Mitochondrial COI Sequence Variations within and among Geographic Samples of the Hemp Pest Psylliodes attenuata from China

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Identification of Psylliodes attenuata
2.2. DNA Sequencing
2.3. Data Analyses
3. Results
3.1. Identification and Characterization of Mitochondrial COI Sequences
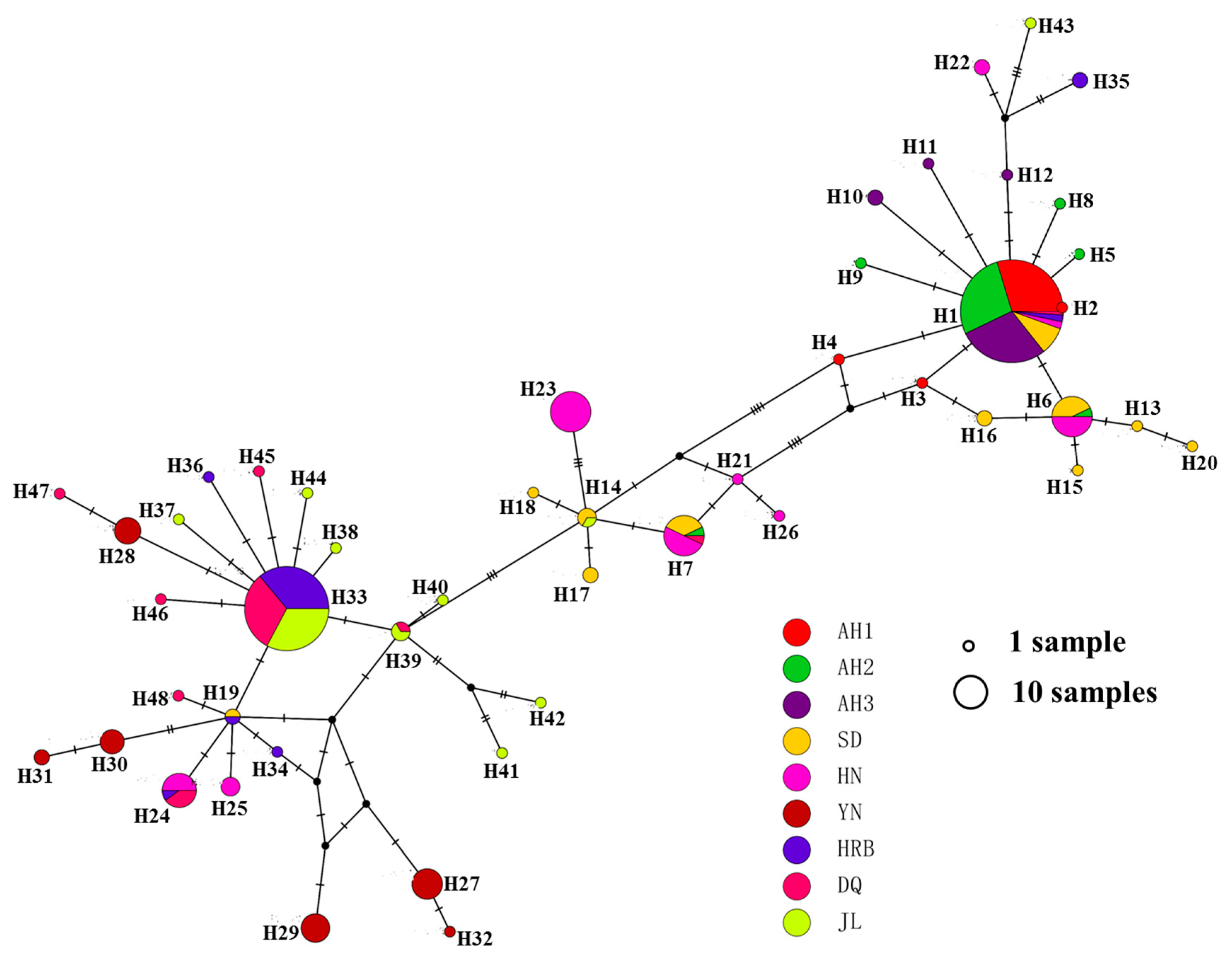
3.2. COI Haplotype Distributions
3.3. Nucleotide and Haplotype Diversities within Local Populations of Psylliodes attenuata
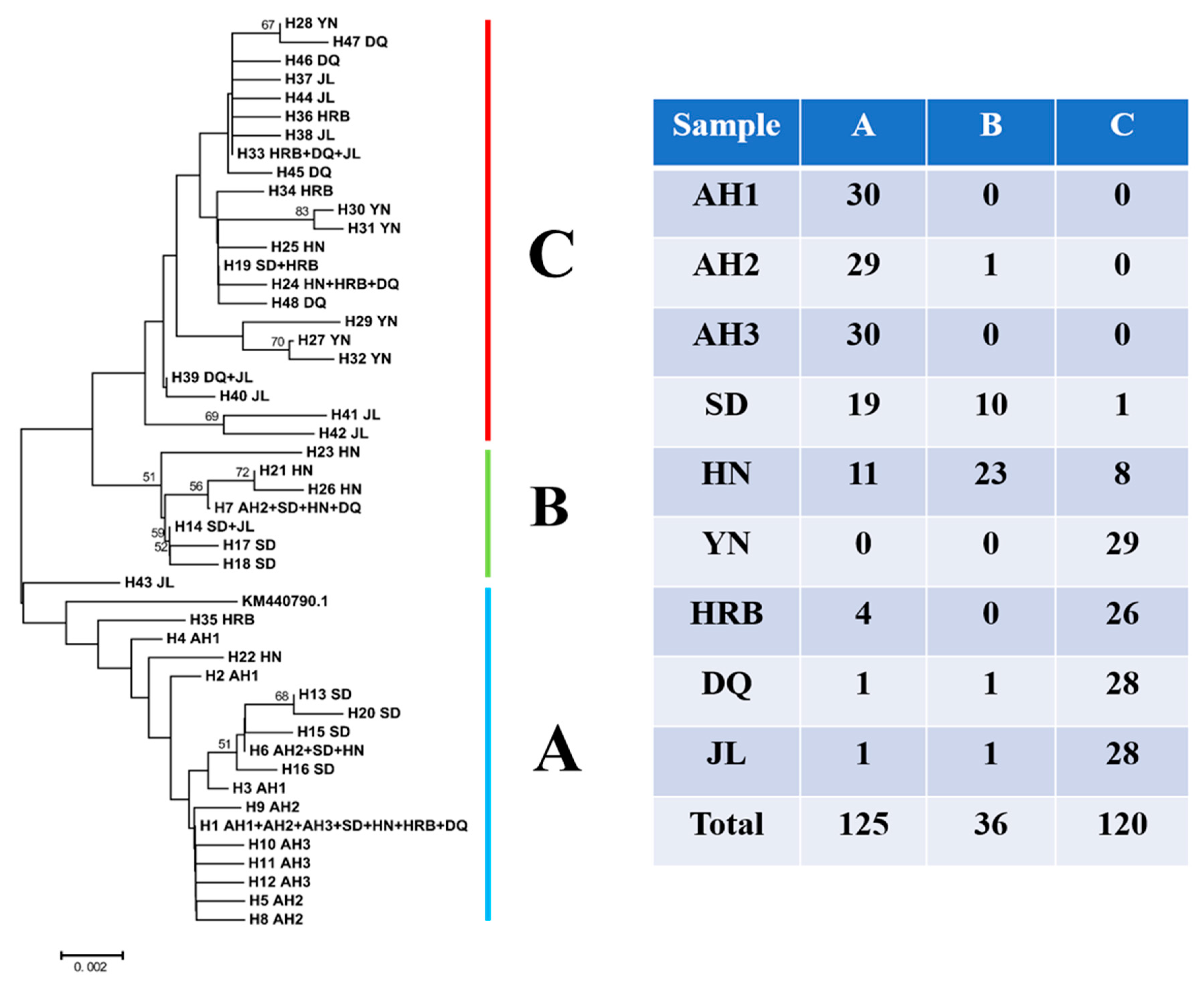
3.4. Phylogenetic Relationships among Haplotypes
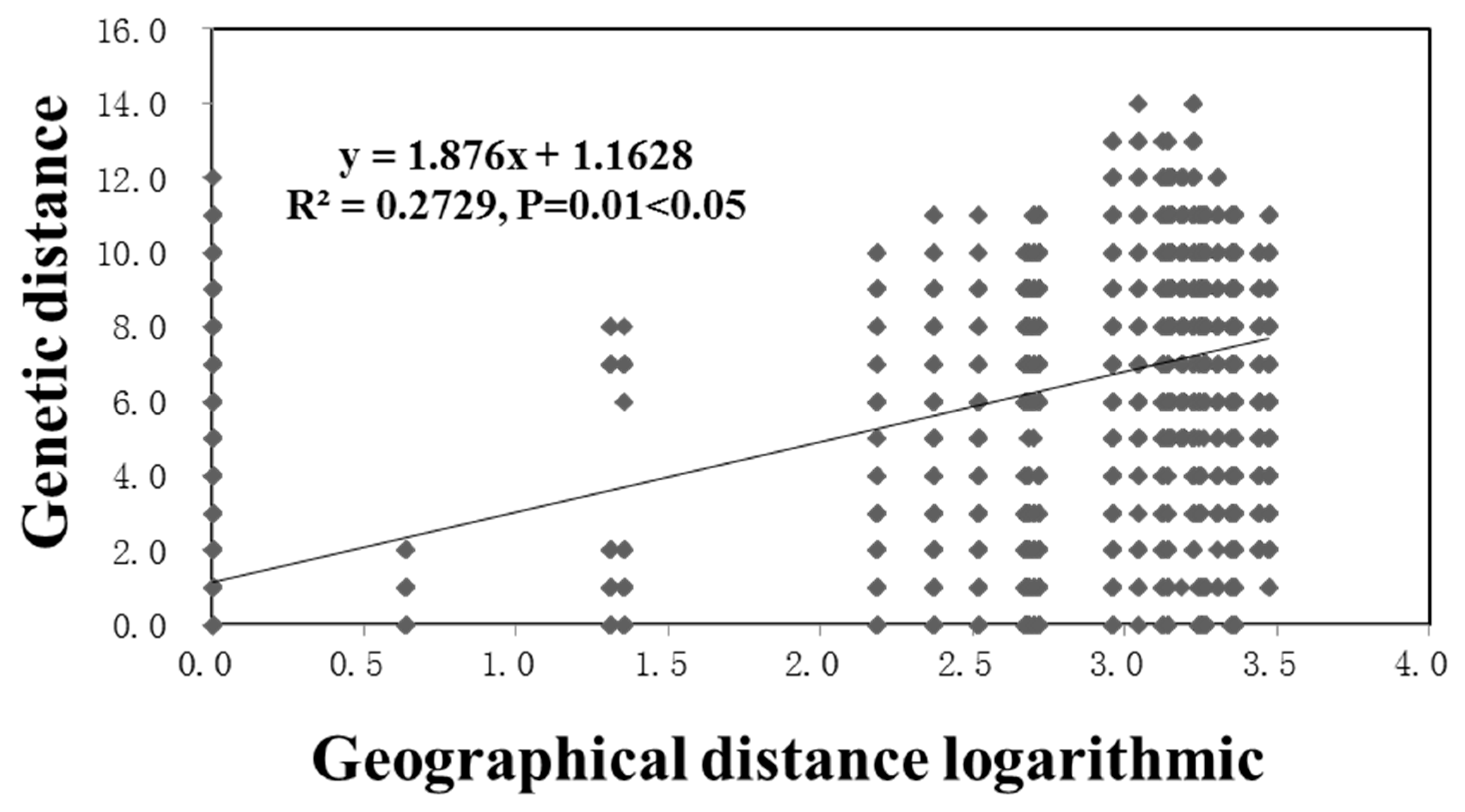
3.5. Population Relationships
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Amaducci, S.; Scordia, D.; Liu, F.H.; Zhang, Q.; Guo, H.; Testa, G.S.; Cosentino, L. Key cultivation techniques for hemp in Europe and China. Ind. Crops Prod. 2015, 68, 2–16. [Google Scholar] [CrossRef]
- Salentijn, E.M.J.; Zhang, Q.; Amaducci, S.; Yang, M.; Trindade, L.M. New developments in fiber hemp (Cannabis sativa L.) breeding. Ind. Crops Prod. 2015, 68, 32–41. [Google Scholar] [CrossRef]
- Andre, C.M.; Hausman, J.F.; Guerriero, G. Cannabis sativa: The plant of the thousand and one molecules. Front. Plant Sci. 2016, 7, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maria, I.; Eleni, T.; Apostolos, K.; Fotios, K.; Eirini, S.; Catherine, M.C. Effect of Genotype and Growing Year on the Nutritional, Phytochemical, and Antioxidant Properties of Industrial Hemp (Cannabis sativa L.) Seeds. Antioxidants 2019, 8, 491. [Google Scholar]
- Izzo, A.A.; Borrelli, F.; Capasso, R.; Di Marzo, V.; Mechoulam, R. Non-psychotropic plant cannabinoids: New therapeutic opportunities from an ancient herb. Trends Pharmacol. Sci. 2009, 30, 515–527. [Google Scholar] [CrossRef]
- Goncalves, J.; Rosado, T.; Soares, S.; Simão, A.Y.; Caramelo, D.; Luís, Â.; Fernández, N.; Barroso, M.; Gallardo, E.; Duarte, A.P. Cannabis and Its Secondary Metabolites: Their Use as Therapeutic Drugs, Toxicological Aspects, and Analytical Determination. Medicines 2019, 6, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michihito, D.; Daniel, B.; Hannah, W.; Zachary, S.; Shobha, P.; Takumasa, K.; Zhan, Y.; Zhang, J.; Sairam, R. Establishment and optimization of a hemp (Cannabis sativa L.) agroinfiltration system for gene expression and silencing studies. Sci. Rep. 2020, 10, 3504. [Google Scholar]
- McPartland, J.M.; Clarke, R.C.; Watson, D.P. Hemp diseases and pest management and biological control. Australas. Plant Pathol. 2000, 30, 1. [Google Scholar]
- Durnovo, Z.P. Results of work on the maize moth and other pests of newly cultivated annual fibre plants. In Bolyezni Vredit. Nov. Lubyan. Kul.; [Diseases and Pests of newly cultivated Fibre Plants]; Institut Novogo Lubianogo Syriia [Institute of New Bast Raw Materials]: Moscow, Russia, 1933; pp. 85–106. [Google Scholar]
- Silantyev, A. Results of investigations of the Hemp (Hop, Flax) and Beet Flea; Minist. Agric. Gov.: Estates, St. Petersburg, Russia, 1897; p. 7. [Google Scholar]
- Ragazzi, G. Nemici vegetali ed animali della canapa. Humus 1954, 10, 27–29. [Google Scholar]
- Dempsey, J.M. Hemp. In Fiber Crops; University of Florida Press: Gainesville, FL, USA, 1975; pp. 46–89. [Google Scholar]
- Bocsa, I.; Karus, M. The Cultivation of Hemp: Botany, Varieties, Cultivation and Harvesting; Hemptech: Sebastopol, CA, USA, 1997; p. 173. [Google Scholar]
- Mohyuddin, A.l.; Scheibelreiter, G.K. Investigations on the Fauna of Papaver spp. and Cannabis sativa; Annual Report; Commonwealth Institute of Biological Control, Switzerland Station: Delemont, Switzerland, 1973; pp. 32–33. [Google Scholar]
- Sun, X.; Tao, J.; Alain, R.; Luo, Y. Invasion History of Sirex noctilio Based on COI Sequence: The First Six Years in China. Insects 2020, 11, 111. [Google Scholar] [CrossRef] [Green Version]
- Hebert, P.D.N.; Cywinska, A.; Ball, S.L.; De Waard, J.R. Biological identifications through DNA barcodes. Proc. R. Soc. Lond. 2003, 270, 313–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moritz, C.; Cicero, C. DNA Barcoding: Promise and Pitfalls. PLoS Biol. 2004, 2, e354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malausa, T.; Fenis, A.; Warot, S.; Germain, J.; Ris, N.; Prado, E.; Botton, M.; Vanlerberghe-Masutti, F.; Sforza, R.; Cruaud, C. DNA markers to disentangle complexes of cryptic taxa in mealybugs (Hemiptera: Pseudococcidae). J. Appl. Entomol. 2011, 135, 142–155. [Google Scholar] [CrossRef] [Green Version]
- Palraju, M.; Paulchamy, R.; Sundaraman, S. Population genetic structure and molecular diversity of Leucinodes orbonalis based on mitochondrial COI gene sequences. Mitochondrial DNA 2018, 29, 1231–1239. [Google Scholar] [CrossRef] [PubMed]
- Cieniewicz, E.; Poplaski, V.; Brunelli, M.; Dombroskie, J.; Fuchs, M. Two Distinct Genotypes of Spissistilus festinus (Say, 1830) (Hemiptera, Membracidae) in the United States Revealed by Phylogenetic and Morphological Analyses. Insects 2020, 11, 80. [Google Scholar] [CrossRef] [Green Version]
- Folmer, O.; Black, M.; Hoeh, W.; Lutz, R.; Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit 1 from diverse metazoan invertebrates. Mol. Mar. Biol. Biotech. 1994, 3, 294–299. [Google Scholar]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl. Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. Bioinform. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [Green Version]
- Leigh, J.; Bryant, D. POPART: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Beckenbach, A. Evolution of the mitochondrial cytochrome oxidase II gene among 10 orders of insects. Mol. Phylogenet. Evol. 1992, 1, 41–52. [Google Scholar] [CrossRef]
- Simon, C.; Frati, F.; Beckenbach, A.; Crespi, B.; Liu, H.; Flook, P. Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann. Entomol. Soc. Am. 1994, 87, 651–701. [Google Scholar] [CrossRef]
- Sun, W.; Zhang, Z.T.; Dong, H.; Qian, H.T.; Shi, Y.; Xie, L.N.; Cong, B. Analysis of genetic differentiation and gene flow among different geographic populations of Oedaleus infernalis (Orthoptera: Acrididae) based on mtDNA COI gene sequences. Acta Entomol. Sin. 2013, 56, 907–916. [Google Scholar]
- Xu, J. The inheritance of organelle genes and genomes: Patterns and mechanisms. Genome 2005, 48, 951–958. [Google Scholar] [CrossRef]
- Sandor, S.; Zhang, Y.J.; Xu, J. Fungal mitochondrial genomes and genetic polymorphisms. Appl. Microbiol. Biotech. 2018, 102, 9433–9448. [Google Scholar] [CrossRef]
- Breton, S.; Stewart, D.T. Atypical mitochondrial inheritance patterns in eukaryotes. Genome 2015, 58, 423–431. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Zhou, G.Y.; Liu, J.A.; Xu, J. Population genetic analyses of the fungal pathogen Colletotrichum fructicola on tea-oil trees in China. PLoS ONE 2016, 11, e0156841. [Google Scholar] [CrossRef] [Green Version]
- Bebber, D.P.; Holmes, T.; Gurr, S.J. The global spread of crop pests and pathogens. Glob. Ecol. Biogeogr. 2014, 23, 1398–1407. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.Y.; Xu, W.; Gao, Y.; Cui, J.; Ou, S.Q.; Shi, S.S. Analysis of genetic differentiation of geographic populations of Leguminivora glycinivorella (Lepidoptera: Olethreutidae) based on mitochondrial DNA COII gene sequences in China. Acta Entomol. Sin. 2017, 60, 475–486. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Province | Site | Latitude/Longitude | Host Cultivar | Date Collected | Sample Collected | Population Code | Number of Samples |
|---|---|---|---|---|---|---|---|
| Anhui | Wangqiao Village | 31.67/116.357 | Wan Cannabis No. 1 | July 2019 | Yes | AH1 | 30 |
| Lu’an | 31.806/116.519 | Wan Cannabis No. 1 | July 2019 | Yes | AH2 | 30 | |
| Zhangwan Village | 31.70/116.358 | Wan Cannabis No. 1 | July 2019 | Yes | AH3 | 30 | |
| Shandong | Tai’an | 35.97/116.97 | Zhong Cannabis No. 5 | August 2019 | Yes | SD | 30 |
| Hunan | Shijihu | 28.8/112.36 | Zhong Cannabis No. 1 | August 2019 | Yes | HN | 42 |
| Yuanjiang | 28.79/112.34 | Zhong Cannabis No. 5 | August 2019 | No | |||
| Chenpo Village | 28.78/112.35 | Wan Cannabis No. 1 | August 2019 | No | |||
| Yunnan | Xishuangbanna | 21.99/100.41 | Yunma No. 1 and 7 | September 2019 | No | ||
| Chuxiong | 25.14/101.55 | Yunma No. 7 | September 2019 | No | |||
| Qujing | 25.85/103.75 | Yunma No. 7 | September 2019 | Yes | YN | 29 | |
| Heilongjiang | Daoli | 45.59/126.44 | Long Cannabis No. 5 | September 2019 | No | ||
| Harbin | 45.59/126.44 | Long Cannabis No. 3 | September 2019 | Yes | HRB | 30 | |
| Daqing | 46.67/125.23 | Qingma No. 1 and 2 | September 2019 | Yes | DQ | 30 | |
| Jilin | Changchun | 43.72/125.09 | Fenma No. 3 | September 2019 | Yes | JL | 30 |
| Haplotype | AH1 (30) | AH2 (30) | AH3 (30) | SD (30) | HN (42) | YN (29) | HRB (30) | DQ (30) | JL (30) | Total (281) | GenBank Accession No. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| H1 | 27 | 25 | 26 | 8 | 2 | 0 | 2 | 1 | 0 | 91 | MT447350 |
| H2 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | MT447351 |
| H3 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | MT447352 |
| H4 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | MT447353 |
| H5 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | MT447354 |
| H6 | 0 | 1 | 0 | 6 | 7 | 0 | 0 | 0 | 0 | 14 | MT447355 |
| H7 | 0 | 1 | 0 | 5 | 7 | 0 | 0 | 1 | 0 | 14 | MT447356 |
| H8 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | MT447357 |
| H9 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | MT447358 |
| H10 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | MT447359 |
| H11 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | MT447360 |
| H12 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | MT447361 |
| H13 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | MT447362 |
| H14 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 1 | 3 | MT447363 |
| H15 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | MT447364 |
| H16 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 2 | MT447365 |
| H17 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 2 | MT447366 |
| H18 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | MT447367 |
| H19 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 2 | MT447368 |
| H20 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | MT447369 |
| H21 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | MT447370 |
| H22 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 2 | MT447371 |
| H23 | 0 | 0 | 0 | 0 | 14 | 0 | 0 | 0 | 0 | 14 | MT447372 |
| H24 | 0 | 0 | 0 | 0 | 5 | 0 | 1 | 4 | 0 | 10 | MT447373 |
| H25 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 3 | MT447374 |
| H26 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | MT447375 |
| H27 | 0 | 0 | 0 | 0 | 0 | 8 | 0 | 0 | 0 | 8 | MT447376 |
| H28 | 0 | 0 | 0 | 0 | 0 | 6 | 0 | 0 | 0 | 6 | MT447377 |
| H29 | 0 | 0 | 0 | 0 | 0 | 7 | 0 | 0 | 0 | 7 | MT447378 |
| H30 | 0 | 0 | 0 | 0 | 0 | 5 | 0 | 0 | 0 | 5 | MT447379 |
| H31 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 2 | MT447380 |
| H32 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | MT447381 |
| H33 | 0 | 0 | 0 | 0 | 0 | 0 | 22 | 19 | 20 | 61 | MT447382 |
| H34 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | MT447383 |
| H35 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 2 | MT447384 |
| H36 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | MT447385 |
| H37 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | MT447386 |
| H38 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | MT447387 |
| H39 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 2 | 3 | MT447388 |
| H40 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | MT447389 |
| H41 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | MT447390 |
| H42 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | MT447391 |
| H43 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | MT447392 |
| H44 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | MT447393 |
| H45 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | MT447394 |
| H46 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | MT447395 |
| H47 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | MT447396 |
| H48 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | MT447397 |
| Population Code | Number of Haplotypes (No. of Private Haplotypes) | Haplotype Diversity (Hd) | Nucleotide Diversity (π) | Tajima’s D |
|---|---|---|---|---|
| AH1 | 4 (3) | 0.193 | 0.00031 | −1.73178 NS |
| AH2 | 6 (3) | 0.310 | 0.00114 | −2.36834 ** |
| AH3 | 4 (3) | 0.251 | 0.00041 | −1.53889 NS |
| SD | 11 (6) | 0.871 | 0.00688 | 0.11613 NS |
| HN | 9 (5) | 0.828 | 0.00982 | 1.41773 NS |
| YN | 6 (6) | 0.815 | 0.00600 | 1.23395 NS |
| HRB | 7 (3) | 0.464 | 0.00342 | −1.08139 NS |
| DQ | 9 (4) | 0.593 | 0.00274 | −2.01006 * |
| JL | 10 (7) | 0.561 | 0.00260 | −1.98693 * |
| Total | 48 (40) | 0.839 | 0.00871 | −0.74006 NS |
| AH2 | AH3 | SD | HN | YN | HRB | JL | DQ | |
|---|---|---|---|---|---|---|---|---|
| AH1 | −0.01205 | 0.00985 | 0.27120 ** | 0.50895 ** | 0.78838 *** | 0.83614 *** | 0.88233 *** | 0.87890 ** |
| AH2 | 0.00460 | 0.22032 ** | 0.47284 ** | 0.76146 *** | 0.80146 *** | 0.84955 *** | 0.84681 *** | |
| AH3 | 0.28079 ** | 0.51317 ** | 0.78834 *** | 0.83467 *** | 0.88037 *** | 0.87709 *** | ||
| SD | 0.16657 * | 0.52979 ** | 0.52815 ** | 0.58310 ** | 0.58559 ** | |||
| HN | 0.32704 ** | 0.33311 ** | 0.37098 ** | 0.36893 ** | ||||
| YN | 0.28706 ** | 0.31985 ** | 0.28882 ** | |||||
| HRB | 0.02170 | 0.00353 | ||||||
| JL | 0.02226 | |||||||
| DQ |
| Source of Variation | df | Sum of Squares | Variance Components | Variation Proportion |
|---|---|---|---|---|
| Among populations | 8 | 475.802 | 1.866 | 58% |
| Within populations | 272 | 363.586 | 1.337 | 42% |
| Total | 280 | 839.388 | 3.202 | 100% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, L.; Gao, F.; Cheng, Y.; Gao, C.; Chen, J.; Li, Z.; Wang, T.; Xu, J. Mitochondrial COI Sequence Variations within and among Geographic Samples of the Hemp Pest Psylliodes attenuata from China. Insects 2020, 11, 370. https://doi.org/10.3390/insects11060370
Guo L, Gao F, Cheng Y, Gao C, Chen J, Li Z, Wang T, Xu J. Mitochondrial COI Sequence Variations within and among Geographic Samples of the Hemp Pest Psylliodes attenuata from China. Insects. 2020; 11(6):370. https://doi.org/10.3390/insects11060370
Chicago/Turabian StyleGuo, Litao, Feng Gao, Yi Cheng, Chunsheng Gao, Jia Chen, Zhimin Li, Tuhong Wang, and Jianping Xu. 2020. "Mitochondrial COI Sequence Variations within and among Geographic Samples of the Hemp Pest Psylliodes attenuata from China" Insects 11, no. 6: 370. https://doi.org/10.3390/insects11060370