Identification of Lead Compounds against Scm (fms10) in Enterococcus faecium Using Computer Aided Drug Designing


 ,
,  ,
,
Abstract
:1. Introduction
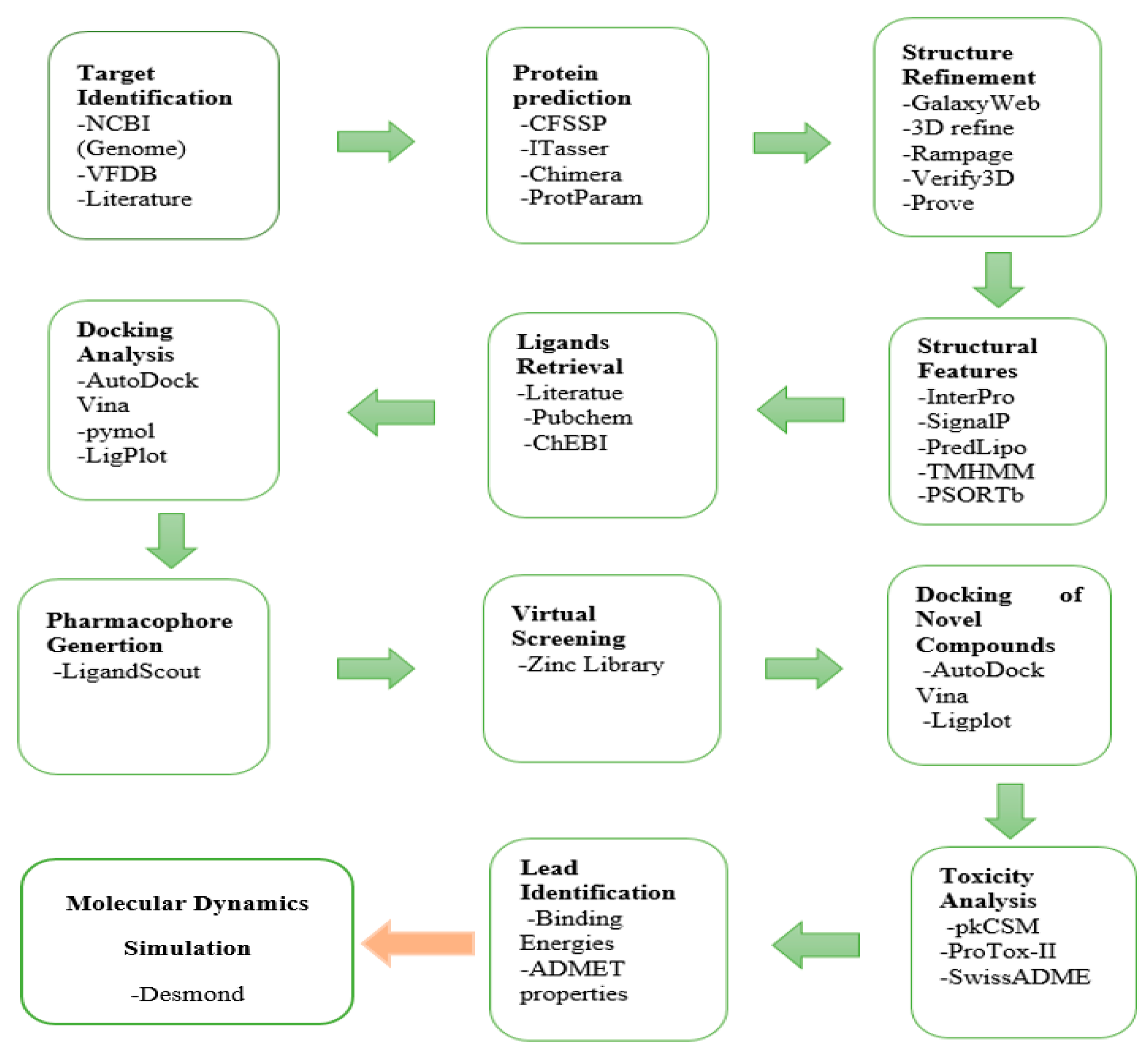
2. Materials and Methods
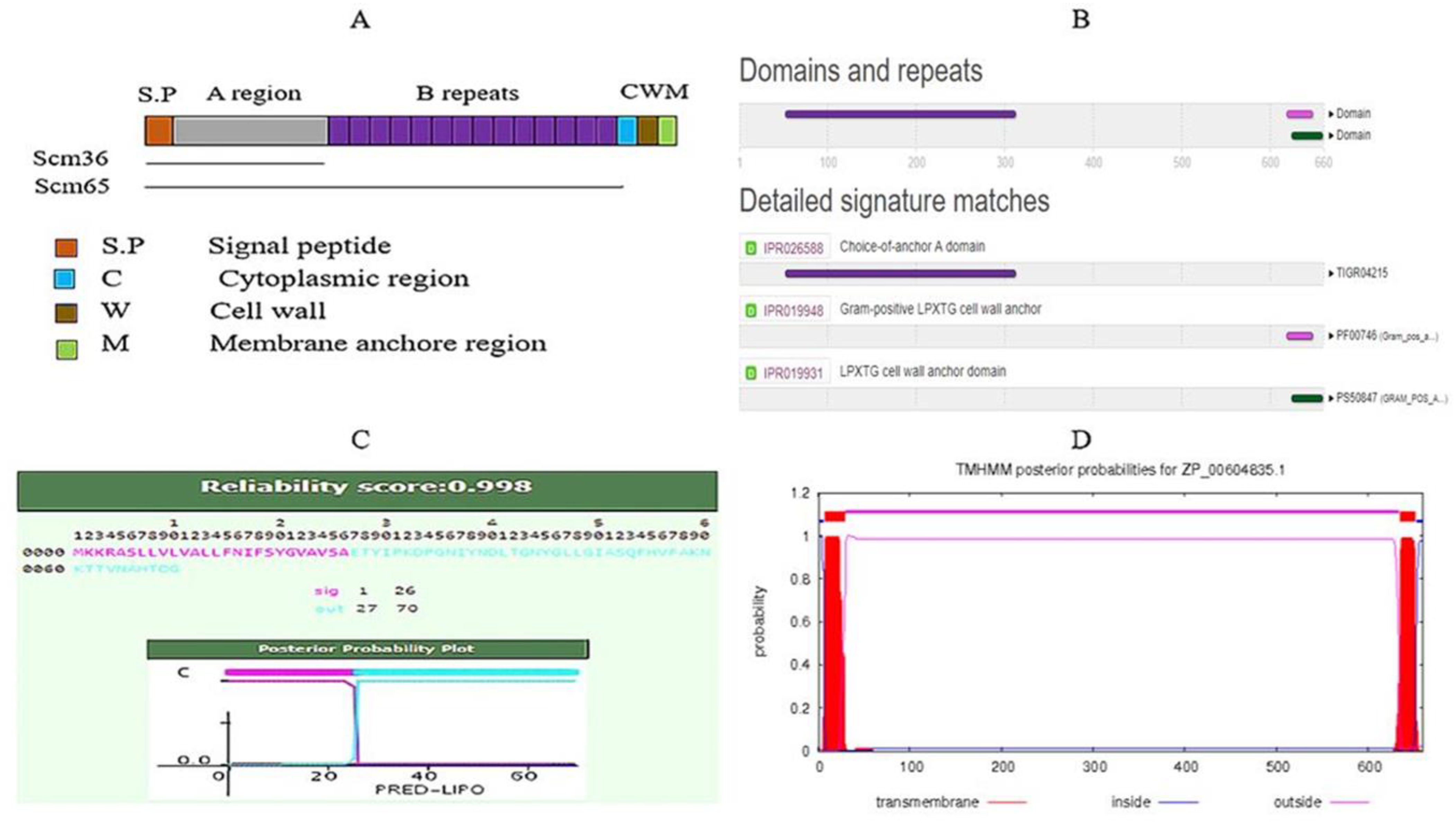
2.1. Target Identification
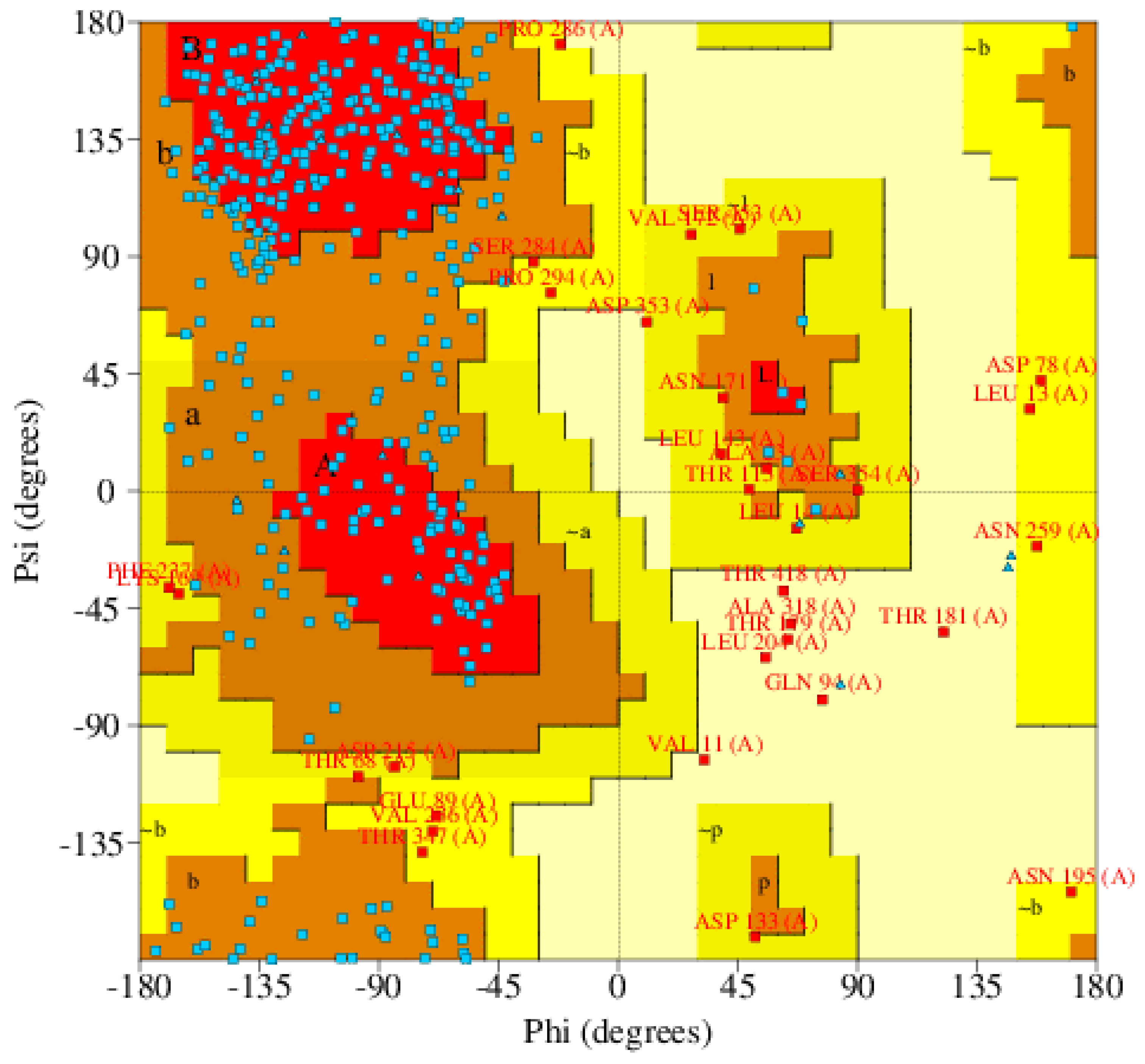
2.2. Protein Selection and Structural Refinement
2.3. Protein Properties
2.4. Selection and Retrieval of Ligands
2.5. Docking Analysis
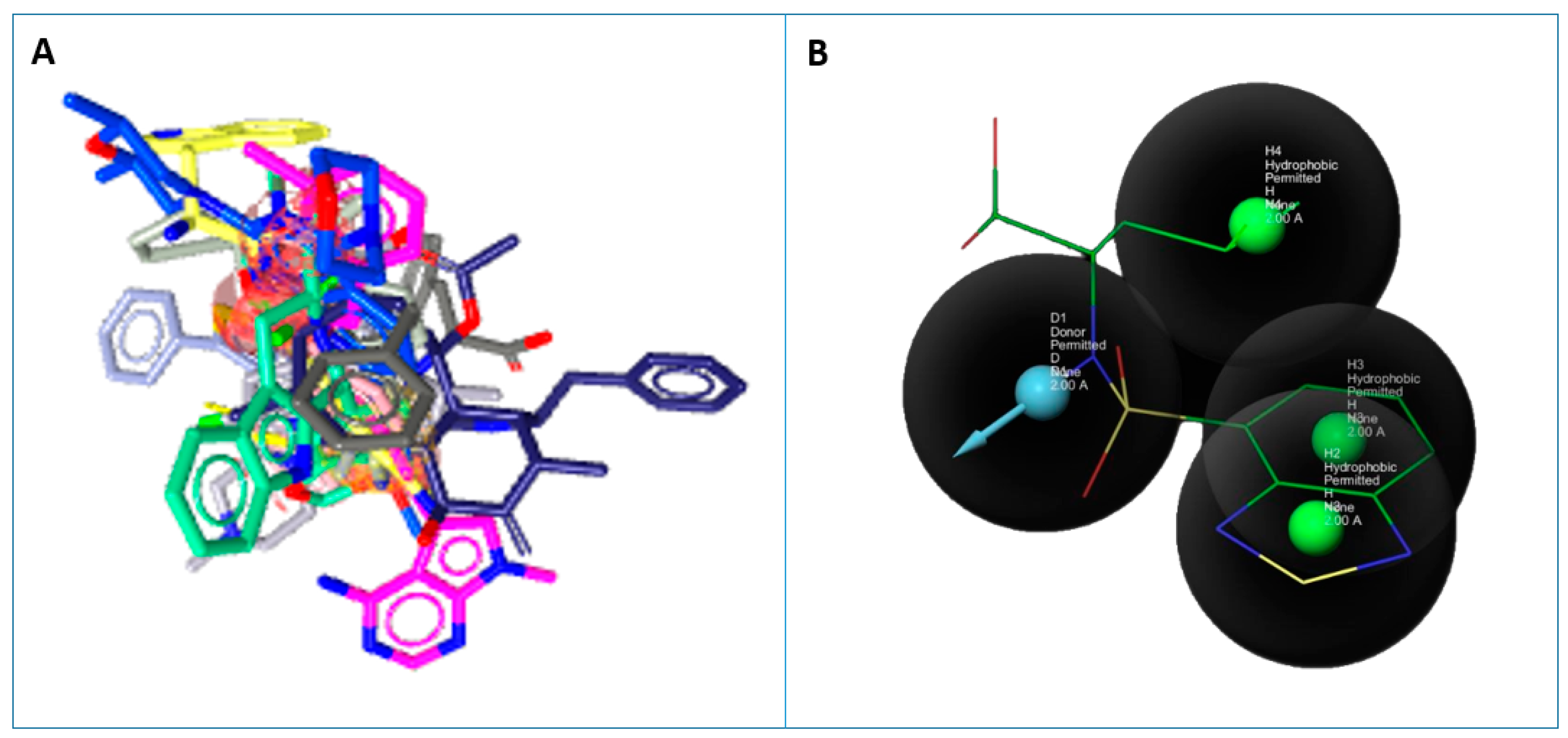
2.6. Pharmacophore Generation
2.7. Docking of Novel Compounds
2.8. Toxicity Analysis
2.9. Lead Identification
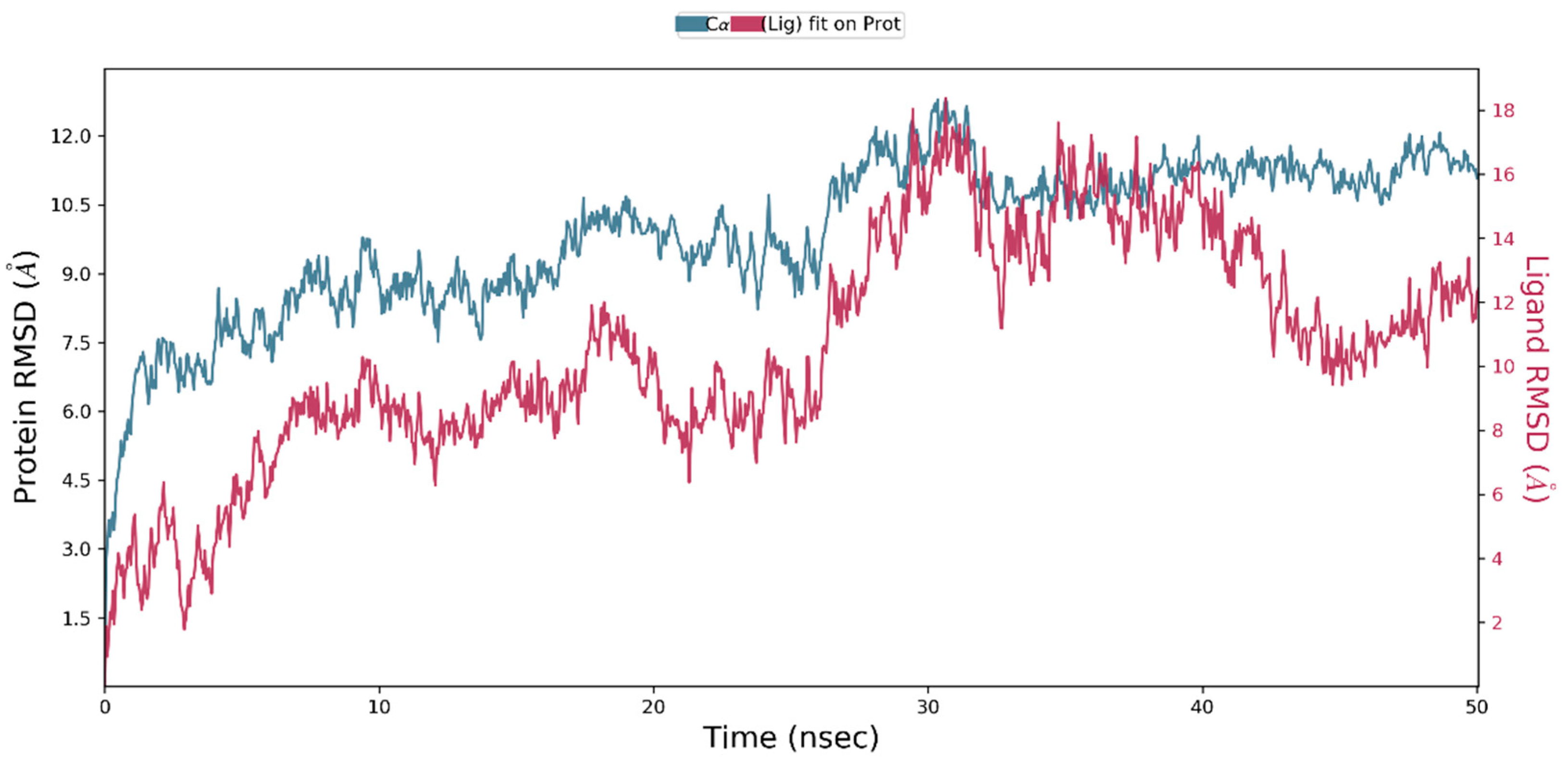
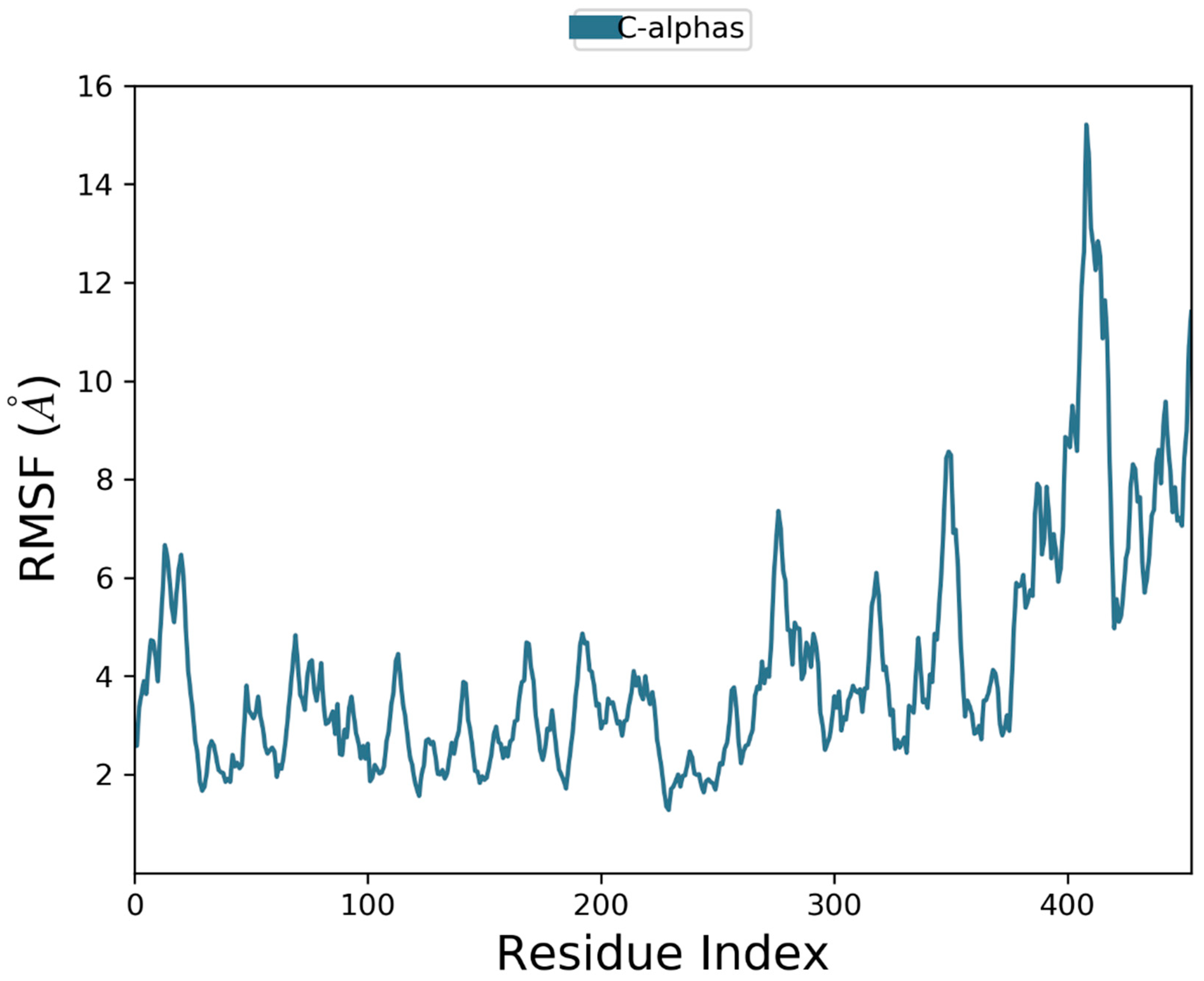
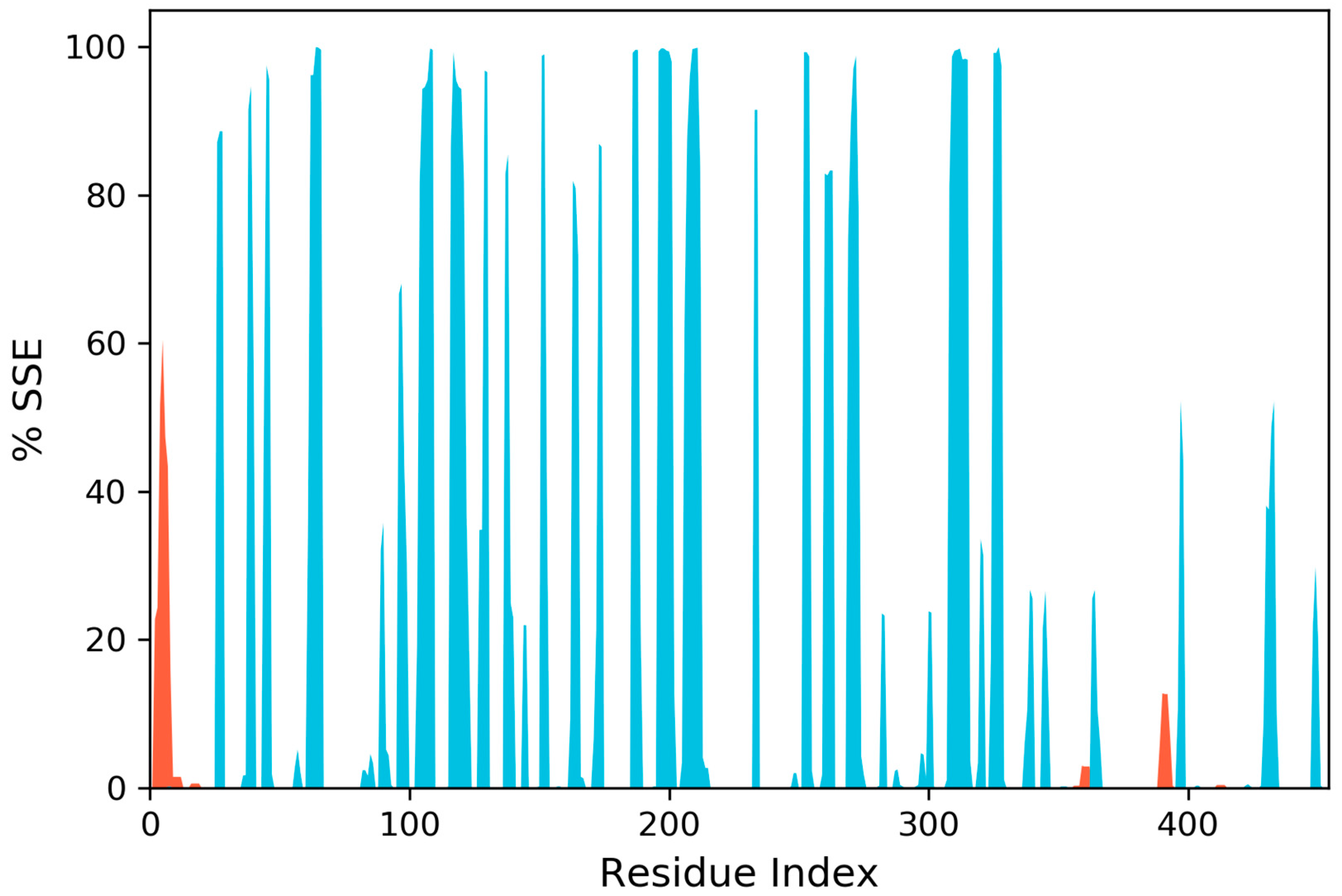
2.10. Molecular Dynamics Simulation
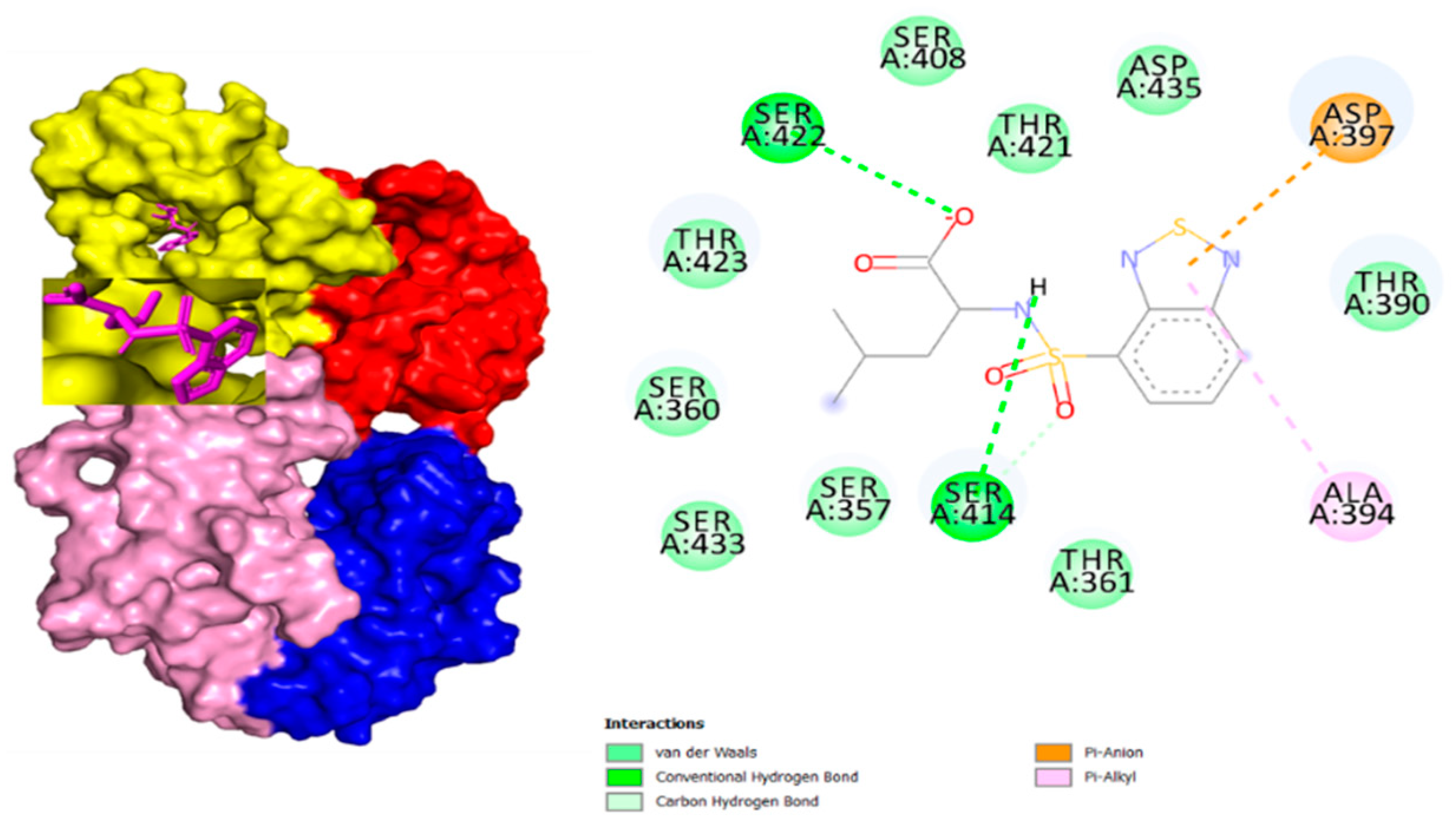
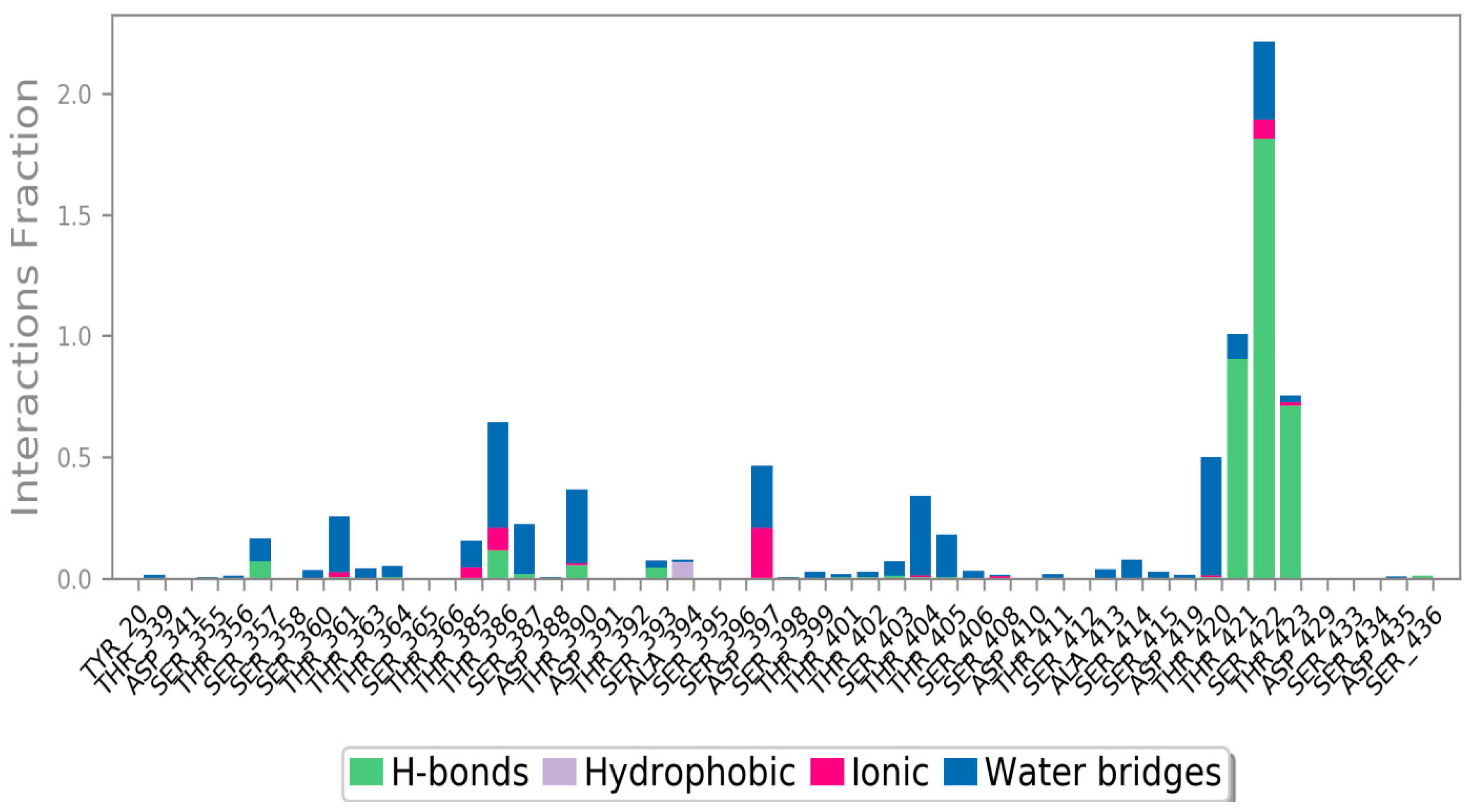
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Dubin, K.; Pamer, E.G. Enterococci and Their Interactions with the Intestinal Microbiome. Microbiol. Spectr. 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Huycke, M.M.; Sahm, D.F.; Gilmore, M.S. Multiple-drug resistant enterococci: The nature of the problem and an agenda for the future. Emerg Infect. Dis 1998, 4, 239–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gülhan, T.; Aksakal, A.; Ekin, I.H.; Savaşan, S.; Boynukara, B. Virulence factors of Enterococcus faecium and Enterococcus faecalis strains isolated from humans and pets. Turk. J. Vet. Anim. Sci. 2007, 30, 477–482. [Google Scholar]
- Bharti, A.K.; Farooq, U.; Singh, S.; Kaur, N.; Ahmed, R.; Singh, K. Incidence of Enterococcal Urinary Tract Infection and it’s Sensitivity Pattern among Patients Attending Teerthanker Mahaveer Medical College and Research Centre, Moradabad, India. Int. J. Sci. Study 2016, 3, 112–116. [Google Scholar]
- Vankerckhoven, V.; Van Autgaerden, T.; Vael, C.; Lammens, C.; Chapelle, S.; Rossi, R.; Jabes, D.; Goossens, H. Development of a multiplex PCR for the detection of asa1, gelE, cylA, esp, and hyl genes in enterococci and survey for virulence determinants among European hospital isolates of Enterococcus faecium. J. Clin. Microbiol. 2004, 42, 4473–4479. [Google Scholar] [CrossRef] [Green Version]
- Munita, J.M.; Arias, C.A. Mechanisms of Antibiotic Resistance. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [Green Version]
- Enterococcus Faecalis and Faecium. Available online: https://www.msdsonline.com/resources/sds-resources/free-safety-data-sheet-index/enterococcus-faecalis-and-faecium/ (accessed on 7 July 2019).
- Bortolaia, V.; Espinosa-Gongora, C.; Guardabassi, L. Human health risks associated with antimicrobial-resistant enterococci and Staphylococcus aureus on poultry meat. Clin. Microbiol. Infect. 2016, 22, 130–140. [Google Scholar] [CrossRef] [Green Version]
- Arias, C.A.; Contreras, G.A.; Murray, B.E. Management of multidrug-resistant enterococcal infections. Clin. Microbiol. Infect. 2010, 16, 555–562. [Google Scholar] [CrossRef] [Green Version]
- Pradeepkiran, J.A.; Reddy, P.H. Structure Based Design and Molecular Docking Studies for Phosphorylated Tau Inhibitors in Alzheimer’s Disease. Cells 2019, 8, 260. [Google Scholar] [CrossRef] [Green Version]
- Recommendations for preventing the spread of vancomycin resistance: Recommendations of the Hospital Infection Control Practices Advisory Committee (HICPAC). Am. J. Infect. Control. 1995, 23, 87–94. [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sillanpaa, J.; Nallapareddy, S.R.; Prakash, V.P.; Qin, X.; Hook, M.; Weinstock, G.M.; Murray, B.E. Identification and phenotypic characterization of a second collagen adhesin, Scm, and genome-based identification and analysis of 13 other predicted MSCRAMMs, including four distinct pilus loci, in Enterococcus faecium. Microbiology 2008, 154, 3199–3211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Zheng, D.; Jin, Q.; Chen, L.; Yang, J. VFDB 2019: A comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 2019, 47, D687–D692. [Google Scholar] [CrossRef] [PubMed]
- UniProt, C. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef] [Green Version]
- Kumar, T.A. CFSSP: Chou and Fasman secondary structure prediction server. Wide Spectr. 2013, 1, 15–19. [Google Scholar]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [Green Version]
- Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Huang, C.C.; Ferrin, T.E. Tools for integrated sequence-structure analysis with UCSF Chimera. BMC Bioinform. 2006, 7, 339. [Google Scholar]
- Ko, J.; Park, H.; Heo, L.; Seok, C. GalaxyWEB server for protein structure prediction and refinement. Nucleic Acids Res. 2012, 40, W294–W297. [Google Scholar] [CrossRef]
- Bhattacharya, D.; Nowotny, J.; Cao, R.; Cheng, J. 3Drefine: An interactive web server for efficient protein structure refinement. Nucleic Acids Res. 2016, 44, W406–W409. [Google Scholar] [CrossRef]
- Ho, B.K.; Brasseur, R. The Ramachandran plots of glycine and pre-proline. BMC Struct. Biol. 2005, 5, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benkert, P.; Biasini, M.; Schwede, T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 2011, 27, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Pontius, J.; Richelle, J.; Wodak, S.J. Deviations from standard atomic volumes as a quality measure for protein crystal structures. J. Mol. Biol. 1996, 264, 121–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luthy, R.; Bowie, J.U.; Eisenberg, D. Assessment of protein models with three-dimensional profiles. Nature 1992, 356, 83–85. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, A.L.; Attwood, T.K.; Babbitt, P.C.; Blum, M.; Bork, P.; Bridge, A.; Brown, S.D.; Chang, H.Y.; El-Gebali, S.; Fraser, M.I.; et al. InterPro in 2019: Improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 2019, 47, D351–D360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagos, P.G.; Tsirigos, K.D.; Liakopoulos, T.D.; Hamodrakas, S.J. Prediction of lipoprotein signal peptides in Gram-positive bacteria with a Hidden Markov Model. J. Proteome Res. 2008, 7, 5082–5093. [Google Scholar] [CrossRef] [PubMed]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, N.Y.; Wagner, J.R.; Laird, M.R.; Melli, G.; Rey, S.; Lo, R.; Dao, P.; Sahinalp, S.C.; Ester, M.; Foster, L.J.; et al. PSORTb 3.0: Improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 2010, 26, 1608–1615. [Google Scholar] [CrossRef]
- Zhang, C.; Freddolino, P.L.; Zhang, Y. COFACTOR: Improved protein function prediction by combining structure, sequence and protein-protein interaction information. Nucleic Acids Res. 2017, 45, W291–W299. [Google Scholar] [CrossRef]
- Liang, J.; Edelsbrunner, H.; Woodward, C. Anatomy of protein pockets and cavities: Measurement of binding site geometry and implications for ligand design. Protein Sci. A Publ. Protein Soc. 1998, 7, 1884–1897. [Google Scholar] [CrossRef] [Green Version]
- Irwin, J.J.; Shoichet, B.K. ZINC—A free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Pradeepkiran, J.A.; Kumar, K.K.; Kumar, Y.N.; Bhaskar, M. Modeling, molecular dynamics, and docking assessment of transcription factor rho: A potential drug target in Brucella melitensis 16M. Drug Des. Devel. 2015, 9, 1897–1912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Molecular dynamics—Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing—SC’06, Tampa, FL, USA, 11–17 November 2006. [Google Scholar]
- Shivakumar, D.; Williams, J.; Wu, Y.; Damm, W.; Shelley, J.; Sherman, W. Prediction of Absolute Solvation Free Energies using Molecular Dynamics Free Energy Perturbation and the OPLS Force Field. J. Chem. Theory Comput. 2010, 6, 1509–1519. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| UniProt Ids | I3U5K9 |
|---|---|
| GalaxyWEB and 3Drefine | |
| GDT-HA | 0.9992 |
| RMSD | 0.448 |
| MolProbity | 3.054 |
| 3Drefine Score | 42,972.1 |
| Rama favored | 89.7 |
| Refinement Databases Score | |
| Qmean | −6.87 |
| Prove | 6.9 |
| Rampage (favored region) | 90.1 |
| Verify 3D | 60.70 |
| Subcellular Localization | |
| Cell Wall | 9.98 |
| Cytoplasmic Membrane | 0.01 |
| Extracellular | 0.001 |
| Cytoplasmic | 0.0001 |
| Final Prediction | Cell Wall |
| Ligand ID’s | Binding Energies | No. of H-Bonds | 2D Structures |
|---|---|---|---|
| 71940 | −10 | 1 |  |
| 85572 | −9.4 | 2 |  |
| 131686 | −9.3 | 1 |  |
| 139047 | −9.1 | 2 |  |
| 140296 | −9.1 | 1 |  |
| 63598 | −8.6 | 1 |  |
| 104872 | −8.2 | 1 |  |
| 137693 | −8.2 | 1 |  |
| 43541 | −8.1 | 1 |  |
| 529996 | −8.1 | 1 |  |
| Zinc IDs | Pharma-Cophore Score | Binding Affinity | HBA | HBD | Rings | RBs | M.W | logP | TPSA |
|---|---|---|---|---|---|---|---|---|---|
| zinc_5347917 | 43.62 | −8.8 | 5 | 2 | 4 | 8 | 406.39 | 1.43 | 124.88 |
| zinc_5223045 | 43.96 | −8.2 | 8 | 2 | 5 | 8 | 452.49 | 2.08 | 93.06 |
| zinc_12603547 | 43.66 | −7.8 | 6 | 5 | 3 | 9 | 417.44 | 0.86 | 119.92 |
| zinc_49590510 | 43.78 | −7.2 | 5 | 0 | 4 | 8 | 433.53 | 1.21 | 82.03 |
| zinc_77264335 | 43.71 | −7.2 | 5 | 3 | 3 | 7 | 493.71 | 4.19 | 102.76 |
| zinc_1440812 | 43.62 | −7.1 | 5 | 2 | 3 | 9 | 358.35 | 0.93 | 124.88 |
| zinc_72321048 | 43.62 | −7.1 | 5 | 3 | 3 | 6 | 362.4 | 1.83 | 104.73 |
| zinc_57855 | 43.67 | −7 | 4 | 1 | 3 | 4 | 270.28 | 2.21 | 55.76 |
| zinc_12603524 | 43.79 | −7 | 7 | 4 | 3 | 9 | 420.41 | 0.37 | 107.89 |
| zinc_35425432 | 43.62 | −6.7 | 7 | 1 | 4 | 9 | 382.44 | 0.95 | 86.4 |
| zinc_57662 | 43.89 | −6.6 | 3 | 0 | 3 | 5 | 282.29 | 3.53 | 48.67 |
| zinc_48942 | 43.78 | −6.4 | 6 | 1 | 2 | 8 | 328.39 | 0.14 | 112.08 |
| zinc_3897410 | 43.97 | −6.1 | 6 | 4 | 2 | 7 | 268.24 | −3.69 | 122.08 |
| zinc_12603765 | 43.77 | −5.6 | 6 | 5 | 2 | 8 | 350.35 | −0.97 | 107.89 |
| zinc_3839734 | 44.2 | −4.7 | 5 | 3 | 2 | 8 | 399.49 | 1.07 | 118.8 |
| Compounds ID | Zinc_48942 |
|---|---|
| Pharmacophore Score | 43.67 |
| Molecular Weight | 270.28 |
| H-bond Donor | 4 |
| H-bond Acceptor | 1 |
| Rotatable bonds | 4 |
| Rings | 3 |
| Toxicity | Non-Toxic |
| Carcinogenetic | Non-Carcinogen |
| Binding Energy | −7 |
| No. of H-Bonds | 2 |
| Interacting Residues | NDI-His67:O15; N-Ala66:O15 |
| Distance | 2.99 2.89 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rasheed, M.A.; Iqbal, M.N.; Saddick, S.; Ali, I.; Khan, F.S.; Kanwal, S.; Ahmed, D.; Ibrahim, M.; Afzal, U.; Awais, M. Identification of Lead Compounds against Scm (fms10) in Enterococcus faecium Using Computer Aided Drug Designing. Life 2021, 11, 77. https://doi.org/10.3390/life11020077
Rasheed MA, Iqbal MN, Saddick S, Ali I, Khan FS, Kanwal S, Ahmed D, Ibrahim M, Afzal U, Awais M. Identification of Lead Compounds against Scm (fms10) in Enterococcus faecium Using Computer Aided Drug Designing. Life. 2021; 11(2):77. https://doi.org/10.3390/life11020077
Chicago/Turabian StyleRasheed, Muhammad Asif, Muhammad Nasir Iqbal, Salina Saddick, Iqra Ali, Falak Sher Khan, Sumaira Kanwal, Dawood Ahmed, Muhammad Ibrahim, Umara Afzal, and Muhammad Awais. 2021. "Identification of Lead Compounds against Scm (fms10) in Enterococcus faecium Using Computer Aided Drug Designing" Life 11, no. 2: 77. https://doi.org/10.3390/life11020077