Follicle-Stimulating Hormone (FSH) Action on Spermatogenesis: A Focus on Physiological and Therapeutic Roles

 , ,
, ,  , , and
, , and
Abstract
:1. Introduction
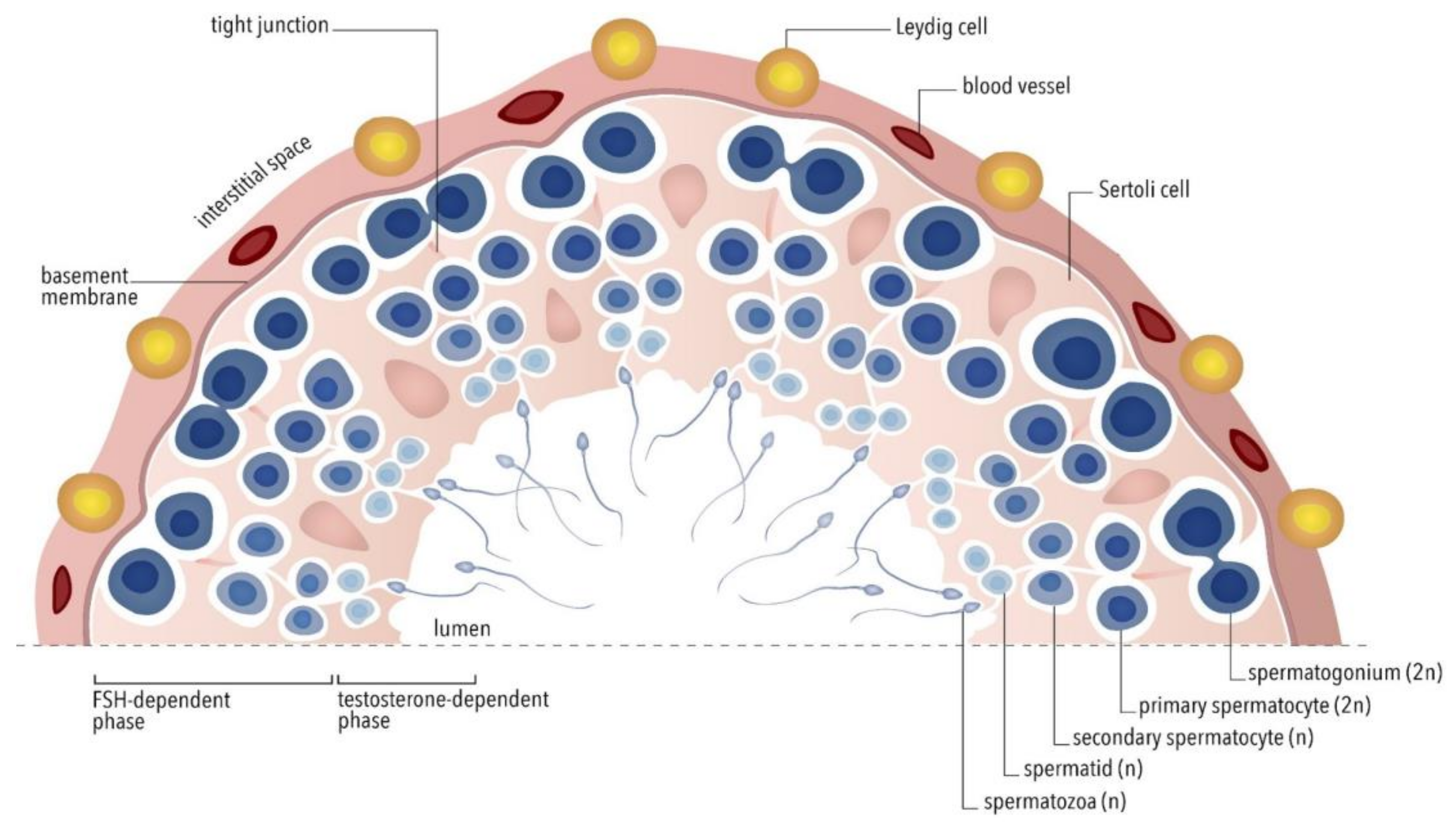
2. Physiological Control of Spermatogenesis
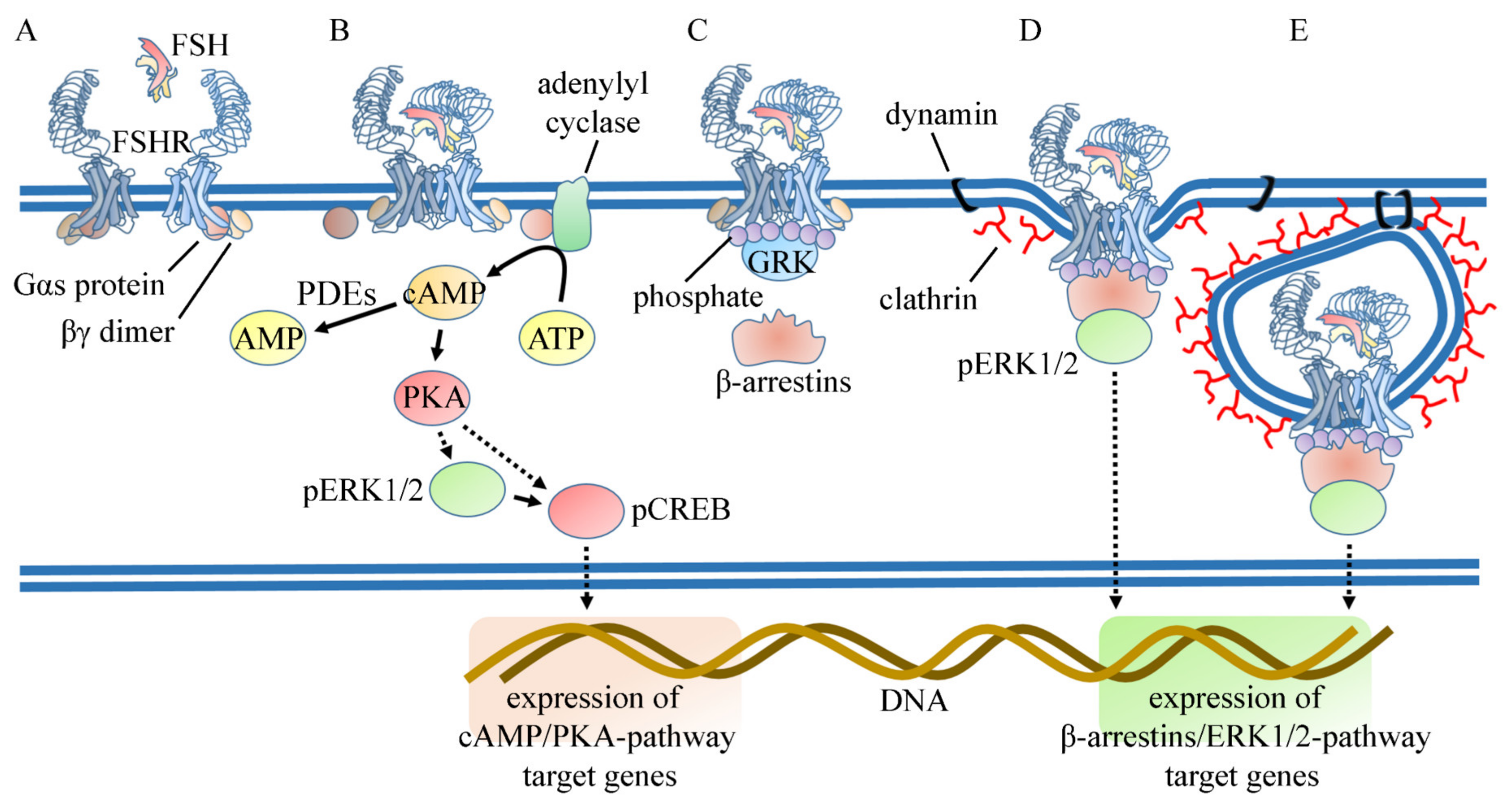
3. Mechanism of Action of FSH in the Sertoli Cell
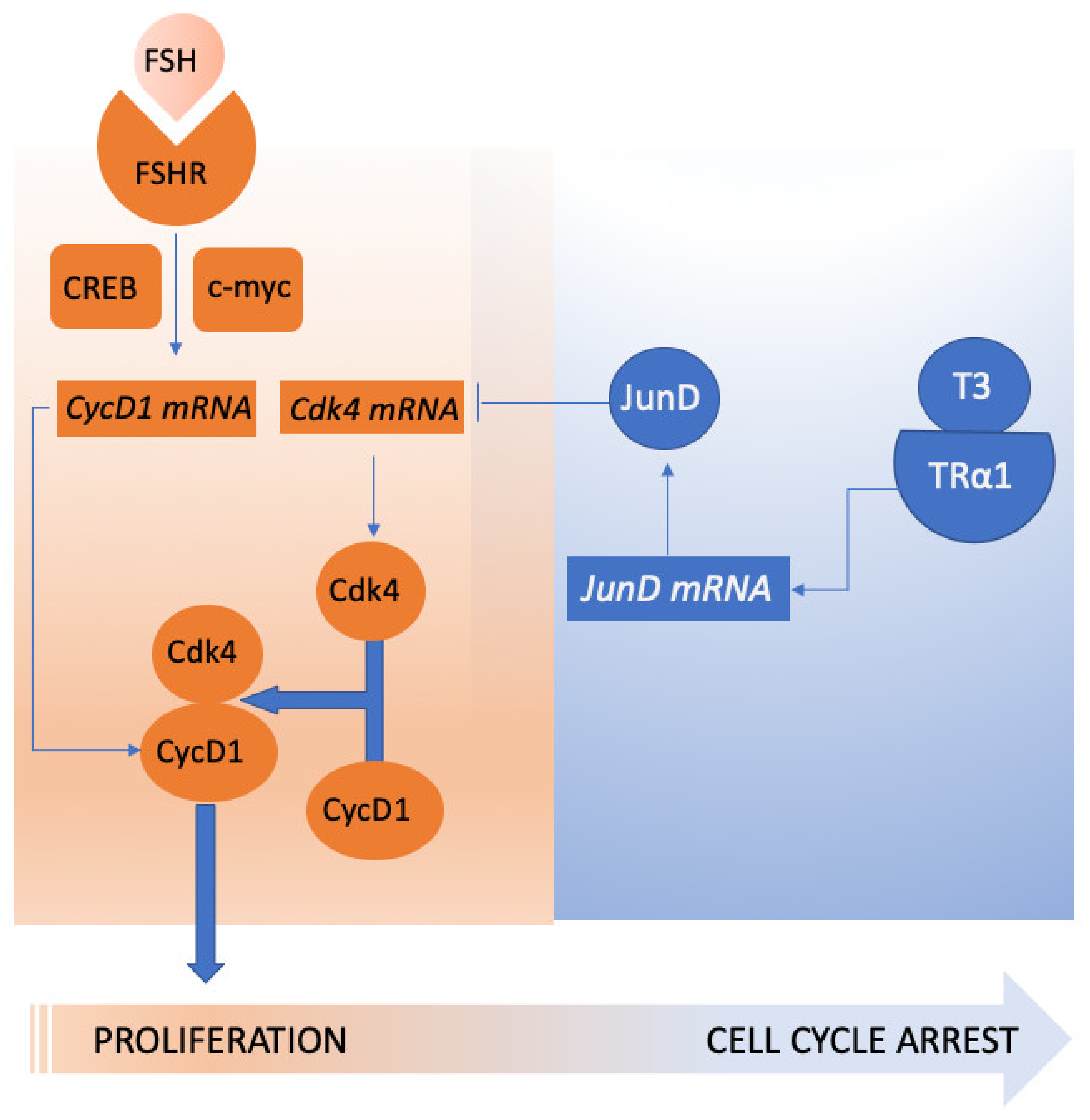
4. Genetic Regulation of FSH Action on Spermatogenesis
5. Therapeutic Options to Improve Sperm Production
6. Future Perspectives
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rastrelli, G.; Corona, G.; Mannucci, E.; Maggi, M. Factors affecting spermatogenesis upon gonadotropin-replacement therapy: A meta-analytic study. Andrology 2014, 2, 794–808. [Google Scholar] [CrossRef] [PubMed]
- Casarini, L.; Santi, D.; Brigante, G.; Simoni, M. Two Hormones for One Receptor: Evolution, Biochemistry, Actions, and Pathophysiology of LH and hCG. Endocr. Rev. 2018, 39, 549–592. [Google Scholar] [CrossRef] [PubMed]
- Santi, D.; Poti, F.; Simoni, M.; Casarini, L. Pharmacogenetics of G-protein-coupled receptors variants: FSH receptor and infertility treatment. Best Pract. Res. Clin. Endocrinol. Metab. 2018, 32, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Rohayem, J.; Sinthofen, N.; Nieschlag, E.; Kliesch, S.; Zitzmann, M. Causes of hypogonadotropic hypogonadism predict response to gonadotropin substitution in adults. Andrology 2016, 4, 87–94. [Google Scholar] [CrossRef]
- Santi, D.; De Vincentis, S.; Alfano, P.; Balercia, G.; Calogero, A.E.; Cargnelutti, F.; Coccia, M.E.; Condorelli, R.A.; Dal Lago, A.; De Angelis, C.; et al. Use of follicle-stimulating hormone (FSH) for the male partner of idiopathic infertile couples in Italy: Results from a multicentre, observational, clinical practice survey. Andrology 2019. [Google Scholar] [CrossRef]
- Jan, S.Z.; Hamer, G.; Repping, S.; de Rooij, D.G.; van Pelt, A.M.; Vormer, T.L. Molecular control of rodent spermatogenesis. Biochim. Biophys. Acta 2012, 1822, 1838–1850. [Google Scholar] [CrossRef] [Green Version]
- Oduwole, O.O.; Peltoketo, H.; Huhtaniemi, I.T. Role of Follicle-Stimulating Hormone in Spermatogenesis. Front. Endocrinol. 2018, 9, 763. [Google Scholar] [CrossRef] [Green Version]
- Watkins, P.C.; Eddy, R.; Beck, A.K.; Vellucci, V.; Leverone, B.; Tanzi, R.E.; Gusella, J.F.; Shows, T.B. DNA sequence and regional assignment of the human follicle-stimulating hormone beta-subunit gene to the short arm of human chromosome 11. DNA 1987, 6, 205–212. [Google Scholar] [CrossRef]
- Kangasniemi, M.; Kaipia, A.; Toppari, J.; Perheentupa, A.; Huhtaniemi, I.; Parvinen, M. Cellular regulation of follicle-stimulating hormone (FSH) binding in rat seminiferous tubules. J. Androl. 1990, 11, 336–343. [Google Scholar]
- Mruk, D.D.; Cheng, C.Y. The Mammalian Blood-Testis Barrier: Its Biology and Regulation. Endocr. Rev. 2015, 36, 564–591. [Google Scholar] [CrossRef]
- Sharpe, R.M. Sperm counts and fertility in men: A rocky road ahead. Science & Society Series on Sex and Science. EMBO Rep. 2012, 13, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Roth, M.Y.; Lin, K.; Amory, J.K.; Matsumoto, A.M.; Anawalt, B.D.; Snyder, C.N.; Kalhorn, T.F.; Bremner, W.J.; Page, S.T. Serum LH correlates highly with intratesticular steroid levels in normal men. J. Androl. 2010, 31, 138–145. [Google Scholar] [CrossRef] [Green Version]
- Saez, J.M. Leydig cells: Endocrine, paracrine, and autocrine regulation. Endocr. Rev. 1994, 15, 574–626. [Google Scholar] [CrossRef] [PubMed]
- Mruk, D.D.; Cheng, C.Y. Sertoli-Sertoli and Sertoli-germ cell interactions and their significance in germ cell movement in the seminiferous epithelium during spermatogenesis. Endocr. Rev. 2004, 25, 747–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharpe, R.M.; McKinnell, C.; Kivlin, C.; Fisher, J.S. Proliferation and functional maturation of Sertoli cells, and their relevance to disorders of testis function in adulthood. Reproduction 2003, 125, 769–784. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, C.; Stevant, I.; Borel, C.; Conne, B.; Pitetti, J.L.; Calvel, P.; Kaessmann, H.; Jegou, B.; Chalmel, F.; Nef, S. Research resource: The dynamic transcriptional profile of sertoli cells during the progression of spermatogenesis. Mol. Endocrinol. 2015, 29, 627–642. [Google Scholar] [CrossRef] [Green Version]
- Huhtaniemi, I.T.; Yamamoto, M.; Ranta, T.; Jalkanen, J.; Jaffe, R.B. Follicle-stimulating hormone receptors appear earlier in the primate fetal testis than in the ovary. J. Clin. Endocrinol. Metab. 1987, 65, 1210–1214. [Google Scholar] [CrossRef]
- Cheng, C.Y.; Mruk, D.D. Cell junction dynamics in the testis: Sertoli-germ cell interactions and male contraceptive development. Physiol. Rev. 2002, 82, 825–874. [Google Scholar] [CrossRef] [Green Version]
- Regueira, M.; Riera, M.F.; Galardo, M.N.; Camberos Mdel, C.; Pellizzari, E.H.; Cigorraga, S.B.; Meroni, S.B. FSH and bFGF regulate the expression of genes involved in Sertoli cell energetic metabolism. Gen. Comp. Endocrinol. 2015, 222, 124–133. [Google Scholar] [CrossRef]
- Guo, J.; Shi, Y.Q.; Yang, W.; Li, Y.C.; Hu, Z.Y.; Liu, Y.X. Testosterone upregulation of tissue type plasminogen activator expression in Sertoli cells: tPA expression in Sertoli cells. Endocrine 2007, 32, 83–89. [Google Scholar] [CrossRef]
- Allan, C.M.; Garcia, A.; Spaliviero, J.; Zhang, F.P.; Jimenez, M.; Huhtaniemi, I.; Handelsman, D.J. Complete Sertoli cell proliferation induced by follicle-stimulating hormone (FSH) independently of luteinizing hormone activity: Evidence from genetic models of isolated FSH action. Endocrinology 2004, 145, 1587–1593. [Google Scholar] [CrossRef] [PubMed]
- Ruwanpura, S.M.; McLachlan, R.I.; Meachem, S.J. Hormonal regulation of male germ cell development. J. Endocrinol. 2010, 205, 117–131. [Google Scholar] [CrossRef] [PubMed]
- Ruwanpura, S.M.; McLachlan, R.I.; Stanton, P.G.; Loveland, K.L.; Meachem, S.J. Pathways involved in testicular germ cell apoptosis in immature rats after FSH suppression. J. Endocrinol. 2008, 197, 35–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abel, M.H.; Wootton, A.N.; Wilkins, V.; Huhtaniemi, I.; Knight, P.G.; Charlton, H.M. The effect of a null mutation in the follicle-stimulating hormone receptor gene on mouse reproduction. Endocrinology 2000, 141, 1795–1803. [Google Scholar] [CrossRef]
- Kumar, T.R.; Wang, Y.; Lu, N.; Matzuk, M.M. Follicle stimulating hormone is required for ovarian follicle maturation but not male fertility. Nat. Genet. 1997, 15, 201–204. [Google Scholar] [CrossRef]
- Franca, M.M.; Lerario, A.M.; Funari, M.F.A.; Nishi, M.Y.; Narcizo, A.M.; de Mello, M.P.; Guerra-Junior, G.; Maciel-Guerra, A.T.; Mendonca, B.B. A Novel Homozygous Missense FSHR Variant Associated with Hypergonadotropic Hypogonadism in Two Siblings from a Brazilian Family. Sex. Dev. Genet. Mol. Biol. Evol. Endocrinol. Embryol. Pathol. Sex. Determ. Differ. 2017, 11, 137–142. [Google Scholar] [CrossRef]
- Siegel, E.T.; Kim, H.G.; Nishimoto, H.K.; Layman, L.C. The molecular basis of impaired follicle-stimulating hormone action: Evidence from human mutations and mouse models. Reprod. Sci. 2013, 20, 211–233. [Google Scholar] [CrossRef] [Green Version]
- Tapanainen, J.S.; Aittomaki, K.; Min, J.; Vaskivuo, T.; Huhtaniemi, I.T. Men homozygous for an inactivating mutation of the follicle-stimulating hormone (FSH) receptor gene present variable suppression of spermatogenesis and fertility. Nat. Genet. 1997, 15, 205–206. [Google Scholar] [CrossRef]
- Lei, Z.M.; Mishra, S.; Zou, W.; Xu, B.; Foltz, M.; Li, X.; Rao, C.V. Targeted disruption of luteinizing hormone/human chorionic gonadotropin receptor gene. Mol. Endocrinol. 2001, 15, 184–200. [Google Scholar] [CrossRef]
- Oduwole, O.O.; Vydra, N.; Wood, N.E.; Samanta, L.; Owen, L.; Keevil, B.; Donaldson, M.; Naresh, K.; Huhtaniemi, I.T. Overlapping dose responses of spermatogenic and extragonadal testosterone actions jeopardize the principle of hormonal male contraception. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2014, 28, 2566–2576. [Google Scholar] [CrossRef]
- Sluka, P.; O’Donnell, L.; Bartles, J.R.; Stanton, P.G. FSH regulates the formation of adherens junctions and ectoplasmic specialisations between rat Sertoli cells in vitro and in vivo. J. Endocrinol. 2006, 189, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Stanton, P.G. Regulation of the blood-testis barrier. Semin. Cell Dev. Biol. 2016, 59, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Abel, M.H.; Baker, P.J.; Charlton, H.M.; Monteiro, A.; Verhoeven, G.; De Gendt, K.; Guillou, F.; O’Shaughnessy, P.J. Spermatogenesis and sertoli cell activity in mice lacking sertoli cell receptors for follicle-stimulating hormone and androgen. Endocrinology 2008, 149, 3279–3285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oduwole, O.O.; Peltoketo, H.; Poliandri, A.; Vengadabady, L.; Chrusciel, M.; Doroszko, M.; Samanta, L.; Owen, L.; Keevil, B.; Rahman, N.A.; et al. Constitutively active follicle-stimulating hormone receptor enables androgen-independent spermatogenesis. J. Clin. Investig. 2018, 128, 1787–1792. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.; Chen, Y.T.; Yeh, S.D.; Xu, Q.; Wang, R.S.; Guillou, F.; Lardy, H.; Yeh, S. Infertility with defective spermatogenesis and hypotestosteronemia in male mice lacking the androgen receptor in Sertoli cells. Proc. Natl. Acad. Sci. USA 2004, 101, 6876–6881. [Google Scholar] [CrossRef] [Green Version]
- De Gendt, K.; Swinnen, J.V.; Saunders, P.T.; Schoonjans, L.; Dewerchin, M.; Devos, A.; Tan, K.; Atanassova, N.; Claessens, F.; Lecureuil, C.; et al. A Sertoli cell-selective knockout of the androgen receptor causes spermatogenic arrest in meiosis. Proc. Natl. Acad. Sci. USA 2004, 101, 1327–1332. [Google Scholar] [CrossRef] [Green Version]
- Casarini, L.; Crepieux, P. Molecular Mechanisms of Action of FSH. Front. Endocrinol. 2019, 10, 305. [Google Scholar] [CrossRef]
- Zheng, J.; Mao, J.; Cui, M.; Liu, Z.; Wang, X.; Xiong, S.; Nie, M.; Wu, X. Novel FSHbeta mutation in a male patient with isolated FSH deficiency and infertility. Eur. J. Med. Genet. 2017, 60, 335–339. [Google Scholar] [CrossRef]
- Rivero-Muller, A.; Potorac, I.; Pintiaux, A.; Daly, A.F.; Thiry, A.; Rydlewski, C.; Nisolle, M.; Parent, A.S.; Huhtaniemi, I.; Beckers, A. A novel inactivating mutation of the LH/chorionic gonadotrophin receptor with impaired membrane trafficking leading to Leydig cell hypoplasia type 1. Eur. J. Endocrinol. Eur. Fed. Endocr. Soc. 2015, 172, K27–K36. [Google Scholar] [CrossRef] [Green Version]
- Achard, C.; Courtillot, C.; Lahuna, O.; Meduri, G.; Soufir, J.C.; Liere, P.; Bachelot, A.; Benyounes, H.; Schumacher, M.; Kuttenn, F.; et al. Normal spermatogenesis in a man with mutant luteinizing hormone. New Engl. J. Med. 2009, 361, 1856–1863. [Google Scholar] [CrossRef]
- Bruysters, M.; Christin-Maitre, S.; Verhoef-Post, M.; Sultan, C.; Auger, J.; Faugeron, I.; Larue, L.; Lumbroso, S.; Themmen, A.P.; Bouchard, P. A new LH receptor splice mutation responsible for male hypogonadism with subnormal sperm production in the propositus, and infertility with regular cycles in an affected sister. Hum. Reprod. (Oxf. Engl.) 2008, 23, 1917–1923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casas-Gonzalez, P.; Scaglia, H.E.; Perez-Solis, M.A.; Durand, G.; Scaglia, J.; Zarinan, T.; Dias, J.A.; Reiter, E.; Ulloa-Aguirre, A. Normal testicular function without detectable follicle-stimulating hormone. A novel mutation in the follicle-stimulating hormone receptor gene leading to apparent constitutive activity and impaired agonist-induced desensitization and internalization. Mol. Cell. Endocrinol. 2012, 364, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Gromoll, J.; Simoni, M.; Nieschlag, E. An activating mutation of the follicle-stimulating hormone receptor autonomously sustains spermatogenesis in a hypophysectomized man. J. Clin. Endocrinol. Metab. 1996, 81, 1367–1370. [Google Scholar] [CrossRef] [PubMed]
- Huhtaniemi, I. A short evolutionary history of FSH-stimulated spermatogenesis. Horm. 2015, 14, 468–478. [Google Scholar] [CrossRef] [Green Version]
- Bercovici, J.P.; Nahoul, K.; Ducasse, M.; Tater, D.; Kerlan, V.; Scholler, R. Leydig cell tumor with gynecomastia: Further studies--the recovery after unilateral orchidectomy. J. Clin. Endocrinol. Metab. 1985, 61, 957–962. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.L.; Schoenemann, H.M.; Chakraborty, P.K.; Stuart, L.D.; Dahl, K.D. Increased bioactivity of serum follicle-stimulating hormone, but not luteinizing hormone, following hemicastration in ram lambs. Biol. Reprod. 1990, 43, 548–553. [Google Scholar] [CrossRef]
- Cunningham, G.R.; Tindall, D.J.; Huckins, C.; Means, A.R. Mechanisms for the testicular hypertrophy which follows hemicastration. Endocrinology 1978, 102, 16–23. [Google Scholar] [CrossRef]
- Johnson, L.; Neaves, W.B. Enhanced daily sperm production in the remaining testis of aged rats following hemicastration. J. Androl. 1983, 4, 162–166. [Google Scholar] [CrossRef]
- Ramaswamy, S.; Marshall, G.R.; McNeilly, A.S.; Plant, T.M. Dynamics of the follicle-stimulating hormone (FSH)-inhibin B feedback loop and its role in regulating spermatogenesis in the adult male rhesus monkey (Macaca mulatta) as revealed by unilateral orchidectomy. Endocrinology 2000, 141, 18–27. [Google Scholar] [CrossRef]
- Dahlqvist, P.; Koskinen, L.O.; Brannstrom, T.; Hagg, E. Testicular enlargement in a patient with a FSH-secreting pituitary adenoma. Endocrine 2010, 37, 289–293. [Google Scholar] [CrossRef]
- Dabaja, A.A.; Schlegel, P.N. Medical treatment of male infertility. Transl. Androl. Urol. 2014, 3, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Simoni, M.; Santi, D. FSH Treatment of male idiopathic infertility: Time for a paradigm change. Andrology 2019. [Google Scholar] [CrossRef]
- Dorrington, J.H.; Roller, N.F.; Fritz, I.B. Effects of follicle-stimulating hormone on cultures of Sertoli cell preparations. Mol. Cell. Endocrinol. 1975, 3, 57–70. [Google Scholar] [CrossRef]
- Conti, M.; Kasson, B.G.; Hsueh, A.J. Hormonal regulation of 3’,5’-adenosine monophosphate phosphodiesterases in cultured rat granulosa cells. Endocrinology 1984, 114, 2361–2368. [Google Scholar] [CrossRef] [PubMed]
- Conti, M.; Toscano, M.V.; Petrelli, L.; Geremia, R.; Stefanini, M. Regulation of follicle-stimulating hormone and dibutyryl adenosine 3’,5’-monophosphate of a phosphodiesterase isoenzyme of the Sertoli cell. Endocrinology 1982, 110, 1189–1196. [Google Scholar] [CrossRef] [PubMed]
- Swinnen, J.V.; Tsikalas, K.E.; Conti, M. Properties and hormonal regulation of two structurally related cAMP phosphodiesterases from the rat Sertoli cell. J. Biol. Chem. 1991, 266, 18370–18377. [Google Scholar] [PubMed]
- Marion, S.; Kara, E.; Crepieux, P.; Piketty, V.; Martinat, N.; Guillou, F.; Reiter, E. G protein-coupled receptor kinase 2 and beta-arrestins are recruited to FSH receptor in stimulated rat primary Sertoli cells. J. Endocrinol. 2006, 190, 341–350. [Google Scholar] [CrossRef]
- Marion, S.; Robert, F.; Crepieux, P.; Martinat, N.; Troispoux, C.; Guillou, F.; Reiter, E. G protein-coupled receptor kinases and beta arrestins are relocalized and attenuate cyclic 3’,5’-adenosine monophosphate response to follicle-stimulating hormone in rat primary Sertoli cells. Biol. Reprod. 2002, 66, 70–76. [Google Scholar] [CrossRef] [Green Version]
- Crepieux, P.; Marion, S.; Martinat, N.; Fafeur, V.; Vern, Y.L.; Kerboeuf, D.; Guillou, F.; Reiter, E. The ERK-dependent signalling is stage-specifically modulated by FSH, during primary Sertoli cell maturation. Oncogene 2001, 20, 4696–4709. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, I.; Gautam, M.; Sarkar, H.; Shukla, M.; Majumdar, S.S. Advantages of pulsatile hormone treatment for assessing hormone-induced gene expression by cultured rat Sertoli cells. Cell Tissue Res. 2017, 368, 389–396. [Google Scholar] [CrossRef]
- Musnier, A.; Leon, K.; Morales, J.; Reiter, E.; Boulo, T.; Costache, V.; Vourc’h, P.; Heitzler, D.; Oulhen, N.; Poupon, A.; et al. mRNA-selective translation induced by FSH in primary Sertoli cells. Mol. Endocrinol. 2012, 26, 669–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meroni, S.B.; Riera, M.F.; Pellizzari, E.H.; Cigorraga, S.B. Regulation of rat Sertoli cell function by FSH: Possible role of phosphatidylinositol 3-kinase/protein kinase B pathway. J. Endocrinol. 2002, 174, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Tang, Z.; Li, Y.; Liu, W.; Zhang, S.; Wang, B.; Tian, Y.; Zhao, Y.; Ran, H.; Liu, W.; et al. Deletion of the tyrosine phosphatase Shp2 in Sertoli cells causes infertility in mice. Sci. Rep. 2015, 5, 12982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiter, E.; Lefkowitz, R.J. GRKs and beta-arrestins: Roles in receptor silencing, trafficking and signaling. Trends Endocrinol. Metab. 2006, 17, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Kara, E.; Crepieux, P.; Gauthier, C.; Martinat, N.; Piketty, V.; Guillou, F.; Reiter, E. A phosphorylation cluster of five serine and threonine residues in the C-terminus of the follicle-stimulating hormone receptor is important for desensitization but not for beta-arrestin-mediated ERK activation. Mol. Endocrinol. 2006, 20, 3014–3026. [Google Scholar] [CrossRef] [PubMed]
- Heitzler, D.; Durand, G.; Gallay, N.; Rizk, A.; Ahn, S.; Kim, J.; Violin, J.D.; Dupuy, L.; Gauthier, C.; Piketty, V.; et al. Competing G protein-coupled receptor kinases balance G protein and beta-arrestin signaling. Mol. Syst. Biol. 2012, 8, 590. [Google Scholar] [CrossRef] [PubMed]
- Luttrell, L.M.; Wang, J.; Plouffe, B.; Smith, J.S.; Yamani, L.; Kaur, S.; Jean-Charles, P.Y.; Gauthier, C.; Lee, M.H.; Pani, B.; et al. Manifold roles of beta-arrestins in GPCR signaling elucidated with siRNA and CRISPR/Cas9. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Trefier, A.; Musnier, A.; Landomiel, F.; Bourquard, T.; Boulo, T.; Ayoub, M.A.; Leon, K.; Bruneau, G.; Chevalier, M.; Durand, G.; et al. G protein-dependent signaling triggers a beta-arrestin-scaffolded p70S6K/rpS6 module that controls 5’TOP mRNA translation. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2018, 32, 1154–1169. [Google Scholar] [CrossRef] [Green Version]
- Tranchant, T.; Durand, G.; Gauthier, C.; Crepieux, P.; Ulloa-Aguirre, A.; Royere, D.; Reiter, E. Preferential beta-arrestin signalling at low receptor density revealed by functional characterization of the human FSH receptor A189 V mutation. Mol. Cell. Endocrinol. 2011, 331, 109–118. [Google Scholar] [CrossRef] [Green Version]
- Musnier, A.; Heitzler, D.; Boulo, T.; Tesseraud, S.; Durand, G.; Lecureuil, C.; Guillou, H.; Poupon, A.; Reiter, E.; Crepieux, P. Developmental regulation of p70 S6 kinase by a G protein-coupled receptor dynamically modelized in primary cells. Cell Mol. Life Sci. 2009, 66, 3487–3503. [Google Scholar] [CrossRef]
- Dupont, J.; Musnier, A.; Decourtye, J.; Boulo, T.; Lecureuil, C.; Guillou, H.; Valet, S.; Fouchecourt, S.; Pitetti, J.L.; Nef, S.; et al. FSH-stimulated PTEN activity accounts for the lack of FSH mitogenic effect in prepubertal rat Sertoli cells. Mol. Cell. Endocrinol. 2010, 315, 271–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riera, M.F.; Regueira, M.; Galardo, M.N.; Pellizzari, E.H.; Meroni, S.B.; Cigorraga, S.B. Signal transduction pathways in FSH regulation of rat Sertoli cell proliferation. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E914–E923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLean, D.J.; Friel, P.J.; Pouchnik, D.; Griswold, M.D. Oligonucleotide microarray analysis of gene expression in follicle-stimulating hormone-treated rat Sertoli cells. Mol. Endocrinol. 2002, 16, 2780–2792. [Google Scholar] [CrossRef] [PubMed]
- Sadate-Ngatchou, P.I.; Pouchnik, D.J.; Griswold, M.D. Follicle-stimulating hormone induced changes in gene expression of murine testis. Mol. Endocrinol. 2004, 18, 2805–2816. [Google Scholar] [CrossRef] [Green Version]
- Abel, M.H.; Baban, D.; Lee, S.; Charlton, H.M.; O’Shaughnessy, P.J. Effects of FSH on testicular mRNA transcript levels in the hypogonadal mouse. J. Mol. Endocrinol. 2009, 42, 291–303. [Google Scholar] [CrossRef] [Green Version]
- Gautam, M.; Bhattacharya, I.; Rai, U.; Majumdar, S.S. Hormone induced differential transcriptome analysis of Sertoli cells during postnatal maturation of rat testes. PLoS ONE 2018, 13, e0191201. [Google Scholar] [CrossRef] [Green Version]
- Santos, N.C.; Kim, K.H. Activity of retinoic acid receptor-alpha is directly regulated at its protein kinase A sites in response to follicle-stimulating hormone signaling. Endocrinology 2010, 151, 2361–2372. [Google Scholar] [CrossRef]
- Sanz, E.; Evanoff, R.; Quintana, A.; Evans, E.; Miller, J.A.; Ko, C.; Amieux, P.S.; Griswold, M.D.; McKnight, G.S. RiboTag analysis of actively translated mRNAs in Sertoli and Leydig cells in vivo. PLoS ONE 2013, 8, e66179. [Google Scholar] [CrossRef] [Green Version]
- Papaioannou, M.D.; Pitetti, J.L.; Ro, S.; Park, C.; Aubry, F.; Schaad, O.; Vejnar, C.E.; Kuhne, F.; Descombes, P.; Zdobnov, E.M.; et al. Sertoli cell Dicer is essential for spermatogenesis in mice. Dev. Biol. 2009, 326, 250–259. [Google Scholar] [CrossRef] [Green Version]
- Nicholls, P.K.; Harrison, C.A.; Walton, K.L.; McLachlan, R.I.; O’Donnell, L.; Stanton, P.G. Hormonal regulation of sertoli cell micro-RNAs at spermiation. Endocrinology 2011, 152, 1670–1683. [Google Scholar] [CrossRef] [Green Version]
- Griswold, M.; Mably, E.; Fritz, I.B. Stimulation by follicle stimulating hormone and dibutyryl cyclic AMP of incorporation of 3H-thymidine into nuclear DNA of cultured Sertoli cell-enriched preparations from immature rats. Curr Top. Mol. Endocrinol. 1975, 2, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Meachem, S.J.; McLachlan, R.I.; de Kretser, D.M.; Robertson, D.M.; Wreford, N.G. Neonatal exposure of rats to recombinant follicle stimulating hormone increases adult Sertoli and spermatogenic cell numbers. Biol. Reprod. 1996, 54, 36–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Haaster, L.H.; De Jong, F.H.; Docter, R.; De Rooij, D.G. The effect of hypothyroidism on Sertoli cell proliferation and differentiation and hormone levels during testicular development in the rat. Endocrinology 1992, 131, 1574–1576. [Google Scholar] [CrossRef] [PubMed]
- Fumel, B.; Roy, S.; Fouchecourt, S.; Livera, G.; Parent, A.S.; Casas, F.; Guillou, F. Depletion of the p43 mitochondrial T3 receptor increases Sertoli cell proliferation in mice. PLoS ONE 2013, 8, e74015. [Google Scholar] [CrossRef] [PubMed]
- Holsberger, D.R.; Kiesewetter, S.E.; Cooke, P.S. Regulation of neonatal Sertoli cell development by thyroid hormone receptor alpha1. Biol. Reprod. 2005, 73, 396–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quignodon, L.; Vincent, S.; Winter, H.; Samarut, J.; Flamant, F. A point mutation in the activation function 2 domain of thyroid hormone receptor alpha1 expressed after CRE-mediated recombination partially recapitulates hypothyroidism. Mol. Endocrinol 2007, 21, 2350–2360. [Google Scholar] [CrossRef]
- Borland, K.; Mita, M.; Oppenheimer, C.L.; Blinderman, L.A.; Massague, J.; Hall, P.F.; Czech, M.P. The actions of insulin-like growth factors I and II on cultured Sertoli cells. Endocrinology 1984, 114, 240–246. [Google Scholar] [CrossRef]
- Pitetti, J.L.; Calvel, P.; Zimmermann, C.; Conne, B.; Papaioannou, M.D.; Aubry, F.; Cederroth, C.R.; Urner, F.; Fumel, B.; Crausaz, M.; et al. An essential role for insulin and IGF1 receptors in regulating sertoli cell proliferation, testis size, and FSH action in mice. Mol. Endocrinol. 2013, 27, 814–827. [Google Scholar] [CrossRef]
- Marx, J. How p53 suppresses cell growth. Science 1993, 262, 1644–1645. [Google Scholar] [CrossRef]
- Froment, P.; Vigier, M.; Negre, D.; Fontaine, I.; Beghelli, J.; Cosset, F.L.; Holzenberger, M.; Durand, P. Inactivation of the IGF-I receptor gene in primary Sertoli cells highlights the autocrine effects of IGF-I. J. Endocrinol. 2007, 194, 557–568. [Google Scholar] [CrossRef] [Green Version]
- Fumel, B.; Guerquin, M.J.; Livera, G.; Staub, C.; Magistrini, M.; Gauthier, C.; Flamant, F.; Guillou, F.; Fouchecourt, S. Thyroid hormone limits postnatal Sertoli cell proliferation in vivo by activation of its alpha1 isoform receptor (TRalpha1) present in these cells and by regulation of Cdk4/JunD/c-myc mRNA levels in mice. Biol. Reprod. 2012, 87, 16. 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, P.; Roy, S.K. Follicle stimulating hormone-induced DNA synthesis in the granulosa cells of hamster preantral follicles involves activation of cyclin-dependent kinase-4 rather than cyclin d2 synthesis. Biol. Reprod. 2004, 70, 509–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hazra, R.; Corcoran, L.; Robson, M.; McTavish, K.J.; Upton, D.; Handelsman, D.J.; Allan, C.M. Temporal role of Sertoli cell androgen receptor expression in spermatogenic development. Mol. Endocrinol. 2013, 27, 12–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buzzard, J.J.; Wreford, N.G.; Morrison, J.R. Thyroid hormone, retinoic acid, and testosterone suppress proliferation and induce markers of differentiation in cultured rat sertoli cells. Endocrinology 2003, 144, 3722–3731. [Google Scholar] [CrossRef] [Green Version]
- Simoni, M.; Gromoll, J.; Nieschlag, E. The follicle-stimulating hormone receptor: Biochemistry, molecular biology, physiology, and pathophysiology. Endocr. Rev. 1997, 18, 739–773. [Google Scholar] [CrossRef]
- Gloaguen, P.; Crepieux, P.; Heitzler, D.; Poupon, A.; Reiter, E. Mapping the follicle-stimulating hormone-induced signaling networks. Front. Endocrinol. 2011, 2, 45. [Google Scholar] [CrossRef] [Green Version]
- Simoni, M.; Casarini, L. Mechanisms in endocrinology: Genetics of FSH action: A 2014-and-beyond view. Eur. J. Endocrinol. Eur. Fed. Endocr. Soc. 2014, 170, R91–R107. [Google Scholar] [CrossRef] [Green Version]
- Simoni, M.; Gromoll, J.; Hoppner, W.; Kamischke, A.; Krafft, T.; Stahle, D.; Nieschlag, E. Mutational analysis of the follicle-stimulating hormone (FSH) receptor in normal and infertile men: Identification and characterization of two discrete FSH receptor isoforms. J. Clin. Endocrinol. Metab. 1999, 84, 751–755. [Google Scholar] [CrossRef] [Green Version]
- Perez Mayorga, M.; Gromoll, J.; Behre, H.M.; Gassner, C.; Nieschlag, E.; Simoni, M. Ovarian response to follicle-stimulating hormone (FSH) stimulation depends on the FSH receptor genotype. J. Clin. Endocrinol. Metab. 2000, 85, 3365–3369. [Google Scholar] [CrossRef]
- Casarini, L.; Moriondo, V.; Marino, M.; Adversi, F.; Capodanno, F.; Grisolia, C.; La Marca, A.; La Sala, G.B.; Simoni, M. FSHR polymorphism p.N680S mediates different responses to FSH in vitro. Mol. Cell. Endocrinol. 2014, 393, 83–91. [Google Scholar] [CrossRef]
- Alviggi, C.; Conforti, A.; Santi, D.; Esteves, S.C.; Andersen, C.Y.; Humaidan, P.; Chiodini, P.; De Placido, G.; Simoni, M. Clinical relevance of genetic variants of gonadotrophins and their receptors in controlled ovarian stimulation: A systematic review and meta-analysis. Hum. Reprod. Update 2018, 24, 599–614. [Google Scholar] [CrossRef] [PubMed]
- Behre, H.M.; Greb, R.R.; Mempel, A.; Sonntag, B.; Kiesel, L.; Kaltwasser, P.; Seliger, E.; Ropke, F.; Gromoll, J.; Nieschlag, E.; et al. Significance of a common single nucleotide polymorphism in exon 10 of the follicle-stimulating hormone (FSH) receptor gene for the ovarian response to FSH: A pharmacogenetic approach to controlled ovarian hyperstimulation. Pharm. Genom. 2005, 15, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Grigorova, M.; Punab, M.; Poolamets, O.; Sober, S.; Vihljajev, V.; Zilaitiene, B.; Erenpreiss, J.; Matulevicius, V.; Tsarev, I.; Laan, M. Study in 1790 Baltic men: FSHR Asn680Ser polymorphism affects total testes volume. Andrology 2013, 1, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Tuttelmann, F.; Laan, M.; Grigorova, M.; Punab, M.; Sober, S.; Gromoll, J. Combined effects of the variants FSHB -211G>T and FSHR 2039A>G on male reproductive parameters. J. Clin. Endocrinol. Metab. 2012, 97, 3639–3647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simoni, M.; Santi, D.; Negri, L.; Hoffmann, I.; Muratori, M.; Baldi, E.; Cambi, M.; Marcou, M.; Greither, T.; Baraldi, E.; et al. Treatment with human, recombinant FSH improves sperm DNA fragmentation in idiopathic infertile men depending on the FSH receptor polymorphism p.N680S: A pharmacogenetic study. Hum. Reprod. (Oxf. Engl.) 2016, 31, 1960–1969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, S.S.; Achrekar, S.K.; Pathak, B.R.; Desai, S.K.; Mangoli, V.S.; Mangoli, R.V.; Mahale, S.D. Follicle-stimulating hormone receptor polymorphism (G-29A) is associated with altered level of receptor expression in Granulosa cells. J. Clin. Endocrinol. Metab. 2011, 96, 2805–2812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wunsch, A.; Ahda, Y.; Banaz-Yasar, F.; Sonntag, B.; Nieschlag, E.; Simoni, M.; Gromoll, J. Single-nucleotide polymorphisms in the promoter region influence the expression of the human follicle-stimulating hormone receptor. Fertil. Steril. 2005, 84, 446–453. [Google Scholar] [CrossRef]
- Achrekar, S.K.; Modi, D.N.; Desai, S.K.; Mangoli, V.S.; Mangoli, R.V.; Mahale, S.D. Poor ovarian response to gonadotrophin stimulation is associated with FSH receptor polymorphism. Reprod. Biomed. Online 2009, 18, 509–515. [Google Scholar] [CrossRef]
- Grigorova, M.; Punab, M.; Punab, A.M.; Poolamets, O.; Vihljajev, V.; Zilaitiene, B.; Erenpreiss, J.; Matulevicius, V.; Laan, M. Reproductive physiology in young men is cumulatively affected by FSH-action modulating genetic variants: FSHR -29G/A and c.2039 A/G, FSHB -211G/T. PLoS ONE 2014, 9, e94244. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Zhang, J.; Zhu, P.; Jiang, W.; Liu, S.; Ni, M.; Zhang, M.; Li, W.; Zhou, Q.; Cui, Y.; et al. The susceptibility of FSHB -211G > T and FSHR G-29A, 919A > G, 2039A > G polymorphisms to men infertility: An association study and meta-analysis. BMC Med. Genet. 2017, 18, 81. [Google Scholar] [CrossRef]
- Bianco, S.D.; Kaiser, U.B. The genetic and molecular basis of idiopathic hypogonadotropic hypogonadism. Nat. Rev. Endocrinol. 2009, 5, 569–576. [Google Scholar] [CrossRef] [Green Version]
- Mao, J.F.; Liu, Z.X.; Nie, M.; Wang, X.; Xu, H.L.; Huang, B.K.; Zheng, J.J.; Min, L.; Kaiser, U.B.; Wu, X.Y. Pulsatile gonadotropin-releasing hormone therapy is associated with earlier spermatogenesis compared to combined gonadotropin therapy in patients with congenital hypogonadotropic hypogonadism. Asian J. Androl. 2017, 19, 680–685. [Google Scholar] [CrossRef]
- Salenave, S.; Trabado, S.; Maione, L.; Brailly-Tabard, S.; Young, J. Male acquired hypogonadotropic hypogonadism: Diagnosis and treatment. Ann. D’endocrinologie 2012, 73, 141–146. [Google Scholar] [CrossRef]
- Salonia, A.; Rastrelli, G.; Hackett, G.; Seminara, S.B.; Huhtaniemi, I.T.; Rey, R.A.; Hellstrom, W.J.G.; Palmert, M.R.; Corona, G.; Dohle, G.R.; et al. Paediatric and adult-onset male hypogonadism. Nat. Rev. Dis. Primers 2019, 5, 38. [Google Scholar] [CrossRef]
- Corona, G.; Rastrelli, G.; Vignozzi, L.; Maggi, M. Emerging medication for the treatment of male hypogonadism. Expert Opin. Emerg. Drugs 2012, 17, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Young, J.; Xu, C.; Papadakis, G.E.; Acierno, J.S.; Maione, L.; Hietamaki, J.; Raivio, T.; Pitteloud, N. Clinical Management of Congenital Hypogonadotropic Hypogonadism. Endocr. Rev. 2019, 40, 669–710. [Google Scholar] [CrossRef] [Green Version]
- Delemarre-Van de Waal, H.A.; Odink, R.J. Pulsatile GnRH treatment in boys and girls with idiopathic hypogonadotrophic hypogonadism. Hum. Reprod. (Oxf. Engl.) 1993, 8 (Suppl. S2), 180–183. [Google Scholar] [CrossRef]
- Pitteloud, N.; Hayes, F.J.; Dwyer, A.; Boepple, P.A.; Lee, H.; Crowley, W.F., Jr. Predictors of outcome of long-term GnRH therapy in men with idiopathic hypogonadotropic hypogonadism. J. Clin. Endocrinol. Metab. 2002, 87, 4128–4136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sykiotis, G.P.; Hoang, X.H.; Avbelj, M.; Hayes, F.J.; Thambundit, A.; Dwyer, A.; Au, M.; Plummer, L.; Crowley, W.F., Jr.; Pitteloud, N. Congenital idiopathic hypogonadotropic hypogonadism: Evidence of defects in the hypothalamus, pituitary, and testes. J. Clin. Endocrinol. Metab. 2010, 95, 3019–3027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, J.; Mao, J.; Xu, H.; Wang, X.; Huang, B.; Liu, Z.; Cui, M.; Xiong, S.; Ma, W.; Min, L.; et al. Pulsatile GnRH Therapy May Restore Hypothalamus-Pituitary-Testis Axis Function in Patients With Congenital Combined Pituitary Hormone Deficiency: A Prospective, Self-Controlled Trial. J. Clin. Endocrinol. Metab. 2017, 102, 2291–2300. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.Y.; Baker, H.W.; Jayadev, V.; Zacharin, M.; Conway, A.J.; Handelsman, D.J. Induction of spermatogenesis and fertility during gonadotropin treatment of gonadotropin-deficient infertile men: Predictors of fertility outcome. J. Clin. Endocrinol. Metab. 2009, 94, 801–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chua, M.E.; Escusa, K.G.; Luna, S.; Tapia, L.C.; Dofitas, B.; Morales, M. Revisiting oestrogen antagonists (clomiphene or tamoxifen) as medical empiric therapy for idiopathic male infertility: A meta-analysis. Andrology 2013, 1, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Vandekerckhove, P.; Lilford, R.; Vail, A.; Hughes, E. Clomiphene or tamoxifen for idiopathic oligo/asthenospermia. Cochrane Database Syst. Rev. 2000, Cd000151. [Google Scholar] [CrossRef] [PubMed]
- Barbonetti, A.; Calogero, A.E.; Balercia, G.; Garolla, A.; Krausz, C.; La Vignera, S.; Lombardo, F.; Jannini, E.A.; Maggi, M.; Lenzi, A.; et al. The use of follicle stimulating hormone (FSH) for the treatment of the infertile man: Position statement from the Italian Society of Andrology and Sexual Medicine (SIAMS). J. Endocrinol. Investig. 2018, 41, 1107–1122. [Google Scholar] [CrossRef] [PubMed]
- Colpi, G.M.; Francavilla, S.; Haidl, G.; Link, K.; Behre, H.M.; Goulis, D.G.; Krausz, C.; Giwercman, A. European Academy of Andrology guideline Management of oligo-astheno-teratozoospermia. Andrology 2018, 6, 513–524. [Google Scholar] [CrossRef] [Green Version]
- van Alphen, M.M.; van de Kant, H.J.; de Rooij, D.G. Follicle-stimulating hormone stimulates spermatogenesis in the adult monkey. Endocrinology 1988, 123, 1449–1455. [Google Scholar] [CrossRef]
- Simorangkir, D.R.; Ramaswamy, S.; Marshall, G.R.; Pohl, C.R.; Plant, T.M. A selective monotropic elevation of FSH, but not that of LH, amplifies the proliferation and differentiation of spermatogonia in the adult rhesus monkey (Macaca mulatta). Hum. Reprod. (Oxf. Engl.) 2009, 24, 1584–1595. [Google Scholar] [CrossRef] [Green Version]
- Foresta, C.; Bettella, A.; Merico, M.; Garolla, A.; Ferlin, A.; Rossato, M. Use of recombinant human follicle-stimulating hormone in the treatment of male factor infertility. Fertil. Steril. 2002, 77, 238–244. [Google Scholar] [CrossRef]
- Ding, Y.M.; Zhang, X.J.; Li, J.P.; Chen, S.S.; Zhang, R.T.; Tan, W.L.; Shi, X.J. Treatment of idiopathic oligozoospermia with recombinant human follicle-stimulating hormone: A prospective, randomized, double-blind, placebo-controlled clinical study in Chinese population. Clin. Endocrinol. 2015, 83, 866–871. [Google Scholar] [CrossRef]
- Foresta, C.; Bettella, A.; Garolla, A.; Ambrosini, G.; Ferlin, A. Treatment of male idiopathic infertility with recombinant human follicle-stimulating hormone: A prospective, controlled, randomized clinical study. Fertil. Steril. 2005, 84, 654–661. [Google Scholar] [CrossRef]
- Colacurci, N.; Monti, M.G.; Fornaro, F.; Izzo, G.; Izzo, P.; Trotta, C.; Mele, D.; De Franciscis, P. Recombinant human FSH reduces sperm DNA fragmentation in men with idiopathic oligoasthenoteratozoospermia. J. Androl. 2012, 33, 588–593. [Google Scholar] [CrossRef]
- Foresta, C.; Selice, R.; Ferlin, A.; Garolla, A. Recombinant FSH in the treatment of oligozoospermia. Expert Opin. Biol. Ther. 2009, 9, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Ben-Rafael, Z.; Farhi, J.; Feldberg, D.; Bartoov, B.; Kovo, M.; Eltes, F.; Ashkenazi, J. Follicle-stimulating hormone treatment for men with idiopathic oligoteratoasthenozoospermia before in vitro fertilization: The impact on sperm microstructure and fertilization potential. Fertil. Steril. 2000, 73, 24–30. [Google Scholar] [CrossRef]
- Matorras, R.; Perez, C.; Corcostegui, B.; Pijoan, J.I.; Ramon, O.; Delgado, P.; Rodriguez-Escudero, F.J. Treatment of the male with follicle-stimulating hormone in intrauterine insemination with husband’s spermatozoa: A randomized study. Hum. Reprod. (Oxf. Engl.) 1997, 12, 24–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selice, R.; Garolla, A.; Pengo, M.; Caretta, N.; Ferlin, A.; Foresta, C. The response to FSH treatment in oligozoospermic men depends on FSH receptor gene polymorphisms. Int. J. Androl. 2011, 34, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Knuth, U.A.; Honigl, W.; Bals-Pratsch, M.; Schleicher, G.; Nieschlag, E. Treatment of severe oligospermia with human chorionic gonadotropin/human menopausal gonadotropin: A placebo-controlled, double blind trial. J. Clin. Endocrinol. Metab. 1987, 65, 1081–1087. [Google Scholar] [CrossRef] [PubMed]
- Kamischke, A.; Behre, H.M.; Bergmann, M.; Simoni, M.; Schafer, T.; Nieschlag, E. Recombinant human follicle stimulating hormone for treatment of male idiopathic infertility: A randomized, double-blind, placebo-controlled, clinical trial. Hum. Reprod. (Oxf. Engl.) 1998, 13, 596–603. [Google Scholar] [CrossRef] [Green Version]
- Paradisi, R.; Busacchi, P.; Seracchioli, R.; Porcu, E.; Venturoli, S. Effects of high doses of recombinant human follicle-stimulating hormone in the treatment of male factor infertility: Results of a pilot study. Fertil. Steril. 2006, 86, 728–731. [Google Scholar] [CrossRef]
- Caroppo, E.; Niederberger, C.; Vizziello, G.M.; D’Amato, G. Recombinant human follicle-stimulating hormone as a pretreatment for idiopathic oligoasthenoteratozoospermic patients undergoing intracytoplasmic sperm injection. Fertil. Steril. 2003, 80, 1398–1403. [Google Scholar] [CrossRef]
- Foresta, C.; Bettella, A.; Merico, M.; Garolla, A.; Plebani, M.; Ferlin, A.; Rossato, M. FSH in the treatment of oligozoospermia. Mol. Cell. Endocrinol. 2000, 161, 89–97. [Google Scholar] [CrossRef]
- Ashkenazi, J.; Bar-Hava, I.; Farhi, J.; Levy, T.; Feldberg, D.; Orvieto, R.; Ben-Rafael, Z. The role of purified follicle stimulating hormone therapy in the male partner before intracytoplasmic sperm injection. Fertil. Steril. 1999, 72, 670–673. [Google Scholar] [CrossRef]
- Bartoov, B.; Eltes, F.; Lunenfeld, E.; Har-Even, D.; Lederman, H.; Lunenfeld, B. Sperm quality of subfertile males before and after treatment with human follicle-stimulating hormone. Fertil. Steril. 1994, 61, 727–734. [Google Scholar] [CrossRef]
- Baccetti, B.; Piomboni, P.; Bruni, E.; Capitani, S.; Gambera, L.; Moretti, E.; Sterzik, K.; Strehler, E. Effect of follicle-stimulating hormone on sperm quality and pregnancy rate. Asian J. Androl. 2004, 6, 133–137. [Google Scholar] [PubMed]
- Attia, A.M.; Abou-Setta, A.M.; Al-Inany, H.G. Gonadotrophins for idiopathic male factor subfertility. Cochrane Database Syst. Rev. 2013, CD005071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attia, A.M.; Al-Inany, H.G.; Farquhar, C.; Proctor, M. Gonadotrophins for idiopathic male factor subfertility. Cochrane Database Syst. Rev. 2007, Cd005071. [Google Scholar] [CrossRef] [Green Version]
- Santi, D.; Granata, A.R.; Simoni, M. Follicle-stimulating hormone treatment of male idiopathic infertility improves pregnancy rate: A meta-analysis. Endocr. Connect. 2015. [Google Scholar] [CrossRef] [Green Version]
- Cannarella, R.; La Vignera, S.; Condorelli, R.A.; Mongioi, L.M.; Calogero, A.E. FSH dosage effect on conventional sperm parameters: A meta-analysis of randomized controlled studies. Asian J. Androl. 2019. [Google Scholar] [CrossRef]
- Foresta, C.; Bettella, A.; Ferlin, A.; Garolla, A.; Rossato, M. Evidence for a stimulatory role of follicle-stimulating hormone on the spermatogonial population in adult males. Fertil. Steril. 1998, 69, 636–642. [Google Scholar] [CrossRef]
- Hussein, A.; Ozgok, Y.; Ross, L.; Rao, P.; Niederberger, C. Optimization of spermatogenesis-regulating hormones in patients with non-obstructive azoospermia and its impact on sperm retrieval: A multicentre study. BJU Int. 2013, 111, E110–E114. [Google Scholar] [CrossRef]
- Shiraishi, K.; Ohmi, C.; Shimabukuro, T.; Matsuyama, H. Human chorionic gonadotrophin treatment prior to microdissection testicular sperm extraction in non-obstructive azoospermia. Hum. Reprod. (Oxf. Engl.) 2012, 27, 331–339. [Google Scholar] [CrossRef] [Green Version]
- Aydos, K.; Unlu, C.; Demirel, L.C.; Evirgen, O.; Tolunay, O. The effect of pure FSH administration in non-obstructive azoospermic men on testicular sperm retrieval. Eur. J. Obstet. Gynecol. Reprod. Biol. 2003, 108, 54–58. [Google Scholar] [CrossRef]
- Cocci, A.; Cito, G.; Russo, G.I.; Falcone, M.; Capece, M.; Timpano, M.; Della Camera, P.A.; Morselli, S.; Tasso, G.; Morelli, G.; et al. Effectiveness of highly purified urofollitropin treatment in patients with idiopathic azoospermia before testicular sperm extraction. Urologia 2018, 85, 19–21. [Google Scholar] [CrossRef] [PubMed]
- Jungwirth, A.; Giwercman, A.; Tournaye, H.; Diemer, T.; Kopa, Z.; Dohle, G.; Krausz, C. European Association of Urology guidelines on Male Infertility: The 2012 update. Eur. Urol. 2012, 62, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Casarini, L.; Lispi, M.; Longobardi, S.; Milosa, F.; La Marca, A.; Tagliasacchi, D.; Pignatti, E.; Simoni, M. LH and hCG action on the same receptor results in quantitatively and qualitatively different intracellular signalling. PLoS ONE 2012, 7, e46682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO. WHO Laboratory Manual for the Examination and Processing of Human Semen, 5th ed.; World Health Organization: Geneva, Switzerland, 2010. [Google Scholar]
- Anawalt, B.D. Approach to male infertility and induction of spermatogenesis. J. Clin. Endocrinol. Metab. 2013, 98, 3532–3542. [Google Scholar] [CrossRef] [Green Version]
- Bouloux, P.; Warne, D.W.; Loumaye, E. Efficacy and safety of recombinant human follicle-stimulating hormone in men with isolated hypogonadotropic hypogonadism. Fertil. Steril. 2002, 77, 270–273. [Google Scholar] [CrossRef]
- Bouloux, P.M.; Nieschlag, E.; Burger, H.G.; Skakkebaek, N.E.; Wu, F.C.; Handelsman, D.J.; Baker, G.H.; Ochsenkuehn, R.; Syska, A.; McLachlan, R.I.; et al. Induction of spermatogenesis by recombinant follicle-stimulating hormone (puregon) in hypogonadotropic azoospermic men who failed to respond to human chorionic gonadotropin alone. J. Androl. 2003, 24, 604–611. [Google Scholar] [CrossRef]
- Burris, A.S.; Clark, R.V.; Vantman, D.J.; Sherins, R.J. A low sperm concentration does not preclude fertility in men with isolated hypogonadotropic hypogonadism after gonadotropin therapy. Fertil. Steril. 1988, 50, 343–347. [Google Scholar] [CrossRef]
- Han, T.S.; Bouloux, P.M. What is the optimal therapy for young males with hypogonadotropic hypogonadism? Clin. Endocrinol. 2010, 72, 731–737. [Google Scholar] [CrossRef]
- Matsumoto, A.M.; Snyder, P.J.; Bhasin, S.; Martin, K.; Weber, T.; Winters, S.; Spratt, D.; Brentzel, J.; O’Dea, L. Stimulation of spermatogenesis with recombinant human follicle-stimulating hormone (follitropin alfa; GONAL-f): Long-term treatment in azoospermic men with hypogonadotropic hypogonadism. Fertil. Steril. 2009, 92, 979–990. [Google Scholar] [CrossRef]
- Herbison, A.E. The Gonadotropin-Releasing Hormone Pulse Generator. Endocrinology 2018, 159, 3723–3736. [Google Scholar] [CrossRef] [Green Version]
- Boehm, U.; Bouloux, P.M.; Dattani, M.T.; de Roux, N.; Dode, C.; Dunkel, L.; Dwyer, A.A.; Giacobini, P.; Hardelin, J.P.; Juul, A.; et al. Expert consensus document: European Consensus Statement on congenital hypogonadotropic hypogonadism--pathogenesis, diagnosis and treatment. Nat. Rev. Endocrinol. 2015, 11, 547–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vicari, E.; Mongioi, A.; Calogero, A.E.; Moncada, M.L.; Sidoti, G.; Polosa, P.; D’Agata, R. Therapy with human chorionic gonadotrophin alone induces spermatogenesis in men with isolated hypogonadotrophic hypogonadism--long-term follow-up. Int. J. Androl. 1992, 15, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Rohayem, J.; Hauffa, B.P.; Zacharin, M.; Kliesch, S.; Zitzmann, M. Testicular growth and spermatogenesis: New goals for pubertal hormone replacement in boys with hypogonadotropic hypogonadism? -a multicentre prospective study of hCG/rFSH treatment outcomes during adolescence. Clin. Endocrinol. 2017, 86, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Zacharin, M.; Sabin, M.A.; Nair, V.V.; Dabadghao, P. Addition of recombinant follicle-stimulating hormone to human chorionic gonadotropin treatment in adolescents and young adults with hypogonadotropic hypogonadism promotes normal testicular growth and may promote early spermatogenesis. Fertil. Steril. 2012, 98, 836–842. [Google Scholar] [CrossRef] [PubMed]
- Fauser, B.C.; Mannaerts, B.M.; Devroey, P.; Leader, A.; Boime, I.; Baird, D.T. Advances in recombinant DNA technology: Corifollitropin alfa, a hybrid molecule with sustained follicle-stimulating activity and reduced injection frequency. Hum. Reprod. Update 2009, 15, 309–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devroey, P.; Boostanfar, R.; Koper, N.P.; Mannaerts, B.M.; Ijzerman-Boon, P.C.; Fauser, B.C. A double-blind, non-inferiority RCT comparing corifollitropin alfa and recombinant FSH during the first seven days of ovarian stimulation using a GnRH antagonist protocol. Hum. Reprod. (Oxf. Engl.) 2009, 24, 3063–3072. [Google Scholar] [CrossRef] [Green Version]
- Nieschlag, E.; Bouloux, P.G.; Stegmann, B.J.; Shankar, R.R.; Guan, Y.; Tzontcheva, A.; McCrary Sisk, C.; Behre, H.M. An open-label clinical trial to investigate the efficacy and safety of corifollitropin alfa combined with hCG in adult men with hypogonadotropic hypogonadism. Reprod. Biol. Endocrinol. 2017, 15, 17. [Google Scholar] [CrossRef] [Green Version]
- Uchiyama, Y.; Hosoe, M.; Sato, T.; Takahashi, T. Biological and immunological characteristics of porcine follicle-stimulating hormone chemically modified with a polyethylene glycol derivative. Vet. J. 2010, 184, 208–211. [Google Scholar] [CrossRef]
- DeFrees, S.; Bayer, R.; Bowe, C. Glycopegylated Follicle Stimulating Hormone. U.S. Patent 2008/0015142 A1, 17 January 2008. [Google Scholar]
- Hashimoto, S.; Kimura, K.; Kuramochi, T.; Aoyagi, K.; Hirako, M.; Kawaguchi, M.; Iwata, H.; Hirao, M.; Kitada, K.; Hirasawa, K.; et al. Responsiveness of rabbits to superovulation treatment by a single injection of follicle-stimulating hormone with aluminum hydroxide gel. Mol. Reprod. Dev. 2007, 74, 1208–1212. [Google Scholar] [CrossRef]
- Kimura, K.; Hirako, M.; Iwata, H.; Aoki, M.; Kawaguchi, M.; Seki, M. Successful superovulation of cattle by a single administration of FSH in aluminum hydroxide gel. Theriogenology 2007, 68, 633–639. [Google Scholar] [CrossRef]
- Anderson, R.C.; Newton, C.L.; Anderson, R.A.; Millar, R.P. Gonadotropins and Their Analogs: Current and Potential Clinical Applications. Endocr. Rev. 2018, 39, 911–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugahara, T.; Pixley, M.R.; Minami, S.; Perlas, E.; Ben-Menahem, D.; Hsueh, A.J.; Boime, I. Biosynthesis of a biologically active single peptide chain containing the human common alpha and chorionic gonadotropin beta subunits in tandem. Proc. Natl. Acad. Sci. USA 1995, 92, 2041–2045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Campayo, V.; Sato, A.; Hirsch, B.; Sugahara, T.; Muyan, M.; Hsueh, A.J.; Boime, I. Design of stable biologically active recombinant lutropin analogs. Nat. Biotechnol. 1997, 15, 663–667. [Google Scholar] [CrossRef] [PubMed]
- Lemke, E.P.; Adams, B.M.; Jablonka-Shariff, A.; Boime, I.; Adams, T.E. Single-chain human gonadotropin analogs induce follicle development in sheep. J. Endocrinol. 2008, 196, 593–600. [Google Scholar] [CrossRef] [Green Version]
- Sugahara, T.; Grootenhuis, P.D.; Sato, A.; Kudo, M.; Ben-Menahem, D.; Pixley, M.R.; Hsueh, A.J.; Boime, I. Expression of biologically active fusion genes encoding the common alpha subunit and either the CG beta or FSH beta subunits: Role of a linker sequence. Mol. Cell. Endocrinol. 1996, 125, 71–77. [Google Scholar] [CrossRef]
- Klein, J.; Lobel, L.; Pollak, S.; Ferin, M.; Xiao, E.; Sauer, M.; Lustbader, J.W. Pharmacokinetics and pharmacodynamics of single-chain recombinant human follicle-stimulating hormone containing the human chorionic gonadotropin carboxyterminal peptide in the rhesus monkey. Fertil. Steril. 2002, 77, 1248–1255. [Google Scholar] [CrossRef]
- Kanda, M.; Jablonka-Shariff, A.; Sato, A.; Pixley, M.R.; Bos, E.; Hiro’oka, T.; Ben-Menahem, D.; Boime, I. Genetic fusion of an alpha-subunit gene to the follicle-stimulating hormone and chorionic gonadotropin-beta subunit genes: Production of a bifunctional protein. Mol. Endocrinol. 1999, 13, 1873–1881. [Google Scholar] [CrossRef] [Green Version]
- Rutigliano, H.M.; Adams, B.M.; Jablonka-Shariff, A.; Boime, I.; Adams, T.E. Effect of single-chain ovine gonadotropins with dual activity on ovarian function in sheep. Reprod 2014, 148, 129–136. [Google Scholar] [CrossRef] [Green Version]
- Low, S. Heterodimeric Follicle Stimulating Hormone-Fc (FSH-Fc) Fusion Proteins for the Treatment of Infertility. U.S. Patent 8293707, 21 April 2009. [Google Scholar]
- Zhang, Y.L.; Guo, K.P.; Ji, S.Y.; Liu, X.M.; Wang, P.; Wu, J.; Gao, L.; Jiang, T.Q.; Xu, T.; Fan, H.Y. Development and characterization of a novel long-acting recombinant follicle stimulating hormone agonist by fusing Fc to an FSH-beta subunit. Hum. Reprod. (Oxf. Engl.) 2016, 31, 169–182. [Google Scholar] [CrossRef] [Green Version]
- Maclean, D.; Holden, F.; Davis, A.M.; Scheuerman, R.A.; Yanofsky, S.; Holmes, C.P.; Fitch, W.L.; Tsutsui, K.; Barrett, R.W.; Gallop, M.A. Agonists of the follicle stimulating hormone receptor from an encoded thiazolidinone library. J. Comb. Chem. 2004, 6, 196–206. [Google Scholar] [CrossRef]
- Yanofsky, S.D.; Shen, E.S.; Holden, F.; Whitehorn, E.; Aguilar, B.; Tate, E.; Holmes, C.P.; Scheuerman, R.; MacLean, D.; Wu, M.M.; et al. Allosteric activation of the follicle-stimulating hormone (FSH) receptor by selective, nonpeptide agonists. J. Biol. Chem. 2006, 281, 13226–13233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sriraman, V.; Denis, D.; de Matos, D.; Yu, H.; Palmer, S.; Nataraja, S. Investigation of a thiazolidinone derivative as an allosteric modulator of follicle stimulating hormone receptor: Evidence for its ability to support follicular development and ovulation. Biochem. Pharm. 2014, 89, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Arey, B.J.; Yanofsky, S.D.; Claudia Perez, M.; Holmes, C.P.; Wrobel, J.; Gopalsamy, A.; Stevis, P.E.; Lopez, F.J.; Winneker, R.C. Differing pharmacological activities of thiazolidinone analogs at the FSH receptor. Biochem. Biophys. Res. Commun. 2008, 368, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.N.; Richardson, T.E.; Nataraja, S.; Fischer, D.J.; Sriraman, V.; Jiang, X.; Bharathi, P.; Foglesong, R.J.; Haxell, T.F.; Heasley, B.H.; et al. Discovery of substituted benzamides as follicle stimulating hormone receptor allosteric modulators. Bioorg Med. Chem. Lett. 2014, 24, 2168–2172. [Google Scholar] [CrossRef]
- van Koppen, C.J.; Verbost, P.M.; van de Lagemaat, R.; Karstens, W.J.; Loozen, H.J.; van Achterberg, T.A.; van Amstel, M.G.; Brands, J.H.; van Doornmalen, E.J.; Wat, J.; et al. Signaling of an allosteric, nanomolar potent, low molecular weight agonist for the follicle-stimulating hormone receptor. Biochem. Pharm. 2013, 85, 1162–1170. [Google Scholar] [CrossRef]
- van Straten, N.C.; van Berkel, T.H.; Demont, D.R.; Karstens, W.J.; Merkx, R.; Oosterom, J.; Schulz, J.; van Someren, R.G.; Timmers, C.M.; van Zandvoort, P.M. Identification of substituted 6-amino-4-phenyltetrahydroquinoline derivatives: Potent antagonists for the follicle-stimulating hormone receptor. J. Med. Chem 2005, 48, 1697–1700. [Google Scholar] [CrossRef]
- Dias, J.A.; Bonnet, B.; Weaver, B.A.; Watts, J.; Kluetzman, K.; Thomas, R.M.; Poli, S.; Mutel, V.; Campo, B. A negative allosteric modulator demonstrates biased antagonism of the follicle stimulating hormone receptor. Mol. Cell. Endocrinol. 2011, 333, 143–150. [Google Scholar] [CrossRef] [Green Version]
- Dias, J.A.; Campo, B.; Weaver, B.A.; Watts, J.; Kluetzman, K.; Thomas, R.M.; Bonnet, B.; Mutel, V.; Poli, S.M. Inhibition of follicle-stimulating hormone-induced preovulatory follicles in rats treated with a nonsteroidal negative allosteric modulator of follicle-stimulating hormone receptor. Biol. Reprod. 2014, 90, 19. [Google Scholar] [CrossRef]
- Ayoub, M.A.; Yvinec, R.; Jegot, G.; Dias, J.A.; Poli, S.M.; Poupon, A.; Crepieux, P.; Reiter, E. Profiling of FSHR negative allosteric modulators on LH/CGR reveals biased antagonism with implications in steroidogenesis. Mol. Cell. Endocrinol. 2016, 436, 10–22. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| FSH Doses | Treatment Duration | FSH Total Amount | Authors | FSH Doses | Treatment Duration | FSH Total Amount | Authors |
|---|---|---|---|---|---|---|---|
| RCT | |||||||
| Recombinant FSH | Urinary-derived FSH | ||||||
| 50 IU on alternate days | 12 weeks | 2250 IU | Foresta et al. (2002) [128] | 50 IU on alternate days | 12 weeks | 2250 IU | Ding et al. (2015) [129] |
| 100 IU on alternate days | 12 weeks | 4500 IU | Foresta et al. (2002), Foresta et al. (2005) [128,130] | 100 IU on alternate days | 12 weeks | 4500 IU | Ding et al. (2015) [129] |
| 6750 IU | Colacurci et al. (2012) [131] | ||||||
| 150 IU on alternate days | 12 weeks | 6750 IU | Simoni et al. (2016), Foresta et al. (2009) [105,132] | 75 IU daily (150 IU on alternate days) | Not reported | Not reported | Ben-Rafael et al. (2000), Matorras et al. (1997) [133,134] |
| 5400 IU | Selice et al. (2011) [135] | 13 weeks | 6825 IU | Knuth et al. (1987)*§ [136] | |||
| 150 IU daily (300 IU on alternate days) | 12 weeks | 12600 IU | Kamischke et al. (1998)§ [137] | 200 IU on alternate days | 12 weeks | 9000 IU | Ding et al. (2015) [129] |
| 16 weeks | 18000 IU | Paradisi et al. (2006)§ [138] | 150 IU daily (300 IU on alternate days) | 12 weeks | 12600 IU | Ding et al. (2015), Ben-Rafael et al. (2000) [129,133] | |
| Non randomized, retrospective trials | |||||||
| Recombinant FSH | Urinary-derived FSH | ||||||
| 150 IU on alternate days | 12 weeks | 5400 IU | Caroppo et al. (2003) [139] | 75IU on alternate days | 12 weeks | 3375 IU | Foresta et al. (2000) [140] |
| 75 IU daily | 12 weeks | 6300 IU | Ashkenazi et al. (1999) [141] | ||||
| 4 weeks | 2100 IU | Bartoov et al. (1994) [142] | |||||
| 150 IU daily | 12 weeks | 12600 IU | Baccetti et al. (2004) [143] | ||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santi, D.; Crépieux, P.; Reiter, E.; Spaggiari, G.; Brigante, G.; Casarini, L.; Rochira, V.; Simoni, M. Follicle-Stimulating Hormone (FSH) Action on Spermatogenesis: A Focus on Physiological and Therapeutic Roles. J. Clin. Med. 2020, 9, 1014. https://doi.org/10.3390/jcm9041014
Santi D, Crépieux P, Reiter E, Spaggiari G, Brigante G, Casarini L, Rochira V, Simoni M. Follicle-Stimulating Hormone (FSH) Action on Spermatogenesis: A Focus on Physiological and Therapeutic Roles. Journal of Clinical Medicine. 2020; 9(4):1014. https://doi.org/10.3390/jcm9041014
Chicago/Turabian StyleSanti, Daniele, Pascale Crépieux, Eric Reiter, Giorgia Spaggiari, Giulia Brigante, Livio Casarini, Vincenzo Rochira, and Manuela Simoni. 2020. "Follicle-Stimulating Hormone (FSH) Action on Spermatogenesis: A Focus on Physiological and Therapeutic Roles" Journal of Clinical Medicine 9, no. 4: 1014. https://doi.org/10.3390/jcm9041014