
Yield-Related QTL Clusters and the Potential Candidate Genes in Two Wheat DH Populations

 ,
,  , ,
, , 
 and add
Show full author list
and add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
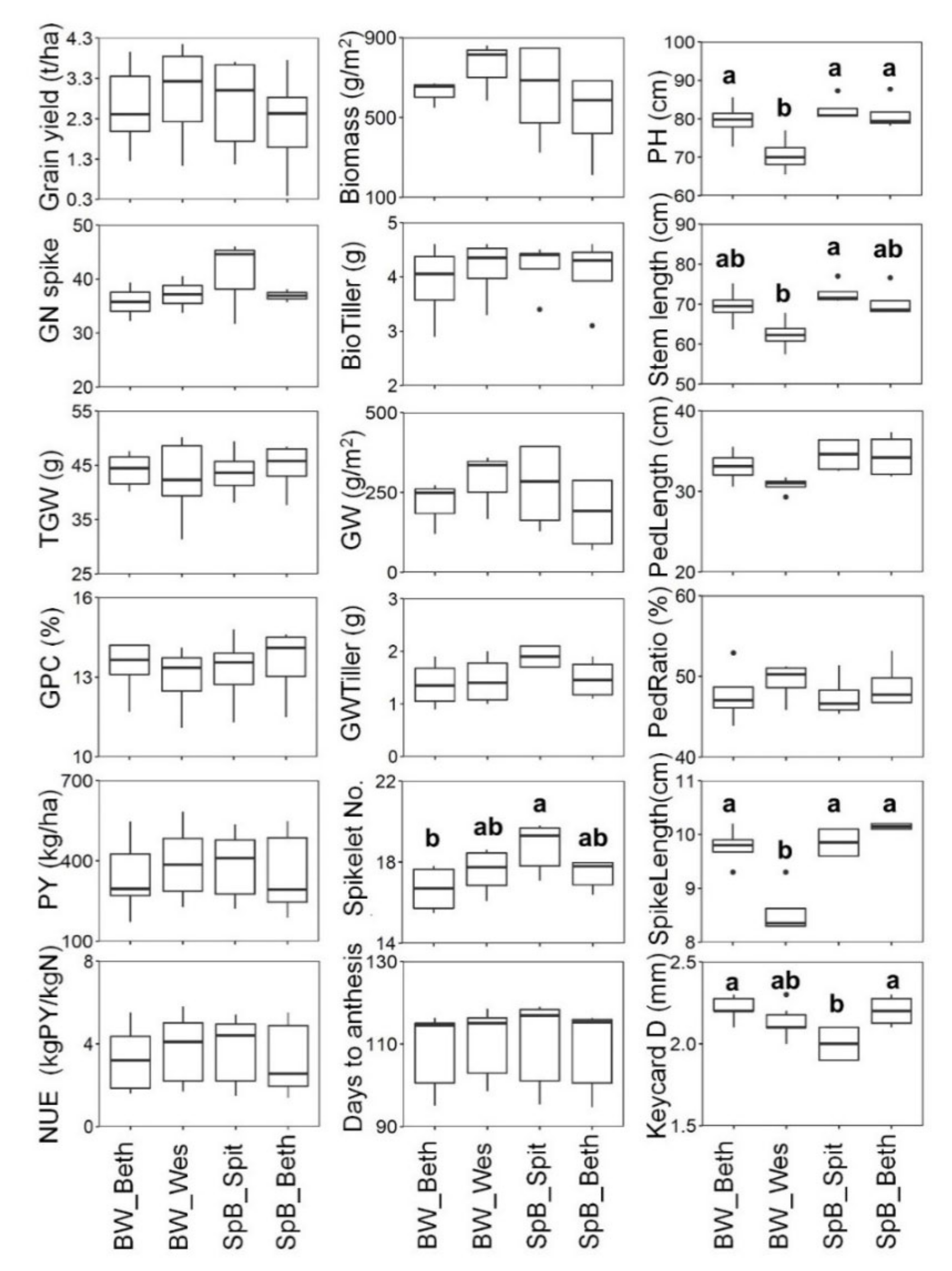
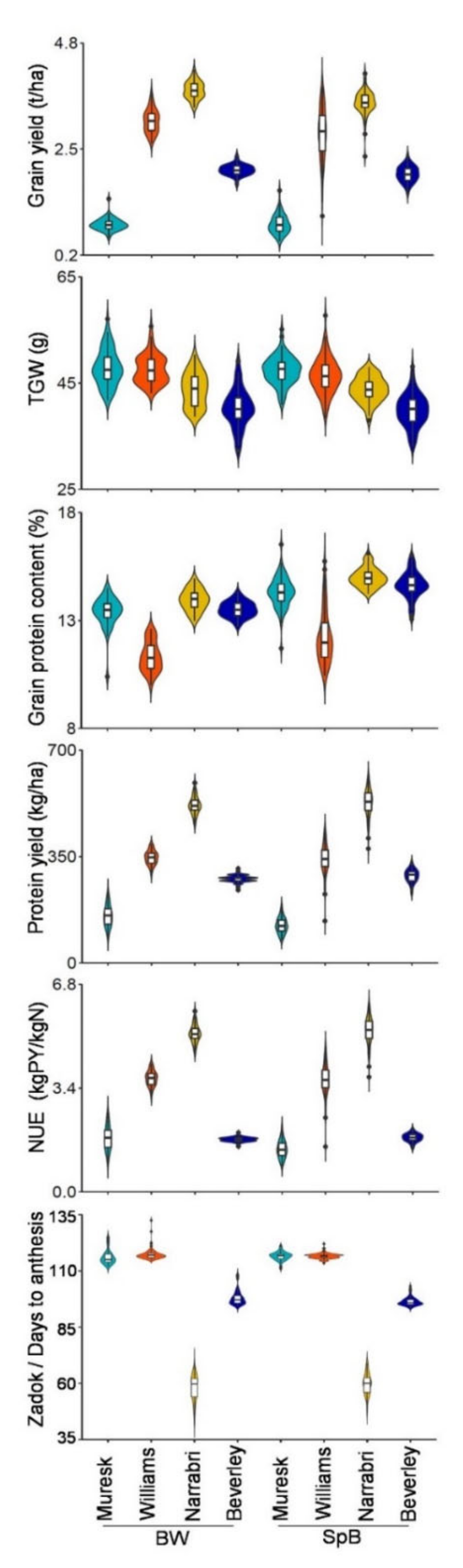
2.1. Phenotypic Variations
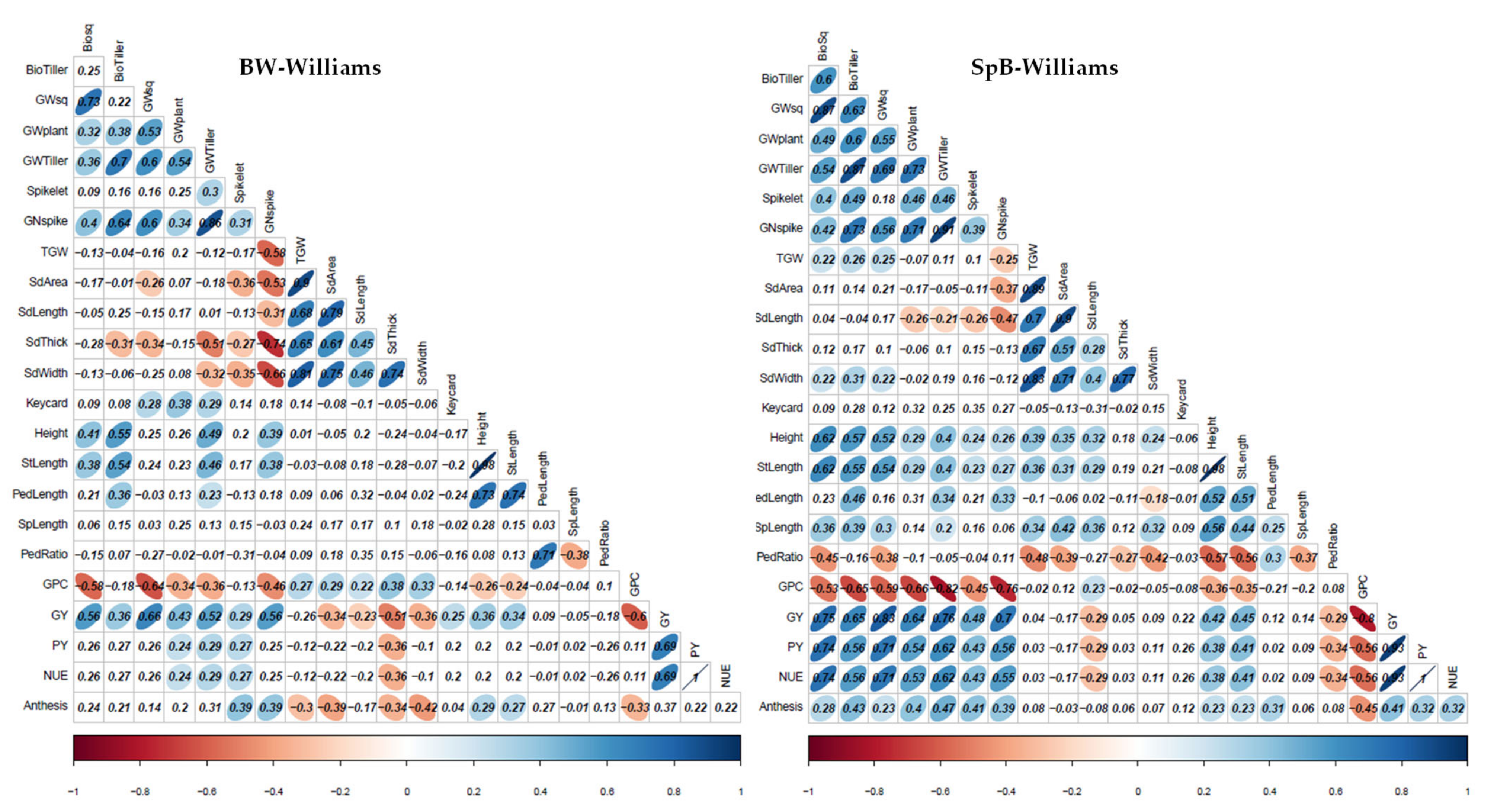
2.2. Phenotypic Correlations in Each Environment
2.3. Consensus Map Construction
2.4. Robust QTL Identification in Two Populations
2.4.1. QTL for Anthesis
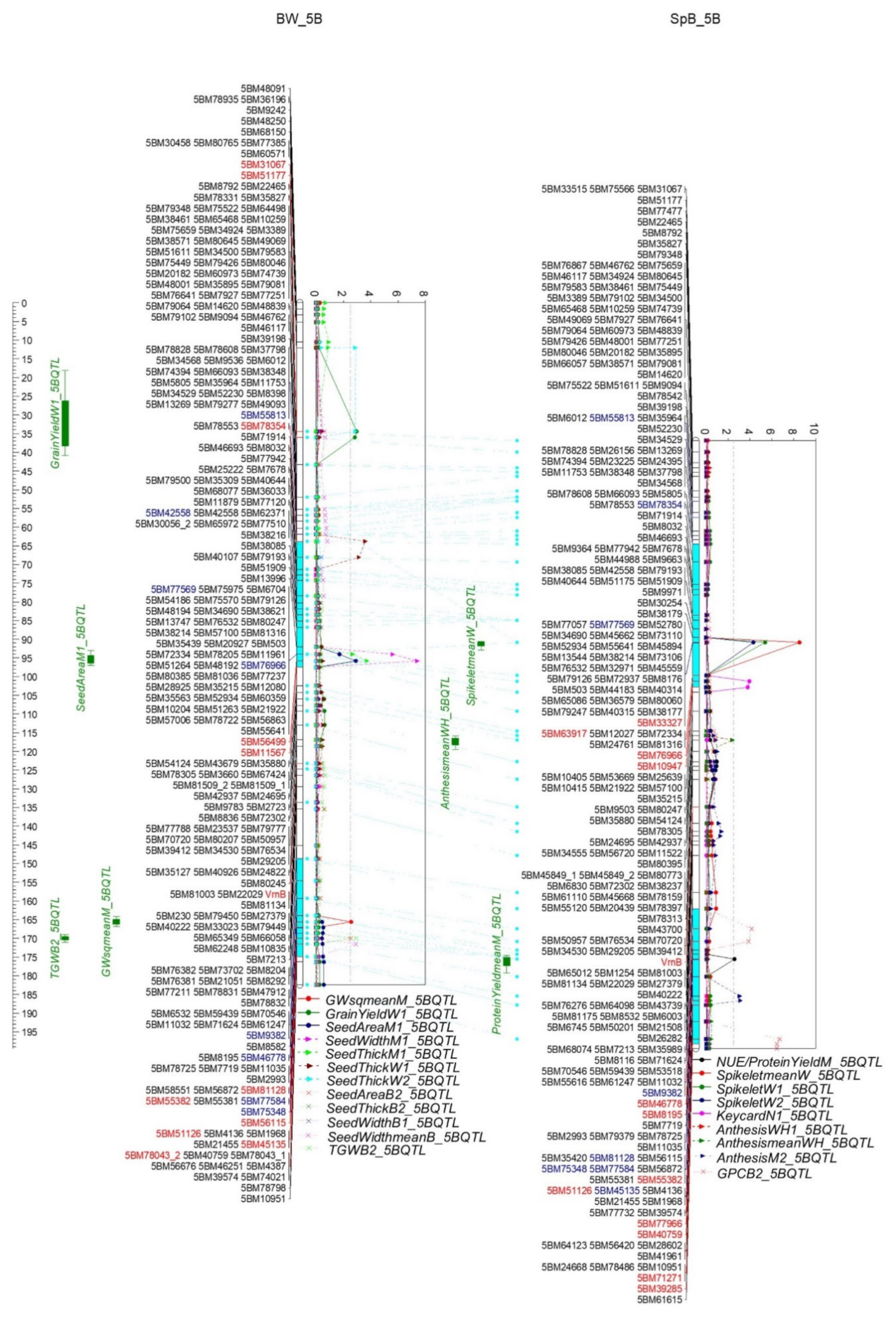
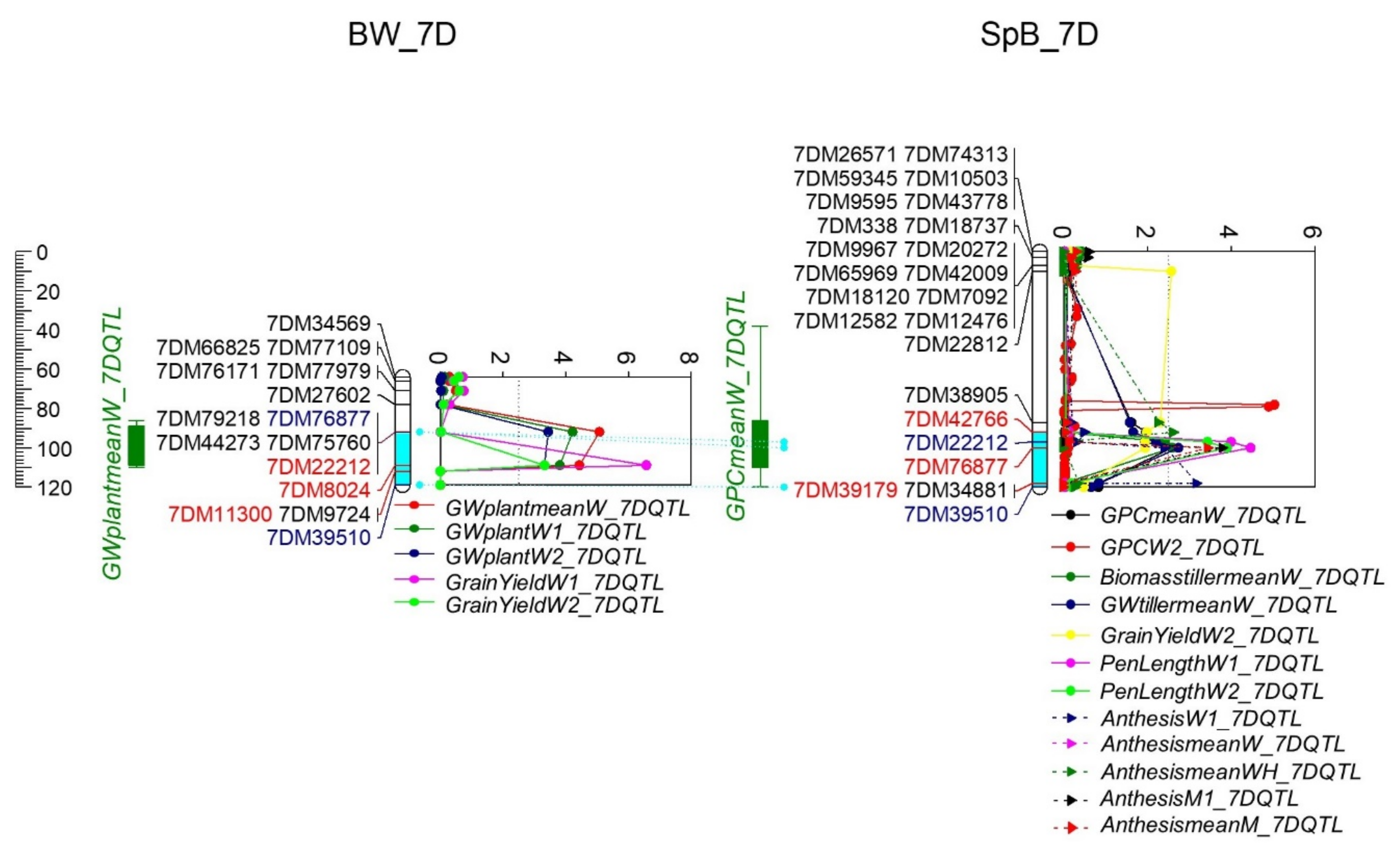
2.4.2. QTL for Grain Yield and Grain Weight
2.4.3. QTL for GPC, NUE and Protein Yield
2.4.4. QTL for Biomass
2.4.5. QTL for GN and Spikelet Number per Spike
2.4.6. QTL for TGW and Seed Parameters
2.4.7. QTL for Plant Height and Stem Length
2.4.8. QTL for Peduncle Length and Peduncle Ratio
2.4.9. QTL for Spike Length and Keycard Diameter
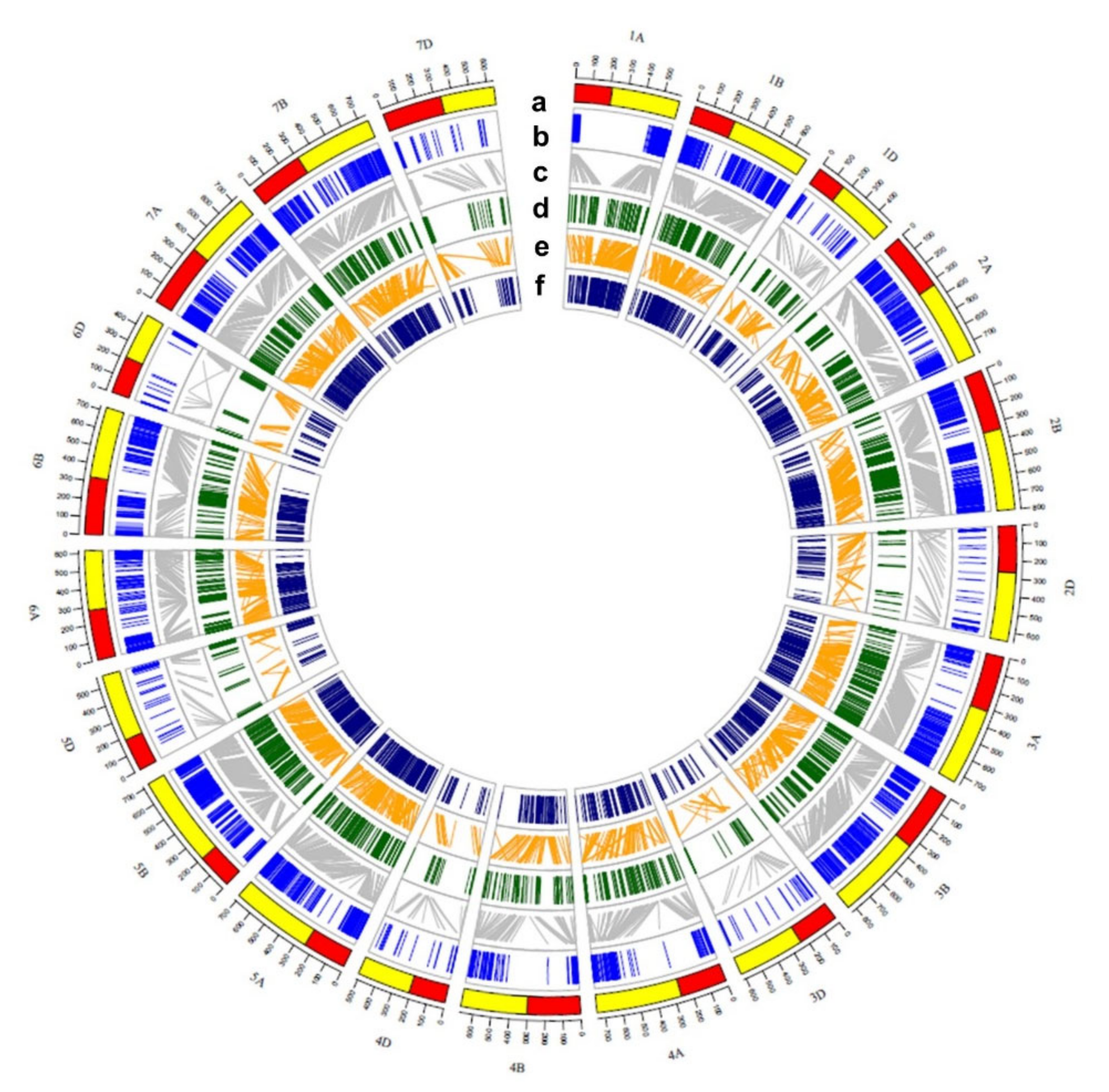
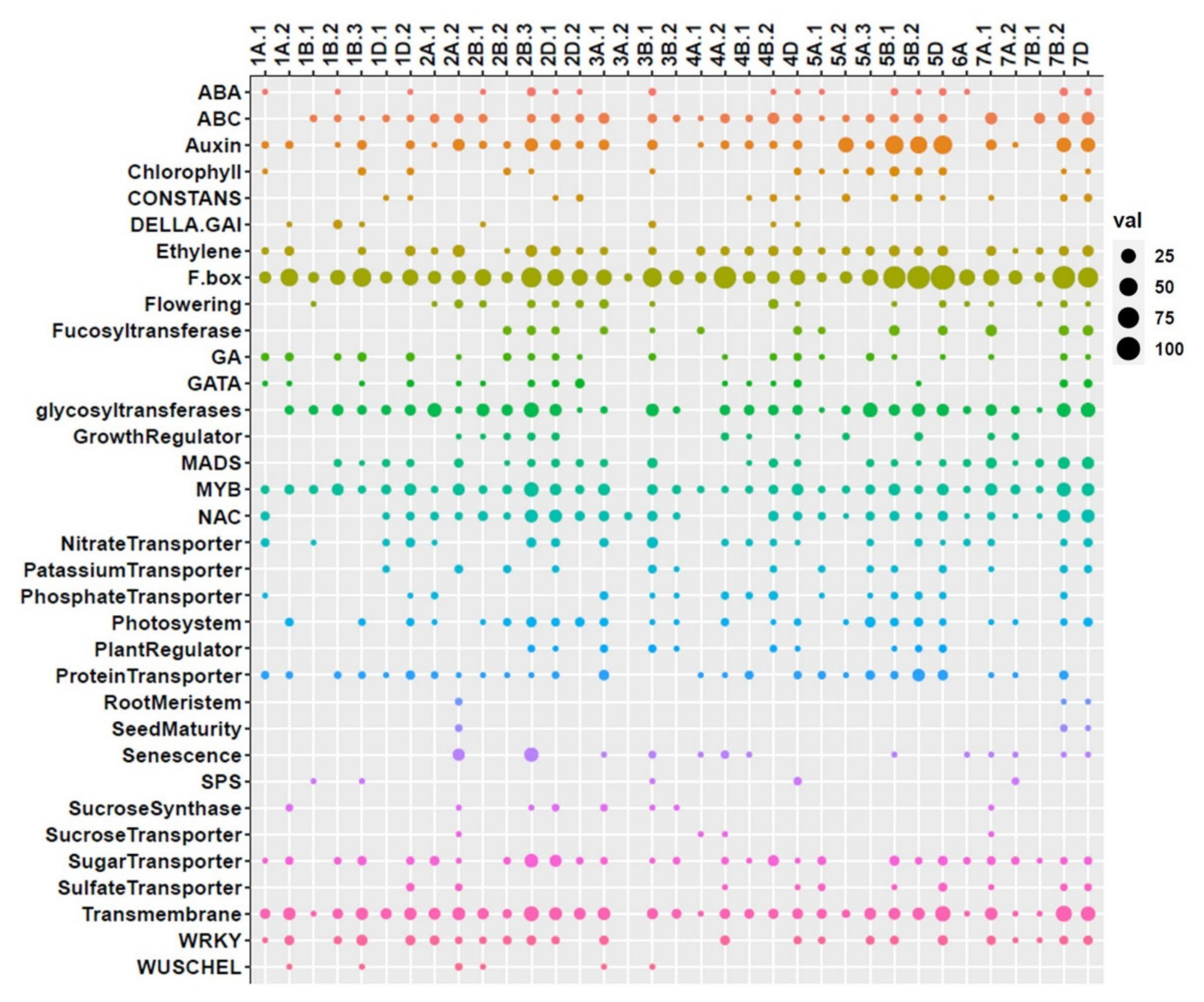
2.5. QTL Clusters and Related Potential Candidate Genes
3. Discussion
3.1. Consensus Map Used for QTL Clusters
3.2. Confirmation of QTL Clusters and Reported Genes
3.3. Trait Correlations within QTL Clusters and Applications in Breeding
4. Materials and Methods
4.1. Plant Materials
4.2. Field and Glasshouse Experiments
4.3. Core Phenotype Measurements
4.4. Linkage Map Construction
4.5. QTL Mapping
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pang, Y.; Liu, C.; Wang, D.; Amand, P.S.; Bernardo, A.; Li, W.; He, F.; Li, L.; Wang, L.; Yuan, X.; et al. High-Resolution Genome-Wide Association Study Identifies Genomic Regions and Candidate Genes for Important Agronomic Traits in Wheat. Mol. Plant 2020, 13, 1311–1327. [Google Scholar] [CrossRef] [PubMed]
- Shiferaw, B.; Smale, M.; Braun, H.-J.; Duveiller, E.; Reynolds, M.; Muricho, G. Crops that feed the world 10. Past successes and future challenges to the role played by wheat in global food security. Food Secur. 2013, 5, 291–317. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Wang, X.; Zhang, G.; Jiang, P.; Chen, W.; Hao, Y.; Ma, X.; Xu, S.; Jia, J.; Kong, L.; et al. QTL mapping for yield-related traits in wheat based on four RIL populations. Theor. Appl. Genet. 2020, 133, 917–933. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Wen, W.; Fu, L.; Li, F.; Li, J.; Xie, L.; Xia, X.; Ni, Z.; He, Z.; Cao, S. Genetic dissection of a major QTL for kernel weight spanning the Rht-B1 locus in bread wheat. Theor. Appl. Genet. 2019, 132, 3191–3200. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Ding, P.; Liu, J.; Li, T.; Zou, Y.; Habib, A.; Mu, Y.; Tang, H.; Jiang, Q.; Liu, Y.; et al. Identification and validation of a major and stably expressed QTL for spikelet number per spike in bread wheat. Theor. Appl. Genet. 2019, 132, 3155–3167. [Google Scholar] [CrossRef] [PubMed]
- Kuzay, S.; Xu, Y.; Zhang, J.; Katz, A.; Pearce, S.; Su, Z.; Fraser, M.; Anderson, J.A.; Brown-Guedira, G.; DeWitt, N.; et al. Identification of a candidate gene for a QTL for spikelet number per spike on wheat chromosome arm 7AL by high-resolution genetic mapping. Theor. Appl. Genet. 2019, 132, 2689–2705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Sidhu, H.S.; Kaviani, M.; McElroy, M.S.; Pozniak, C.J.; Navabi, A. Application of image-based phenotyping tools to identify QTL for in-field winter survival of winter wheat (Triticum aestivum L.). Theor. Appl. Genet. 2019, 132, 2591–2604. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Islam, S.; Zhao, Y.; Anwar, M.; Alhabbar, Z.; She, M.; Yang, R.; Juhasz, A.; Tang, G.; Chen, J.; et al. Non-escaping frost tolerant QTL linked genetic loci at reproductive stage in six wheat DH populations. Crop. J. 2021. [Google Scholar] [CrossRef]
- Xiong, H.; Li, Y.; Guo, H.; Xie, Y.; Zhao, L.; Gu, J.; Zhao, S.; Ding, Y.; Liu, L. Genetic Mapping by Integration of 55K SNP Array and KASP Markers Reveals Candidate Genes for Important Agronomic Traits in Hexaploid Wheat. Front. Plant Sci. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Ren, T.; Fan, T.; Chen, S.; Li, C.; Chen, Y.; Ou, X.; Jiang, Q.; Ren, Z.; Tan, F.; Luo, P.; et al. Utilization of a Wheat55K SNP array-derived high-density genetic map for high-resolution mapping of quantitative trait loci for important kernel-related traits in common wheat. Theor. Appl. Genet. 2021, 134, 807–821. [Google Scholar] [CrossRef]
- Hu, X.; Zhang, Y.; Zhang, J.; Islam, S.; She, M.; Zhao, Y.; Tang, G.; Jiang, Y.; Rong, J.; Ma, W. Consensus Genetic Linkage Map Construction Based on One Common Parental Line for QTL Mapping in Wheat. Agronomy 2021, 11, 227. [Google Scholar] [CrossRef]
- Wen, W.; He, Z.; Gao, F.; Liu, J.; Jin, H.; Zhai, S.; Qu, Y.; Xia, X. A High-Density Consensus Map of Common Wheat Integrating Four Mapping Populations Scanned by the 90K SNP Array. Front. Plant Sci. 2017, 8, 1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galeano, C.H.; Fernandez, A.C.; Franco-Herrera, N.; Cichy, K.A.; McClean, P.E.; Vanderleyden, J.; Blair, M.W. Saturation of an Intra-Gene Pool Linkage Map: Towards a Unified Consensus Linkage Map for Fine Mapping and Synteny Analysis in Common Bean. PLoS ONE 2011, 6, e28135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tshikunde, N.M.; Mashilo, J.; Shimelis, H.; Odindo, A.O. Agronomic and Physiological Traits, and Associated Quantitative Trait Loci (QTL) Affecting Yield Response in Wheat (Triticum aestivum L.): A Review. Front. Plant Sci. 2019, 10, 1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondal, S.; Singh, R.; Mason, E.; Huerta-Espino, J.; Autrique, E.; Joshi, A. Grain yield, adaptation and progress in breeding for early-maturing and heat-tolerant wheat lines in South Asia. Field Crop. Res. 2016, 192, 78–85. [Google Scholar] [CrossRef] [Green Version]
- Distelfeld, A.; Li, C.; Dubcovsky, J. Regulation of flowering in temperate cereals. Curr. Opin. Plant Biol. 2009, 12, 178–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, L.; Loukoianov, A.; Blechl, A.; Tranquilli, G.; Ramakrishna, W.; SanMiguel, P.; Bennetzen, J.L.; Echenique, V.; Dubcovsky, J. The Wheat VRN2 Gene Is a Flowering Repressor Down-Regulated by Vernalization. Science 2004, 303, 1640–1644. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.; Fu, D.; Li, C.; Blechl, A.; Tranquilli, G.; Bonafede, M.; Sanchez, A.; Valárik, M.; Yasuda, S.; Dubcovsky, J. The wheat and barley vernalization gene VRN3 is an orthologue of FT. Proc. Natl. Acad. Sci. USA 2006, 103, 19581–19586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kippes, N.; Debernardi, J.; Vasquez-Gross, H.A.; Akpinar, B.A.; Budak, H.; Kato, K.; Chao, S.; Akhunov, E.; Dubcovsky, J. Identification of the vernalization 4gene reveals the origin of spring growth habit in ancient wheats from South Asia. Proc. Natl. Acad. Sci. USA 2015, 112, E5401–E5410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, L.; Zhang, Y.; Wang, K.; Luo, X.; Xu, D.; Tian, X.; Li, L.; Ye, X.; Xia, X.; Li, W.; et al. TaVrt2, an SVP-like gene, cooperates with TaVrn1 to regulate vernalization-induced flowering in wheat. New Phytol. 2021, 231, 834–848. [Google Scholar] [CrossRef]
- Kane, N.A.; Danyluk, J.; Tardif, G.; Ouellet, F.; Laliberteé, J.-F.; Limin, A.E.; Fowler, D.B.; Sarhan, F. TaVRT-2, a Member of the StMADS-11 Clade of Flowering Repressors, Is Regulated by Vernalization and Photoperiod in Wheat. Plant Physiol. 2005, 138, 2354–2363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beales, J.; Turner, A.; Griffiths, S.; Snape, J.W.; Laurie, D.A. A Pseudo-Response Regulator is misexpressed in the photoperiod insensitive Ppd-D1a mutant of wheat (Triticum aestivum L.). Theor. Appl. Genet. 2007, 115, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Worland, A.; Börner, A.; Korzun, V.; Li, W.; Petrovíc, S.; Sayers, E. The influence of photoperiod genes on the adaptability of European winter wheats. Euphytica 1998, 100, 385–394. [Google Scholar] [CrossRef]
- Würschum, T.; Rapp, M.; Miedaner, T.; Longin, C.F.H.; Leiser, W.L. Copy number variation of Ppd-B1 is the major determinant of heading time in durum wheat. BMC Genet. 2019, 20, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Arjona, J.; Royo, C.; Dreisigacker, S.; Ammar, K.; Villegas, D. Effect of Ppd-A1 and Ppd-B1 Allelic Variants on Grain Number and Thousand Kernel Weight of Durum Wheat and Their Impact on Final Grain Yield. Front. Plant Sci. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Zikhali, M.; Griffiths, S. The Effect of Earliness per se (Eps) Genes on Flowering Time in Bread Wheat; Springer: Tokyo, Japan, 2015; pp. 339–345. [Google Scholar]
- Alvarez, M.A.; Tranquilli, G.; Lewis, S.; Kippes, N.; Dubcovsky, J. Genetic and physical mapping of the earliness per se locus Eps-A m 1 in Triticum monococcum identifies EARLY FLOWERING 3 (ELF3) as a candidate gene. Funct. Integr. Genom. 2016, 16, 365–382. [Google Scholar] [CrossRef] [Green Version]
- Ochagavia, H.; Prieto, P.; Zikhali, M.; Griffiths, S.; Slafer, G.A. Earliness Per Se by Temperature Interaction on Wheat Development. Sci. Rep. 2019, 9, 25841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoogendoorn, J. A reciprocal F1 monosomic analysis of the genetic control of time of ear emergence, number of leaves and number of spikelets in wheat (Triticum aestivum L.). Euphytica 1985, 34, 545–558. [Google Scholar] [CrossRef]
- Zikhali, M.; Leverington-Waite, M.; Fish, L.; Simmonds, J.; Orford, S.; Wingen, L.U.; Goram, R.; Gosman, N.; Bentley, A.; Griffiths, S. Validation of a 1DL earliness per se (eps) flowering QTL in bread wheat (Triticum aestivum). Mol. Breed. 2014, 34, 1023–1033. [Google Scholar] [CrossRef] [Green Version]
- Scarth, R.; Law, C.N. The location of the photoperiod gene, Ppd2 and an additional genetic factor for ear-emergence time on chromosome 2B of wheat. Heredity 1983, 51, 607–619. [Google Scholar] [CrossRef]
- Wang, J.; Wen, W.; Hanif, M.; Xia, X.; Wang, H.; Liu, S.; Liu, J.; Yang, L.; Cao, S.; He, Z. TaELF3-1DL, a homolog of ELF3, is associated with heading date in bread wheat. Mol. Breed. 2016, 36, 161. [Google Scholar] [CrossRef]
- Zikhali, M.; Wingen, L.U.; Griffiths, S. Delimitation of theEarliness per se D1(Eps-D1) flowering gene to a subtelomeric chromosomal deletion in bread wheat (Triticum aestivum). J. Exp. Bot. 2016, 67, 287–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, L.M.; Li, C.; Woods, D.P.; Alvarez, M.A.; Lin, H.; Lau, M.Y.; Chen, A.; Dubcovsky, J. Epistatic interactions between PHOTOPERIOD1, CONSTANS1 and CONSTANS2 modulate the photoperiodic response in wheat. PLoS Genet. 2020, 16, e1008812. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.Y.; Liu, M.S.; Li, J.R.; Guan, C.M.; Zhang, X.S. The wheat TaGI1, involved in photoperiodic flowering, encodesan Arabidopsis GI ortholog. Plant Mol. Biol. 2005, 58, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Shitsukawa, N.; Ikari, C.; Shimada, S.; Kitagawa, S.; Sakamoto, K.; Saito, H.; Ryuto, H.; Fukunishi, N.; Abe, T.; Takumi, S.; et al. The einkorn wheat (Triticum monococcum) mutant, maintained vegetative phase, is caused by a deletion in the VRN1 gene. Genes Genet. Syst. 2007, 82, 167–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zikhali, M.; Wingen, L.U.; Leverington-Waite, M.; Specel, S.; Griffiths, S. The identification of new candidate genes Triticum aestivum FLOWERING LOCUS T3-B1 (TaFT3-B1) and TARGET OF EAT1 (TaTOE1-B1) controlling the short-day photoperiod response in bread wheat. Plant Cell Environ. 2017, 40, 2678–2690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, C.; Zhao, L.; Zhang, X.; Lv, G.; Pan, Y.; Chen, F. Gene regulatory network and abundant genetic variation play critical roles in heading stage of polyploidy wheat. BMC Plant Biol. 2019, 19, 1–16. [Google Scholar] [CrossRef]
- Nemoto, Y.; Kisaka, M.; Fuse, T.; Yano, M.; Ogihara, Y. Characterization and functional analysis of three wheat genes with homology to the CONSTANS flowering time gene in transgenic rice. Plant J. 2003, 36, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Mohan, A.; Grant, N.P.; Schillinger, W.F.; Gill, K.S. Characterizing reduced height wheat mutants for traits affecting abiotic stress and photosynthesis during seedling growth. Physiol. Plant. 2021, 172, 233–246. [Google Scholar] [CrossRef]
- Worland, A.J.; Law, C.N.; Shakoor, A. The genetical analysis of an induced height mutant in wheat. Heredity 1980, 45, 61–71. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Xing, L.; Xing, S.; Hu, P.; Cui, C.; Zhang, M.; Xiao, J.; Wang, H.; Zhang, R.; Wang, X.; et al. Characterization of a Putative New Semi-Dominant Reduced Height Gene, Rht_NM9, in Wheat (Triticum aestivum L.). J. Genet. Genom. 2015, 42, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Ellis, M.H.; Rebetzke, G.; Azanza, F.; Richards, R.A.; Spielmeyer, W. Molecular mapping of gibberellin-responsive dwarfing genes in bread wheat. Theor. Appl. Genet. 2005, 111, 423–430. [Google Scholar] [CrossRef]
- Chai, S.; Yao, Q.; Zhang, X.; Xiao, X.; Fan, X.; Zeng, J.; Sha, L.; Kang, H.; Zhang, H.; Li, J.; et al. The semi-dwarfing gene Rht-dp from dwarf polish wheat (Triticum polonicum L.) is the “Green Revolution” gene Rht-B1b. BMC Genom. 2021, 22, 63. [Google Scholar] [CrossRef] [PubMed]
- Gale, M.; Youssefian, S. Dwarfing genes in wheat. In Proceedings of the Progress in Plant Breeding–1; Butterworths: London, UK, 1985; pp. 1–35. [Google Scholar]
- Worland, A.J.; Petrovic, S. The gibberellic acid insensitive dwarfing gene from the wheat variety Saitama 27. Euphytica 1988, 38, 55–63. [Google Scholar] [CrossRef]
- Mo, Y.; Vanzetti, L.S.; Hale, I.; Spagnolo, E.J.; Guidobaldi, F.; Al-Oboudi, J.; Odle, N.; Pearce, S.; Helguera, M.; Dubcovsky, J. Identification and characterization of Rht25, a locus on chromosome arm 6AS affecting wheat plant height, heading time, and spike development. Theor. Appl. Genet. 2018, 131, 2021–2035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haque, M.A.; Martinek, P.; Kobayashi, S.; Kita, I.; Ohwaku, K.; Watanabe, N.; Kuboyama, T. Microsatellite mapping of genes for semi-dwarfism and branched spike in Triticum durum Desf. var. ramosoobscurum Jakubz “Vetvistokoloskaya.”. Genet. Resour. Crop. Evol. 2012, 59, 831–837. [Google Scholar] [CrossRef]
- Bazhenov, M.; Divashuk, M.; Amagai, Y.; Watanabe, N.; Karlov, G.I. Isolation of the dwarfing Rht-B1p (Rht17) gene from wheat and the development of an allele-specific PCR marker. Mol. Breed. 2015, 35, 213. [Google Scholar] [CrossRef]
- Börner, A.; Mettin, D. The genetic control of gibberellic acid insensitivity of the wheat variety Ai-Bain. Proceedings of Seventh International Wheat Genetics Symposium, Cambridge, UK, 1 January 1988; Bath Press: Avon, OH, USA, 1988; pp. 489–492. [Google Scholar]
- Börner, A.; Lehmann, C.O.; Mettin, D.; Plaschke, J.; Schlegel, G.; Schlegel, R.; Melz, G.; Thiele, V. GA-insensitivity of ‘Aibain 1a’/Pleiotropic effects of isogenic Rht-lines. Ann. Wheat Newsl. 1991, 37, 59–60. [Google Scholar]
- Chen, S.; Gao, R.; Wang, H.; Wen, M.; Xiao, J.; Bian, N.; Zhang, R.; Hu, W.; Cheng, S.; Bie, T.; et al. Characterization of a novel reduced height gene (Rht23) regulating panicle morphology and plant architecture in bread wheat. Euphytica 2015, 203, 583–594. [Google Scholar] [CrossRef]
- Würschum, T.; Langer, S.M.; Longin, C.F.H.; Tucker, M.R.; Leiser, W.L. A modern Green Revolution gene for reduced height in wheat. Plant J. 2017, 92, 892–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Z.; Li, X.; Yang, Z.; Liao, M. A new reduced height gene found in the tetraploid semi-dwarf wheat landrace Aiganfanmai. Genet. Mol. Res. 2011, 10, 2349–2357. [Google Scholar] [CrossRef] [PubMed]
- Arif, M.A.R.; Shokat, S.; Plieske, J.; Ganal, M.; Lohwasser, U.; Chesnokov, Y.V.; Kocherina, N.V.; Kulwal, P.; Kumar, N.; McGuire, P.M.; et al. A SNP-based genetic dissection of versatile traits in bread wheat (Triticum aestivum L.). Plant J. 2021. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, J.; Scott, P.; Brinton, J.; Mestre, T.C.; Bush, M.; Del Blanco, A.; Dubcovsky, J.; Uauy, C. A splice acceptor site mutation in TaGW2-A1 increases thousand grain weight in tetraploid and hexaploid wheat through wider and longer grains. Theor. Appl. Genet. 2016, 129, 1099–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Z.; Hao, C.; Wang, L.; Dong, Y.; Zhang, X. Identification and development of a functional marker of TaGW2 associated with grain weight in bread wheat (Triticum aestivum L.). Theor. Appl. Genet. 2011, 122, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Li, T.; Hao, C.; Wang, Y.; Chen, X.; Zhang, X. TaGS5-3A, a grain size gene selected during wheat improvement for larger kernel and yield. Plant Biotechnol. J. 2016, 14, 25. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.-J.; Zhang, H.-P.; Cao, J.-J.; Zhu, X.-F.; Wang, S.-X.; Jiang, H.; Wu, Z.Y.; Lu, J.; Chang, C.; Sun, G.-L.; et al. Characterization of an IAA-glucose hydrolase gene TaTGW6 associated with grain weight in common wheat (Triticum aestivum L.). Mol. Breed. 2016, 36, 1–11. [Google Scholar] [CrossRef]
- Hu, M.-J.; Zhang, H.-P.; Liu, K.; Cao, J.-J.; Wang, S.-X.; Jiang, H.; Wu, Z.-Y.; Lu, J.; Zhu, X.F.; Xia, X.-C.; et al. Cloning and Characterization of TaTGW-7A Gene Associated with Grain Weight in Wheat via SLAF-seq-BSA. Front. Plant Sci. 2016, 7, 1902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Zhou, Y.; Wu, Q.; Chen, Y.; Zhang, P.; Zhang, Y.; Hu, W.; Wang, X.; Zhao, H.; Dong, L.; et al. Molecular characterization of a novel TaGL3-5A allele and its association with grain length in wheat (Triticum aestivum L.). Theor. Appl. Genet. 2019, 132, 1799–1814. [Google Scholar] [CrossRef] [PubMed]
- Pearce, S.; Tabbita, F.; Cantu, D.; Buffalo, V.; Avni, R.; Vazquez-Gross, H.; Zhao, R.; Conley, C.J.; Distelfeld, A.; Dubcovksy, J. Regulation of Zn and Fe transporters by the GPC1gene during early wheat monocarpic senescence. BMC Plant Biol. 2014, 14, 368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avivi, L. High grain protein content in wild tetraploid wheat Triticum dicoccoides KORN. Proceedings of Fifth International Wheat Genetics Symposium, New Delhi, India, 23–28 February 1978; pp. 372–380. [Google Scholar]
- Uauy, C.; Distelfeld, A.; Fahima, T.; Blechl, A.; Dubcovsky, J. A NAC Gene Regulating Senescence Improves Grain Protein, Zinc, and Iron Content in Wheat. Science 2006, 314, 1298–1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cormier, F.; Throude, M.; Ravel, C.; Le Gouis, J.; Leveugle, M.; Lafarge, S.; Exbrayat-Vinson, F.; Duranton, N.; Praud, S. Detection of NAM-A1 Natural Variants in Bread Wheat Reveals Differences in Haplotype Distribution between a Worldwide Core Collection and European Elite Germplasm. Agronomy 2015, 5, 143–151. [Google Scholar] [CrossRef]
- Yang, R.; Juhasz, A.; Zhang, Y.; Chen, X.; Zhang, Y.; She, M.; Zhang, J.; Maddern, R.; Edwards, I.; Diepeveen, D.; et al. Molecular characterisation of the NAM-1 genes in bread wheat in Australia. Crop. Pasture Sci. 2018, 69, 1173–1181. [Google Scholar] [CrossRef]
- Nigro, D.; Fortunato, S.; Giove, S.L.; Mazzucotelli, E.; Gadaleta, A. Functional Validation of Glutamine synthetase and Glutamate synthase Genes in Durum Wheat near Isogenic Lines with QTL for High GPC. Int. J. Mol. Sci. 2020, 21, 9253. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wong, D.; Forrest, K.; Allen, A.; Chao, S.; Huang, B.E.; Maccaferri, M.; Salvi, S.; Milner, S.G.; Cattivelli, L.; et al. Characterization of polyploid wheat genomic diversity using a high-density 90,000 single nucleotide polymorphism array. Plant Biotechnol. J. 2014, 12, 787–796. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.-X.; Barbier, H.; Rouse, M.N.; Singh, S.; Singh, R.P.; Bhavani, S.; Huerta-Espino, J.; Sorrells, M.E. A consensus map for Ug99 stem rust resistance loci in wheat. Theor. Appl. Genet. 2014, 127, 1561–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramya, P.; Chaubal, A.; Kulkarni, K.; Gupta, L.; Kadoo, N.; Dhaliwal, H.S.; Chhuneja, P.; Lagu, M.; Gupt, V. QTL mapping of 1000-kernel weight, kernel length, and kernel width in bread wheat (Triticum aestivum L.). J. Appl. Genet. 2010, 51, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.; Zhang, X.; Zhang, W.; Zhang, N.; Song, L.; Liu, L.; Xue, X.; Liu, G.; Liu, J.; Meng, D.; et al. QTL Detection for Kernel Size and Weight in Bread Wheat (Triticum aestivum L.) Using a High-Density SNP and SSR-Based Linkage Map. Front. Plant Sci. 2018, 9, 1484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Igrejas, G.; Branlard, G.; Carnide, V.; Gateau, I.; Pinto, G.H. Storage protein diversity within the old Portuguese bread wheat Barbela population. J. Genet. Breed. 1997, 51, 167–173. [Google Scholar]
- Corsi, B.; Obinu, L.; Zanella, C.M.; Cutrupi, S.; Day, R.; Geyer, M.; Lillemo, M.; Lin, M.; Mazza, L.; Percival-Alwyn, L.; et al. Identification of eight QTL controlling multiple yield components in a German multi-parental wheat population, including Rht24, WAPO-A1, WAPO-B1 and genetic loci on chromosomes 5A and 6A. Theor. Appl. Genet. 2021, 134, 1435–1454. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Pandey, S.; Assmann, S.M. Arabidopsis extra-large G proteins (XLGs) regulate root morphogenesis. Plant J. 2007, 53, 248–263. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y. Genetic Dissection of Wheat Nitrogen Use Efficiency Related Traits; Murdoch University: Perth, Australia, 2019. [Google Scholar]
- Guan, P.; Lu, L.; Jia, L.; Kabir, M.R.; Zhang, J.; Lan, T.; Zhao, Y.; Xin, M.; Hu, Z.; Yao, Y.; et al. Global QTL Analysis Identifies Genomic Regions on Chromosomes 4A and 4B Harboring Stable Loci for Yield-Related Traits Across Different Environments in Wheat (Triticum aestivum L.). Front. Plant Sci. 2018, 9, 529. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Wang, L.; Meng, Y.; Hao, Y.; Xu, H.; Hao, M.; Lan, S.; Zhang, Y.; Lv, L.; Zhang, K.; et al. Dissection of Genetic Basis Underpinning Kernel Weight-Related Traits in Common Wheat. Plants 2021, 10, 713. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhao, X.; He, X.; Zhao, G.; Li, B.; Liu, D.; Zhang, A.; Zhang, X.; Tong, Y.; Li, Z. Haplotype analysis of the genes encoding glutamine synthetase plastic isoforms and their association with nitrogen-use- and yield-related traits in bread wheat. New Phytol. 2011, 189, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, S.; Golan, G.; Guo, Z.; Ogawa, T.; Tagiri, A.; Sugimoto, K.; Bernhardt, N.; Brassac, J.; Mascher, M.; Hensel, G.; et al. Unleashing floret fertility in wheat through the mutation of a homeobox gene. Proc. Natl. Acad. Sci. USA 2019, 116, 5182–5187. [Google Scholar] [CrossRef] [Green Version]
- Worland, A.J.; Korzun, V.; Röder, M.S.; Ganal, M.W.; Law, C.N. Genetic analysis of the dwarfing gene Rht8 in wheat. Part II. The distribution and adaptive significance of allelic variants at the Rht8 locus of wheat as revealed by microsatellite screening. Theor. Appl. Genet. 1998, 96, 1110–1120. [Google Scholar] [CrossRef]
- Börner, A.; Worland, A.J. Does the Chinese dwarf wheat variety ‘XN0004’ carry Rht21? Cereal Res. Commun. 2002, 30, 25–29. [Google Scholar] [CrossRef]
- Nigro, D.; Blanco, A.; Anderson, O.D.; Gadaleta, A. Characterization of Ferredoxin-Dependent Glutamine-Oxoglutarate Amidotransferase (Fd-GOGAT) Genes and Their Relationship with Grain Protein Content QTL in Wheat. PLoS ONE 2014, 9, e103869. [Google Scholar] [CrossRef] [Green Version]
- Hou, J.; Jiang, Q.; Hao, C.; Wang, Y.; Zhang, H.; Zhang, X. Global Selection on Sucrose Synthase Haplotypes during a Century of Wheat Breeding. Plant Physiol. 2014, 164, 1918–1929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.-F.; Zhang, H.-P.; Hu, M.-J.; Wu, Z.-Y.; Jiang, H.; Cao, J.-J.; Xia, X.-C.; Ma, C.-X.; Chang, C. Cloning and characterization of Tabas1-B1 gene associated with flag leaf chlorophyll content and thousand-grain weight and development of a gene-specific marker in wheat. Mol. Breed. 2016, 36, 142. [Google Scholar] [CrossRef]
- Wang, N.; Xie, Y.Z.; Li, Y.Z.; Wu, S.N.; Wei, H.S.; Wang, C.S. Molecular mapping of a novel early leaf-senescence gene Els2 in common wheat by SNP genotyping arrays. Crop. Pasture Sci. 2020, 71, 356–367. [Google Scholar] [CrossRef]
- Hanif, M.; Gao, F.; Liu, J.; Wen, W.; Zhang, Y.; Rasheed, A.; Xia, X.; He, Z.; Cao, S. TaTGW6-A1, an ortholog of rice TGW6, is associated with grain weight and yield in bread wheat. Mol. Breed. 2015, 36, 1–8. [Google Scholar] [CrossRef]
- Sawa, M.; Nusinow, D.A.; Kay, S.A.; Imaizumi, T. FKF1 and GIGANTEA Complex Formation Is Required for Day-Length Measurement in Arabidopsis. Science 2007, 318, 261–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gawroński, P.; Ariyadasa, R.; Himmelbach, A.; Poursarebani, N.; Kilian, B.; Stein, N.; Steuernagel, B.; Hensel, G.; Kumlehn, J.; Sehgal, S.K.; et al. A Distorted Circadian Clock Causes Early Flowering and Temperature-Dependent Variation in Spike Development in the Eps-3Am Mutant of Einkorn Wheat. Genetics 2014, 196, 1253–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearce, S.; Saville, R.; Vaughan, S.P.; Chandler, P.M.; Wilhelm, E.P.; Sparks, C.A.; Al-Kaff, N.; Korolev, A.; Boulton, M.I.; Phillips, A.L.; et al. Molecular Characterization of Rht-1 Dwarfing Genes in Hexaploid Wheat. Plant Physiol. 2011, 157, 1820–1831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernard, S.M.; Møller, A.L.B.; Dionisio, G.; Kichey, T.; Jahn, T.P.; Dubois, F.; Baudo, M.; Lopes, M.S.; Tercé-Laforgue, T.; Foyer, C.H.; et al. Gene expression, cellular localisation and function of glutamine synthetase isozymes in wheat (Triticum aestivum L.). Plant Mol. Biol. 2008, 67, 89–105. [Google Scholar] [CrossRef]
- Sun, L.; Yang, W.; Li, Y.; Shan, Q.; Ye, X.; Wang, D.; Yu, K.; Lu, W.; Xin, P.; Pei, Z.; et al. A wheat dominant dwarfing line withRht12, which reduces stem cell length and affects gibberellic acid synthesis, is a 5AL terminal deletion line. Plant J. 2019, 97, 887–900. [Google Scholar] [CrossRef]
- Vikhe, P.; Venkatesan, S.; Chavan, A.; Tamhankar, S.; Patil, R. Mapping of dwarfing gene Rht14 in durum wheat and its effect on seedling vigor, internode length and plant height. Crop. J. 2019, 7, 187–197. [Google Scholar] [CrossRef]
- Yang, Z.; Zheng, J.; Liu, C.; Wang, Y.; Condon, A.G.; Chen, Y.; Hu, Y.-G. Effects of the GA-responsive dwarfing gene Rht18 from tetraploid wheat on agronomic traits of common wheat. Field Crop. Res. 2015, 183, 92–101. [Google Scholar] [CrossRef]
- Haque, M.A.; Martinek, P.; Watanabe, N.; Kuboyama, T. Genetic mapping of gibberellic acid-sensitive genes for semi-dwarfism in durum wheat. Cereal Res. Commun. 2011, 39, 171–178. [Google Scholar] [CrossRef]
- Nishimura, K.; Moriyama, R.; Katsura, K.; Saito, H.; Takisawa, R.; Kitajima, A.; Nakazaki, T. The early flowering trait of an emmer wheat accession (Triticum turgidum L. ssp. dicoccum) is associated with the cis-element of the Vrn-A3 locus. Theor. Appl. Genet. 2018, 131, 2037–2053. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Wang, F.; Liu, T.; Dong, Z.; Li, A.; Jing, R.; Mao, L.; Li, Y.; Liu, X.; Zhang, K.; et al. Natural variation of TaGASR7-A1 affects grain length in common wheat under multiple cultivation conditions. Mol. Breed. 2014, 34, 937–947. [Google Scholar] [CrossRef]
- Chang, J.; Zhang, J.; Mao, X.; Li, A.; Jia, J.; Jing, R. Polymorphism of TaSAP1-A1 and its association with agronomic traits in wheat. Planta 2013, 237, 1495–1508. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, J.; Xia, X.; He, Z. TaGS-D1, an ortholog of rice OsGS3, is associated with grain weight and grain length in common wheat. Mol. Breed. 2014, 34, 1097–1107. [Google Scholar] [CrossRef]
- Xie, Q.; Mayes, S.; Sparkes, D.L. Carpel size, grain filling, and morphology determine individual grain weight in wheat. J. Exp. Bot. 2015, 66, 6715–6730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondal, S.; Rutkoski, J.E.; Velu, G.; Singh, P.K.; Crespo-Herrera, L.A.; Guzman, C.G.; Bhavani, S.; Lan, C.; He, X.; Singh, R.P. Harnessing Diversity in Wheat to Enhance Grain Yield, Climate Resilience, Disease and Insect Pest Resistance and Nutrition Through Conventional and Modern Breeding Approaches. Front. Plant Sci. 2016, 7, 991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coombes, N. DiGGer: DiGGer Design Generator under Correlation and Blocking, 1.0.3 ed; NSW Department of Primary Industries, Australia: Albury, NSW, Australia, 2018. [Google Scholar]
- Zhang, J.; Dell, B.; Biddulph, B.; Drake-Brockman, F.; Walker, E.; Khan, N.; Wong, D.; Hayden, M.; Appels, R. Wild-type alleles of Rht-B1 and Rht-D1 as independent determinants of thousand-grain weight and kernel number per spike in wheat. Mol. Breed. 2013, 32, 771–783. [Google Scholar] [CrossRef]
- Zhang, J.; Huang, S.; Fosu-Nyarko, J.; Dell, B.; McNeil, M.; Waters, I.; Moolhuijzen, P.; Conocono, E.; Appels, R. The genome structure of the 1-FEH genes in wheat (Triticum aestivum L.): New markers to track stem carbohydrates and grain filling QTLs in breeding. Mol. Breed. 2008, 22, 339–351. [Google Scholar] [CrossRef]
- Clarke, K.R.; Gorley, R.N. Plymouth Routines in Multivariate Ecological Research, v6 ed.; PRIMER-E Ltd.: Plymouth, UK, 2006. [Google Scholar]
- Rohlf, F.J. Numerical Taxonomy and Multivariate Analysis System, 2.2 ed.; Applied Biostatistics Inc.: New York, NY, USA, 2009. [Google Scholar]
- Manly, K.F.; Cudmore, R.; Meer, J.M. Map Manager QTX, cross-platform software for genetic mapping. Mamm. Genome 2001, 12, 930–932. [Google Scholar] [CrossRef]
- Broman, K.W.; Sen, S. A Guide to QTL Mapping with R/qtl; Springer: New York, NY, USA, 2009; pp. 75–282. [Google Scholar]
- Zhang, J.; Dell, B.; Biddulph, B.; Khan, N.; Xu, Y.; Luo, H.; Appels, R. Vernalization gene combination to maximize grain yield in bread wheat (Triticum aestivum L.) in diverse environments. Euphytica 2014, 198, 439–454. [Google Scholar] [CrossRef]
- Li, S.; Wang, J.; Zhang, L. Inclusive Composite Interval Mapping of QTL by Environment Interactions in Biparental Populations. PLoS ONE 2015, 10, e0132414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Ye, G.; Wang, J. A Modified Algorithm for the Improvement of Composite Interval Mapping. Genetics 2007, 175, 361–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, D.G.; Cullis, B.R.; Gilmour, A.R.; Gogel, B.J.; Thompson, R. ASReml-R Reference Manual Version 4; VSN International Ltd.: Hemel Hempstead, UK, 2018. [Google Scholar]
- Patterson, H.D.; Thompson, R. Recovery of inter-block information when block sizes are unequal. Biometrika 1971, 58, 545–554. [Google Scholar] [CrossRef]
- Harrell, J.F.E. Package ‘Hmisc’, Harrel Miscellaneous, 4.5-0; CRAN, 2021, Computer Software. Available online: https://hbiostat.org/R/Hmisc/ (accessed on 13 February 2020).










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; She, M.; Yang, R.; Jiang, Y.; Qin, Y.; Zhai, S.; Balotf, S.; Zhao, Y.; Anwar, M.; Alhabbar, Z.; et al. Yield-Related QTL Clusters and the Potential Candidate Genes in Two Wheat DH Populations. Int. J. Mol. Sci. 2021, 22, 11934. https://doi.org/10.3390/ijms222111934
Zhang J, She M, Yang R, Jiang Y, Qin Y, Zhai S, Balotf S, Zhao Y, Anwar M, Alhabbar Z, et al. Yield-Related QTL Clusters and the Potential Candidate Genes in Two Wheat DH Populations. International Journal of Molecular Sciences. 2021; 22(21):11934. https://doi.org/10.3390/ijms222111934
Chicago/Turabian StyleZhang, Jingjuan, Maoyun She, Rongchang Yang, Yanjie Jiang, Yebo Qin, Shengnan Zhai, Sadegh Balotf, Yun Zhao, Masood Anwar, Zaid Alhabbar, and et al. 2021. "Yield-Related QTL Clusters and the Potential Candidate Genes in Two Wheat DH Populations" International Journal of Molecular Sciences 22, no. 21: 11934. https://doi.org/10.3390/ijms222111934