrossRef] [PubMed]
- Nadolsky, K.Z.; Hurley, D.L.; Garvey, W.T. Covid-19 & Obesity: Beyond Bmi. Endocr. Pract. 2020, 26, 923–925. [Google Scholar] [CrossRef] [PubMed]
- Costagliola, G.; Spada, E.; Consolini, R. Age-Related Differences in the Immune Response Could Contribute to Determine the Spectrum of Severity of COVID-19. Immun. Inflamm. Dis. 2021, 9, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Hesamirostami, M.; Nazarian, R.; Asghari, H.; Jafarirad, A.; Khosravi, A.; Nouranibaladezaei, S.; Radfar, A. A Case Series of Concomitant Burn and COVID-19. Burns Open 2021, 5, 34–38. [Google Scholar] [CrossRef]
- De Luca, M.; Sartori, A.; Vitiello, A.; Piatto, G.; Noaro, G.; Olmi, S.; Foschi, D.; De Re, L.; Zappa, M.; Sarro, G.; et al. Complications and Mortality in a Cohort of Patients Undergoing Emergency and Elective Surgery with Perioperative SARS-CoV-2 Infection: An Italian Multicenter Study. Teachings of Phase 1 to Be Brought in Phase 2 Pandemic. Updates Surg. 2021, 73, 745–752. [Google Scholar] [CrossRef]
- Li, J.; Guo, M.; Tian, X.; Wang, X.; Yang, X.; Wu, P.; Liu, C.; Xiao, Z.; Qu, Y.; Yin, Y.; et al. Virus-Host Interactome and Proteomic Survey Reveal Potential Virulence Factors Influencing SARS-CoV-2. Pathog. Med. 2021, 2, 99–112. [Google Scholar] [CrossRef]
- Yap, J.K.Y.; Moriyama, M.; Iwasaki, A. Inflammasomes and Pyroptosis as Therapeutic Targets for COVID-19. J. Immunol. 2020, 205, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Luo, S.; Qin, R.; Yang, M.; Wang, X.; Yang, Q.; Zhang, Y.; Wang, Q.; Zhu, R.; Fan, H.; et al. Long-Term Infection of SARS-CoV-2 Changed the Body’s Immune Status. Clin. Immunol. 2020, 218, 108524. [Google Scholar] [CrossRef]
- Antushevich, H. Interplays between Inflammasomes and Viruses, Bacteria (Pathogenic and Probiotic), Yeasts and Parasites. Immunol. Lett. 2020, 228, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.G.; Simpson, L.J.; Ferreira, A.-M.; Rustagi, A.; Roque, J.; Asuni, A.; Ranganath, T.; Grant, P.M.; Subramanian, A.; Rosenberg-Hasson, Y.; et al. Cytokine Profile in Plasma of Severe COVID-19 Does Not Differ from ARDS and Sepsis. JCI Insight 2020, 5, e140289. [Google Scholar] [CrossRef]
- Coperchini, F.; Chiovato, L.; Croce, L.; Magri, F.; Rotondi, M. The Cytokine Storm in COVID-19: An Overview of the Involvement of the Chemokine/Chemokine-Receptor System. Cytokine Growth Factor Rev. 2020, 53, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Hammock, B.D.; Wang, W.; Gilligan, M.M.; Panigrahy, D. Eicosanoids: The Overlooked Storm in Coronavirus Disease 2019 (COVID-19)? Am. J. Pathol. 2020, 190, 1782–1788. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Qin, L.; Zhang, P.; Li, K.; Liang, L.; Sun, J.; Xu, B.; Dai, Y.; Li, X.; Zhang, C.; et al. Longitudinal COVID-19 Profiling Associates IL-1RA and IL-10 with Disease Severity and RANTES with Mild Disease. JCI Insight 2020, 5, e139834. [Google Scholar] [CrossRef]
- Chen, L.; Liu, H.G.; Liu, W.; Liu, J.; Liu, K.; Shang, J.; Deng, Y.; Wei, S. Analysis of clinical features of 29 patients with 2019 novel coronavirus pneumonia. Chin. J. Tuberc. Respir. Dis. 2020, 43, E005. [Google Scholar] [CrossRef]
- Diao, B.; Wang, C.; Tan, Y.; Chen, X.; Liu, Y.; Ning, L.; Chen, L.; Li, M.; Liu, Y.; Wang, G.; et al. Reduction and Functional Exhaustion of T Cells in Patients with Coronavirus Disease 2019 (COVID-19). Front. Immunol. 2020, 11, 827. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical Features of Patients Infected with 2019 Novel Coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Han, H.; Ma, Q.; Li, C.; Liu, R.; Zhao, L.; Wang, W.; Zhang, P.; Liu, X.; Gao, G.; Liu, F.; et al. Profiling Serum Cytokines in COVID-19 Patients Reveals IL-6 and IL-10 Are Disease Severity Predictors. Emerg. Microbes Infect. 2020, 9, 1123–1130. [Google Scholar] [CrossRef]
- Ruan, Q.; Yang, K.; Wang, W.; Jiang, L.; Song, J. Clinical Predictors of Mortality Due to COVID-19 Based on an Analysis of Data of 150 Patients from Wuhan, China. Intensive Care Med. 2020, 46, 846–848. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Y.; Liu, Y.; Cao, L.; Wang, D.; Guo, M.; Jiang, A.; Guo, D.; Hu, W.; Yang, J.; Tang, Z.; et al. Transcriptomic Characteristics of Bronchoalveolar Lavage Fluid and Peripheral Blood Mononuclear Cells in COVID-19 Patients. Emerg. Microbes Infect. 2020, 9, 761–770. [Google Scholar] [CrossRef]
- Wang, J.; Jiang, M.; Chen, X.; Montaner, L.J. Cytokine Storm and Leukocyte Changes in Mild versus Severe SARS-CoV-2 Infection: Review of 3939 COVID-19 Patients in China and Emerging Pathogenesis and Therapy Concepts. J. Leukoc. Biol. 2020, 108, 17–41. [Google Scholar] [CrossRef]
- Qin, C.; Zhou, L.; Hu, Z.; Zhang, S.; Yang, S.; Tao, Y.; Xie, C.; Ma, K.; Shang, K.; Wang, W.; et al. Dysregulation of Immune Response in Patients with Coronavirus 2019 (COVID-19) in Wuhan, China. Clin. Infect. Dis. 2020, 71, 762–768. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Liu, Y.; Zhou, R.; Deng, X.; Li, F.; Liang, K.; Shi, Y. Immunopathological Characteristics of Coronavirus Disease 2019 Cases in Guangzhou, China. Immunology 2020, 160, 261–268. [Google Scholar] [CrossRef]
- Gao, Y.; Li, T.; Han, M.; Li, X.; Wu, D.; Xu, Y.; Zhu, Y.; Liu, Y.; Wang, X.; Wang, L. Diagnostic Utility of Clinical Laboratory Data Determinations for Patients with the Severe COVID-19. J. Med. Virol. 2020, 92, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Li, L.; Xu, M.; Wu, J.; Luo, D.; Zhu, Y.; Li, B.; Song, X.; Zhou, X. Prognostic Value of Interleukin-6, C-Reactive Protein, and Procalcitonin in Patients with COVID-19. J. Clin. Virol. 2020, 127, 104370. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Wu, D.; Guo, W.; Cao, Y.; Huang, D.; Wang, H.; Wang, T.; Zhang, X.; Chen, H.; Yu, H.; et al. Clinical and Immunological Features of Severe and Moderate Coronavirus Disease 2019. J. Clin. Investig. 2020, 130, 2620–2629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Li, S.; Liu, J.; Liang, B.; Wang, X.; Wang, H.; Li, W.; Tong, Q.; Yi, J.; Zhao, L.; et al. Longitudinal Characteristics of Lymphocyte Responses and Cytokine Profiles in the Peripheral Blood of SARS-CoV-2 Infected Patients. EBioMedicine 2020, 55, 102763. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, J.; Yang, Y.; Ma, H.; Li, Z.; Zhang, J.; Cheng, J.; Zhang, X.; Zhao, Y.; Xia, Z.; et al. The Role of Interleukin-6 in Monitoring Severe Case of Coronavirus Disease 2019. EMBO Mol. Med. 2020, 12, e12421. [Google Scholar] [CrossRef]
- Gong, J.; Dong, H.; Xia, Q.-S.; Huang, Z.-Y.; Wang, D.-K.; Zhao, Y.; Liu, W.-H.; Tu, S.-H.; Zhang, M.-M.; Wang, Q.; et al. Correlation Analysis between Disease Severity and Inflammation-Related Parameters in Patients with COVID-19: A Retrospective Study. BMC Infect. Dis. 2020, 20, 963. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, B.; Li, Q.; Wen, L.; Zhang, R. Clinical Features of 69 Cases with Coronavirus Disease 2019 in Wuhan, China. Clin. Infect. Dis. 2020, 71, 769–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Zhao, B.; Qu, Y.; Chen, Y.; Xiong, J.; Feng, Y.; Men, D.; Huang, Q.; Liu, Y.; Yang, B.; et al. Detectable Serum Severe Acute Respiratory Syndrome Coronavirus 2 Viral Load (RNAemia) Is Closely Correlated with Drastically Elevated Interleukin 6 Level in Critically Ill Patients with Coronavirus Disease 2019. Clin. Infect. Dis. 2020, 71, 1937–1942. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Y.; Yin, J.; Wang, W.; Shi, H.; Shi, Y.; Xu, B.; Qiao, L.; Feng, Y.; Pang, L.; Wei, F.; et al. Downregulated Gene Expression Spectrum and Immune Responses Changed During the Disease Progression in Patients With COVID-19. Clin. Infect. Dis. 2020, 71, 2052–2060. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Zhao, X.; Qi, Y.; Li, H.; Ye, G.; Liu, Y.; Zhang, Y.; Gou, J. Sepsis-Associated Severe Interleukin-6 Storm in Critical Coronavirus Disease 2019. Cell. Mol. Immunol. 2020, 17, 1092–1094. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xu, S.; Yu, M.; Wang, K.; Tao, Y.; Zhou, Y.; Shi, J.; Zhou, M.; Wu, B.; Yang, Z.; et al. Risk Factors for Severity and Mortality in Adult COVID-19 Inpatients in Wuhan. J. Allergy Clin. Immunol. 2020, 146, 110–118. [Google Scholar] [CrossRef]
- Yang, Y.; Shen, C.; Li, J.; Yuan, J.; Wei, J.; Huang, F.; Wang, F.; Li, G.; Li, Y.; Xing, L.; et al. Plasma IP-10 and MCP-3 Levels Are Highly Associated with Disease Severity and Predict the Progression of COVID-19. J. Allergy Clin. Immunol. 2020, 146, 119–127. [Google Scholar] [CrossRef]
- Rovas, A.; Osiaevi, I.; Buscher, K.; Sackarnd, J.; Tepasse, P.-R.; Fobker, M.; Kühn, J.; Braune, S.; Göbel, U.; Thölking, G.; et al. Microvascular Dysfunction in COVID-19: The MYSTIC Study. Angiogenesis 2021, 24, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Guo, W.; Dong, Y.; Wang, X.; Dai, D.; Liu, X.; Wu, Y.; Li, M.; Zhang, W.; Zhou, H.; et al. Elevated Exhaustion Levels of NK and CD8+ T Cells as Indicators for Progression and Prognosis of COVID-19 Disease. Front. Immunol. 2020, 11, 580237. [Google Scholar] [CrossRef]
- Zotova, N.V.; Zhuravleva, Y.V.; Zubova, T.E.; Gusev, E.Y. Integral Estimation of Systemic Inflammatory Response under Sepsis. Gen. Physiol. Biophys. 2020, 39, 13–26. [Google Scholar] [CrossRef]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef] [PubMed]
- Olmos, G.; Lladó, J. Tumor Necrosis Factor Alpha: A Link between Neuroinflammation and Excitotoxicity. Mediat. Inflamm. 2014, 2014, 861231. [Google Scholar] [CrossRef]
- Miller, A.H.; Maletic, V.; Raison, C.L. Inflammation and Its Discontents: The Role of Cytokines in the Pathophysiology of Major Depression. Biol. Psychiatry 2009, 65, 732–741. [Google Scholar] [CrossRef] [Green Version]
- Simi, A.; Tsakiri, N.; Wang, P.; Rothwell, N.J. Interleukin-1 and Inflammatory Neurodegeneration. Biochem. Soc. Trans. 2007, 35, 1122–1126. [Google Scholar] [CrossRef]
- de la Rica, R.; Borges, M.; Gonzalez-Freire, M. COVID-19: In the Eye of the Cytokine Storm. Front. Immunol. 2020, 11, 558898. [Google Scholar] [CrossRef]
- Chauhan, A.; Kaur, R.; Chakrbarti, P.; Pal, A. “Silent Hypoxemia” Leads to Vicious Cycle of Infection, Coagulopathy and Cytokine Storm in COVID-19: Can Prophylactic Oxygen Therapy Prevent It? Indian J. Clin. Biochem. 2021, 1–5. [Google Scholar] [CrossRef]
- Xie, J.; Covassin, N.; Fan, Z.; Singh, P.; Gao, W.; Li, G.; Kara, T.; Somers, V.K. Association Between Hypoxemia and Mortality in Patients With COVID-19. Mayo Clin. Proc. 2020, 95, 1138–1147. [Google Scholar] [CrossRef]
- AbdelMassih, A.; Yacoub, E.; Husseiny, R.J.; Kamel, A.; Hozaien, R.; El Shershaby, M.; Rajab, M.; Yacoub, S.; Eid, M.A.; Elahmady, M.; et al. Hypoxia-Inducible Factor (HIF): The Link between Obesity and COVID-19. Obes. Med. 2021, 22, 100317. [Google Scholar] [CrossRef]
- Somers, V.K.; Kara, T.; Xie, J. Progressive Hypoxia: A Pivotal Pathophysiologic Mechanism of COVID-19 Pneumonia. Mayo Clin. Proc. 2020, 95, 2339–2342. [Google Scholar] [CrossRef]
- Dengler, V.L.; Galbraith, M.; Espinosa, J.M. Transcriptional Regulation by Hypoxia Inducible Factors. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, W.; Zhou, J.; Zhou, N.; Liu, Q.; Cui, J.; Zou, W.; Zhang, W. Hypoxia-Increased RAGE Expression Regulates Chemotaxis and pro-Inflammatory Cytokines Release through Nuclear Translocation of NF-κ B and HIF1α in THP-1 cells. Biochem. Biophys. Res. Commun. 2018, 495, 2282–2288. [Google Scholar] [CrossRef] [PubMed]
- Winning, S.; Splettstoesser, F.; Fandrey, J.; Frede, S. Acute Hypoxia Induces HIF-Independent Monocyte Adhesion to Endothelial Cells through Increased Intercellular Adhesion Molecule-1 Expression: The Role of Hypoxic Inhibition of Prolyl Hydroxylase Activity for the Induction of NF-Kappa, B. J. Immunol. 2010, 185, 1786–1793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madjdpour, C.; Jewell, U.R.; Kneller, S.; Ziegler, U.; Schwendener, R.; Booy, C.; Kläusli, L.; Pasch, T.; Schimmer, R.C.; Beck-Schimmer, B. Decreased Alveolar Oxygen Induces Lung Inflammation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 284, L360–L367. [Google Scholar] [CrossRef] [Green Version]
- Ertel, W.; Morrison, M.H.; Ayala, A.; Chaudry, I.H. Hypoxemia in the Absence of Blood Loss or Significant Hypotension Causes Inflammatory Cytokine Release. Am. J. Physiol. 1995, 269, R160–R166. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Sahu, A.; Prabhakar, A.; Chatterjee, T.; Tyagi, T.; Kumari, B.; Khan, N.; Nair, V.; Bajaj, N.; Sharma, M.; et al. Activation of NLRP3 Inflammasome Complex Potentiates Venous Thrombosis in Response to Hypoxia. Proc. Natl. Acad. Sci. USA 2017, 114, 4763–4768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zonneveld, M.I.; Keulers, T.G.H.; Rouschop, K.M.A. Extracellular Vesicles as Transmitters of Hypoxia Tolerance in Solid Cancers. Cancers 2019, 11, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamakawa, M.; Liu, L.X.; Date, T.; Belanger, A.J.; Vincent, K.A.; Akita, G.Y.; Kuriyama, T.; Cheng, S.H.; Gregory, R.J.; Jiang, C. Hypoxia-Inducible Factor-1 Mediates Activation of Cultured Vascular Endothelial Cells by Inducing Multiple Angiogenic Factors. Circ. Res. 2003, 93, 664–673. [Google Scholar] [CrossRef] [Green Version]
- Serebrovska, Z.O.; Chong, E.Y.; Serebrovska, T.V.; Tumanovska, L.V.; Xi, L. Hypoxia, HIF-1α, and COVID-19: From Pathogenic Factors to Potential Therapeutic Targets. Acta Pharmacol. Sin. 2020, 41, 1539–1546. [Google Scholar] [CrossRef]
- Chu, X.; Wen, J.; Raju, R.P. Rapid Senescence-like Response after Acute Injury. Aging Cell 2020, 19, e13201. [Google Scholar] [CrossRef]
- Cannon, J.W. Hemorrhagic Shock. N. Engl. J. Med. 2018, 378, 370–379. [Google Scholar] [CrossRef]
- Frede, S.; Stockmann, C.; Winning, S.; Freitag, P.; Fandrey, J. Hypoxia-Inducible Factor (HIF) 1alpha Accumulation and HIF Target Gene Expression Are Impaired after Induction of Endotoxin Tolerance. J. Immunol. 2009, 182, 6470–6476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández, A.; Geng, Y.; Sepúlveda, R.; Solís, N.; Torres, J.; Arab, J.P.; Barrera, F.; Cabrera, D.; Moshage, H.; Arrese, M. Chemical Hypoxia Induces Pro-Inflammatory Signals in Fat-Laden Hepatocytes and Contributes to Cellular Crosstalk with Kupffer Cells through Extracellular Vesicles. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165753. [Google Scholar] [CrossRef]
- Snodgrass, R.G.; Boß, M.; Zezina, E.; Weigert, A.; Dehne, N.; Fleming, I.; Brüne, B.; Namgaladze, D. Hypoxia Potentiates Palmitate-Induced Pro-Inflammatory Activation of Primary Human Macrophages. J. Biol. Chem. 2016, 291, 413–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.S.; Rajagopal, V.; Gonsalves, C.; Johnson, C.; Kalra, V.K. A Novel Role of Hypoxia-Inducible Factor in Cobalt Chloride- and Hypoxia-Mediated Expression of IL-8 Chemokine in Human Endothelial Cells. J. Immunol. 2006, 177, 7211–7224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabryelska, A.; Karuga, F.F.; Szmyd, B.; Białasiewicz, P. HIF-1α as a Mediator of Insulin Resistance, T2DM, and Its Complications: Potential Links With Obstructive Sleep Apnea. Front. Physiol. 2020, 11, 1035. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern Recognition Receptors and Inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [Green Version]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-Sensing Receptors in Sterile Inflammation and Inflammatory Diseases. Nat. Rev. Immunol. 2020, 20, 95–112. [Google Scholar] [CrossRef]
- Zhu, Y.; Deng, J.; Nan, M.-L.; Zhang, J.; Okekunle, A.; Li, J.-Y.; Yu, X.-Q.; Wang, P.-H. The Interplay Between Pattern Recognition Receptors and Autophagy in Inflammation. Adv. Exp. Med. Biol. 2019, 1209, 79–108. [Google Scholar] [CrossRef]
- Amarante-Mendes, G.P.; Adjemian, S.; Branco, L.M.; Zanetti, L.C.; Weinlich, R.; Bortoluci, K.R. Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front. Immunol. 2018, 9, 2379. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.L.; Wagner, D.D. Peptidylarginine Deiminase 4: A Nuclear Button Triggering Neutrophil Extracellular Traps in Inflammatory Diseases and Aging. FASEB J. 2018, 32, 6258–6370. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Hall, S.; Vitetta, L. Altered Gut Microbial Metabolites Could Mediate the Effects of Risk Factors in Covid-19. Rev. Med. Virol. 2021, e2211. [Google Scholar] [CrossRef]
- Devaux, C.A.; Lagier, J.-C.; Raoult, D. New Insights into the Physiopathology of COVID-19: SARS-CoV-2-Associated Gastrointestinal Illness. Front. Med. 2021, 8, 99. [Google Scholar] [CrossRef] [PubMed]
- Aboudounya, M.M.; Heads, R.J. COVID-19 and Toll-Like Receptor 4 (TLR4): SARS-CoV-2 May Bind and Activate TLR4 to Increase ACE2 Expression, Facilitating Entry and Causing Hyperinflammation. Mediat. Inflamm. 2021, 2021, 8874339. [Google Scholar] [CrossRef]
- Choudhury, A.; Mukherjee, S. In Silico Studies on the Comparative Characterization of the Interactions of SARS-CoV-2 Spike Glycoprotein with ACE-2 Receptor Homologs and Human TLRs. J. Med. Virol. 2020, 92, 2105–2113. [Google Scholar] [CrossRef] [PubMed]
- Gusev, E.Y.; Zotova, N.V.; Zhuravleva, Y.A.; Chereshnev, V.A. Physiological and pathogenic role of scavenger receptors in humans. Med. Immunol. 2020, 22, 7–48. [Google Scholar] [CrossRef]
- PrabhuDas, M.R.; Baldwin, C.L.; Bollyky, P.L.; Bowdish, D.M.E.; Drickamer, K.; Febbraio, M.; Herz, J.; Kobzik, L.; Krieger, M.; Loike, J.; et al. A Consensus Definitive Classification of Scavenger Receptors and Their Roles in Health and Disease. J. Immunol. 2017, 198, 3775–3789. [Google Scholar] [CrossRef] [Green Version]
- McEwen, B.S.; Wingfield, J.C. The Concept of Allostasis in Biology and Biomedicine. Horm. Behav. 2003, 43, 2–15. [Google Scholar] [CrossRef]
- Amraie, R.; Napoleon, M.A.; Yin, W.; Berrigan, J.; Suder, E.; Zhao, G.; Olejnik, J.; Gummuluru, S.; Muhlberger, E.; Chitalia, V.; et al. CD209L/L-SIGN and CD209/DC-SIGN Act as Receptors for SARS-CoV-2 and Are Differentially Expressed in Lung and Kidney Epithelial and Endothelial Cells. bioRxiv 2020. [Google Scholar] [CrossRef]
- Wei, C.; Wan, L.; Yan, Q.; Wang, X.; Zhang, J.; Yang, X.; Zhang, Y.; Fan, C.; Li, D.; Deng, Y.; et al. HDL-Scavenger Receptor B Type 1 Facilitates SARS-CoV-2 Entry. Nat. Metab. 2020, 2, 1391–1400. [Google Scholar] [CrossRef]
- Al-Banna, N.; Lehmann, C. Oxidized LDL and LOX-1 in Experimental Sepsis. Mediat. Inflamm. 2013, 2013, 761789. [Google Scholar] [CrossRef] [Green Version]
- Sparvero, L.J.; Asafu-Adjei, D.; Kang, R.; Tang, D.; Amin, N.; Im, J.; Rutledge, R.; Lin, B.; Amoscato, A.A.; Zeh, H.J.; et al. RAGE (Receptor for Advanced Glycation Endproducts), RAGE Ligands, and Their Role in Cancer and Inflammation. J. Transl. Med. 2009, 7, 17. [Google Scholar] [CrossRef] [Green Version]
- Liliensiek, B.; Weigand, M.A.; Bierhaus, A.; Nicklas, W.; Kasper, M.; Hofer, S.; Plachky, J.; Gröne, H.-J.; Kurschus, F.C.; Schmidt, A.M.; et al. Receptor for Advanced Glycation End Products (RAGE) Regulates Sepsis but Not the Adaptive Immune Response. J. Clin. Investig. 2004, 113, 1641–1650. [Google Scholar] [CrossRef]
- Riehl, A.; Németh, J.; Angel, P.; Hess, J. The Receptor RAGE: Bridging Inflammation and Cancer. Cell Commun. Signal. 2009, 7, 12. [Google Scholar] [CrossRef] [Green Version]
- Zou, Q.; Wen, W.; Zhang, X.-C. Presepsin as a Novel Sepsis Biomarker. World J. Emerg. Med. 2014, 5, 16–19. [Google Scholar] [CrossRef] [Green Version]
- Feng, L.; Zhou, X.; Su, L.-X.; Feng, D.; Jia, Y.-H.; Xie, L.-X. Clinical Significance of Soluble Hemoglobin Scavenger Receptor CD163 (SCD163) in Sepsis, a Prospective Study. PLoS ONE 2012, 7, e38400. [Google Scholar] [CrossRef]
- Rødgaard-Hansen, S.; Rafique, A.; Christensen, P.A.; Maniecki, M.B.; Sandahl, T.D.; Nexø, E.; Møller, H.J. A Soluble Form of the Macrophage-Related Mannose Receptor (MR/CD206) Is Present in Human Serum and Elevated in Critical Illness. Clin. Chem. Lab. Med. 2014, 52, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Fukada, A.; Kitagawa, Y.; Matsuoka, M.; Sakai, J.; Imai, K.; Tarumoto, N.; Orihara, Y.; Kawamura, R.; Takeuchi, S.; Maesaki, S.; et al. Presepsin as a Predictive Biomarker of Severity in COVID-19: A Case Series. J. Med. Virol. 2021, 93, 99–101. [Google Scholar] [CrossRef] [PubMed]
- Zaninotto, M.; Mion, M.M.; Cosma, C.; Rinaldi, D.; Plebani, M. Presepsin in Risk Stratification of SARS-CoV-2 Patients. Clin. Chim. Acta 2020, 507, 161–163. [Google Scholar] [CrossRef]
- Zingaropoli, M.A.; Nijhawan, P.; Carraro, A.; Pasculli, P.; Zuccalà, P.; Perri, V.; Marocco, R.; Kertusha, B.; Siccardi, G.; Del Borgo, C.; et al. Increased SCD163 and SCD14 Plasmatic Levels and Depletion of Peripheral Blood Pro-Inflammatory Monocytes, Myeloid and Plasmacytoid Dendritic Cells in Patients with Severe COVID-19 Pneumonia. Front. Immunol. 2021, 12, 627548. [Google Scholar] [CrossRef]
- Etzerodt, A.; Moestrup, S.K. CD163 and Inflammation: Biological, Diagnostic, and Therapeutic Aspects. Antioxid. Redox Signal. 2013, 18, 2352–2363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Møller, H.J. Soluble CD163. Scand. J. Clin. Lab. Investig. 2012, 72, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, K.; Suzuki, Y.; Yoshimura, K.; Yasui, H.; Karayama, M.; Hozumi, H.; Furuhashi, K.; Enomoto, N.; Fujisawa, T.; Nakamura, Y.; et al. Macrophage Mannose Receptor CD206 Predicts Prognosis in Community-Acquired Pneumonia. Sci. Rep. 2019, 9, 18750. [Google Scholar] [CrossRef]
- Stewart, C.R.; Stuart, L.M.; Wilkinson, K.; van Gils, J.M.; Deng, J.; Halle, A.; Rayner, K.J.; Boyer, L.; Zhong, R.; Frazier, W.A.; et al. CD36 Ligands Promote Sterile Inflammation through Assembly of a Toll-like Receptor 4 and 6 Heterodimer. Nat. Immunol. 2010, 11, 155–161. [Google Scholar] [CrossRef] [Green Version]
- Silverstein, R.L.; Febbraio, M. CD36, a Scavenger Receptor Involved in Immunity, Metabolism, Angiogenesis, and Behavior. Sci. Signal. 2009, 2, re3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goyal, T.; Mitra, S.; Khaidakov, M.; Wang, X.; Singla, S.; Ding, Z.; Liu, S.; Mehta, J.L. Current Concepts of the Role of Oxidized LDL Receptors in Atherosclerosis. Curr. Atheroscler. Rep. 2012, 14, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Kholmukhamedov, A.; Schulte, M.L.; Cooley, B.C.; Scoggins, N.O.; Wood, J.P.; Cameron, S.J.; Morrell, C.N.; Jobe, S.M.; Silverstein, R.L. Platelet CD36 Signaling through ERK5 Promotes Caspase-Dependent Procoagulant Activity and Fibrin Deposition in Vivo. Blood Adv. 2018, 2, 2848–2861. [Google Scholar] [CrossRef] [PubMed]
- Deitch, E.A.; Condon, M.; Feketeova, E.; Machiedo, G.W.; Mason, L.; Vinluan, G.M.; Alli, V.A.; Neal, M.D.; Tomaio, J.N.; Fishman, J.E.; et al. Trauma-Hemorrhagic Shock Induces a CD36-Dependent RBC Endothelial-Adhesive Phenotype. Crit. Care Med. 2014, 42, e200–e210. [Google Scholar] [CrossRef] [PubMed]
- Fain, J.N. Release of Interleukins and Other Inflammatory Cytokines by Human Adipose Tissue Is Enhanced in Obesity and Primarily Due to the Nonfat Cells. Vitam Horm. 2006, 74, 443–477. [Google Scholar] [CrossRef] [PubMed]
- Bastard, J.-P.; Maachi, M.; Lagathu, C.; Kim, M.J.; Caron, M.; Vidal, H.; Capeau, J.; Feve, B. Recent Advances in the Relationship between Obesity, Inflammation, and Insulin Resistance. Eur. Cytokine Netw. 2006, 17, 4–12. [Google Scholar]
- Fain, J.N.; Nesbit, A.S.; Sudlow, F.F.; Cheema, P.; Peeples, J.M.; Madan, A.K.; Tichansky, D.S. Release in Vitro of Adipsin, Vascular Cell Adhesion Molecule 1, Angiotensin 1-Converting Enzyme, and Soluble Tumor Necrosis Factor Receptor 2 by Human Omental Adipose Tissue as Well as by the Nonfat Cells and Adipocytes. Metabolism 2007, 56, 1583–1590. [Google Scholar] [CrossRef]
- Smith, U.; Kahn, B.B. Adipose Tissue Regulates Insulin Sensitivity: Role of Adipogenesis, de Novo Lipogenesis and Novel Lipids. J. Intern. Med. 2016, 280, 465–475. [Google Scholar] [CrossRef] [Green Version]
- Galic, S.; Oakhill, J.S.; Steinberg, G.R. Adipose Tissue as an Endocrine Organ. Mol. Cell. Endocrinol. 2010, 316, 129–139. [Google Scholar] [CrossRef]
- Marra, F.; Svegliati-Baroni, G. Lipotoxicity and the Gut-Liver Axis in NASH Pathogenesis. J. Hepatol. 2018, 68, 280–295. [Google Scholar] [CrossRef]
- Ye, J. Mechanisms of Insulin Resistance in Obesity. Front. Med. 2013, 7, 14–24. [Google Scholar] [CrossRef] [Green Version]
- Ferrara, D.; Montecucco, F.; Dallegri, F.; Carbone, F. Impact of Different Ectopic Fat Depots on Cardiovascular and Metabolic Diseases. J. Cell. Physiol. 2019, 234, 21630–21641. [Google Scholar] [CrossRef]
- Tumova, J.; Andel, M.; Trnka, J. Excess of Free Fatty Acids as a Cause of Metabolic Dysfunction in Skeletal Muscle. Physiol. Res. 2016, 65, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Engin, A. Eat and Death: Chronic Over-Eating. Adv. Exp. Med. Biol. 2017, 960, 53–80. [Google Scholar] [CrossRef]
- Schaffer, J.E. Lipotoxicity: Many Roads to Cell Dysfunction and Cell Death: Introduction to a Thematic Review Series. J. Lipid Res. 2016, 57, 1327–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chabowski, A.; Zmijewska, M.; Górski, J.; Bonen, A.; Kamiński, K.; Winnicka, M.M. Effect of IL-6 Deficiency on Myocardial Expression of Fatty Acid Transporters and Intracellular Lipid Deposits. J. Physiol. Pharmacol. 2007, 58, 73–82. [Google Scholar] [PubMed]
- Rada, P.; González-Rodríguez, Á.; García-Monzón, C.; Valverde, Á.M. Understanding Lipotoxicity in NAFLD Pathogenesis: Is CD36 a Key Driver? Cell Death Dis. 2020, 11, 802. [Google Scholar] [CrossRef] [PubMed]
- Hathaway, Q.A.; Pinti, M.V.; Durr, A.J.; Waris, S.; Shepherd, D.L.; Hollander, J.M. Regulating MicroRNA Expression: At the Heart of Diabetes Mellitus and the Mitochondrion. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H293–H310. [Google Scholar] [CrossRef]
- Römer, A.; Linn, T.; Petry, S.F. Lipotoxic Impairment of Mitochondrial Function in β-Cells: A Review. Antioxidants 2021, 10, 293. [Google Scholar] [CrossRef]
- Nogueira, A.C.; Kawabata, V.; Biselli, P.; Lins, M.H.; Valeri, C.; Seckler, M.; Hoshino, W.; Júnior, L.G.; Bernik, M.M.S.; de Andrade Machado, J.B.; et al. Changes in Plasma Free Fatty Acid Levels in Septic Patients Are Associated with Cardiac Damage and Reduction in Heart Rate Variability. Shock 2008, 29, 342–348. [Google Scholar] [CrossRef] [Green Version]
- Erol, A. Role of Oxidized LDL-Induced “Trained Macrophages” in the Pathogenesis of COVID-19 and Benefits of Pioglitazone: A Hypothesis. Diabetes Metab. Syndr. 2020, 14, 713–714. [Google Scholar] [CrossRef]
- Cho, K.-H.; Kim, J.-R.; Lee, I.-C.; Kwon, H.-J. Native High-Density Lipoproteins (HDL) with Higher Paraoxonase Exerts a Potent Antiviral Effect against SARS-CoV-2 (COVID-19), While Glycated HDL Lost the Antiviral Activity. Antioxidants 2021, 10, 209. [Google Scholar] [CrossRef]
- Thomas, T.; Stefanoni, D.; Reisz, J.A.; Nemkov, T.; Bertolone, L.; Francis, R.O.; Hudson, K.E.; Zimring, J.C.; Hansen, K.C.; Hod, E.A.; et al. COVID-19 Infection Alters Kynurenine and Fatty Acid Metabolism, Correlating with IL-6 Levels and Renal Status. JCI Insight 2020, 5, e140327. [Google Scholar] [CrossRef]
- Rivas, A.M.; Nugent, K. Hyperglycemia, Insulin, and Insulin Resistance in Sepsis. Am. J. Med. Sci. 2021, 361, 297–302. [Google Scholar] [CrossRef]
- Green, P.; Theilla, M.; Singer, P. Lipid Metabolism in Critical Illness. Curr. Opin. Clin. Nutr. Metab. Care 2016, 19, 111–115. [Google Scholar] [CrossRef]
- Lee, S.H.; Park, M.S.; Park, B.H.; Jung, W.J.; Lee, I.S.; Kim, S.Y.; Kim, E.Y.; Jung, J.Y.; Kang, Y.A.; Kim, Y.S.; et al. Prognostic Implications of Serum Lipid Metabolism over Time during Sepsis. BioMed Res. Int. 2015, 2015, 789298. [Google Scholar] [CrossRef] [Green Version]
- Nabavi, A.; Allami, A.; QasemiBarqi, R. Changes in Plasma Lipid and In-Hospital Deaths in Patients with Sepsis. Med. J. Islam. Repub. Iran 2020, 34, 45. [Google Scholar] [CrossRef]
- Lekkou, A.; Mouzaki, A.; Siagris, D.; Ravani, I.; Gogos, C.A. Serum Lipid Profile, Cytokine Production, and Clinical Outcome in Patients with Severe Sepsis. J. Crit. Care 2014, 29, 723–727. [Google Scholar] [CrossRef]
- Das, U.N. Serum Adipocyte Fatty Acid-Binding Protein in the Critically Ill. Crit. Care 2013, 17, 121. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.-L.; Wu, Y.-W.; Hsieh, A.-R.; Hung, Y.-H.; Chen, W.-J.; Yang, W.-S. Serum Adipocyte Fatty Acid-Binding Protein Levels in Patients with Critical Illness Are Associated with Insulin Resistance and Predict Mortality. Crit. Care 2013, 17, R22. [Google Scholar] [CrossRef] [Green Version]
- Furuhashi, M.; Hotamisligil, G.S. Fatty Acid-Binding Proteins: Role in Metabolic Diseases and Potential as Drug Targets. Nat. Rev. Drug Discov. 2008, 7, 489–503. [Google Scholar] [CrossRef] [Green Version]
- Rival, T.; Cinq-Frais, C.; Silva-Sifontes, S.; Garcia, J.; Riu, B.; Salvayre, R.; Genestal, M.; Caspar-Bauguil, S. Alteration of Plasma Phospholipid Fatty Acid Profile in Patients with Septic Shock. Biochimie 2013, 95, 2177–2181. [Google Scholar] [CrossRef]
- Cappi, S.B.; Noritomi, D.T.; Velasco, I.T.; Curi, R.; Loureiro, T.C.A.; Soriano, F.G. Dyslipidemia: A Prospective Controlled Randomized Trial of Intensive Glycemic Control in Sepsis. Intensive Care Med. 2012, 38, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Behnes, M.; Brueckmann, M.; Liebe, V.; Liebetrau, C.; Lang, S.; Putensen, C.; Borggrefe, M.; Hoffmann, U. Levels of Oxidized Low-Density Lipoproteins Are Increased in Patients with Severe Sepsis. J. Crit. Care 2008, 23, 537–541. [Google Scholar] [CrossRef]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 Links Innate Immunity and Fatty Acid-Induced Insulin Resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef] [PubMed]
- Spruss, A.; Kanuri, G.; Wagnerberger, S.; Haub, S.; Bischoff, S.C.; Bergheim, I. Toll-like Receptor 4 Is Involved in the Development of Fructose-Induced Hepatic Steatosis in Mice. Hepatology 2009, 50, 1094–1104. [Google Scholar] [CrossRef] [PubMed]
- Pal, D.; Dasgupta, S.; Kundu, R.; Maitra, S.; Das, G.; Mukhopadhyay, S.; Ray, S.; Majumdar, S.S.; Bhattacharya, S. Fetuin-A Acts as an Endogenous Ligand of TLR4 to Promote Lipid-Induced Insulin Resistance. Nat. Med. 2012, 18, 1279–1285. [Google Scholar] [CrossRef]
- Shen, X.; Yang, L.; Yan, S.; Zheng, H.; Liang, L.; Cai, X.; Liao, M. Fetuin A Promotes Lipotoxicity in β Cells through the TLR4 Signaling Pathway and the Role of Pioglitazone in Anti-Lipotoxicity. Mol. Cell. Endocrinol. 2015, 412, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.-M.; Xu, G.; Wang, B.; Liu, B.-C. Cytokine Storm Syndrome in Coronavirus Disease 2019: A Narrative Review. J. Intern. Med. 2021, 289, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Hay, K.A.; Hanafi, L.-A.; Li, D.; Gust, J.; Liles, W.C.; Wurfel, M.M.; López, J.A.; Chen, J.; Chung, D.; Harju-Baker, S.; et al. Kinetics and Biomarkers of Severe Cytokine Release Syndrome after CD19 Chimeric Antigen Receptor–Modified T-Cell Therapy. Blood 2017, 130, 2295–2306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.W.; Gardner, R.; Porter, D.L.; Louis, C.U.; Ahmed, N.; Jensen, M.; Grupp, S.A.; Mackall, C.L. Current Concepts in the Diagnosis and Management of Cytokine Release Syndrome. Blood 2014, 124, 188–195. [Google Scholar] [CrossRef] [Green Version]
- Shimabukuro-Vornhagen, A.; Gödel, P.; Subklewe, M.; Stemmler, H.J.; Schlößer, H.A.; Schlaak, M.; Kochanek, M.; Böll, B.; von Bergwelt-Baildon, M.S. Cytokine Release Syndrome. J. Immunother. Cancer 2018, 6, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prilutskiy, A.; Kritselis, M.; Shevtsov, A.; Yambayev, I.; Vadlamudi, C.; Zhao, Q.; Kataria, Y.; Sarosiek, S.R.; Lerner, A.; Sloan, J.M.; et al. SARS-CoV-2 Infection–Associated Hemophagocytic Lymphohistiocytosis. Am. J. Clin. Pathol. 2020, 154, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Weaver, L.K.; Behrens, E.M. Hyperinflammation, rather than Hemophagocytosis, Is the Common Link between Macrophage Activation Syndrome and Hemophagocytic Lymphohistiocytosis. Curr. Opin. Rheumatol. 2014, 26, 562–569. [Google Scholar] [CrossRef]
- Crayne, C.B.; Albeituni, S.; Nichols, K.E.; Cron, R.Q. The Immunology of Macrophage Activation Syndrome. Front. Immunol. 2019, 10, 119. [Google Scholar] [CrossRef] [Green Version]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J.; HLH Across Speciality Collaboration, UK. COVID-19: Consider Cytokine Storm Syndromes and Immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Soy, M.; Atagündüz, P.; Atagündüz, I.; Sucak, G.T. Hemophagocytic Lymphohistiocytosis: A Review Inspired by the COVID-19 Pandemic. Rheumatol. Int. 2021, 41, 7–18. [Google Scholar] [CrossRef]
- Henter, J.-I.; Horne, A.; Aricó, M.; Egeler, R.M.; Filipovich, A.H.; Imashuku, S.; Ladisch, S.; McClain, K.; Webb, D.; Winiarski, J.; et al. HLH-2004: Diagnostic and Therapeutic Guidelines for Hemophagocytic Lymphohistiocytosis. Pediatr. Blood Cancer. 2007, 48, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, G.; Moog, P.; Bachmann, Q.; La Rosée, P.; Schneider, H.; Schlegl, M.; Spinner, C.; Heemann, U.; Schmid, R.M.; Algül, H.; et al. Title: Cytokine Release Syndrome Is Not Usually Caused by Secondary Hemophagocytic Lymphohistiocytosis in a Cohort of 19 Critically Ill COVID-19 Patients. Sci. Rep. 2020, 10, 18277. [Google Scholar] [CrossRef]
- Yongzhi, X. COVID-19-Associated Cytokine Storm Syndrome and Diagnostic Principles: An Old and New Issue. Emerg. Microbes Infect. 2021, 10, 266–276. [Google Scholar] [CrossRef]
- Ombrello, M.J.; Schulert, G.S. COVID-19 and Cytokine Storm Syndrome: Are There Lessons from Macrophage Activation Syndrome? Transl. Res. 2021, 232, 1–12. [Google Scholar] [CrossRef]
- Colafrancesco, S.; Alessandri, C.; Conti, F.; Priori, R. COVID-19 Gone Bad: A New Character in the Spectrum of the Hyperferritinemic Syndrome? Autoimmun. Rev. 2020, 19, 102573. [Google Scholar] [CrossRef]
- Stone, J.H.; Frigault, M.J.; Serling-Boyd, N.J.; Fernandes, A.D.; Harvey, L.; Foulkes, A.S.; Horick, N.K.; Healy, B.C.; Shah, R.; Bensaci, A.M.; et al. Efficacy of Tocilizumab in Patients Hospitalized with Covid-19. N. Engl. J. Med. 2020, 383, 2333–2344. [Google Scholar] [CrossRef]
- CORIMUNO-19 Collaborative group Effect of Anakinra versus Usual Care in Adults in Hospital with COVID-19 and Mild-to-Moderate Pneumonia (CORIMUNO-ANA-1): A Randomised Controlled Trial. Lancet Respir. Med. 2021, 9, 295–304. [CrossRef]
- Salvarani, C.; Dolci, G.; Massari, M.; Merlo, D.F.; Cavuto, S.; Savoldi, L.; Bruzzi, P.; Boni, F.; Braglia, L.; Turrà, C.; et al. Effect of Tocilizumab vs Standard Care on Clinical Worsening in Patients Hospitalized with COVID-19 Pneumonia: A Randomized Clinical Trial. JAMA Intern. Med. 2021, 181, 24–31. [Google Scholar] [CrossRef]
- Arnaldez, F.I.; O’Day, S.J.; Drake, C.G.; Fox, B.A.; Fu, B.; Urba, W.J.; Montesarchio, V.; Weber, J.S.; Wei, H.; Wigginton, J.M.; et al. The Society for Immunotherapy of Cancer Perspective on Regulation of Interleukin-6 Signaling in COVID-19-Related Systemic Inflammatory Response. J. Immunother. Cancer 2020, 8, L596–L602. [Google Scholar] [CrossRef]
- Minnich, D.J.; Moldawer, L.L. Anti-Cytokine and Anti-Inflammatory Therapies for the Treatment of Severe Sepsis: Progress and Pitfalls. Proc. Nutr. Soc. 2004, 63, 437–441. [Google Scholar] [CrossRef] [Green Version]
- Chýlek, V.; Kula, R.; Holub, M.; Szturz, P.; Sklienka, P.; Máca, J. Anticytokine therapy in sepsis and why it fails. Klin. Mikrobiol. Infekc. Lek. 2008, 14, 209–212. [Google Scholar]
- Caricchio, R.; Gallucci, M.; Dass, C.; Zhang, X.; Gallucci, S.; Fleece, D.; Bromberg, M.; Criner, G.J. Temple University COVID-19 Research Group Preliminary Predictive Criteria for COVID-19 Cytokine Storm. Ann. Rheum. Dis. 2021, 80, 88–95. [Google Scholar] [CrossRef]
- Wang, C.; Xie, J.; Zhao, L.; Fei, X.; Zhang, H.; Tan, Y.; Nie, X.; Zhou, L.; Liu, Z.; Ren, Y.; et al. Alveolar Macrophage Dysfunction and Cytokine Storm in the Pathogenesis of Two Severe COVID-19 Patients. EBioMedicine 2020, 57, 102833. [Google Scholar] [CrossRef]
- Bone, R.C.; Balk, R.A.; Cerra, F.B.; Dellinger, R.P.; Fein, A.M.; Knaus, W.A.; Schein, R.M.; Sibbald, W.J. Definitions for Sepsis and Organ Failure and Guidelines for the Use of Innovative Therapies in Sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest 1992, 101, 1644–1655. [Google Scholar] [CrossRef] [Green Version]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.-D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef]
- Martin, H.; Olander, B.; Norman, M. Reactive Hyperemia and Interleukin 6, Interleukin 8, and Tumor Necrosis Factor-Alpha in the Diagnosis of Early-Onset Neonatal Sepsis. Pediatrics 2001, 108, E61. [Google Scholar] [CrossRef] [Green Version]
- Damas, P.; Ledoux, D.; Nys, M.; Vrindts, Y.; De Groote, D.; Franchimont, P.; Lamy, M. Cytokine Serum Level during Severe Sepsis in Human IL-6 as a Marker of Severity. Ann. Surg. 1992, 215, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Molano Franco, D.; Arevalo-Rodriguez, I.; Roqué, I.; Figuls, M.; Montero Oleas, N.G.; Nuvials, X.; Zamora, J. Plasma Interleukin-6 Concentration for the Diagnosis of Sepsis in Critically Ill Adults. Cochrane Database Syst. Rev. 2019, 4, CD011811. [Google Scholar] [CrossRef]
- Gorham, J.; Moreau, A.; Corazza, F.; Peluso, L.; Ponthieux, F.; Talamonti, M.; Izzi, A.; Nagant, C.; Ndieugnou Djangang, N.; Garufi, A.; et al. Interleukine-6 in Critically Ill COVID-19 Patients: A Retrospective Analysis. PLoS ONE 2020, 15, e0244628. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R. Endothelial Activation and Dysfunction in Metabolic Syndrome, Type 2 Diabetes and Coronavirus Disease 2019. J. Int. Med. Res. 2020, 48, 300060520939746. [Google Scholar] [CrossRef] [PubMed]
- Fosse, J.H.; Haraldsen, G.; Falk, K.; Edelmann, R. Endothelial Cells in Emerging Viral Infections. Front. Cardiovasc. Med. 2021, 8, 619690. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Lv, J.; Lin, L. Coagulopathy in COVID-19: Focus on Vascular Thrombotic Events. J. Mol. Cell. Cardiol. 2020, 146, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Becker, R.C. COVID-19 Update: Covid-19-Associated Coagulopathy. J. Thromb. Thrombolysis 2020, 50, 54–67. [Google Scholar] [CrossRef]
- Tang, N.; Li, D.; Wang, X.; Sun, Z. Abnormal Coagulation Parameters Are Associated with Poor Prognosis in Patients with Novel Coronavirus Pneumonia. J. Thromb. Haemost. 2020, 18, 844–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- José, R.J.; Williams, A.E.; Chambers, R.C. Proteinase-Activated Receptors in Fibroproliferative Lung Disease. Thorax 2014, 69, 190–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goshua, G.; Pine, A.B.; Meizlish, M.L.; Chang, C.-H.; Zhang, H.; Bahel, P.; Baluha, A.; Bar, N.; Bona, R.D.; Burns, A.J.; et al. Endotheliopathy in COVID-19-Associated Coagulopathy: Evidence from a Single-Centre, Cross-Sectional Study. Lancet Haematol. 2020, 7, e575–e582. [Google Scholar] [CrossRef]
- Zhang, J.; Tecson, K.M.; McCullough, P.A. Endothelial Dysfunction Contributes to COVID-19-Associated Vascular Inflammation and Coagulopathy. Rev. Cardiovasc. Med. 2020, 21, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Sahai, A.; Bhandari, R.; Koupenova, M.; Freedman, J.; Godwin, M.; McIntyre, T.; Chung, M.; Iskandar, J.-P.; Kamran, H.; Aggarwal, A.; et al. SARS-CoV-2 Receptors Are Expressed on Human Platelets and the Effect of Aspirin on Clinical Outcomes in COVID-19 Patients. Res. Sq. 2020. [Google Scholar] [CrossRef]
- Yamaoka-Tojo, M. Endothelial Glycocalyx Damage as a Systemic Inflammatory Microvascular Endotheliopathy in COVID-19. Biomed. J. 2020, 43, 399–413. [Google Scholar] [CrossRef]
- Sorop, O.; Olver, T.D.; van de Wouw, J.; Heinonen, I.; van Duin, R.W.; Duncker, D.J.; Merkus, D. The Microcirculation: A Key Player in Obesity-Associated Cardiovascular Disease. Cardiovasc. Res. 2017, 113, 1035–1045. [Google Scholar] [CrossRef]
- Zhao, L.; Fu, Z.; Wu, J.; Aylor, K.W.; Barrett, E.J.; Cao, W.; Liu, Z. Inflammation-Induced Microvascular Insulin Resistance Is an Early Event in Diet-Induced Obesity. Clin. Sci. 2015, 129, 1025–1036. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.M.; Ko, C.B.; Park, Y.P.; Kim, Y.J.; Paik, S.G. Octamer Motif Is Required for the NF-KappaB-Mediated Induction of the Inducible Nitric Oxide Synthase Gene Expression in RAW 264.7 Macrophages. Mol. Cells 1999, 9, 99–109. [Google Scholar]
- Zhou, Z.; Gengaro, P.; Wang, W.; Wang, X.; Li, C.; Faubel, S.; Rivard, C.; Schrier, R.W. Role of NF-KappaB and PI 3-Kinase/Akt in TNF-Alpha-Induced Cytotoxicity in Microvascular Endothelial Cells. Am. J. Physiol. Ren. Physiol. 2008, 295, F932–F941. [Google Scholar] [CrossRef]
- Hollenberg, S.M.; Guglielmi, M.; Parrillo, J.E. Discordance between Microvascular Permeability and Leukocyte Dynamics in Septic Inducible Nitric Oxide Synthase Deficient Mice. Crit. Care 2007, 11, R125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eyenga, P.; Lhuillier, F.; Morel, J.; Roussel, D.; Sibille, B.; Letexier, D.; Cespuglio, R.; Duchamp, C.; Goudable, J.; Bricca, G.; et al. Time Course of Liver Nitric Oxide Concentration in Early Septic Shock by Cecal Ligation and Puncture in Rats. Nitric Oxide 2010, 23, 194–198. [Google Scholar] [CrossRef]
- Mario, L.; Roberto, M.; Marta, L.; Teresa, C.M.; Laura, M. Hypothesis of COVID-19 Therapy with Sildenafil. Int. J. Prev. Med. 2020, 11, 76. [Google Scholar] [CrossRef] [PubMed]
- Karki, R.; Sharma, B.R.; Tuladhar, S.; Williams, E.P.; Zalduondo, L.; Samir, P.; Zheng, M.; Sundaram, B.; Banoth, B.; Malireddi, R.K.S.; et al. COVID-19 Cytokines and the Hyperactive Immune Response: Synergism of TNF-α and IFN-γ in Triggering Inflammation, Tissue Damage, and Death. bioRxiv 2020. [Google Scholar] [CrossRef]
- Kan, W.-H.; Hsu, J.-T.; Schwacha, M.G.; Choudhry, M.A.; Raju, R.; Bland, K.I.; Chaudry, I.H. Selective Inhibition of INOS Attenuates Trauma-Hemorrhage/Resuscitation-Induced Hepatic Injury. J. Appl. Physiol. 2008, 105, 1076–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, Y.; Deitch, E.A.; Mishima, S.; Duràn, W.N.; Xu, D.Z. Endotoxin-Induced Mesenteric Microvascular Changes Involve INOS-Derived Nitric Oxide: Results from a Study Using INOS Knock out Mice. Shock 2000, 13, 397–403. [Google Scholar] [CrossRef] [Green Version]
- Harkin, D.W.; Rubin, B.B.; Romaschin, A.; Lindsay, T.F. Selective Inducible Nitric Oxide Synthase (INOS) Inhibition Attenuates Remote Acute Lung Injury in a Model of Ruptured Abdominal Aortic Aneurysm. J. Surg. Res. 2004, 120, 230–241. [Google Scholar] [CrossRef]
- Korkmaz, H.I.; Ulrich, M.M.W.; Vogels, S.; de Wit, T.; van Zuijlen, P.P.M.; Krijnen, P.A.J.; Niessen, H.W.M. Neutrophil Extracellular Traps Coincide with a Pro-Coagulant Status of Microcirculatory Endothelium in Burn Wounds. Wound Repair Regen. 2017, 25, 609–617. [Google Scholar] [CrossRef]
- Schönrich, G.; Raftery, M.J.; Samstag, Y. Devilishly Radical NETwork in COVID-19: Oxidative Stress, Neutrophil Extracellular Traps (NETs), and T Cell Suppression. Adv. Biol. Regul. 2020, 77, 100741. [Google Scholar] [CrossRef]
- Leppkes, M.; Knopf, J.; Naschberger, E.; Lindemann, A.; Singh, J.; Herrmann, I.; Stürzl, M.; Staats, L.; Mahajan, A.; Schauer, C.; et al. Vascular Occlusion by Neutrophil Extracellular Traps in COVID-19. EBioMedicine 2020, 58, 102925. [Google Scholar] [CrossRef]
- Middleton, E.A.; He, X.-Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil Extracellular Traps Contribute to Immunothrombosis in COVID-19 Acute Respiratory Distress Syndrome. Blood 2020, 136, 1169–1179. [Google Scholar] [CrossRef]
- Barnes, B.J.; Adrover, J.M.; Baxter-Stoltzfus, A.; Borczuk, A.; Cools-Lartigue, J.; Crawford, J.M.; Daßler-Plenker, J.; Guerci, P.; Huynh, C.; Knight, J.S.; et al. Targeting Potential Drivers of COVID-19: Neutrophil Extracellular Traps. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Weber, A.; Barnes, B.J.; Egeblad, M.; et al. Neutrophil Extracellular Traps (NETs) as Markers of Disease Severity in COVID-19. medRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, L.S.; Migliari Branco, L.; Franklin, B.S. Regulation of Innate Immune Responses by Platelets. Front. Immunol. 2019, 10, 1320. [Google Scholar] [CrossRef]
- Schrottmaier, W.C.; Kral, J.B.; Badrnya, S.; Assinger, A. Aspirin and P2Y12 Inhibitors in Platelet-Mediated Activation of Neutrophils and Monocytes. Thromb. Haemost. 2015, 114, 478–489. [Google Scholar] [CrossRef] [PubMed]
- Finsterbusch, M.; Schrottmaier, W.C.; Kral-Pointner, J.B.; Salzmann, M.; Assinger, A. Measuring and Interpreting Platelet-Leukocyte Aggregates. Platelets 2018, 29, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, A.; Sapra, L.; Saini, C.; Azam, Z.; Mishra, P.K.; Verma, B.; Mishra, G.C.; Srivastava, R.K. COVID-19: Immunology, Immunopathogenesis and Potential Therapies. Int. Rev. Immunol. 2021, 1–36. [Google Scholar] [CrossRef]
- Cugno, M.; Meroni, P.L.; Gualtierotti, R.; Griffini, S.; Grovetti, E.; Torri, A.; Panigada, M.; Aliberti, S.; Blasi, F.; Tedesco, F.; et al. Complement Activation in Patients with COVID-19: A Novel Therapeutic Target. J. Allergy Clin. Immunol. 2020, 146, 215–217. [Google Scholar] [CrossRef]
- Curran, C.S.; Rivera, D.R.; Kopp, J.B. COVID-19 Usurps Host Regulatory Networks. Front. Pharmacol. 2020, 11, 1278. [Google Scholar] [CrossRef]
- Grobler, C.; Maphumulo, S.C.; Grobbelaar, L.M.; Bredenkamp, J.C.; Laubscher, G.J.; Lourens, P.J.; Steenkamp, J.; Kell, D.B.; Pretorius, E. Covid-19: The Rollercoaster of Fibrin(Ogen), D-Dimer, Von Willebrand Factor, P-Selectin and Their Interactions with Endothelial Cells, Platelets and Erythrocytes. Int. J. Mol. Sci. 2020, 21, 5168. [Google Scholar] [CrossRef]
- Evans, P.C.; Rainger, G.E.; Mason, J.C.; Guzik, T.J.; Osto, E.; Stamataki, Z.; Neil, D.; Hoefer, I.E.; Fragiadaki, M.; Waltenberger, J.; et al. Endothelial Dysfunction in COVID-19: A Position Paper of the ESC Working Group for Atherosclerosis and Vascular Biology, and the ESC Council of Basic Cardiovascular Science. Cardiovasc. Res. 2020, 116, 2177–2184. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial Cell Infection and Endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Hamming, I.; Timens, W.; Bulthuis, M.L.C.; Lely, A.T.; Navis, G.J.; van Goor, H. Tissue Distribution of ACE2 Protein, the Functional Receptor for SARS Coronavirus. A First Step in Understanding SARS Pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef]
- Siddiqi, H.K.; Libby, P.; Ridker, P.M. COVID-19—A Vascular Disease. Trends Cardiovasc. Med. 2021, 31, 1–5. [Google Scholar] [CrossRef]
- Vanarsdall, A.L.; Pritchard, S.R.; Wisner, T.W.; Liu, J.; Jardetzky, T.S.; Johnson, D.C. CD147 Promotes Entry of Pentamer-Expressing Human Cytomegalovirus into Epithelial and Endothelial Cells. mBio 2018, 9, e00781–e00818. [Google Scholar] [CrossRef] [Green Version]
- Pons, S.; Fodil, S.; Azoulay, E.; Zafrani, L. The Vascular Endothelium: The Cornerstone of Organ Dysfunction in Severe SARS-CoV-2 Infection. Crit. Care 2020, 24, 353. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, S.; Mendes, M.C.; Camargo Martins, A.P.; Borges, N.H.; Godoy, T.M.; Miggiolaro, A.F.R.D.S.; da Silva Dezidério, F.; Machado-Souza, C.; de Noronha, L. Endothelial Dysfunction and Thrombosis in Patients With COVID-19-Brief Report. Arterioscler. Thromb. Vasc Biol. 2020, 40, 2404–2407. [Google Scholar] [CrossRef] [PubMed]
- Wazny, V.; Siau, A.; Wu, K.X.; Cheung, C. Vascular Underpinning of COVID-19. Open Biol. 2020, 10, 200208. [Google Scholar] [CrossRef]
- Lowenstein, C.J.; Solomon, S.D. Severe COVID-19 Is a Microvascular Disease. Circulation 2020, 142, 1609–1611. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, L.C.; Gonçalves, T.L.; de Araujo, L.P.; Rosario, L.V. de O.; Ferrer, V.P. Endothelial Cells and SARS-CoV-2: An Intimate Relationship. Vascul. Pharmacol. 2021, 137, 106829. [Google Scholar] [CrossRef]
- Selye, H. The Stress Concept. Can. Med. Assoc. J. 1976, 115, 718. [Google Scholar]
- Tan, T.; Khoo, B.; Mills, E.G.; Phylactou, M.; Patel, B.; Eng, P.C.; Thurston, L.; Muzi, B.; Meeran, K.; Prevost, A.T.; et al. Association between High Serum Total Cortisol Concentrations and Mortality from COVID-19. Lancet Diabetes Endocrinol. 2020, 8, 659–660. [Google Scholar] [CrossRef]
- Pal, R. COVID-19, Hypothalamo-Pituitary-Adrenal Axis and Clinical Implications. Endocrine 2020, 68, 251–252. [Google Scholar] [CrossRef]
- Ramezani, M.; Simani, L.; Karimialavijeh, E.; Rezaei, O.; Hajiesmaeili, M.; Pakdaman, H. The Role of Anxiety and Cortisol in Outcomes of Patients with Covid-19. Basic Clin. Neurosci. 2020, 11, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Alzahrani, A.S.; Mukhtar, N.; Aljomaiah, A.; Aljamei, H.; Bakhsh, A.; Alsudani, N.; Elsayed, T.; Alrashidi, N.; Fadel, R.; Alqahtani, E.; et al. The Impact of COVID-19 Viral Infection on the Hypothalamic-Pituitary-Adrenal Axis. Endocr. Pract. 2021, 27, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Xu, B.; Guan, W.; Xu, D.; Li, F.; Ren, R.; Zhu, X.; Gao, Y.; Jiang, L. The Adrenal Cortex, an Underestimated Site of SARS-CoV-2 Infection. Front. Endocrinol. 2020, 11, 593179. [Google Scholar] [CrossRef]
- Russell, C.D.; Millar, J.E.; Baillie, J.K. Clinical Evidence Does Not Support Corticosteroid Treatment for 2019-NCoV Lung Injury. Lancet 2020, 395, 473–475. [Google Scholar] [CrossRef] [Green Version]
- Granger, D.N.; Senchenkova, E. Inflammation and the Microcirculation; Integrated Systems Physiology—From Cell to Function; Morgan & Claypool Life Sciences: San Rafael, CA, USA, 2010. [Google Scholar]
- Majno, G.; Joris, I. Cells, Tissues and Disease: Principles of General Pathology; Oxford University Press: Oxford, UK, 2004; p. 1005. [Google Scholar]
- Gottesman, S. Trouble Is Coming: Signaling Pathways That Regulate General Stress Responses in Bacteria. J. Biol. Chem. 2019, 294, 11685–11700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khitun, A.; Ness, T.J.; Slavoff, S.A. Small Open Reading Frames and Cellular Stress Responses. Mol. Omics 2019, 15, 108–116. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Brocker, C.; Koppaka, V.; Chen, Y.; Jackson, B.C.; Matsumoto, A.; Thompson, D.C.; Vasiliou, V. Aldehyde Dehydrogenases in Cellular Responses to Oxidative/Electrophilic Stress. Free Radic. Biol. Med. 2013, 56, 89–101. [Google Scholar] [CrossRef] [Green Version]
- Owttrim, G.W. RNA Helicases: Diverse Roles in Prokaryotic Response to Abiotic Stress. RNA Biol. 2013, 10, 96–110. [Google Scholar] [CrossRef] [Green Version]
- Bardwell, L.; Zou, X.; Nie, Q.; Komarova, N.L. Mathematical Models of Specificity in Cell Signaling. Biophys. J. 2007, 92, 3425–3441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindahl, T.; Barnes, D.E. Repair of Endogenous DNA Damage. Cold Spring Harb. Symp. Quant. Biol. 2000, 65, 127–133. [Google Scholar] [CrossRef]
- Webster, G.A.; Perkins, N.D. Transcriptional Cross Talk between NF-KappaB and P53. Mol. Cell. Biol. 1999, 19, 3485–3495. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Tsuchiya, R.; Hozumi, Y.; Nakano, T.; Okada, M.; Goto, K. Reciprocal Regulation of P53 and NF-ΚB by Diacylglycerol Kinase ζ. Adv. Biol. Regul. 2016, 60, 15–21. [Google Scholar] [CrossRef]
- Chittiboyina, S.; Bai, Y.; Lelièvre, S.A. Microenvironment-Cell Nucleus Relationship in the Context of Oxidative Stress. Front. Cell Dev. Biol. 2018, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Kostov, R.V.; Canning, P. Keap1, the Cysteine-Based Mammalian Intracellular Sensor for Electrophiles and Oxidants. Arch. Biochem. Biophys. 2017, 617, 84–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porta, C.; Paglino, C.; Mosca, A. Targeting PI3K/Akt/MTOR Signaling in Cancer. Front. Oncol. 2014, 4, 64. [Google Scholar] [CrossRef] [Green Version]
- Owonikoko, T.K.; Khuri, F.R. Targeting the PI3K/AKT/MTOR Pathway: Biomarkers of Success and Tribulation. Am. Soc. Clin. Oncol. Educ. Book 2013. [Google Scholar] [CrossRef]
- Rafalski, V.A.; Brunet, A. Energy Metabolism in Adult Neural Stem Cell Fate. Prog. Neurobiol. 2011, 93, 182–203. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, S.; Zhai, A.; Zhang, B.; Tian, G. AMPK-Mediated Regulation of Lipid Metabolism by Phosphorylation. Biol. Pharm. Bull. 2018, 41, 985–993. [Google Scholar] [CrossRef] [Green Version]
- Maiese, K. The Mechanistic Target of Rapamycin (MTOR): Novel Considerations as an Antiviral Treatment. Curr. Neurovasc. Res. 2020, 17, 332–337. [Google Scholar] [CrossRef]
- Pietzner, M.; Kaul, A.; Henning, A.-K.; Kastenmüller, G.; Artati, A.; Lerch, M.M.; Adamski, J.; Nauck, M.; Friedrich, N. Comprehensive Metabolic Profiling of Chronic Low-Grade Inflammation among Generally Healthy Individuals. BMC Med. 2017, 15, 210. [Google Scholar] [CrossRef] [Green Version]
- Zmora, N.; Levy, M.; Pevsner-Fishcer, M.; Elinav, E. Inflammasomes and Intestinal Inflammation. Mucosal Immunol. 2017, 10, 865–883. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Sheng, B.; Yang, K.; Sun, L.; Xiao, W.; Yang, H. The Protective Roles of NLRP6 in Intestinal Epithelial Cells. Cell Prolif. 2019, 52, e12555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, B.K. Muscle as a Secretory Organ. Compr. Physiol. 2013, 3, 1337–1362. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.K.; Febbraio, M.A. Muscle as an Endocrine Organ: Focus on Muscle-Derived Interleukin-6. Physiol. Rev. 2008, 88, 1379–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeung, Y.T.; Aziz, F.; Guerrero-Castilla, A.; Arguelles, S. Signaling Pathways in Inflammation and Anti-Inflammatory Therapies. Curr. Pharm. Des. 2018, 24, 1449–1484. [Google Scholar] [CrossRef] [PubMed]
- Naito, T.; Tanaka, H.; Naoe, Y.; Taniuchi, I. Transcriptional Control of T-Cell Development. Int. Immunol. 2011, 23, 661–668. [Google Scholar] [CrossRef]
- Basso, K.; Dalla-Favera, R. Germinal Centres and B Cell Lymphomagenesis. Nat. Rev. Immunol. 2015, 15, 172–184. [Google Scholar] [CrossRef]
- Baumeister, D.; Akhtar, R.; Ciufolini, S.; Pariante, C.M.; Mondelli, V. Childhood Trauma and Adulthood Inflammation: A Meta-Analysis of Peripheral C-Reactive Protein, Interleukin-6 and Tumour Necrosis Factor-α. Mol. Psychiatry 2016, 21, 642–649. [Google Scholar] [CrossRef] [Green Version]
- Heilbronn, L.K.; Campbell, L.V. Adipose Tissue Macrophages, Low Grade Inflammation and Insulin Resistance in Human Obesity. Curr. Pharm. Des. 2008, 14, 1225–1230. [Google Scholar] [CrossRef]
- De Nigris, V.; Prattichizzo, F.; Iijima, H.; Ceriello, A. DPP-4 Inhibitors Have Different Effects on Endothelial Low-Grade Inflammation and on the M1-M2 Macrophage Polarization Under Hyperglycemic Conditions. Diabetes Metab. Syndr. Obes. 2021, 14, 1519–1531. [Google Scholar] [CrossRef]
- Giannotti, G.; Landmesser, U. Endothelial Dysfunction as an Early Sign of Atherosclerosis. Herz 2007, 32, 568–572. [Google Scholar] [CrossRef] [PubMed]
- Versari, D.; Daghini, E.; Virdis, A.; Ghiadoni, L.; Taddei, S. The Ageing Endothelium, Cardiovascular Risk and Disease in Man. Exp. Physiol. 2009, 94, 317–321. [Google Scholar] [CrossRef] [Green Version]
- Lockhart, C.J.; Hamilton, P.K.; Quinn, C.E.; McVeigh, G.E. End-Organ Dysfunction and Cardiovascular Outcomes: The Role of the Microcirculation. Clin. Sci. 2009, 116, 175–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Querfeld, U.; Mak, R.H.; Pries, A.R. Microvascular Disease in Chronic Kidney Disease: The Base of the Iceberg in Cardiovascular Comorbidity. Clin. Sci. 2020, 134, 1333–1356. [Google Scholar] [CrossRef] [PubMed]
- Yamanouchi, M.; Mori, M.; Hoshino, J.; Kinowaki, K.; Fujii, T.; Ohashi, K.; Furuichi, K.; Wada, T.; Ubara, Y. Retinopathy Progression and the Risk of End-Stage Kidney Disease: Results from a Longitudinal Japanese Cohort of 232 Patients with Type 2 Diabetes and Biopsy-Proven Diabetic Kidney Disease. BMJ Open Diabetes Res. Care 2019, 7, e000726. [Google Scholar] [CrossRef] [Green Version]
- Zeng, L.-F.; Xiao, Y.; Sun, L. A Glimpse of the Mechanisms Related to Renal Fibrosis in Diabetic Nephropathy. Adv. Exp. Med. Biol. 2019, 1165, 49–79. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Mou, S. Role of Immune Cells in Diabetic Kidney Disease. Curr. Gene Ther. 2017, 17, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Rinella, M.E. Nonalcoholic Fatty Liver Disease: A Systematic Review. JAMA 2015, 313, 2263–2273. [Google Scholar] [CrossRef] [PubMed]
- Sheka, A.C.; Adeyi, O.; Thompson, J.; Hameed, B.; Crawford, P.A.; Ikramuddin, S. Nonalcoholic Steatohepatitis: A Review. JAMA 2020, 323, 1175–1183. [Google Scholar] [CrossRef]
- Westfall, E.; Jeske, R.; Bader, A.R. Nonalcoholic Fatty Liver Disease: Common Questions and Answers on Diagnosis and Management. Am. Fam. Physician 2020, 102, 603–612. [Google Scholar]
- Abbate, M.; Mascaró, C.M.; Montemayor, S.; Casares, M.; Gómez, C.; Ugarriza, L.; Tejada, S.; Abete, I.; Zulet, M.A.; Sureda, A.; et al. Non-Alcoholic Fatty Liver Disease Is Associated with Kidney Glomerular Hyperfiltration in Adults with Metabolic Syndrome. J. Clin. Med. 2021, 10, 1717. [Google Scholar] [CrossRef]
- Laforge, M.; Elbim, C.; Frère, C.; Hémadi, M.; Massaad, C.; Nuss, P.; Benoliel, J.-J.; Becker, C. Tissue Damage from Neutrophil-Induced Oxidative Stress in COVID-19. Nat. Rev. Immunol. 2020, 20, 515–516. [Google Scholar] [CrossRef]
- Suhail, S.; Zajac, J.; Fossum, C.; Lowater, H.; McCracken, C.; Severson, N.; Laatsch, B.; Narkiewicz-Jodko, A.; Johnson, B.; Liebau, J.; et al. Role of Oxidative Stress on SARS-CoV (SARS) and SARS-CoV-2 (COVID-19) Infection: A Review. Protein J. 2020, 39, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Lüscher, T. COVID-19 Is, in the End, an Endothelial Disease. Eur. Heart J. 2020, 41, 3038–3044. [Google Scholar] [CrossRef] [PubMed]
- Beltrán-García, J.; Osca-Verdegal, R.; Pallardó, F.V.; Ferreres, J.; Rodríguez, M.; Mulet, S.; Sanchis-Gomar, F.; Carbonell, N.; García-Giménez, J.L. Oxidative Stress and Inflammation in COVID-19-Associated Sepsis: The Potential Role of Anti-Oxidant Therapy in Avoiding Disease Progression. Antioxidants 2020, 9, 936. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, S. Coronavirus (Covid-19) Sepsis: Revisiting Mitochondrial Dysfunction in Pathogenesis, Aging, Inflammation, and Mortality. Inflamm. Res. 2020, 69, 1077–1085. [Google Scholar] [CrossRef]
- Cardozo, C.M.; Hainaut, P. Viral Strategies for Circumventing P53: The Case of Severe Acute Respiratory Syndrome Coronavirus. Curr. Opin. Oncol. 2021, 33, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Xu, W.; Reed, J.C. Cell Death and Endoplasmic Reticulum Stress: Disease Relevance and Therapeutic Opportunities. Nat. Rev. Drug Discov. 2008, 7, 1013–1030. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Czinn, S.J.; Reiter, R.J.; Blanchard, T.G. Crosstalk between Endoplasmic Reticulum Stress and Anti-Viral Activities: A Novel Therapeutic Target for COVID-19. Life Sci. 2020, 255, 117842. [Google Scholar] [CrossRef]
- Khomari, F.; Nabi-Afjadi, M.; Yarahmadi, S.; Eskandari, H.; Bahreini, E. Effects of Cell Proteostasis Network on the Survival of SARS-CoV-2. Biol. Proced. Online 2021, 23, 8. [Google Scholar] [CrossRef]
- Ramos, C.H.I.; Ayinde, K.S. Are Hsp90 Inhibitors Good Candidates against Covid-19? Curr. Protein Pept. Sci 2020, 22, 192–200. [Google Scholar] [CrossRef]
- Bartoszewski, R.; Dabrowski, M.; Jakiela, B.; Matalon, S.; Harrod, K.S.; Sanak, M.; Collawn, J.F. SARS-CoV-2 May Regulate Cellular Responses through Depletion of Specific Host MiRNAs. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 319, L444–L455. [Google Scholar] [CrossRef] [PubMed]
- Centa, A.; Fonseca, A.S.; Ferreira, S.G. da S.; Azevedo, M.L.V.; Vaz de Paula, C.B.; Nagashima, S.; Machado-Souza, C.; Miggiolaro, A.F.R.D.S.; Baena, C.P.; de Noronha, L.; et al. Deregulated MiRNA Expression Is Associated with Endothelial Dysfunction in Post-Mortem Lung Biopsies of COVID-19 Patients. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 320, L405–L412. [Google Scholar] [CrossRef]
- Randhawa, P.K.; Scanlon, K.; Rappaport, J.; Gupta, M.K. Modulation of Autophagy by SARS-CoV-2: A Potential Threat for Cardiovascular System. Front. Physiol. 2020, 11, 611275. [Google Scholar] [CrossRef]
- Gorshkov, K.; Chen, C.Z.; Bostwick, R.; Rasmussen, L.; Xu, M.; Pradhan, M.; Tran, B.N.; Zhu, W.; Shamim, K.; Huang, W.; et al. The SARS-CoV-2 Cytopathic Effect Is Blocked with Autophagy Modulators. bioRxiv 2020, 2020.05.16.091520. [Google Scholar] [CrossRef]
- Benvenuto, D.; Angeletti, S.; Giovanetti, M.; Bianchi, M.; Pascarella, S.; Cauda, R.; Ciccozzi, M.; Cassone, A. Evolutionary Analysis of SARS-CoV-2: How Mutation of Non-Structural Protein 6 (NSP6) Could Affect Viral Autophagy. J. Infect. 2020, 81, e24–e27. [Google Scholar] [CrossRef]
- Shojaei, S.; Suresh, M.; Klionsky, D.J.; Labouta, H.I.; Ghavami, S. Autophagy and SARS-CoV-2 Infection: A possible Smart Targeting of the Autophagy Pathway. Virulence 2020, 11, 805–810. [Google Scholar] [CrossRef]
- Rodrigues, T.S.; de Sá, K.S.G.; Ishimoto, A.Y.; Becerra, A.; Oliveira, S.; Almeida, L.; Gonçalves, A.V.; Perucello, D.B.; Andrade, W.A.; Castro, R.; et al. Inflammasomes Are Activated in Response to SARS-CoV-2 Infection and Are Associated with COVID-19 Severity in Patients. J. Exp. Med. 2021, 218, e20201707. [Google Scholar] [CrossRef]
- Ferreira, A.C.; Soares, V.C.; de Azevedo-Quintanilha, I.G.; Dias, S.d.S.G.; Fintelman-Rodrigues, N.; Sacramento, C.Q.; Mattos, M.; de Freitas, C.S.; Temerozo, J.R.; Teixeira, L.; et al. SARS-CoV-2 Engages Inflammasome and Pyroptosis in Human Primary Monocytes. Cell Death Discov. 2021, 7, 43. [Google Scholar] [CrossRef] [PubMed]
- Freeman, T.L.; Swartz, T.H. Targeting the NLRP3 Inflammasome in Severe COVID-19. Front. Immunol. 2020, 11, 1518. [Google Scholar] [CrossRef] [PubMed]
- Bordea, I.R.; Candrea, S.; Sălăgean, T.; Pop, I.D.; Lucaciu, O.; Ilea, A.; Manole, M.; Băbțan, A.-M.; Sirbu, A.; Hanna, R. Impact of COVID-19 Pandemic on Healthcare Professionals and Oral Care Operational Services: A Systemic Review. Risk Manag. Healthc. Policy 2021, 14, 453–463. [Google Scholar] [CrossRef]
- Vernuccio, F.; Lombardo, F.P.; Cannella, R.; Panzuto, F.; Giambelluca, D.; Arzanauskaite, M.; Midiri, M.; Cabassa, P. Thromboembolic Complications of COVID-19: The Combined Effect of a pro-Coagulant Pattern and an Endothelial Thrombo-Inflammatory Syndrome. Clin. Radiol. 2020, 75, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Ciceri, F.; Beretta, L.; Scandroglio, A.M.; Colombo, S.; Landoni, G.; Ruggeri, A.; Peccatori, J.; D’Angelo, A.; De Cobelli, F.; Rovere-Querini, P.; et al. Microvascular COVID-19 Lung Vessels Obstructive Thromboinflammatory Syndrome (MicroCLOTS): An Atypical Acute Respiratory Distress Syndrome Working Hypothesis. Crit. Care Resusc. 2020, 22, 95–97. [Google Scholar] [PubMed]
- Blaschke, T.; Bajorath, J. Fine-Tuning of a Generative Neural Network for Designing Multi-Target Compounds. J. Comput. Aided Mol. Des. 2021, 1–9. [Google Scholar] [CrossRef]
- Ramsay, R.R.; Popovic-Nikolic, M.R.; Nikolic, K.; Uliassi, E.; Bolognesi, M.L. A Perspective on Multi-Target Drug Discovery and Design for Complex Diseases. Clin. Transl. Med. 2018, 7, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overington, J.P.; Al-Lazikani, B.; Hopkins, A.L. How Many Drug Targets Are There? Nat. Rev. Drug Discov. 2006, 5, 993–996. [Google Scholar] [CrossRef] [PubMed]
- Hanna, R.; Dalvi, S.; Sălăgean, T.; Pop, I.D.; Bordea, I.R.; Benedicenti, S. Understanding COVID-19 Pandemic: Molecular Mechanisms and Potential Therapeutic Strategies. An Evidence-Based Review. J. Inflamm. Res. 2021, 14, 13–56. [Google Scholar] [CrossRef]
- Sarapultsev, A.P.; Vassiliev, P.M.; Sarapultsev, P.A.; Chupakhin, O.N.; Ianalieva, L.R.; Sidorova, L.P. Immunomodulatory Action of Substituted 1,3,4-Thiadiazines on the Course of Myocardial Infarction. Molecules 2018, 23, E1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldmann, C.; Bajorath, J. Compounds with Multitarget Activity: Structure-Based Analysis and Machine Learning. Future Drug Discov. 2020, 2, FDD44. [Google Scholar] [CrossRef]
- Yumura, M.; Nagano, T.; Nishimura, Y. Novel Multitarget Therapies for Lung Cancer and Respiratory Disease. Molecules 2020, 25, E3987. [Google Scholar] [CrossRef]
- Naik, B.; Gupta, N.; Ojha, R.; Singh, S.; Prajapati, V.K.; Prusty, D. High Throughput Virtual Screening Reveals SARS-CoV-2 Multi-Target Binding Natural Compounds to Lead Instant Therapy for COVID-19 Treatment. Int. J. Biol. Macromol. 2020, 160, 1–17. [Google Scholar] [CrossRef]
- López-Cortés, A.; Guevara-Ramírez, P.; Kyriakidis, N.C.; Barba-Ostria, C.; León Cáceres, Á.; Guerrero, S.; Ortiz-Prado, E.; Munteanu, C.R.; Tejera, E.; Cevallos-Robalino, D.; et al. In Silico Analyses of Immune System Protein Interactome Network, Single-Cell RNA Sequencing of Human Tissues, and Artificial Neural Networks Reveal Potential Therapeutic Targets for Drug Repurposing Against COVID-19. Front. Pharmacol. 2021, 12, 598925. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.-T.; Hsu, W.-C.; Lin, C.-C. Antiviral Natural Products and Herbal Medicines. J. Tradit. Complement. Med. 2014, 4, 24–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurung, A.B.; Ali, M.A.; Lee, J.; Farah, M.A.; Al-Anazi, K.M. Unravelling Lead Antiviral Phytochemicals for the Inhibition of SARS-CoV-2 Mpro Enzyme through in Silico Approach. Life Sci. 2020, 255, 117831. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.L.D.; Lam, W.C.; Yang, W.; Chan, K.W.; Sze, S.C.W.; Miao, J.; Yung, K.K.L.; Bian, Z.; Wong, V.T. Potential Targets for Treatment of Coronavirus Disease 2019 (COVID-19): A Review of Qing-Fei-Pai-Du-Tang and Its Major Herbs. Am. J. Chin. Med. 2020, 48, 1051–1071. [Google Scholar] [CrossRef] [PubMed]
- Borse, S.; Joshi, M.; Saggam, A.; Bhat, V.; Walia, S.; Marathe, A.; Sagar, S.; Chavan-Gautam, P.; Girme, A.; Hingorani, L.; et al. Ayurveda Botanicals in COVID-19 Management: An in Silico Multi-Target Approach. PLoS ONE 2021, 16, e0248479. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, Z.; Li, S.; Ye, X.; Li, X.; He, K. Synergy Effects of Herb Extracts: Pharmacokinetics and Pharmacodynamic Basis. Fitoterapia 2014, 92, 133–147. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.-Q.; Xu, J.; Chen, S.-D. In Silico Screening of Potential Chinese Herbal Medicine Against COVID-19 by Targeting SARS-CoV-2 3CLpro and Angiotensin Converting Enzyme II Using Molecular Docking. Chin. J. Integr. Med. 2020, 26, 527–532. [Google Scholar] [CrossRef]
- Joshi, R.S.; Jagdale, S.S.; Bansode, S.B.; Shankar, S.S.; Tellis, M.B.; Pandya, V.K.; Chugh, A.; Giri, A.P.; Kulkarni, M.J. Discovery of Potential Multi-Target-Directed Ligands by Targeting Host-Specific SARS-CoV-2 Structurally Conserved Main Protease. J. Biomol. Struct. Dyn. 2021, 39, 3099–3114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, M.; Luo, G.; Ye, D.; She, M.; Sun, N.; Lu, Y.-J.; Zheng, J. Network Pharmacology, Molecular Docking Integrated Surface Plasmon Resonance Technology Reveals the Mechanism of Toujie Quwen Granules against Coronavirus Disease 2019 Pneumonia. Phytomedicine 2021, 85, 153401. [Google Scholar] [CrossRef] [PubMed]
- Tao, Q.; Du, J.; Li, X.; Zeng, J.; Tan, B.; Xu, J.; Lin, W.; Chen, X.-L. Network Pharmacology and Molecular Docking Analysis on Molecular Targets and Mechanisms of Huashi Baidu Formula in the Treatment of COVID-19. Drug Dev. Ind. Pharm. 2020, 46, 1345–1353. [Google Scholar] [CrossRef]
- de Oliveira, P.G.; Termini, L.; Durigon, E.L.; Lepique, A.P.; Sposito, A.C.; Boccardo, E. Diacerein: A Potential Multi-Target Therapeutic Drug for COVID-19. Med. Hypotheses 2020, 144, 109920. [Google Scholar] [CrossRef]
- Shree, P.; Mishra, P.; Selvaraj, C.; Singh, S.K.; Chaube, R.; Garg, N.; Tripathi, Y.B. Targeting COVID-19 (SARS-CoV-2) Main Protease through Active Phytochemicals of Ayurvedic Medicinal Plants—Withania Somnifera (Ashwagandha), Tinospora Cordifolia (Giloy) and Ocimum Sanctum (Tulsi)—A Molecular Docking Study. J. Biomol. Struct. Dyn. 2020, 1–14. [Google Scholar] [CrossRef]
- Rabie, A.M. Two Antioxidant 2,5-Disubstituted-1,3,4-Oxadiazoles (CoViTris2020 and ChloViD2020): Successful Repurposing against COVID-19 as the First Potent Multitarget Anti-SARS-CoV-2 Drugs. New J. Chem. 2021, 45, 761–771. [Google Scholar] [CrossRef]
- Di Micco, S.; Musella, S.; Scala, M.C.; Sala, M.; Campiglia, P.; Bifulco, G.; Fasano, A. In Silico Analysis Revealed Potential Anti-SARS-CoV-2 Main Protease Activity by the Zonulin Inhibitor Larazotide Acetate. Front. Chem. 2020, 8, 628609. [Google Scholar] [CrossRef]
- Chakraborty, C.; Bhattacharya, M.; Mallick, B.; Sharma, A.R.; Lee, S.-S.; Agoramoorthy, G. SARS-CoV-2 Protein Drug Targets Landscape: A Potential Pharmacological Insight View for the New Drug Development. Expert Rev. Clin. Pharmacol. 2021, 14, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Schultz, I.C.; Bertoni, A.P.S.; Wink, M.R. Mesenchymal Stem Cell-Derived Extracellular Vesicles Carrying MiRNA as a Potential Multi Target Therapy to COVID-19: An in Silico Analysis. Stem Cell Rev. Rep. 2021, 17, 341–356. [Google Scholar] [CrossRef] [PubMed]



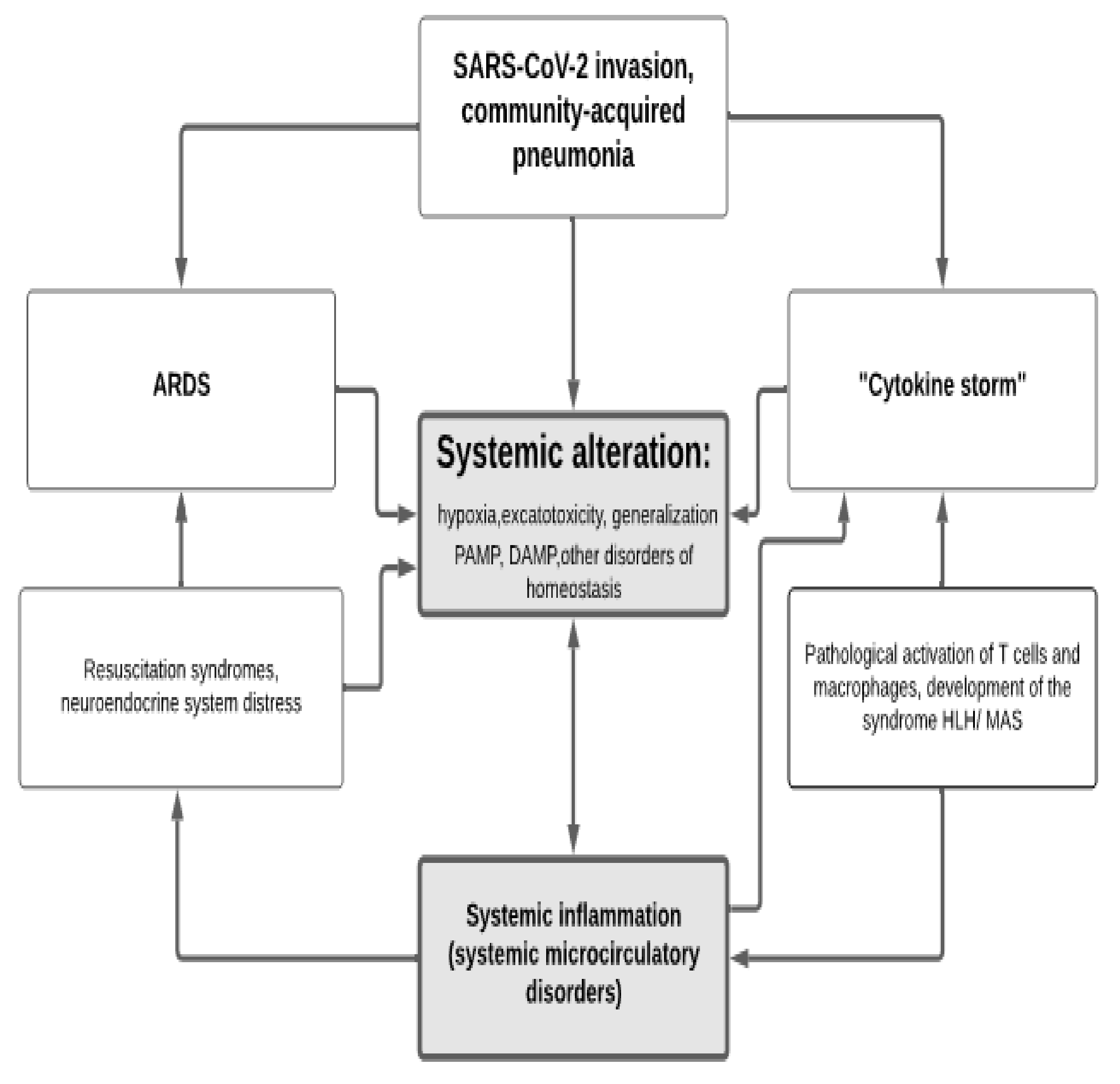{kind=link}
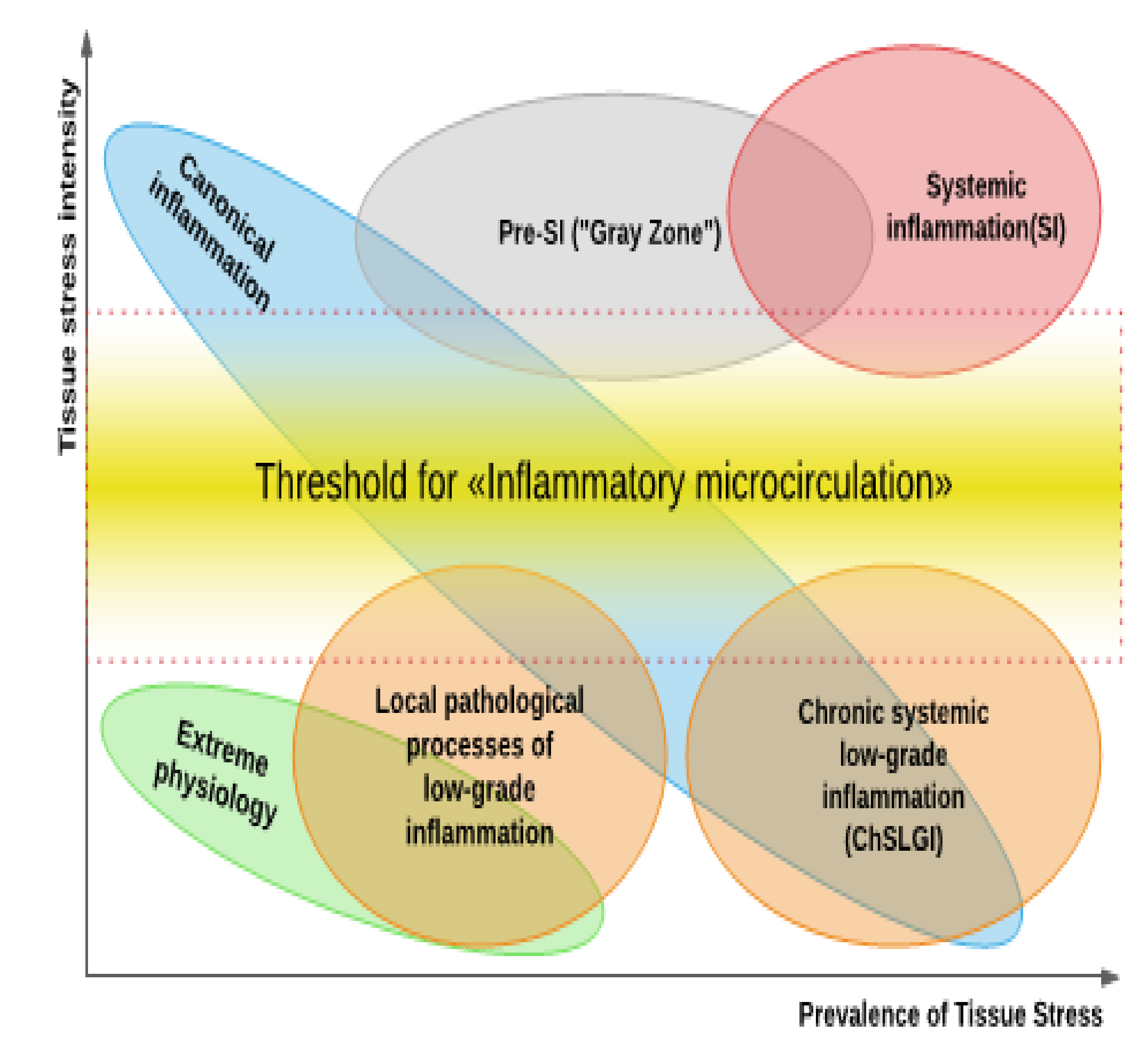{kind=link}
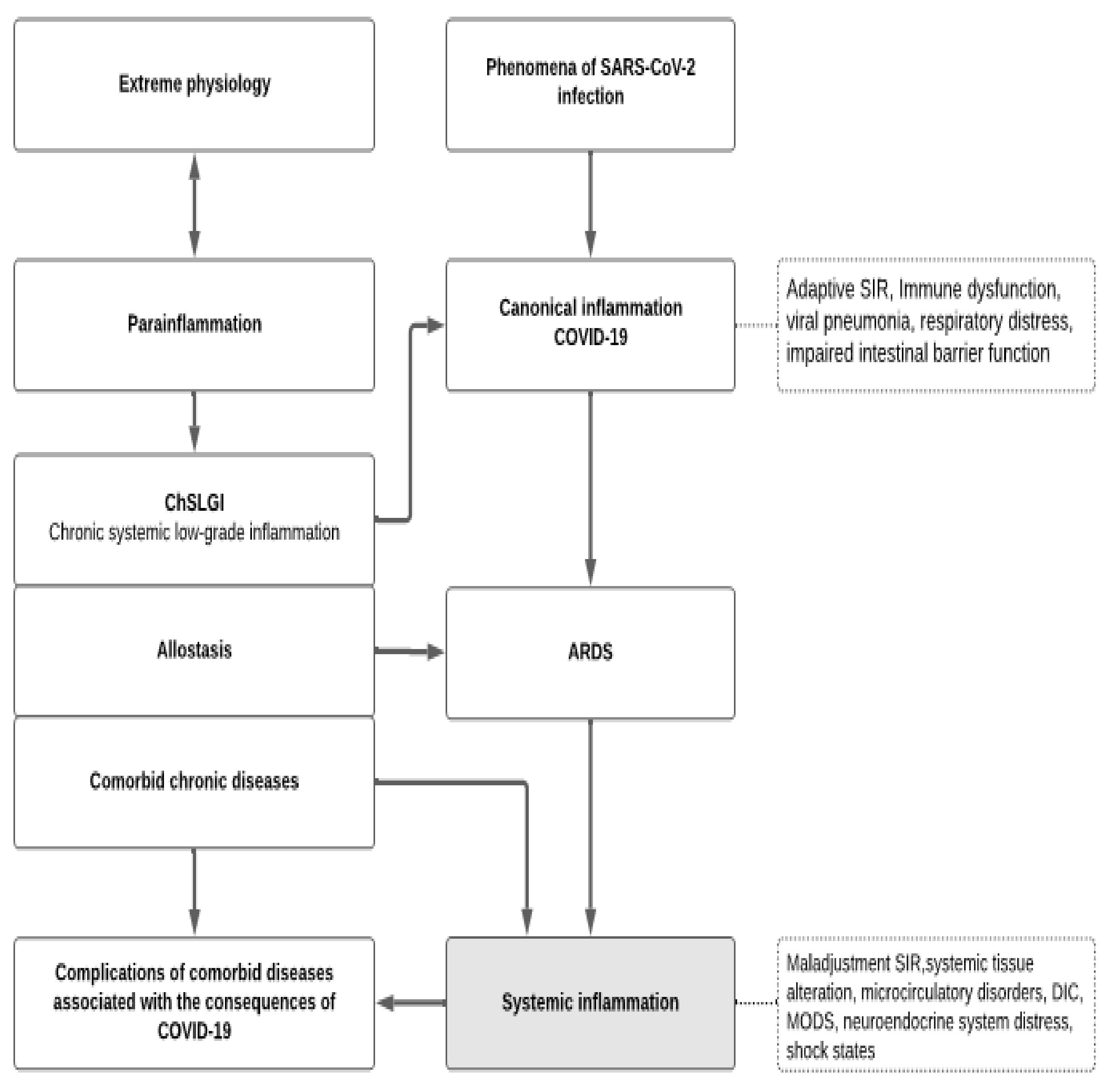{kind=link}
| Significant Differences (p < 0.05) | Unreliable (p > 0.05) or Insignificant * | Sources |
|---|---|---|
| IL2, IL7, IL10, IP10 (CXCL10), MCP-1 (CCL2), MIP-1α (CCL3), TNF-α | IL4, IL5, IL13, Eotaxin, RANTES, IL9, IL12p70, IL15, IL17A, IL1β, IFN-γ | [36] |
| sIL-2R (sCD25) and IL-6 | TNF-α, IL-1β, IL-8, IL-10, CRP | [34,42] |
| IL-6 and IL-10 | TNF-α, IFN-γ, IL-2, IL-4 [36] | [37,49] |
| IL-6 and IFN-γ (in the later), IL-10, IL-1RA | RANTES (CCL5), GM-CSF, TNF-α | [33] |
| IP10 (CXCL10) and MCP-1(CCL2) | [39] | |
| TNF-α, IL-10 and IL-6, IP-10, MCP-3 (CCL7), IL-1ra, IL-17A | [40] | |
| CRP, IL-6, Ferritin | IL1β [40], low differences Me *: TNF-α, IL-8, IL-10, sIL-2R [40]; PCT [43] | [41,43,44] |
| sIL-2R, TNF-α and IL-10, IL-6 | [28,35,45,53] | |
| IL-6, IL-10, IL-2, IFN-γ | [46] | |
| IL-6, IL-10 | IL-2, IL-4, IFN-γ, TNF-α | [42,47,50,51,52] |
| IL-6, sIL-2R, IL-8, IL-10, and TNF-α, | [48] | |
| IP-10, MCP-3, IL-1ra | [54] | |
| PCT, TNF-α, VEGF-A, Angpt-2 | CRP, IL-6, Ferritin | [55] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gusev, E.; Sarapultsev, A.; Hu, D.; Chereshnev, V. Problems of Pathogenesis and Pathogenetic Therapy of COVID-19 from the Perspective of the General Theory of Pathological Systems (General Pathological Processes). Int. J. Mol. Sci. 2021, 22, 7582. https://doi.org/10.3390/ijms22147582
Gusev E, Sarapultsev A, Hu D, Chereshnev V. Problems of Pathogenesis and Pathogenetic Therapy of COVID-19 from the Perspective of the General Theory of Pathological Systems (General Pathological Processes). International Journal of Molecular Sciences. 2021; 22(14):7582. https://doi.org/10.3390/ijms22147582
Chicago/Turabian StyleGusev, Evgenii, Alexey Sarapultsev, Desheng Hu, and Valeriy Chereshnev. 2021. "Problems of Pathogenesis and Pathogenetic Therapy of COVID-19 from the Perspective of the General Theory of Pathological Systems (General Pathological Processes)" International Journal of Molecular Sciences 22, no. 14: 7582. https://doi.org/10.3390/ijms22147582