Study of PtOx/TiO2 Photocatalysts in the Photocatalytic Reforming of Glycerol: The Role of Co-Catalyst Formation

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
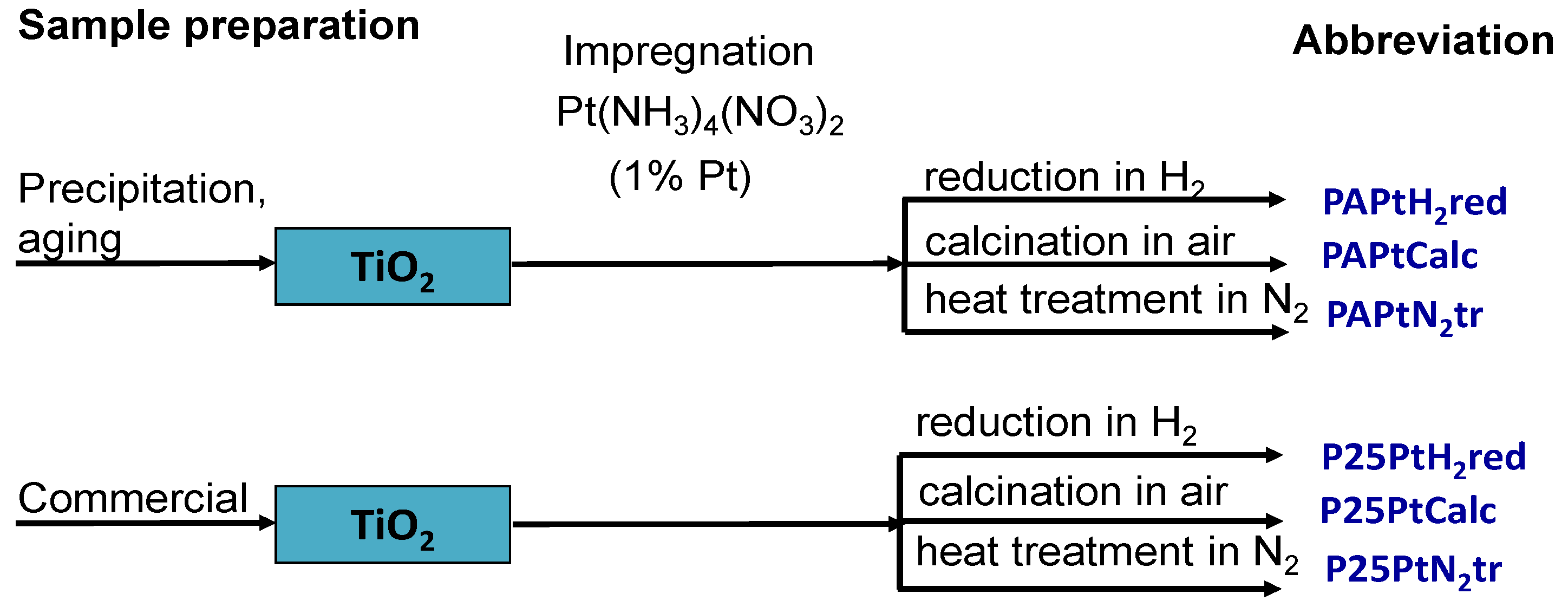
2.2. Synthesis of Photocatalysts
2.3. Photocatalytic Hydrogen Generation
2.4. Chemico-Physical Characterization of Photocatalysts
3. Results
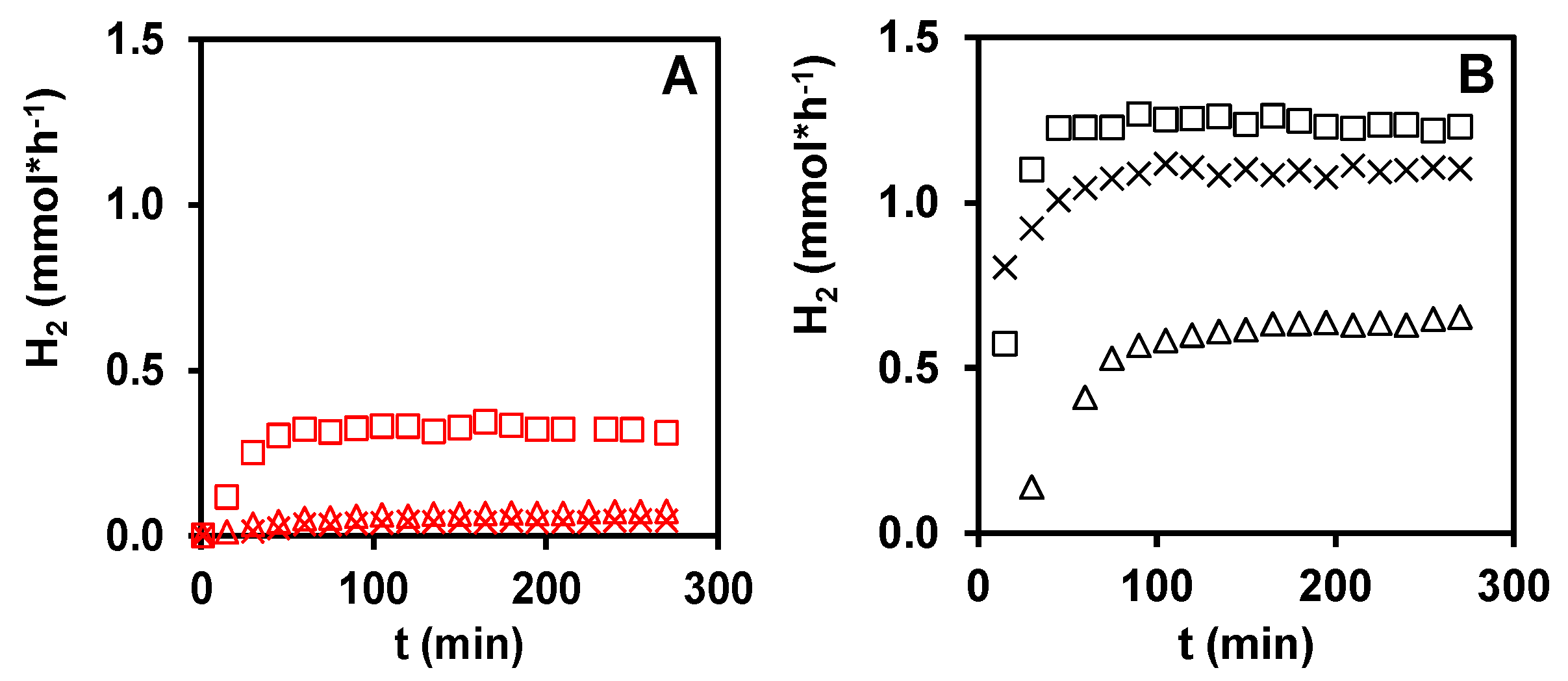
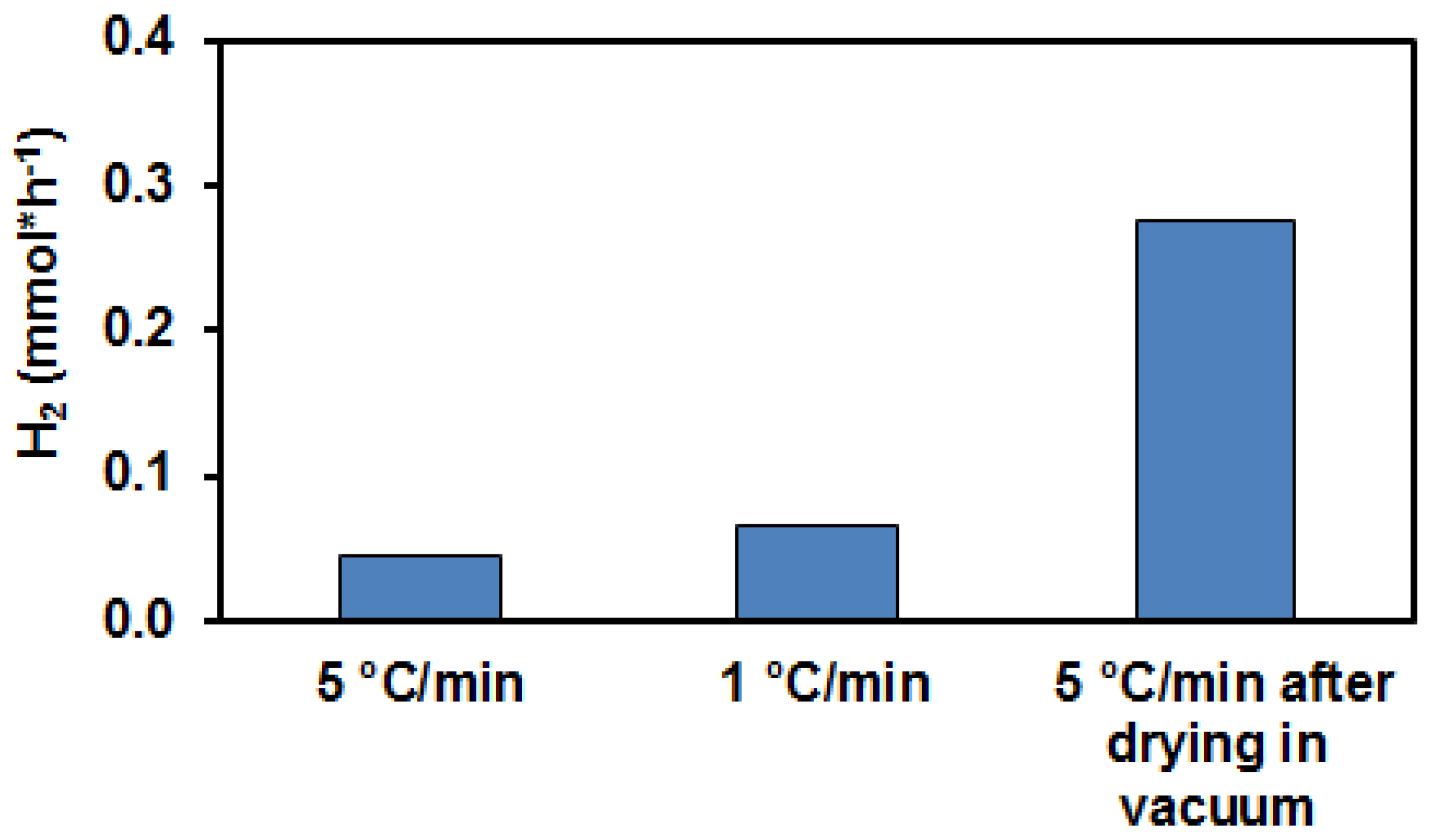
3.1. Photocatalytic Hydrogen Production from Glycerol over PtOx/TiO2 Catalyst Systems
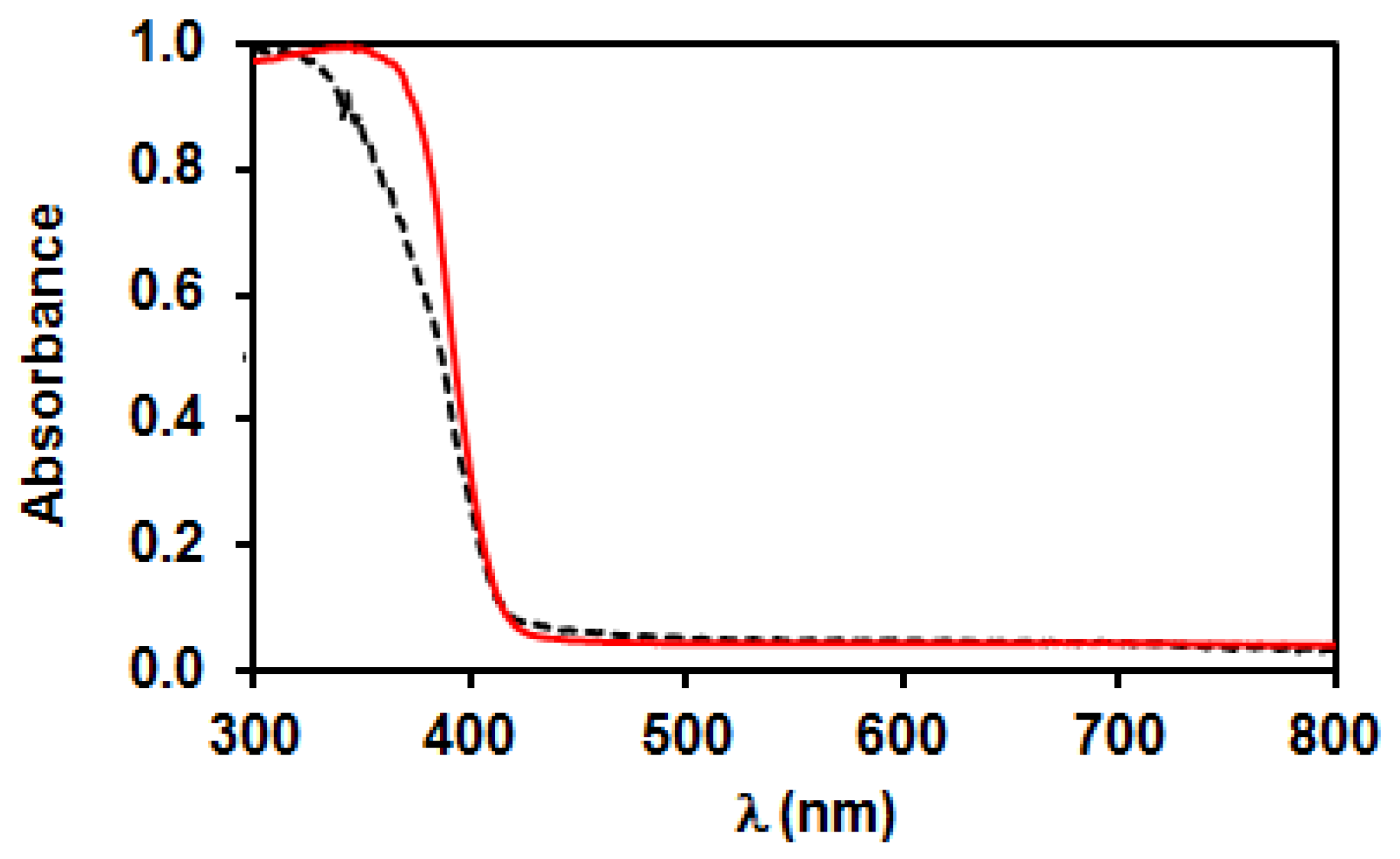
3.2. Characterization of Bare TiO2
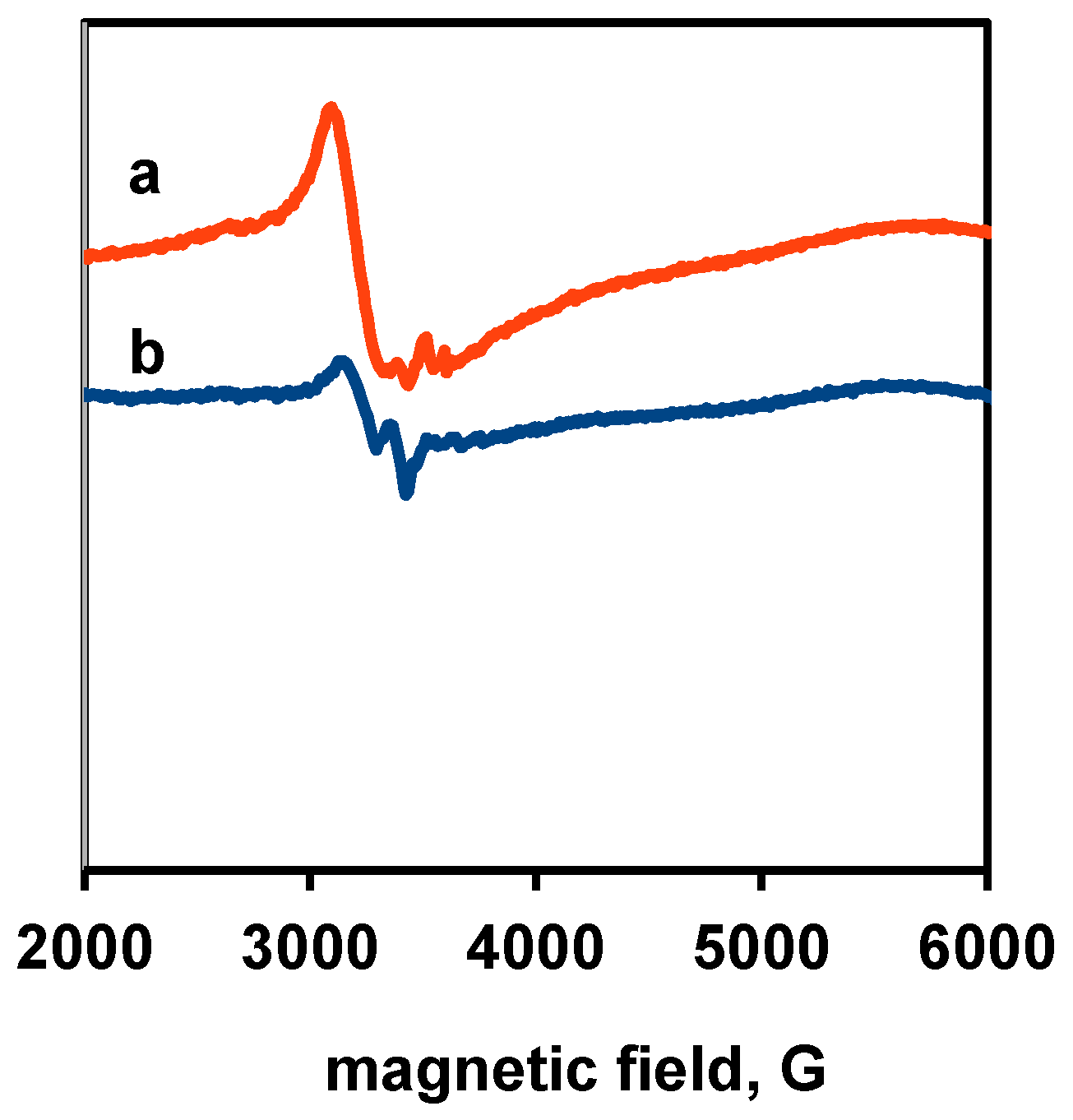
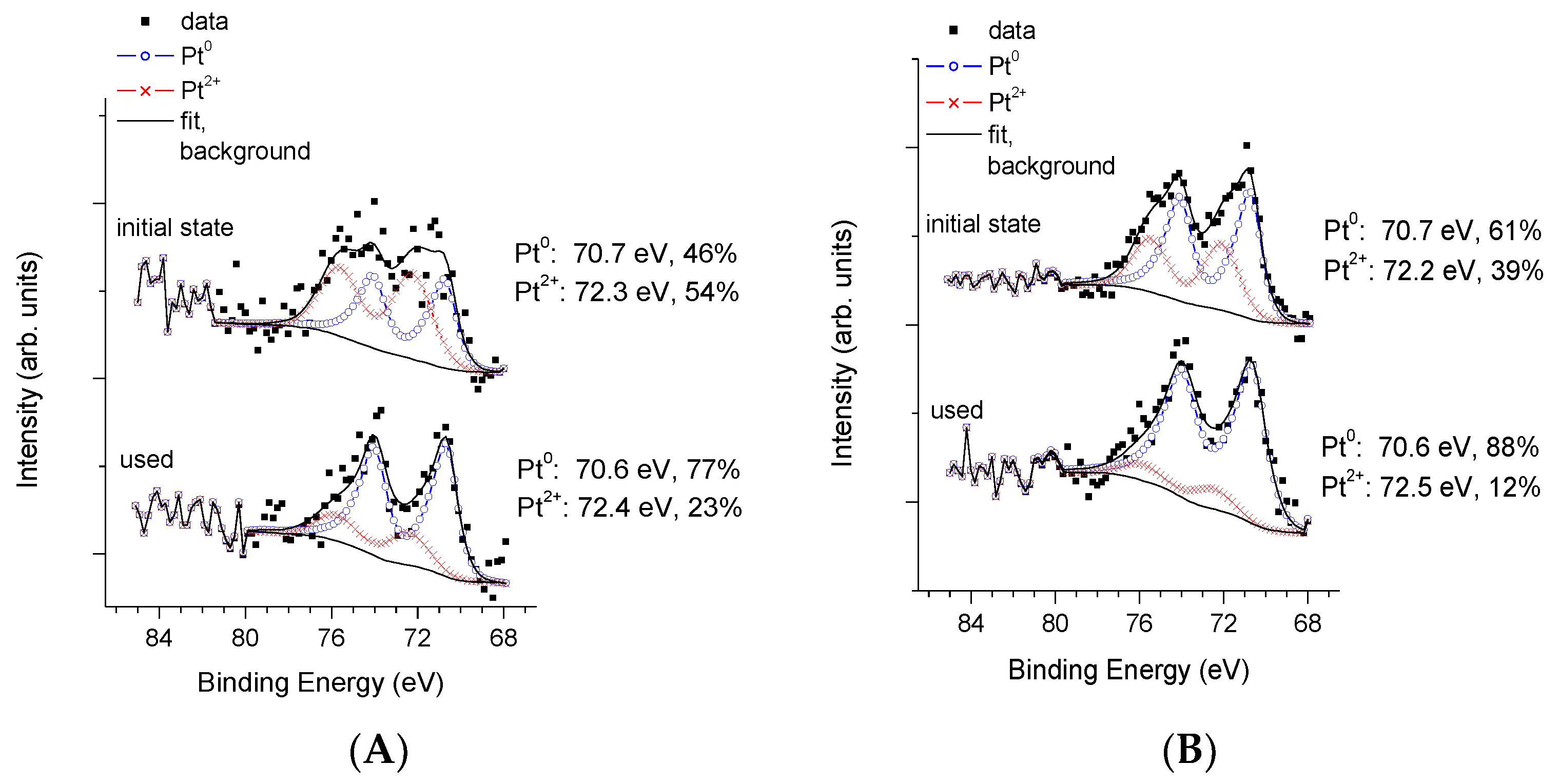
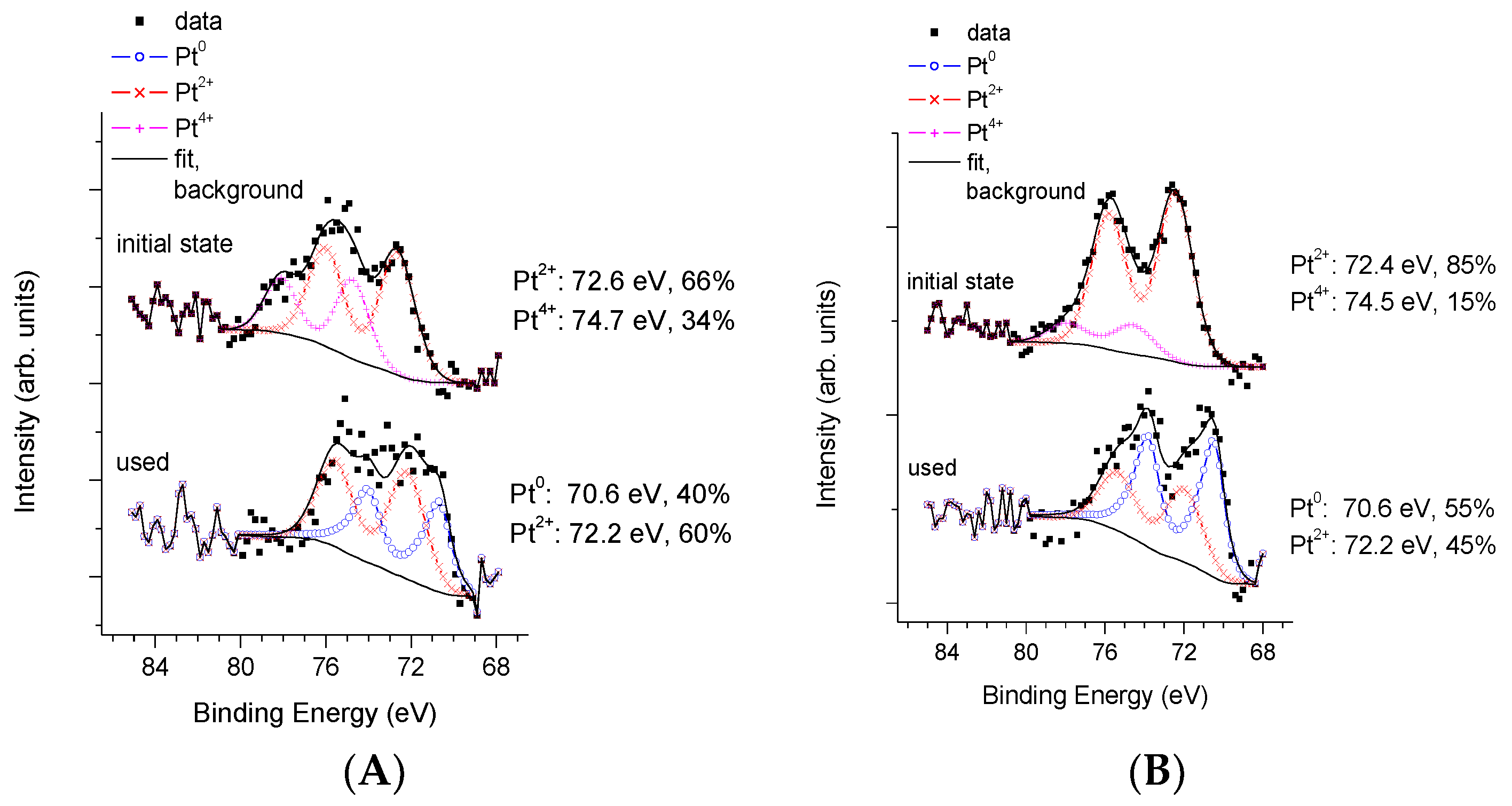
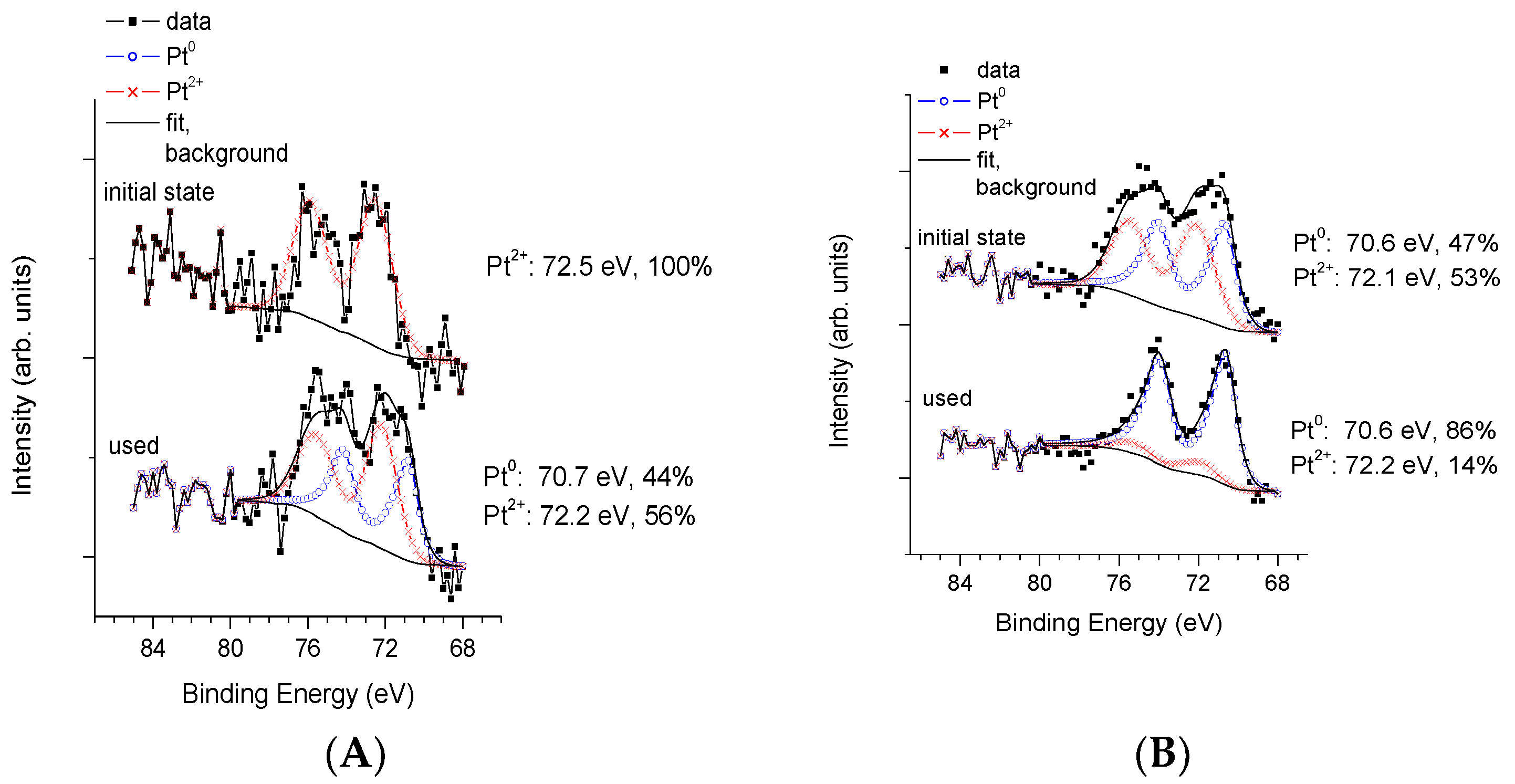
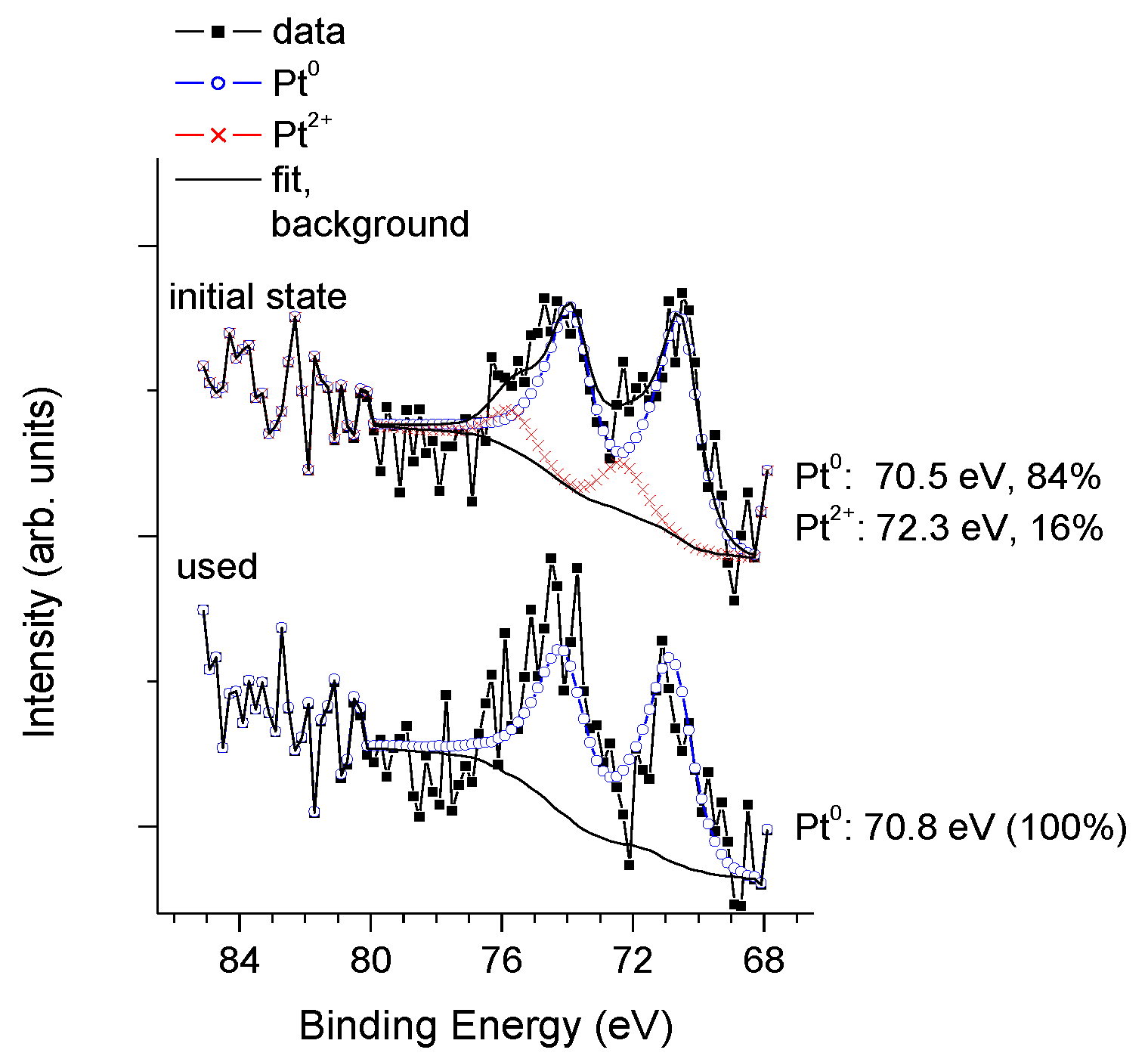
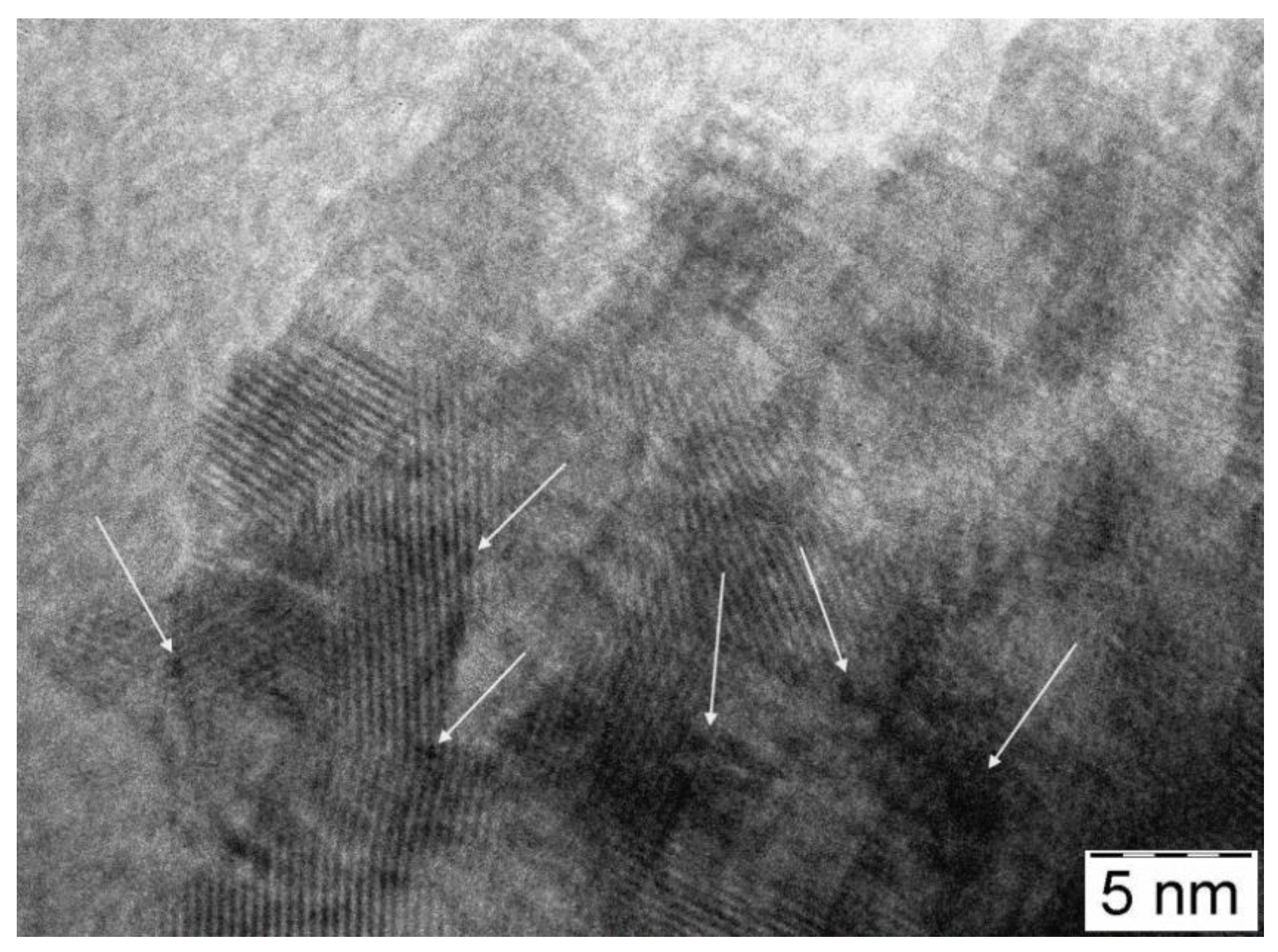
3.3. Physico-Chemical and Structural Characterization of Platinum Co-Catalysts
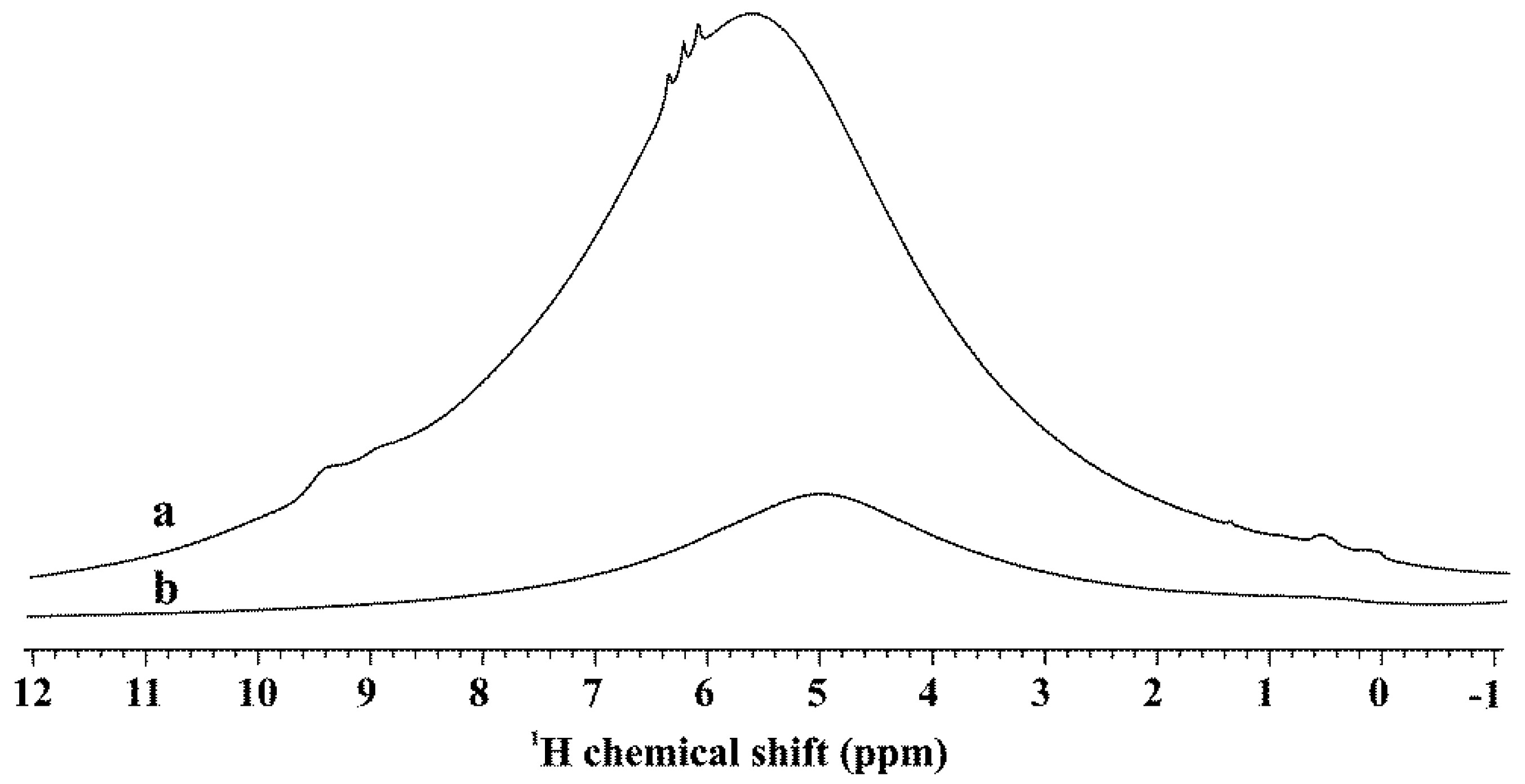
3.4. Effect of Water on the Autoreduction of Pt(NH3)4(NO3)2
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mazloomi, K.; Gomes, C. Hydrogen as an energy carrier: Prospects and challenges. Renew. Sustain. Energy Rev. 2012, 16, 3024–3033. [Google Scholar] [CrossRef]
- Cipriani, G.; Di Dio, V.; Genduso, F.; Cascia, D.; Liga, R.; Miceli, R.; Galluzzo, G.R. Perspective on hydrogen energy carrier and its automotive applications. Int. J. Hydrogen Energy 2014, 39, 8482–8494. [Google Scholar] [CrossRef]
- Sharma, S.; Ghoshal, S.K. Hydrogen the future transportation fuel: From production to applications. Renew. Sustain. Energy Rev. 2015, 43, 1151–1158. [Google Scholar] [CrossRef]
- Hwang, J.J. Review on development and demonstration of hydrogen fuel cell scooters. Renew. Sustain. Energy Rev. 2012, 16, 3803–3815. [Google Scholar] [CrossRef]
- Gurz, M.; Baltacioglu, E.; Hames, Y.; Kaya, K. The meeting of hydrogen and automotive: A review. Int. J. Hydrogen Energy 2017, 42, 23334–23346. [Google Scholar] [CrossRef]
- Behling, N.; Williams, M.C.; Manag, S. Fuel cells and the hydrogen revolution: Analysis of a strategic plan in Japan. Econ. Anal. Policy 2015, 48, 204–221. [Google Scholar] [CrossRef]
- Colón, G. Towards the hydrogen production by photocatalysis. Appl. Catal. A 2016, 518, 48–59. [Google Scholar] [CrossRef]
- Montini, T.; Monai, M.; Beltram, A.; Romero-Ocaña, I.; Fornasiero, P. H2 production by photocatalytic reforming of oxygenated compounds using TiO2-based materials. Mater. Sci. Semicond. Process. 2016, 42, 122–130. [Google Scholar] [CrossRef]
- Puga, A.V. Photocatalytic production of hydrogen from biomass-derived feedstocks. Coord. Chem. Rev. 2016, 315, 1–66. [Google Scholar] [CrossRef]
- Tran, N.H.; Kannangara, G.S.K. Conversion of glycerol to hydrogen rich gas. Chem. Soc. Rev. 2013, 42, 9454–9479. [Google Scholar] [CrossRef] [PubMed]
- Daskalaki, V.M.; Kondarides, D.I. Efficient production of hydrogen by photo-induced reforming of glycerol at ambient conditions. Catal. Today 2009, 144, 75–80. [Google Scholar] [CrossRef]
- Melián, E.P.; López, C.R.; Santiago, D.E.; Quesada-Cabrera, R.; Méndez, J.A.O.; Rodríguez, J.M.D.; Díaz, O.G. Study of the photocatalytic activity of Pt-modified commercial TiO2 for hydrogen production in the presence of common organic sacrificial agents. Appl. Catal. A 2016, 518, 189–197. [Google Scholar] [CrossRef]
- Gu, Q.; Long, J.; Fan, L.; Chen, L.; Zhao, L.; Lin, H.; Wang, X. Single-site Sn-grafted Ru/TiO2 photocatalysts for biomass reforming: Synergistic effect of dual co-catalysts and molecular mechanism. J. Catal. 2013, 303, 141–155. [Google Scholar] [CrossRef]
- Yang, J.; Wang, D.; Han, H.; Li, C. Roles of cocatalysts in photocatalysis and photoelectrocatalysis. Acc. Chem. Res. 2013, 46, 1900–1909. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.X.; Mao, S.S. Titanium dioxide nanomaterials: Synthesis, properties, modifications, and applications. Chem. Rev. 2007, 107, 2891–2959. [Google Scholar] [CrossRef] [PubMed]
- Ni, M.; Leung, M.K.H.; Leung, D.Y.C.; Sumathy, K. A review and recent developments in photocatalytic water-splitting using TiO2 for hydrogen production. Renew. Sustain. Energy Rev. 2007, 11, 401–425. [Google Scholar] [CrossRef]
- Fujishima, A.; Zhang, X.; Tryk, D.A. TiO2 photocatalysis and related surface phenomena. Surf. Sci. Rep. 2008, 36, 515–582. [Google Scholar] [CrossRef]
- Singh, R.; Dutta, S. A review on H2 production through photocatalytic reactions using TiO2/TiO2-assisted catalysts. Fuel 2018, 220, 607–620. [Google Scholar] [CrossRef]
- Akpan, U.G.; Hameed, B.H. The advancements in sol–gel method of doped-TiO2 photocatalysts. Appl. Catal. A 2010, 375, 1–11. [Google Scholar] [CrossRef]
- Wang, H.; Liu, P.; Cheng, X.; Shui, A.; Zeng, L. Effect of surfactants on synthesis of TiO2 nano-particles by homogeneous precipitation method. Powder Technol. 2008, 188, 52–54. [Google Scholar] [CrossRef]
- Chiarello, G.L.; Selli, E.; Forni, L. Photocatalytic hydrogen production over flame spray pyrolysis-synthesised TiO2 and Au/TiO2. Appl. Catal. B 2008, 84, 332–339. [Google Scholar] [CrossRef]
- Jiang, Y.; Scott, J.; Amal, R. Exploring the relationship between surface structure and photocatalytic activity of flame-made TiO2-based catalysts. Appl. Catal. B 2012, 126, 290–297. [Google Scholar] [CrossRef]
- Montes-Navajas, P.; Serra, M.; Corma, A.; Garcia, H. Contrasting photocatalytic activity of commercial TiO2 samples for hydrogen generation. Catal. Today 2014, 225, 52–54. [Google Scholar] [CrossRef]
- Chen, W.T.; Chan, A.; Al-Azri, Z.H.N.; Dosado, A.G.; Nadeem, M.A.; Sun-Waterhouse, D.; Idriss, H.; Waterhouse, G.I.N. Effect of TiO2 polymorph and alcohol sacrificial agent on the activity of Au/TiO2 photocatalysts for H2 production in alcohol–water mixtures. J. Catal. 2015, 329, 499–513. [Google Scholar] [CrossRef]
- Aprile, C.; Corma, A.; Garcia, H. Enhancement of the photocatalytic activity of TiO2 through spatial structuring and particle size control: From subnanometric to submillimetric length scale. Phys. Chem. Chem. Phys. 2008, 10, 769–783. [Google Scholar] [CrossRef] [PubMed]
- Pap, Z.; Karácsonyi, É.; Baia, L.; Pop, L.C.; Danciu, V.; Hernádi, K.; Mogyorósi, K.; Dombi, A. TiO2/WO3/Au/MWCNT composite materials for photocatalytic hydrogen production: Advantages and draw-backs. Phys. Status Solidi B 2012, 249, 2592–2595. [Google Scholar] [CrossRef]
- Sun, W.; Zhang, S.; Liu, Z.; Wang, C.; Mao, Z. Studies on the enhanced photocatalytic hydrogen evolution over Pt/PEG-modified TiO2 photocatalysts. Int. J. Hydrogen Energy 2008, 33, 1112–1117. [Google Scholar] [CrossRef]
- Beltram, A.; Romero-Ocaña, I.; Jaen, J.J.D.; Montini, T.; Fornasiero, P. Photocatalytic valorization of ethanol and glycerol over TiO2 polymorphs for sustainable hydrogen production. Appl. Catal. A 2016, 518, 167–175. [Google Scholar] [CrossRef]
- Tálas, E.; Pászti, Z.; Korecz, L.; Domján, A.; Németh, P.; Szijjártó, G.P.; Mihály, J.; Tompos, A. PtOx-SnOx-TiO2 catalyst system for methanol photocatalytic reforming: Influence of cocatalysts on the hydrogen production. Catal. Today 2018, 306, 71–80. [Google Scholar] [CrossRef]
- Ding, Z.; Lu, G.Q.; Greenfield, P.F. Role of the crystallite phase of TiO2 in heterogeneous photocatalysis for phenol oxidation in water. J. Phys. Chem. B 2000, 104, 4815–4820. [Google Scholar] [CrossRef]
- Al-Mazroai, L.S.; Bowker, M.; Davies, P.; Dickinson, A.; Greaves, J.; James, D.; Millard, L. The photocatalytic reforming of methanol. Catal. Today 2007, 122, 46–50. [Google Scholar] [CrossRef]
- Cui, W.; Feng, L.; Xu, C.; Lü, S.; Qiu, F. Hydrogen production by photocatalytic decomposition of methanol gas on Pt/TiO2 nano-film. Catal. Commun. 2004, 5, 533–536. [Google Scholar] [CrossRef]
- Lin, W.C.; Yang, W.D.; Huang, I.L.; Wu, T.S.; Chung, Z.J. Hydrogen production from methanol/water photocatalytic decomposition using Pt/TiO2−xNx catalyst. Energy Fuels 2009, 23, 2192–2196. [Google Scholar] [CrossRef]
- Linsebigler, A.L.; Lu, G.; Yates, J.T., Jr. Photocatalysis on TiO2 surfaces: Principles, mechanisms, and selected results. Chem. Rev. 1995, 95, 735–758. [Google Scholar] [CrossRef]
- Halasi, G.; Schubert, G.; Solymosi, F. Comparative study on the photocatalytic decomposition of methanol on TiO2 modified by N and promoted by metals. J. Catal. 2012, 294, 199–206. [Google Scholar] [CrossRef]
- Kraeutler, B.; Bard, A.J. Heterogeneous photocatalytic preparation of supported catalysts. Photodeposition of platinum on titanium dioxide powder and other substrates. J. Am. Chem. Soc. 1978, 100, 4317–4318. [Google Scholar] [CrossRef]
- Wenderich, K.; Mul, G. Methods, mechanism, and applications of photodeposition in photocatalysis: A review. Chem. Rev. 2016, 116, 14587–14619. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Shangguan, W. Rational removal of stabilizer-ligands from platinum nanoparticles supported on photocatalysts by self-photocatalysis degradation. Catal. Today 2015, 242, 372–380. [Google Scholar] [CrossRef]
- Schafer, S.; Wyrzgol, S.A.; Caterino, R.; Jentys, A.; Schoell, S.J.; Havecker, M.; Knop-Gericke, A.; Lercher, J.A.; Sharp, I.D.; Stutzmann, M. Platinum nanoparticles on gallium nitride surfaces: Effect of semiconductor doping on nanoparticle reactivity. J. Am. Chem. Soc. 2012, 134, 12528–12535. [Google Scholar] [CrossRef] [PubMed]
- Che, M.; Clause, O.; Marcilly, C. 4.1 Deposition of active component. In Preparation of Solid Catalysts; Ertl, G., Knözinger, H., Weitkamp, J., Eds.; Wiley-VCH: Weinheim, Germany, 1999; pp. 315–371. ISBN 9783527298266. [Google Scholar]
- Maeda, K.; Teramura, K.; Saito, N.; Inoue, Y.; Domen, K. Improvement of photocatalytic activity of (Ga1−xZnx)(N1−xOx) solid solution for overall water splitting by co-loading Cr and another transition metal. J. Catal. 2006, 243, 303–308. [Google Scholar] [CrossRef]
- Vass, Á.; Pászti, Z.; Bálint, S.; Németh, P.; Szijjártó, G.P.; Tompos, A.; Tálas, E. Structural evolution in Pt/Ga-Zn-oxynitride catalysts for photocatalytic reforming of methanol. Mater. Res. Bull. 2016, 83, 65–76. [Google Scholar] [CrossRef] [Green Version]
- Margitfalvi, J.; Kern-Tálas, E.; Szedlacsek, P. Contradictions in the interpretation of experimental results obtained upon high-temperature hydrogen treatment of supported platinum catalysts. J. Catal. 1985, 92, 193–195. [Google Scholar] [CrossRef]
- Reyes, P.; Oportus, M.; Pecchi, G.; Fréty, R.; Moraweck, B. Influence of the nature of the platinum precursor on the surface properties and catalytic activity of alumina-supported catalysts. Catal. Lett. 1996, 37, 193–197. [Google Scholar] [CrossRef]
- Mondloch, J.E.; Yan, X.; Finke, R.G. Monitoring supported-nanocluster heterogeneous catalyst formation: Product and kinetic evidence for a 2-step, nucleation and autocatalytic growth mechanism of Pt(0)n formation from H2PtCl6 on Al2O3 or TiO2. J. Am. Chem. Soc. 2009, 131, 6389–6396. [Google Scholar] [CrossRef] [PubMed]
- Westerberg, B.; Fridell, E. A transient FTIR study of species formed during NOx storage in the Pt/BaO/Al2O3 system. J. Mol. Catal. A 2001, 165, 249–263. [Google Scholar] [CrossRef]
- Meng, F.; Li, J.; Hong, Z.; Zhi, M.; Sakla, A.; Xiang, C.; Wu, N. Photocatalytic generation of hydrogen with visible-light nitrogen-doped lanthanum titanium oxides. Catal. Today 2013, 199, 48–52. [Google Scholar] [CrossRef]
- Fiddy, S.G.; Newton, M.A.; Dent, A.J.; Salvini, G.; Corker, J.M.; Turin, S.; Campbell, T.; Evans, J. In situ energy dispersive EXAFS (EDE) of low loaded Pt(acac)2/HI SiO2 catalyst precursors on a timescale of seconds and below. Chem. Commun. 1999, 851–852. [Google Scholar] [CrossRef]
- Janampelli, S.; Darbha, S. Selective and reusable Pt-WOx/Al2O3 catalyst for deoxygenation of fatty acids and their esters to diesel-range hydrocarbons. Catal. Today 2018, 309, 219–226. [Google Scholar] [CrossRef]
- Pereda-Ayo, B.; De La Torre, U.; González-Marcos, M.P.; González-Velasco, J.R. Influence of ceria loading on the NOx storage and reduction performance of model Pt–Ba/Al2O3 NSR catalyst. Catal. Today 2015, 241, 133–142. [Google Scholar] [CrossRef]
- Pendyala, V.R.R.; Jacobs, G.; Ma, W.; Shafer, W.D.; Sparks, D.E.; MacLennan, A.; Hu, Y.; Davis, B.H. Fischer-Tropsch synthesis: Effect of carbonyl sulfide poison over a Pt promoted Co/alumina catalyst. Catal. Today 2018, 299, 14–19. [Google Scholar] [CrossRef]
- García-Fernández, S.; Gandarias, I.; Requies, J.; Soulimani, F.; Arias, P.L.; Weckhuysen, B.M. The role of tungsten oxide in the selective hydrogenolysis of glycerol to 1,3-propanediol over Pt/WOx/Al2O3. Appl. Catal. B 2017, 204, 260–272. [Google Scholar] [CrossRef]
- Jentoft, R.E.; Tsapatsis, M.; Davis, M.E.; Gates, B.C. Platinum clusters supported in zeolite LTL: Influence of catalyst morphology on performance in n-hexane reforming. J. Catal. 1998, 179, 565–580. [Google Scholar] [CrossRef]
- Jeon, S.; Park, Y.M.; Saravanan, K.; Han, G.H.; Kim, B.W.; Lee, J.B.; Bae, J.W. Aqueous phase reforming of ethylene glycol over bimetallic platinum-cobalt on ceria-zirconia mixed oxide. Int. J. Hydrogen Energy 2017, 42, 9892–9902. [Google Scholar] [CrossRef]
- Özkara-Aydınoğlu, Ş.; Özensoy, E.; Aksoylu, E. The effect of impregnation strategy on methane dry reforming activity of Ce promoted Pt/ZrO2. Int. J. Hydrogen Energy 2009, 34, 9711–9722. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Arroyo-Ramírez, L.; Wei, J.; Yun, H.; Murray, C.B.; Gorte, R.J. Comparison of HMF hydrodeoxygenation over different metalcatalysts in a continuous flow reactor. Appl. Catal. A 2015, 50, 86–93. [Google Scholar] [CrossRef]
- Liang, B.; Duan, H.; Su, X.; Chen, X.; Huang, Y.; Chen, X.; Delgado, J.J.; Zhang, T. Promoting role of potassium in the reverse water gas shift reaction on Pt/mullite catalyst. Catal. Today 2017, 281, 319–326. [Google Scholar] [CrossRef]
- Hernandez, J.O.; Choren, E.A. Thermal stability of some platinum complexes. Thermochim. Acta 1983, 71, 265–272. [Google Scholar] [CrossRef]
- Reagan, W.J.; Chester, A.W.; Kerr, G.T. Studies of the thermal decomposition and catalytic properties of some platinum and palladium ammine zeolites. J. Catal. 1981, 69, 89–100. [Google Scholar] [CrossRef]
- Park, S.H.; Tzou, M.S.; Sachtler, I.M.H. Temperature programmed reduction and re-oxidation of platinum in Y-zeolites. Appl. Catal. 1986, 24, 85–98. [Google Scholar] [CrossRef]
- Keegan, M.B.T.; Dent, A.J.; Blake, A.B.; Conyers, L.; Moyes, R.B.; Wells, P.B.; Whan, D.A. An in situ energy dispersive EXAFS study of the calcination and reduction of a PtZSM-5 catalyst precursor. Catal. Today 1991, 9, 183–188. [Google Scholar] [CrossRef]
- Van den Broek, A.C.M.; van Grondelle, J.; van Santen, R.A. Preparation of highly dispersed platinum particles in HZSM-5 Zeolite: A study of the pretreatment process of [Pt(NH3)4]2+. J. Catal. 1997, 167, 417–424. [Google Scholar] [CrossRef]
- Exner, D.; Jaeger, N.; Möller, K.; Schulz-Ekloff, G. Thermal analysis of the decomposition mechanism of platinum and palladium tetrammine faujasite X. J. Chem. Soc. Faraday Trans. 1 1982, 78, 3537–3544. [Google Scholar] [CrossRef]
- Homeyer, S.T.; Sachtler, W.M.H. Elementary steps in the formation of highly dispersed palladium in NaY I. Pd ion coordination and migration. J. Catal. 1989, 117, 91–101. [Google Scholar] [CrossRef]
- Sauvage, A.; Massiani, P.; Briend, M.; Barthomeuf, D.; Bozon-Verduraz, F. Comparison of Pd(NH3)42+ decomposition and Pd2+reduction in zeolites NaY, NaX and CsX. J. Chem. Soc. Faraday Trans. 1 1995, 91, 3291–3297. [Google Scholar] [CrossRef]
- De Graaf, J.; van Dillen, A.J.; de Jong, K.P.; Koningsberger, D.C. Preparation of highly dispersed Pt particles in zeolite Y with a narrow particle size distribution: Characterization by hydrogen chemisorption, TEM, EXAFS spectroscopy, and particle modeling. J. Catal. 2001, 203, 307–321. [Google Scholar] [CrossRef]
- Muñoz-Páez, A.; Koningsberger, D.C. Decomposition of the precursor [Pt(NH3)4](OH)2, genesis and structure of the metal-support interface of alumina supported platinum particles: A structural study using TPR, MS, and XAFS spectroscopy. J. Phys. Chem. 1995, 99, 4193–4204. [Google Scholar] [CrossRef]
- Miller, J.T.; Schreier, M.; Kropf, J.A.; Regalbuto, J.R. A fundamental study of platinum tetraammine impregnation of silica 2. The effect of method of preparation, loading, and calcination temperature on (reduced) particle size. J. Catal. 2004, 225, 190–202. [Google Scholar] [CrossRef]
- Oudenhuijzen, M.K.; Kooyman, P.J.; Tappel, B.; van Bokhoven, J.A.; Koningsberger, D.C. Understanding the influence of the pretreatment procedure on platinum particle size and particle-size distribution for SiO2 impregnated with [Pt2+(NH3)4](NO3−)2: A combination of HRTEM, Mass Spectrometry, and Quick EXAFS. J. Catal. 2002, 205, 135–146. [Google Scholar] [CrossRef]
- Kaluža, L.; Larsen, M.J.; Zdražil, M.; Gulková, D.; Víta, Z.; Šolcová, O.; Soukup, K.; Koštejn, M.; Bonde, J.L.; Maixnerová, L.; et al. Highly loaded carbon black supported Pt catalysts for fuel cells. Catal. Today 2015, 256, 375–383. [Google Scholar] [CrossRef]
- Moulder, J.F.; Stickle, W.F.; Sobol, P.E.; Bomben, K.D. Handbook of X-ray Photoelectron Spectroscopy; Perkin-Elmer Corp.: Eden Prairie, MN, USA, 1992. [Google Scholar]
- Naumkin, A.V.; Kraut-Vass, A.; Gaarenstroom, S.W.; Powell, C.J. NIST X-ray Photoelectron Spectroscopy Database, Version 4.1, National Institute of Standards and Technology, Gaithersburg, MD, USA. 2012. Available online: http://srdata.nist.gov/xps/ (accessed on 8 October 2018).
- Fairley, N. CasaXPS: Processing Software for XPS, AES, SIMS and More. Available online: www.casaxps.com/ (accessed on 8 October 2018).
- Mohai, M. XPS MultiQuant: Multimodel XPS quantification software. Surf. Interface Anal. 2004, 36, 828–832. [Google Scholar] [CrossRef]
- Mohai, M. “XPS MultiQuant: Multi-Model X-ray Photoelectron Spectroscopy Quantification Program. Version 7.00.92. 2011. Available online: http://www.chemres.hu/aki/XMQpages/XMQhome.htm/ (accessed on 8 October 2018).
- Nosaka, A.Y.; Fujiwara, T.; Yagi, H.; Akutsu, H.; Nosaka, Y. Characteristics of water adsorbed on TiO2 photocatalytic systems with increasing temperature as studied by solid-state 1H NMR spectroscopy. J. Phys. Chem. B 2004, 108, 9121–9125. [Google Scholar] [CrossRef]
- Ray, K.J.A.; Wiswanathan, B. Effect of surface area, pore volume and particle size of P25 titania on the phase transformation of anatase to rutile. Indian J. Chem. 2009, 48A, 1378–1382. [Google Scholar]
- Mills, A.; Le Hunte, S. An overview of semiconductor photocatalysis. J. Photochem. Photobiol. A 1997, 108, 1–35. [Google Scholar] [CrossRef]
- Huang, B.S.; Chang, F.Y.; Wey, M.Y. Photocatalytic properties of redox-treated Pt/TiO2 photocatalysts for H2 production from an aqueous methanol solution. Int. J. Hydrogen Energy 2010, 35, 7699–7705. [Google Scholar] [CrossRef]
- Hilaire, L.; Guerrero, G.D.; Légaré, P.; Maire, G.; Krill, G. A photoemission study of the oxidation of platinum in Pt-based alloys: Pt-Pd, Pt-Ru, Pt-Ir. Surf. Sci. 1984, 146, 569–582. [Google Scholar] [CrossRef]
- Mueller, R.; Kammler, H.K.; Wegner, K.; Pratsinis, S.E. OH Surface density of SiO2 and TiO2 by thermogravimetric analysis. Langmuir 2003, 19, 160–165. [Google Scholar] [CrossRef]
- Shalvoy, R.B.; Reucrofty, P.J. Quantitative analysis of ESCA signal intensifies from coprecipitated nickel on alumina catalysts. J. Electron. Spectrosc. Relat. Phenom. 1977, 12, 351–356. [Google Scholar] [CrossRef]
- Luttrel, T.; Halpegamage, S.; Tao, J.; Kramer, A.; Sutter, E.; Batzill, M. Why is anatase a better photocatalyst than rutile? Model studies on epitaxial TiO2 films. Sci. Rep. 2014. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, B.; Prieto-Mahaney, O.O.; Li, D.; Abe, R. What is Degussa (Evonik) P25? Crystalline composition analysis, reconstruction from isolated pure particles and photocatalytic activity test. J. Photochem. Photobiol. A 2010, 216, 179–182. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Wei, Z.S.; Ohtani, B.; Kowalska, E. Interparticle electron transfer in methanol dehydrogenation on platinum loaded titania particles prepared from P25. Catal. Today 2018, 303, 327–333. [Google Scholar] [CrossRef]
- Rivera, A.P.; Tanaka, K.; Hisanaga, T. Photocatalytic degradation of pollutant over TiO2 in different crystal structures. Appl. Cata. B 1993, 3, 37–44. [Google Scholar] [CrossRef]
- Jiang, X.; Fu, X.; Zhang, L.; Meng, S.; Chen, S. Photocatalytic reforming of glycerol for H2 evolution on Pt/TiO2: Fundamental understanding the effect of co-catalyst Pt and the Pt deposition route. J. Mater. Chem. A 2015, 3, 2271–2282. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of TiO2 | SSA 1, m2 g−1 | Maximum Pore Diameter, nm | Pore Volume, cm3 g−1 | Crystalline Phases 2, % | Average Particle Size, nm | ||||
|---|---|---|---|---|---|---|---|---|---|
| A 3 | B 4 | R 5 | A 3 | B 4 | R 5 | ||||
| PA | 133.8 | 3.71 | 0.1064 | 23 | 23 | 54 | 6 2 | 7 2 | 19 2 |
| P25 | 52.5 | 31.55 | 0.3674 | 82 | - | 18 | 24 2 | - | 45 2 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Majrik, K.; Pászti, Z.; Korecz, L.; Trif, L.; Domján, A.; Bonura, G.; Cannilla, C.; Frusteri, F.; Tompos, A.; Tálas, E. Study of PtOx/TiO2 Photocatalysts in the Photocatalytic Reforming of Glycerol: The Role of Co-Catalyst Formation. Materials 2018, 11, 1927. https://doi.org/10.3390/ma11101927
Majrik K, Pászti Z, Korecz L, Trif L, Domján A, Bonura G, Cannilla C, Frusteri F, Tompos A, Tálas E. Study of PtOx/TiO2 Photocatalysts in the Photocatalytic Reforming of Glycerol: The Role of Co-Catalyst Formation. Materials. 2018; 11(10):1927. https://doi.org/10.3390/ma11101927
Chicago/Turabian StyleMajrik, Katalin, Zoltán Pászti, László Korecz, László Trif, Attila Domján, Giuseppe Bonura, Catia Cannilla, Francesco Frusteri, András Tompos, and Emília Tálas. 2018. "Study of PtOx/TiO2 Photocatalysts in the Photocatalytic Reforming of Glycerol: The Role of Co-Catalyst Formation" Materials 11, no. 10: 1927. https://doi.org/10.3390/ma11101927