
Insulin Resistance and Cancer: In Search for a Causal Link


 , , , ,
, , , ,  and
and 
Abstract
:1. Introduction
2. Insulin Receptor Signaling and Cancer
3. Diabetes and Cancer
4. Adipose Tissue, Obesity, Inflammation, and Cancer
5. Epigenetic Modifications
6. Gut Microbiota and Cancer
7. Antidiabetic Medications as Potential Anticancer Agents
7.1. Metformin
7.2. Thiazolidinediones
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Reaven, G.M. Banting Lecture. Role of insulin resistance in human disease. Diabetes. 1988, 37, 1595–1607. [Google Scholar] [CrossRef] [PubMed]
- Defronzo, R.A. Banting Lecture. From the triumvirate to the ominous octet: A new paradigm for the treatment of type 2 diabetes mellitus. Diabetes. 2009, 58, 773–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alwahsh, S.M.; Ramadori, G. How Does Bariatric Surgery Improve Type II Diabetes? The ‘‘Neglected’’ Importance of the Liver in Clearing Glucose and Insulin from the Portal Blood. J. Obes. Weight Loss Ther. 2015, 5, 280. [Google Scholar] [CrossRef] [Green Version]
- Alwahsh, S.M.; Dwyer, B.J.; Forbes, S.; Thiel, D.H.; Lewis, P.J.; Ramadori, G. Insulin Production and Resistance in Different Models of Diet-Induced Obesity and Metabolic Syndrome. Int. J. Mol. Sci. 2017, 18, 285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberti, K.G.; Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z.; Cleeman, J.I.; Donato, K.A.; Fruchart, J.C.; James, W.P.; Loria, C.M.; Smith, S.C., Jr. International Diabetes Federation Task Force on Epidemiology and Prevention. Harmonizing the metabolic syndrome: A joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 2009, 120, 640–1645. [Google Scholar] [CrossRef] [Green Version]
- Brunetti, A.; Chiefari, E.; Foti, D. Recent advances in the molecular genetics of type 2 diabetes mellitus. World J Diabetes. 2014, 5, 128–140. [Google Scholar] [CrossRef] [PubMed]
- Bansal, D.; Bhansali, A.; Kapil, G.; Undela, K.; Tiwari, P. Type 2 diabetes and risk of prostate cancer: A meta-analysis of observational studies. Prostate Cancer Prostatic Dis. 2013, 16, 151–158. [Google Scholar] [CrossRef]
- Orgel, E.; Mittelman, S.D. The links between insulin resistance, diabetes, and cancer. Curr. Diabates Rep. 2013, 13, 213–222. [Google Scholar] [CrossRef] [Green Version]
- Fathi Dizaji, B. The investigations of genetic determinants of the metabolic syndrome. Diabetes Metab. Syndr. 2018, 12, 783–789. [Google Scholar] [CrossRef]
- Mirabelli, M.; Chiefari, E.; Arcidiacono, B.; Corigliano, D.M.; Brunetti, F.S.; Maggisano, V.; Russo, D.; Foti, D.P.; Brunetti, A. Mediterranean Diet Nutrients to Turn the Tide against Insulin Resistance and Related Diseases. Nutrients 2020, 12, 1066. [Google Scholar] [CrossRef] [Green Version]
- Greco, M.; Chiefari, E.; Montalcini, T.; Accattato, F.; Costanzo, F.S.; Pujia, A.; Foti, D.; Brunetti, A.; Gulletta, E. Early effects of a hypocaloric, Mediterranean diet on laboratory parameters in obese individuals. Mediat. Inflamm. 2014, 2014, 750860. [Google Scholar] [CrossRef] [Green Version]
- Schenk, S.; Harber, M.P.; Shrivastava, C.R.; Burant, C.F.; Horowitz, J.F. Improved insulin sensitivity after weight loss and exercise training is mediated by a reduction in plasma fatty acid mobilization, not enhanced oxidative capacity. J. Physiol. 2009, 587, 4949–4961. [Google Scholar] [CrossRef]
- Greco, A.V.; Mingrone, G.; Giancaterini, A.; Manco, M.; Morroni, M.; Cinti, S.; Granzotto, M.; Vettor, R.; Camastra, S.; Ferrannini, E. Insulin resistance in morbid obesity: Reversal with intramyocellular fat depletion. Diabetes 2002, 51, 144–151. [Google Scholar] [CrossRef] [Green Version]
- Mari, A.; Manco, M.; Guidone, C.; Nanni, G.; Castagneto, M.; Mingrone, G.; Ferrannini, E. Restoration of normal glucose tolerance in severely obese patients after bilio-pancreatic diversion: Role of insulin sensitivity and beta cell function. Diabetologia 2006, 49, 2136–2143. [Google Scholar] [CrossRef] [Green Version]
- Rector, R.S.; Warner, S.O.; Liu, Y.; Hinton, P.S.; Sun, G.Y.; Cox, R.H.; Stump, C.S.; Laughlin, M.H.; Dellsperger, K.C.; Thomas, T.R. Exercise and diet induced weight loss improves measures of oxidative stress and insulin sensitivity in adults with characteristics of the metabolic syndrome. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E500–E506. [Google Scholar] [CrossRef] [Green Version]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef] [Green Version]
- Goldfine, I.D.; Kahn, C.R.; Neville, D.M., Jr.; Roth, J.; Garrison, M.M.; Bates, R.W. Decreased binding of insulin to its receptors in rats with hormone induced insulin resistance. Biochem. Biophys. Res. Commun. 1973, 53, 852–857. [Google Scholar] [CrossRef]
- Kolterman, O.G.; Insel, J.; Saekow, M.; Olefsky, J.M. Mechanisms of insulin resistance in human obesity: Evidence for receptor and postreceptor defects. J. Clin. Investig. 1980, 65, 1272–1284. [Google Scholar] [CrossRef] [Green Version]
- Soll, A.H.; Kahn, C.R.; Neville, D.M., Jr. Insulin binding to liver plasm membranes in the obese hyperglycemic (ob/ob) mouse. Demonstration of a decreased number of functionally normal receptors. J. Biol Chem. 1975, 250, 4702–4707. [Google Scholar] [CrossRef]
- Arcidiacono, B.; Chiefari, E.; Foryst-Ludwig, A.; Currò, G.; Navarra, G.; Brunetti, F.S.; Mirabelli, M.; Corigliano, D.M.; Kintscher, U.; Britti, D.; et al. Obesity-related hypoxia via miR-128 decreases insulin-receptor expression in human and mouse adipose tissue promoting systemic insulin resistance. EBioMedicine. 2020, 59, 102912. [Google Scholar] [CrossRef]
- Ebina, Y.; Edery, M.; Ellis, L.; Standring, D.; Beaudoin, J.; Roth, R.A.; Rutter, W.J. Expression of a functional human insulin receptor from a cloned cDNA in Chinese hamster ovary cells. Proc. Natl. Acad. Sci. USA 1985, 82, 8014–8018. [Google Scholar] [CrossRef] [Green Version]
- Seino, S.; Seino, M.; Nishi, S.; Bell, G.I. Structure of the human insulin receptor gene and characterization of its promoter. Proc. Natl. Acad. Sci. USA 1989, 86, 114–118. [Google Scholar] [CrossRef] [Green Version]
- Ullrich, A.; Bell, J.R.; Chen, E.Y.; Herrera, R.; Petruzzelli, L.M.; Dull, T.J.; Gray, A.; Coussens, L.; Liao, Y.C.; Tsubokawa, M.; et al. Human insulin receptor and its relationship to the tyrosine kinase family of oncogenes. Nature 1985, 313, 756–761. [Google Scholar] [CrossRef]
- Malaguarnera, R.; Sacco, A.; Voci, C.; Pandini, G.; Vigneri, R.; Belfiore, A. Proinsulin binds with high affinity the insulin receptor isoform A and predominantly activates the mitogenic pathway. Endocrinology 2012, 153, 2152–2163. [Google Scholar] [CrossRef] [Green Version]
- Sciacca, L.; Costantino, A.; Pandini, G.; Mineo, R.; Frasca, F.; Scalia, P.; Sbraccia, P.; Goldfine, I.D.; Vigneri, R.; Belfiore, A. Insulin receptor activation by IGF-II in breast cancers: Evidence for a new autocrine/paracrine mechanism. Oncogene 1999, 18, 2471–2479. [Google Scholar] [CrossRef]
- Belfiore, A.; Frasca, F.; Pandini, G.; Sciacca, L.; Vigneri, R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr. Rev. 2009, 30, 586–623. [Google Scholar] [CrossRef] [Green Version]
- Mosthaf, L.; Grako, K.; Dull, T.J.; Coussens, L.; Ullrich, A.; McClain, D.A. Functionally distinct insulin receptors generated by tissue-specific alternative splicing. EMBO J. 1990, 9, 2409–2413. [Google Scholar] [CrossRef]
- Vogt, B.; Carrascosa, J.M.; Ermel, B.; Ullrich, A.; Häring, H.U. The two isotypes of the human insulin receptor (HIR-A and HIR-B) follow different internalization kinetics. Biochem. Biophys. Res. Commun. 1991, 177, 1013–1018. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Flier, J.S.; Yokota, A.; Benecke, H.; Backer, J.M.; Moller, D.E. Functional properties of two naturally occurring isoforms of the human insulin receptor in Chinese hamster ovary cells. Endocrinology 1991, 129, 2058–2066. [Google Scholar] [CrossRef]
- Pandini, G.; Medico, E.; Conte, E.; Sciacca, L.; Vigneri, R.; Belfiore, A. Differential gene expression induced by insulin and insulin-like growth factor-II through the insulin receptor isoform A. J. Biol. Chem. 2003, 278, 42178–42189. [Google Scholar] [CrossRef] [Green Version]
- Sciacca, L.; Prisco, M.; Wu, A.; Belfiore, A.; Vigneri, R.; Baserga, R. Signaling differences from the A and B isoforms of the insulin receptor (IR) in 32D cells in the presence or absence of IR substrate-1. Endocrinology 2003, 144, 2650–2658. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hua, S.; Tian, W.; Zhang, L.; Zhao, J.; Zhang, H.; Zhang, W.; Xue, F. Mitogenic and anti-apoptotic effects of insulin in endometrial cancer are phosphatidylinositol 3-kinase/Akt dependent. Gynecol. Oncol. 2012, 125, 734–741. [Google Scholar] [CrossRef] [PubMed]
- Mamula, P.W.; McDonald, A.R.; Brunetti, A.; Okabayashi, Y.; Wong, K.Y.; Maddux, B.A.; Logsdon, C.; Goldfine, I.D. Regulating insulin-receptor-gene expression by differentiation and hormones. Diabetes Care. 1990, 13, 288–301. [Google Scholar] [CrossRef] [PubMed]
- Araki, E.; Shimada, F.; Uzawa, H.; Mori, M.; Ebina, Y. Characterization of the promoter region of the human insulin receptor gene. Evidence for promoter activity. J. Biol. Chem. 1987, 262, 16186–16191. [Google Scholar] [CrossRef]
- Brunetti, A.; Foti, D.; Goldfine, I.D. Identification of unique nuclear regulatory proteins for the insulin receptor gene, which appear during myocyte and adipocyte differentiation. J. Clin. Investig. 1993, 92, 1288–1295. [Google Scholar] [CrossRef] [Green Version]
- Iiritano, S.; Chiefari, E.; Ventura, V.; Possidente, K.; Nocera, A.; Nevolo, M.T.; Fedele, M.; Greco, A.; Greco, M.; Brunetti, G.; et al. The HMGA1-IGF-I/IGFBP system: A novel pathway for modulating glucose uptake. Mol. Endocrinol. 2012, 26, 1578–1589. [Google Scholar] [CrossRef] [Green Version]
- Arcidiacono, B.; Iiritano, S.; Chiefari, E.; Brunetti, F.S.; Gu, G.; Foti, D.P.; Brunetti, A. Cooperation between HMGA1, PDX-1, and MafA is Essential for Glucose-Induced Insulin Transcription in Pancreatic Beta Cells. Front. Endocrinol. 2015, 5, 237. [Google Scholar] [CrossRef] [Green Version]
- Brunetti, A.; Manfioletti, G.; Chiefari, E.; Goldfine, I.D.; Foti, D. Transcriptional regulation of human insulin receptor gene by the high-mobility group protein HMGI(Y). FASEB J. 2001, 15, 492–500. [Google Scholar] [CrossRef]
- Foti, D.; Iuliano, R.; Chiefari, E.; Brunetti, A. A nucleoprotein complex containing Sp1, C/EBP beta, and HMGI-Y controls human insulin receptor gene transcription. Mol. Cell Biol. 2003, 23, 2720–2732. [Google Scholar] [CrossRef] [Green Version]
- Chiefari, E.; Nevolo, M.T.; Arcidiacono, B.; Maurizio, E.; Nocera, A.; Iiritano, S.; Sgarra, R.; Possidente, K.; Palmieri, C.; Paonessa, F.; et al. HMGA1 is a novel downstream nuclear target of the insulin receptor signaling pathway. Sci. Rep. 2012, 2, 251. [Google Scholar] [CrossRef] [Green Version]
- Arnoldo, L.; Sgarra, R.; Chiefari, E.; Maurizio, E.; Nocera, A.; Iiritano, S.; Sgarra, R.; Possidente, K.; Palmieri, C.; Paonessa, F.; et al. A novel mechanism of post-translational modulation of HMGA functions by the histone chaperone nucleophosmin. Sci. Rep. 2015, 5, 8552. [Google Scholar] [CrossRef] [Green Version]
- Chiefari, E.; Foti, D.P.; Sgarra, R.; Pegoraro, S.; Arcidiacono, B.; Brunetti, F.S.; Greco, M.; Manfioletti, G.; Brunetti, A. Transcriptional Regulation of Glucose Metabolism: The Emerging Role of the HMGA1 Chromatin Factor. Front. Endocrinol. 2018, 9, 357. [Google Scholar] [CrossRef] [Green Version]
- Foti, D.; Chiefari, E.; Fedele, M.; Iuliano, R.; Brunetti, L.; Paonessa, F.; Manfioletti, G.; Barbetti, F.; Brunetti, A.; Croce, C.M.; et al. Lack of the architectural factor HMGA1 causes insulin resistance and diabetes in humans and mice. Nat. Med. 2005, 11, 765–773. [Google Scholar] [CrossRef]
- Chiefari, E.; Tanyolaç, S.; Paonessa, F.; Pullinger, C.R.; Capula, C.; Iiritano, S.; Mazza, T.; Forlin, M.; Fusco, A.; Durlach, V.; et al. Functional variants of the HMGA1 gene and type 2 diabetes mellitus. JAMA 2011, 305, 903–912. [Google Scholar] [CrossRef] [Green Version]
- Chiefari, E.; Tanyolaç, S.; Iiritano, S.; Sciacqua, A.; Capula, C.; Arcidiacono, B.; Nocera, A.; Possidente, K.; Baudi, F.; Ventura, V.; et al. A polymorphism of HMGA1 is associated with increased risk of metabolic syndrome and related components. Sci. Rep. 2013, 3, 1491. [Google Scholar] [CrossRef]
- Chiefari, E.; Iiritano, S.; Paonessa, F.; Le Pera, I.; Arcidiacono, B.; Filocamo, M.; Foti, D.; Liebhaber, S.A.; Brunetti, A. Pseudogene-mediated posttranscriptional silencing of HMGA1 can result in insulin resistance and type 2 diabetes. Nat. Commun. 2010, 1, 40. [Google Scholar] [CrossRef] [Green Version]
- Sgarra, R.; Pegoraro, S.; Ros, G.; Penzo, C.; Chiefari, E.; Foti, D.; Brunetti, A.; Manfioletti, G. High Mobility Group A (HMGA) proteins: Molecular instigators of breast cancer onset and progression. Biochim. Biophys. Acta Rev. Cancer 2018, 1869, 216–229. [Google Scholar] [CrossRef]
- Osborne, C.K.; Bolan, G.; Monaco, M.E.; Lippman, M.E. Hormone responsive human breast cancer in long-term tissue culture: Effect of insulin. Proc. Natl. Acad. Sci. USA 1976, 73, 4536–4540. [Google Scholar] [CrossRef] [Green Version]
- Giovannucci, E.; Harlan, D.M.; Archer, M.C.; Bergenstal, R.M.; Gapstur, S.M.; Habel, L.A.; Pollak, M.; Regensteiner, J.G.; Yee, D. Diabetes and cancer: A consensus report. Diabetes Care 2010, 33, 1674–1685. [Google Scholar] [CrossRef] [Green Version]
- Nead, K.T.; Sharp, S.J.; Thompson, D.J.; Painter, J.N.; Savage, D.B.; Semple, R.K.; Barker, A.; Australian National Endometrial Cancer Study Group; Perry, J.R.; Attia, J.; et al. Evidence of a causal association between insulinemia and endometrial cancer: A mendelian randomization analysis. J. Natl. Cancer Inst. 2015, 107, djv178. [Google Scholar] [CrossRef]
- Semple, R.K.; Savage, D.B.; Cochran, E.K.; Gorden, P.; O’Rahilly, S. Genetic syndromes of severe insulin resistance. Endocr. Rev. 2011, 32, 498–514. [Google Scholar] [CrossRef] [Green Version]
- Weber, D.R.; Stanescu, D.E.; Semple, R.; Holland, C.; Magge, S.N. Continuous subcutaneous IGF-1 therapy via insulin pump in a patient with Donohue syndrome. J. Pediatr. Endocrinol. Metab. 2014, 27, 1237–1241. [Google Scholar] [CrossRef] [Green Version]
- Brisigotti, M.; Fabbretti, G.; Pesce, F.; Gatti, R.; Cohen, A.; Parenti, G.; Callea, F. Congenital bilateral juvenile granulosa cell tumor of the ovary in leprechaunism: A case report. Pediatric Pathol. 1993, 13, 549–558. [Google Scholar] [CrossRef]
- Paonessa, F.; Foti, D.; Costa, V.; Chiefari, E.; Brunetti, G.; Leone, F.; Luciano, F.; Wu, F.; Lee, A.S.; Gulletta, E.; et al. Activator protein-2 overexpression accounts for increased insulin receptor expression in human breast cancer. Cancer Res. 2006, 66, 5085–5093. [Google Scholar] [CrossRef] [Green Version]
- Chettouh, H.; Fartoux, L.; Aoudjehane, L.; Wendum, D.; Clapéron, A.; Chrétien, Y.; Rey, C.; Scatton, O.; Soubrane, O.; Conti, F.; et al. Mitogenic insulin receptor-A is overexpressed in human hepatocellular carcinoma due to EGFR-mediated dysregulation of RNA splicing factors. Cancer Res. 2013, 73, 3974–3986. [Google Scholar] [CrossRef] [Green Version]
- Poloz, Y.; Stambolic, V. Obesity and cancer, a case for insulin signaling. Cell Death Dis. 2015, 6, e2037. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.J.; Rothenberg, P.; Kahn, C.R.; Backer, J.M.; Araki, E.; Wilden, P.A.; Cahill, D.A.; Goldstein, B.J.; White, M.F. Structure of the insulin receptor substrate IRS-1 defines a unique signal transduction protein. Nature 1991, 352, 73–77. [Google Scholar] [CrossRef]
- Sun, X.J.; Wang, L.M.; Zhang, Y.; Yenush, L.; Myers, M.G., Jr.; Glasheen, E.; Lane, W.S.; Pierce, J.H.; White, M.F. Role of IRS-2 in insulin and cytokine signalling. Nature 1995, 377, 173–177. [Google Scholar] [CrossRef]
- Hawkins, P.T.; Jackson, T.R.; Stephens, L.R. Platelet-derived growth factor stimulates synthesis of PtdIns(3,4,5)P3 by activating a PtdIns(4,5)P2 3-OH kinase. Nature 1992, 358, 157–159. [Google Scholar] [CrossRef]
- Alessi, D.R.; James, S.R.; Downes, C.P.; Holmes, A.B.; Gaffney, P.R.; Reese, C.B.; Cohen, P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr. Biol. 1997, 7, 261–269. [Google Scholar] [CrossRef] [Green Version]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polak, P.; Hall, M.N. mTOR and the control of whole body metabolism. Curr. Opin. Cell Biol. 2009, 21, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.S. mTOR as a Key Regulator in Maintaining Skeletal Muscle Mass. Front. Physiol. 2017, 8, 788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martini, M.; De Santis, M.C.; Braccini, L.; Gulluni, F.; Hirsch, E. PI3K/AKT signaling pathway and cancer: An updated review. Ann. Med. 2014, 46, 372–383. [Google Scholar] [CrossRef]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [Green Version]
- Lien, E.C.; Lyssiotis, C.A.; Cantley, L.C. Metabolic Reprogramming by the PI3K-Akt-mTOR Pathway in Cancer. Recent Results Cancer Res. 2016, 207, 39–72. [Google Scholar] [CrossRef]
- Dibble, C.C.; Cantley, L.C. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 2015, 25, 545–555. [Google Scholar] [CrossRef] [Green Version]
- Fruman, D.A.; Rommel, C. PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.; Fernandez de Mattos, S.; van der Horst, A.; Klompmaker, R.; Kops, G.J.; Lam, E.W.; Burgering, B.M.; Medema, R.H. Cell cycle inhibition by FoxO forkhead transcription factors involves downregulation of cyclin D. Mol. Cell Biol. 2002, 22, 7842–7852. [Google Scholar] [CrossRef] [Green Version]
- Kong, L.; Wang, Q.; Jin, J.; Xiang, Z.; Chen, T.; Shen, S.; Wang, H.; Gao, Q.; Wang, Y. Insulin resistance enhances the mitogen-activated protein kinase signaling pathway in ovarian granulosa cells. PLoS ONE 2017, 12, e0188029. [Google Scholar] [CrossRef] [Green Version]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef] [Green Version]
- Frasca, F.; Pandini, G.; Scalia, P.; Sciacca, L.; Mineo, R.; Costantino, A.; Goldfine, I.D.; Belfiore, A.; Vigneri, R. Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol. Cell Biol. 1999, 19, 3278–3288. [Google Scholar] [CrossRef] [Green Version]
- Denley, A.; Bonython, E.R.; Booker, G.W.; Cosgrove, L.J.; Forbes, B.E.; Ward, C.W.; Wallace, J.C. Structural determinants for high-affinity binding of insulin-like growth factor II to insulin receptor (IR)-A, the exon 11 minus isoform of the IR. Mol. Endocrinol. 2004, 18, 2502–2512. [Google Scholar] [CrossRef] [Green Version]
- Yakar, S.; Pennisi, P.; Zhao, H.; Zhang, Y.; LeRoith, D. Circulating IGF-1 and its role in cancer: Lessons from the IGF-1 gene deletion (LID) mouse. In Biology of IGF-1: Its Interaction with Insulin in Health and Malignant States; Volume 262 of Novartis Foundation Symposia; John Wiley & Sons: Hoboken, NJ, USA, 2005; p. 288. [Google Scholar]
- Arcidiacono, B.; Iiritano, S.; Nocera, A.; Possidente, K.; Nevolo, M.T.; Ventura, V.; Foti, D.; Chiefari, E.; Brunetti, A. Insulin resistance and cancer risk: An overview of the pathogenetic mechanisms. Exp. Diabetes Res. 2012, 2012, 789174. [Google Scholar] [CrossRef] [Green Version]
- Bowers, L.W.; Rossi, E.L.; O’Flanagan, C.H.; de Graffenried, L.A.; Hursting, S.D. The Role of the Insulin/IGF System in Cancer: Lessons Learned from Clinical Trials and the Energy Balance-Cancer Link. Front. Endocrinol. 2015, 6, 77. [Google Scholar] [CrossRef] [Green Version]
- Renehan, A.G.; Zwahlen, M.; Minder, C.; O’Dwyer, S.T.; Shalet, S.M.; Egger, M. Insulin-like growth factor (IGF)-I, IGF binding protein-3, and cancer risk: Systematic review and meta-regression analysis. Lancet 2004, 363, 1346–1353. [Google Scholar] [CrossRef]
- Catsburg, C.; Gunter, M.J.; Chen, C.; Cote, M.L.; Kabat, G.C.; Nassir, R.; Tinker, L.; Wactawski-Wende, J.; Page, D.L.; Rohan, T.E. Insulin, estrogen, inflammatory markers, and risk of benign proliferative breast disease. Cancer Res. 2014, 74, 3248–3258. [Google Scholar] [CrossRef] [Green Version]
- Rose, D.P.; Vona-Davis, L. The cellular and molecular mechanisms by which insulin influences breast cancer risk and progression. Endocr. Relat. Cancer. 2012, 19, R225–R241. [Google Scholar] [CrossRef]
- American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care 2004, 27 (Suppl. 1), S5–S10. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization (WHO). Health Topics: Diabetes. Available online: https://www.who.int/health-topics/diabetes (accessed on 29 September 2020).
- Pearson-Stuttard, J.; Bennett, J.; Cheng, Y.J.; Vamos, E.P.; Cross, A.J.; Ezzati, M.; Gregg, E.W. Trends in predominant causes of death in individuals with and without diabetes in England from 2001 to 2018: An epidemiological analysis of linked primary care records. Lancet Diabetes Endocrinol. 2021, 9, 165–173. [Google Scholar] [CrossRef]
- Sona, M.F.; Myung, S.K.; Park, K.; Jargalsaikhan, G. Type 1 diabetes mellitus and risk of cancer: A meta-analysis of observational studies. Jpn. J. Clin. Oncol. 2018, 48, 426–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noto, H.; Tsujimoto, T.; Sasazuki, T.; Noda, M. Significantly increased risk of cancer in patients with diabetes mellitus: A systematic review and meta-analysis. Endocr. Pract. 2011, 17, 616–628. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.B.; Yu, T.; Liu, C.; Li, Y.Q. Diabetes mellitus and increased risk of biliary tract cancer: Systematic review and meta-analysis. Cancer Causes Control 2011, 22, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Zhang, X.; Shen, Z.; Zhong, S.; Wang, X.; Lu, Y.; Xu, C. Diabetes mellitus and risk of bladder cancer: A meta-analysis of cohort studies. PLoS ONE 2013, 8, e56662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsson, S.C.; Mantzoros, C.S.; Wolk, A. Diabetes mellitus and risk of breast cancer: A meta-analysis. Int. J. Cancer 2007, 121, 856–862. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.B.; Ren, G.S. Diabetes mellitus and prognosis in women with breast cancer: A systematic review and meta-analysis. Medicine 2016, 95, e5602. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, X.; Gu, C.; Xia, J. Diabetes mellitus is associated with breast cancer: Systematic review, meta-analysis, and in silico reproduction. Panminerva Med. 2015, 57, 101–108. [Google Scholar] [PubMed]
- Chen, S.; Tao, M.; Zhao, L.; Zhang, X. The association between diabetes/hyperglycemia and the prognosis of cervical cancer patients: A systematic review and meta-analysis. Medicine 2017, 96, e7981. [Google Scholar] [CrossRef]
- Jiang, Y.; Ben, Q.; Shen, H.; Lu, W.; Zhang, Y.; Zhu, J. Diabetes mellitus and incidence and mortality of colorectal cancer: A systematic review and meta-analysis of cohort studies. Eur. J. Epidemiol. 2011, 26, 863–876. [Google Scholar] [CrossRef]
- Zhu, B.; Wu, X.; Wu, B.; Pei, D.; Zhang, L.; Wei, L. The relationship between diabetes and colorectal cancer prognosis: A meta-analysis based on the cohort studies. PLoS ONE 2017, 12, e0176068. [Google Scholar] [CrossRef] [Green Version]
- Ling, S.; Brown, K.; Miksza, J.K.; Howells, L.; Morrison, A.; Issa, E.; Yates, T.; Khunti, K.; Davies, M.J.; Zaccardi, F. Association of Type 2 Diabetes With Cancer: A Meta-analysis With Bias Analysis for Unmeasured Confounding in 151 Cohorts Comprising 32 Million People. Diabetes Care 2020, 43, 2313–2322. [Google Scholar] [CrossRef]
- Tsilidis, K.K.; Kasimis, J.C.; Lopez, D.S.; Ntzani, E.E.; Ioannidis, J.P. Type 2 diabetes and cancer: Umbrella review of meta-analyses of observational studies. BMJ 2015, 2, 7607. [Google Scholar] [CrossRef] [Green Version]
- Carstensen, B.; Read, S.H.; Friis, S.; Sund, R.; Keskimäki, I.; Svensson, A.M.; Ljung, R.; Wild, S.H.; Kerssens, J.J.; Harding, J.L.; et al. Cancer incidence in persons with type 1 diabetes: A five-country study of 9,000 cancers in type 1 diabetic individuals. Diabetologia 2016, 59, 980–988. [Google Scholar] [CrossRef] [Green Version]
- Castillo, J.J.; Mull, N.; Reagan, J.L.; Nemr, S.; Mitri, J. Increased incidence of non-Hodgkin lymphoma, leukemia, and myeloma in patients with diabetes mellitus type 2: A meta-analysis of observational studies. Blood 2012, 119, 4845–4850. [Google Scholar] [CrossRef] [Green Version]
- Bao, C.; Yang, X.; Xu, W.; Luo, H.; Xu, Z.; Su, C.; Qi, X. Diabetes mellitus and incidence and mortality of kidney cancer: A meta-analysis. J. Diabetes Complications. 2013, 27, 357–364. [Google Scholar] [CrossRef]
- Jing, W.; Jin, G.; Zhou, X.; Zhou, Y.; Zhang, Y.; Shao, C.; Liu, R.; Hu, X. Diabetes mellitus and increased risk of cholangiocarcinoma: A meta-analysis. Eur. J. Cancer Prev. 2012, 21, 24–31. [Google Scholar] [CrossRef]
- Adami, H.O.; Chow, W.H.; Nyrén, O.; Berne, C.; Linet, M.S.; Ekbom, A.; Wolk, A.; McLaughlin, J.K.; Fraumeni, J.F., Jr. Excess risk of primary liver cancer in patients with diabetes mellitus. J Natl Cancer Inst. 1996, 88, 1472–1477. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Jeon, I.; Lee, J.M.; Yoon, J.M.; Park, S.M. Diabetes mellitus as an independent risk factor for lung cancer: A meta-analysis of observational studies. Eur J. Cancer 2013, 49, 2411–2423. [Google Scholar] [CrossRef]
- Zhu, L.; Cao, H.; Zhang, T.; Shen, H.; Dong, W.; Wang, L.; Du, J. The Effect of Diabetes Mellitus on Lung Cancer Prognosis: A PRISMA-compliant Meta-analysis of Cohort Studies. Medicine 2016, 95, e3528. [Google Scholar] [CrossRef]
- Lee, J.Y.; Jeon, I.; Kim, J.W.; Song, Y.S.; Yoon, J.M.; Park, S.M. Diabetes mellitus and ovarian cancer risk: A systematic review and meta-analysis of observational studies. Int. J. Gynecol Cancer 2013, 23, 402–412. [Google Scholar] [CrossRef]
- Wang, L.; Wang, L.; Zhang, J.; Wang, B.; Liu, H. Association between diabetes mellitus and subsequent ovarian cancer in women: A systematic review and meta-analysis of cohort studies. Medicine 2017, 96, e6396. [Google Scholar] [CrossRef] [PubMed]
- Ben, Q.; Xu, M.; Ning, X.; Liu, J.; Hong, S.; Huang, W.; Zhang, H.; Li, Z. Diabetes mellitus and risk of pancreatic cancer: A meta-analysis of cohort studies. Eur. J. Cancer 2011, 47, 1928–1937. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Xu, Z.; Xu, T.; Yu, B.; Zou, Q. Diabetes mellitus is associated with elevated risk of mortality amongst patients with prostate cancer: A meta-analysis of 11 cohort studies. Diabetes Metab Res. Rev. 2015, 31, 336–343. [Google Scholar] [CrossRef]
- Tian, T.; Zhang, L.Q.; Ma, X.H.; Zhou, J.N.; Shen, J. Diabetes mellitus and incidence and mortality of gastric cancer: A meta-analysis. Exp. Clin. Endocrinol. Diabetes 2012, 120, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.; Ben, Q.; Qian, J.; Wang, Y.; Li, Y. Diabetes mellitus and risk of gastric cancer: A systematic review and meta-analysis of observational studies. Eur. J. Gastroenterol. Hepatol. 2011, 23, 1127–1135. [Google Scholar] [CrossRef] [PubMed]
- Schmid, D.; Behrens, G.; Jochem, C.; Keimling, M.; Leitzmann, M. Physical activity, diabetes, and risk of thyroid cancer: A systematic review and meta-analysis. Eur. J. Epidemiol. 2013, 28, 945–958. [Google Scholar] [CrossRef] [PubMed]
- Cignarelli, A.; Genchi, V.A.; Caruso, I.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Diabetes and cancer: Pathophysiological fundamentals of a ‘dangerous affair’. Diabetes Res. Clin. Pract. 2018, 143, 378–388. [Google Scholar] [CrossRef] [PubMed]
- Carstensen, B.; Witte, D.R.; Friis, S. Cancer occurrence in Danish diabetic patients: Duration and insulin effects. Diabetologia 2012, 55, 948–958. [Google Scholar] [CrossRef] [Green Version]
- Scharf, J.G.; Ramadori, G.; Dombrowski, F. Analysis of the IGF axis in preneoplastic hepatic foci and hepatocellular neoplasms developing after low-number pancreatic islet transplantation into the livers of streptozotocin diabetic rats. Lab. Investig. 2000, 80, 1399–1411. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, E.J.; LeRoith, D. Hyperinsulinaemia in cancer. Nat. Rev. Cancer 2020, 20, 629–644. [Google Scholar] [CrossRef]
- Vigneri, R.; Sciacca, L.; Vigneri, P. Rethinking the Relationship between Insulin and Cancer. Trends Endocrinol. Metab. 2020, 31, 551–560. [Google Scholar] [CrossRef]
- Hidaka, A.; Sasazuki, S.; Goto, A.; Sawada, N.; Shimazu, T.; Yamaji, T.; Iwasaki, M.; Inoue, M.; Noda, M.; Tajiri, H.; et al. Plasma insulin, C-peptide and blood glucose and the risk of gastric cancer: The Japan Public Health Center-based prospective study. Int. J. Cancer 2015, 136, 1402–1410. [Google Scholar] [CrossRef]
- Loftfield, E.; Freedman, N.D.; Lai, G.Y.; Weinstein, S.J.; McGlynn, K.A.; Taylor, P.R.; Männistö, S.; Albanes, D.; Stolzenberg-Solomon, R.Z. Higher Glucose and Insulin Levels Are Associated with Risk of Liver Cancer and Chronic Liver Disease Mortality among Men without a History of Diabetes. Cancer Prev. Res. 2016, 9, 866–874. [Google Scholar] [CrossRef] [Green Version]
- Tsujimoto, T.; Kajio, H.; Sugiyama, T. Association between hyperinsulinemia and increased risk of cancer death in nonobese and obese people: A population-based observational study. Int. J. Cancer 2017, 141, 102–111. [Google Scholar] [CrossRef]
- Yuan, S.; Kar, S.; Carter, P.; Vithayathil, M.; Mason, A.M.; Burgess, S.; Larsson, S.C. Is Type 2 Diabetes Causally Associated with Cancer Risk? Evidence from a Two-Sample Mendelian Randomization Study. Diabetes 2020, 69, 1588–1596. [Google Scholar] [CrossRef]
- Balkau, B.; Kahn, H.S.; Courbon, D.; Eschwège, E.; Ducimetière, P.; Paris Prospective Study. Hyperinsulinemia predicts fatal liver cancer but is inversely associated with fatal cancer at some other sites: The Paris Prospective Study. Diabetes Care 2001, 24, 843–849. [Google Scholar] [CrossRef] [Green Version]
- Irwin, M.L.; Duggan, C.; Wang, C.Y.; Smith, A.W.; McTiernan, A.; Baumgartner, R.N.; Baumgartner, K.B.; Bernstein, L.; Ballard-Barbash, R. Fasting C-peptide levels and death resulting from all causes and breast cancer: The health, eating, activity, and lifestyle study. J. Clin. Oncol. 2011, 29, 47–53. [Google Scholar] [CrossRef]
- Pan, K.; Nelson, R.A.; Wactawski-Wende, J.; Lee, D.J.; Manson, J.E.; Aragaki, A.K.; Mortimer, J.E.; Phillips, L.S.; Rohan, T.; Ho, G.Y.F.; et al. Insulin Resistance and Cancer-Specific and All-Cause Mortality in Postmenopausal Women: The Women’s Health Initiative. J. Natl. Cancer Inst. 2020, 112, 170–178. [Google Scholar] [CrossRef]
- Currie, C.J.; Poole, C.D.; Gale, E.A. The influence of glucose-lowering therapies on cancer risk in type 2 diabetes. Diabetologia. 2009, 52, 1766–1777. [Google Scholar] [CrossRef] [Green Version]
- Tseng, C.H. Prolonged use of human insulin increases breast cancer risk in Taiwanese women with type 2 diabetes. BMC Cancer 2015, 15, 846. [Google Scholar] [CrossRef] [Green Version]
- Hensley, C.T.; Faubert, B.; Yuan, Q.; Lev-Cohain, N.; Jin, E.; Kim, J.; Jiang, L.; Ko, B.; Skelton, R.; Loudat, L.; et al. Metabolic Heterogeneity in Human Lung Tumors. Cell 2016, 164, 681–694. [Google Scholar] [CrossRef] [Green Version]
- Birts, C.N.; Banerjee, A.; Darley, M.; Dunlop, C.R.; Nelson, S.; Nijjar, S.K.; Parker, R.; West, J.; Tavassoli, A.; Rose-Zerilli, M.J.J.; et al. p53 is regulated by aerobic glycolysis in cancer cells by the CtBP family of NADH-dependent transcriptional regulators. Sci. Signal. 2020, 13, eaau9529. [Google Scholar] [CrossRef] [PubMed]
- Kalyanaraman, B. Teaching the basics of cancer metabolism: Developing antitumor strategies by exploiting the differences between normal and cancer cell metabolism. Redox Biol. 2017, 12, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Stocks, T.; Rapp, K.; Bjørge, T.; Manjer, J.; Ulmer, H.; Selmer, R.; Lukanova, A.; Johansen, D.; Concin, H.; Tretli, S.; et al. Blood glucose and risk of incident and fatal cancer in the metabolic syndrome and cancer project (me-can): Analysis of six prospective cohorts. PLoS Med. 2009, 6, e1000201. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; LeRoith, D.; Gallagher, E.J. Diabetes, Obesity, and Breast Cancer. Endocrinology 2018, 159, 3801–3812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, S.Y.; Mancuso, N.; Papp, J.; Sobel, E.; Zhang, Z.F. Post genome-wide gene-environment interaction study: The effect of genetically driven insulin resistance on breast cancer risk using Mendelian randomization. PLoS ONE 2019, 14, e0218917. [Google Scholar] [CrossRef] [Green Version]
- Carreras-Torres, R.; Johansson, M.; Gaborieau, V.; Haycock, P.C.; Wade, K.H.; Relton, C.L.; Martin, R.M.; Davey Smith, G.; Brennan, P. The Role of Obesity, Type 2 Diabetes, and Metabolic Factors in Pancreatic Cancer: A Mendelian Randomization Study. J. Natl. Cancer Inst. 2017, 109, djx012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO. Obesity and Overweight Fact Sheet. 2020. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 29 September 2020).
- American Cancer Society. Cancer Facts and Figures. 2011. Available online: https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2011.html (accessed on 29 September 2020).
- Renehan, A.G.; Tyson, M.; Egger, M.; Heller, R.F.; Zwahlen, M. Body-mass index and incidence of cancer: A systematic review and meta-analysis of prospective observational studies. Lancet 2008, 371, 569–578. [Google Scholar] [CrossRef]
- Calle, E.E.; Rodriguez, C.; Walker-Thurmond, K.; Thun, M.J. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N. Engl. J. Med. 2003, 348, 1625–1638. [Google Scholar] [CrossRef] [Green Version]
- Goodwin, P.J.; Stambolic, V. Impact of the obesity epidemic on cancer. Annu Rev. Med. 2015, 66, 281–296. [Google Scholar] [CrossRef]
- World Cancer Research Fund/American Institute for Cancer Research. Body Fatness and Weight Gain and Risk of Cancer. Available online: https://www.wcrf.org/sites/default/files/Body-fatness-and-weight-gain_0.pdf (accessed on 29 September 2020).
- Murphy, T.K.; Calle, E.E.; Rodriguez, C.; Kahn, H.S.; Thun, M.J. Body mass index and colon cancer mortality in a large prospective study. Am. J. Epidemiol. 2000, 152, 847–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pischon, T.; Lahmann, P.H.; Boeing, H.; Friedenreich, C.; Norat, T.; Tjønneland, A.; Halkjaer, J.; Overvad, K.; Clavel-Chapelon, F.; Boutron-Ruault, M.C.; et al. Body size and risk of colon and rectal cancer in the European Prospective Investigation into Cancer and Nutrition (EPIC). J. Natl. Cancer Inst. 2006, 98, 920–931. [Google Scholar] [CrossRef] [PubMed]
- Nam, G.E.; Baek, S.J.; Choi, H.B.; Han, K.; Kwak, J.M.; Kim, J.; Kim, S.H. Association between Abdominal Obesity and Incident Colorectal Cancer: A Nationwide Cohort Study in Korea. Cancers 2020, 12, 1368. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Ness-Jensen, E.; Martling, A.; Hveem, K. Anthropometry-based Obesity Phenotypes and Risk of Colorectal Adenocarcinoma: A Large Prospective Cohort Study in Norway. Epidemiology 2016, 27, 423–432. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.H.; Wu, K.; Ng, K.; Zauber, A.G.; Nguyen, L.H.; Song, M.; He, X.; Fuchs, C.S.; Ogino, S.; Willett, W.C.; et al. Association of Obesity with Risk of Early-Onset Colorectal Cancer Among Women. JAMA Oncol. 2019, 5, 37–44. [Google Scholar] [CrossRef] [Green Version]
- Blair, C.K.; Wiggins, C.L.; Nibbe, A.M.; Storlie, C.B.; Prossnitz, E.R.; Royce, M.; Lomo, L.C.; Hill, D.A. Obesity and survival among a cohort of breast cancer patients is partially mediated by tumor characteristics. NPJ Breast Cancer 2019, 5, 33. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, L.; Zhou, Q.; Imam, M.U.; Cai, J.; Wang, Y.; Qi, M.; Sun, P.; Ping, Z.; Fu, X. Body mass index had different effects on premenopausal and postmenopausal breast cancer risks: A dose-response meta-analysis with 3,318,796 subjects from 31 cohort studies. BMC Public Health 2017, 17, 936. [Google Scholar] [CrossRef]
- Trayhurn, P.; Alomar, S.Y. Oxygen deprivation and the cellular response to hypoxia in adipocytes - perspectives on white and brown adipose tissues in obesity. Front. Endocrinol. 2015, 6, 19. [Google Scholar] [CrossRef] [Green Version]
- Foti, D.P.; Brunetti, A. Editorial: “Linking Hypoxia to Obesity”. Front. Endocrinol. 2017, 8, 34. [Google Scholar] [CrossRef]
- Laria, A.E.; Messineo, S.; Arcidiacono, B.; Varano, M.; Chiefari, E.; Semple, R.K.; Rocha, N.; Russo, D.; Cuda, G.; Gaspari, M.; et al. Secretome Analysis of Hypoxia-Induced 3T3-L1 Adipocytes Uncovers Novel Proteins Potentially Involved in Obesity. Proteomics 2018, 18, e1700260. [Google Scholar] [CrossRef] [Green Version]
- Messineo, S.; Laria, A.E.; Arcidiacono, B.; Chiefari, E.; Luque Huertas, R.M.; Foti, D.P.; Brunetti, A. Cooperation between HMGA1 and HIF-1 Contributes to Hypoxia-Induced VEGF and Visfatin Gene Expression in 3T3-L1 Adipocytes. Front. Endocrinol. 2016, 7, 73. [Google Scholar] [CrossRef] [PubMed]
- D’Ignazio, L.; Bandarra, D.; Rocha, S. NF-κB and HIF crosstalk in immune responses. FEBS J. 2016, 283, 413–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, T.W.; McPherson, M.; Gail Darlington, L. Obesity and Cancer: Existing and New Hypotheses for a Causal Connection. EBioMedicine 2018, 30, 14–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Li, B.; Li, Z.; Li, J.; Sun, S.; Sun, S. Cancer-associated adipocytes: Key players in breast cancer progression. J. Hematol. Oncol. 2019, 12, 95. [Google Scholar] [CrossRef]
- D’Esposito, V.; Ambrosio, M.R.; Giuliano, M.; Cabaro, S.; Miele, C.; Beguinot, F.; Formisano, P. Mammary Adipose Tissue Control of Breast Cancer Progression: Impact of Obesity and Diabetes. Front. Oncol. 2020, 10, 1554. [Google Scholar] [CrossRef]
- Wang, Q.A.; Song, A.; Chen, W.; Schwalie, P.C.; Zhang, F.; Vishvanath, L.; Jiang, L.; Ye, R.; Shao, M.; Tao, C.; et al. Reversible De-differentiation of Mature White Adipocytes into Preadipocyte-like Precursors during Lactation. Cell Metab. 2018, 28, 282–288.e3. [Google Scholar] [CrossRef] [Green Version]
- Duong, M.N.; Cleret, A.; Matera, E.L.; Chettab, K.; Mathé, D.; Valsesia-Wittmann, S.; Clémenceau, B.; Dumontet, C. Adipose cells promote resistance of breast cancer cells to trastuzumab-mediated antibody-dependent cellular cytotoxicity. Breast Cancer Res. 2015, 17, 57. [Google Scholar] [CrossRef] [Green Version]
- Rybinska, I.; Agresti, R.; Trapani, A.; Tagliabue, E.; Triulzi, T. Adipocytes in Breast Cancer, the Thick and the Thin. Cells 2020, 9, 560. [Google Scholar] [CrossRef] [Green Version]
- Naimo, G.D.; Gelsomino, L.; Catalano, S.; Mauro, L.; Andò, S. Interfering Role of ERα on Adiponectin Action in Breast Cancer. Front. Endocrinol. 2020, 11, 66. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.L.; Heist, K.; DePaoli, A.M.; Veldhuis, J.D.; Mantzoros, C.S. The role of falling leptin levels in the neuroendocrine and metabolic adaptation to short-term starvation in healthy men. J. Clin. Investig. 2003, 111, 1409–1421. [Google Scholar] [CrossRef] [Green Version]
- Thomas, T.; Burguera, B.; Melton, L.J., 3rd; Atkinson, E.J.; O’Fallon, W.M.; Riggs, B.L.; Khosla, S. Relationship of serum leptin levels with body composition and sex steroid and insulin levels in men and women. Metabolism 2000, 49, 1278–1284. [Google Scholar] [CrossRef]
- Zhao, L.; Shen, Z.X.; Luo, H.S.; Shen, L. Possible involvement of leptin and leptin receptor in developing gastric adenocarcinoma. World J. Gastroenterol. 2005, 11, 7666–7670. [Google Scholar] [CrossRef]
- Ishikawa, M.; Kitayama, J.; Nagawa, H. Enhanced expression of leptin and leptin receptor (OB-R) in human breast cancer. Clin. Cancer Res. 2004, 10, 4325–4331. [Google Scholar] [CrossRef] [Green Version]
- Yuan, S.S.; Tsai, K.B.; Chung, Y.F.; Chan, T.F.; Yeh, Y.T.; Tsai, L.Y.; Su, J.H. Aberrant expression and possible involvement of the leptin receptor in endometrial cancer. Gynecol. Oncol. 2004, 92, 769–775. [Google Scholar] [CrossRef]
- Bowers, L.W.; Rossi, E.L.; McDonell, S.B.; Doerstling, S.S.; Khatib, S.A.; Lineberger, C.G.; Albright, J.E.; Tang, X.; de Graffenried, L.A.; Hursting, S.D. Leptin Signaling Mediates Obesity-Associated CSC Enrichment and EMT in Preclinical TNBC Models. Mol. Cancer Res. 2018, 16, 869–879. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Wu, M.; Zeng, N.; Xiong, M.; Hu, W.; Lv, W.; Yi, Y.; Zhang, Q.; Wu, Y. Cancer-associated adipocytes: Emerging supporters in breast cancer. J. Exp. Clin. Cancer Res. 2020, 39, 156. [Google Scholar] [CrossRef]
- Rui, L.; Yuan, M.; Frantz, D.; Shoelson, S.; White, M.F. SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. J. Biol. Chem. 2002, 277, 42394–42398. [Google Scholar] [CrossRef] [Green Version]
- Barbieri, I.; Pensa, S.; Pannellini, T.; Quaglino, E.; Maritano, D.; Demaria, M.; Voster, A.; Turkson, J.; Cavallo, F.; Watson, C.J.; et al. Constitutively active Stat3 enhances neu-mediated migration and metastasis in mammary tumors via upregulation of Cten. Cancer Res. 2010, 70, 2558–2567. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Lee, H.; Herrmann, A.; Buettner, R.; Jove, R. Revisiting STAT3 signalling in cancer: New and unexpected biological functions. Nat. Rev. Cancer 2014, 14, 736–746. [Google Scholar] [CrossRef]
- Wang, C.H.; Wang, P.J.; Hsieh, Y.C.; Lo, S.; Lee, Y.C.; Chen, Y.C.; Tsai, C.H.; Chiu, W.C.; Chu-Sung Hu, S.; Lu, C.W.; et al. Resistin facilitates breast cancer progression via TLR4-mediated induction of mesenchymal phenotypes and stemness properties. Oncogene 2018, 37, 589–600. [Google Scholar] [CrossRef]
- Lee, J.O.; Kim, N.; Lee, H.J.; Lee, Y.W.; Kim, S.J.; Park, S.H.; Kim, H.S. Resistin, a fat-derived secretory factor, promotes metastasis of MDA-MB-231 human breast cancer cells through ERM activation. Sci. Rep. 2016, 6, 18923. [Google Scholar] [CrossRef] [Green Version]
- Grossmann, M.E.; Nkhata, K.J.; Mizuno, N.K.; Ray, A.; Cleary, M.P. Effects of adiponectin on breast cancer cell growth and signaling. Br. J. Cancer 2008, 98, 370–379. [Google Scholar] [CrossRef] [Green Version]
- Taliaferro-Smith, L.; Nagalingam, A.; Zhong, D.; Zhou, W.; Saxena, N.K.; Sharma, D. LKB1 is required for adiponectin-mediated modulation of AMPK-S6K axis and inhibition of migration and invasion of breast cancer cells. Oncogene 2009, 28, 2621–2633. [Google Scholar] [CrossRef] [Green Version]
- Taliaferro-Smith, L.; Nagalingam, A.; Knight, B.B.; Oberlick, E.; Saxena, N.K.; Sharma, D. Integral role of PTP1B in adiponectin-mediated inhibition of oncogenic actions of leptin in breast carcinogenesis. Neoplasia 2013, 15, 23–38. [Google Scholar] [CrossRef] [Green Version]
- Bulló, M.; García-Lorda, P.; Peinado-Onsurbe, J.; Hernández, M.; Del Castillo, D.; Argilés, J.M.; Salas-Salvadó, J. TNFalpha expression of subcutaneous adipose tissue in obese and morbid obese females: Relationship to adipocyte LPL activity and leptin synthesis. Int. J. Obes. Relat. Metab. Disord. 2002, 26, 652–658. [Google Scholar] [CrossRef] [Green Version]
- Uysal, K.T.; Wiesbrock, S.M.; Marino, M.W.; Hotamisligil, G.S. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature 1997, 389, 610–614. [Google Scholar] [CrossRef]
- Pfalzer, A.C.; Leung, K.; Crott, J.W.; Kim, S.J.; Tai, A.K.; Parnell, L.D.; Kamanu, F.K.; Liu, Z.; Rogers, G.; Shea, M.K.; et al. Incremental Elevations in TNFα and IL6 in the Human Colon and Procancerous Changes in the Mucosal Transcriptome Accompany Adiposity. Cancer Epidemiol. Biomark. Prev. 2018, 27, 1416–1423. [Google Scholar] [CrossRef] [Green Version]
- Kershaw, E.E.; Flier, J.S. Adipose tissue as an endocrine organ. J. Clin. Endocrinol. Metab. 2004, 89, 2548–2556. [Google Scholar] [CrossRef]
- Vozarova, B.; Weyer, C.; Hanson, K.; Tataranni, P.A.; Bogardus, C.; Pratley, R.E. Circulating interleukin-6 in relation to adiposity, insulin action, and insulin secretion. Obes. Res. 2001, 9, 414–417. [Google Scholar] [CrossRef]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]
- Gonullu, G.; Ersoy, C.; Ersoy, A.; Evrensel, T.; Basturk, B.; Kurt, E.; Oral, B.; Gokgoz, S.; Manavoglu, O. Relation between insulin resistance and serum concentrations of IL-6 and TNF-alpha in overweight or obese women with early stage breast cancer. Cytokine 2005, 31, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Stark, J.R.; Li, H.; Kraft, P.; Giovannucci, E.L.; Stampfer, M.J.; Ma, J.; Mucci, L.A. Circulating prediagnostic interleukin-6 and C-reactive protein and prostate cancer incidence and mortality. Int. J. Cancer 2009, 124, 2683–2689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sartipy, P.; Loskutoff, D.J. Monocyte chemoattractant protein 1 in obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2003, 100, 7265–7270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curat, C.A.; Miranville, A.; Sengenès, C.; Diehl, M.; Tonus, C.; Busse, R.; Bouloumié, A. From blood monocytes to adipose tissue-resident macrophages: Induction of diapedesis by human mature adipocytes. Diabetes 2004, 53, 1285–1292. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef]
- Arkan, M.C.; Hevener, A.L.; Greten, F.R.; Maeda, S.; Li, Z.W.; Long, J.M.; Wynshaw-Boris, A.; Poli, G.; Olefsky, J.; Karin, M. IKK-beta links inflammation to obesity-induced insulin resistance. Nat. Med. 2005, 11, 191–198. [Google Scholar] [CrossRef]
- Hirosumi, J.; Tuncman, G.; Chang, L.; Gorgun, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336. [Google Scholar] [CrossRef]
- Solinas, G.; Vilcu, C.; Neels, J.G.; Maeda, S.; Li, Z.W.; Long, J.M.; Wynshaw-Boris, A.; Poli, G.; Olefsky, J.; Karin, M. JNK1 in hematopoietically derived cells contributes to diet-induced inflammation and insulin resistance without affecting obesity. Cell Metab. 2007, 6, 386–397. [Google Scholar] [CrossRef] [Green Version]
- Unger, R.H. Lipid overload and overflow: Metabolic trauma and the metabolic syndrome. Trends Endocrinol. Metab. 2003, 14, 398–403. [Google Scholar] [CrossRef]
- Yuan, M.; Konstantopoulos, N.; Lee, J.; Hansen, L.; Li, Z.W.; Karin, M.; Shoelson, S.E. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science 2001, 293, 1673–1677. [Google Scholar] [CrossRef]
- Hammes, H.P.; Martin, S.; Federlin, K.; Geisen, K.; Brownlee, M. Aminoguanidine treatment inhibits the development of experimental diabetic retinopathy. Proc. Natl. Acad. Sci. USA 1991, 88, 11555–11558. [Google Scholar] [CrossRef] [Green Version]
- Giardino, I.; Edelstein, D.; Brownlee, M. Nonenzymatic glycosylation in vitro and in bovine endothelial cells alters basic fibroblast growth factor activity. A model for intracellular glycosylation in diabetes. J. Clin. Investig. 1994, 94, 110–117. [Google Scholar] [CrossRef]
- Egger, G.; Liang, G.; Aparicio, A.; Jones, P.A. Epigenetics in human disease and prospects for epigenetic therapy. Nature 2004, 429, 457–463. [Google Scholar] [CrossRef]
- Sharma, M.; Li, Y.; Stoll, M.L.; Tollefsbol, T.O. The Epigenetic Connection Between the Gut Microbiome in Obesity and Diabetes. Front. Genet. 2020, 10, 1329. [Google Scholar] [CrossRef] [Green Version]
- Crujeiras, A.B.; Casanueva, F.F. Obesity and the reproductive system disorders: Epigenetics as a potential bridge. Hum. Reprod. Update 2015, 21, 249–261. [Google Scholar] [CrossRef] [Green Version]
- Kwak, S.H.; Park, K.S. Recent progress in genetic and epigenetic research on type 2 diabetes. Exp. Mol. Med. 2016, 48, e220. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef] [Green Version]
- Ling, C.; Rönn, T. Epigenetics in Human Obesity and Type 2 Diabetes. Cell Metab. 2019, 29, 1028–1044. [Google Scholar] [CrossRef] [Green Version]
- Ramos-Lopez, O.; Riezu-Boj, J.I.; Milagro, F.I.; Martinez, J.A.; MENA Project. DNA methylation signatures at endoplasmic reticulum stress genes are associated with adiposity and insulin resistance. Mol. Genet. Metab. 2018, 123, 50–58. [Google Scholar] [CrossRef]
- You, D.; Nilsson, E.; Tenen, D.E.; Lyubetskaya, A.; Lo, J.C.; Jiang, R.; Deng, J.; Dawes, B.A.; Vaag, A.; Ling, C.; et al. Dnmt3a is an epigenetic mediator of adipose insulin resistance. Elife 2017, 6, e30766. [Google Scholar] [CrossRef]
- Walaszczyk, E.; Luijten, M.; Spijkerman, A.M.W.; Bonder, M.J.; Lutgers, H.L.; Snieder, H.; Wolffenbuttel, B.H.R.; van Vliet-Ostaptchouk, J.V. DNA methylation markers associated with type 2 diabetes, fasting glucose and HbA1c levels: A systematic review and replication in a case-control sample of the Lifelines study. Diabetologia 2018, 61, 354–368. [Google Scholar] [CrossRef] [Green Version]
- Pedroso, J.A.B.; Ramos-Lobo, A.M.; Donato, J., Jr. SOCS3 as a future target to treat metabolic disorders. Hormones 2019, 18, 127–136. [Google Scholar] [CrossRef]
- Robinson, N.; Brown, H.; Antoun, E.; Godfrey, K.M.; Hanson, M.A.; Lillycrop, K.A.; Crozier, S.R.; Murray, R.; Pearce, M.S.; Relton, C.L.; et al. Childhood DNA methylation as a marker of early life rapid weight gain and subsequent overweight. Clin. Epigenetics 2021, 13, 8. [Google Scholar] [CrossRef]
- Orouji, E.; Utikal, J. Tackling malignant melanoma epigenetically: Histone lysine methylation. Clin. Epigenetics 2018, 10, 145. [Google Scholar] [CrossRef]
- Audia, J.E.; Campbell, R.M. Histone Modifications and Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019521. [Google Scholar] [CrossRef]
- Paneni, F.; Costantino, S.; Battista, R.; Castello, L.; Capretti, G.; Chiandotto, S.; Scavone, G.; Villano, A.; Pitocco, D.; Lanza, G.; et al. Adverse epigenetic signatures by histone methyltransferase Set7 contribute to vascular dysfunction in patients with type 2 diabetes mellitus. Circ. Cardiovasc. Genet. 2015, 8, 150–158. [Google Scholar] [CrossRef]
- Miao, F.; Wu, X.; Zhang, L.; Yuan, Y.C.; Riggs, A.D.; Natarajan, R. Genome-wide analysis of histone lysine methylation variations caused by diabetic conditions in human monocytes. J. Biol. Chem. 2007, 282, 13854–13863. [Google Scholar] [CrossRef] [Green Version]
- Greco, M.; Chiefari, E.; Accattato, F.; Corigliano, D.M.; Arcidiacono, B.; Mirabelli, M.; Liguori, R.; Brunetti, F.S.; Pullano, S.A.; Scorcia, V.; et al. MicroRNA-1281 as a Novel Circulating Biomarker in Patients with Diabetic Retinopathy. Front. Endocrinol. 2020, 11, 528. [Google Scholar] [CrossRef]
- Chen, B.; Li, J.; Chi, D.; Sahnoune, I.; Calin, S.; Girnita, L.; Calin, G.A. Non-Coding RNAs in IGF-1R Signaling Regulation: The Underlying Pathophysiological Link between Diabetes and Cancer. Cells 2019, 8, 1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Statello, L.; Guo, C.J.; Chen, L.L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, M.; Garzon, R.; Andreeff, M.; Kantarjian, H.M.; Garcia-Manero, G.; Calin, G.A. MicroRNAs and noncoding RNAs in hematological malignancies: Molecular, clinical and therapeutic implications. Leukemia 2008, 22, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- Trajkovski, M.; Hausser, J.; Soutschek, J.; Bhat, B.; Akin, A.; Zavolan, M.; Heim, M.H.; Stoffel, M. MicroRNAs 103 and 107 regulate insulin sensitivity. Nature 2011, 474, 649–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martello, G.; Rosato, A.; Ferrari, F.; Manfrin, A.; Cordenonsi, M.; Dupont, S.; Enzo, E.; Guzzardo, V.; Rondina, M.; Spruce, T.; et al. A MicroRNA targeting dicer for metastasis control. Cell 2010, 141, 1195–1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.Y.; Lang, Y.D.; Lin, H.N.; Liu, Y.R.; Liao, C.C.; Nana, A.W.; Yen, Y.; Chen, R.H. miR-103/107 prolong Wnt/β-catenin signaling and colorectal cancer stemness by targeting Axin2. Sci. Rep. 2019, 9, 9687. [Google Scholar] [CrossRef] [Green Version]
- Yu, Q.F.; Liu, P.; Li, Z.Y.; Zhang, C.F.; Chen, S.Q.; Li, Z.H.; Zhang, G.Y.; Li, J.C. MiR-103/107 induces tumorigenicity in bladder cancer cell by suppressing PTEN. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 8616–8623. [Google Scholar] [CrossRef]
- Bulger, D.A.; Conley, J.; Conner, S.H.; Majumdar, G.; Solomon, S.S. Role of PTEN in TNFα induced insulin resistance. Biochem. Biophys. Res. Commun. 2015, 461, 533–536. [Google Scholar] [CrossRef] [Green Version]
- Pal, A.; Barber, T.M.; Van de Bunt, M.; Rudge, S.A.; Zhang, Q.; Lachlan, K.L.; Cooper, N.S.; Linden, H.; Levy, J.C.; Wakelam, M.J.; et al. PTEN mutations as a cause of constitutive insulin sensitivity and obesity. N. Engl. J. Med. 2012, 367, 1002–1011. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Naizabekov, S.; Chen, Z.; Tokay, T. Power of PTEN/AKT: Molecular switch between tumor suppressors and oncogenes. Oncol. Lett. 2016, 12, 375–378. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Sobenin, I.A.; Orekhov, A.N.; Bobryshev, Y.V. Human miR-221/222 in Physiological and Atherosclerotic Vascular Remodeling. Biomed. Res. Int. 2015, 2015, 354517. [Google Scholar] [CrossRef]
- Meerson, A.; Traurig, M.; Ossowski, V.; Fleming, J.M.; Mullins, M.; Baier, L.J. Human adipose microRNA-221 is upregulated in obesity and affects fat metabolism downstream of leptin and TNF-α. Diabetologia 2013, 56, 1971–1979. [Google Scholar] [CrossRef] [Green Version]
- Cochrane, D.R.; Cittelly, D.M.; Howe, E.N.; Spoelstra, N.S.; McKinsey, E.L.; LaPara, K.; Elias, A.; Yee, D.; Richer, J.K. MicroRNAs link estrogen receptor alpha status and Dicer levels in breast cancer. Horm. Cancer 2010, 1, 306–319. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Yang, H.; Lu, X.Q.; Xu, F.; Li, J.; Qian, J. ARHI suppresses pancreatic cancer by regulating MAPK/ERK 1/2 pathway. Pancreas 2015, 44, 342–343. [Google Scholar] [CrossRef]
- Chen, Y.; Zaman, M.S.; Deng, G.; Majid, S.; Saini, S.; Liu, J.; Tanaka, Y.; Dahiya, R. MicroRNAs 221/222 and genistein-mediated regulation of ARHI tumor suppressor gene in prostate cancer. Cancer Prev. Res. 2011, 4, 76–86. [Google Scholar] [CrossRef] [Green Version]
- Mari, E.; Zicari, A.; Fico, F.; Massimi, I.; Martina, L.; Mardente, S. Action of HMGB1 on miR-221/222 cluster in neuroblastoma cell lines. Oncol. Lett. 2016, 12, 2133–2138. [Google Scholar] [CrossRef] [Green Version]
- Di Martino, M.T.; Rossi, M.; Caracciolo, D.; Gullà, A.; Tagliaferri, P.; Tassone, P. Mir-221/222 are promising targets for innovative anticancer therapy. Expert Opin. Ther. Targets 2016, 20, 1099–1108. [Google Scholar] [CrossRef]
- Howe, E.N.; Cochrane, D.R.; Richer, J.K. The miR-200 and miR-221/222 microRNA families: Opposing effects on epithelial identity. J. Mammary Gland Biol. Neoplasia. 2012, 17, 65–77. [Google Scholar] [CrossRef] [Green Version]
- Ślusarz, A.; Pulakat, L. The two faces of miR-29. J. Cardiovasc. Med. 2015, 16, 480–490. [Google Scholar] [CrossRef]
- Baran-Gale, J.; Fannin, E.E.; Kurtz, C.L.; Sethupathy, P. Beta cell 5’-shifted isomiRs are candidate regulatory hubs in type 2 diabetes. PLoS ONE 2013, 8, e73240. [Google Scholar] [CrossRef]
- He, A.; Zhu, L.; Gupta, N.; Chang, Y.; Fang, F. Overexpression of micro ribonucleic acid 29, highly up-regulated in diabetic rats, leads to insulin resistance in 3T3-L1 adipocytes. Mol. Endocrinol. 2007, 21, 2785–2794. [Google Scholar] [CrossRef]
- Rottiers, V.; Näär, A.M. MicroRNAs in metabolism and metabolic disorders. Nat. Rev. Mol. Cell Biol. 2012, 13, 239–250. [Google Scholar] [CrossRef]
- Vivacqua, A.; De Marco, P.; Belfiore, A.; Maggiolini, M. Recent Advances on the Role of microRNAs in both Insulin Resistance and Cancer. Curr. Pharm. Des. 2017, 23, 3658–3666. [Google Scholar] [CrossRef]
- Kwon, J.J.; Factora, T.D.; Dey, S.; Kota, J. A Systematic Review of miR-29 in Cancer. Mol. Ther. Oncolytics 2018, 12, 173–194. [Google Scholar] [CrossRef] [Green Version]
- Pei, Y.F.; Lei, Y.; Liu, X.Q. MiR-29a promotes cell proliferation and EMT in breast cancer by targeting ten eleven translocation 1. Biochim. Biophys. Acta 2016, 1862, 2177–2185. [Google Scholar] [CrossRef]
- Yoshihara, H.; Fukushima, T.; Hakuno, F.; Saeki, Y.; Tanaka, K.; Ito, A.; Yoshida, M.; Iemura, S.; Natsume, T.; Asano, T.; et al. Insulin/insulin-like growth factor (IGF) stimulation abrogates an association between a deubiquitinating enzyme USP7 and insulin receptor substrates (IRSs) followed by proteasomal degradation of IRSs. Biochem. Biophys. Res. Commun. 2012, 423, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Shyh-Chang, N.; Segrè, A.V.; Shinoda, G.; Shah, S.P.; Einhorn, W.S.; Takeuchi, A.; Engreitz, J.M.; Hagan, J.P.; Kharas, M.G.; et al. The Lin28/let-7 axis regulates glucose metabolism. Cell 2011, 147, 81–94. [Google Scholar] [CrossRef] [Green Version]
- Pérez, L.M.; Bernal, A.; San Martín, N.; Lorenzo, M.; Fernández-Veledo, S.; Gálvez, B.G. Metabolic rescue of obese adipose-derived stem cells by Lin28/Let7 pathway. Diabetes 2013, 62, 2368–2379. [Google Scholar] [CrossRef] [Green Version]
- Frost, R.J.; Olson, E.N. Control of glucose homeostasis and insulin sensitivity by the Let-7 family of microRNAs. Proc. Natl. Acad. Sci USA 2011, 108, 21075–21080. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Ren, Y.; Shi, E.; Tan, Z.; Xiong, J.; Yan, L.; Jiang, X. Inhibition of the Let-7 Family MicroRNAs Induces Cardioprotection Against Ischemia-Reperfusion Injury in Diabetic Rats. Ann. Thorac. Surg. 2016, 102, 829–835. [Google Scholar] [CrossRef] [Green Version]
- Baldeón, R.L.; Weigelt, K.; de Wit, H.; Ozcan, B.; van Oudenaren, A.; Sempértegui, F.; Sijbrands, E.; Grosse, L.; van Zonneveld, A.J.; Drexhage, H.A.; et al. Type 2 Diabetes Monocyte MicroRNA and mRNA Expression: Dyslipidemia Associates with Increased Differentiation-Related Genes but Not Inflammatory Activation. PLoS ONE 2015, 10, e0129421. [Google Scholar] [CrossRef] [Green Version]
- Deiuliis, J.A.; Syed, R.; Duggineni, D.; Rutsky, J.; Rengasamy, P.; Zhang, J.; Huang, K.; Needleman, B.; Mikami, D.; Perry, K.; et al. Visceral Adipose MicroRNA 223 Is Upregulated in Human and Murine Obesity and Modulates the Inflammatory Phenotype of Macrophages. PLoS ONE 2016, 11, e0165962. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Pan, J.; Zhao, D.; Liu, F. MicroRNA-223 inhibits deposition of the extracellular matrix by airway smooth muscle cells through targeting IGF-1R in the PI3K/Akt pathway. Am. J. Transl. Res. 2018, 10, 744–752. [Google Scholar] [PubMed]
- Josse, C.; Bouznad, N.; Geurts, P.; Irrthum, A.; Huynh-Thu, V.A.; Servais, L.; Hego, A.; Delvenne, P.; Bours, V.; Oury, C. Identification of a microRNA landscape targeting the PI3K/Akt signaling pathway in inflammation-induced colorectal carcinogenesis. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, G229–G243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Xie, L.; He, X.; Li, J.; Tu, K.; Wei, L.; Wu, J.; Guo, Y.; Ma, X.; Zhang, P.; et al. Diagnostic and prognostic implications of microRNAs in human hepatocellular carcinoma. Int. J. Cancer 2008, 123, 1616–1622. [Google Scholar] [CrossRef]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef] [Green Version]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [Green Version]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [Green Version]
- Jordan, S.D.; Krüger, M.; Willmes, D.M.; Redemann, N.; Wunderlich, F.T.; Brönneke, H.S.; Merkwirth, C.; Kashkar, H.; Olkkonen, V.M.; Böttger, T.; et al. Obesity-induced overexpression of miRNA-143 inhibits insulin-stimulated AKT activation and impairs glucose metabolism. Nat. Cell Biol. 2011, 13, 434–446. [Google Scholar] [CrossRef]
- Maroulakou, I.G.; Struhl, K.; Tsichlis, P.N.; Iliopoulos, D.; Polytarchou, C.; Hatziapostolou, M.; Kottakis, F. MicroRNAs differentially regulated by Akt isoforms control EMT and stem cell renewal in cancer cells. Sci. Signal. 2009, 2, ra62. [Google Scholar] [CrossRef] [Green Version]
- Nagaraja, A.K.; Creighton, C.J.; Yu, Z.; Zhu, H.; Gunaratne, P.H.; Reid, J.G.; Olokpa, E.; Itamochi, H.; Ueno, N.T.; Hawkins, S.M.; et al. A link between mir-100 and FRAP1/mTOR in clear cell ovarian cancer. Mol. Endocrinol. 2010, 24, 447–463. [Google Scholar] [CrossRef] [Green Version]
- Fornari, F.; Milazzo, M.; Chieco, P.; Negrini, M.; Calin, G.A.; Grazi, G.L.; Pollutri, D.; Croce, C.M.; Bolondi, L.; Gramantieri, L. MiR-199a-3p regulates mTOR and c-Met to influence the doxorubicin sensitivity of human hepatocarcinoma cells. Cancer Res. 2010, 70, 5184–5193. [Google Scholar] [CrossRef] [Green Version]
- Matsha, T.E.; Kengne, A.P.; Hector, S.; Mbu, D.L.; Yako, Y.Y.; Erasmus, R.T. MicroRNA profiling and their pathways in South African individuals with prediabetes and newly diagnosed type 2 diabetes mellitus. Oncotarget 2018, 9, 30485–30498. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Li, X.; Quan, X.; Yang, X.; Zheng, C.; Hao, X.; Qu, R.; Zhou, B. MiR-486 as an effective biomarker in cancer diagnosis and prognosis: A systematic review and meta-analysis. Oncotarget 2018, 9, 13948–13958. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Dai, Y.; Hitchcock, C.; Yang, X.; Kassis, E.S.; Liu, L.; Luo, Z.; Sun, H.L.; Cui, R.; Wei, H.; et al. Insulin growth factor signaling is regulated by microRNA-486, an underexpressed microRNA in lung cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 15043–15048. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.J.; Yuan, W.D.; Yuan, J.Q.; Yuan, K.; Wang, Y. miR-486-5p functions as an oncogene by targeting PTEN in non-small cell lung cancer. Pathol. Res. Pract. 2018, 214, 700–705. [Google Scholar] [CrossRef]
- Wang, J.; Tian, X.; Han, R.; Zhang, X.; Wang, X.; Shen, H.; Xue, L.; Liu, Y.; Yan, X.; Shen, J.; et al. Downregulation of miR-486-5p contributes to tumor progression and metastasis by targeting protumorigenic ARHGAP5 in lung cancer. Oncogene 2014, 33, 1181–1189. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Cui, C.; Xiao, F.; Wang, H.; Xu, J.; Shi, X.; Yang, Y.; Zhang, Q.; Zheng, X.; Yang, X.; et al. miR-486 regulates metastasis and chemosensitivity in hepatocellular carcinoma by targeting CLDN10 and CITRON. Hepatol. Res. 2015, 45, 1312–1322. [Google Scholar] [CrossRef]
- Huang, X.P.; Hou, J.; Shen, X.Y.; Huang, C.Y.; Zhang, X.H.; Xie, Y.A.; Luo, X.L. MicroRNA-486-5p, which is downregulated in hepatocellular carcinoma, suppresses tumor growth by targeting PIK3R1. FEBS J. 2015, 282, 579–594. [Google Scholar] [CrossRef] [Green Version]
- Lang, H.; Xiang, Y.; Lin, N.; Ai, Z.; You, Z.; Xiao, J.; Liu, D.; Yang, Y. Identification of a Panel of MiRNAs as Positive Regulators of Insulin Release in Pancreatic Β-Cells. Cell Physiol. Biochem. 2018, 48, 185–193. [Google Scholar] [CrossRef]
- Xu, Y.; Chen, J.; Gao, C.; Zhu, D.; Xu, X.; Wu, C.; Jiang, J. MicroRNA-497 inhibits tumor growth through targeting insulin receptor substrate 1 in colorectal cancer. Oncol. Lett. 2017, 14, 6379–6386. [Google Scholar] [CrossRef] [Green Version]
- Luo, M.; Shen, D.; Zhou, X.; Chen, X.; Wang, W. MicroRNA-497 is a potential prognostic marker in human cervical cancer and functions as a tumor suppressor by targeting the insulin-like growth factor 1 receptor. Surgery 2013, 153, 836–847. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Kang, Y.; Ning, L.; Tan, J.; Wang, H.; Ying, Y. Identification of microRNAs involved in gefitinib resistance of non-small-cell lung cancer through the insulin-like growth factor receptor 1 signaling pathway. Exp. Ther. Med. 2017, 14, 2853–2862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, D.; Tu, M.; Zeng, B.; Cai, L.; Zheng, W.; Su, Z.; Yu, Z. Up-regulation of miR-497 confers resistance to temozolomide in human glioma cells by targeting mTOR/Bcl-2. Cancer Med. 2017, 6, 452–462. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Guo, Y.; Wang, X.J.; Duan, B.H.; Li, L. HOTAIR participates in hepatic insulin resistance via regulating SIRT1. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 7883–7890. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Wu, Y.B.; Zhou, J.; Kang, D.M. Upregulation of lncRNA MEG3 promotes hepatic insulin resistance via increasing FoxO1 expression. Biochem. Biophys. Res. Commun. 2016, 469, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Li, J.; Feng, S.; Li, Y.; Tan, L. Long noncoding RNA Gomafu upregulates Foxo1 expression to promote hepatic insulin resistance by sponging miR-139-5p. Cell Death Dis. 2018, 9, 289. [Google Scholar] [CrossRef] [PubMed]
- Carter, G.; Miladinovic, B.; Patel, A.A.; Deland, L.; Mastorides, S.; Patel, N.A. Circulating long noncoding RNA GAS5 levels are correlated to prevalence of type 2 diabetes mellitus. BBA Clin. 2015, 4, 102–107. [Google Scholar] [CrossRef] [Green Version]
- Sathishkumar, C.; Prabu, P.; Mohan, V.; Balasubramanyam, M. Linking a role of lncRNAs (long non-coding RNAs) with insulin resistance, accelerated senescence, and inflammation in patients with type 2 diabetes. Hum. Genom. 2018, 12, 41. [Google Scholar] [CrossRef]
- Shi, X.; Sun, M.; Liu, H.; Yao, Y.; Kong, R.; Chen, F.; Song, Y. A critical role for the long non-coding RNA GAS5 in proliferation and apoptosis in non-small-cell lung cancer. Mol. Carcinog. 2015, 54 (Suppl. 1), E1–E12. [Google Scholar] [CrossRef]
- Mourtada-Maarabouni, M.; Pickard, M.R.; Hedge, V.L.; Farzaneh, F.; Williams, G.T. GAS5, a non-protein-coding RNA, controls apoptosis and is downregulated in breast cancer. Oncogene 2009, 28, 195–208. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Li, W.; Sun, Y.; Yu, D.; Wen., X.; Wang, H.; Cui, J.; Wang, G.; Hoffman, A.R.; Hu, J.F. A novel antisense long noncoding RNA within the IGF1R gene locus is imprinted in hematopoietic malignancies. Nucleic Acids Res. 2014, 42, 9588–9601. [Google Scholar] [CrossRef] [Green Version]
- Kang, L.; Sun, J.; Wen, X.; Cui, J.; Wang, G.; Hoffman, A.; Hu, J.F.; Li, W. Aberrant allele-switch imprinting of a novel IGF1R intragenic antisense non-coding RNA in breast cancers. Eur. J. Cancer 2015, 51, 260–270. [Google Scholar] [CrossRef]
- Pian, L.; Wen, X.; Kang, L.; Li, Z.; Nie, Y.; Du, Z.; Yu, D.; Zhou, L.; Jia, L.; Chen, N.; et al. Targeting the IGF1R Pathway in Breast Cancer Using Antisense lncRNA-Mediated Promoter cis Competition. Mol. Ther. Nucleic Acids 2018, 12, 105–117. [Google Scholar] [CrossRef]
- Ji, P.; Diederichs, S.; Wang, W.; Böing, S.; Metzger, R.; Schneider, P.M.; Tidow, N.; Brandt, B.; Buerger, H.; Bulk, E.; et al. MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene 2003, 22, 8031–8041. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.Y.; Yao, J.; Li, X.M.; Song, Y.C.; Wang, X.Q.; Li, Y.J.; Yan, B.; Jiang, Q. Pathogenic role of lncRNA-MALAT1 in endothelial cell dysfunction in diabetes mellitus. Cell Death Dis. 2014, 5, e1506. [Google Scholar] [CrossRef] [Green Version]
- Kong, Y.; Hsieh, C.H.; Alonso, L.C. ANRIL: A lncRNA at the CDKN2A/B Locus with Roles in Cancer and Metabolic Disease. Front. Endocrinol. 2018, 9, 405. [Google Scholar] [CrossRef] [Green Version]
- Broadbent, H.M.; Peden, J.F.; Lorkowski, S.; Goel, A.; Ongen, H.; Green, F.; Clarke, R.; Collins, R.; Franzosi, M.G.; Tognoni, G.; et al. Susceptibility to coronary artery disease and diabetes is encoded by distinct, tightly linked SNPs in the ANRIL locus on chromosome 9p. Hum. Mol. Genet. 2008, 17, 806–814. [Google Scholar] [CrossRef]
- Sherwood, L.; Willey, J.; Woolverton, C. Prescott’s Microbiology, 9th ed.; Mc Graw Hill: New York, NY, USA, 2013. [Google Scholar]
- Rowland, I.R. Toxicological implications of the normal microflora. In Medical importance of the normal microflora; Tannock, G.W., Ed.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1999. [Google Scholar]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef]
- Vrieze, A.; Van Nood, E.; Holleman, F.; Salojärvi, J.; Kootte, R.S.; Bartelsman, J.F.; Dallinga-Thie, G.M.; Ackermans, M.T.; Serlie, M.J.; Oozeer, R.; et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 2012, 143, 913–916.e7. [Google Scholar] [CrossRef]
- Zhou, H.; Zhao, X.; Sun, L.; Liu, Y.; Lv, Y.; Gang, X.; Wang, G. Gut Microbiota Profile in Patients with Type 1 Diabetes Based on 16S rRNA Gene Sequencing: A Systematic Review. Dis Markers 2020, 2020, 3936247. [Google Scholar] [CrossRef]
- Lee, C.J.; Sears, C.L.; Maruthur, N. Gut microbiome and its role in obesity and insulin resistance. Ann. N. Y. Acad. Sci. 2020, 1461, 37–52. [Google Scholar] [CrossRef]
- Parida, S.; Sharma, D. The Microbiome and Cancer: Creating Friendly Neighborhoods and Removing the Foes Within. Cancer Res. 2021, 81, 790–800. [Google Scholar] [CrossRef]
- Heianza, Y.; Sun, D.; Li, X.; DiDonato, J.A.; Bray, G.A.; Sacks, F.M.; Qi, L. Gut microbiota metabolites, amino acid metabolites and improvements in insulin sensitivity and glucose metabolism: The POUNDS Lost trial. Gut 2019, 68, 263–270. [Google Scholar] [CrossRef]
- Everard, A.; Belzer, C.; Geurts, L.; Ouwerkerk, J.P.; Druart, C.; Bindels, L.B.; Guiot, Y.; Derrien, M.; Muccioli, G.G.; Delzenne, N.M.; et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc. Natl. Acad. Sci. USA 2013, 110, 9066–9071. [Google Scholar] [CrossRef] [Green Version]
- Den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, Y.; Adachi, K.; Sugiyama, T.; Shimozato, A.; Ebi, M.; Ogasawara, N.; Funaki, Y.; Goto, C.; Sasaki, M.; Kasugai, K. Association of Intestinal Microbiota with Metabolic Markers and Dietary Habits in Patients with Type 2 Diabetes. Digestion 2016, 94, 66–72. [Google Scholar] [CrossRef]
- Whitt, J.; Woo, V.; Lee, P.; Moncivaiz, J.; Haberman, Y.; Denson, L.; Tso, P.; Alenghat, T. Disruption of Epithelial HDAC3 in Intestine Prevents Diet-Induced Obesity in Mice. Gastroenterology 2018, 155, 501–513. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Fan, C.; Li, P.; Lu, Y.; Chang, X.; Qi, K. Short Chain Fatty Acids Prevent High-fat-diet-induced Obesity in Mice by Regulating G Protein-coupled Receptors and Gut Microbiota. Sci. Rep. 2016, 6, 37589. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, J.; Su, W.; Rahat-Rozenbloom, S.; Wolever, T.M.; Comelli, E.M. Adiposity, gut microbiota and faecal short chain fatty acids are linked in adult humans. Nutr Diabetes 2014, 4, e121. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef]
- Baker, J.M.; Al-Nakkash, L.; Herbst-Kralovetz, M.M. Estrogen-gut microbiome axis: Physiological and clinical implications. Maturitas 2017, 103, 45–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwa, M.; Plottel, C.S.; Blaser, M.J.; Adams, S. The Intestinal Microbiome and Estrogen Receptor-Positive Female Breast Cancer. J. Natl. Cancer Inst. 2016, 108, djw029. [Google Scholar] [CrossRef]
- Mirabelli, M.; Chiefari, E.; Tocci, V.; Caroleo, P.; Giuliano, S.; Greco, E.; Luque, R.M.; Puccio, L.; Foti, D.P.; Aversa, A.; et al. Clinical Effectiveness and Safety of Once-Weekly GLP-1 Receptor Agonist Dulaglutide as Add-On to Metformin or Metformin Plus Insulin Secretagogues in Obesity and Type 2 Diabetes. J. Clin. Med. 2021, 10, 985. [Google Scholar] [CrossRef] [PubMed]
- Schiel, R.; Müller, U.A.; Braun, A.; Stein, G.; Kath, R. Risk of malignancies in patients with insulin-treated diabetes mellitus: Results of a population-based trial with 10-year follow-up (JEVIN). Eur. J. Med. Res. 2005, 10, 339–344. [Google Scholar]
- Chang, C.H.; Lin, J.W.; Wu, L.C.; Lai, M.S.; Chuang, L.M. Oral insulin secretagogues, insulin, and cancer risk in type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 2012, 97, E1170–E1175. [Google Scholar] [CrossRef]
- American Diabetes Association. 9. Pharmacologic Approaches to Glycemic Treatment: Standards of Medical Care in Diabetes-2020. Diabetes Care. 2020, 43, S98–S110. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.S.; Choi, E.A.; Lee, J.W.; Kim, Y.; You, H.S.; Han, Y.E.; Kim, H.S.; Bae, Y.J.; Kang, H.T.; Kim, J. Metformin use reduced the overall risk of cancer in diabetic patients: A study based on the Korean NHIS-HEALS cohort. Nutr. Metab. Cardiovasc Dis. 2020, 30, 1714–1722. [Google Scholar] [CrossRef]
- Heckman-Stoddard, B.M.; DeCensi, A.; Sahasrabuddhe, V.V.; Ford, L.G. Repurposing metformin for the prevention of cancer and cancer recurrence. Diabetologia 2017, 60, 1639–1647. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Chen, K.; Jia, X.; Tian, Y.; Dai, Y.; Li, D.; Xie, J.; Tao, M.; Mao, Y. Metformin Use Is Associated with Better Survival of Breast Cancer Patients With Diabetes: A Meta-Analysis. Oncologist 2015, 20, 1236–1244. [Google Scholar] [CrossRef] [Green Version]
- Chlebowski, R.T.; McTiernan, A.; Wactawski-Wende, J.; Manson, J.E.; Aragaki, A.K.; Rohan, T.; Ipp, E.; Kaklamani, V.G.; Vitolins, M.; Wallace, R. ; et al. Diabetes, metformin, and breast cancer in postmenopausal women. J. Clin. Oncol. 2012, 30, 2844–2852. [Google Scholar] [CrossRef]
- Samuel, S.M.; Varghese, E.; Kubatka, P.; Triggle, C.R.; Büsselberg, D. Metformin: The Answer to Cancer in a Flower? Current Knowledge and Future Prospects of Metformin as an Anti-Cancer Agent in Breast Cancer. Biomolecules 2019, 9, 846. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Chen, J.B.; Cui, Y.; Zhu, Y.W.; Ren, W.B.; Zhou, X.; Liu, L.F.; Chen, H.Q.; Zu, X.B. Association of metformin intake with bladder cancer risk and oncologic outcomes in type 2 diabetes mellitus patients: A systematic review and meta-analysis. Medicine 2018, 97, e11596. [Google Scholar] [CrossRef]
- Peng, M.; Su, Q.; Zeng, Q.; Li, L.; Liu, Z.; Xue, L.; Cheng, Y.; Huang, Y.; Tao, T.; Lv, H.; et al. High efficacy of intravesical treatment of metformin on bladder cancer in preclinical model. Oncotarget 2016, 7, 9102–9117. [Google Scholar] [CrossRef] [Green Version]
- Molenaar, R.J.; van Hattum, J.W.; Brummelhuis, I.S.; Oddens, J.R.; Savci-Heijink, C.D.; Boevé, E.R.; van der Meer, S.A.; Witjes, J.F.; Pollak, M.N.; de Reijke, T.M.; et al. Study protocol of a phase II clinical trial of oral metformin for the intravesical treatment of non-muscle invasive bladder cancer. BMC Cancer 2019, 19, 1133. [Google Scholar] [CrossRef] [Green Version]
- Nayan, M.; Bhindi, B.; Yu, J.L.; Hermanns, T.; Mohammed, A.; Hamilton, R.J.; Finelli, A.; Jewett, M.A.; Zlotta, A.R.; Fleshner, N.E.; et al. The effect of metformin on cancer-specific survival outcomes in diabetic patients undergoing radical cystectomy for urothelial carcinoma of the bladder. Urol. Oncol. 2015, 33, 386. [Google Scholar] [CrossRef]
- Yu, H.; Yin, L.; Jiang, X.; Sun, X.; Wu, J.; Tian, H.; Gao, X.; He, X. Effect of metformin on cancer risk and treatment outcome of prostate cancer: A meta-analysis of epidemiological observational studies. PLoS ONE 2014, 9, e116327. [Google Scholar] [CrossRef] [Green Version]
- He, K.; Hu, H.; Ye, S.; Wang, H.; Cui, R.; Yi, L. The effect of metformin therapy on incidence and prognosis in prostate cancer: A systematic review and meta-analysis. Sci. Rep. 2019, 9, 2218. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Tong, D.; Liu, G.; Gao, J.; Wang, L.A.; Xu, J.; Yang, X.; Xie, Q.; Huang, Y.; Pang, J.; et al. Metformin Inhibits Prostate Cancer Progression by Targeting Tumor-Associated Inflammatory Infiltration. Clin. Cancer Res. 2018, 24, 5622–5634. [Google Scholar] [CrossRef] [Green Version]
- Mark, M.; Klingbiel, D.; Mey, U.; Winterhalder, R.; Rothermundt, C.; Gillessen, S.; von Moos, R.; Pollak, M.; Manetsch, G.; Strebel, R.; et al. Impact of Addition of Metformin to Abiraterone in Metastatic Castration-Resistant Prostate Cancer Patients with Disease Progressing While Receiving Abiraterone Treatment (MetAb-Pro): Phase 2 Pilot Study. Clin. Genitourin. Cancer 2019, 17, e323–e328. [Google Scholar] [CrossRef]
- Gong, T.T.; Wu, Q.J.; Lin, B.; Ruan, S.K.; Kushima, M.; Takimoto, M. Observational Studies on the Association Between Post-diagnostic Metformin Use and Survival in Ovarian Cancer: A Systematic Review and Meta-Analysis. Front. Oncol. 2019, 9, 458. [Google Scholar] [CrossRef]
- Chu, D.; Wu, J.; Wang, K.; Zhao, M.; Wang, C.; Li, L.; Guo, R. Effect of metformin use on the risk and prognosis of endometrial cancer: A systematic review and meta-analysis. BMC Cancer 2018, 18, 438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, Q.; Zhao, Z.; Wen, J.; Zhou, J.; Wu, J.; Lei, S.; Miao, Y. The association between metformin therapy and risk of gynecological cancer in patients: Two meta-analyses. Eur. J. Obstet. Gynecol. Reprod. Biol. 2019, 237, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Liu, M.; Huang, Y.; Wu, K.; Huang, X.; Zhao, Y.; He, W.; Zhang, R. Metformin Use and Lung Cancer Risk in Diabetic Patients: A Systematic Review and Meta-Analysis. Dis. Mark. 2019, 2019, 6230162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, K.; Liu, F.; Liu, J.; Xu, J.; Wu, Q.; Li, X. The effect of metformin on lung cancer risk and survival in patients with type 2 diabetes mellitus: A meta-analysis. J. Clin. Pharm. Ther. 2020, 45, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Chen, X.; Wang, L.; Yang, B.; Cai, S. Metformin Adjunct with Antineoplastic Agents for the Treatment of Lung Cancer: A Meta-Analysis of Randomized Controlled Trials and Observational Cohort Studies. Front. Pharmacol. 2021, 12, 639016. [Google Scholar] [CrossRef]
- Cao, X.; Wu, Y.; Wang, J.; Liu, K.; Wang, X. The Effect of Metformin on Mortality Among Diabetic Cancer Patients: A Systematic Review and Meta-analysis. JNCI Cancer Spectr. 2017, 1, pkx007. [Google Scholar] [CrossRef] [Green Version]
- Shuai, Y.; Li, C.; Zhou, X. The effect of metformin on gastric cancer in patients with type 2 diabetes: A systematic review and meta-analysis. Clin. Transl. Oncol. 2020, 22, 1580–1590. [Google Scholar] [CrossRef]
- Xin, W.; Fang, L.; Fang, Q.; Zheng, X.; Huang, P. Effects of metformin on survival outcomes of pancreatic cancer patients with diabetes: A meta-analysis. Mol. Clin. Oncol. 2018, 8, 483–488. [Google Scholar] [CrossRef]
- Wang, Y.; Xiao, J.; Zhao, Y.; Du, S.; Du, J. Effect of metformin on the mortality of colorectal cancer patients with T2DM: Meta-analysis of sex differences. Int. J. Colorectal Dis. 2020, 35, 827–835. [Google Scholar] [CrossRef]
- Vancura, A.; Bu, P.; Bhagwat, M.; Zeng, J.; Vancurova, I. Metformin as an Anticancer Agent. Trends Pharmacol. Sci. 2018, 39, 867–878. [Google Scholar] [CrossRef]
- Ling, S.; Tian, Y.; Zhang, H.; Jia, K.; Feng, T.; Sun, D.; Gao, Z.; Xu, F.; Hou, Z.; Li, Y.; et al. Metformin reverses multidrug resistance in human hepatocellular carcinoma Bel-7402/5-fluorouracil cells. Mol. Med. Rep. 2014, 10, 2891–2897. [Google Scholar] [CrossRef] [Green Version]
- Messineo, S.; Arcidiacono, B.; Corigliano, D.M.; Foti, D.P.; Castano, J.P.; Luque, R.M.; Brunetti, A. Metformin inhibits vistatin gene expression via HIF1 in PC3 prostate cancer cells: A potential role for visfatin as a non-invasive biomarker. Abstract #1226. Diabetologia 2017, 60, S564–S565. [Google Scholar]
- Semenza, G.L. Hypoxia-inducible factor 1 (HIF-1) pathway. Sci STKE 2007, 2007, cm8. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factors: Mediators of cancer progression and targets for cancer therapy. Trends Pharmacol. Sci. 2012, 33, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Viollet, B.; Guigas, B.; Sanz Garcia, N.; Leclerc, J.; Foretz, M.; Andreelli, F. Cellular and molecular mechanisms of metformin: An overview. Clin. Sci. 2012, 122, 253–270. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Shu, X.O.; Li, B.D.; Dai, Q.; Gao, Y.T.; Jin, F.; Zheng, W. Joint effect of insulin-like growth factors and sex steroids on breast cancer risk. Cancer Epidemiol. Biomark. Prev. 2003, 12, 1067–1073. [Google Scholar]
- Campagnoli, C.; Berrino, F.; Venturelli, E.; Abbà, C.; Biglia, N.; Brucato, T.; Cogliati, P.; Danese, S.; Donadio, M.; Zito, G.; et al. Metformin decreases circulating androgen and estrogen levels in nondiabetic women with breast cancer. Clin. Breast Cancer 2013, 13, 433–438. [Google Scholar] [CrossRef]
- Wallach, J.D.; Wang, K.; Zhang, A.D.; Cheng, D.; Grossetta Nardini, H.K.; Lin, H.; Bracken, M.B.; Desai, M.; Krumholz, H.M.; Ross, J.S. Updating insights into rosiglitazone and cardiovascular risk through shared data: Individual patient and summary level meta-analyses. BMJ 2020, 368, l7078. [Google Scholar] [CrossRef] [Green Version]
- Monami, M.; Dicembrini, I.; Mannucci, E. Thiazolidinediones and cancer: Results of a meta-analysis of randomized clinical trials. Acta Diabetol. 2014, 51, 91–101. [Google Scholar] [CrossRef]
- Corigliano, D.M.; Syed, R.; Messineo, S.; Lupia, A.; Patel, R.; Reddy, C.V.R.; Dubey, P.K.; Colica, C.; Amato, R.; De Sarro, G.; et al. Indole and 2,4-Thiazolidinedione conjugates as potential anticancer modulators. PeerJ 2018, 6, e5386. [Google Scholar] [CrossRef]
- Tseng, C.H. Pioglitazone and bladder cancer: A population-based study of Taiwanese. Diabetes Care 2012, 35, 278–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuccori, M.; Filion, K.B.; Yin, H.; Yu, O.H.; Platt, R.W.; Azoulay, L. Pioglitazone use and risk of bladder cancer: Population based cohort study. BMJ 2016, 352, i1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piccinni, C.; Motola, D.; Marchesini, G.; Poluzzi, E. Assessing the association of pioglitazone use and bladder cancer through drug adverse event reporting. Diabetes Care 2011, 34, 1369–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palakurthi, S.S.; Aktas, H.; Grubissich, L.M.; Mortensen, R.M.; Halperin, J.A. Anticancer effects of thiazolidinediones are independent of peroxisome proliferator-activated receptor gamma and mediated by inhibition of translation initiation. Cancer Res. 2001, 61, 6213–6218. [Google Scholar]
- Costa, V.; Foti, D.; Paonessa, F.; Chiefari, E.; Palaia, L.; Brunetti, G.; Gulletta, E.; Fusco, A.; Brunetti, A. The insulin receptor: A new anticancer target for peroxisome proliferator-activated receptor-gamma (PPARgamma) and thiazolidinedione-PPARgamma agonists. Endocr. Relat. Cancer 2008, 15, 325–335. [Google Scholar] [CrossRef]
- Du, R.; Lin, L.; Cheng, D.; Xu, Y.; Xu, M.; Chen, Y.; Wang, W.; Bi, Y.; Li, D.; Lu, J. Thiazolidinedione therapy and breast cancer risk in diabetic women: A systematic review and meta-analysis. Diabetes Metab. Res. Rev. 2018, 34, e2961. [Google Scholar] [CrossRef]
- Colin-Cassin, C.; Yao, X.; Cerella, C.; Chbicheb, S.; Kuntz, S.; Mazerbourg, S.; Boisbrun, M.; Chapleur, Y.; Diederich, M.; Flament, S.; et al. PPARγ-inactive Δ2-troglitazone independently triggers ER stress and apoptosis in breast cancer cells. Mol. Carcinog. 2015, 54, 393–404. [Google Scholar] [CrossRef]
- Grillier-Vuissoz, I.; Kuntz, S.; Chapleur, Y.; Flament, S. PPARγ-independent Activity of Thiazolidinediones: A Promising Mechanism of Action for New Anticancer Drugs? J. Carcinogene. Mutagene. 2012, S8, 002. [Google Scholar] [CrossRef]






{kind=link}
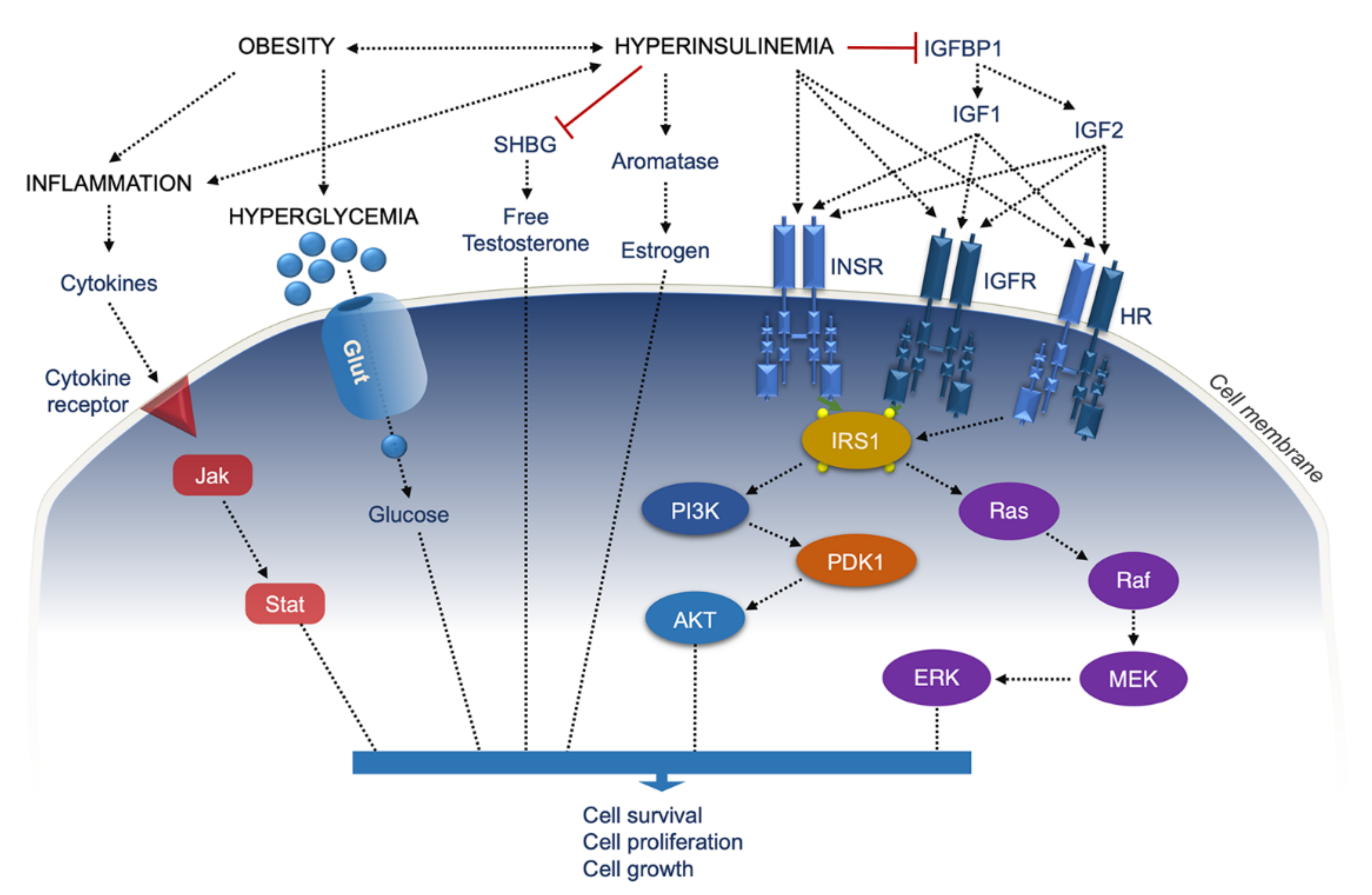{kind=link}
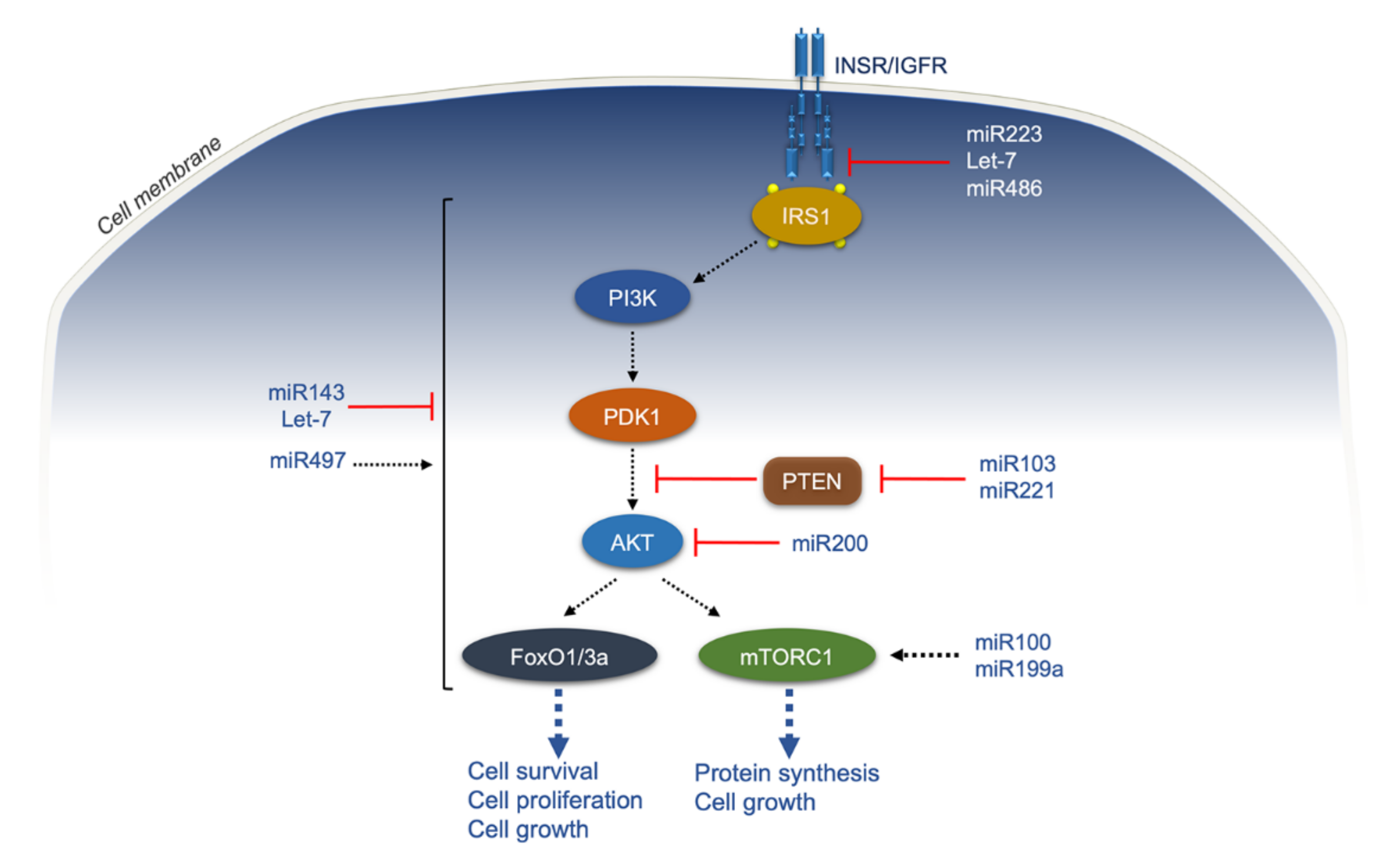{kind=link}
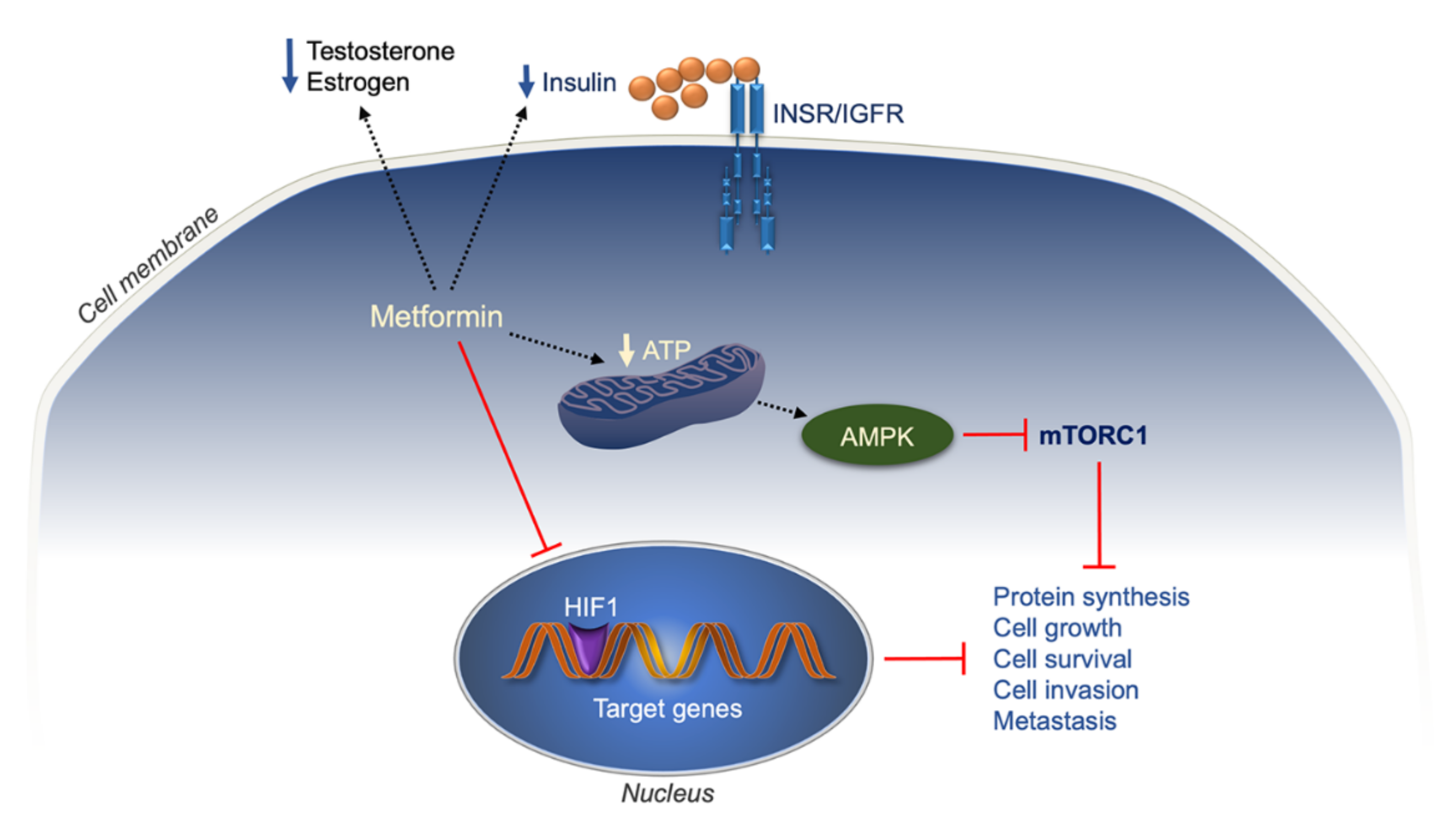{kind=link}
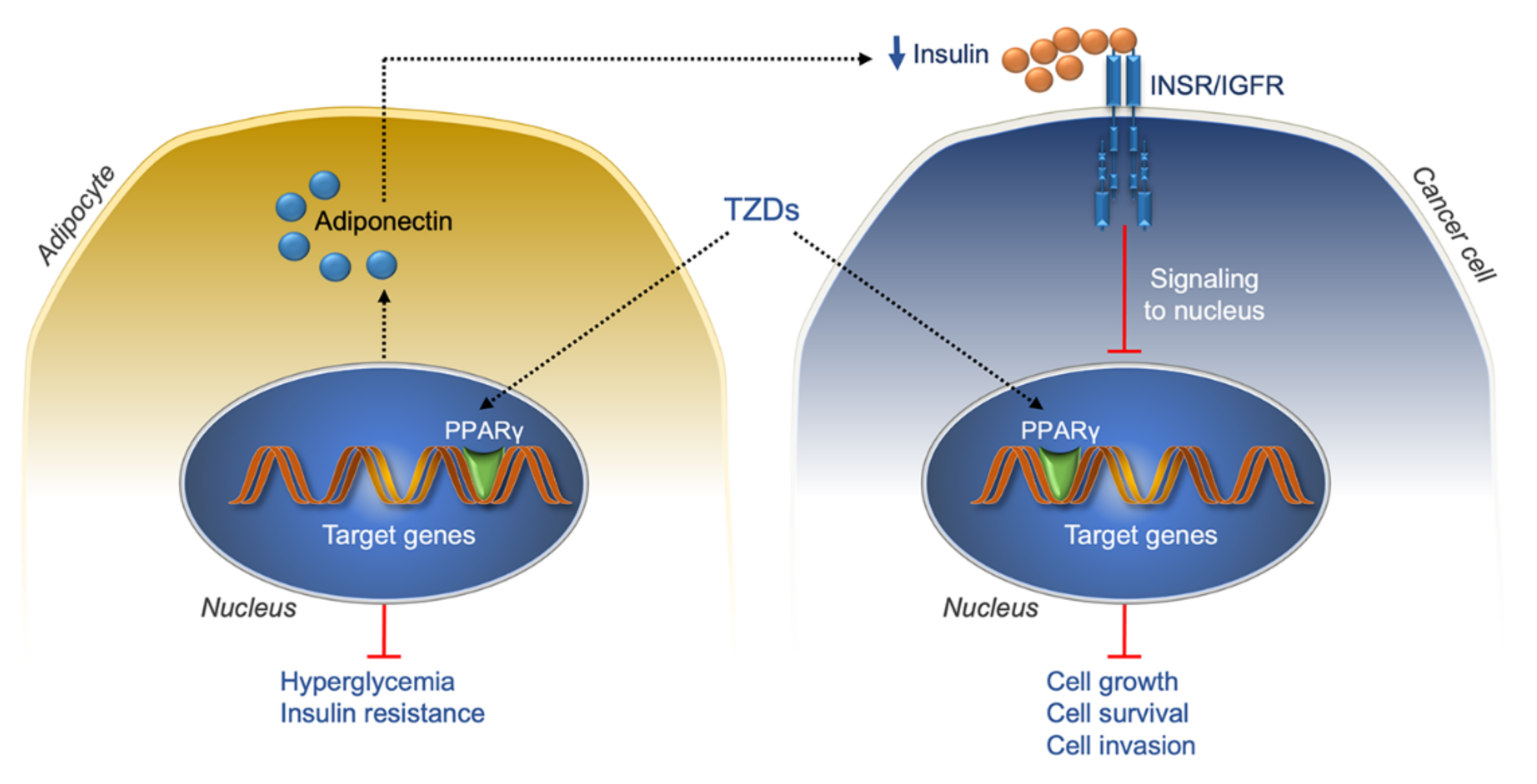{kind=link}
| Cancer Site | Type 1 Diabetes | Type 2 Diabetes | |
|---|---|---|---|
| Incidence | Incidence | Mortality | |
| All sites | ▲ RR = 1.29 (1.09–1.52) [83] | ▲ RR = 1.10 (1.04–1.17) [84] | ▲ RR = 1.16 (1.03–1.30) [84] |
| Biliary tract | ▲ RR = 1.43 (1.18–1.72) [85] | ||
| Bladder | ⬄ RR = 0.98 (0.88–1.09) [83] | ▲ RR = 1.35 (1.17–1.56) [86] | |
| Breast | ⬄ RR = 0.91 (0.86–0.95) [83] | ▲ RR = 1.20 (1.12–1.28) [87] | ▲ HR = 1.51 (1.34–1.70) [88] HR = 1.37 (1.34–1.41) [89] |
| Cervix | ⬄ RR = 1.24 (0.99–1.56) [83] | ▲▲ HR = 1.59 (1.35–1.87) [90] | |
| Colon-rectum | ⬄ RR = 0.90 (0.61–1.31) [83] | ▲ RR = 1.27 (1.21–1.34) [91] | ▲ HR = 1.18 (1.12–1.24) [92] |
| Endometrium | ▲▲ RR = 1.67 (1.22–2.30) [83] ▲ RR = 1.42 (1.27–1.58) [95] | ▲▲ RR = 1.63 (1.41–1.88) [93] | ⬄ RR = 1.23 (0.78–1.93) [94] |
| Esophagus | ▲ RR = 1.06 (1.02–2.42) [83] | ▲ RR = 1.30 (1.12–1.50) [94] | |
| Hematological malignancies | ⬄ RR = 1.01 (0.88–1.16) [83] | ▲ OR 1.22 (1.03–1.44) [96] | |
| Kidney | ▲ RR = 1.37 (1.23–1.52) [83] | ▲ RR = 1.40 (1.16–1.69) [97] | ⬄ RR = 1.12 (0.99–1.20) [97] |
| Liver | ▲▲▲ RR = 2.35 (2.12–2.61) [83] | ▲▲ RR = 1.60 (1.38–1.87)] [98] ▲ RR = 1.49 (1.32–1.70) [85,99] | |
| Lung | ▲ RR = 1.09 (1.02–1.17) [83] | ▲ HR = 1.14 (1.09–1.20) [100] | ▲ HR = 1.33 (0.87–2.03) [101] |
| Ovary | ▲ RR = 1.17 (1.04–1.32) [83] | ▲ RR = 1.17 (1.02–1.33) [102,103] | |
| Pancreas | ▲ RR = 1.34 (1.18–1.52) [83] | ▲▲ RR = 1.94 (1.66–2.27) [104] | ▲ RR = 1.67 (1.30–2.14) [93] |
| Prostate | ⬄ RR = 1.15 (0.30–4.41) [83] ▼ RR = 0.56 (0.51–0.61) [95] | ▼ RR = 0.83 (0.79–0.88) [93] | ▲ HR = 1.50 (1.25–1.79) [105] |
| Stomach | ▲ RR = 1.44 (1.29–1.61) [83] | ▲ RR = 1.19 (1.07–1.32) [106] ⬄ RR = 1.9 (0.98–1.22) [107] | ▲ RR = RR 1.29 (1.04–1.59) [106] |
| Thyroid | ▲ RR = 1.40 (1.19–1.66) [83] | ▲ RR = 1.17 (0.99–1.39) [108] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiefari, E.; Mirabelli, M.; La Vignera, S.; Tanyolaç, S.; Foti, D.P.; Aversa, A.; Brunetti, A. Insulin Resistance and Cancer: In Search for a Causal Link. Int. J. Mol. Sci. 2021, 22, 11137. https://doi.org/10.3390/ijms222011137
Chiefari E, Mirabelli M, La Vignera S, Tanyolaç S, Foti DP, Aversa A, Brunetti A. Insulin Resistance and Cancer: In Search for a Causal Link. International Journal of Molecular Sciences. 2021; 22(20):11137. https://doi.org/10.3390/ijms222011137
Chicago/Turabian StyleChiefari, Eusebio, Maria Mirabelli, Sandro La Vignera, Sinan Tanyolaç, Daniela Patrizia Foti, Antonio Aversa, and Antonio Brunetti. 2021. "Insulin Resistance and Cancer: In Search for a Causal Link" International Journal of Molecular Sciences 22, no. 20: 11137. https://doi.org/10.3390/ijms222011137