
The Disruption of the Endothelial Barrier Contributes to Acute Lung Injury Induced by Coxsackievirus A2 Infection in Mice

 , , and
, , and
Abstract
:1. Introduction
2. Results
2.1. CVA2 Infection Led to Acute Lung Injury in Mice
2.2. Histopathological Changes of Lung Tissues after CVA2 Infection
2.3. The Degradation of Tight Junction Proteins in Lung Tissues after CVA2 Infection
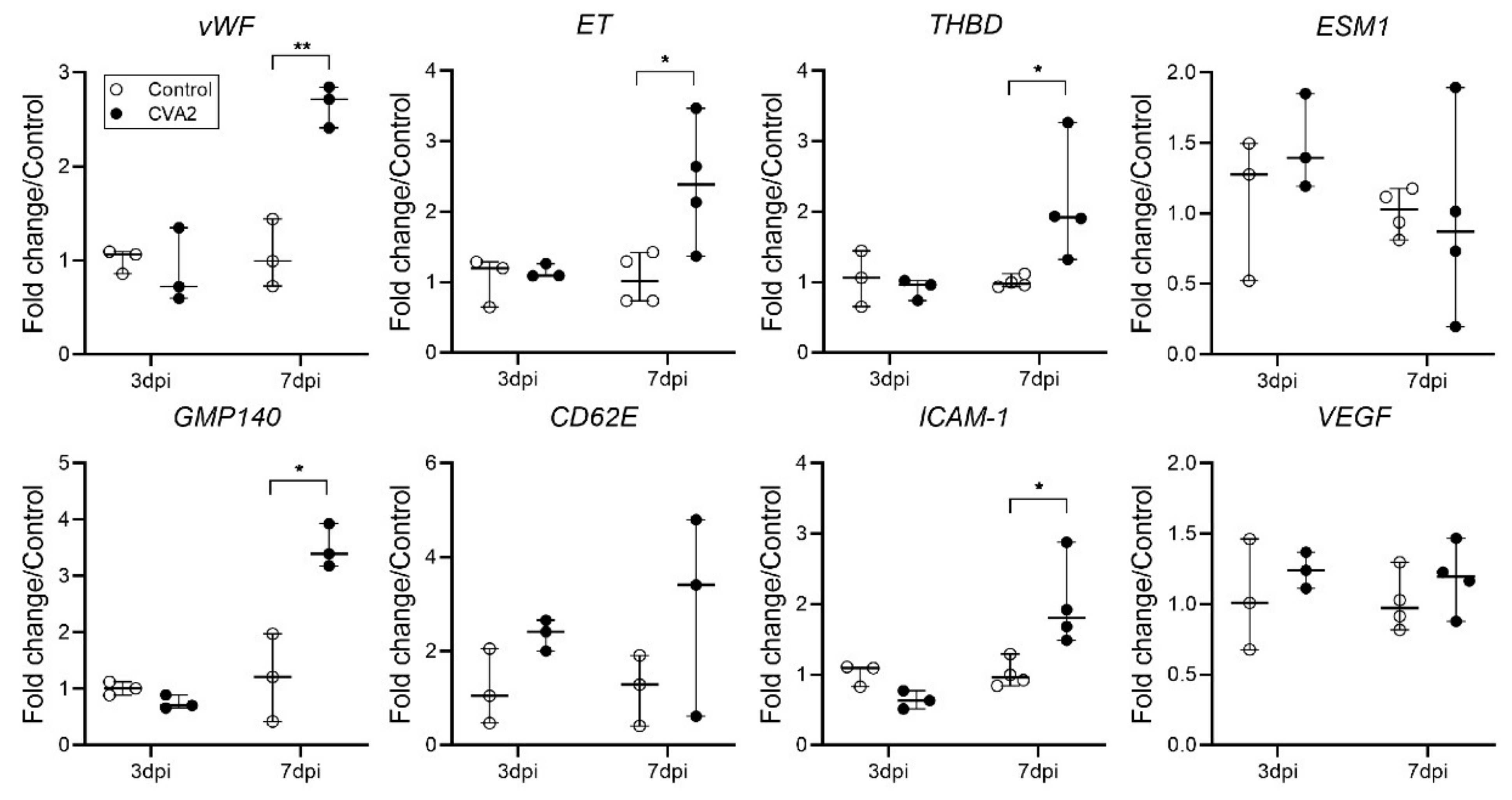
2.4. The Effect of CVA2 Infection on Pulmonary Endothelial Function
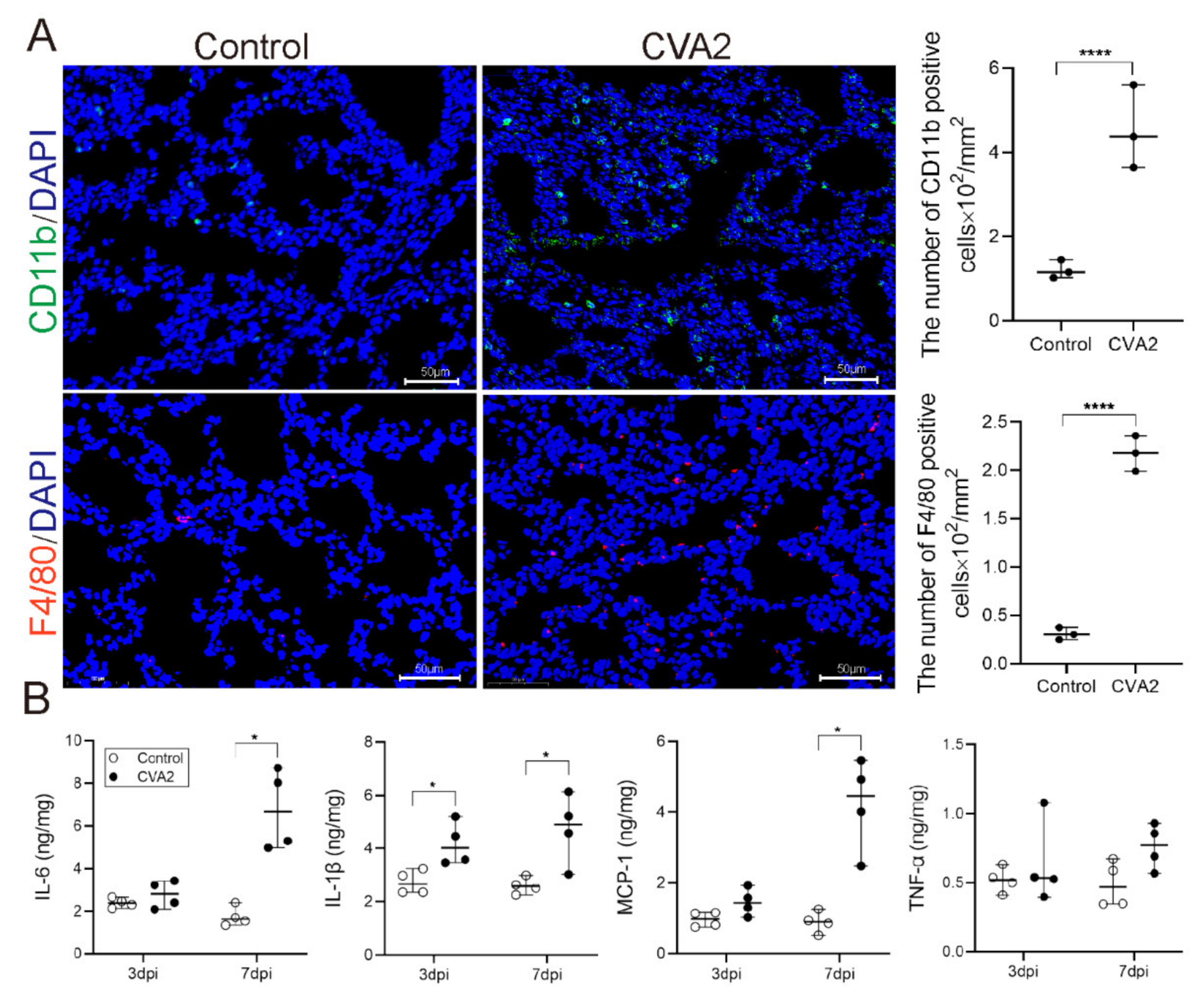
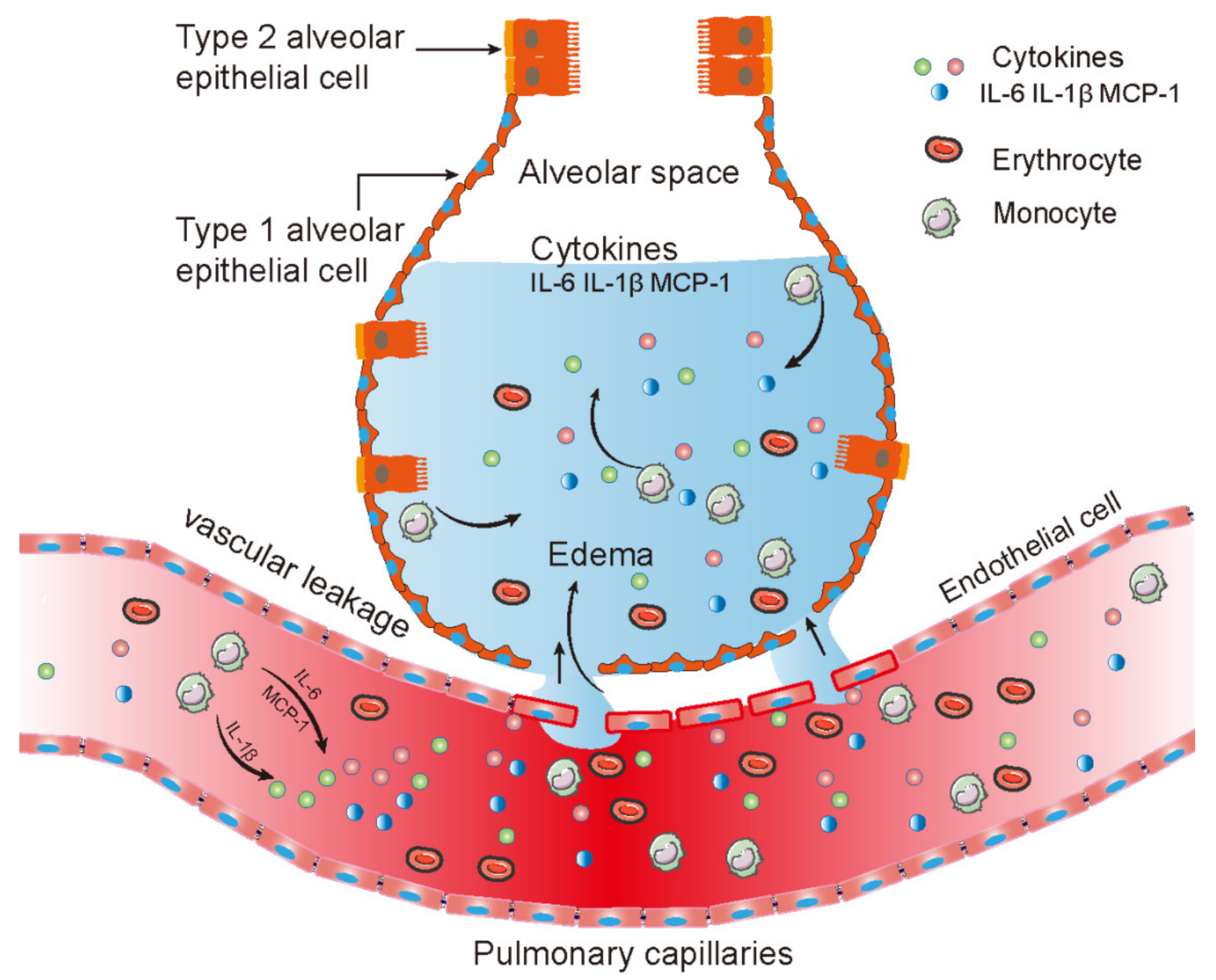
2.5. Monocytes Infiltration and Cytokines Expression Induced by CVA2 Infection
2.6. CVA2 Infection Activated p38-MAPK Signal Pathway in Lung Tissues
3. Discussion
4. Materials and Methods
4.1. Cells and Viruses
4.2. Mice and Infection
4.3. Detection of Pulmonary Vascular Permeability
4.4. Transmission Electron Microscopy
4.5. Histopathological and Immunohistochemical Analysis
4.6. Immunofluorescent Staining
4.7. Quantitative PCR
4.8. TUNEL Staining
4.9. Analysis of Tissue Lysates
4.10. Western Blotting
4.11. Antibodies
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ooi, M.H.; Wong, S.C.; Lewthwaite, P.; Cardosa, M.J.; Solomon, T. Clinical features, diagnosis, and management of enterovirus 71. Lancet Neurol. 2010, 9, 1097–1105. [Google Scholar] [CrossRef]
- Chen, L.; Xu, S.J.; Yao, X.J.; Yang, H.; Zhang, H.L.; Meng, J.; Zeng, H.R.; Huang, X.H.; Zhang, R.L.; He, Y.Q. Molecular epidemiology of enteroviruses associated with severe hand, foot and mouth disease in Shenzhen, China, 2014–2018. Arch. Virol. 2020, 165, 2213–2227. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Gu, X.; Zhang, Y.; Wei, H.; Li, Q.; Fan, H.; Xu, Y.; Li, J.; Tan, Z.; Song, Y.; et al. Persistent circulation of genotype D coxsackievirus A2 in mainland of China since 2008. PLoS ONE 2018, 13, e0204359. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.Y.; Chiang, P.S.; Luo, S.T.; Lin, T.Y.; Tsao, K.C.; Lee, M.S. A Molecular Approach Applied to Enteroviruses Surveillance in Northern Taiwan, 2008–2012. PLoS ONE 2016, 11, e0167532. [Google Scholar] [CrossRef]
- Chansaenroj, J.; Auphimai, C.; Puenpa, J.; Mauleekoonphairoj, J.; Wanlapakorn, N.; Vuthitanachot, V.; Vongpunsawad, S.; Poovorawan, Y. High prevalence of coxsackievirus A2 in children with herpangina in Thailand in 2015. Virusdisease 2017, 28, 111–114. [Google Scholar] [CrossRef] [Green Version]
- Molet, L.; Saloum, K.; Marque-Juillet, S.; Garbarg-Chenon, A.; Henquell, C.; Schuffenecker, I.; Peigue-Lafeuille, H.; Rozenberg, F.; Mirand, A. Enterovirus infections in hospitals of Ile de France region over 2013. J. Clin. Virol. Off. Publ. Pan Am. Soc. Clin. Virol. 2016, 74, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Sousa, I.P., Jr.; Oliveira, M.L.A.; Burlandy, F.M.; Machado, R.S.; Oliveira, S.S.; Tavares, F.N.; Gomes-Neto, F.; da Costa, E.V.; da Silva, E.E. Molecular characterization and epidemiological aspects of non-polio enteroviruses isolated from acute flaccid paralysis in Brazil: A historical series (2005-2017). Emerg. Microbes Infect. 2020, 9, 2536–2546. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Lee, B.; Baek, K.; Cheon, D.; Yeo, S.; Park, J.; Soh, J.; Cheon, H.; Yoon, K.; Choi, Y. Enteroviruses isolated from herpangina and hand-foot-and-mouth disease in Korean children. Virol. J. 2012, 9, 205. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Nicholls, J.M.; Liu, F.; Wang, J.; Feng, Z.; Liu, D.; Sun, Y.; Zhou, C.; Li, Y.; Li, H.; et al. Pulmonary and central nervous system pathology in fatal cases of hand foot and mouth disease caused by enterovirus A71 infection. Pathology 2016, 48, 267–274. [Google Scholar] [CrossRef]
- Lum, L.C.; Wong, K.T.; Lam, S.K.; Chua, K.B.; Goh, A.Y. Neurogenic pulmonary oedema and enterovirus 71 encephalomyelitis. Lancet 1998, 352, 1391. [Google Scholar] [CrossRef]
- Solomon, T.; Lewthwaite, P.; Perera, D.; Cardosa, M.J.; McMinn, P.; Ooi, M.H. Virology, epidemiology, pathogenesis, and control of enterovirus 71. Lancet Infect. Dis. 2010, 10, 778–790. [Google Scholar] [CrossRef]
- Wu, J.M.; Wang, J.N.; Tsai, Y.C.; Liu, C.C.; Huang, C.C.; Chen, Y.J.; Yeh, T.F. Cardiopulmonary manifestations of fulminant enterovirus 71 infection. Pediatrics 2002, 109, e26. [Google Scholar] [CrossRef] [Green Version]
- Lin, T.Y.; Hsia, S.H.; Huang, Y.C.; Wu, C.T.; Chang, L.Y. Proinflammatory cytokine reactions in enterovirus 71 infections of the central nervous system. Clin Infect. Dis. 2003, 36, 269–274. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.M.; Lei, H.Y.; Huang, K.J.; Wu, J.M.; Wang, J.R.; Yu, C.K.; Su, I.J.; Liu, C.C. Pathogenesis of enterovirus 71 brainstem encephalitis in pediatric patients: Roles of cytokines and cellular immune activation in patients with pulmonary edema. J Infect. Dis. 2003, 188, 564–570. [Google Scholar] [CrossRef] [Green Version]
- Duan, G.; Yang, H.; Shi, L.; Sun, W.; Sui, M.; Zhang, R.; Wang, X.; Wang, F.; Zhang, W.; Xi, Y.; et al. Serum inflammatory cytokine levels correlate with hand-foot-mouth disease severity: A nested serial case-control study. PLoS ONE 2014, 9, e112676. [Google Scholar] [CrossRef]
- Wang, S.M.; Lei, H.Y.; Huang, M.C.; Su, L.Y.; Lin, H.C.; Yu, C.K.; Wang, J.L.; Liu, C.C. Modulation of cytokine production by intravenous immunoglobulin in patients with enterovirus 71-associated brainstem encephalitis. J. Clin. Virol. 2006, 37, 47–52. [Google Scholar] [CrossRef]
- Chanthick, C.; Suttitheptumrong, A.; Rawarak, N.; Pattanakitsakul, S.N. Transcytosis Involvement in Transport System and Endothelial Permeability of Vascular Leakage during Dengue Virus Infection. Viruses 2018, 10, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dejana, E.; Tournier-Lasserve, E.; Weinstein, B.M. The control of vascular integrity by endothelial cell junctions: Molecular basis and pathological implications. Dev. Cell. 2009, 16, 209–221. [Google Scholar] [CrossRef] [Green Version]
- Schneeberger, E.E. Structure of intercellular junctions in different segments of the intrapulmonary vasculature. Ann. N. Y. Acad. Sci. 1982, 384, 54–63. [Google Scholar] [CrossRef]
- Schnittler, H.J. Structural and functional aspects of intercellular junctions in vascular endothelium. Basic Res. Cardiol. 1998, 93 (Suppl. S3), 30–39. [Google Scholar] [CrossRef]
- Jin, Y.; Ji, W.; Yang, H.; Chen, S.; Zhang, W.; Duan, G. Endothelial activation and dysfunction in COVID-19: From basic mechanisms to potential therapeutic approaches. Signal. Transduct. Target. Ther. 2020, 5, 293. [Google Scholar] [CrossRef]
- Hu, Y.; Song, J.; Liu, L.; Zhang, Y.; Wang, L.; Li, Q. microRNA-4516 Contributes to Different Functions of Epithelial Permeability Barrier by Targeting Poliovirus Receptor Related Protein 1 in Enterovirus 71 and Coxsackievirus A16 Infections. Front. Cell Infect. Microbiol. 2018, 8, 110. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Hu, Y.; Li, H.; Huang, X.; Zheng, H.; Hu, Y.; Wang, J.; Jiang, X.; Li, J.; Yang, Z.; et al. miR-1303 regulates BBB permeability and promotes CNS lesions following CA16 infections by directly targeting MMP9. Emerg. Microbes Infect. 2018, 7, 155. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Yang, X.; Sun, J.; Sun, Z.; Ma, Q.; Wang, Z.; Chen, Z.; Wang, Z.; Hu, F.; Wang, H.; et al. Neutrophil extracellular traps induced by VP1 contribute to pulmonary edema during EV71 infection. Cell Death Discov. 2019, 5, 111. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Sun, J.; Wang, N.; Sun, Z.; Ma, Q.; Li, J.; Zhang, M.; Xu, J. Enterovirus A71 capsid protein VP1 increases blood-brain barrier permeability and virus receptor vimentin on the brain endothelial cells. J. Neurovirol. 2020, 26, 84–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yip, C.C.; Lau, S.K.; Woo, P.C.; Wong, S.S.; Tsang, T.H.; Lo, J.Y.; Lam, W.K.; Tsang, C.C.; Chan, K.H.; Yuen, K.Y. Recombinant coxsackievirus A2 and deaths of children, Hong Kong, 2012. Emerg. Infect Dis. 2013, 19, 1285–1288. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Nesbitt, W.S.; Kulkarni, S. Signaling events underlying thrombus formation. J. Thromb. Haemost. JTH 2003, 1, 1602–1612. [Google Scholar] [CrossRef]
- Kawecki, C.; Lenting, P.J.; Denis, C.V. von Willebrand factor and inflammation. J. Thromb. Haemost. JTH 2017, 15, 1285–1294. [Google Scholar] [CrossRef]
- Szmitko, P.E.; Wang, C.H.; Weisel, R.D.; de Almeida, J.R.; Anderson, T.J.; Verma, S. New markers of inflammation and endothelial cell activation: Part I. Circulation 2003, 108, 1917–1923. [Google Scholar] [CrossRef]
- Kozuka, K.; Kohriyama, T.; Nomura, E.; Ikeda, J.; Kajikawa, H.; Nakamura, S. Endothelial markers and adhesion molecules in acute ischemic stroke--sequential change and differences in stroke subtype. Atherosclerosis 2002, 161, 161–168. [Google Scholar] [CrossRef]
- Mehta, D.; Malik, A.B. Signaling mechanisms regulating endothelial permeability. Physiol. Rev. 2006, 86, 279–367. [Google Scholar] [CrossRef]
- Bazzoni, G.; Dejana, E. Endothelial cell-to-cell junctions: Molecular organization and role in vascular homeostasis. Physiol. Rev. 2004, 84, 869–901. [Google Scholar] [CrossRef] [Green Version]
- Dejana, E.; Giampietro, C. Vascular endothelial-cadherin and vascular stability. Curr. Opin. Hematol. 2012, 19, 218–223. [Google Scholar] [CrossRef]
- Taddei, A.; Giampietro, C.; Conti, A.; Orsenigo, F.; Breviario, F.; Pirazzoli, V.; Potente, M.; Daly, C.; Dimmeler, S.; Dejana, E. Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat. Cell Biol. 2008, 10, 923–934. [Google Scholar] [CrossRef]
- Morita, K.; Sasaki, H.; Furuse, M.; Tsukita, S. Endothelial claudin: Claudin-5/TMVCF constitutes tight junction strands in endothelial cells. J. Cell Biol. 1999, 147, 185–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Günzel, D.; Yu, A.S. Claudins and the modulation of tight junction permeability. Physiol. Rev. 2013, 93, 525–569. [Google Scholar] [CrossRef] [Green Version]
- Fanning, A.S.; Anderson, J.M. Zonula occludens-1 and -2 are cytosolic scaffolds that regulate the assembly of cellular junctions. Ann. N. Y. Acad. Sci. 2009, 1165, 113–120. [Google Scholar] [CrossRef] [Green Version]
- Tsukita, S.; Yamazaki, Y.; Katsuno, T.; Tamura, A.; Tsukita, S. Tight junction-based epithelial microenvironment and cell proliferation. Oncogene 2008, 27, 6930–6938. [Google Scholar] [CrossRef] [Green Version]
- Blasig, I.E.; Haseloff, R.F. Tight junctions and tissue barriers. Antioxid. Redox Signal. 2011, 15, 1163–1166. [Google Scholar] [CrossRef]
- Van Itallie, C.M.; Anderson, J.M. Architecture of tight junctions and principles of molecular composition. Semin. Cell Dev. Biol. 2014, 36, 157–165. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, S.M.; Wang, C.; Tigdi, J.; Si, X.; Dumpit, C.; Charles, S.; Gamage, A.; Moraes, T.J.; Lee, W.L. Influenza infects lung microvascular endothelium leading to microvascular leak: Role of apoptosis and claudin-5. PLoS ONE 2012, 7, e47323. [Google Scholar]
- Cui, N.; Hu, M.; Khalil, R.A. Biochemical and Biological Attributes of Matrix Metalloproteinases. Prog. Mol. Biol. Transl. Sci. 2017, 147, 1–73. [Google Scholar] [PubMed] [Green Version]
- Yang, Y.; Estrada, E.Y.; Thompson, J.F.; Liu, W.; Rosenberg, G.A. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2007, 27, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Cen, J.; Huang, Y.; Shen, H.; Yao, L.; Wang, Y.; Chen, Z. Matrix metalloproteinase-2 and -9 secreted by leukemic cells increase the permeability of blood-brain barrier by disrupting tight junction proteins. PLoS ONE 2011, 6, e20599. [Google Scholar] [CrossRef]
- Liang, Z.; Pan, H.; Wang, X.; Zhu, Y.; Dang, Y.; Fan, X.; Gao, L.; Zhang, Z. Histopathological Features and Viral Antigen Distribution in the Lung of Fatal Patients with Enterovirus 71 Infection. VirologicaSinica 2018, 33, 278–281. [Google Scholar] [CrossRef] [Green Version]
- Müller-Redetzky, H.C.; Suttorp, N.; Witzenrath, M. Dynamics of pulmonary endothelial barrier function in acute inflammation: Mechanisms and therapeutic perspectives. Cell Tissue Res. 2014, 355, 657–673. [Google Scholar] [CrossRef]
- Libby, P.; Lüscher, T. COVID-19 is, in the end, an endothelial disease. Eur. Heart J. 2020, 41, 3038–3044. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, M.; Moochhala, S. Role of inflammatory mediators in the pathophysiology of acute respiratory distress syndrome. J. Pathol. 2004, 202, 145–156. [Google Scholar] [CrossRef]
- Huang, X.; Hussain, B.; Chang, J. Peripheral inflammation and blood-brain barrier disruption: Effects and mechanisms. CNS Neurosci. Ther. 2021, 27, 36–47. [Google Scholar] [CrossRef]
- Zhang, W.; Huang, Z.; Huang, M.; Zeng, J. Predicting Severe Enterovirus 71-Infected Hand, Foot, and Mouth Disease: Cytokines and Chemokines. Mediat. Inflamm. 2020, 2020, 9273241. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.F.; Li, H.L.; Sun, B.X. Correlation analysis on serum inflammatory cytokine level and neurogenic pulmonary edema for children with severe hand-foot-mouth disease. Eur. J. Med. Res. 2018, 23, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Li, R.; Xie, Z.; Huang, G.; Yuan, Q.; Zeng, J. IL-6, IL-10 and IL-13 are associated with pathogenesis in children with Enterovirus 71 infection. Int. J. Clin. Exp. Med. 2014, 7, 2718–2723. [Google Scholar]
- Cerutti, C.; Ridley, A.J. Endothelial cell-cell adhesion and signaling. Exp. Cell Res. 2017, 358, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Wettschureck, N.; Strilic, B.; Offermanns, S. Passing the Vascular Barrier: Endothelial Signaling Processes Controlling Extravasation. Physiol. Rev. 2019, 99, 1467–1525. [Google Scholar] [CrossRef]
- Geissmann, F.; Manz, M.G.; Jung, S.; Sieweke, M.H.; Merad, M.; Ley, K. Development of monocytes, macrophages, and dendritic cells. Science 2010, 327, 656–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Xiu, H.; Zhang, S.; Zhang, G. The Role of Macrophages in the Pathogenesis of ALI/ARDS. Mediat. Inflamm. 2018, 2018, 1264913. [Google Scholar] [CrossRef] [PubMed]
- Johnston, L.K.; Rims, C.R.; Gill, S.E.; McGuire, J.K.; Manicone, A.M. Pulmonary macrophage subpopulations in the induction and resolution of acute lung injury. Am. J. Respir. Cell Mol. Biol. 2012, 47, 417–426. [Google Scholar] [CrossRef]
- Kevil, C.G.; Patel, R.P.; Bullard, D.C. Essential role of ICAM-1 in mediating monocyte adhesion to aortic endothelial cells. Am. J. Physiol. Cell Physiol. 2001, 281, C1442–C1447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, X.Q.; Chen, X.Q.; Gao, Y.; Fu, M.; Chen, Y.P.; Xu, D.P.; Lin, A.; Yan, W.H. Elevation of human leukocyte antigen-G expression is associated with the severe encephalitis associated with neurogenic pulmonary edema caused by Enterovirus 71. Clin. Exp. Med. 2014, 14, 161–167. [Google Scholar] [CrossRef]
- Guan, Z.; Buckman, S.Y.; Pentland, A.P.; Templeton, D.J.; Morrison, A.R. Induction of cyclooxygenase-2 by the activated MEKK1 --> SEK1/MKK4 --> p38 mitogen-activated protein kinase pathway. J. Biol. Chem. 1998, 273, 12901–12908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yong, H.Y.; Koh, M.S.; Moon, A. The p38 MAPK inhibitors for the treatment of inflammatory diseases and cancer. Expert Opin. Investig. Drugs. 2009, 18, 1893–1905. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Han, J. The p38 signal transduction pathway: Activation and function. Cell. Signal. 2000, 12, 1–13. [Google Scholar] [CrossRef]
- Song, H.; Ki, S.H.; Kim, S.G.; Moon, A. Activating transcription factor 2 mediates matrix metalloproteinase-2 transcriptional activation induced by p38 in breast epithelial cells. Cancer Res. 2006, 66, 10487–10496. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Hu, J.; He, T.; Zhang, Q.; Yang, X.; Lan, X.; Zhang, D.; Mei, H.; Chen, B.; Huang, Y. P38/MAPK contributes to endothelial barrier dysfunction via MAP4 phosphorylation-dependent microtubule disassembly in inflammation-induced acute lung injury. Sci. Rep. 2015, 5, 8895. [Google Scholar] [CrossRef] [Green Version]
- Ju, H.; Behm, D.J.; Nerurkar, S.; Eybye, M.E.; Haimbach, R.E.; Olzinski, A.R.; Douglas, S.A.; Willette, R.N. p38 MAPK inhibitors ameliorate target organ damage in hypertension: Part 1. p38 MAPK-dependent endothelial dysfunction and hypertension. J. Pharmacol. Exp. Ther. 2003, 307, 932–938. [Google Scholar] [CrossRef] [Green Version]
- He, T.; Zhao, L.; Zhang, D.; Zhang, Q.; Jia, J.; Hu, J.; Huang, Y. Pigment Epithelium-Derived Factor Induces Endothelial Barrier Dysfunction via p38/MAPK Phosphorylation. BioMed Res. Int. 2015, 2015, 791825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widder, J.; Behr, T.; Fraccarollo, D.; Hu, K.; Galuppo, P.; Tas, P.; Angermann, C.E.; Ertl, G.; Bauersachs, J. Vascular endothelial dysfunction and superoxide anion production in heart failure are p38 MAP kinase-dependent. Cardiovasc. Res. 2004, 63, 161–167. [Google Scholar] [CrossRef] [Green Version]
- Weerackody, R.P.; Welsh, D.J.; Wadsworth, R.M.; Peacock, A.J. Inhibition of p38 MAPK reverses hypoxia-induced pulmonary artery endothelial dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1312–H1320. [Google Scholar] [CrossRef] [Green Version]
- Mu, S.; Liu, Y.; Jiang, J.; Ding, R.; Li, X.; Li, X. Unfractionated heparin ameliorates pulmonary microvascular endothelial barrier dysfunction via microtubule stabilization in acute lung injury. Respir. Res. 2018, 19, 220. [Google Scholar] [CrossRef]
- Leng, B.; Li, C.; Sun, Y.; Zhao, K.; Zhang, L.; Lu, M.L.; Wang, H.X. Protective Effect of Astragaloside IV on High Glucose-Induced Endothelial Dysfunction via Inhibition of P2X7R Dependent P38 MAPK Signaling Pathway. Oxidative Med. Cell. Longev. 2020, 2020, 5070415. [Google Scholar] [CrossRef]
- Reed, L.J.M.H. A simple method of estimating 50 percent end-points. Am. J. Epidemiol. 1938, 27, 493–497. [Google Scholar] [CrossRef]
- Ji, W.; Qin, L.; Tao, L.; Zhu, P.; Liang, R.; Zhou, G.; Chen, S.; Zhang, W.; Yang, H.; Duan, G.; et al. Neonatal Murine Model of Coxsackievirus A2 Infection for the Evaluation of Antiviral Therapeutics and Vaccination. Front. Microbiol. 2021, 12, 658093. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zhao, Y.Y. Transgenic expression of FoxM1 promotes endothelial repair following lung injury induced by polymicrobial sepsis in mice. PLoS ONE 2012, 7, e50094. [Google Scholar] [CrossRef] [Green Version]
- Inagawa, R.; Okada, H.; Takemura, G.; Suzuki, K.; Takada, C.; Yano, H.; Ando, Y.; Usui, T.; Hotta, Y.; Miyazaki, N.; et al. Ultrastructural Alteration of Pulmonary Capillary Endothelial Glycocalyx During Endotoxemia. Chest 2018, 154, 317–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Dong, Z.; Li, J.; Carr, M.J.; Zhuang, D.; Wang, J.; Zhang, Y.; Ding, S.; Tong, Y.; Li, D.; et al. Protective Efficacies of Formaldehyde-Inactivated Whole-Virus Vaccine and Antivirals in a Murine Model of Coxsackievirus A10 Infection. J. Virol. 2017, 91, e00333-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Y.; Sun, T.; Zhou, G.; Li, D.; Chen, S.; Zhang, W.; Li, X.; Zhang, R.; Yang, H.; Duan, G. Pathogenesis Study of Enterovirus 71 Using a Novel Human SCARB2 Knock-In Mouse Model. Msphere 2021, 6, e01048-20. [Google Scholar] [CrossRef] [PubMed]
- Liou, A.T.; Wu, S.Y.; Liao, C.C.; Chang, Y.S.; Chang, C.S.; Shih, C. A new animal model containing human SCARB2 and lacking stat-1 is highly susceptible to EV71. Sci. Rep. 2016, 6, 31151. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward | Reverse | Product Lengths (bp) |
|---|---|---|---|
| CVA2-VP1 | TCAGTCCCATTCATGTCGCC | AATGCGTTGTTGGGGCATTG | 118 |
| ESM1 | CAAGTCTCTTTGCATTCCATCC | GGCTGAAGTGTCACTTTTACAG | 106 |
| VEGF | TAGAGTACATCTTCAAGCCGTC | CTTTCTTTGGTCTGCATTCACA | 198 |
| ICAM-1 | CTGAAAGATGAGCTCGAGAGTG | AAACGAATACACGGTGATGGTA | 141 |
| vWF | GTGATTTTAACATCTTCGCGGA | CAGGAGTTGGCAAAATCATAGG | 85 |
| THBD | GCTGTGAGTACTTGTGCAATAG | TCACACATACAGGAGTAAGAGC | 181 |
| ET | TTTTCCCGTGATCTTCTCTCTG | CAGAAGTAGACACACTCCTTGT | 192 |
| GMP140 | TGGGAGCAAGTGTGATAAGATG | GAACTGGCATGTGGATTTGTAG | 161 |
| CD62E | ACATTCACCGAGTTACTACTGG | GAGCCAGCTTCTTTTTGTTACA | 218 |
| ZO-1 | CTGGTGAAGTCTCGGAAAAATG | CATCTCTTGCTGCCAAACTATC | 97 |
| Occludin | TGCTTCATCGCTTCCTTAGTAA | GGGTTCACTCCCATTATGTACA | 155 |
| Claudin-5 | GTGGCACTCTTTGTTACCTTG | GATCATAGAACTCGCGGACAA | 172 |
| Mouse β-actin | GTGCTATGTTGCTCTAGACTTCG | ATGCCACAGGATTCCATACC | 174 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, W.; Hu, Q.; Zhang, M.; Zhang, C.; Chen, C.; Yan, Y.; Zhang, X.; Chen, S.; Tao, L.; Zhang, W.; et al. The Disruption of the Endothelial Barrier Contributes to Acute Lung Injury Induced by Coxsackievirus A2 Infection in Mice. Int. J. Mol. Sci. 2021, 22, 9895. https://doi.org/10.3390/ijms22189895
Ji W, Hu Q, Zhang M, Zhang C, Chen C, Yan Y, Zhang X, Chen S, Tao L, Zhang W, et al. The Disruption of the Endothelial Barrier Contributes to Acute Lung Injury Induced by Coxsackievirus A2 Infection in Mice. International Journal of Molecular Sciences. 2021; 22(18):9895. https://doi.org/10.3390/ijms22189895
Chicago/Turabian StyleJi, Wangquan, Qiang Hu, Mengdi Zhang, Chuwen Zhang, Chen Chen, Yujie Yan, Xue Zhang, Shuaiyin Chen, Ling Tao, Weiguo Zhang, and et al. 2021. "The Disruption of the Endothelial Barrier Contributes to Acute Lung Injury Induced by Coxsackievirus A2 Infection in Mice" International Journal of Molecular Sciences 22, no. 18: 9895. https://doi.org/10.3390/ijms22189895