
Current Concepts on Genetic Aspects of Mitochondrial Dysfunction in Amyotrophic Lateral Sclerosis

 ,
,
Abstract
:1. Introduction
2. Phenotype Variability
3. Mitochondrial (Dys)Function
4. Mitochondrial Genome Variations
5. Genes Involved in Mitochondrial Dysfunction
6. Novel Biomarkers and Therapies for ALS: All Roads Lead to Mitochondria?
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Brotman, R.G.; Moreno-Escobar, M.C.; Joseph, J.; Pawar, G. Amyotrophic Lateral Sclerosis. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Logroscino, G.; Piccininni, M. Amyotrophic Lateral Sclerosis Descriptive Epidemiology: The Origin of Geographic Difference. Neuroepidemiology 2019, 52, 93–103. [Google Scholar] [CrossRef]
- Verde, F.; Silani, V.; Otto, M. Neurochemical biomarkers in amyotrophic lateral sclerosis. Curr. Opin. Neurol. 2019, 32, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Saberi, S.; Stauffer, J.E.; Schulte, D.J.; Ravits, J. Neuropathology of Amyotrophic Lateral Sclerosis and Its Variants. Neurol. Clin. 2015, 33, 855–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longinetti, E.; Fang, F. Epidemiology of amyotrophic lateral sclerosis: An update of recent literature. Curr. Opin. Neurol. 2019, 32, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Kadena, K.; Vlamos, P. Amyotrophic Lateral Sclerosis: Current Status in Diagnostic Biomarkers. Adv. Exp. Med. Biol. 2020, 1195, 179–187. [Google Scholar]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers 2017, 3, 17071. [Google Scholar] [CrossRef] [PubMed]
- Morgan, S.; Orrell, R.W. Pathogenesis of amyotrophic lateral sclerosis. Br. Med. Bull. 2016, 119, 87–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manjaly, Z.R.; Scott, K.M.; Abhinav, K.; Wijesekera, L.; Ganesalingam, J.; Goldstein, L.H.; Janssen, A.; Dougherty, A.; Willey, E.; Stanton, B.R.; et al. The sex ratio in amyotrophic lateral sclerosis: A population based study. Amyotroph. Lateral Scler. 2010, 11, 439–442. [Google Scholar] [CrossRef]
- Chiò, A.; Logroscino, G.; Traynor, B.J.; Collins, J.; Simeone, J.C.; Goldstein, L.A.; White, L.A. Global epidemiology of amyotrophic lateral sclerosis: A systematic review of the published literature. Neuroepidemiology 2013, 41, 118–130. [Google Scholar] [CrossRef] [Green Version]
- Hulisz, D. Amyotrophic lateral sclerosis: Disease state overview. Am. J. Manag. Care 2018, 24 (Suppl. 15), S320–S326. [Google Scholar]
- Norris, S.P.; Likanje, M.N.; Andrews, J.A. Amyotrophic lateral sclerosis: Update on clinical management. Curr. Opin. Neurol. 2020, 33, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Oskarsson, B.; Gendron, T.F.; Staff, N.P. Amyotrophic Lateral Sclerosis: An Update for 2018. Mayo Clin. Proc. 2018, 93, 1617–1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valko, K.; Ciesla, L. Amyotrophic lateral sclerosis. Prog. Med. Chem. 2019, 58, 63–117. [Google Scholar] [PubMed]
- Tard, C.; Defebvre, L.; Moreau, C.; Devos, D.; Danel-Brunaud, V. Clinical features of amyotrophic lateral sclerosis and their prognostic value. Rev. Neurol. 2017, 173, 263–272. [Google Scholar] [CrossRef] [PubMed]
- van Es, M.A.; Hardiman, O.; Chio, A.; Al-Chalabi, A.; Pasterkamp, R.J.; Veldink, J.H.; van den Berg, L.H. Amyotrophic lateral sclerosis. Lancet 2017, 390, 2084–2098. [Google Scholar] [CrossRef]
- Takeda, T.; Kitagawa, K.; Arai, K. Phenotypic variability and its pathological basis in amyotrophic lateral sclerosis. Neuropathology 2020, 40, 40–56. [Google Scholar] [CrossRef]
- Swinnen, B.; Robberecht, W. The phenotypic variability of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2014, 10, 661–670. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, S.; Iwata, M. Mitochondrial alterations in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2007, 66, 10–16. [Google Scholar] [CrossRef] [Green Version]
- Corcia, P.; Lumbroso, S.; Cazeneuve, C.; Mouzat, K.; Camu, W.; Vourc’h, P.; on Behalf the FILSLAN network. Pre-symptomatic diagnosis in ALS. Rev. Neurol. 2020, 176, 166–169. [Google Scholar]
- Ranganathan, R.; Haque, S.; Coley, K.; Shepheard, S.; Cooper-Knock, J.; Kirby, J. Multifaceted Genes in Amyotrophic Lateral Sclerosis-Frontotemporal Dementia. Front. Neurosci. 2020, 14, 684. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Lattante, S.; Marangi, G.; Doronzio, P.N.; Conte, A.; Bisogni, G.; Zollino, M.; Sabatelli, M. High-Throughput Genetic Testing in ALS: The Challenging Path of Variant Classification Considering the ACMG Guidelines. Genes 2020, 11, 1123. [Google Scholar] [CrossRef]
- Weishaupt, J.H.; Hyman, T.; Dikic, I. Common Molecular Pathways in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Trends Mol. Med. 2016, 22, 769–783. [Google Scholar] [CrossRef]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef]
- Pizzuti, A.; Petrucci, S. Mitochondrial disfunction as a cause of ALS. Arch. Ital. Biol. 2011, 149, 113–119. [Google Scholar]
- Cho, B.; Choi, S.Y.; Cho, H.M.; Kim, H.J.; Sun, W. Physiological and pathological significance of dynamin-related protein 1 (drp1)-dependent mitochondrial fission in the nervous system. Exp. Neurobiol. 2013, 22, 149–157. [Google Scholar] [CrossRef]
- Liu, W.; Yamashita, T.; Tian, F.; Morimoto, N.; Ikeda, Y.; Deguchi, K.; Abe, K. Mitochondrial fusion and fission proteins expression dynamically change in a murine model of amyotrophic lateral sclerosis. Curr. Neurovasc. Res. 2013, 10, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.Y.; Lee, J.H.; Chung, A.Y.; Jo, Y.; Shin, J.H.; Park, H.C.; Kim, H.; Lopez-Gonzalez, R.; Ryu, J.R.; Sun, W. Prevention of mitochondrial impairment by inhibition of protein phosphatase 1 activity in amyotrophic lateral sclerosis. Cell Death Dis. 2020, 11, 888. [Google Scholar] [CrossRef]
- Rangaraju, V.; Lauterbach, M.; Schuman, E.M. Spatially stable mitochondrial compartments fuel local translation during plasticity. Cell 2019, 176, 73–84.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, P.; Wei, Y.; Zhang, J.; Gal, J.; Zhu, H. Mitochondrial dysfunction is a converging point of multiple pathological pathways in amyotrophic lateral sclerosis. J. Alzheimers. Dis. 2010, 20 (Suppl. 2), S311–S324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borthwick, G.M.; Johnson, M.A.; Ince, P.G.; Shaw, P.J.; Turnbull, D.M. Mitochondrial enzyme activity in amyotrophic lateral sclerosis: Implications for the role of mitochondria in neuronal cell death. Ann. Neurol. 1999, 46, 787–790. [Google Scholar] [CrossRef]
- Sharma, P.; Sampath, H. Mitochondrial DNA Integrity: Role in Health and Disease. Cells 2019, 8, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef] [Green Version]
- Stoccoro, A.; Mosca, L.; Carnicelli, V.; Cavallari, U.; Lunetta, C.; Marocchi, A.; Migliore, L.; Coppedè, F. Mitochondrial DNA copy number and D-loop region methylation in carriers of amyotrophic lateral sclerosis gene mutations. Epigenomics 2018, 10, 1431–1443. [Google Scholar] [CrossRef]
- Stoccoro, A.; Smith, A.R.; Mosca, L.; Marocchi, A.; Gerardi, F.; Lunetta, C.; Cereda, C.; Gagliardi, S.; Lunnon, K.; Migliore, L.; et al. Reduced mitochondrial D-loop methylation levels in sporadic amyotrophic lateral sclerosis. Clin. Epigenetics. 2020, 12, 137. [Google Scholar] [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62, Erratum in 1993, 364, 362. [Google Scholar] [CrossRef]
- Kim, G.; Gautier, O.; Tassoni-Tsuchida, E.; Ma, X.R.; Gitler, A.D. ALS Genetics: Gains, Losses, and Implications for Future Therapies. Neuron 2020, 108, 822–842. [Google Scholar] [CrossRef]
- Bunton-Stasyshyn, R.K.; Saccon, R.A.; Fratta, P.; Fisher, E.M. SOD1 Function and Its Implications for Amyotrophic Lateral Sclerosis Pathology: New and Renascent Themes. Neuroscientist 2015, 21, 519–529. [Google Scholar] [CrossRef] [Green Version]
- Pickles, S.; Destroismaisons, L.; Peyrard, S.L.; Cadot, S.; Rouleau, G.A.; Brown, R.H., Jr.; Julien, J.P.; Arbour, N.; Vande Velde, C. Mitochondrial damage revealed by immunoselection for ALS-linked misfolded SOD1. Hum. Mol. Genet. 2013, 22, 3947–3959. [Google Scholar] [CrossRef]
- Tak, Y.J.; Park, J.H.; Rhim, H.; Kang, S. ALS-Related Mutant SOD1 Aggregates Interfere with Mitophagy by Sequestering the Autophagy Receptor Optineurin. Int. J. Mol. Sci. 2020, 21, 7525. [Google Scholar] [CrossRef] [PubMed]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [Green Version]
- Smeyers, J.; Banchi, E.G.; Latouche, M. C9ORF72: What It Is, What It Does, and Why It Matters. Front. Cell. Neurosci. 2021, 15, 661447. [Google Scholar] [CrossRef]
- Cook, C.N.; Wu, Y.; Odeh, H.M.; Gendron, T.F.; Jansen-West, K.; Del Rosso, G.; Yue, M.; Jiang, P.; Gomes, E.; Tong, J.; et al. C9orf72 poly(GR) aggregation induces TDP-43 proteinopathy. Sci. Transl. Med. 2020, 12, eabb3774. [Google Scholar] [CrossRef] [PubMed]
- Onesto, E.; Colombrita, C.; Gumina, V.; Borghi, M.O.; Dusi, S.; Doretti, A.; Fagiolari, G.; Invernizzi, F.; Moggio, M.; Tiranti, V.; et al. Gene-specific mitochondria dysfunctions in human TARDBP and C9ORF72 fibroblasts. Acta Neuropathol. Commun. 2016, 4, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Gonzalez, R.; Lu, Y.; Gendron, T.F.; Karydas, A.; Tran, H.; Yang, D.; Petrucelli, L.; Miller, B.L.; Almeida, S.; Gao, F.B. Poly(GR) in C9ORF72-Related ALS/FTD Compromises Mitochondrial Function and Increases Oxidative Stress and DNA Damage in iPSC-Derived Motor Neurons. Neuron 2016, 92, 383–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Wu, Z.; Tantray, I.; Li, Y.; Chen, S.; Dong, J.; Glynn, S.; Vogel, H.; Snyder, M.; Lu, B. Quality-control mechanisms targeting translationally stalled and C-terminally extended poly(GR) associated with ALS/FTD. Proc. Natl. Acad. Sci. USA 2020, 117, 25104–25115. [Google Scholar] [CrossRef] [PubMed]
- Dafinca, R.; Barbagallo, P.; Farrimond, L.; Candalija, A.; Scaber, J.; Ababneh, N.A.; Sathyaprakash, C.; Vowles, J.; Cowley, S.A.; Talbot, K. Impairment of Mitochondrial Calcium Buffering Links Mutations in C9ORF72 and TARDBP in iPS-Derived Motor Neurons from Patients with ALS/FTD. Stem Cell Rep. 2020, 14, 892–908. [Google Scholar] [CrossRef]
- Dafinca, R.; Barbagallo, P.; Talbot, K. The Role of Mitochondrial Dysfunction and ER Stress in TDP-43 and C9ORF72 ALS. Front. Cell. Neurosci. 2021, 15, 653688. [Google Scholar] [CrossRef]
- Mehta, A.R.; Gregory, J.M.; Dando, O.; Carter, R.N.; Burr, K.; Nanda, J.; Story, D.; McDade, K.; Smith, C.; Morton, N.M.; et al. Mitochondrial bioenergetic deficits in C9orf72 amyotrophic lateral sclerosis motor neurons cause dysfunctional axonal homeostasis. Acta Neuropathol. 2021, 141, 257–279. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Liu, H.; Itoh, K.; Oh, S.; Zhao, L.; Murata, D.; Sesaki, H.; Hartung, T.; Na, C.H.; Wang, J. C9orf72 regulates energy homeostasis by stabilizing mitochondrial complex I assembly. Cell. Metab. 2021, 33, 531–546.e9. [Google Scholar] [CrossRef]
- Choi, S.Y.; Lopez-Gonzalez, R.; Krishnan, G.; Phillips, H.L.; Li, A.N.; Seeley, W.W.; Yao, W.D.; Almeida, S.; Gao, F.B. C9ORF72-ALS/FTD-associated poly(GR) binds Atp5a1 and compromises mitochondrial function in vivo. Nat. Neurosci. 2019, 22, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Yang, B.; Zhang, D. Activation of interferon signaling pathways in spinal cord astrocytes from an ALS mouse model. Glia 2011, 59, 946–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swarup, V.; Phaneuf, D.; Dupré, N.; Petri, S.; Strong, M.; Kriz, J.; Julien, J.P. Deregulation of TDP-43 in amyotrophic lateral sclerosis triggers nuclear factor κB-mediated pathogenic pathways. J. Exp. Med. 2011, 208, 2429–2447. [Google Scholar] [CrossRef] [PubMed]
- Lucini, C.B.; Braun, R.J. Mitochondrion-Dependent Cell Death in TDP-43 Proteinopathies. Biomedicines 2021, 9, 376. [Google Scholar] [CrossRef]
- Yu, C.H.; Davidson, S.; Harapas, C.R.; Hilton, J.B.; Mlodzianoski, M.J.; Laohamonthonkul, P.; Louis, C.; Low, R.R.J.; Moecking, J.; De Nardo, D.; et al. TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell 2020, 183, 636–649.e18. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Sun, L.; Chen, Z.J. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 2016, 17, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef]
- Sliter, D.A.; Martinez, J.; Hao, L.; Chen, X.; Sun, N.; Fischer, T.D.; Burman, J.L.; Li, Y.; Zhang, Z.; Narendra, D.P.; et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature 2018, 561, 258–262. [Google Scholar] [CrossRef]
- Dormann, D.; Rodde, R.; Edbauer, D.; Bentmann, E.; Fischer, I.; Hruscha, A.; Than, M.E.; Mackenzie, I.R.; Capell, A.; Schmid, B.; et al. ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J. 2010, 29, 2841–2857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, Y.L.; Coady, T.H.; Lu, L.; Zheng, D.; Alland, I.; Tian, B.; Shneider, N.A.; Manley, J.L. ALS/FTD-associated protein FUS induces mitochondrial dysfunction by preferentially sequestering respiratory chain complex mRNAs. Genes Dev. 2020, 34, 785–805. [Google Scholar] [CrossRef] [PubMed]
- Ho, W.Y.; Agrawal, I.; Tyan, S.H.; Sanford, E.; Chang, W.T.; Lim, K.; Ong, J.; Tan, B.S.Y.; Moe, A.A.K.; Yu, R.; et al. Dysfunction in nonsense-mediated decay, protein homeostasis, mitochondrial function, and brain connectivity in ALS-FUS mice with cognitive deficits. Acta Neuropathol. Commun. 2021, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Bassot, A.; Giuliani, F.; Simmen, T. Amyotrophic Lateral Sclerosis (ALS): Stressed by Dysfunctional Mitochondria-Endoplasmic Reticulum Contacts (MERCs). Cells 2021, 10, 1789. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Simmen, T. Mechanistic Connections between Endoplasmic Reticulum (ER) Redox Control and Mitochondrial Metabolism. Cells 2019, 8, 1071. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, A.L.; Mitne-Neto, M.; Silva, H.C.; Richieri-Costa, A.; Middleton, S.; Cascio, D.; Kok, F.; Oliveira, J.R.; Gillingwater, T.; Webb, J.; et al. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am. J. Hum. Genet. 2004, 75, 822–831. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, P.; Guo, H.; Dreser, A.; Yamoah, A.; Sechi, A.; Jesse, C.M.; Katona, I.; Doukas, P.; Nikolin, S.; Ernst, S.; et al. Pathomechanisms of ALS8: Altered autophagy and defective RNA binding protein (RBP) homeostasis due to the VAPB P56S mutation. Cell Death Dis. 2021, 12, 466. [Google Scholar] [CrossRef]
- Gitcho, M.A.; Strider, J.; Carter, D.; Taylor-Reinwald, L.; Forman, M.S.; Goate, A.M.; Cairns, N.J. VCP mutations causing frontotemporal lobar degeneration disrupt localization of TDP-43 and induce cell death. J. Biol. Chem. 2009, 284, 12384–12398. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.O.; Mandrioli, J.; Benatar, M.; Abramzon, Y.; Van Deerlin, V.M.; Trojanowski, J.Q.; Gibbs, J.R.; Brunetti, M.; Gronka, S.; Wuu, J.; et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 2010, 68, 857–864. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.C.; Tresse, E.; Kolaitis, R.M.; Molliex, A.; Thomas, R.E.; Alami, N.H.; Wang, B.; Joshi, A.; Smith, R.B.; Ritson, G.P.; et al. VCP is essential for mitochondrial quality control by PINK1/Parkin and this function is impaired by VCP mutations. Neuron 2013, 78, 65–80, Erratum in 2013, 78, 403. [Google Scholar] [CrossRef] [Green Version]
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H.; et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010, 465, 223–226. [Google Scholar] [CrossRef]
- Moore, A.S.; Holzbaur, E.L. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc. Natl. Acad. Sci. USA 2016, 113, E3349–E3358. [Google Scholar] [CrossRef] [Green Version]
- Cirulli, E.T.; Lasseigne, B.N.; Petrovski, S.; Sapp, P.C.; Dion, P.A.; Leblond, C.S.; Couthouis, J.; Lu, Y.F.; Wang, Q.; Krueger, B.J.; et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 2015, 347, 1436–1441. [Google Scholar] [CrossRef] [Green Version]
- Freischmidt, A.; Wieland, T.; Richter, B.; Ruf, W.; Schaeffer, V.; Müller, K.; Marroquin, N.; Nordin, F.; Hübers, A.; Weydt, P.; et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 2015, 18, 631–636. [Google Scholar] [CrossRef]
- Wong, Y.C.; Holzbaur, E.L. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. USA 2014, 111, E4439–E4448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fecto, F.; Yan, J.; Vemula, S.P.; Liu, E.; Yang, Y.; Chen, W.; Zheng, J.G.; Shi, Y.; Siddique, N.; Arrat, H.; et al. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch. Neurol. 2011, 68, 1440–1446. [Google Scholar] [CrossRef] [PubMed]
- Layfield, R.; Hocking, L.J. SQSTM1 and Paget’s disease of bone. Calcif. Tissue Int. 2004, 75, 347–357. [Google Scholar] [CrossRef]
- Haack, T.B.; Ignatius, E.; Calvo-Garrido, J.; Iuso, A.; Isohanni, P.; Maffezzini, C.; Lönnqvist, T.; Suomalainen, A.; Gorza, M.; Kremer, L.S.; et al. Absence of the Autophagy Adaptor SQSTM1/p62 Causes Childhood-Onset Neurodegeneration with Ataxia, Dystonia, and Gaze Palsy. Am. J. Hum. Genet. 2016, 99, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Foster, A.D.; Rea, S.L. The role of sequestosome 1/p62 protein in amyotrophic lateral sclerosis and frontotemporal dementia pathogenesis. Neural Regen Res. 2020, 15, 2186–2194. [Google Scholar]
- Gautam, M.; Jara, J.H.; Sekerkova, G.; Yasvoina, M.V.; Martina, M.; Özdinler, P.H. Absence of alsin function leads to corticospinal motor neuron vulnerability via novel disease mechanisms. Hum. Mol. Genet. 2016, 25, 1074–1087. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Hentati, A.; Deng, H.X.; Dabbagh, O.; Sasaki, T.; Hirano, M.; Hung, W.Y.; Ouahchi, K.; Yan, J.; Azim, A.C.; et al. The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat. Genet. 2001, 29, 160–165. [Google Scholar] [CrossRef]
- Hadano, S.; Hand, C.K.; Osuga, H.; Yanagisawa, Y.; Otomo, A.; Devon, R.S.; Miyamoto, N.; Showguchi-Miyata, J.; Okada, Y.; Singaraja, R.; et al. A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2. Nat. Genet. 2001, 29, 166–173. [Google Scholar] [CrossRef]
- Al-Saif, A.; Al-Mohanna, F.; Bohlega, S. A mutation in sigma-1 receptor causes juvenile amyotrophic lateral sclerosis. Ann. Neurol. 2011, 70, 913–919. [Google Scholar] [CrossRef]
- Watanabe, S.; Ilieva, H.; Tamada, H.; Nomura, H.; Komine, O.; Endo, F.; Jin, S.; Mancias, P.; Kiyama, H.; Yamanaka, K. Mitochondria-associated membrane collapse is a common pathomechanism in SIGMAR1- and SOD1-linked ALS. EMBO Mol. Med. 2016, 8, 1421–1437. [Google Scholar] [CrossRef] [PubMed]
- Bannwarth, S.; Ait-El-Mkadem, S.; Chaussenot, A.; Genin, E.C.; Lacas-Gervais, S.; Fragaki, K.; Berg-Alonso, L.; Kageyama, Y.; Serre, V.; Moore, D.G.; et al. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 2014, 137 Pt 8, 2329–2345. [Google Scholar] [CrossRef] [Green Version]
- Straub, I.R.; Weraarpachai, W.; Shoubridge, E.A. Multi-OMICS study of a CHCHD10 variant causing ALS demonstrates metabolic rewiring and activation of endoplasmic reticulum and mitochondrial unfolded protein responses. Hum. Mol. Genet. 2021, 30, 687–705. [Google Scholar] [CrossRef] [PubMed]
- Lehmer, C.; Schludi, M.H.; Ransom, L.; Greiling, J.; Junghänel, M.; Exner, N.; Riemenschneider, H.; van der Zee, J.; Van Broeckhoven, C.; Weydt, P.; et al. A novel CHCHD10 mutation implicates a Mia40-dependent mitochondrial import deficit in ALS. EMBO Mol. Med. 2018, 10, e8558. [Google Scholar] [CrossRef]
- Woo, J.A.; Liu, T.; Trotter, C.; Fang, C.C.; De Narvaez, E.; LePochat, P.; Maslar, D.; Bukhari, A.; Zhao, X.; Deonarine, A.; et al. Loss of function CHCHD10 mutations in cytoplasmic TDP-43 accumulation and synaptic integrity. Nat. Commun. 2017, 8, 15558. [Google Scholar] [CrossRef]
- Brenner, D.; Yilmaz, R.; Müller, K.; Grehl, T.; Petri, S.; Meyer, T.; Grosskreutz, J.; Weydt, P.; Ruf, W.; Neuwirth, C.; et al. Hot-spot KIF5A mutations cause familial ALS. Brain 2018, 141, 688–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolas, A.; Kenna, K.P.; Renton, A.E.; Ticozzi, N.; Faghri, F.; Chia, R.; Dominov, J.A.; Kenna, B.J.; Nalls, M.A.; Keagle, P.; et al. Genome-wide Analyses Identify KIF5A as a Novel ALS Gene. Neuron 2018, 97, 1268–1283.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duis, J.; Dean, S.; Applegate, C.; Harper, A.; Xiao, R.; He, W.; Dollar, J.D.; Sun, L.R.; Waberski, M.B.; Crawford, T.O.; et al. KIF5A mutations cause an infantile onset phenotype including severe myoclonus with evidence of mitochondrial dysfunction. Ann. Neurol. 2016, 80, 633–637. [Google Scholar] [CrossRef] [Green Version]
- Konovalova, J.; Gerasymchuk, D.; Parkkinen, I.; Chmielarz, P.; Domanskyi, A. Interplay between MicroRNAs and Oxidative Stress in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 6055. [Google Scholar] [CrossRef] [Green Version]
- Aschrafi, A.; Kar, A.N.; Natera-Naranjo, O.; MacGibeny, M.A.; Gioio, A.E.; Kaplan, B.B. MicroRNA-338 regulates the axonal expression of multiple nuclear-encoded mitochondrial mRNAs encoding subunits of the oxidative phosphorylation machinery. Cell Mol. Life Sci. 2012, 69, 4017–4027. [Google Scholar] [CrossRef] [PubMed]
- Vaz, A.R.; Vizinha, D.; Morais, H.; Colaço, A.R.; Loch-Neckel, G.; Barbosa, M.; Brites, D. Overexpression of miR-124 in Motor Neurons Plays a Key Role in ALS Pathological Processes. Int. J. Mol. Sci. 2021, 22, 6128. [Google Scholar] [CrossRef] [PubMed]
- Yardeni, T.; Fine, R.; Joshi, Y.; Gradus-Pery, T.; Kozer, N.; Reichenstein, I.; Yanowski, E.; Nevo, S.; Weiss-Tishler, H.; Eisenberg-Bord, M.; et al. High content image analysis reveals function of miR-124 upstream of Vimentin in regulating motor neuron mitochondria. Sci. Rep. 2018, 8, 59. [Google Scholar] [CrossRef] [Green Version]
- Russell, A.P.; Wada, S.; Vergani, L.; Hock, M.B.; Lamon, S.; Léger, B.; Ushida, T.; Cartoni, R.; Wadley, G.D.; Hespel, P.; et al. Disruption of skeletal muscle mitochondrial network genes and miRNAs in amyotrophic lateral sclerosis. Neurobiol. Dis. 2013, 49, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Gagliardi, S.; Zucca, S.; Pandini, C.; Diamanti, L.; Bordoni, M.; Sproviero, D.; Arigoni, M.; Olivero, M.; Pansarasa, O.; Ceroni, M.; et al. Long non-coding and coding RNAs characterization in Peripheral Blood Mononuclear Cells and Spinal Cord from Amyotrophic Lateral Sclerosis patients. Sci. Rep. 2018, 8, 2378. [Google Scholar] [CrossRef]
- Harley, J.; Clarke, B.E.; Patani, R. The Interplay of RNA Binding Proteins, Oxidative Stress and Mitochondrial Dysfunction in ALS. Antioxidants 2021, 10, 552. [Google Scholar] [CrossRef] [PubMed]
- Marrone, L.; Drexler, H.C.A.; Wang, J.; Tripathi, P.; Distler, T.; Heisterkamp, P.; Anderson, E.N.; Kour, S.; Moraiti, A.; Maharana, S.; et al. FUS pathology in ALS is linked to alterations in multiple ALS-associated proteins and rescued by drugs stimulating autophagy. Acta Neuropathol. 2019, 138, 67–84. [Google Scholar] [CrossRef] [Green Version]
- Bordet, T.; Buisson, B.; Michaud, M.; Drouot, C.; Galéa, P.; Delaage, P.; Akentieva, N.P.; Evers, A.S.; Covey, D.F.; Ostuni, M.A.; et al. Identification andcharacterization of cholest-4-en-3-one, oxime (TRO19622), a novel drug candidate for amyotrophic lateral sclerosis. J. Pharmacol. Exp. Ther. 2007, 322, 709–720. [Google Scholar] [CrossRef] [Green Version]
- Sunyach, C.; Michaud, M.; Arnoux, T.; Bernard-Marissal, N.; Aebischer, J.; Latyszenok, V.; Gouarné, C.; Raoul, C.; Pruss, R.M.; Bordet, T.; et al. Olesoxime delays muscle denervation, astrogliosis, microglial activation and motoneuron death in an ALS mouse model. Neuropharmacology 2012, 62, 2346–2352. [Google Scholar] [CrossRef]
- Lenglet, T.; Lacomblez, L.; Abitbol, J.L.; Ludolph, A.; Mora, J.S.; Robberecht, W.; Shaw, P.J.; Pruss, R.M.; Cuvier, V.; Meininger, V.; et al. A phase II-III trial of olesoxime in subjects with amyotrophic lateral sclerosis. Eur. J. Neurol. 2014, 21, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Fancelli, D.; Wong, M.; Niedzwiecki, M.; Ballarini, M.; Plyte, S.; Chang, Q. GNX-4728, a novel small molecule drug inhibitor of mitochondrial permeability transition, is therapeutic in a mouse model of amyotrophic lateral sclerosis. Front. Cell. Neurosci. 2014, 8, 433. [Google Scholar] [CrossRef] [Green Version]
- Ono, Y.; Tanaka, H.; Takata, M.; Nagahara, Y.; Noda, Y.; Tsuruma, K.; Shimazawa, M.; Hozumi, I.; Hara, H. SA4503, a sigma-1 receptor agonist, suppresses motor neuron damage in in vitro and in vivo amyotrophic lateral sclerosis models. Neurosci. Lett. 2014, 559, 174–178. [Google Scholar] [CrossRef]
- Smith, R.; Pioro, E.; Myers, K.; Sirdofsky, M.; Goslin, K.; Meekins, G.; Yu, H.; Wymer, J.; Cudkowicz, M.; Macklin, E.A.; et al. Enhanced bulbar function in amyotrophic lateral sclerosis: The nuedexta treatment trial. Neurotherapeutics 2017, 14, 762–772. [Google Scholar] [CrossRef] [Green Version]
- Taylor, C.P.; Traynelis, S.F.; Siffert, J.; Pope, L.E.; Matsumoto, R.R. Pharmacology of dextromethorphan: Relevance to dextromethorphan/quinidine (Nuedexta®) clinical use. Pharmacol. Ther. 2016, 164, 170–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.; Li, M.; Li, J.C.; Wei, E.Q. Edaravone protects PC12 cells from ischemic-like injury via attenuating the damage to mitochondria. J. Zhejiang Univ. Sci. B 2006, 7, 749–756. [Google Scholar] [CrossRef] [Green Version]
- Writing Group; Edaravone (MCI-186) ALS 19 Study Group. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: A randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2017, 16, 505–512. [Google Scholar] [CrossRef]
- Cappella, M.; Ciotti, C.; Cohen-Tannoudji, M.; Biferi, M.G. Gene Therapy for ALS-A Perspective. Int. J. Mol. Sci. 2019, 20, 4388. [Google Scholar] [CrossRef] [Green Version]
- Cappella, M.; Pradat, P.F.; Querin, G.; Biferi, M.G. Beyond the Traditional Clinical Trials for Amyotrophic Lateral Sclerosis and The Future Impact of Gene Therapy. J. Neuromuscul. Dis. 2021, 8, 25–38. [Google Scholar] [CrossRef]
- McCampbell, A.; Cole, T.; Wegener, A.J.; Tomassy, G.S.; Setnicka, A.; Farley, B.J.; Schoch, K.M.; Hoye, M.L.; Shabsovich, M.; Sun, L.; et al. Antisense oligonucleotides extend survival and reverse decrement in muscle response in ALS models. J. Clin. Investig. 2018, 128, 3558–3567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, T.; Cudkowicz, M.; Shaw, P.J.; Andersen, P.M.; Atassi, N.; Bucelli, R.C.; Genge, A.; Glass, J.; Ladha, S.; Ludolph, A.L.; et al. Phase 1-2 Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N. Engl. J. Med. 2020, 383, 109–119. [Google Scholar] [CrossRef] [PubMed]
- NCT03626012. A Study to Assess the Safety, Tolerability, and Pharmacokinetics of BIIB078 in Adults With C9ORF72-Associated Amyotrophic Lateral Sclerosis. Available online: https://clinicaltrials.gov/ct2/show/NCT03626012 (accessed on 4 September 2021).
- Corrales, L.; Glickman, L.H.; McWhirter, S.M.; Kanne, D.B.; Sivick, K.E.; Katibah, G.E.; Woo, S.R.; Lemmens, E.; Banda, T.; Leong, J.J.; et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep. 2015, 11, 1018–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramanjulu, J.M.; Pesiridis, G.S.; Yang, J.; Concha, N.; Singhaus, R.; Zhang, S.Y.; Tran, J.L.; Moore, P.; Lehmann, S.; Eberl, H.C.; et al. Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature 2018, 564, 439–443. [Google Scholar] [CrossRef] [PubMed]
- Haag, S.M.; Gulen, M.F.; Reymond, L.; Gibelin, A.; Abrami, L.; Decout, A.; Heymann, M.; van der Goot, F.G.; Turcatti, G.; Behrendt, R.; et al. Targeting STING with covalent small-molecule inhibitors. Nature 2018, 559, 269–273. [Google Scholar] [CrossRef] [PubMed]



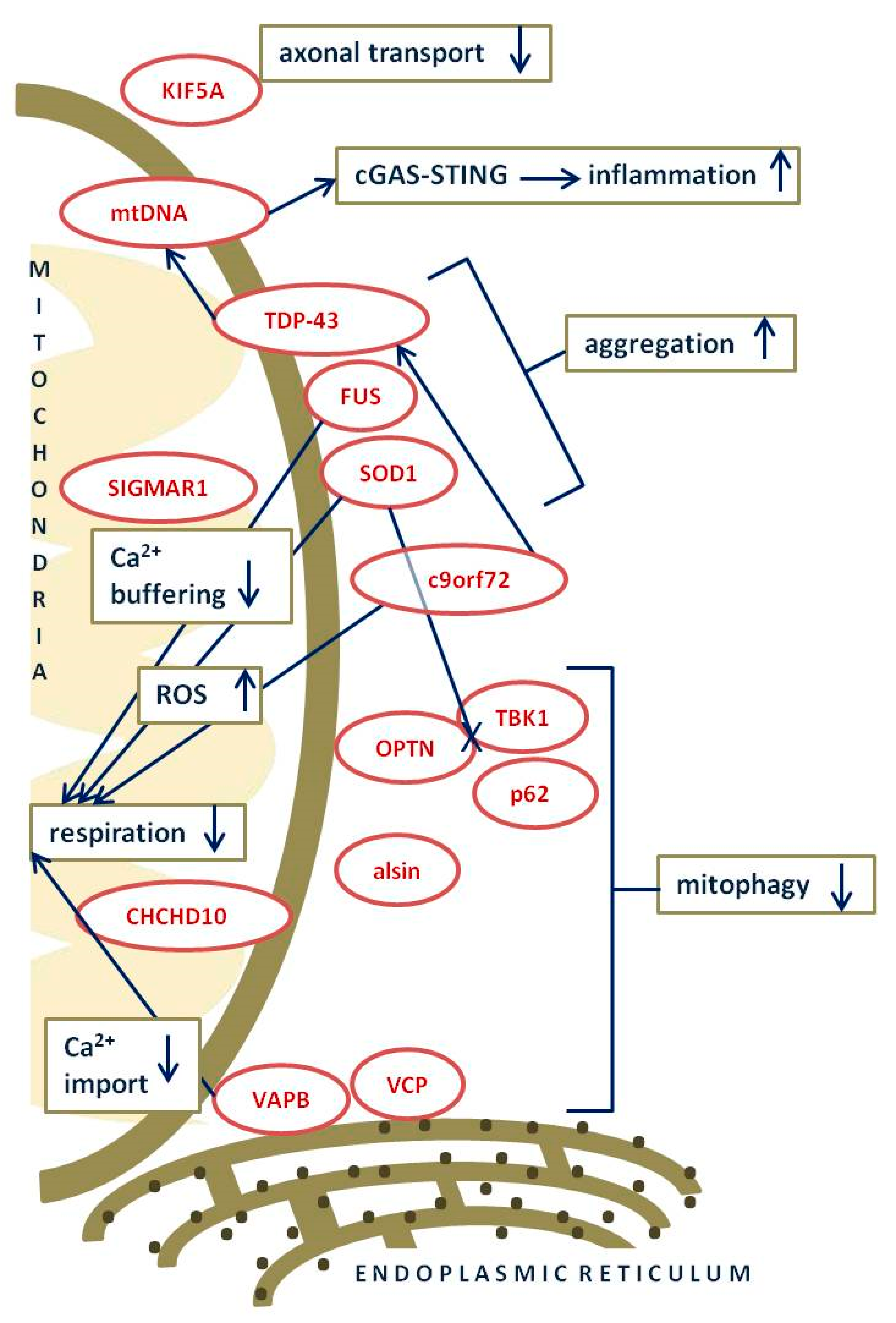{kind=link}
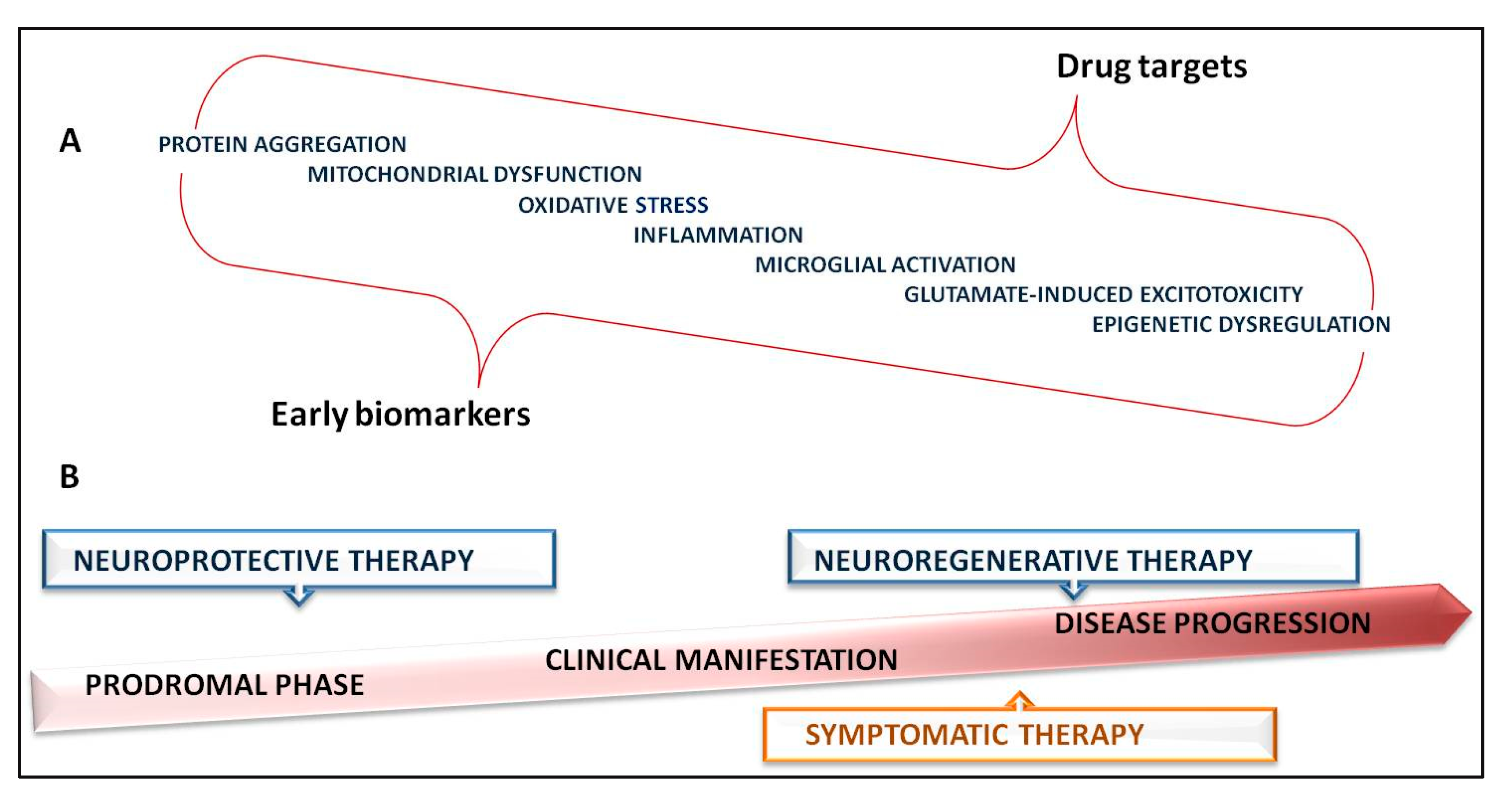{kind=link}
| Amyotrophic lateral sclerosis | References | |
|---|---|---|
| Epidemiological facts |
| [5,9,10,11,12] |
| Clinical presentation |
| [12,13,14,15] |
| Type |
| [11,15] |
| Diagnostics |
| [12,13,16] |
| Potential Drug | Structure | Target | Proposed Mechanism | Outcome | Ref. |
|---|---|---|---|---|---|
| Olesoxime | SM | Mitochondrial permeability | Inhibiting permeability of mPTP | Failure in 18-month survival clinical study (phase 2/3) | [101,102,103] |
| GNX-4728 | SM | Mitochondrial permeability | Modulating permeability mPTP | Efficient disease modifying in a preclinical ALS mouse model | [104] |
| Cutamesine (SA4503) | SM | sigma-1 receptor | Control of Ca2+ release from ER into mitochondria | Reducing oxidative stress and regulating calcium flux | [105] |
| Nuedexta (dextromethorphan and quinidine) | SM | sigma-1 receptor and P450 cytochrome | Control of Ca2+ release from ER into mitochondria | Improving bulbar physiology and function in ALS (approved and recruiting novel trials) | [106,107] |
| Edaravone | SM | ROS | free radical scavenger | Approved | [108,109] |
| Tofersen (BIIB067) | ASO | Mutant SOD1 | Reduces levels of mutant SOD1 | Ongoing clinical trials phase 1 | [113] |
| BIIBO78 | ASO | Mutant C9orf72 | Reduces levels of mutant C9orf72 | Clinical trial phase 1 | [114] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jankovic, M.; Novakovic, I.; Gamil Anwar Dawod, P.; Gamil Anwar Dawod, A.; Drinic, A.; Abdel Motaleb, F.I.; Ducic, S.; Nikolic, D. Current Concepts on Genetic Aspects of Mitochondrial Dysfunction in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2021, 22, 9832. https://doi.org/10.3390/ijms22189832
Jankovic M, Novakovic I, Gamil Anwar Dawod P, Gamil Anwar Dawod A, Drinic A, Abdel Motaleb FI, Ducic S, Nikolic D. Current Concepts on Genetic Aspects of Mitochondrial Dysfunction in Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences. 2021; 22(18):9832. https://doi.org/10.3390/ijms22189832
Chicago/Turabian StyleJankovic, Milena, Ivana Novakovic, Phepy Gamil Anwar Dawod, Ayman Gamil Anwar Dawod, Aleksandra Drinic, Fayda I. Abdel Motaleb, Sinisa Ducic, and Dejan Nikolic. 2021. "Current Concepts on Genetic Aspects of Mitochondrial Dysfunction in Amyotrophic Lateral Sclerosis" International Journal of Molecular Sciences 22, no. 18: 9832. https://doi.org/10.3390/ijms22189832