
Ferroptosis Induction in Multiple Myeloma Cells Triggers DNA Methylation and Histone Modification Changes Associated with Cellular Senescence

 , ,
, ,  , , , , , and
, , , , , and
Abstract
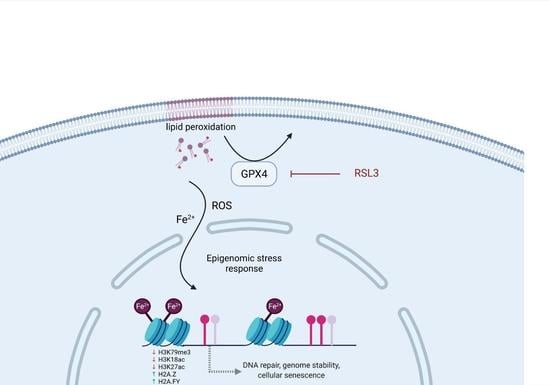
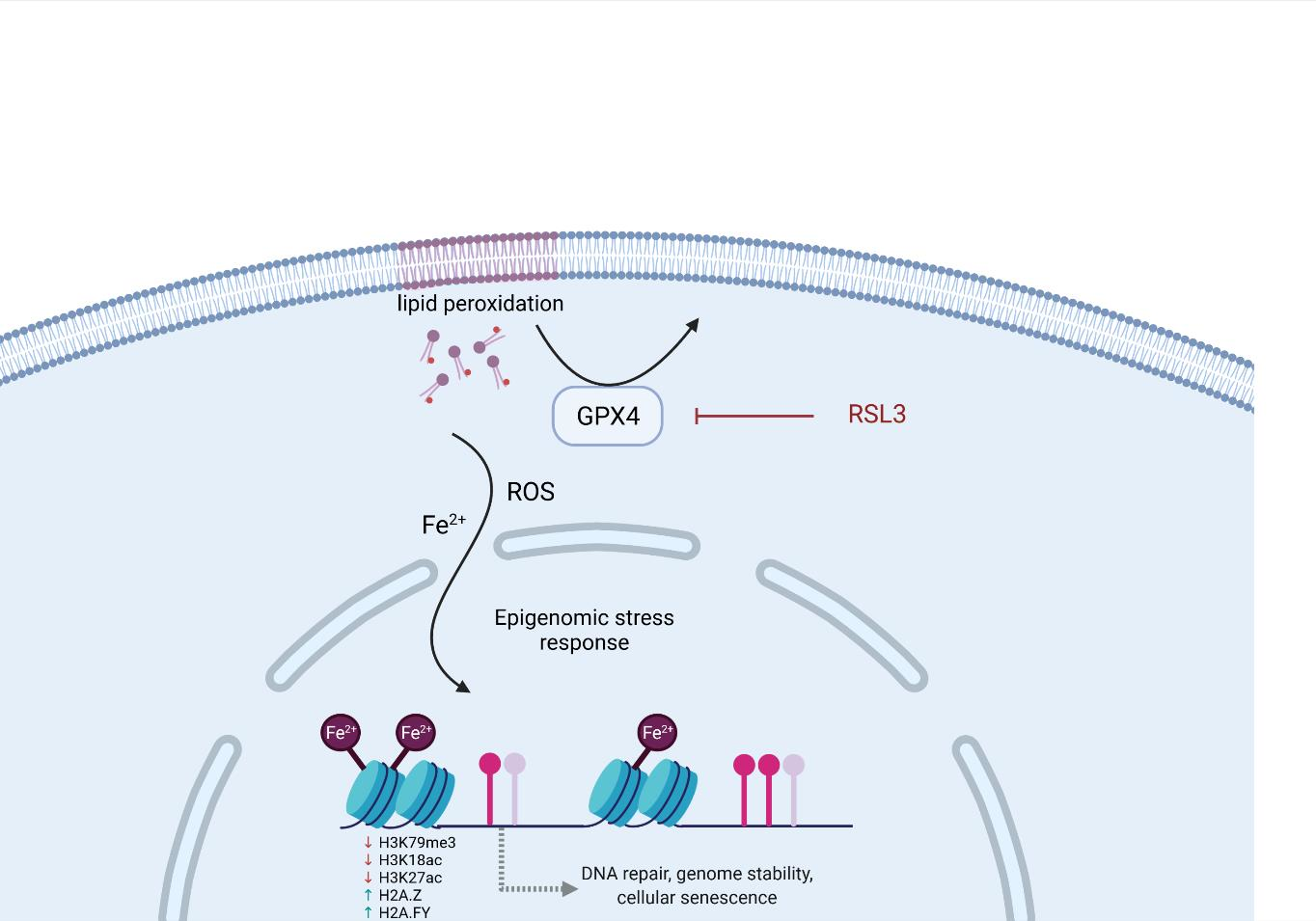
:
1. Introduction
2. Results
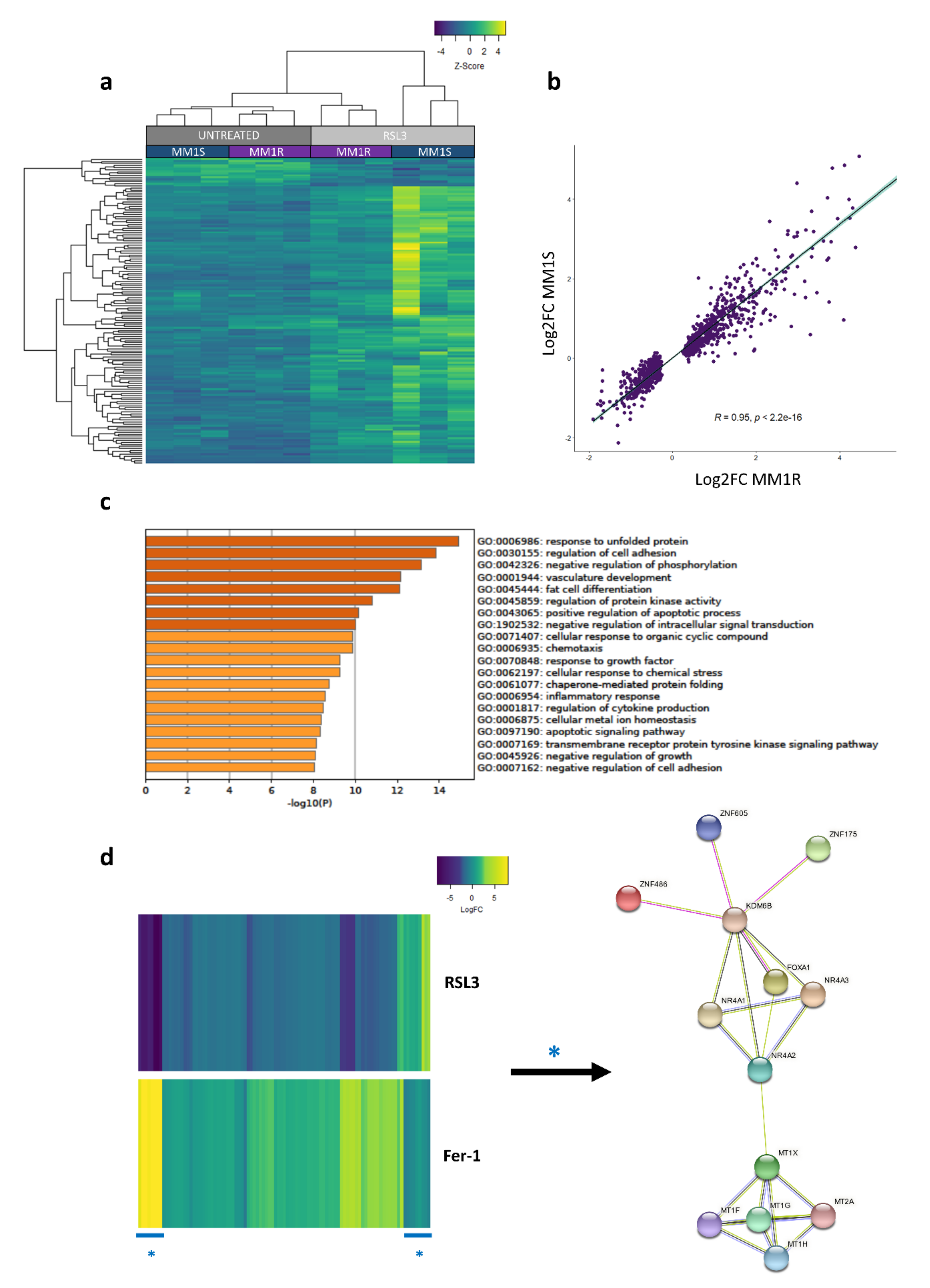
2.1. Glucocorticoid-Sensitive and -Resistant MM1 Cells Exhibit a Similar Transcriptome Stress Response upon Ferroptotic Cell Death Induction
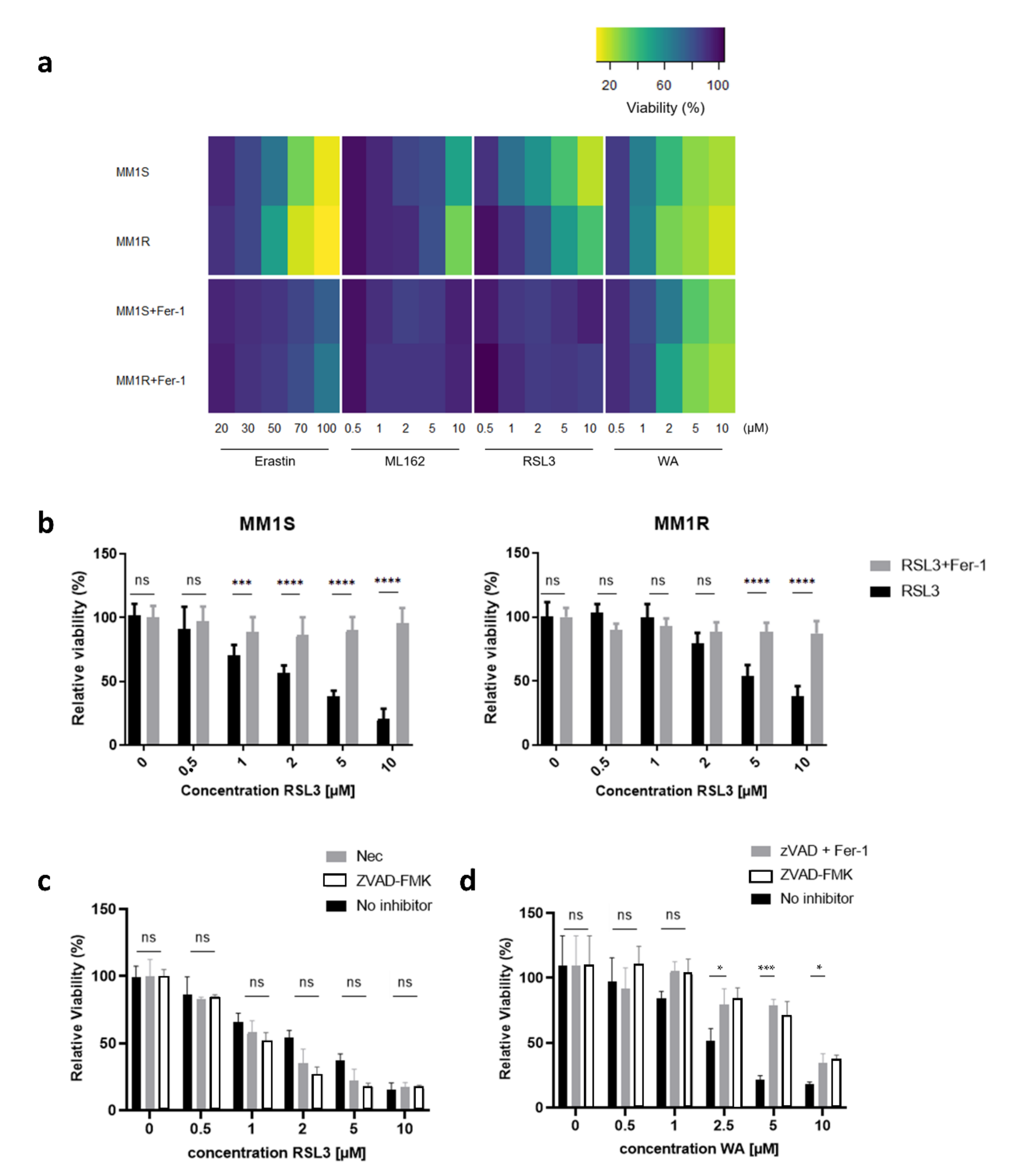
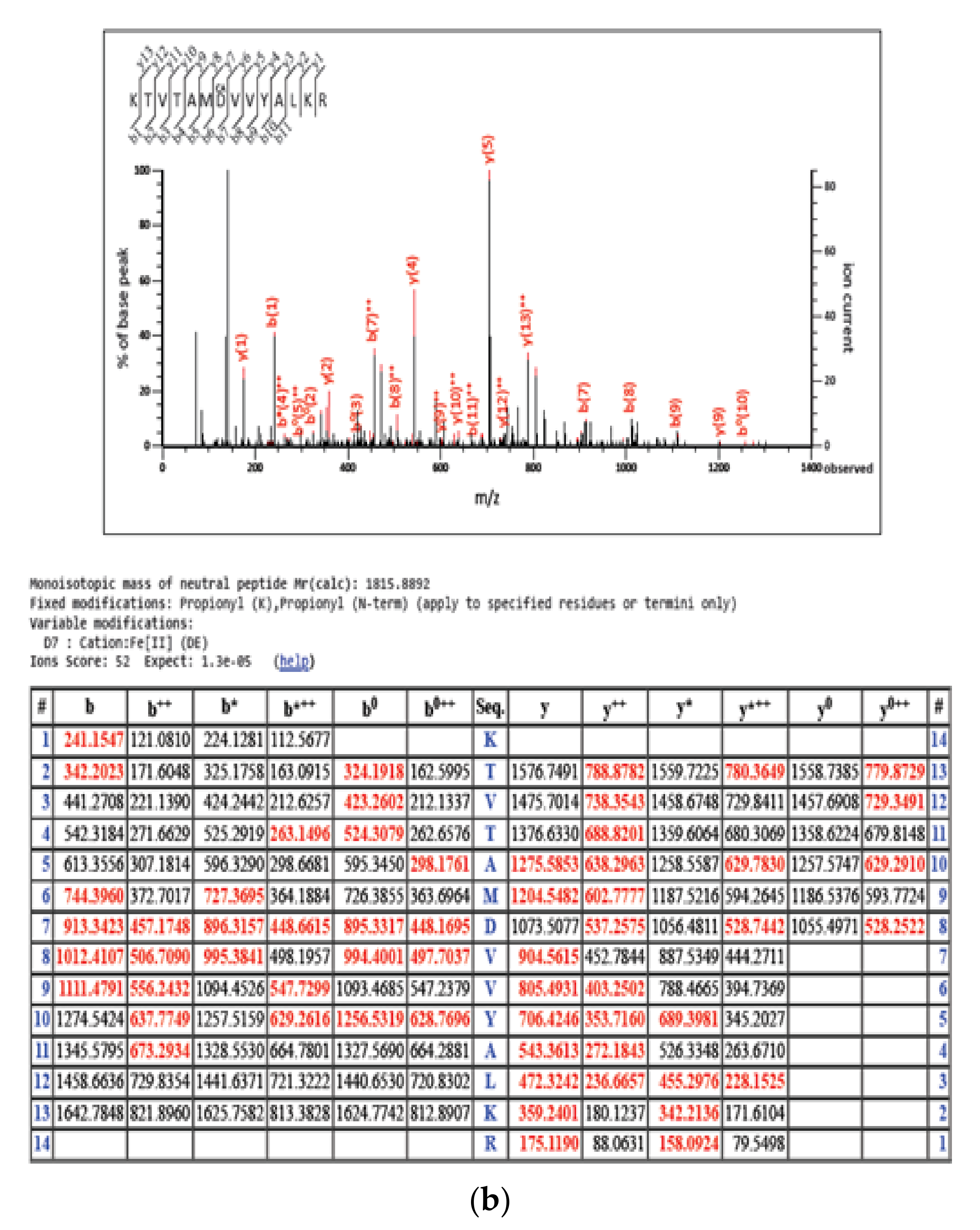
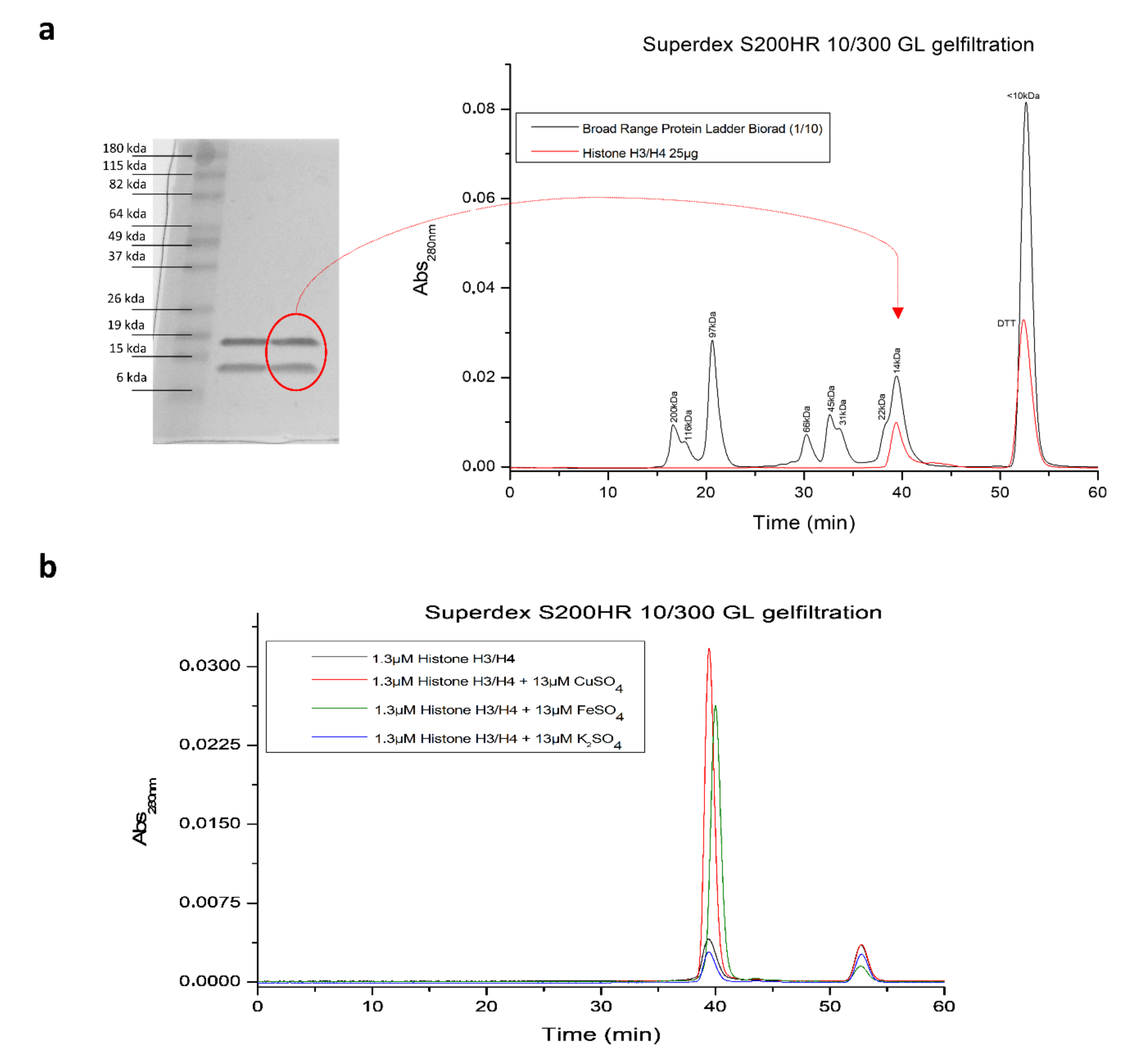
2.2. RSL3-Mediated GPX4 Inhibition Triggers the Formation of Non-Heme Iron-Histone Complexes and DNA Damage Responses
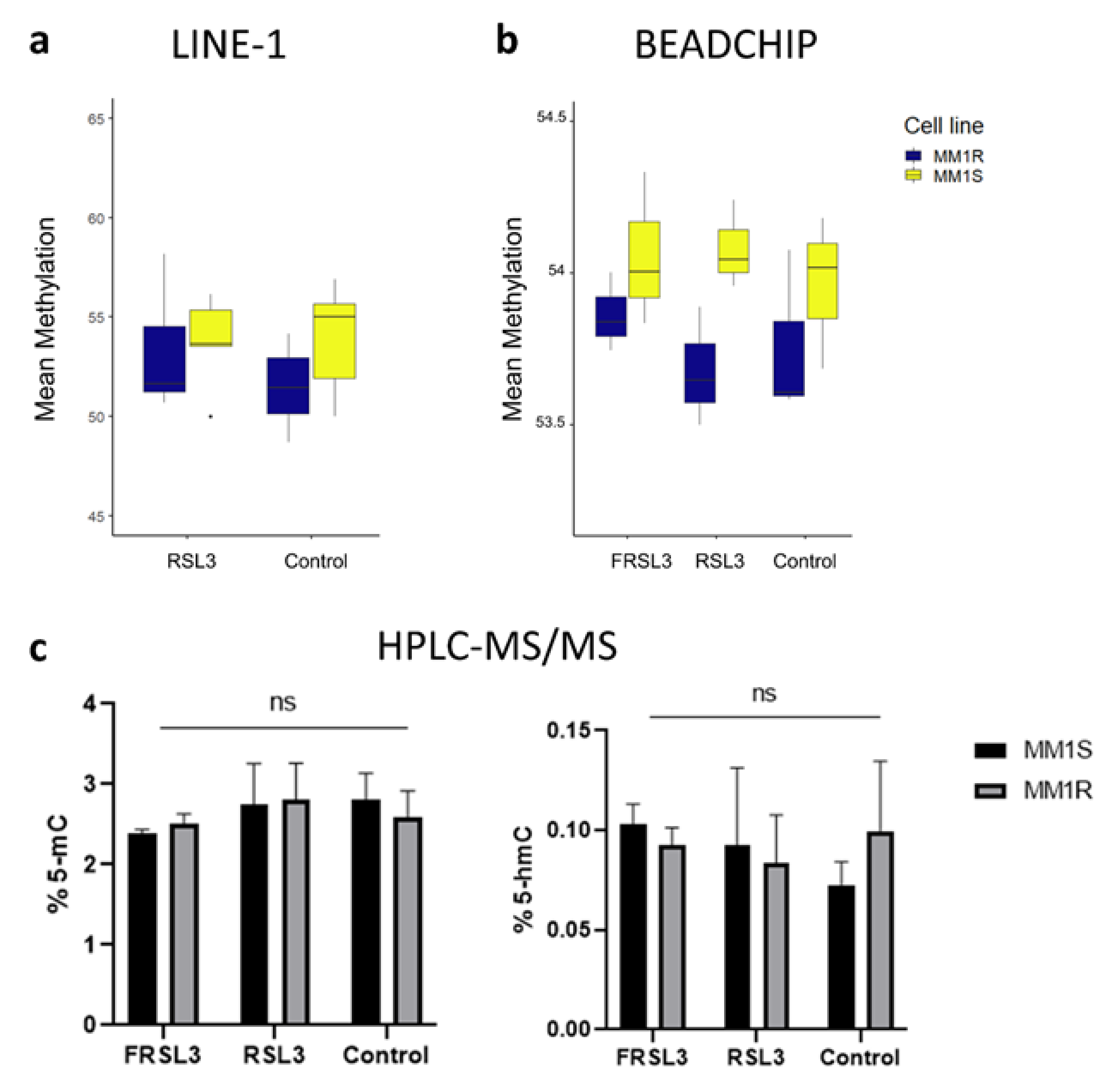
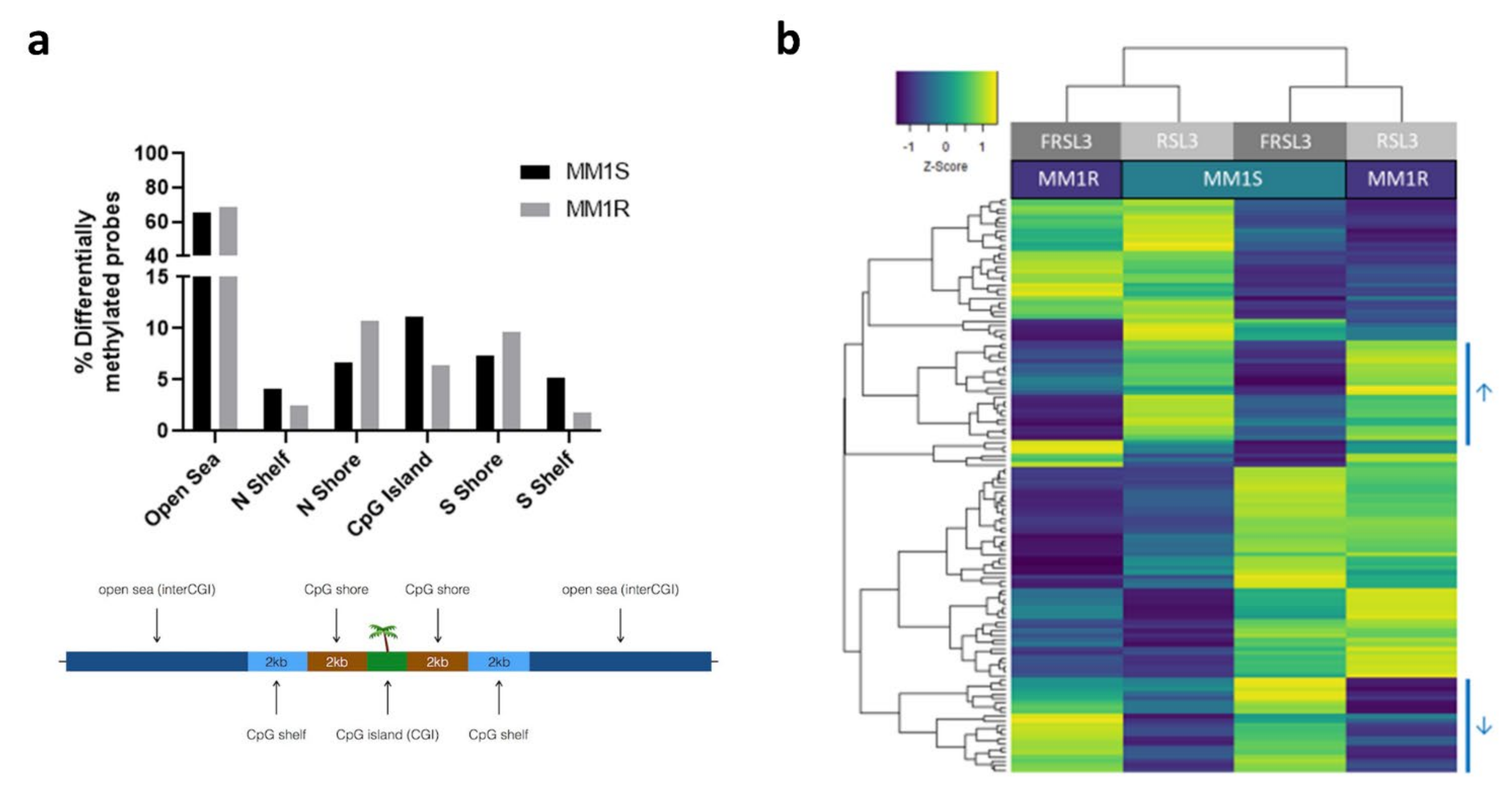
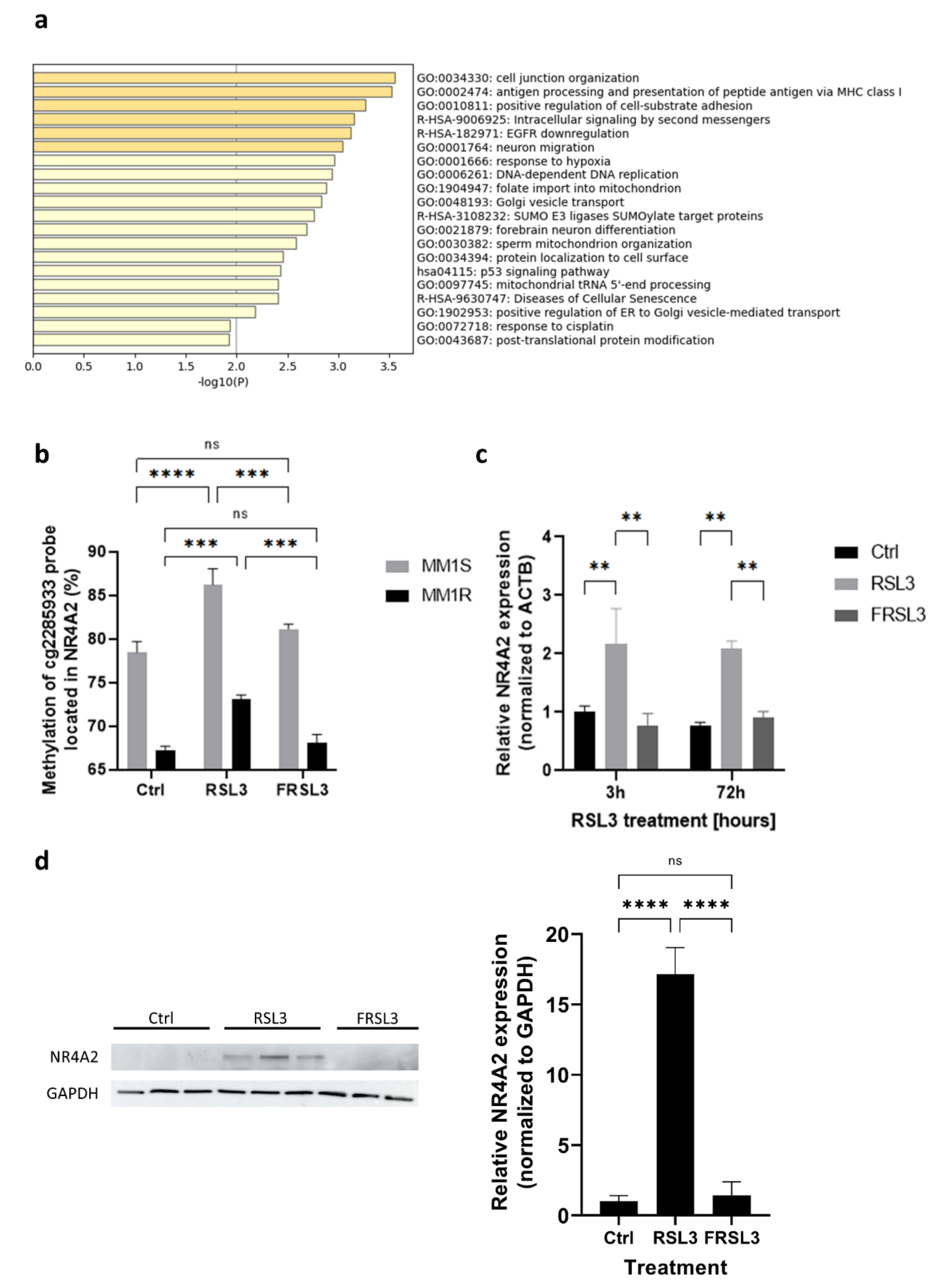
2.3. Prolonged Exposure of MM1 Cells to RSL3 Results in Local DNA Methylation Changes and Promotes Expression of DNA Damage Repair Protein NR4A2
3. Discussion
4. Materials and Methods
4.1. Antibodies and Reagents
4.2. Cell Culture and Cell Viability Assays
4.3. Lipid Peroxidation Assay
4.4. RNA Extraction, RNA Sequencing, and RNA Sequencing Data Analysis
4.5. cDNA Synthesis and qPCR Analysis
4.6. Histone Extraction and MS Sample Preparation
4.7. LC-MS Method and Data Analysis
4.8. Protein Extraction and Western Blot Analysis
4.9. Gel Filtration Chromatography
4.10. DNA Extraction and Bisulfite Conversion
4.11. LINE-1 Pyrosequencing
4.12. Infinium Methylation EPIC BeadChip Analysis
4.13. Global DNA Methylation Analysis by LC-MS/MS
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kazandjian, D. Multiple myeloma epidemiology and survival: A unique malignancy. Semin. Oncol. 2016, 43, 676–681. [Google Scholar] [CrossRef] [Green Version]
- Nunnelee, J.; Zhao, Q.; Benson, N.M.; Rosko, A.E.; Chaudhry, M.; Bumma, N.; Khan, A.M.; Devarakonda, S.; Efebera, Y.A.; Sharma, N. Improvement in Survival of Multiple Myeloma Patients: A Long-Term Institutional Experience. Blood 2019, 134, 4573. [Google Scholar] [CrossRef]
- Martinez-Baños, D.; Hernandez, B.S.; Jiménez, G.; Barrera-Lumbreras, G.; Barrales-Benitez, O. Global methylation and promoter-specific methylation of the P16, SOCS-1, E-cadherin, P73 and SHP-1 genes and their expression in patients with multiple myeloma during active disease and remission. Exp. Ther. Med. 2017, 13, 2442–2450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Hu, W.-X.; Luo, S.-Q.; Xiong, D.-H.; Sun, S.; Wang, Y.-P.; Bu, X.-F.; Liu, J.; Hu, J. Promoter methylation induced epigenetic silencing of DAZAP2, a downstream effector of p38/MAPK pathway, in multiple myeloma cells. Cell. Signal. 2019, 60, 136–145. [Google Scholar] [CrossRef]
- Popovic, R.; Martinez-Garcia, E.; Giannopoulou, E.G.; Zhang, Q.; Zhang, Q.; Ezponda, T.; Shah, M.Y.; Zheng, Y.; Will, C.M.; Small, E.C.; et al. Histone Methyltransferase MMSET/NSD2 Alters EZH2 Binding and Reprograms the Myeloma Epigenome through Global and Focal Changes in H3K36 and H3K27 Methylation. PLoS Genet. 2014, 10, e1004566. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Ahn, J.H.; Liu, H.; Tsai, Y.-H.; Bhanu, N.V.; Koss, B.; Allison, D.F.; Ma, A.; Storey, A.J.; Wang, P.; et al. PHF19 promotes multiple myeloma tumorigenicity through PRC2 activation and broad H3K27me3 domain formation. Blood 2019, 134, 1176–1189. [Google Scholar] [CrossRef] [PubMed]
- Handa, H.; Murakami, Y.; Ishihara, R.; Kimura-Masuda, K.; Masuda, Y. The Role and Function of microRNA in the Pathogenesis of Multiple Myeloma. Cancers 2019, 11, 1738. [Google Scholar] [CrossRef] [Green Version]
- Tatekawa, S.; Chinen, Y.; Ri, M.; Narita, T.; Shimura, Y.; Matsumura-Kimoto, Y.; Tsukamoto, T.; Kobayashi, T.; Kawata, E.; Uoshima, N.; et al. Epigenetic repression of miR-375 is the dominant mechanism for constitutive activation of the PDPK1/RPS6KA3 signalling axis in multiple myeloma. Br. J. Haematol. 2017, 178, 534–546. [Google Scholar] [CrossRef]
- Dimopoulos, K.; Helbo, A.S.; Munch-Petersen, H.F.; Sjö, L.; Christensen, J.; Kristensen, L.S.; Asmar, F.; Hermansen, N.E.U.; O’Connel, C.; Gimsing, P.; et al. Dual inhibition of DNMTs and EZH2 can overcome both intrinsic and acquired resistance of myeloma cells to IMiDs in a cereblon-independent manner. Mol. Oncol. 2018, 12, 180–195. [Google Scholar] [CrossRef]
- Hentati-Kallel, M.; LE Jan, S.; Bernard, P.; Antonicelli, F.; Trussardi-Régnier, A. Histone deacetylases meet microRNA-associated MMP-9 expression regulation in glucocorticoid-sensitive and -resistant cell lines. Int. J. Oncol. 2016, 50, 717–726. [Google Scholar] [CrossRef] [Green Version]
- Qin, Y.; Zhang, S.; Deng, S.; An, G.; Qin, X.; Li, F.; Xu, Y.; Hao, M.; Yang, Y.; Zhou, W.; et al. Epigenetic silencing of miR-137 induces drug resistance and chromosomal instability by targeting AURKA in multiple myeloma. Leukemia 2017, 31, 1123–1135. [Google Scholar] [CrossRef]
- Issa, M.E.; Takhsha, F.S.; Chirumamilla, C.S.; Perez-Novo, C.; Vanden Berghe, W.; Cuendet, M. Epigenetic strategies to reverse drug resistance in heterogeneous multiple myeloma. Clin. Epigenetics 2017, 9, 17. [Google Scholar] [CrossRef] [Green Version]
- Herviou, L.; Kassambara, A.; Boireau, S.; Robert, N.; Requirand, G.; Müller-Tidow, C.; Vincent, L.; Seckinger, A.; Goldschmidt, H.; Cartron, G.; et al. PRC2 targeting is a therapeutic strategy for EZ score defined high-risk multiple myeloma patients and overcome resistance to IMiDs. Clin. Epigenetics 2018, 10, 121. [Google Scholar] [CrossRef]
- Zhou, J.; Shen, Q.; Lin, H.; Hu, L.; Li, G.; Zhang, X. Decitabine shows potent anti-myeloma activity by depleting monocytic myeloid-derived suppressor cells in the myeloma microenvironment. J. Cancer Res. Clin. Oncol. 2019, 145, 329–336. [Google Scholar] [CrossRef]
- Che, F.; Chen, J.; Dai, J.; Liu, X. Inhibition of Multiple Myeloma Using 5-Aza-2ʹ-Deoxycytidine and Bortezomib-Loaded Self-Assembling Nanoparticles. Cancer Manag. Res. 2020, 12, 6969–6976. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Shen, L.; Li, X.; Liu, J. Efficacy and toxicity of histone deacetylase inhibitors in relapsed/refractory multiple myeloma: Systematic review and meta-analysis of clinical trials. Exp. Ther. Med. 2019, 18, 1057–1068. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, N.; Zeng, G.-Z.; Yin, J.-L.; Bian, Z.-X. Artesunate activates the ATF4-CHOP-CHAC1 pathway and affects ferroptosis in Burkitt’s Lymphoma. Biochem. Biophys. Res. Commun. 2019, 519, 533–539. [Google Scholar] [CrossRef]
- Zhang, Y.; Tan, H.; Daniels, J.D.; Zandkarimi, F.; Liu, H.; Brown, L.M.; Uchida, K.; O’Connor, O.A.; Stockwell, B.R. Imidazole Ketone Erastin Induces Ferroptosis and Slows Tumor Growth in a Mouse Lymphoma Model. Cell Chem. Biol. 2019, 26, 623–633.e9. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Cort, A.; Ozben, T.; Saso, L.; De Luca, C.; Korkina, L. Redox Control of Multidrug Resistance and Its Possible Modulation by Antioxidants. Oxidative Med. Cell. Longev. 2016, 2016, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Yang, L.; Zhang, X.; Cui, W.; Liu, Y.; Sun, Q.; He, Q.; Zhao, S.; Zhang, G.; Wang, Y.; et al. Epigenetic regulation of ferroptosis by H2B monoubiquitination and p53. EMBO Rep. 2019, 20, e47563. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Mao, C.; Yang, R.; Yan, B.; Shi, Y.; Liu, X.; Lai, W.; Liu, Y.; Wang, X.; Xiao, D.; et al. EGLN1/c-Myc Induced Lymphoid-Specific Helicase Inhibits Ferroptosis through Lipid Metabolic Gene Expression Changes. Theranostics 2017, 7, 3293–3305. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Koppula, P.; Gan, B. Regulation of H2A ubiquitination and SLC7A11 expression by BAP1 and PRC1. Cell Cycle 2019, 18, 773–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhao, Y.; Wang, H.; Zhang, C.; Wang, M.; Yang, Y.; Xu, X.; Hu, Z. Histone demethylase KDM3B protects against ferroptosis by upregulating SLC7A11. FEBS Open Bio 2020, 10, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Li, Q.; Sun, X.; Sun, X.; Tang, Y.; Qu, Y.; Liu, D.; Yu, T.; Li, G.; Tong, T.; et al. CRL4DCAF8 dependent opposing stability control over the chromatin remodeler LSH orchestrates epigenetic dynamics in ferroptosis. Cell Death Differ. 2021, 28, 1593–1609. [Google Scholar] [CrossRef] [PubMed]
- Rroji, O.; Kumar, A.; Karuppagounder, S.S.; Ratan, R.R. Epigenetic regulators of neuronal ferroptosis identify novel therapeutics for neurological diseases: HDACs, transglutaminases, and HIF prolyl hydroxylases. Neurobiol. Dis. 2021, 147, 105145. [Google Scholar] [CrossRef]
- Wang, M.; Mao, C.; Ouyang, L.; Liu, Y.; Lai, W.; Liu, N.; Shi, Y.; Chen, L.; Xiao, D.; Yu, F.; et al. Long noncoding RNA LINC00336 inhibits ferroptosis in lung cancer by functioning as a competing endogenous RNA. Cell Death Differ. 2019, 26, 2329–2343. [Google Scholar] [CrossRef] [PubMed]
- Cyr, A.R.; Domann, F.E. The Redox Basis of Epigenetic Modifications: From Mechanisms to Functional Consequences. Antioxidants Redox Signal. 2011, 15, 551–589. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Yu, L.; Ding, J.; Chen, Y. Iron Metabolism in Cancer. Int. J. Mol. Sci. 2018, 20, 95. [Google Scholar] [CrossRef] [Green Version]
- Cao, L.-L.; Liu, H.; Yue, Z.; Liu, L.; Pei, L.; Gu, J.; Wang, H.; Jia, M. Iron chelation inhibits cancer cell growth and modulates global histone methylation status in colorectal cancer. BioMetals 2018, 31, 797–805. [Google Scholar] [CrossRef]
- Shi, Z.; Zhang, L.; Zheng, J.; Sun, H.; Shao, C. Ferroptosis: Biochemistry and Biology in Cancers. Front. Oncol. 2021, 11. [Google Scholar] [CrossRef]
- Kawai, K.; Li, Y.-S.; Song, M.-F.; Kasai, H. DNA methylation by dimethyl sulfoxide and methionine sulfoxide triggered by hydroxyl radical and implications for epigenetic modifications. Bioorgan. Med. Chem. Lett. 2010, 20, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Lin, X.; Huang, C. Activation of the reverse transsulfuration pathway through NRF2/CBS confers erastin-induced ferroptosis resistance. Br. J. Cancer 2020, 122, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Lertratanangkoon, K.; Wu, C.J.; Savaraj, N.; Thomas, M.L. Alterations of DNA methylation by glutathione depletion. Cancer Lett. 1997, 120, 149–156. [Google Scholar] [CrossRef]
- Ulrey, C.L.; Liu, L.; Andrews, L.G.; Tollefsbol, T.O. The impact of metabolism on DNA methylation. Hum. Mol. Genet. 2005, 14, R139–R147. [Google Scholar] [CrossRef]
- Pinto, V.; Bergantim, R.; Caires, H.R.; Seca, H.; Guimarães, J.E.; Vasconcelos, M.H. Multiple Myeloma: Available Therapies and Causes of Drug Resistance. Cancers 2020, 12, 407. [Google Scholar] [CrossRef] [Green Version]
- Hassannia, B.; Logie, E.; Vandenabeele, P.; Berghe, T.V.; Berghe, W.V. Withaferin A: From ayurvedic folk medicine to preclinical anti-cancer drug. Biochem. Pharmacol. 2020, 173, 113602. [Google Scholar] [CrossRef] [PubMed]
- Rai, M.; Jogee, P.S.; Agarkar, G.; Dos Santos, C.A. Anticancer activities ofWithania somnifera: Current research, formulations, and future perspectives. Pharm. Biol. 2015, 54, 189–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassannia, B.; Wiernicki, B.; Ingold, I.; Qu, F.; Van Herck, S.; Tyurina, Y.Y.; Bayır, H.; Abhari, B.A.; Angeli, J.P.F.; Choi, S.M.; et al. Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J. Clin. Investig. 2018, 128, 3341–3355. [Google Scholar] [CrossRef]
- Chen, M.-S.; Wang, S.-F.; Hsu, C.-Y.; Yin, P.-H.; Yeh, T.-S.; Lee, H.-C.; Tseng, L.-M. CHAC1 degradation of glutathione enhances cystine-starvation-induced necroptosis and ferroptosis in human triple negative breast cancer cells via the GCN2-eIF2α-ATF4 pathway. Oncotarget 2017, 8, 114588–114602. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Ou, Z.; Xie, M.; Kang, R.; Fan, Y.; Niu, X.; Wang, H.; Cao, L.; Tang, D. HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene 2015, 34, 5617–5625. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.; Slusher, B.S.; Stockwell, B.R.; et al. Data from: Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 2014, 3, e02523. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Verhelst, S.; De Clerck, L.; Willems, S.; Van Puyvelde, B.; Daled, S.; Deforce, D.; Dhaenens, M. Comprehensive histone epigenetics: A mass spectrometry based screening assay to measure epigenetic toxicity. MethodsX 2020, 7, 101055. [Google Scholar] [CrossRef] [PubMed]
- Govaert, E.; Van Steendam, K.; Scheerlinck, E.; Vossaert, L.; Meert, P.; Stella, M.; Willems, S.; De Clerck, L.; Dhaenens, M.; Deforce, D. Extracting histones for the specific purpose of label-free MS. Proteomics 2016, 16, 2937–2944. [Google Scholar] [CrossRef]
- Licht, J.D. Epigenetic Regulation and Therapeutic Targeting in Myeloma. Blood 2018, 132, SCI-37. [Google Scholar] [CrossRef]
- Ontoso, D.; Kauppi, L.; Keeney, S.; San-Segundo, P.A. Dynamics of DOT1L localization and H3K79 methylation during meiotic prophase I in mouse spermatocytes. Chromosoma 2014, 123, 147–164. [Google Scholar] [CrossRef] [Green Version]
- Altaf, M.; Utley, R.T.; Lacoste, N.; Tan, S.; Briggs, S.D.; Côté, J. Interplay of Chromatin Modifiers on a Short Basic Patch of Histone H4 Tail Defines the Boundary of Telomeric Heterochromatin. Mol. Cell 2007, 28, 1002–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, H.H.; Feng, Q.; Wang, H.; Erdjument-Bromage, H.; Tempst, P.; Zhang, Y.; Struhl, K. Lysine methylation within the globular domain of histone H3 by Dot1 is important for telomeric silencing and Sir protein association. Genes Dev. 2002, 16, 1518–1527. [Google Scholar] [CrossRef] [Green Version]
- Lazzaro, F.; Sapountzi, V.; Granata, M.; Pellicioli, A.; Vaze, M.; Haber, J.; Plevani, P.; Lydall, D.; Muzi-Falconi, M. Histone methyltransferase Dot1 and Rad9 inhibit single-stranded DNA accumulation at DSBs and uncapped telomeres. EMBO J. 2008, 27, 1502–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bostelman, L.J.; Keller, A.M.; Albrecht, A.M.; Arat, A.; Thompson, J.S. Methylation of histone H3 lysine-79 by Dot1p plays multiple roles in the response to UV damage in Saccharomyces cerevisiae. DNA Repair 2007, 6, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.L.; Chen, L.; Ma, L.; Ding, X.J.; Tang, S.F.; Zhang, A.H.; Li, J. Role of H3K18ac-regulated nucleotide excision repair-related genes in arsenic-induced DNA damage and repair of HaCaT cells. Hum. Exp. Toxicol. 2020, 39, 1168–1177. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.-Y.; Li, G.; Deng, Z.-J.; Liu, L.-Y.; Chen, L.; Tang, J.-Z.; Wang, Y.-Q.; Cao, S.-T.; Fang, Y.-X.; Wen, F.; et al. Dicer interacts with SIRT7 and regulates H3K18 deacetylation in response to DNA damaging agents. Nucleic Acids Res. 2016, 44, 3629–3642. [Google Scholar] [CrossRef]
- Ma, L.; Li, J.; Zhan, Z.; Chen, L.; Li, D.; Bai, Q.; Gao, C.; Li, J.; Zeng, X.; He, Z.; et al. Specific histone modification responds to arsenic-induced oxidative stress. Toxicol. Appl. Pharmacol. 2016, 302, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, D.R.; Deckard, C.E.; Zeng, Y.; Sczepanski, J.T. Acetylation of the histone H3 tail domain regulates base excision repair on higher-order chromatin structures. Sci. Rep. 2019, 9, 15972. [Google Scholar] [CrossRef] [PubMed]
- Tasselli, L.; Xi, Y.; Zheng, W.; Tennen, R.I.; Odrowaz, Z.; Simeoni, F.; Li, Y.X.W.; Chua, K.F. SIRT6 deacetylates H3K18ac at pericentric chromatin to prevent mitotic errors and cellular senescence. Nat. Struct. Mol. Biol. 2016, 23, 434–440. [Google Scholar] [CrossRef] [Green Version]
- Schick, S.; Fournier, D.; Thakurela, S.; Sahu, S.K.; Garding, A.; Tiwari, V.K. Dynamics of chromatin accessibility and epigenetic state in response to UV damage. J. Cell Sci. 2015, 128, 4380–4394. [Google Scholar] [CrossRef] [Green Version]
- Ito, T.; Teo, Y.V.; Evans, S.A.; Neretti, N.; Sedivy, J.M. Regulation of Cellular Senescence by Polycomb Chromatin Modifiers through Distinct DNA Damage- and Histone Methylation-Dependent Pathways. Cell Rep. 2018, 22, 3480–3492. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; So, K.K.; Li, Y.; Li, Y.; Yuan, J.; Ding, Y.; Chen, F.; Huang, Y.; Liu, J.; Lee, W.; et al. Elevated H3K27ac in aged skeletal muscle leads to increase in extracellular matrix and fibrogenic conversion of muscle satellite cells. Aging Cell 2019, 18, e12996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, Y.; Zhang, C.; Lyu, G.; Huang, X.; Zhang, X.; Zhuang, T.; Jia, L.; Zhang, L.; Zhang, C.; Li, C.; et al. Senescence-activated enhancer landscape orchestrates the senescence-associated secretory phenotype in murine fibroblasts. Nucleic Acids Res. 2020, 48, 10909–10923. [Google Scholar] [CrossRef] [PubMed]
- Nishibuchi, I.; Suzuki, H.; Kinomura, A.; Sun, J.; Liu, N.-A.; Horikoshi, Y.; Shima, H.; Kusakabe, M.; Harata, M.; Fukagawa, T.; et al. Reorganization of Damaged Chromatin by the Exchange of Histone Variant H2A.Z-2. Int. J. Radiat. Oncol. 2014, 89, 736–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalocsay, M.; Hiller, N.J.; Jentsch, S. Chromosome-wide Rad51 Spreading and SUMO-H2A.Z-Dependent Chromosome Fixation in Response to a Persistent DNA Double-Strand Break. Mol. Cell 2009, 33, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ayrapetov, M.K.; Xu, C.; Gursoy-Yuzugullu, O.; Hu, Y.; Price, B.D. Histone H2A.Z Controls a Critical Chromatin Remodeling Step Required for DNA Double-Strand Break Repair. Mol. Cell 2012, 48, 723–733. [Google Scholar] [CrossRef] [Green Version]
- Rangasamy, D.; Berven, L.; Ridgway, P.; Tremethick, D. Pericentric heterochromatin becomes enriched with H2A.Z during early mammalian development. EMBO J. 2003, 22, 1599–1607. [Google Scholar] [CrossRef] [Green Version]
- Meneghini, M.D.; Wu, M.; Madhani, H.D. Conserved Histone Variant H2A.Z Protects Euchromatin from the Ectopic Spread of Silent Heterochromatin. Cell 2003, 112, 725–736. [Google Scholar] [CrossRef] [Green Version]
- Hou, H.; Wang, Y.; Kallgren, S.; Thompson, J.; Yates, J.R.; Jia, S. Histone Variant H2A.Z Regulates Centromere Silencing and Chromosome Segregation in Fission Yeast. J. Biol. Chem. 2010, 285, 1909–1918. [Google Scholar] [CrossRef] [Green Version]
- Greaves, I.; Rangasamy, D.; Ridgway, P.; Tremethick, D.J. H2A.Z contributes to the unique 3D structure of the centromere. Proc. Natl. Acad. Sci. USA 2007, 104, 525–530. [Google Scholar] [CrossRef] [Green Version]
- Ávila-López, P.A.; Guerrero, G.; Nuñez-Martínez, H.N.; Peralta-Alvarez, C.A.; Hernández-Montes, G.; Álvarez-Hilario, L.G.; Herrera-Goepfert, R.; Albores-Saavedra, J.; Villegas-Sepúlveda, N.; Cedillo-Barrón, L.; et al. H2A.Z overexpression suppresses senescence and chemosensitivity in pancreatic ductal adenocarcinoma. Oncogene 2021, 40, 2065–2080. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, P.D.; Gamble, M.J. MacroH2A1 chromatin specification requires its docking domain and acetylation of H2B lysine 20. Nat. Commun. 2018, 9, 5143. [Google Scholar] [CrossRef] [PubMed]
- Re, O.L.; Vinciguerra, M. Histone MacroH2A1: A Chromatin Point of Intersection between Fasting, Senescence and Cellular Regeneration. Genes 2017, 8, 367. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, P.D.; Hamilton, G.; Park, J.W.; Gamble, M.J. MacroH2A1 Regulation of Poly(ADP-Ribose) Synthesis and Stability Prevents Necrosis and Promotes DNA Repair. Mol. Cell. Biol. 2019, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutta, A.; Abmayr, S.M.; Workman, J.L. Diverse Activities of Histone Acylations Connect Metabolism to Chromatin Function. Mol. Cell 2016, 63, 547–552. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-Y.; Kim, W.; Bae, K.-H.; Lee, S.; Lee, E.-W. Lipid Metabolism and Ferroptosis. Biology 2021, 10, 184. [Google Scholar] [CrossRef]
- Flynn, E.M.; Huang, O.W.; Poy, F.; Oppikofer, M.; Bellon, S.F.; Tang, Y.; Cochran, A.G. A Subset of Human Bromodomains Recognizes Butyryllysine and Crotonyllysine Histone Peptide Modifications. Structure 2015, 23, 1801–1814. [Google Scholar] [CrossRef] [Green Version]
- Goudarzi, A.; Zhang, D.; Huang, H.; Barral, S.; Kwon, O.K.; Qi, S.; Tang, Z.; Buchou, T.; Vitte, A.-L.; He, T.; et al. Dynamic Competing Histone H4 K5K8 Acetylation and Butyrylation Are Hallmarks of Highly Active Gene Promoters. Mol. Cell 2016, 62, 169–180. [Google Scholar] [CrossRef] [Green Version]
- Tan, M.; Luo, H.; Lee, S.; Jin, F.; Yang, J.S.; Montellier, E.; Buchou, T.; Cheng, Z.; Rousseaux, S.; Rajagopal, N.; et al. Identification of 67 Histone Marks and Histone Lysine Crotonylation as a New Type of Histone Modification. Cell 2011, 146, 1016–1028. [Google Scholar] [CrossRef] [Green Version]
- Attar, N.; Campos, O.A.; Vogelauer, M.; Cheng, C.; Xue, Y.; Schmollinger, S.; Salwinski, L.; Mallipeddi, N.V.; Boone, B.A.; Yen, L.; et al. The histone H3-H4 tetramer is a copper reductase enzyme. Science 2020, 369, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Pogribny, I.P.; Tryndyak, V.P.; Pogribna, M.; Shpyleva, S.; Surratt, G.; Da Costa, G.G.; Beland, F.A. Modulation of intracellular iron metabolism by iron chelation affects chromatin remodeling proteins and corresponding epigenetic modifications in breast cancer cells and increases their sensitivity to chemotherapeutic agents. Int. J. Oncol. 2013, 42, 1822–1832. [Google Scholar] [CrossRef] [Green Version]
- Ye, Q.; Trivedi, M.; Zhang, Y.; Böhlke, M.; Alsulimani, H.; Chang, J.; Maher, T.; Deth, R.; Kim, J. Brain iron loading impairs DNA methylation and alters GABAergic function in mice. FASEB J. 2019, 33, 2460–2471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bockmühl, Y.; Patchev, A.V.; Madejska, A.; Hoffmann, A.; Sousa, J.C.; Sousa, N.; Holsboer, F.; Almeida, O.F.X.; Spengler, D. Methylation at the CpG island shore region upregulates Nr3c1 promoter activity after early-life stress. Epigenetics 2015, 10, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Pistritto, G.; Trisciuoglio, D.; Ceci, C.; Garufi, A.; D’Orazi, G. Apoptosis as anticancer mechanism: Function and dysfunction of its modulators and targeted therapeutic strategies. Aging 2016, 8, 603–619. [Google Scholar] [CrossRef] [Green Version]
- Löffler, D.; Brocke-Heidrich, K.; Pfeifer, G.; Stocsits, C.; Hackermüller, J.; Kretzschmar, A.K.; Burger, R.; Gramatzki, M.; Blumert, C.; Bauer, K.; et al. Interleukin-6–dependent survival of multiple myeloma cells involves the Stat3-mediated induction of microRNA-21 through a highly conserved enhancer. Blood 2007, 110, 1330–1333. [Google Scholar] [CrossRef] [Green Version]
- Hurt, E.M.; Thomas, S.B.; Peng, B.; Farrar, W.L. Reversal of p53 epigenetic silencing in multiple myeloma permits apoptosis by a p53 activator. Cancer Biol. Ther. 2006, 5, 1154–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enright, H.U.; Miller, W.J.; Hebbel, R.P. Nucleosomal histone protein protects DNA from iron-mediated damage. Nucleic Acids Res. 1992, 20, 3341–3346. [Google Scholar] [CrossRef] [Green Version]
- Gallipoli, P.; Huntly, B.J.P. Histone modifiers are oxygen sensors. Science 2019, 363, 1148–1149. [Google Scholar] [CrossRef]
- Campit, S.E.; Meliki, A.; Youngson, N.A.; Chandrasekaran, S. Nutrient Sensing by Histone Marks: Reading the Metabolic Histone Code Using Tracing, Omics, and Modeling. BioEssays 2020, 42, 2000083. [Google Scholar] [CrossRef] [PubMed]
- Jo, C.; Park, S.; Oh, S.; Choi, J.; Kim, E.-K.; Youn, H.-D.; Cho, E.-J. Histone acylation marks respond to metabolic perturbations and enable cellular adaptation. Exp. Mol. Med. 2020, 52, 2005–2019. [Google Scholar] [CrossRef] [PubMed]
- Camarena, V.; Sant, D.W.; Huff, T.C.; Mustafi, S.; Muir, R.K.; Aron, A.T.; Chang, C.J.; Renslo, A.R.; Monje, P.V.; Wang, G. cAMP signaling regulates DNA hydroxymethylation by augmenting the intracellular labile ferrous iron pool. eLife 2017, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schachtschneider, K.M.; Liu, Y.; Rund, L.A.; Madsen, O.; Johnson, R.W.; Groenen, M.A.M.; Schook, L.B. Impact of neonatal iron deficiency on hippocampal DNA methylation and gene transcription in a porcine biomedical model of cognitive development. BMC Genom. 2016, 17, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.G.; Luk, N.; Newton, R.A.; Roberts, D.W.; Sturm, R.A.; Muscat, G.E.O. Melanocortin-1 Receptor Signaling Markedly Induces the Expression of the NR4A Nuclear Receptor Subgroup in Melanocytic Cells. J. Biol. Chem. 2008, 283, 12564–12570. [Google Scholar] [CrossRef] [Green Version]
- Beard, J.; Tenga, A.; Hills, J.; Hoyer, J.D.; Cherian, M.T.; Wang, Y.-D.; Chen, T. The orphan nuclear receptor NR4A2 is part of a p53–microRNA-34 network. Sci. Rep. 2016, 6, 25108. [Google Scholar] [CrossRef] [PubMed]
- Paillasse, M.R.; de Medina, P. The NR4A nuclear receptors as potential targets for anti-aging interventions. Med Hypotheses 2015, 84, 135–140. [Google Scholar] [CrossRef]
- Sharpless, N.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef]
- Chen, P.-H.; Tseng, W.H.-S.; Chi, J.-T. The Intersection of DNA Damage Response and Ferroptosis—A Rationale for Combination Therapeutics. Biology 2020, 9, 187. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, M.; Wang, Y.; Guo, Y.; Tao, X.; Wang, X.; Cao, Y.; Tian, S.; Li, Q. p53-mediated ferroptosis is required for 1-methyl-4-phenylpyridinium-induced senescence of PC12 cells. Toxicol. Vitr. 2021, 73, 105146. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Hao, C.; Huangfu, J.; Srinivasagan, R.; Zhang, X.; Fan, X. Aging lens epithelium is susceptible to ferroptosis. Free. Radic. Biol. Med. 2021, 167, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zheng, Y.; Wang, C.; Liu, Y. Glutathione depletion induces ferroptosis, autophagy, and premature cell senescence in retinal pigment epithelial cells. Cell Death Dis. 2018, 9, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Wu, B.; Shen, D.; Chen, J.; Yu, Z.; Chen, C. Ferroptosis in a sarcopenia model of senescence accelerated mouse prone 8 (SAMP8). Int. J. Biol. Sci. 2021, 17, 151–162. [Google Scholar] [CrossRef]
- Masaldan, S.; Clatworthy, S.A.; Gamell, C.; Meggyesy, P.M.; Rigopoulos, A.-T.; Haupt, S.; Haupt, Y.; Denoyer, D.; Adlard, P.A.; Bush, A.; et al. Iron accumulation in senescent cells is coupled with impaired ferritinophagy and inhibition of ferroptosis. Redox Biol. 2018, 14, 100–115. [Google Scholar] [CrossRef]
- Borrelli, C.; Ricci, B.; Vulpis, E.; Fionda, C.; Ricciardi, M.R.; Petrucci, M.T.; Masuelli, L.; Peri, A.; Cippitelli, M.; Zingoni, A.; et al. Drug-Induced Senescent Multiple Myeloma Cells Elicit NK Cell Proliferation by Direct or Exosome-Mediated IL15 Trans-Presentation. Cancer Immunol. Res. 2018, 6, 860–869. [Google Scholar] [CrossRef] [Green Version]
- Go, S.; Kang, M.; Kwon, S.P.; Jung, M.; Jeon, O.H.; Kim, B. The Senolytic Drug JQ1 Removes Senescent Cells via Ferroptosis. Tissue Eng. Regen. Med. 2021, 18, 841–850. [Google Scholar] [CrossRef]
- Domann, F.E.; Hitchler, M.J. Aberrant redox biology and epigenetic reprogramming: Co-conspirators across multiple human diseases. Free. Radic. Biol. Med. 2021, 170, 2–5. [Google Scholar] [CrossRef] [PubMed]
- Lamadema, N.; Burr, S.; Brewer, A.C. Dynamic regulation of epigenetic demethylation by oxygen availability and cellular redox. Free. Radic. Biol. Med. 2019, 131, 282–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Li, W.; Wang, Y.; Leng, Y.; Xia, Z. Inhibition of DNMT-1 alleviates ferroptosis through NCOA4 mediated ferritinophagy during diabetes myocardial ischemia/reperfusion injury. Cell Death Discov. 2021, 7, 1–14. [Google Scholar] [CrossRef]
- Tost, J. Engineering of the epigenome: Synthetic biology to define functional causality and develop innovative therapies. Epigenomics 2016, 8, 153–156. [Google Scholar] [CrossRef] [Green Version]
- Brown, C.W.; Chhoy, P.; Mukhopadhyay, D.; Karner, E.R.; Mercurio, A.M. Targeting prominin2 transcription to overcome ferroptosis resistance in cancer. EMBO Mol. Med. 2021, 13, e13792. [Google Scholar] [CrossRef]
- Logie, E.; Chirumamilla, C.; Perez-Novo, C.; Shaw, P.; Declerck, K.; Palagani, A.; Rangarajan, S.; Cuypers, B.; De Neuter, N.; Turabe, F.M.H.U.; et al. Covalent Cysteine Targeting of Bruton’s Tyrosine Kinase (BTK) Family by Withaferin-A Reduces Survival of Glucocorticoid-Resistant Multiple Myeloma MM1 Cells. Cancers 2021, 13, 1618. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 28 September 2021).
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warnes, G.R.; Bolker, B.; Bonebakker, L.; Gentleman, R.; Huber, W.; Liaw, A.; Lumley, T.; Maechler, M.; Magnusson, A.; Moeller, S.; et al. Gplots: Various R Programming Tools for Plotting Data. R Package Version 2.17. 0. Computer Software. 2015. Available online: https://rdrr.io/cran/gplots/ (accessed on 28 September 2021).
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Szic, K.S.V.; Declerck, K.; Crans, R.; Diddens, J.; Scherf, D.B.; Gerhauser, C.; Berghe, W.V. Epigenetic silencing of triple negative breast cancer hallmarks by Withaferin A. Oncotarget 2017, 8, 40434–40453. [Google Scholar] [CrossRef] [Green Version]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Maksimovic, J.; Phipson, B.; Oshlack, A. A cross-package Bioconductor workflow for analysing methylation array data. F1000Research 2016, 5, 1281. [Google Scholar] [CrossRef] [PubMed]
- Du, P.; Zhang, X.; Huang, C.-C.; Jafari, N.; A Kibbe, W.; Hou, L.; Lin, S.M. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinform. 2010, 11, 587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godderis, L.; Schouteden, C.; Tabish, A.M.; Poels, K.; Hoet, P.; Baccarelli, A.; Van Landuyt, K. Global Methylation and Hydroxymethylation in DNA from Blood and Saliva in Healthy Volunteers. BioMed Res. Int. 2015, 2015, 1–8. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
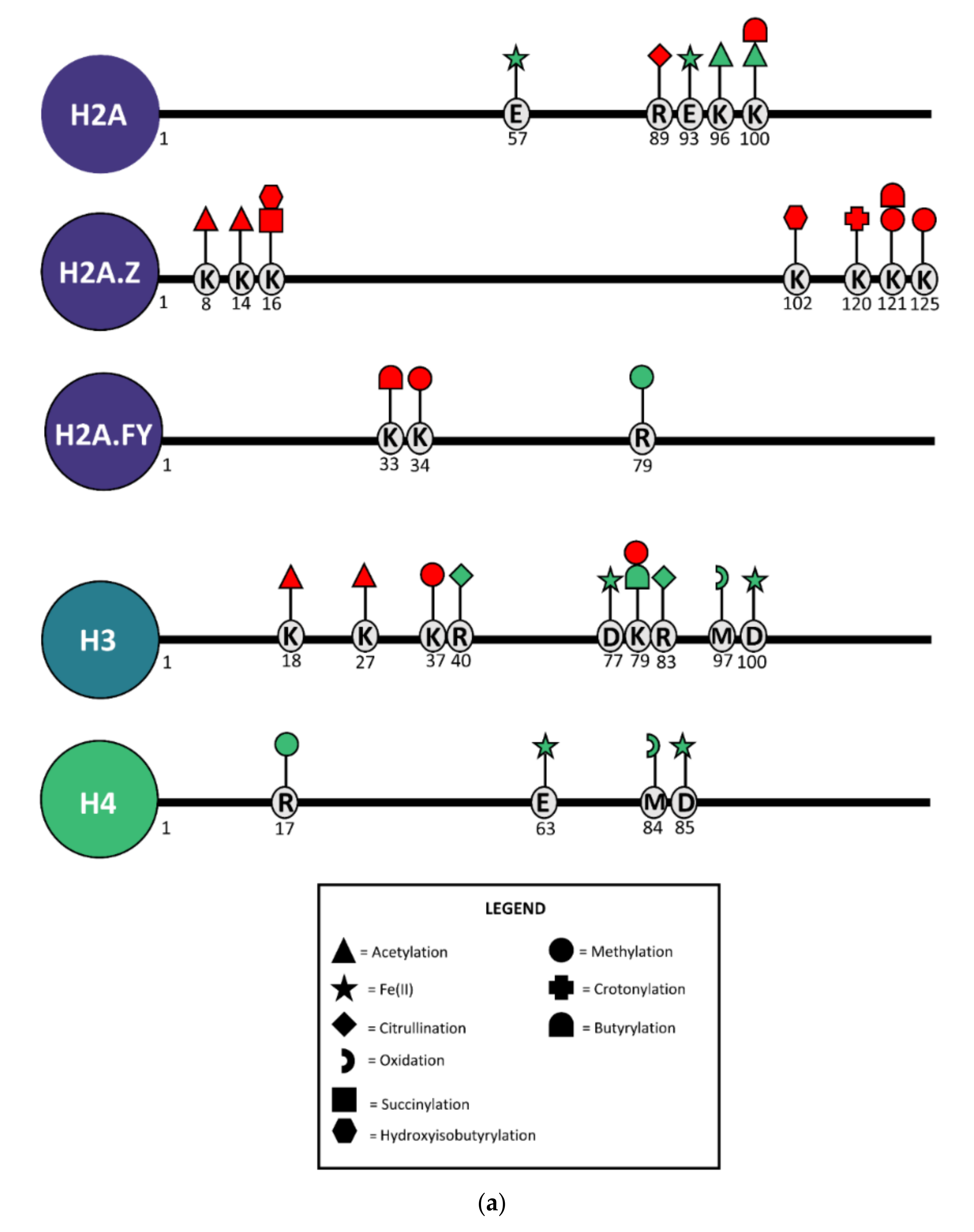
| Histone PTM or Variant | Expression after RSL3 Treatment | Biological Process | Functional Role | References |
|---|---|---|---|---|
| H3K79me3 | ↓ | Genome stability | Enriched in heterochromatic centromeric and telomeric regions; prevents spreading of heterochromatin | [48,49,50] |
| DNA repair | Controls DNA resection at damaged sites during homologous recombination; crucial in repairing UV-induced DNA damage | [51,52] | ||
| H3K18ac | ↓ | DNA repair | Regulates the expression of antioxidant genes; recruits DNA repair enzymes to damaged sites; regulates expression of nucleotide excision repair-related genes | [53,54,55,56] |
| Cellular senescence | H3K18 deacetylation protects cells against mitotic errors and cellular senescence | [57] | ||
| Genome stability | Maintains pericentric hetero-chromatin silencing | [57] | ||
| H3K27ac | ↓ | DNA repair | Regulates the expression of nucleotide excision repair-related genes | [56,58] |
| Cellular senescence | Enriched in enhancers of senescence-associated secretory phenotype (SASP) genes | [59,60,61] | ||
| H2A.Z | ↑ | DNA repair | Facilitates chromatin decondensation to allow for loading of DNA repair proteins to DNA breaks | [62,63,64] |
| Genome stability | Required for chromosome segregation and cytokinesis; prevents spreading of hetero-chromatin; preserves integrity of centromeres and telomeres | [65,66,67,68] | ||
| Cellular senescence | Overexpression alters regulation of cell cycle and DNA damage repair enzymes and suppresses cellular senescence | [69] | ||
| H2A.FY | ↑ | Cellular senescence | Regulates downstream acetylation to regulate senescence transcription programs | [70,71] |
| DNA repair | Alters the kinetics of PAR polymerases during DNA damage responses | [72] |
| Common Pathway | GO Term MM1S Cells | GO Term MM1R Cells | ||
|---|---|---|---|---|
| GO Term | −log p-Value | GO Term | −log p-Value | |
| Cell cycle | Negative regulation of cell cycle process | 10 | DNA replication | 5 |
| Negative regulation of nuclear division | 8 | Meiotic cell cycle | 3.8 | |
| S-phase | 4.5 | Cell cycle | 2.4 | |
| Negative regulation of meiotic nuclear division | 3.8 | Regulation of transcription involved in G1/S transition of mitotic cell cycle | 2.1 | |
| Autophagy | Regulation of autophagy | 4.4 | Autophagy | 2.2 |
| Insulin Signaling Pathway | Insulin signaling pathway | 3.8 | Insulin processing | 2.4 |
| Insulin-like growth factor receptor signaling pathway | 2.0 | |||
| CtBP core complex | CtBP complex | 3.7 | CtBP core complex | 3.9 |
| VEGF Signaling | VEGFA-VEGFR2 signaling pathway | 2.3 | VEGFA-VEGFR2 Signaling Pathway | 4.6 |
| Rett Syndrome | Rett syndrome-causing genes | 3.1 | Rett syndrome-causing genes | 2.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Logie, E.; Van Puyvelde, B.; Cuypers, B.; Schepers, A.; Berghmans, H.; Verdonck, J.; Laukens, K.; Godderis, L.; Dhaenens, M.; Deforce, D.; et al. Ferroptosis Induction in Multiple Myeloma Cells Triggers DNA Methylation and Histone Modification Changes Associated with Cellular Senescence. Int. J. Mol. Sci. 2021, 22, 12234. https://doi.org/10.3390/ijms222212234
Logie E, Van Puyvelde B, Cuypers B, Schepers A, Berghmans H, Verdonck J, Laukens K, Godderis L, Dhaenens M, Deforce D, et al. Ferroptosis Induction in Multiple Myeloma Cells Triggers DNA Methylation and Histone Modification Changes Associated with Cellular Senescence. International Journal of Molecular Sciences. 2021; 22(22):12234. https://doi.org/10.3390/ijms222212234
Chicago/Turabian StyleLogie, Emilie, Bart Van Puyvelde, Bart Cuypers, Anne Schepers, Herald Berghmans, Jelle Verdonck, Kris Laukens, Lode Godderis, Maarten Dhaenens, Dieter Deforce, and et al. 2021. "Ferroptosis Induction in Multiple Myeloma Cells Triggers DNA Methylation and Histone Modification Changes Associated with Cellular Senescence" International Journal of Molecular Sciences 22, no. 22: 12234. https://doi.org/10.3390/ijms222212234