
Distinct Immune Signatures Indicative of Treatment Response and Immune-Related Adverse Events in Melanoma Patients under Immune Checkpoint Inhibitor Therapy

 , , and
, , and
Abstract
:1. Introduction
2. Results
2.1. General Clinical Characteristics
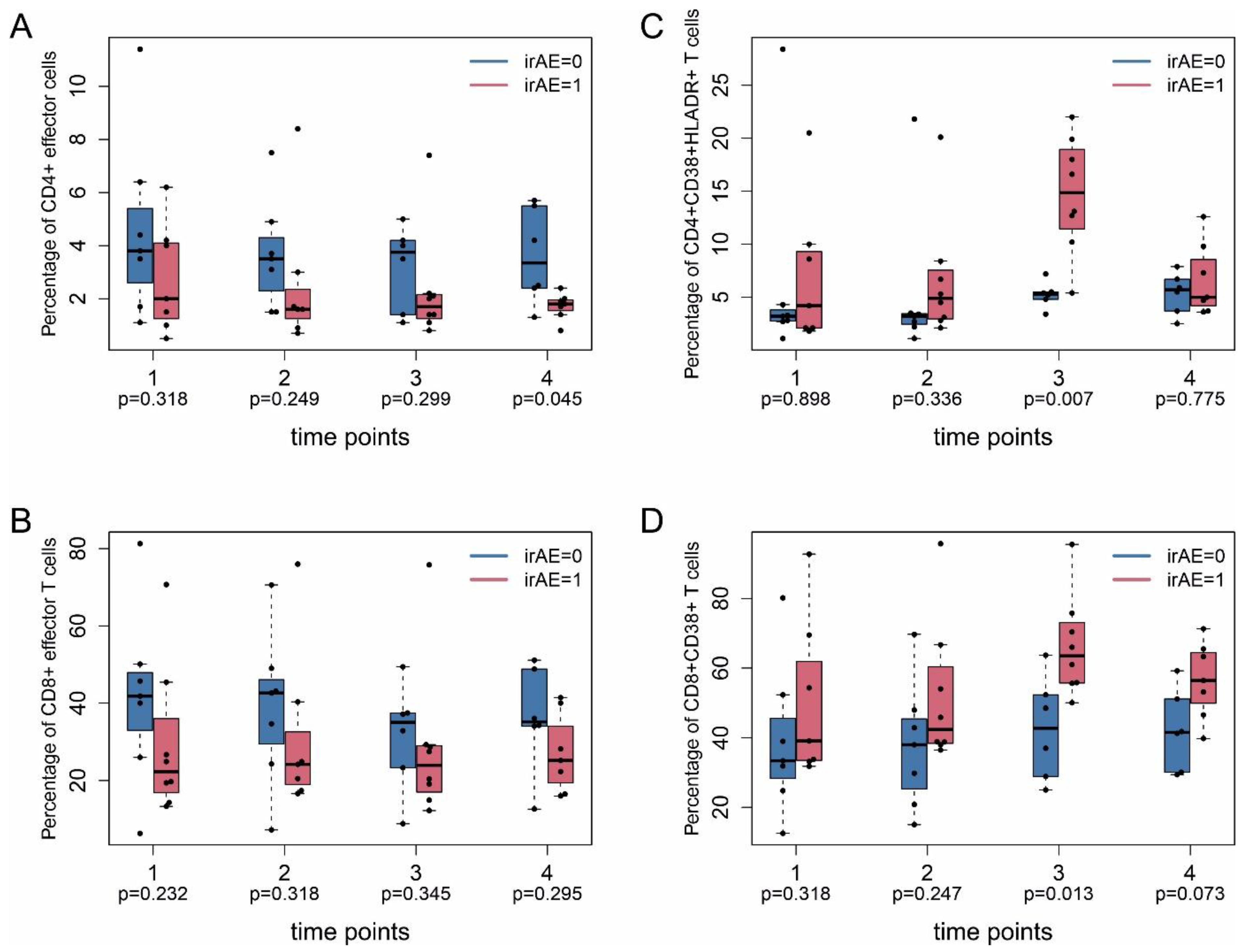
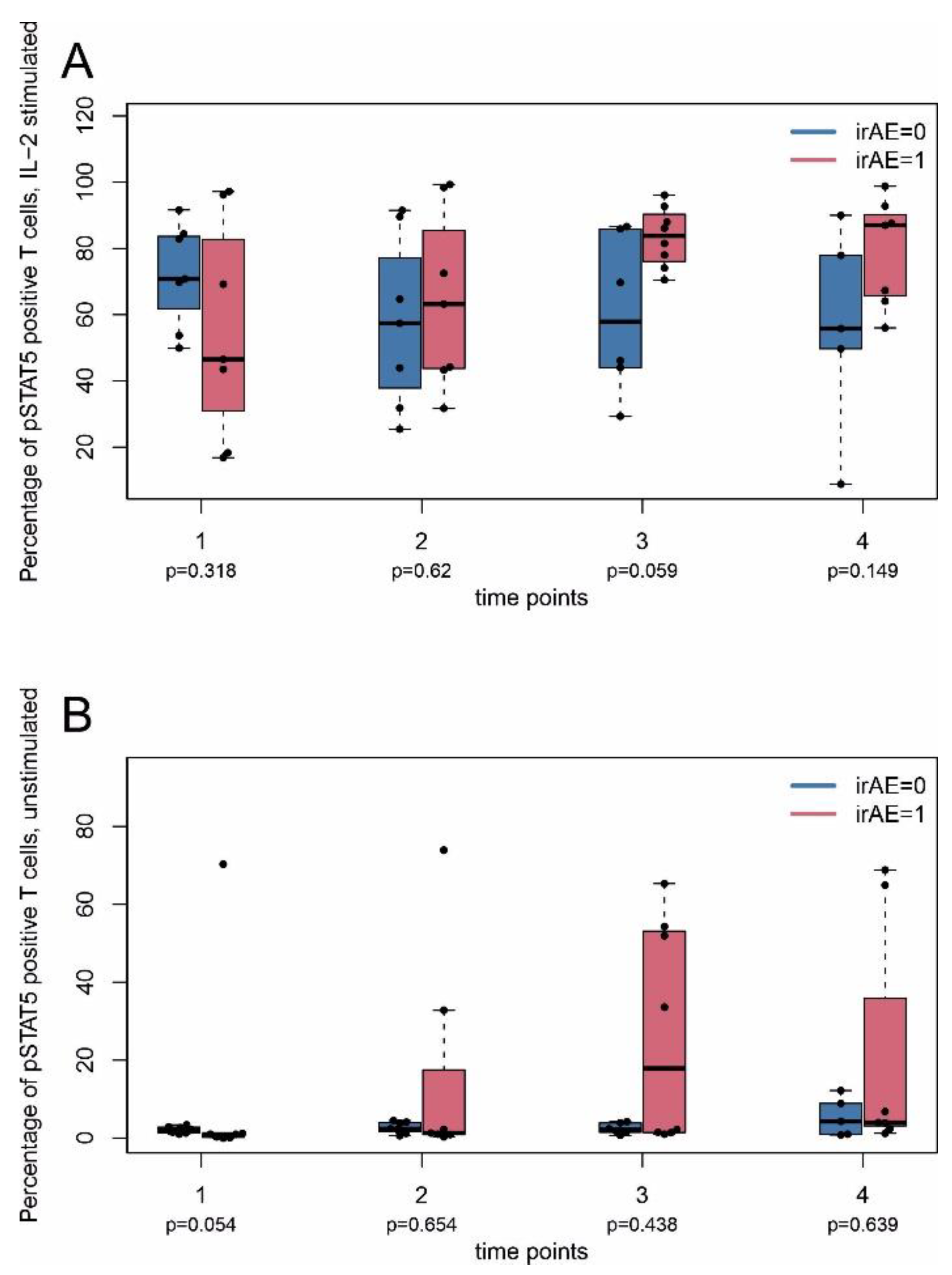
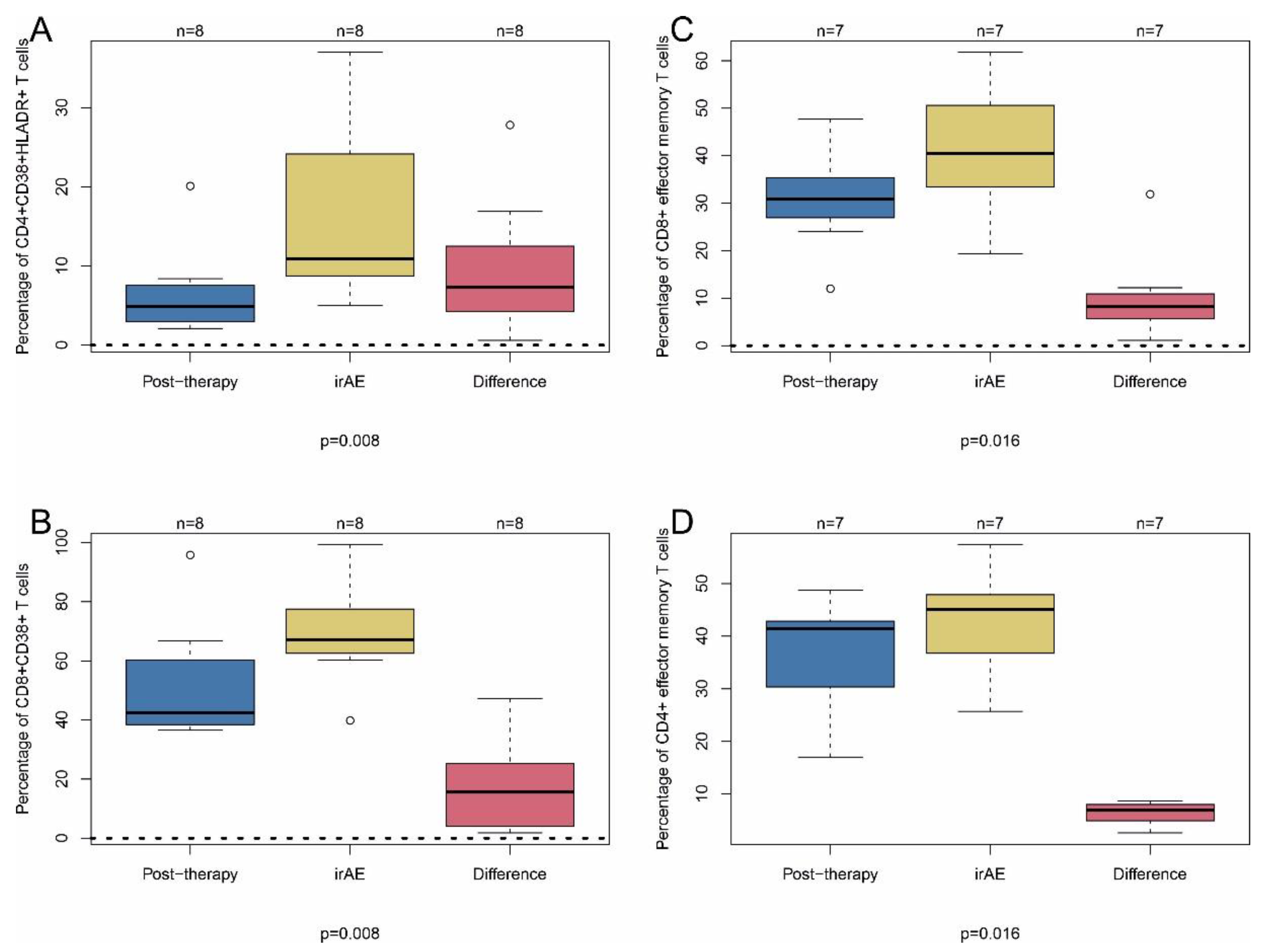
2.2. Immune-Related Adverse Events
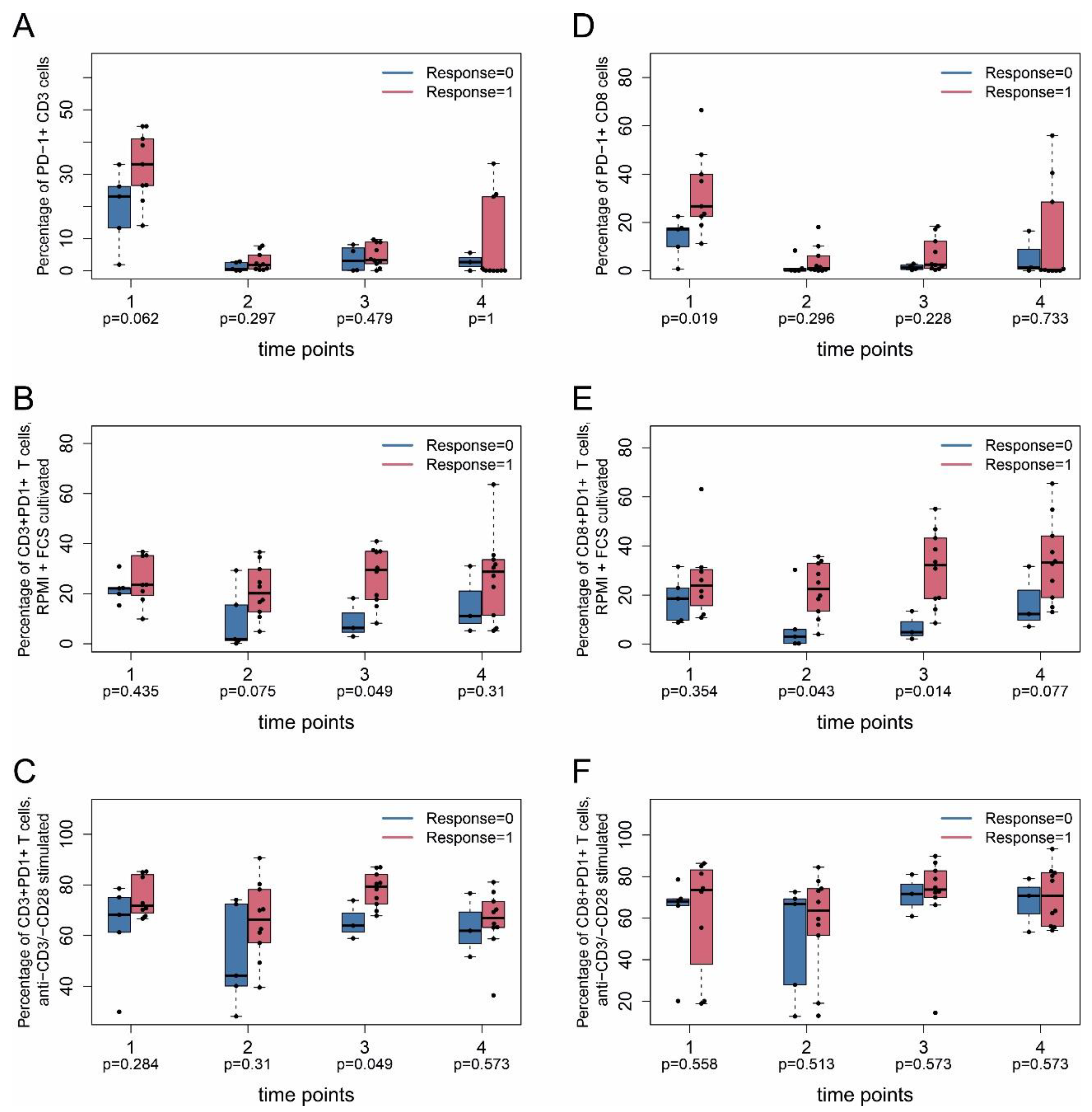
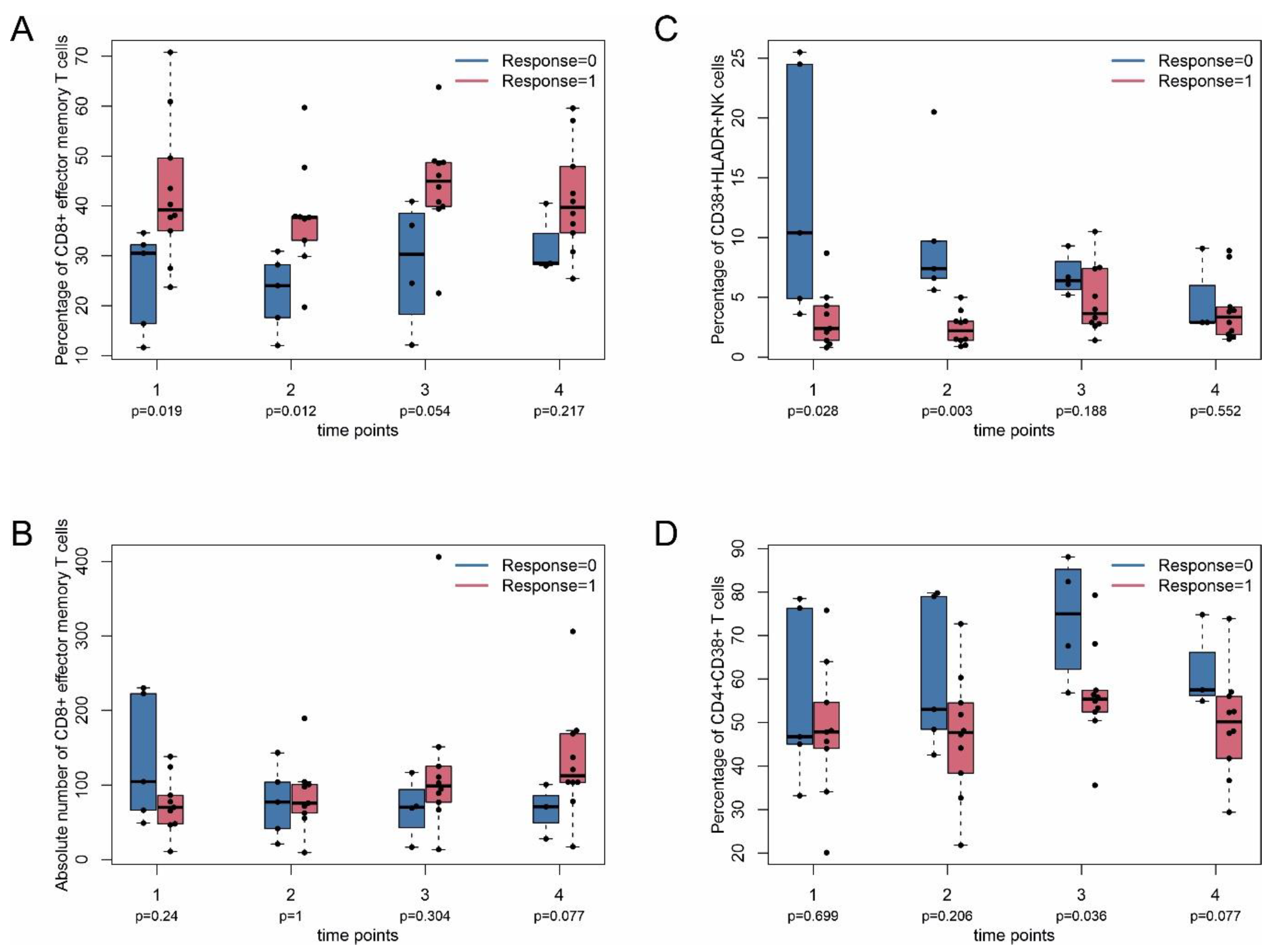
2.3. Tumor Response
2.4. Immune Monitoring
3. Discussion
4. Materials and Methods
4.1. Study Design and Patients
4.2. Flow Cytometry
4.3. Analysis of STAT5-Phosphorylation—IL-2-Stimulation
4.4. Analysis of STAT5-Phosphorylation—CD3/CD28-Stimulation
4.5. Analysis of Lymphocyte Markers and PD-1-Expression
4.6. Analysis of CTLA-4-Expression
4.7. Data Acquisition
4.8. Clinical Data
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hirsch, L.; Zitvogel, L.; Eggermont, A.; Marabelle, A. PD-Loma: A cancer entity with a shared sensitivity to the PD-1/PD-L1 pathway blockade. Br. J. Cancer 2019, 120, 3–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Postow, M.A.; Chesney, J.; Pavlick, A.C.; Robert, C.; Grossmann, K.; McDermott, D. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N. Engl. J. Med. 2015, 372, 2006–2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribas, A.; Puzanov, I.; Dummer, R.; Schadendorf, D.; Hamid, O.; Robert, C.; Hodi, F.S.; Schachter, J.; Pavlick, A.C.; Lewis, K.D. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): A randomised, controlled, phase 2 trial. Lancet Oncol. 2015, 16, 908–918. [Google Scholar] [CrossRef] [Green Version]
- Weber, J.; Mandala, M.; Del Vecchio, M.; Gogas, H.J.; Arance, A.M.; Cowey, C.L.; Arance, A.M.; Cowey, C.L.; Dalle, S.; Schenker, M. Adjuvant Nivolumab versus Ipilimumab in Resected Stage III or IV Melanoma. N. Engl. J. Med. 2017, 377, 1824–1835. [Google Scholar] [CrossRef]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.-X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [Green Version]
- Harlin, H.; Meng, Y.; Peterson, A.C.; Zha, Y.; Tretiakova, M.; Slingluff, C.; McKee, M.; Gajewski, T.F. Chemokine Expression in Melanoma Metastases Associated with CD8 + T-Cell Recruitment. Cancer Res. 2009, 69, 3077–3085. [Google Scholar] [CrossRef] [Green Version]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V. IFN-γ–related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.-L.; Roh, W.; Reuben, A.; Cooper, Z.A.; Spencer, C.N.; Prieto, P.A.; Miller, J.P.; Bassett, R.L.; Gopalakrishnan, V.; Wani, K. Analysis of Immune Signatures in Longitudinal Tumor Samples Yields Insight into Biomarkers of Response and Mechanisms of Resistance to Immune Checkpoint Blockade. Cancer Discov. 2016, 6, 827–837. [Google Scholar] [CrossRef] [Green Version]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.M.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Gajewski, T.F.; Corrales, L.; Williams, J.; Horton, B.; Sivan, A.; Spranger, S. Cancer Immunotherapy Targets Based on Understanding the T Cell-Inflamed Versus Non-T Cell-Inflamed Tumor Microenvironment. Adv. Exp. Med. Biol. 2017, 1036, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.C.; Postow, M.A.; Orlowski, R.J.; Mick, R.; Bengsch, B.; Manne, S.; Xu, W.; Harmon, S.; Giles, J.R.; Wenz, B. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 2017, 545, 60–65. [Google Scholar] [CrossRef] [Green Version]
- Verma, V.; Shrimali, R.K.; Ahmad, S.; Dai, W.; Wang, H.; Lu, S. PD-1 blockade in subprimed CD8 cells induces dysfunctional PD-1+CD38hi cells and anti-PD-1 resistance. Nat. Immunol. 2019, 20, 1231–1243. [Google Scholar] [CrossRef]
- Krieg, C.; Nowicka, M.; Guglietta, S.; Schindler, S.; Hartmann, F.J.; Weber, L.M.; Dummer, R.; Robinson, M.D.; Levesque, M.P.; Becher, B. High-dimensional single-cell analysis predicts response to anti-PD-1 immunotherapy. Nat. Med. 2018, 24, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Gowen, M.F.; Giles, K.M.; Simpson, D.; Tchack, J.; Zhou, H.; Moran, U.; Dawood, Z.; Pavlick, A.C.; Hu, S.; Wilson, M.A. Baseline antibody profiles predict toxicity in melanoma patients treated with immune checkpoint inhibitors. J. Transl. Med. 2018, 16, 82. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.; Khan, S.A.; Luo, X.; Fattah, F.J.; Saltarski, J.; Gloria-McCutchen, Y.; Lu, R.; Xie, Y.; Quan Li, Q.; Wakeland, E. Immune dysregulation in cancer patients developing immune-related adverse events. Br. J. Cancer 2019, 120, 63–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subudhi, S.K.; Aparicio, A.; Gao, J.; Zurita, A.J.; Araujo, J.C.; Logothetis, C.J.; Tahir, S.A.; Korivi, B.R.; Slack, R.S.; Vence, L. Clonal expansion of CD8 T cells in the systemic circulation precedes development of ipilimumab-induced toxicities. Proc. Natl. Acad. Sci. USA 2016, 113, 11919–11924. [Google Scholar] [CrossRef] [Green Version]
- Oh, D.Y.; Cham, J.; Zhang, L.; Fong, G.; Kwek, S.S.; Klinger, M.; Faham, M.; Fong, L. Immune Toxicities Elicted by CTLA-4 Blockade in Cancer Patients Are Associated with Early Diversification of the T-cell Repertoire. Cancer Res. 2017, 77, 1322–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bitar, M.; Boldt, A.; Freitag, M.-T.; Gruhn, B.; Köhl, U.; Sack, U. Evaluating STAT5 Phosphorylation as a Mean to Assess T Cell Proliferation. Front. Immunol. 2019, 10, 722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamphorst, A.O.; Pillai, R.N.; Yang, S.; Nasti, T.H.; Akondy, R.S.; Wieland, A.; Sica, G.L.; Yu, K.; Koenig, L.; Patel, N.T.; et al. Proliferation of PD-1+ CD8 T cells in peripheral blood after PD-1-targeted therapy in lung cancer patients. Proc. Natl. Acad. Sci. USA 2017, 114, 4993–4998. [Google Scholar] [CrossRef] [Green Version]
- Kamphorst, A.O.; Wieland, A.; Nasti, T.; Yang, S.; Zhang, R.; Barber, D.L.; Konieczny, B.T.; Daugherty, C.Z.; Koenig, L.; Yu, K. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science 2017, 355, 1423–1427. [Google Scholar] [CrossRef] [Green Version]
- Bally, A.P.R.; Austin, J.W.; Boss, J.M. Genetic and Epigenetic Regulation of PD-1 Expression. J. Immunol. 2016, 196, 2431–2437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweis, R.F.; Luke, J.J. Mechanistic and pharmacologic insights on immune checkpoint inhibitors. Pharm. Res. 2017, 120, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.-F.; Fouquet, S.; Chapon, M.; Salmon, H.; Regnier, F.; Labroquère, K.; Badoual, C.; Damotte, D.; Validire, P.; Maubec, E. Early T cell signalling is reversibly altered in PD-1+ T lymphocytes infiltrating human tumors. PLoS ONE 2011, 6, e17621. [Google Scholar] [CrossRef] [PubMed]
- Gambichler, T.; Schröter, U.; Höxtermann, S.; Susok, L.; Stockfleth, E.; Becker, J.C. Decline of programmed death-1-positive circulating T regulatory cells predicts more favourable clinical outcome of patients with melanoma under immune checkpoint blockade. Br. J. Derm. 2020, 182, 1214–1220. [Google Scholar] [CrossRef] [PubMed]
- Zelba, H.; Bochem, J.; Pawelec, G.; Garbe, C.; Wistuba-Hamprecht, K.; Weide, B. Accurate quantification of T-cells expressing PD-1 in patients on anti-PD-1 immunotherapy. Cancer Immunol. Immunother. 2018, 67, 1845–1851. [Google Scholar] [CrossRef] [PubMed]
- Fairfax, B.P.; Taylor, C.A.; Watson, R.A.; Nassiri, I.; Danielli, S.; Fang, H.; Mahé, E.A.; Cooper, R.; Woodcock, V.; Traill, Z. Peripheral CD8+ T cell characteristics associated with durable responses to immune checkpoint blockade in patients with metastatic melanoma. Nat. Med. 2020, 26, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Gide, T.N.; Quek, C.; Menzies, A.M.; Tasker, A.T.; Shang, P.; Holst, J.; Madore, J.; Lim, S.Y.; Velickovic, R.; Wongchenko, M. Distinct Immune Cell Populations Define Response to Anti-PD-1 Monotherapy and Anti-PD-1/Anti-CTLA-4 Combined Therapy. Cancer Cell 2019, 35, 238–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sade-Feldman, M.; Yizhak, K.; Bjorgaard, S.L.; Ray, J.P.; de Boer, C.G.; Jenkins, R.W.; Lieb, D.J.; Chen, J.H.; Frederick, D.T.; Barzily-Rokni, M. Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 2018, 175, 998–1013. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, S.; Daenthanasanmak, A.; Chakraborty, P.; Wyatt, M.W.; Dhar, P.; Selvam, S.P.; Fu, J.; Zhang, J.; Nguyen, H.; Kang, I. CD38-NAD+Axis Regulates Immunotherapeutic Anti-Tumor T Cell Response. Cell Metab. 2018, 27, 85–100. [Google Scholar] [CrossRef] [Green Version]
- Raab, M.S.; Engelhardt, M.; Blank, A.; Goldschmidt, H.; Agis, H.; Blau, I.W.; Einsele, H.; Ferstl, B.; Schub, N.; Röllig, C. MOR202, a novel anti-CD38 monoclonal antibody, in patients with relapsed or refractory multiple myeloma: A first-in-human, multicentre, phase 1-2a trial. Lancet Haematol. 2020, 7, e381–e394. [Google Scholar] [CrossRef]
- Saleh, K.; Khalife-Saleh, N.; Kourie, H.R. Do immune-related adverse events correlate with response to immune checkpoint inhibitors? Immunotherapy 2019, 11, 257–259. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, J.A.; Kronenberg, K.; Riquelme, P.; Wenzel, J.J.; Glehr, G.; Schilling, H.-L.; Zeman, F.; Evert, K.; Schmiedel, M.; Mickler, M. Virus-specific memory T cell responses unmasked by immune checkpoint blockade cause hepatitis. Nat. Commun. 2021, 12, 1439. [Google Scholar] [CrossRef]
- Weber, J.S.; Yang, J.C.; Atkins, M.B.; Disis, M.L. Toxicities of Immunotherapy for the Practitioner. J. Clin. Oncol. 2015, 33, 2092–2099. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.X.; Migone, T.S.; Tsang, M.; Friedmann, M.; Weatherbee, J.A.; Zhou, L.; Yamauchi, A.; Bloom, E.T.; Mietz, J.; John, S. The role of shared receptor motifs and common Stat proteins in the generation of cytokine pleiotropy and redundancy by IL-2, IL-4, IL-7, IL-13, and IL-15. Immunity 1995, 2, 331–339. [Google Scholar] [CrossRef] [Green Version]
- Verdeil, G.; Lawrence, T.; Schmitt-Verhulst, A.-M.; Auphan-Anezin, N. Targeting STAT3 and STAT5 in Tumor-Associated Immune Cells to Improve Immunotherapy. Cancers 2019, 11, 1832. [Google Scholar] [CrossRef] [Green Version]
- Zen, Y.; Yeh, M.M. Hepatotoxicity of immune checkpoint inhibitors: A histology study of seven cases in comparison with autoimmune hepatitis and idiosyncratic drug-induced liver injury. Mod. Pathol. 2018, 31, 965–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Läubli, H.; Balmelli, C.; Bossard, M.; Pfister, O.; Glatz, K.; Zippelius, A. Acute heart failure due to autoimmune myocarditis under pembrolizumab treatment for metastatic melanoma. J. Immunother. Cancer 2015, 3, 11. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Yu, D.; Sarnaik, A.A.; Yu, B.; Hall, M.; Morelli, D.; Zhang, Y.; Zhao, X.; Weber, J.S. Biomarkers on melanoma patient T cells associated with ipilimumab treatment. J. Transl. Med. 2012, 10, 146. [Google Scholar] [CrossRef] [Green Version]
- Willsmore, Z.N.; Harris, R.J.; Crescioli, S.; Hussein, K.; Kakkassery, H.; Thapa, D.; Cheung, A.; Chauhan, J.; Bax, H.J.; Chenoweth, A. B Cells in Patients With Melanoma: Implications for Treatment With Checkpoint Inhibitor Antibodies. Front. Immunol. 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Boldt, A.; Borte, S.; Fricke, S.; Kentouche, K.; Emmrich, F.; Borte, M.; Kahlenberg, F.; Sack, U. Eight-color immunophenotyping of T-, B-, and NK-cell subpopulations for characterization of chronic immunodeficiencies. Cytom. Part B Clin. Cytom. 2014, 86, 191–206. [Google Scholar] [CrossRef] [PubMed]
- Boldt, A.; Kahlenberg, F.; Fricke, S.; Rasche, F.M.; Sack, U. Flow cytometric phenotyping of lymphocytes in patients with systemic lupus erythematosus. Cytom. Part A 2014, 85, 567–569. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Gender/Age a | Melanoma Subtype/Site/Breslow in mm b | BRAF/PD-L1 c | ICI d | Pre-Treatment e | Tumor Burden in cm f | Involved Sites g | Response h | irAE/Grade i | S100/LDH j |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F/78 | uNM/HN/1.8 | WT/5% | 4NIVO | RT | 1.0 | PUL | CR | none | -/+ |
| 2 | M/76 | uNM/T/1.4 | WT/30% | 1IPI/NIVO, 6NIVO | none | 20.6 | CER, LN, MUS, PER, SC | PR | irFAT/G3 irTHY/G2 | -/- |
| 3 | F/70 | NM/LE/3.9 | WT/20% | 4PEMBRO | aIFN | 6.7 | LN, PUL | PD | irMUC/G2 | -/+ |
| 4 | M/67 | uALM/LE/3.1 | WT/5% | 4PEMBRO | none | 3.4 | HEP | PD | irHEP/G1 | -/+ |
| 5 | F/74 | mucM/AN/12.0 | WT/0% (cKIT WT) | 1IPI/NIVO, 1NIVO | naRT | 20.8 | AN, HEP, PUL | PD | irHEP/G2 | -/+ |
| 6 | F/91 | uNM/T/3.5 | V600E/30% | 4PEMBRO | none | 3.9 | CUT, HEP, PUL | PR | none | -/+ |
| 7 | M/78 | NM/T/9.6 | WT/10% | 2IPI/NIVO | none | 15 | HEP, LN, OSS | PR | irTHY/G2, irHYP/G2, irHEP/G1 | +/+ |
| 8 | M/75 | uNM/UE/3.4 | WT/0% | 4PEMBRO | none | 4.6 | CUT, LN, SC | PR | none | +/- |
| 9 | F/41 | NM/LE/3.1 | V600E/0% | 4IPI/NIVO, 1NIVO | aIFN, aNIVO, aTT | 1.9 | CER, LN | SD | none | +/- |
| 10 | F/57 | SSM/T/0.8 | V600E/5% | 2IPI/NIVO, 4NIVO | TT | 1.0 | CER, PUL | PR | irHEP/G2 | -/- |
| 11 | M/70 | MNOS/T/1.4 | WT/UKN | 3IPI/NIVO, 1NIVO | none | 13.6 | CER, INT, PUL, LN | PR | irFAT/G2, irCOL/G2, irTHY/G1 | +/+ |
| 12 | M/77 | uNM/T/9.0 | V600E/5% | 2NIVO | none | 18.7 | CUT, INT, LN, MUS, OSS, SPLE | n.a. | none | +/+ |
| 13 | M/76 | NM/HN/3.6 | WT/UKN | 1NIVO | none | 16.9 | ADR, BIL, OSS, PUL | PD | none | +/+ |
| 14 | M/61 | uSSM/OE/1.5 | WTUKN | 3PEMBRO | none | 7.6 | CUT, LN, PUL | n.a. | none | -/- |
| 15 | F/95 | uNM/HN/11.0 | WT/10% | 5NIVO | none | 7.5 | ADR, HEP, MUS, PUL | PR | none | -/- |
| 16 | F/52 | CUP/UKN/UKN | WT/2% | 2IPI/NIVO | RT | 2.6 | PAN, MUS | CR | irDER/G1, irTHY/G1, irNEP/G1 | -/- |
| 17 | M/48 | CUP/UKN/UKN | V600E/0% | 4IPI/NIVO | RT, TT | 5.4 | CER | PR | none | +/+ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reschke, R.; Gussek, P.; Boldt, A.; Sack, U.; Köhl, U.; Lordick, F.; Gora, T.; Kreuz, M.; Reiche, K.; Simon, J.-C.; et al. Distinct Immune Signatures Indicative of Treatment Response and Immune-Related Adverse Events in Melanoma Patients under Immune Checkpoint Inhibitor Therapy. Int. J. Mol. Sci. 2021, 22, 8017. https://doi.org/10.3390/ijms22158017
Reschke R, Gussek P, Boldt A, Sack U, Köhl U, Lordick F, Gora T, Kreuz M, Reiche K, Simon J-C, et al. Distinct Immune Signatures Indicative of Treatment Response and Immune-Related Adverse Events in Melanoma Patients under Immune Checkpoint Inhibitor Therapy. International Journal of Molecular Sciences. 2021; 22(15):8017. https://doi.org/10.3390/ijms22158017
Chicago/Turabian StyleReschke, Robin, Philipp Gussek, Andreas Boldt, Ulrich Sack, Ulrike Köhl, Florian Lordick, Thomas Gora, Markus Kreuz, Kristin Reiche, Jan-Christoph Simon, and et al. 2021. "Distinct Immune Signatures Indicative of Treatment Response and Immune-Related Adverse Events in Melanoma Patients under Immune Checkpoint Inhibitor Therapy" International Journal of Molecular Sciences 22, no. 15: 8017. https://doi.org/10.3390/ijms22158017