
Possible Link between Higher Transmissibility of Alpha, Kappa and Delta Variants of SARS-CoV-2 and Increased Structural Stability of Its Spike Protein and hACE2 Affinity


Abstract
:1. Introduction
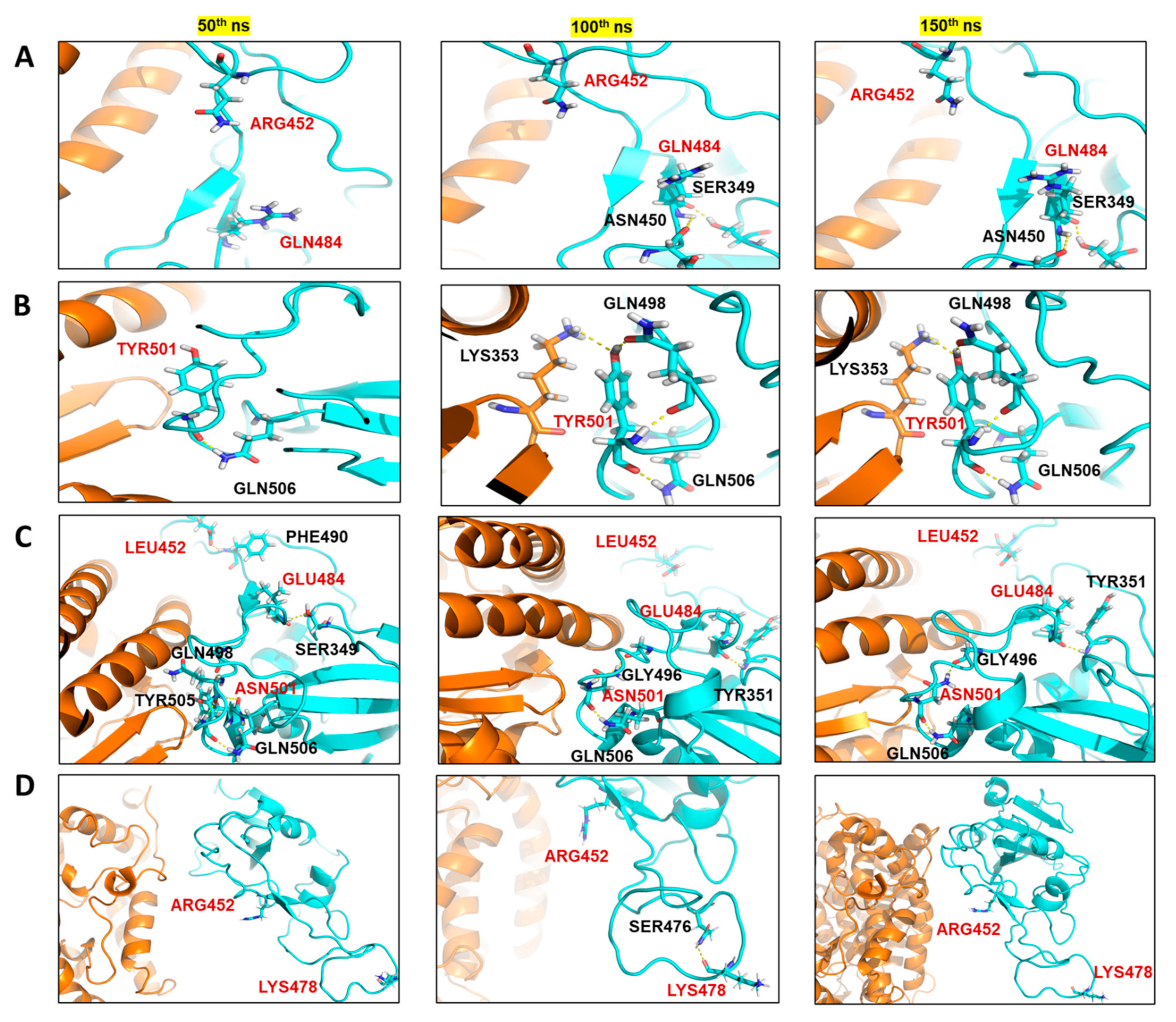
2. Results
The Mutant Spike Proteins Have a Better Binding Affinity with hACE2 in Comparison to Wildtype
3. Discussion
4. Materials and Methods
4.1. MD Simulations
4.2. Analysis of the MD Simulation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Zheng, J. SARS-CoV-2: An Emerging Coronavirus that Causes a Global Threat. Int. J. Biol. Sci. 2020, 16, 1678–1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.; Lian, X.; Su, X.; Wu, W.; Marraro, G.A.; Zeng, Y. From SARS and MERS to COVID-19: A brief summary and comparison of severe acute respiratory infections caused by three highly pathogenic human coronaviruses. Respir. Res. 2020, 21, 224. [Google Scholar] [CrossRef]
- Rahman, S.; Singh, H.; Singh, J.; Khubaib, M.; Jamal, S.; Sheikh, J.; Kohli, S.; Hasnain, S. Mapping the genomic landscape & diversity of COVID-19 based on >3950 clinical isolates of SARS-CoV-2: Likely origin & transmission dynamics of isolates sequenced in India. Indian J. Med Res. 2020, 151, 474–478. [Google Scholar] [CrossRef]
- Naqvi, A.A.T.; Fatima, K.; Mohammad, T.; Fatima, U.; Singh, I.K.; Singh, A.; Atif, S.M.; Hariprasad, G.; Hasan, G.M.; Hassan, I. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: Structural genomics approach. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165878. [Google Scholar] [CrossRef]
- Sironi, M.; Hasnain, S.E.; Rosenthal, B.; Phan, T.; Luciani, F.; Shaw, M.-A.; Sallum, M.A.; Mirhashemi, M.E.; Morand, S.; González-Candelas, F. SARS-CoV-2 and COVID-19: A genetic, epidemiological, and evolutionary perspective. Infect. Genet. Evol. 2020, 84, 104384. [Google Scholar] [CrossRef]
- Sheikh, J.A.; Singh, J.; Singh, H.; Jamal, S.; Khubaib, M.; Kohli, S.; Dobrindt, U.; Rahman, S.A.; Ehtesham, N.Z.; Hasnain, S.E. Emerging genetic diversity among clinical isolates of SARS-CoV-2: Lessons for today. Infect. Genet. Evol. 2020, 84, 104330. [Google Scholar] [CrossRef]
- Li, F. Structure, Function, and Evolution of Coronavirus Spike Proteins. Annu. Rev. Virol. 2016, 3, 237–261. [Google Scholar] [CrossRef] [Green Version]
- Mittal, A.; Manjunath, K.; Ranjan, R.K.; Kaushik, S.; Kumar, S.; Verma, V. COVID-19 pandemic: Insights into structure, function, and hACE2 receptor recognition by SARS-CoV-2. PLoS Pathog. 2020, 16, e1008762. [Google Scholar] [CrossRef]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e278. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yang, C.; Xu, X.-F.; Xu, W.; Liu, S.-W. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef]
- Du, L.; He, Y.; Zhou, Y.; Liu, S.; Zheng, B.-J.; Jiang, S. The spike protein of SARS-CoV—A target for vaccine and therapeutic development. Nat. Rev. Microbiol. 2009, 7, 226–236. [Google Scholar] [CrossRef]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the Treatment of COVID-19—Final Report. N. Engl. J. Med. 2020, 383, 1813–1826. [Google Scholar] [CrossRef]
- Meini, S.; Pagotto, A.; Longo, B.; Vendramin, I.; Pecori, D.; Tascini, C. Role of Lopinavir/Ritonavir in the Treatment of COVID-19: A Review of Current Evidence, Guideline Recommendations, and Perspectives. J. Clin. Med. 2020, 9, 2050. [Google Scholar] [CrossRef]
- Kumar, R.; Sharma, A.; Srivastava, J.K.; Siddiqui, M.H.; Uddin, S.; Aleya, L. Hydroxychloroquine in COVID-19: Therapeutic promises, current status, and environmental implications. Environ. Sci. Pollut. Res. 2021, 28, 40431–40444. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef]
- Singh, J.; Samal, J.; Kumar, V.; Sharma, J.; Agrawal, U.; Ehtesham, N.; Sundar, D.; Rahman, S.; Hira, S.; Hasnain, S. Structure-Function Analyses of New SARS-CoV-2 Variants B.1.1.7, B.1.351 and B.1.1.28.1: Clinical, Diagnostic, Therapeutic and Public Health Implications. Viruses 2021, 13, 439. [Google Scholar] [CrossRef]
- La Rosa, G.; Mancini, P.; Ferraro, G.B.; Veneri, C.; Iaconelli, M.; Lucentini, L.; Bonadonna, L.; Brusaferro, S.; Brandtner, D.; Fasanella, A.; et al. Rapid screening for SARS-CoV-2 variants of concern in clinical and environmental samples using nested RT-PCR assays targeting key mutations of the spike protein. Water Res. 2021, 197, 117104. [Google Scholar] [CrossRef]
- Tang, J.W.; Toovey, O.T.R.; Harvey, K.N.; Hui, D.D.S. Introduction of the South African SARS-CoV-2 variant 501Y.V2 into the UK. J. Infect. 2021, 82, e8–e10. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, J.; Plante, K.S.; Plante, J.A.; Xie, X.; Zhang, X.; Ku, Z.; An, Z.; Scharton, D.; Schindewolf, C. The N501Y spike substitution enhances SARS-CoV-2 transmission. BioRxiv 2021. [Google Scholar] [CrossRef]
- Hirotsu, Y.; Omata, M. Discovery of a SARS-CoV-2 variant 1 from the P.1 lineage harboring K417T/E484K/N501Y mutations in Kofu, Japan. J. Infect. 2021, 82, 276–316. [Google Scholar] [CrossRef]
- Williams, A.H.; Zhan, C.-G. Fast Prediction of Binding Affinities of the SARS-CoV-2 Spike Protein Mutant N501Y (UK Variant) with ACE2 and Miniprotein Drug Candidates. J. Phys. Chem. B 2021, 125, 4330–4336. [Google Scholar] [CrossRef]
- Bernal, J.L.; Andrews, N.; Gower, C.; Gallagher, E.; Simmons, R.; Thelwall, S.; Stowe, J.; Tessier, E.; Groves, N.; Dabrera, G.; et al. Effectiveness of COVID-19 Vaccines against the B.1.617.2 (Delta) Variant. N. Engl. J. Med. 2021, 385, 585–594. [Google Scholar] [CrossRef]
- Sahoo, J.P.; Mishra, A.P.; Samal, K.C. Triple Mutant Bengal Strain (B. 1.618) of Coronavirus and the Worst COVID Outbreak in India. Biot. Res. Today 2021, 3, 261–265. [Google Scholar]
- Mallapaty, S. India’s massive COVID surge puzzles scientists. Nat. Cell Biol. 2021, 592, 667–668. [Google Scholar] [CrossRef]
- Ozono, S.; Zhang, Y.; Ode, H.; Sano, K.; Tan, T.S.; Imai, K.; Miyoshi, K.; Kishigami, S.; Ueno, T.; Iwatani, Y.; et al. SARS-CoV-2 D614G spike mutation increases entry efficiency with enhanced ACE2-binding affinity. Nat. Commun. 2021, 12, 1–9. [Google Scholar] [CrossRef]
- Kannan, S.R.; Spratt, A.N.; Cohen, A.R.; Naqvi, S.H.; Chand, H.S.; Quinn, T.P.; Lorson, C.L.; Byrareddy, S.N.; Singh, K. Evolutionary analysis of the Delta and Delta Plus variants of the SARS-CoV-2 viruses. J. Autoimmun. 2021, 124, 102715. [Google Scholar] [CrossRef]
- Singh, J.; Rahman, S.A.; Ehtesham, N.Z.; Hira, S.; Hasnain, S.E. SARS-CoV-2 variants of concern are emerging in India. Nat. Med. 2021, 27, 1131–1133. [Google Scholar] [CrossRef]
- Dhar, M.S.; Marwal, R.; Radhakrishnan, V.; Ponnusamy, K.; Jolly, B.; Bhoyar, R.C.; Fatihi, S.; Datta, M.; Singh, P.; Sharma, U.; et al. Genomic characterization and Epidemiology of an emerging SARS-CoV-2 variant in Delhi, India. medRxiv 2021. [Google Scholar] [CrossRef]
- Arora, P.; Kempf, A.; Nehlmeier, I.; Sidarovich, A.; Krüger, N.; Graichen, L.; Moldenhauer, A.-S.; Winkler, M.S.; Schulz, S.; Jäck, H.-M.; et al. Increased lung cell entry of B.1.617.2 and evasion of antibodies induced by infection and BNT162b2 vaccination. BioRxiv 2021. [Google Scholar] [CrossRef]
- Schrödinger Protein Preparation Wizard, Epik, Impact, Prime, LigPrep, Glide; Desmond Molecular Dynamics System (Developed by D.E. Shaw Research); Maestro-Desmond Interoperability Tools; Schrödinger, LLC.: New York, NY, USA, 2020.
- Ylilauri, M.; Pentikäinen, O.T. MMGBSA As a Tool to Understand the Binding Affinities of Filamin–Peptide Interactions. J. Chem. Inf. Model. 2013, 53, 2626–2633. [Google Scholar] [CrossRef]
- Pascarella, S.; Ciccozzi, M.; Zella, D.; Bianchi, M.; Benedetti, F.; Benvenuto, D.; Broccolo, F.; Cauda, R.; Caruso, A.; Angeletti, S.; et al. SARS-CoV-2 B.1.617 Indian variants: Are electrostatic potential changes responsible for a higher transmission rate? J. Med. Virol. 2021, 1–6. [Google Scholar] [CrossRef]
- Kar, S.K.; Ransing, R.; Arafat, S.; Menon, V. Second wave of COVID-19 pandemic in India: Barriers to effective governmental response. EClinicalMedicine 2021, 36, 100915. [Google Scholar] [CrossRef]
- Aleem, A.; Akbar Samad, A.B.; Slenker, A.K. Emerging Variants of SARS-CoV-2 and Novel Therapeutics Against Coronavirus (COVID-19); StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Wang, P.; Liu, L.; Iketani, S.; Luo, Y.; Guo, Y.; Wang, M.; Yu, J.; Zhang, B.; Kwong, P.D.; Graham, B.S.; et al. Increased Resistance of SARS-CoV-2 Variants B.1.351 and B.1.1.7 to Antibody Neutralization. BioRxiv 2021. [Google Scholar] [CrossRef]
- Sabino, E.C.; Buss, L.F.; Carvalho, M.P.S.; Prete, C.A.; Crispim, M.A.E.; Fraiji, N.A.; Pereira, R.H.M.; Parag, K.V.; Peixoto, P.D.S.; Kraemer, M.U.G.; et al. Resurgence of COVID-19 in Manaus, Brazil, despite high seroprevalence. Lancet 2021, 397, 452–455. [Google Scholar] [CrossRef]
- Shen, X.; Tang, H.; Pajon, R.; Smith, G.; Glenn, G.M.; Shi, W.; Korber, B.; Montefiori, D.C. Neutralization of SARS-CoV-2 Variants B.1.429 and B.1.351. N. Engl. J. Med. 2021, 384, 2352–2354. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, I.; Datir, R.; Kemp, S.; Papa, G.; Rakshit, P.; Singh, S.; Meng, B.; Pandey, R.; Ponnusamy, K.; Radhakrishnan, V.S.; et al. SARS-CoV-2 B.1.617 emergence and sensitivity to vaccine-elicited antibodies. BioRxiv 2021. [Google Scholar] [CrossRef]
- Yadav, P.D.; Sapkal, G.N.; Abraham, P.; Ella, R.; Deshpande, G.; Patil, D.Y.; A Nyayanit, D.; Gupta, N.; Sahay, R.R.; Shete, A.M.; et al. Neutralization of Variant Under Investigation B.1.617.1 With Sera of BBV152 Vaccinees. Clin. Infect. Dis. 2021, ciab411. [Google Scholar] [CrossRef]
- Winger, A.; Caspari, T. The Spike of Concern—The Novel Variants of SARS-CoV-2. Viruses 2021, 13, 1002. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Greenidge, P.A.; Kramer, C.; Mozziconacci, J.-C.; Wolf, R.M. MM/GBSA Binding Energy Prediction on the PDBbind Data Set: Successes, Failures, and Directions for Further Improvement. J. Chem. Inf. Model. 2013, 53, 201–209. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Kappa (kcal/mol) | Delta (kcal/mol) | WT (kcal/mol) | Alpha (kcal/mol) | |||||
|---|---|---|---|---|---|---|---|---|
| R452 | Q484 | R452 | K478 | L452 | E484 | N501 | T478 | Y501 |
| −47.16 | −51.43 | −48.67 | −7.51 | −23.45 | −47.06 | 3.44 | 3.44 | −31.72 |
| −50.69 | −57.57 | −49.32 | −7.05 | −23.04 | −48.53 | 9.92 | 9.92 | −27.6 |
| −42.13 | −56.15 | −47.15 | −10.92 | −23.89 | −45.52 | 3.81 | 3.81 | −32.73 |
| −52.43 | −56.51 | −45.49 | −11.73 | −20.05 | −48.94 | 2.89 | 2.89 | −31.96 |
| −54.38 | −58.47 | −47.68 | −9.31 | −23.68 | −50.2 | 2.03 | 2.03 | −26.71 |
| −54.05 | −56.86 | −46.76 | −12.17 | −25.88 | −47.91 | 2.60 | 2.60 | −29.47 |
| −56.11 | −53.98 | −50.68 | −7.42 | −21.65 | −47.86 | 4.45 | 4.45 | −37.94 |
| −47.14 | −56.4 | −45.18 | −10.38 | −22.44 | −44.1 | 7.27 | 7.27 | −33.03 |
| −56.47 | −57.21 | −50.03 | −13.46 | −22.87 | −43.55 | −3.05 | −3.05 | −31.87 |
| −54.79 | −53.25 | −48.51 | −10.35 | −22.05 | −48.18 | 3.55 | 3.55 | −33.2 |
| −50.07 | −51.51 | −52.83 | −6.85 | −20.9 | −46.46 | 7.14 | 7.14 | −33.69 |
| −45.38 | −53.04 | −55.77 | −9.25 | −17.67 | −45.08 | 1.14 | 1.14 | −25.84 |
| −49.22 | −59.17 | −50.34 | −11.01 | −25.26 | −49.82 | 4.00 | 4.00 | −34.98 |
| −51.75 | −58.8 | −47.65 | −9.99 | −16.19 | −47.49 | −0.80 | −0.80 | −29.93 |
| −45.95 | −55.64 | −45.07 | −14.89 | −18.58 | −47.46 | 2.51 | 2.51 | −34.11 |
| −48.85 | −56.21 | −45.52 | −11.16 | −20.55 | −49.06 | 4.97 | 4.97 | −34.51 |
| −48.93 | −56.69 | −49.63 | −6.93 | −24.27 | −46.24 | 5.18 | 5.18 | −31.49 |
| −54.01 | −56.35 | −48.91 | −4.89 | −23.78 | −50.55 | 1.66 | 1.66 | −32.64 |
| −55.01 | −59.25 | −54.44 | −7.28 | −21.35 | −41.95 | 0.17 | 0.17 | −32.55 |
| −53.5 | −56.27 | −53.66 | −5.53 | −25.55 | −46.55 | 1.23 | 1.23 | −25.06 |
| −50.90 ± 3.99 | −56.03 ± 2.31 | −49.16 ± 3.11 | −9.40 ± 2.66 | −22.15 ± 2.60 | −47.12 ± 2.25 | −64.31 ± 3.59 | −3.20 ± 2.93 | −31.55 ± 3.26 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, V.; Singh, J.; Hasnain, S.E.; Sundar, D. Possible Link between Higher Transmissibility of Alpha, Kappa and Delta Variants of SARS-CoV-2 and Increased Structural Stability of Its Spike Protein and hACE2 Affinity. Int. J. Mol. Sci. 2021, 22, 9131. https://doi.org/10.3390/ijms22179131
Kumar V, Singh J, Hasnain SE, Sundar D. Possible Link between Higher Transmissibility of Alpha, Kappa and Delta Variants of SARS-CoV-2 and Increased Structural Stability of Its Spike Protein and hACE2 Affinity. International Journal of Molecular Sciences. 2021; 22(17):9131. https://doi.org/10.3390/ijms22179131
Chicago/Turabian StyleKumar, Vipul, Jasdeep Singh, Seyed E. Hasnain, and Durai Sundar. 2021. "Possible Link between Higher Transmissibility of Alpha, Kappa and Delta Variants of SARS-CoV-2 and Increased Structural Stability of Its Spike Protein and hACE2 Affinity" International Journal of Molecular Sciences 22, no. 17: 9131. https://doi.org/10.3390/ijms22179131