Can Adenosine Fight COVID-19 Acute Respiratory Distress Syndrome?

 , , ,
, , ,  , and
, and
Abstract
:1. Introduction
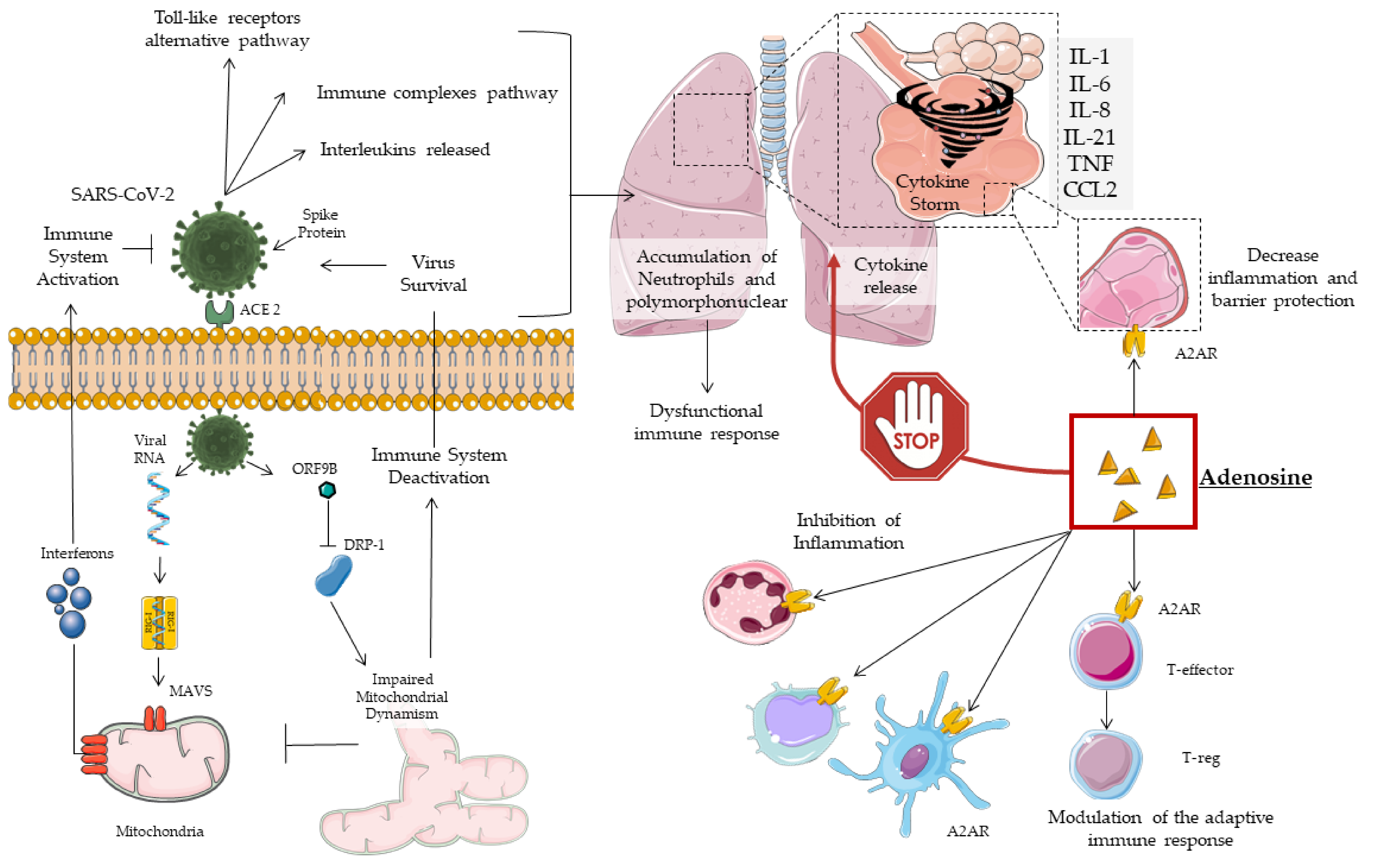
2. COVID19 Pathogenesis, Mitochondrial Dynamism and Immune Response
3. Cytokine Storm
Current COVID-19 and Cytokine Storm Therapeutic Strategies
4. Adenosine
4.1. Subtypes of Adenosine Receptors (AR)
4.2. Adenosine in the Treatment of Lung Injury
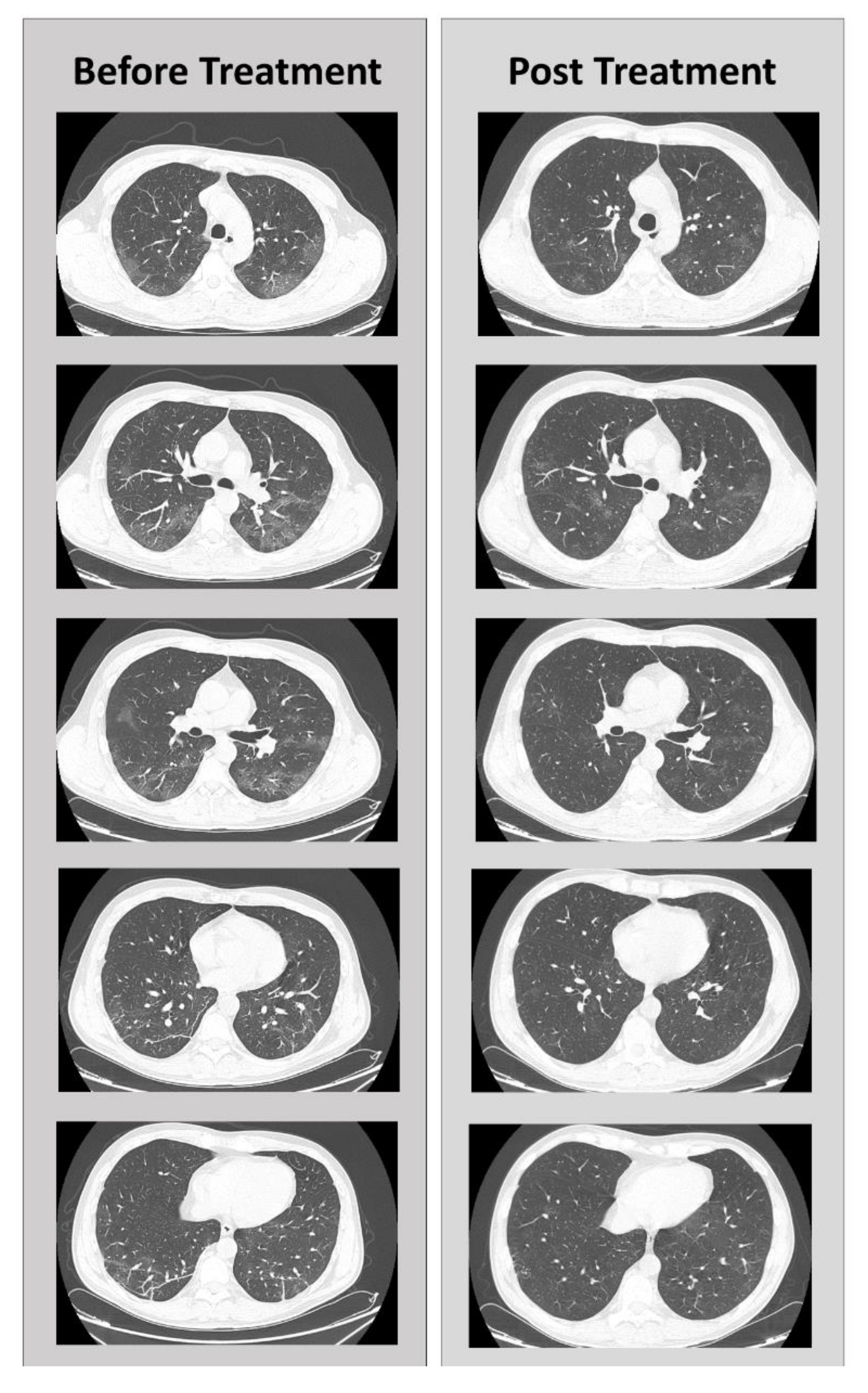
5. Clinical Case
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ARDS | Acute respiratory distress syndrome |
| IL | Interleukin |
| TCZ | Tocilizumab |
| JAK | Janus kinase inhibitors |
| A2AR | A2A receptor |
| MOF | Multi organ failure |
| TNF-α | Tumor necrosis factor-α |
| hCoV | Human coronavirus |
| DRP | Dynamin-related proteins |
| PINK-1 | Phosphatase and tensin homolog - induced kinase 1 |
| RIG-I | Retinoic acid-inducible gene 1 |
| RLRs | RIG-I-like receptors |
| MAVS | Mitochondrial associated antiviral signaling protein |
| CARDs | Caspase recruitment domains |
| NFkB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| ORF | Open reading frame |
| INF | Interferon |
| TRAF | TNF receptor associated factor |
| PMN | Polymorphonuclear leukocytes |
| ADP | Adenosine diphosphate |
| ATP | Adenosine triphosphate |
| AMP | Adenosine monophosphate |
| CD | Cluster of differentiation |
| ALI | Acute lung injury |
| AR | Adenosine receptors |
| CXCL1-3 | Chemokine (C-X-C motif) ligand 3 |
| T Regs | Regulatory T cells |
| PPARγ | Peroxisome proliferator-activated receptor γ |
| ACE2 | Angiotensin-converting enzyme |
| EAT | Epicardial fat |
| GOM | Great Metropolitan Bianchi Melacrino Morelli |
| CT | Computed tomography |
| RUB | Right upper lobe |
References
- WHO Coronavirus Disease (COVID-19) Dashboard. Available online: https://covid19.who.int/?gclid=Cj0KCQjw0rr4BRCtARIsAB0_48NF8a417ap3xz5a6rC5bv4LHq4iaWP5iTQPyvEhFlQLpGa7fyo6R0aAhVTEALw_wcB (accessed on 15 July 2020).
- Ye, Q.; Wang, B.; Mao, J. The pathogenesis and treatment of the ‘Cytokine Storm’ in COVID-19. J. Infect. 2020, 80, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Thiel, M.; Choukèr, A.; Ohta, A.; Jackson, E.; Caldwell, C.C.; Smith, P.; Lukashev, D.; Bittmann, I.; Sitkovsky, M.V. Oxygenation Inhibits the Physiological Tissue-Protecting Mechanism and Thereby Exacerbates Acute Inflammatory Lung Injury. PLoS Boil. 2005, 3, e174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tay, M.Z.; Poh, C.M.; Rénia, L.; Macary, P.A.; Ng, L.F.P. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Gorbalenya, A.E.; Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef] [Green Version]
- Kucirka, L.M.; Lauer, S.A.; Laeyendecker, O.; Boon, D.; Lessler, J. Variation in False-Negative Rate of Reverse Transcriptase Polymerase Chain Reaction-Based SARS-CoV-2 Tests by Time Since Exposure. Ann. Intern. Med. 2020, 173, 262–267. [Google Scholar] [CrossRef]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA 2020, 323, 1061. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef]
- Park, W.B.; Kwon, N.-J.; Choi, S.J.; Kang, C.K.; Choe, P.G.; Kim, J.Y.; Yun, J.; Lee, G.-W.; Seong, M.-W.; Kim, N.J.; et al. Virus Isolation from the First Patient with SARS-CoV-2 in Korea. J. Korean Med. Sci. 2020, 35, e84. [Google Scholar] [CrossRef]
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More Than Just a Powerhouse. Curr. Boil. 2006, 16, R551–R560. [Google Scholar] [CrossRef] [Green Version]
- Anand, S.K.; Tikoo, S.K. Viruses as Modulators of Mitochondrial Functions. Adv. Virol. 2013, 2013, 1–17. [Google Scholar] [CrossRef]
- Khan, M.; Syed, G.H.; Kim, S.-J.; Siddiqui, A. Mitochondrial dynamics and viral infections: A close nexus. Biochim. et Biophys. Acta BBA Bioenerg. 2015, 1853, 2822–2833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Losón, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Boil. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.-F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Boil. 2010, 8, e1000298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Chan, D.C. Mitochondrial dynamics-fusion, fission, movement, and mitophagy-in neurodegenerative diseases. Hum. Mol. Genet. 2009, 18, R169–R176. [Google Scholar] [CrossRef]
- Seth, R.B.; Sun, L.; Ea, C.-K.; Chen, Z.J. Identification and Characterization of MAVS, a Mitochondrial Antiviral Signaling Protein that Activates NF-κB and IRF3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.-S.; Qi, H.-Y.; Boularan, C.; Huang, N.-N.; Abu-Asab, M.; Shelhamer, J.H.; Kehrl, J.H. SARS-Coronavirus Open Reading Frame-9b Suppresses Innate Immunity by Targeting Mitochondria and the MAVS/TRAF3/TRAF6 Signalosome. J. Immunol. 2014, 193, 3080–3089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimoto, F.K. The Proteins of Severe Acute Respiratory Syndrome Coronavirus-2 (SARS CoV-2 or n-COV19), the Cause of COVID-19. Protein J. 2020, 39, 198–216. [Google Scholar] [PubMed]
- Wang, C.; Liu, Z.; Chen, Z.; Huang, X.; Xu, M.; He, T.; Zhang, Z. The establishment of reference sequence for SARS-CoV-2 and variation analysis. J. Med. Virol. 2020, 92, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Law, H.K.W.; Cheung, C.Y.; Ng, H.Y.; Sia, S.F.; Chan, Y.O.; Luk, W.; Nicholls, J.M.; Peiris, J.S.M.; Lau, Y.L. Chemokine up-regulation in SARS-coronavirus–infected, monocyte-derived human dendritic cells. Blood 2005, 106, 2366–2374. [Google Scholar] [CrossRef] [Green Version]
- Cheung, C.Y.; Poon, L.L.M.; Ng, I.H.Y.; Luk, W.; Sia, S.-F.; Wu, M.H.S.; Chan, K.-H.; Yuen, K.-Y.; Gordon, S.; Guan, Y.; et al. Cytokine Responses in Severe Acute Respiratory Syndrome Coronavirus-Infected Macrophages In Vitro: Possible Relevance to Pathogenesis. J. Virol. 2005, 79, 7819–7826. [Google Scholar] [CrossRef] [Green Version]
- Lau, S.K.P.; Lau, C.C.Y.; Chan, K.-H.; Li, C.P.Y.; Chen, H.; Jin, D.-Y.; Chan, J.F.; Woo, P.C.Y.; Yuen, K.-Y. Delayed induction of proinflammatory cytokines and suppression of innate antiviral response by the novel Middle East respiratory syndrome coronavirus: Implications for pathogenesis and treatment. J. Gen. Virol. 2013, 94, 2679–2690. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S.; Choe, P.G.; Park, W.B.; Oh, H.S.; Kim, E.J.; Nam, E.Y.; Na, S.H.; Kim, M.; Song, K.-H.; Bang, J.H.; et al. Clinical Progression and Cytokine Profiles of Middle East Respiratory Syndrome Coronavirus Infection. J. Korean Med. Sci. 2016, 31, 1717–1725. [Google Scholar] [CrossRef] [PubMed]
- Ng, D.; Alhosani, F.; Keating, M.K.; Gerber, S.I.; Jones, T.L.; Metcalfe, M.G.; Tong, S.; Tao, Y.; Alami, N.N.; Haynes, L.M.; et al. Clinicopathologic, Immunohistochemical, and Ultrastructural Findings of a Fatal Case of Middle East Respiratory Syndrome Coronavirus Infection in the United Arab Emirates, April 2014. Am. J. Pathol. 2016, 186, 652–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chien, J.-Y.; Hsueh, P.-R.; Cheng, W.; Yu, C.-J.; Yang, P.-C. Temporal changes in cytokine/chemokine profiles and pulmonary involvement in severe acute respiratory syndrome. Respirology 2006, 11, 715–722. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, J.; Zhan, Y.; Wu, L.; Yu, X.; Zhang, W.; Ye, L.; Xu, S.; Sun, R.; Wang, Y.; et al. Analysis of Serum Cytokines in Patients with Severe Acute Respiratory Syndrome. Infect. Immun. 2004, 72, 4410–4415. [Google Scholar] [CrossRef] [Green Version]
- Channappanavar, R.; Fehr, A.R.; Zheng, J.; Wohlford-Lenane, C.; Abrahante, J.E.; Mack, M.; Sompallae, R.; McCray, P.B.; Meyerholz, D.K.; Perlman, S. IFN-I response timing relative to virus replication determines MERS coronavirus infection outcomes. J. Clin. Investig. 2019, 130, 3625–3639. [Google Scholar] [CrossRef]
- Smits, S.L.; De Lang, A.; Brand, J.M.A.V.D.; Leijten, L.M.; Van Ijcken, W.; Eijkemans, M.J.C.; Van Amerongen, G.; Kuiken, T.; Andeweg, A.C.; Osterhaus, A.D.M.E.; et al. Exacerbated Innate Host Response to SARS-CoV in Aged Non-Human Primates. PLOS Pathog. 2010, 6, e1000756. [Google Scholar] [CrossRef] [Green Version]
- Cameron, M.J.; Bermejo-Martin, J.F.; Danesh, A.; Muller, M.P.; Kelvin, D.J. Human immunopathogenesis of severe acute respiratory syndrome (SARS). Virus Res. 2008, 133, 13–19. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Stefan, N.; Birkenfeld, A.L.; Schulze, M.B.; Ludwig, D.S. Obesity and impaired metabolic health in patients with COVID-19. Nat. Rev. Endocrinol. 2020, 16, 341–342. [Google Scholar] [CrossRef] [Green Version]
- Dietz, W.H.; Santos-Burgoa, C. Obesity and its Implications for COVID-19 Mortality. Obesity 2020, 28, 1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonnet, A.; Chetboun, M.; Poissy, J.; Raverdy, V.; Noulette, J.; Duhamel, A.; Labreuche, J.; Mathieu, D.; Pattou, F.; Jourdain, M.; et al. High Prevalence of Obesity in Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2) Requiring Invasive Mechanical Ventilation. Obesity 2020, 28, 1195–1199. [Google Scholar] [CrossRef] [PubMed]
- Richardson, S.; Hirsch, J.S.; Narasimhan, M.; Crawford, J.M.; McGinn, T.; Davidson, K.W.; Barnaby, D.P.; Becker, L.B.; Chelico, J.D.; Cohen, S.L.; et al. Presenting Characteristics, Comorbidities, and Outcomes Among 5700 Patients Hospitalized With COVID-19 in the New York City Area. JAMA 2020, 323, 2052. [Google Scholar] [CrossRef] [PubMed]
- Finer, N.; Garnett, S.P.; Bruun, J.M. COVID-19 and obesity. Clin. Obes. 2020, 10, 12365. [Google Scholar] [CrossRef] [PubMed]
- Buscemi, S.; Buscemi, C.; Batsis, J.A. There is a relationship between obesity and COVID-19 but more information is needed. Obesity 2020. [Google Scholar] [CrossRef]
- Bloomgarden, Z. Diabetes and COVID-19. J. Diabetes 2020, 12, 347–348. [Google Scholar] [CrossRef] [Green Version]
- Brufsky, A. Hyperglycemia, hydroxychloroquine, and the COVID-19 pandemic. J. Med. Virol. 2020, 92, 770–775. [Google Scholar] [CrossRef] [Green Version]
- De Lucena, T.M.C.; Santos, A.F.D.S.; De Lima, B.R.; Borborema, M.E.D.A.; Silva, J.D.A.; Fabrício, B.R.D.L. Mechanism of inflammatory response in associated comorbidities in COVID-19. Diabetes Metab. Syndr. Clin. Res. Rev. 2020, 14, 597–600. [Google Scholar] [CrossRef]
- Roschewski, M.; Lionakis, M.S.; Sharman, J.P.; Roswarski, J.; Goy, A.; Monticelli, M.A.; Roshon, M.; Wrzesinski, S.H.; Desai, J.V.; Zarakas, M.A.; et al. Inhibition of Bruton tyrosine kinase in patients with severe COVID-19. Sci. Immunol. 2020, 5, 0110. [Google Scholar] [CrossRef]
- Kumari, P.; Rawat, K.; Saha, L. Pipeline Pharmacological Therapies in Clinical Trial for COVID-19 Pandemic: A Recent Update. Curr. Pharmacol. Rep. 2020, 1–13. [Google Scholar] [CrossRef]
- Zhao, J.P.; Hu, Y.; Du, R.H.; Chen, Z.S.; Jin, Y.; Zhou, M.; Zhang, J.; Qu, J.M.; Cao, B. Zhonghua jie he he hu xi za zhi Zhonghua jiehe he huxi zazhi. Chin. J. Tubercul. Resp. Dis. 2020, 43, E007. [Google Scholar]
- Qin, Y.-Y.; Zhou, Y.-H.; Lu, Y.-Q.; Sun, F.; Yang, S.; Harypursat, V.; Chen, Y.; Tang, S.-Q.; Huang, Y.-Q.; He, X.-Q.; et al. Effectiveness of glucocorticoid therapy in patients with severe coronavirus disease 2019. Chin. Med J. 2020, 133, 1080–1086. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Cao, R.; Xu, M.; Wang, X.; Zhang, H.; Hu, H.; Li, Y.; Hu, Z.; Zhong, W.; Wang, M. Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discov. 2020, 6, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Han, M.; Li, T.; Sun, W.; Wang, D.; Fu, B.; Zhou, Y.; Zheng, X.; Yang, Y.; Li, X.; et al. Effective treatment of severe COVID-19 patients with tocilizumab. Proc. Natl. Acad. Sci. USA 2020, 117, 10970–10975. [Google Scholar] [CrossRef]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.-J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Di Castelnuovo, A.; Costanzo, S.; Antinori, A.; Berselli, N.; Blandi, L.; Bruno, R.; Cauda, R.; Guaraldi, G.; Menicanti, L.; My, I.; et al. Use of hydroxychloroquine in hospitalised COVID-19 patients is associated with reduced mortality: Findings from the observational multicentre Italian CORIST study. Eur. J. Intern. Med. 2020, 0953. [Google Scholar] [CrossRef]
- Riva, A.; Conti, F.; Bernacchia, D.; Pezzati, L.; Sollima, S.; Merli, S.; Siano, M.; Lupo, A.; Rusconi, S.; Cattaneo, D.; et al. Darunavir does not prevent SARS-CoV-2 infection in HIV patients. Pharmacol. Res. 2020, 157, 104826. [Google Scholar] [CrossRef]
- Cao, B.; Wang, Y.; Wen, D.; Liu, W.; Wang, J.; Fan, G.; Ruan, L.; Song, B.; Cai, Y.; Wei, M.; et al. A Trial of Lopinavir–Ritonavir in Adults Hospitalized with Severe Covid-19. N. Engl. J. Med. 2020, 382, 1787–1799. [Google Scholar] [CrossRef]
- Gordon, C.J.; Tchesnokov, E.P.; Feng, J.Y.; Porter, D.P.; Gotte, M. The antiviral compound remdesivir potently inhibits RNA-dependent RNA polymerase from Middle East respiratory syndrome coronavirus. J. Boil. Chem. 2020, 295, 4773–4779. [Google Scholar] [CrossRef] [Green Version]
- Cavalli, G.; De Luca, G.; Campochiaro, C.; Della-Torre, E.; Ripa, M.; Canetti, D.; Oltolini, C.; Castiglioni, B.; Din, C.T.; Boffini, N.; et al. Interleukin-1 blockade with high-dose anakinra in patients with COVID-19, acute respiratory distress syndrome, and hyperinflammation: A retrospective cohort study. Lancet Rheumatol. 2020, 2, e325–e331. [Google Scholar] [CrossRef]
- Caracciolo, M.; Macheda, S.; Labate, D.; Tescione, M.; La Scala, S.; Vadalà, E.; Squillaci, R.; D’Aleo, F.; Morabito, A.; Garreffa, C.; et al. Case Report: Canakinumab for the Treatment of a Patient With COVID-19 Acute Respiratory Distress Syndrome. Front. Immunol. 2020, 11, 11. [Google Scholar] [CrossRef]
- Michot, J.-M.; Albiges, L.; Chaput, N.; Saada, V.; Pommeret, F.; Griscelli, F.; Balleyguier, C.; Besse, B.; Marabelle, A.; Netzer, F.; et al. Tocilizumab, an anti-IL-6 receptor antibody, to treat COVID-19-related respiratory failure: A case report. Ann. Oncol. 2020, 31, 961–964. [Google Scholar] [CrossRef]
- AIFA. COVID-19: Studio Randomizzato Italiano, Nessun Beneficio Dal Tocilizumab. Available online: https://www.aifa.gov.it/-/covid-19-studio-randomizzato-italiano-nessun-beneficio-dal-tocilizumab (accessed on 23 June 2020).
- Le, T.-T.T.; Berg, N.K.; Harting, M.T.; Li, X.; Eltzschig, H.K.; Yuan, X. Purinergic Signaling in Pulmonary Inflammation. Front. Immunol. 2019, 10, 1633. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.N.; Batra, V.K. Lipopolysaccharide binds to and activates A1 adenosine receptors on human pulmonary artery endothelial cells. J. Endotoxin Res. 2002, 8, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, M.; Eckle, T.; Eltzschig, H.K. Selective Deletion of the A1 Adenosine Receptor Abolishes Heart-Rate Slowing Effects of Intravascular Adenosine In Vivo. PLoS ONE 2009, 4, e6784. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion—from mechanism to translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef] [Green Version]
- Hart, M.L.; Gorzolla, I.C.; Schittenhelm, J.; Robson, S.C.; Eltzschig, H.K. SP1-dependent induction of CD39 facilitates hepatic ischemic preconditioning. J. Immunol. 2010, 184, 4017–4024. [Google Scholar] [CrossRef] [Green Version]
- Morote–Garcia, J.C.; Rosenberger, P.; Nivillac, N.M.; Coe, I.R.; Eltzschig, H.K. Hypoxia-Inducible Factor–Dependent Repression of Equilibrative Nucleoside Transporter 2 Attenuates Mucosal Inflammation During Intestinal Hypoxia. Gastroenterology 2009, 136, 607–618. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Faigle, M.; Knapp, S.; Karhausen, J.; Ibla, J.; Rosenberger, P.; Odegard, K.C.; Laussen, P.C.; Thompson, L.F.; Colgan, S.P. Endothelial catabolism of extracellular adenosine during hypoxia: The role of surface adenosine deaminase and CD26. Blood 2006, 108, 1602–1610. [Google Scholar] [CrossRef]
- Morote-Garcia, J.C.; Rosenberger, P.; Kuhlicke, J.; Eltzschig, H.K. HIF-1–dependent repression of adenosine kinase attenuates hypoxia-induced vascular leak. Blood 2008, 111, 5571–5580. [Google Scholar] [CrossRef] [Green Version]
- Newton, A.C.; Bootman, M.D.; Scott, J.D. Second Messengers. Cold Spring Harb. Perspect. Boil. 2016, 8, a005926. [Google Scholar] [CrossRef] [PubMed]
- Cronstein, B.N. Adenosine, an endogenous anti-inflammatory agent. J. Appl. Physiol. 1994, 76, 5–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linden, J. Molecularapproach toadenosinereceptors: Receptor-Mediated Mechanisms of Tissue Protection. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Haselton, F.R.; Alexander, J.S.; Mueller, S.N. Adenosine decreases permeability of in vitro endothelial monolayers. J. Appl. Physiol. 1993, 74, 1581–1590. [Google Scholar] [CrossRef]
- Haskó, G. Adenosine: An endogenous regulator of innate immunity. Trends Immunol. 2004, 25, 33–39. [Google Scholar] [CrossRef]
- Salvatore, C.A.; Jacobson, M.A.; Taylor, H.E.; Linden, J.; Johnson, R.G. Molecular cloning and characterization of the human A3 adenosine receptor. Proc. Natl. Acad. Sci. USA 1993, 90, 10365–10369. [Google Scholar] [CrossRef] [Green Version]
- Chunn, J.L.; Young, H.W.J.; Banerjee, S.K.; Colasurdo, G.N.; Blackburn, M. Adenosine-dependent airway inflammation and hyperresponsiveness in partially adenosine deaminase-deficient mice. J. Immunol. 2001, 167, 4676–4685. [Google Scholar] [CrossRef] [Green Version]
- Blackburn, M.R.; Lee, C.G.; Young, H.W.; Zhu, Z.; Chunn, J.L.; Kang, M.J.; Banerjee, S.K.; Elias, J.A. Adenosine mediates IL-13-induced inflammation and remodeling in the lung and interacts in an IL-13-adenosine amplification pathway. J Clin Invest 2003, 112, 332–344. [Google Scholar] [CrossRef] [Green Version]
- Adkins, W.K.; Barnard, J.W.; Moore, T.M.; Allison, R.C.; Prasad, V.R.; Taylor, A.E. Adenosine prevents PMA-induced lung injury via an A2 receptor mechanism. J. Appl. Physiol. 1993, 74, 982–988. [Google Scholar] [CrossRef]
- Yaar, R.; Jones, M.R.; Chen, J.-F.; Ravid, K. Animal models for the study of adenosine receptor function. J. Cell. Physiol. 2004, 202, 9–20. [Google Scholar] [CrossRef]
- Murphree, L.J.; Sullivan, G.W.; Marshall, M.A.; Linden, J. Lipopolysaccharide rapidly modifies adenosine receptor transcripts in murine and human macrophages: Role of NF-κB in A2Aadenosine receptor induction. Biochem. J. 2005, 391, 575–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohta, A.; Sitkovsky, M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature 2001, 414, 916–920. [Google Scholar] [CrossRef] [PubMed]
- Haskó, G.; Pacher, P. A2Areceptors in inflammation and injury: Lessons learned from transgenic animals. J. Leukoc. Boil. 2007, 83, 447–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonneau, O.; Wyss, D.; Ferretti, S.; Blaydon, C.; Stevenson, C.S.; Trifilieff, A. Effect of adenosine A2A receptor activation in murine models of respiratory disorders. Am. J. Physiol. Cell. Mol. Physiol. 2006, 290, L1036–L1043. [Google Scholar] [CrossRef] [Green Version]
- Reutershan, J.; Cagnina, R.E.; Chang, D.; Linden, J.; Ley, K. Therapeutic anti-inflammatory effects of myeloid cell adenosine receptor A2a stimulation in lipopolysaccharide-induced lung injury. J. Immunol. 2007, 179, 1254–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fredholm, B.B. Adenosine, an endogenous distress signal, modulates tissue damage and repair. Cell Death Differ. 2007, 14, 1315–1323. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Zhang, Y.; Nguyen, H.G.; Koupenova, M.; Chauhan, A.K.; Makitalo, M.; Jones, M.R.; Hilaire, C.S.; Seldin, D.C.; Toselli, P.; et al. The A2B adenosine receptor protects against inflammation and excessive vascular adhesion. J. Clin. Investig. 2006, 116, 1913–1923. [Google Scholar] [CrossRef] [Green Version]
- Bouma, M.G.; Jeunhomme, T.M.; Boyle, D.L.; Dentener, M.A.; Voitenok, N.N.; Wildenberg, F.A.V.D.; A. Buurman, W. Adenosine inhibits neutrophil degranulation in activated human whole blood: Involvement of adenosine A2 and A3 receptors. J. Immunol. 1997, 158, 5400–5408. [Google Scholar]
- Avni, I.; Garzozi, H.J.; Barequet, I.S.; Segev, F.; Varssano, D.; Sartani, G.; Chetrit, N.; Bakshi, E.; Zadok, D.; Tomkins-Netzer, O.; et al. Treatment of Dry Eye Syndrome with Orally Administered CF101. Ophthalmology 2010, 117, 1287–1293. [Google Scholar] [CrossRef] [Green Version]
- Layland, J.; Carrick, D.; Lee, M.M.; Oldroyd, K.; Berry, C. Adenosine. JACC: Cardiovasc. Interv. 2014, 7, 581–591. [Google Scholar] [CrossRef] [Green Version]
- Schepp, C.P.; Reutershan, J. Bench-to-bedside review: Adenosine receptors—Promising targets in acute lung injury? Crit. Care 2008, 12, 226. [Google Scholar] [CrossRef] [Green Version]
- Antonioli, L.; Blandizzi, C.; Pacher, P.; Haskó, G. The Purinergic System as a Pharmacological Target for the Treatment of Immune-Mediated Inflammatory Diseases. Pharmacol. Rev. 2019, 71, 345–382. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Mallampalli, R.K. The Role of Surfactant in Lung Disease and Host Defense against Pulmonary Infections. Ann. Am. Thorac. Soc. 2015, 12, 765–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marks, L.R.; Davidson, B.A.; Knight, P.R.; Hakansson, A.P. Interkingdom Signaling Induces Streptococcus pneumoniae Biofilm Dispersion and Transition from Asymptomatic Colonization to Disease. mBio 2013, 4, e00438-13. [Google Scholar] [CrossRef] [Green Version]
- Reutershan, J.; Vollmer, I.; Stark, S.; Wagner, R.; Ngamsri, K.-C.; Eltzschig, H.K. Adenosine and inflammation: CD39 and CD73 are critical mediators in LPS-induced PMN trafficking into the lungs. FASEB J. 2008, 23, 473–482. [Google Scholar] [CrossRef]
- Sharma, A.K.; Linden, J.; Kron, I.L.; Laubach, V.E. Protection from pulmonary ischemia-reperfusion injury by adenosine A2A receptor activation. Respir. Res. 2009, 10, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzales, J.N.; Gorshkov, B.; Varn, M.N.; Zemskova, M.A.; Zemskov, E.A.; Sridhar, S.; Lucas, R.; Verin, A.D. Protective effect of adenosine receptors against lipopolysaccharide-induced acute lung injury. Am. J. Physiol. Cell. Mol. Physiol. 2014, 306, L497–L507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, X.; Hu, J.-L.; Li, J.; Zhao, L.; Zhang, Y.; Zeng, Y.-J.; Dai, S.-S.; He, F.-T. A feedback loop in PPARγ–adenosine A2A receptor signaling inhibits inflammation and attenuates lung damages in a mouse model of LPS-induced acute lung injury. Cell. Signal. 2013, 25, 1913–1923. [Google Scholar] [CrossRef]
- Friebe, D.; Yang, T.; Schmidt, T.; Borg, N.; Steckel, B.; Ding, Z.; Schrader, J. Purinergic Signaling on Leukocytes Infiltrating the LPS-Injured Lung. PLoS ONE 2014, 9, e95382. [Google Scholar] [CrossRef]
- Hoegl, S.; Brodsky, K.S.; Blackburn, M.; Karmouty-Quintana, H.; Zwissler, B.; Eltzschig, H.K. Alveolar Epithelial A2B Adenosine Receptors in Pulmonary Protection during Acute Lung Injury. J. Immunol. 2015, 195, 1815–1824. [Google Scholar] [CrossRef] [Green Version]
- Koscsó, B.; Trepakov, A.; Csóka, B.; Németh, Z.H.; Pacher, P.; Eltzschig, H.K.; Haskó, G. Stimulation of A2B adenosine receptors protects against trauma–hemorrhagic shock-induced lung injury. Purinergic Signal. 2013, 9, 427–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Yang, D.; Carroll, S.H.; Eltzschig, H.K.; Ravid, K. Activation of the macrophage A2b adenosine receptor regulates tumor necrosis factor-alpha levels following vascular injury. Exp. Hematol. 2009, 37, 533–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correale, P.; Caracciolo, M.; Bilotta, F.; Conte, M.; Cuzzola, M.; Falcone, C.; Mangano, C.; Falzea, A.C.; Iuliano, E.; Morabito, A.; et al. Therapeutic effects of Adenosine in high flow 21% oxygen aereosol in patients with Covid19-Pneumonia. PLoS ONE 2020, in press. [Google Scholar]
- Aggarwal, N.R.; D’Alessio, F.R.; Eto, Y.; Chau, E.; Avalos, C.; Waickman, A.T.; Garibaldi, B.T.; Mock, J.R.; Files, D.C.; Sidhaye, V.K.; et al. Macrophage A2A Adenosinergic Receptor Modulates Oxygen-Induced Augmentation of Murine Lung Injury. Am. J. Respir. Cell Mol. Boil. 2013, 48, 635–646. [Google Scholar] [CrossRef]
- Jacobson, K.A.; Tosh, D.K.; Jain, S.; Gao, Z. Historical and Current Adenosine Receptor Agonists in Preclinical and Clinical Development. Front. Cell. Neurosci. 2019, 13, 124. [Google Scholar] [CrossRef] [Green Version]
- Spicuzza, L.; Di Maria, G.; Polosa, R. Adenosine in the airways: Implications and applications. Eur. J. Pharmacol. 2006, 533, 77–88. [Google Scholar] [CrossRef]
- Holgate, S.T.; Cushley, M.J.; Mann, J.S.; Hughes, P.; Church, M.K. The action of purines on human airways. Arch. Int. de Pharmacodyn. et de Ther. 1986, 280, 240–252. [Google Scholar]
- Cushley, M.; Tattersfield, A.; Holgate, S. Inhaled adenosine and guanosine on airway resistance in normal and asthmatic subjects. Br. J. Clin. Pharmacol. 2004, 58, S751–S755. [Google Scholar] [CrossRef]
- Van Der Wiel, E.; Lexmond, A.J.; Berge, M.V.D.; Postma, D.S.; Hagedoorn, P.; Frijlink, H.W.; Farenhorst, M.P.; De Boer, A.H.; Hacken, N.H.T.T. Targeting the small airways with dry powder adenosine: A challenging concept. Eur. Clin. Respir. J. 2017, 4, 1369328. [Google Scholar] [CrossRef]
- Belanger, M.J.; Hill, M.A.; Angelidi, A.M.; Dalamaga, M.; Sowers, J.R.; Mantzoros, C.S. Covid-19 and Disparities in Nutrition and Obesity. New Engl. J. Med. 2020, 383, e69. [Google Scholar] [CrossRef]
- Di Renzo, L.; Gualtieri, P.; Pivari, F.; Soldati, L.; Attinà, A.; Cinelli, G.; Leggeri, C.; Caparello, G.; Barrea, L.; Scerbo, F.; et al. Eating habits and lifestyle changes during COVID-19 lockdown: An Italian survey. J. Transl. Med. 2020, 18, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Romano, L.; Bilotta, F.; Dauri, M.; Macheda, S.; Pujia, A.; De Santis, G.L.; Tarsitano, M.G.; Merra, G.; Di Renzo, L.; Esposito, E.; et al. Short Report—Medical nutrition therapy for critically ill patients with COVID-19. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 4035–4039. [Google Scholar] [PubMed]
- Gualtieri, P.; Falcone, C.; Romano, L.; Macheda, S.; Correale, P.; Arciello, P.; Polimeni, N.; De Lorenzo, A. Body Composition Findings by Computed Tomography in SARS-CoV-2 Patients: Increased Risk of Muscle Wasting in Obesity. Int. J. Mol. Sci. 2020, 21, 4670. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Parameters | Time | ||
|---|---|---|---|
| Admission | Baseline | 120 h | |
| RBC (x106/μL) | 5.12 | 4.8 | 4.54 |
| HGB (g/dL) | 16.2 | 14.8 | 13.8 |
| PLT (x103/μL) | 132 | 301 | 185 |
| WBC (x103/μL) | 2.74 | 4.32 | 3.33 |
| NEU (x103/μL) | 2.14 | 2.93 | 1.85 |
| LYMPH (x103/μL) | 0,39 | 0.9 | 0.95 |
| CRP (μg/dL) | 52.4 | 3.14 | 3.14 |
| D-DIMER (µg/L) | 220 | 290 | 50 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Falcone, C.; Caracciolo, M.; Correale, P.; Macheda, S.; Vadalà, E.G.; La Scala, S.; Tescione, M.; Danieli, R.; Ferrarelli, A.; Tarsitano, M.G.; et al. Can Adenosine Fight COVID-19 Acute Respiratory Distress Syndrome? J. Clin. Med. 2020, 9, 3045. https://doi.org/10.3390/jcm9093045
Falcone C, Caracciolo M, Correale P, Macheda S, Vadalà EG, La Scala S, Tescione M, Danieli R, Ferrarelli A, Tarsitano MG, et al. Can Adenosine Fight COVID-19 Acute Respiratory Distress Syndrome? Journal of Clinical Medicine. 2020; 9(9):3045. https://doi.org/10.3390/jcm9093045
Chicago/Turabian StyleFalcone, Carmela, Massimo Caracciolo, Pierpaolo Correale, Sebastiano Macheda, Eugenio Giuseppe Vadalà, Stefano La Scala, Marco Tescione, Roberta Danieli, Anna Ferrarelli, Maria Grazia Tarsitano, and et al. 2020. "Can Adenosine Fight COVID-19 Acute Respiratory Distress Syndrome?" Journal of Clinical Medicine 9, no. 9: 3045. https://doi.org/10.3390/jcm9093045