
Multiscale Modelling of β-Adrenergic Stimulation in Cardiac Electromechanical Function


Department of Computer Science, BHF Centre of Research Excellence, University of Oxford, Oxford OX1 3QD, UK
*
Author to whom correspondence should be addressed.
Mathematics 2021, 9(15), 1785; https://doi.org/10.3390/math9151785
Submission received: 4 July 2021
/
Revised: 24 July 2021
/
Accepted: 26 July 2021
/
Published: 28 July 2021
(This article belongs to the Special Issue Development and Applications of Multi-Scale Mathematical Models in Cardiology)
Abstract
:β-adrenergic receptor stimulation (β-ARS) is a physiological mechanism that regulates cardiovascular function under stress conditions or physical exercise. Triggered during the so-called “fight-or-flight” response, the activation of the β-adrenergic receptors located on the cardiomyocyte membrane initiates a phosphorylation cascade of multiple ion channel targets that regulate both cellular excitability and recovery and of different proteins involved in intracellular calcium handling. As a result, β-ARS impacts both the electrophysiological and the mechanical response of the cardiomyocyte. β-ARS also plays a crucial role in several cardiac pathologies, greatly modifying cardiac output and potentially causing arrhythmogenic events. Mathematical patient-specific models are nowadays envisioned as an important tool for the personalised study of cardiac disease, the design of tailored treatments, or to inform risk assessment. Despite that, only a reduced number of computational studies of heart disease have incorporated β-ARS modelling. In this review, we describe the main existing multiscale frameworks to equip cellular models of cardiac electrophysiology with a β-ARS response. We also outline various applications of these multiscale frameworks in the study of cardiac pathology. We end with a discussion of the main current limitations and the future steps that need to be taken to adapt these models to a clinical environment and to incorporate them in organ-level simulations.
1. Introduction
β-adrenergic receptor stimulation (β-ARS) is a physiological response mechanism that plays a fundamental role in the regulation of cardiomyocyte activity, producing a positive inotropic (enhanced contraction), lusitropic (faster relaxation), and chronotropic (increased heart rate) effect. Such a multifactorial response is triggered via the activation of the β-adrenergic receptors by the sympathetic nervous system, under either stress conditions or physical exercise, and is also known as the “fight-or-flight” response.
β-adrenergic receptors were first described by Lands et al. in the late 1960s [1,2]. They are situated on the cardiomyocyte membrane and react to different neurotransmitters (norepinephrine, epinephrine) and drugs (isoprenaline). When binding with the adrenergic receptor takes place, it starts a reaction cascade where different cellular substrates become phosphorylated, affecting their individual roles in the overall excitation–contraction coupling. As a result, under healthy physiological conditions, the cardiac action potential shortens, while the intracellular calcium transient exhibits an increased amplitude and a faster decay rate as the main manifestations of β-ARS at the cellular level [3,4]. However, the large number of components involved in the β-adrenergic cascade, and the complexity of these subcellular processes and interactions, make β-ARS signalling highly sensitive to cellular changes and to pathological perturbations. As a result, β-ARS plays a main role in a considerable number of heart diseases [5], and it is well established as an important contributor to cardiomyocyte arrhythmogenicity [6,7,8].
In particular, the β-ARS response has direct effects on the ion channels and pumps of the cell membrane (and, therefore, on intracellular ionic concentrations) regulating calcium intake, intracellular calcium handling, calcium extrusion, and cellular repolarisation. Impairments in the balance between these carefully orchestrated processes can affect heart function and render its constituent cardiomyocytes susceptible to proarrhythmic events, such as early and delayed afterdepolarisations (EADs and DADs, respectively). Such afterdepolarisations under β-ARS are common proarrhythmic manifestations in isolated cardiomyocytes from patients of different pathologies, especially those characterised by action potential and calcium transient abnormalities (such as hypertrophic cardiomyopathy [9], long QT syndrome [10,11], or catecholaminergic polymorphic ventricular tachycardia [12]). An overstimulated β-ARS response is also one of the main mechanisms of cardiac hypertrophy, coronary artery disease, or stroke events [13]. The overexpression of the β-adrenergic response has also been linked to the onset of cardiac hypertrophy or the generation of fibrotic tissue [14,15]. The appearance of these structural changes can lead to the creation of re-entry pathways in myocardial tissue, which also contribute to the generation of self-sustained arrhythmias. Induced arrhythmias are also a common manifestation in heart failure. In chronic heart failure, cardiac remodelling at the structural level can affect the pathways involved in the β-ARS response [16]. As a result, the inotropic response of cardiomyocytes to β-ARS is reduced [17,18], while the propensity to arrhythmogenic events increases. β-adrenergic response is also affected by ageing, and an age-dependent impairment between β-ARS and cardiac function has been demonstrated in both healthy and failing hearts [19,20]. β-ARS is altered as well in severe congenital heart disease patients [21]. Finally, recent studies also suggest a hyperactivation of the positive response of β-ARS patients with coronavirus disease 2019 (COVID-19), potentially leading to life-threatening arrhythmic events [22,23].
Refined knowledge of the role that each cellular component has within the β-ARS cascade, and of the consequences that may arise from disturbing its normal functioning, can therefore lead to a better understanding of different pathologies, as well as to the development of new pharmacological targets for their treatment. In these cases, mathematical modelling and simulation studies of β-ARS can be useful tools for investigating the mechanisms mediating arrhythmic events, assessing their multiscale consequences from the subcellular up to the organ levels, and designing effective treatments [24]. In this paper, we review the main roles of β-ARS in cellular cardiac electrophysiology, placing our emphasis on the description of the existing mathematical frameworks available for its representation and how insights obtained through experimental approaches have been integrated into these mathematical formulations. Then, we focus on how multiscale investigations exploiting mathematical descriptions of β-ARS have been used in different studies to better understand the role of β-ARS in heart disease and arrhythmias. We conclude our review by discussing a number of open questions requiring further combined experimental and computational investigations to improve our current understanding of the processes involved during β-ARS and their implications in cardiac function.
2. Mathematical Models of β-ARS
Several mathematical formulations, with varying degrees of complexity and physiological detail, have been proposed to date to describe different aspects of β-ARS in cardiac myocytes. In particular, special attention has been given to the modelling of β-ARS in mammalian ventricular electrophysiology, while studies on atrial electrophysiology are considerably lagging. Table 1 summarises the main β-ARS computational frameworks discussed in this section as landmark studies underlying the foundation of these modelling efforts. For the sake of simplicity, most models will be referred to by their first authors’ names.
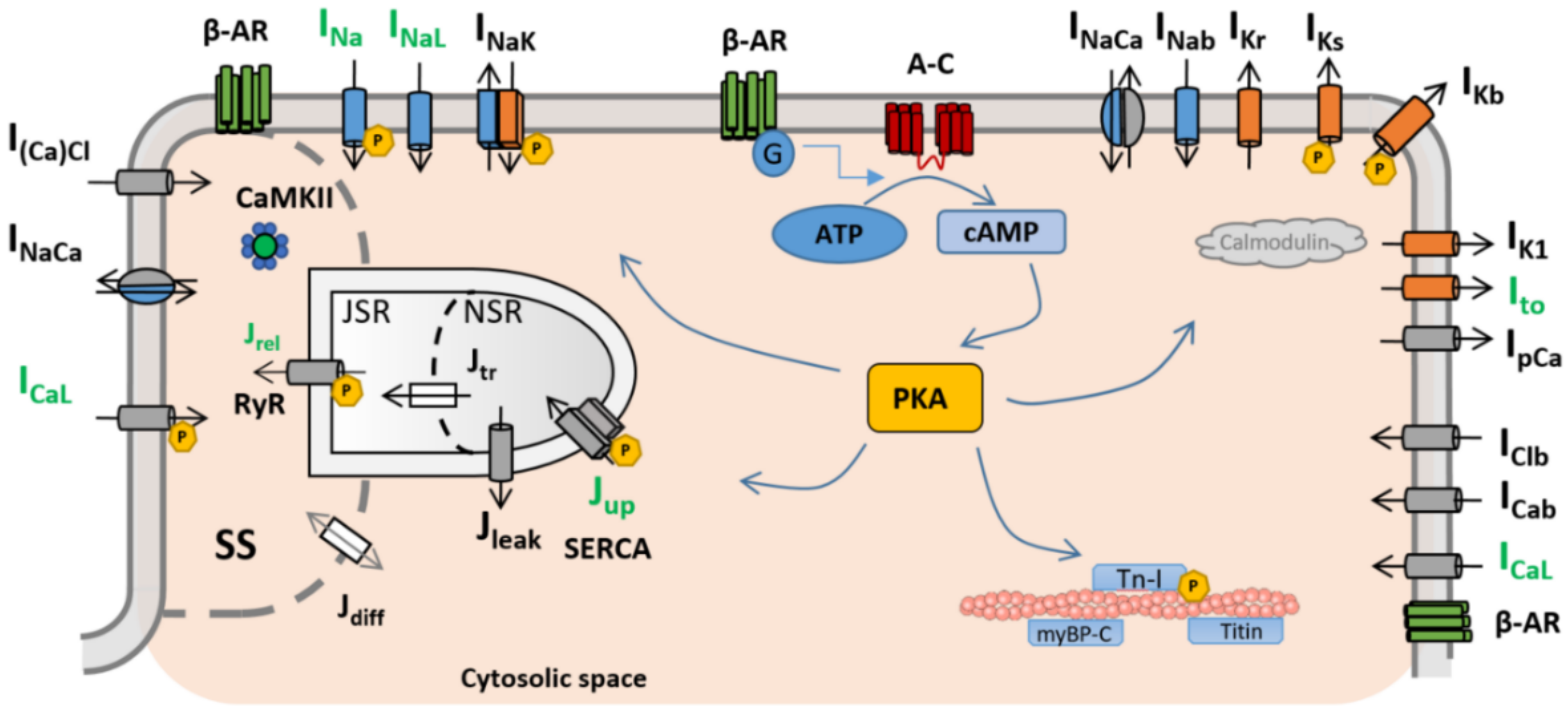
In general terms, cardiac myocytes present three different subtypes of β-adrenergic receptors (β-AR): β1-AR, β2-AR, and β3-AR [25], the former two being the most prevalent and important. Many of the existing mathematical models of β-ARS describe the binding of an agonist, usually isoproterenol (ISO), to β1-AR and β2-AR and the subsequent phosphorylation cascade. In particular, the binding with β-ARs activates the receptor-bound stimulatory G protein, which enhances the production of 3′-5′-cyclic adenosine monophosphate (cAMP), activating protein kinase A (PKA). PKA phosphorylates key cellular substrates that affect the functioning of several channels and ion pumps. The main targets located on the sarcolemma are the L-type calcium channels (ICaL) [26], slow delayed rectifier potassium (IKs) channels [27], fast sodium current (INa) channels [28], the cystic fibrosis transmembrane conductance regulator (ICFTR) [29,30], and the sodium–potassium pump (INaK) [31]. At the subcellular level, PKA also phosphorylates the ryanodine receptors (RyRs) and phospholamban (PLB) [26,32], both located on the sarcoplasmic reticulum, as well as troponin I (TPNI) [33], myosin binding protein-C [33], and titin [34], these located on the myofilaments. A graphical representation of the main PKA phosphorylation cascade is presented in Figure 1.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Landmark studies in the mathematical modelling of β-ARS in cardiac electrophysiology.
| Model (Year) | Species | β-ARS Isoform | Signalling | Substrates | Main Model Advances |
|---|---|---|---|---|---|
| Zeng and Rudy [35] (1995) | Guinea Pig | Generic | None | ICaL; IK; PLB; INaK; INa | Simulation of the isoproterenol effect by increasing conductances and parameter shift |
| Saucerman et al. [36] (2003) | Rat | β1 | cAMP; PKA | ICaL; PLB | Dynamic target phosphorylation integrated with cell signalling |
| Greenstein et al. [37] (2004) | Dog | Generic | None | ICaL; PLB; IKr; IKs | Introduction of a binary population-based approach for target phosphorylation |
| Iancu et al. * [38] (2007) | Guinea Pig | β1 | cAMP | N/A | Cellular signalling compartmentation |
| Soltis & Saucerman [29] (2010) | Rabbit | β1 | cAMP; PKA | ICaL; IKs; PLB; RyR; TnI; ICFTR | Integration with dynamic CaMKII regulation |
| Hiejman et al. [39] (2011) | Dog | β1, β2 | cAMP; PKA | ICaL; IKs; IKur; PLB; INaK; INa; RyR; TnI | Two different β isoforms; population-based approach with four different populations |
| Bondarenko [40] (2014) | Mouse | β1 | cAMP; PKA | ICaL; INa; INaK; RyR; IKur; Ito; IK1; PLB; TnI | Compartmentalised mouse model with new L-type calcium channel subpopulations |
| Khalilimeybodi et al. * [41] (2018) | Mouse | β1, β2 | cAMP; PKA; GSK3β; ERK | N/A | Addition of new molecular signalling pathways (GSK3β and ERK) |
* Models focused only on signalling (not directly considering substrate phosphorylation). N/A: not applicable.
The first modelling approach for the consideration of β-ARS in cardiac electrophysiology described the effects of β-ARS by upscaling the magnitude of the most significantly upregulated ion channels during the β-adrenergic response (notably ICaL and IKs) or by shifting the activation curves of these currents [35,43]. Despite its simplicity, such an approach is sufficient to replicate to a good extent the main steady-state effects of β-ARS at the cellular level, such as action potential shortening, increased calcium transient amplitude, or a potentiated arrhythmogenicity. A more complex model developed by Greenstein et al. [37] accounted for these changes using a population-based approach, i.e., treating phosphorylation as a binary process independent of channel gating. This assumption implies that, at each time step, ionic concentrations and ionic currents or fluxes can be expressed as the sum of their phosphorylated and nonphosphorylated cellular parts. However, the steady-state nature of the abovementioned models imposes limitations for investigating transient behaviours in the activation of β-ARS or in dissecting the contribution to the β-ARS response of subcellular targets of PKA phosphorylation.
Saucerman et al. were the first authors to develop, in the early 2000s, a dynamic formulation of β-ARS [36,44]. The model incorporated a complete phosphorylation cascade of the L-type calcium channel and PLB in a rat ventricular myocyte electrophysiological model. The authors considered an extensive validation against published experimental data, including the temporal response of cAMP to ISO [45], PKA activation levels as a function of the concentration of cAMP [46], and PLB phosphorylation to ISO [47], together with experimental recordings of whole-cell patch-clamp ICaL current, calcium transients, and action potentials [48]. This seminal model of β-ARS has been expanded in multiple subsequent studies. Soltis and Saucerman added dynamic phosphorylation by Ca2+/calmodulin-dependent protein kinase II (CaMKII), in combination with the previously described PKA phosphorylation, to rabbit ventricular cardiomyocytes [29]. Phospholemman phosphorylation was also included in later works [49]. The model was also expanded by Negroni et al. to present an integrated framework of β-ARS and cardiac mechanical contraction [30]. More recently, Meyer et al. investigated a quasi-steady-state approximation of the Soltis and Saucerman model, demonstrating that their reduced-order formulation captured many of the features of the complete model while providing an efficient approximation over a broad range of parameter conditions [50].
The next landmark in β-ARS modelling was provided by Iancu et al., who proposed a new model considering cellular compartmentation of cAMP [38]. This led to a β-ARS model response dependent on local cAMP and PKA concentrations in the different cellular domains. The model response was compared to published measurements of cAMP activity under β-ARS [51]. The compartmentation introduced by the model provided a mechanistic explanation of how the high concentrations of cAMP as measured in experiments can modulate PKA activity.
The ideas described in the Saucerman and Iancu models were further developed by Heijman et al. [39]. In their work, they developed a compartmental model of β-ARS of the canine ventricular myocyte, including a dynamic β-ARS response integrated with CaMKII signalling. The main novelties of the model were the consideration of β1 and β2 isoforms in β-ARS stimulation and the use of a population-based approach with four different populations (phosphorylated by PKA, phosphorylated by CaMKII, phosphorylated by both, and nonphosphorylated) to represent the phosphorylation of its targets. The model was calibrated and validated using a wide range of canine and rodent experimental data, from measurements of molecular signalling (cAMP levels, adenylyl cyclase activity, PKA substrate [52,53]) to the complete cellular response (action potential duration, calcium transients [54]). The Heijman model has also been modified in later works, especially in terms of adjusting its underlying conductances and time constants to mimic the β-ARS response in other species, including humans [55,56].
A later model of β-ARS in mouse myocytes was developed by Bondarenko [40]. The model included some of the elements of the Heijman model, such as the compartmentalisation of the β-ARS system, but considered two types of L-type calcium channels and a different localisation of the RyRs in the compartmentalisation. The model matched experimental data from mice and showed how the different locations of some PKA phosphorylation targets can significantly affect action potential duration and calcium transients.
Recently, Khalilimeybodi et al. [41] formulated a model for β-adrenergic-induced cardiac hypertrophy in rat myocytes. The model included new molecular pathways that ended with the phosphorylation of the GSK3β and ERK molecules, which mediate gene expression in cardiac hypertrophy. The modelling results highlighted the significant contribution of these pathways in regulating cardiac hypertrophy in rats.
3. Differences in Formulation between Leading Mathematical Models of β-ARS
From the aforementioned models, the most widely adopted ones in modelling and simulation studies of β-ARS (either in their original form or with slight modifications) are the Soltis and Saucerman [29] and the Heijman [39] models. In these models, the biochemical reactions are modelled using ordinary differential equations or algebraic equations in cases with very fast reaction dynamics. The concentration of ISO is usually treated as an input to the model. In terms of complexity, the Soltis and Saucerman model is formed of 17 ordinary differential equations, containing 6 different PKA substrates (see Table 1). The Heijman β-ARS signalling model, on the contrary, consists of a coupled system of 57 ordinary differential equations and contains 8 different PKA substrates. The counting on these equations increases after the coupling of the signalling model with the respective cellular electrophysiology model, as illustrated below.
Both models share similar formulations of their phosphorylation targets, following a Michaelis–Menten formalism. To better illustrate differences in mathematical treatment, we consider the case of the inhibitory troponin subunit (TnI) as one of their PKA targets. Physiologically, the phosphorylation of TnI can increase the rate of calcium unbinding to calmodulin by 50% [57], being the binding constant. In the Soltis and Saucerman model, the time evolution of TnI phosphorylation is given by:
where and are Michaelis constants of phosphorylation and dephosphorylation, and and are the corresponding phosphorylation and dephosphorylation rates of TnI. PKAC is the concentration of the PKA molecule, which is calculated by similar differential equations. and represent the concentrations of the phosphorylated and nonphosphorylated TnI, respectively. These are linked by conservation of species as:
where the total TnI concentration, , is constant (70 µM). The phosphorylated PKA fraction is then given by the experimentally informed algebraic relationship:
where , and 0.0031. The phosphorylated fraction by PKA, , can then be used to calculate the effective troponin affinity as:
which will inform the respective differential equations to update the intracellular calcium balance.
The Heijman model also uses a Michaelis–Menten formalism for describing the phosphorylation rate, given by:
which, instead of using concentrations, is written directly in this case in terms of phosphorylated and nonphosphorylated fractions, satisfying . The phosphorylated fraction by PKA is given in this model by the algebraic expression:
with the effective troponin affinity finally calculated as:
More specifically, Equation (7) exemplifies the population-based approach used in the Heijman model for each of its phosphorylation targets. The total contribution of a target is calculated as the weighted sum of two target populations (phosphorylated and nonphosphorylated). In Equation (7), these populations are described as and , each one with different kinetics (or conductances and time constants, in case of ion channels). Another important difference in the formulation between the two models can be seen in Equation (5). Following the compartmental nature of the model, the concentration of the free PKA catalytic subunit differs between the different considered compartments (caveolar, extracaveolar membrane, and cytosol), the concentrations in the latter ( and ) being the ones affecting this particular process.
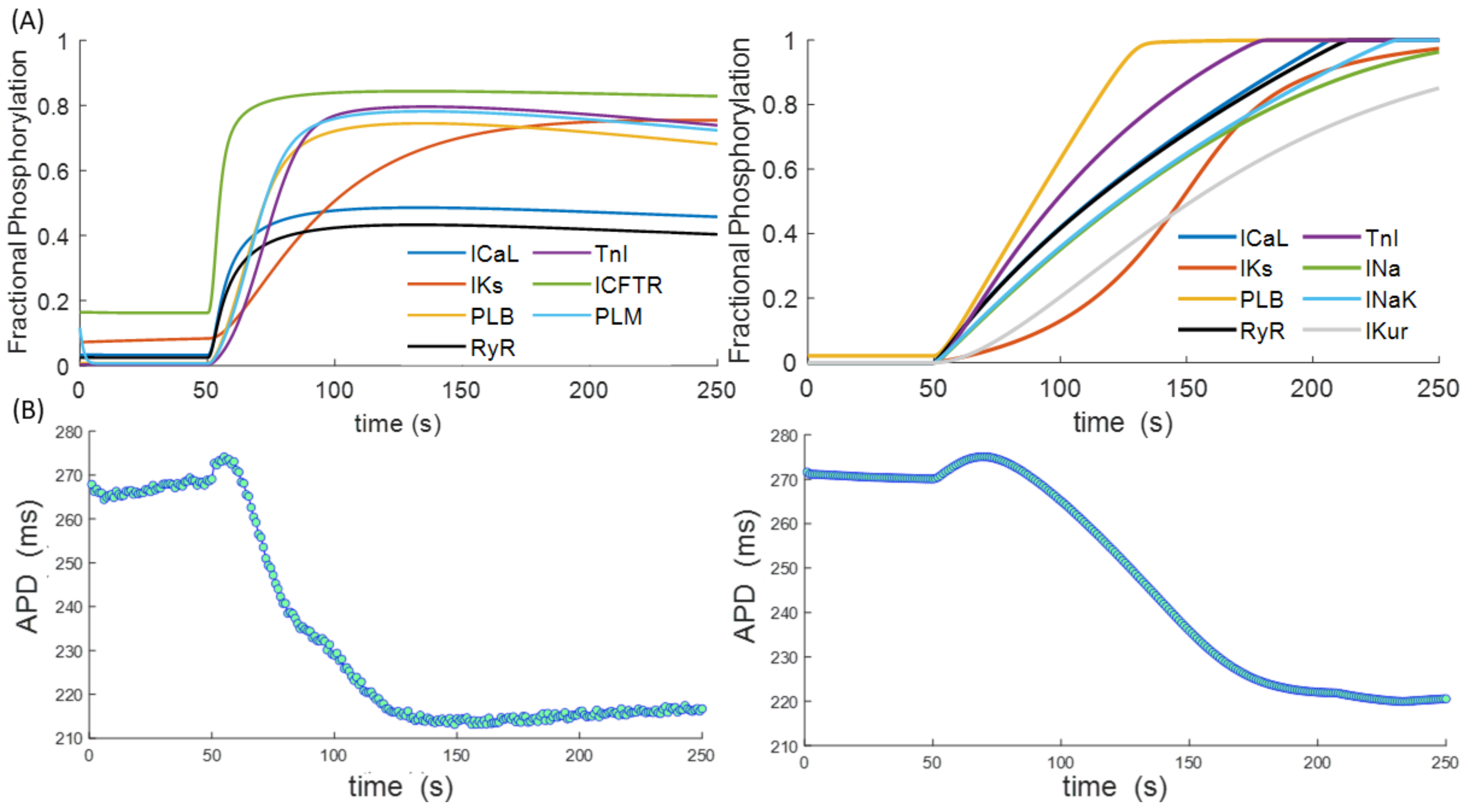
Figure 2A illustrates the time evolution of the different PKA phosphorylation targets between two of the most recent versions of the two described models: the Xie et al. [58] version of the Soltis and Saucerman model and the Gong et al. [56] version of the Heijman model. The Xie version includes a new phosphorylation target, phospholemman (PLM), which affects the INaK current, and a different time constant in IKS current phosphorylation. The Gong version is an adaptation of the Heijman model to human myocytes. Both models were integrated into the O’Hara and Rudy cardiomyocyte model [59] as one of the latest representations of human ventricular electrophysiology and paced at a basic cycle length of 1 Hz for 250 s, with activation of β-ARS by simulated ISO administration after 50 beats. The adaptation of the Xie model to human cardiomyocytes was conducted as in Pueyo et al. [60]. Considerable differences can be appreciated between the two models, the levels of target phosphorylation being higher in the Heijman model. Nevertheless, both models show similar trends in kinetics, with IKS presenting slower phosphorylation than ICaL or PLB and TnI being the ones with faster phosphorylation. The time evolution of the action potential duration exhibited similar behaviour in both models (Figure 2B), with a small increase following ISO administration, followed by a reduction of approximately 50 ms after 250 s.
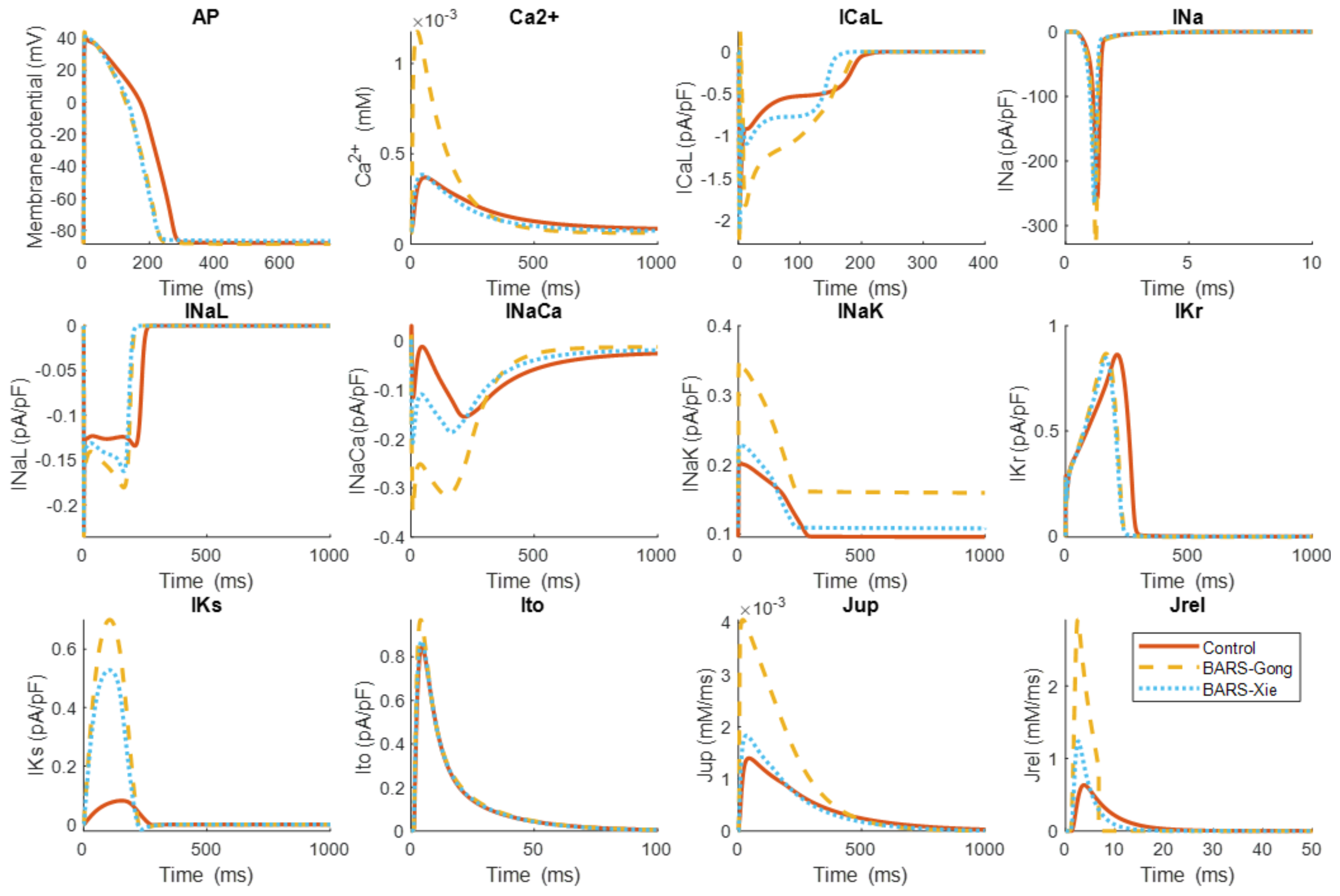
The differences between both β-ARS models at the level of ion channel currents and intracellular calcium fluxes are shown in Figure 3. As can be seen, the action potential shortening is similar in both models. However, the distinct formulations and phosphorylation levels can yield very different effects in specific currents and fluxes. This is for example the case for ICaL, INak, Jrel, or Jup, all exhibiting higher amplitudes when calculated with the Gong model, resulting in a considerably larger amplitude of its calcium transient.
4. Multiscale Studies including β-ARS
The mathematical descriptions of β-ARS discussed before have been integrated into a variety of models of cardiac electrophysiology. Computational studies using the resulting multiscale cellular models have allowed quantitative investigations of the specific role of β-ARS in cardiac electrophysiology and different cardiac pathologies.
As a seminal example in the field, one of the grounds under the development of the earliest models of β-ARS was to resolve the ionic mechanisms underlying the formation of proarrhythmic triggers, such as EADs. The first model of β-ARS, developed by Zheng and Rudy [35], uncovered the nowadays well-established causative link between EADs and the reactivation of the L-type calcium current during the plateau and repolarisation phases of the ventricular action potential. Such an influential finding was made possible not only through mathematical model development but in combination with an exhaustive simulation study evaluating the effects of varying conductance amplitudes of different transmembrane currents, such as the L-type calcium channel or the potassium repolarising channels. A different approach to investigating the generation of EADs was presented by Tanskanen et al. [43], where they evaluated EADs formation using a β-ARS canine model with stochastic gating in the L-type calcium channels. Results suggested that the random fluctuations in channel gating can be the mechanisms underlying EAD generation.
The formulation of more complex models of β-ARS, such as those by Soltis and Saucerman [29] or Heijman et al. [39], allowed for a deeper physiological understanding of the underlying mechanisms. For example, Xie et al. [58] further studied the onset of EADs in rabbit ventricular myocytes using the Soltis and Saucerman β-ARS model, where each PKA target can exhibit different phosphorylation kinetics. As a result, they showed that the formation of EADs can be explained by the difference in phosphorylation levels between the L-type calcium and potassium channels. EAD generation in human myocytes under β-ARS was also studied in later works by Dai et al. [61]. Nevertheless, the range of potential applications in computational studies of β-ARS is certainly not limited to investigations of EADs as proarrhythmic triggers, as demonstrated below.
4.1. Effects of β-ARS in Electromechanical Coupling
Several computational studies have been conducted to evaluate the electrophysiological effects of β-ARS in the excitation–contraction response of cardiac myocytes. In particular, Kuzumoto et al. [62] studied the inotropic response using a guinea-pig ventricular model based on the Soltis and Saucerman model, demonstrating the positive inotropic effect of the ICFTR current under β-ARS. A later study by Negroni et al. [30] also developed a model of β-ARS that included contraction in rabbit myocytes. The model has also been used in [63] to evaluate the role of the PKA phosphorylation in additional myofilament targets and to study their impact on contractility, underscoring the importance of myofilament calcium sensitivity and cross-bridge cycling rate.
Recently, a study by Lyon et al. [64] presented a coupled model of human ventricular cellular electrophysiology [59] with a sarcomere contraction model [65] and investigated the effects of varying degrees of β-ARS in myocyte calcium and developed force during exercise. Among other findings, they identified synergic positive inotropic effects and differential lusitropic effects of β-ARS and sarcomere length on calcium and force dynamics.
4.2. Long QT Syndrome
Mutations in KCNQ1, the gene encoding the α-subunit of IKs channels, are responsible for long QT syndrome type 1, the most common subtype of this congenital cardiac syndrome responsible for up to 30 to 35% of all cases. Most LQT1 patients experience cardiac events during episodes of β-ARS hyperactivity, such as physical exercise or acute emotional reactions [66]. The regulation of the IKs channel activity by β-ARS has been studied in different works. Terrenoire et al. [67] studied it using the Soltis and Saucerman model, showing that alterations of kinetics in IKs are critical in the control of action potential duration. Later studies also studied the role of β-ARS in long QT syndrome using the Heijman model in human myocytes [55,68]. O’Hara and Rudy [55] showed that under the mutation Q357R of the KCNQ1 gene, arrhythmias are caused by the reduced IKs channel expression, whereas Kang et al. [68] found that β-ARS increased IKS contribution to repolarisation. This can lead to an augmentation of the transmural dispersion of repolarisation, which can be arrhythmogenic.
4.3. Heart Failure
Heart failure patients are characterised by a long-term remodelling of multiple cellular processes, including calcium handling, excitation–contraction coupling, and beta-adrenergic signalling [69]. These alterations can lead to arrhythmia, as studied by Morotti et al. [70] through the development of a mouse ventricular model integrating β-ARS based on the Soltis and Saucerman model. The model simulations showed that during heart failure conditions, an increase in sodium concentration can generate a positive feedback loop with CaMKII, increasing phosphorylation of CaMKII targets, thus raising intracellular calcium. Such a positive feedback loop is highly arrhythmogenic, and the study demonstrated it can be potentiated by the β-ARS response. Gong et al. [56] recently adapted the Heijman model to human ventricular electrophysiology and systematically explored what alterations in the cell targets can give place to the phenotypic characteristics of β-ARS in the disease. One additional strength of their work was the consideration of the variability in model parameters, reflecting the heterogeneity between different experimental measurements or individuals, which can greatly affect the simulation outcomes in terms of proarrhythmic findings [71,72]. The model by Gong et al. was also recently used in a study by Mora et al. [73], where they reproduced the electrophysiological response to β-ARS in heart failure and analysed changes in the electrophysiology response. They found that the inotropic effect of β-ARS was reduced in heart failure due to altered calcium handling and that these myocytes have a higher probability of developing EADs. Their simulations also showed that the response to β2-ARs was blunted in heart failure, with β1-ARs being the biggest regulators of most electrophysiological effects under β-ARS.
A different approach in the modelling of heart failure is the one presented by Loucks et al. [74]. In their work, they included the detubulation process (loss of T-tubules of the cell membrane) that is usually concomitant to the remodelling of cardiac myocytes in heart failure. This process affects the distribution of the L-type calcium channels, a target of the β-ARS phosphorylation cascade. The effects of the combination of the detubulation and β-ARS in heart failure and its arrhythmogenicity were also studied in a later study by Sanchez-Alonso et al. [75], showing that redistribution of the L-type calcium channels to the sarcolemma can alter myocyte function and trigger pathological events.
4.4. Other Cardiac Pathologies
Mathematical models of β-ARS have been also used in combined experimental and computational studies to investigate the generation of DADs in catecholaminergic polymorphic ventricular tachycardia [76], the suppression of alternans in the postinfarction border zone [77,78], and the occurrence of re-entrant arrhythmias in Brugada syndrome [79] or to study the possible proarrhythmic effects of two anti-COVID-19 drugs under β-ARS [23]. Finally, the effects that low-frequency oscillatory patterns (e.g., respiration) can have in the sympathetic nervous system, and therefore in the β-ARS response of human heart function, have been experimentally characterised and computationally simulated in several recent works [60,80].
5. Discussion and Perspectives
The importance of β-ARS in regulating cardiac function, as well as its role in promoting arrhythmic events, is nowadays well established. During the last 20 years, a substantial number of modelling and simulation studies have greatly contributed to consolidating the present understanding of the cellular mechanisms underlying the multifactorial response to β-ARS [5,16,20,81,82]. In fact, the changes that β-ARS mobilises at different heart scales can considerably affect organ performance, and the different studies covered in this review paper have underlined the need of including β-ARS to advance the characterisation of (patho-)physiology. It is for these reasons that further advances on β-ARS modelling, and its wider integration in simulation studies of heart function, are expected to considerably advance the potential of these technologies towards precision medicine and the understanding of diseased states, especially in investigations of arrhythmogenicity, exercise, or mechanical response.
5.1. Future Work in β-ARS Modelling
One of the main challenges ahead in the modelling of the β-ARS response and function is to fully understand cAMP and PKA signalling compartmentation. Physiologically, cellular compartmentation is produced by the contribution of different processes that concentrate some molecular transit in confined spaces within the cell [83]. In that way, specific cellular functions can be performed more efficiently. It has been shown that different pathologies such as hypertrophy or heart failure can alter cell compartmentation, affecting cardiac function [84]. In combination with new experimental technologies such as fluorescence resonance energy transfer (FRET) [45,63], additional modelling efforts remain necessary to reliably reflect altered compartmentation in mathematical electrophysiological models and to uncover its functional consequences on cAMP and PKA signalling.
Further experimental characterisation of phosphorylation time dynamics is also expected to notably contribute to the validation and refinement of mathematical models of β-ARS. Both experimental [85] and modelling studies [58,86] have demonstrated that differences in the time course of activation under ISO between different PKA targets can lead to proarrhythmic triggers in the form of EADs. Beyond the studies on compartmentation suggested above, technologies such as FRET also hold great potential to further inform the mathematical modelling of β-ARS in terms of dynamic changes in phosphorylation levels, whose functional consequences are still not clearly understood [87]. Moreover, the extension of β-ARS models to incorporate additional phosphorylation targets, such as titin and myosin binding protein-C, would also qualify such models for quantitative investigations on how their active role in cardiac contraction and electrophysiology is modulated by sympathetic stimulation.
Some of the mechanisms behind how β-ARS affects the normal functioning of ion channels remain unclear. For example, during chronic β-ARS, IKs presents the opposite effect (reduction of IKs current) that under acute β-ARS (IKs increase) [88]. Conditions of chronic β-ARS also pertain to different diseased states and are for instance characteristic of heart failure. Furthermore, several studies [89,90] have suggested a possible mismatch between calcium-dependant and voltage-dependant inactivation of the ICaL channel under β-ARS, potentially leading to arrhythmia formation.
The scarcity of human electrophysiological data implies that the existing mathematical models of β-ARS are largely based on data from other species then carefully reparametrised to reproduce the behaviour observed in human cardiomyocytes [55,56]. Apart from the well-known differences in ion channel conformation and distribution [91], differences have also been reported in the molecular processes involved in β-ARS and in the overall response of human cardiomyocytes to adrenergic stimulation, presenting opposite effects in the shortening of the action potential duration or in the functioning of the sodium–calcium exchanger [90,92,93]. In addition, the density and anatomical location of β-ARS receptors in the heart are different between species [53,94]. Further data acquisition and model developments will allow ascertaining the implications of these interspecies differences in β-ARS in heart function.
5.2. β-ARS and 3D Modelling
The β-ARS response is present at the whole-organ level, including both the ventricles and the atria. For example, many studies have linked β-ARS with the occurrence of episodes of atrial fibrillation [5,95], and in fact, beta-blockers are first-line therapy for rate control in atrial fibrillation patients [96]. However, most modelling efforts of β-ARS to date have been placed on ventricular electrophysiology, while only a small number of recent studies have addressed its modelling in atrial cells, specifically in mice [97,98]. Extending the existing modelling frameworks of β-ARS to human atrial electrophysiology, from the cellular up to the 3D whole-atria level, could lead to significant advances in understanding the involvement of the sympathetic nervous system in atrial arrhythmias, as done in the past in terms of parasympathetic stimulation [99,100,101].
Moreover, β-adrenergic receptors are heterogeneously distributed along different areas of the heart, especially under pathological conditions [81]. Transmural and apicobasal heterogeneities in the sympathetic innervation system have also been described [102,103]. In addition, through its regulation of fast sodium channels, β-ARS also affects the conduction velocity of myocardial tissue [68,104]. The degree to which these spatially distributed regions of heterogeneous action potential and conduction velocity can contribute to providing suitable arrhythmogenic substrates can only be fully realised by upscaling the modelling and simulation efforts of β-ARS to multiscale whole-ventricular, whole-atria, or whole-organ levels. In particular, the resources provided by the “Stimulating Peripheral Activity to Relieve Conditions” (SPARC) programme, including tools and curated and annotated high-resolution datasets in different species for investigations on autonomic nervous stimulation (Figure 4), may prove fundamental in understanding the role of a heterogeneous innervation in heart function [105].
However, there is a lack of computational studies to date implementing β-ARS models in 3D geometries. One of the reasons is the increased complexity of these models, which escalate the size of the state space by splitting the β-adrenergic targets into phosphorylated and unphosphorylated counterparts. Nevertheless, the main reasons underpinning their reduced uptake in 3D studies might actually come from the enormous computational cost needed to conduct simulations in time scales of relevance for β-ARS effects, usually after several tens to hundreds of heartbeats. Current whole-heart human 3D models are often conformed by meshes of millions of elements, and a vast computational power would be required to solve all model equations on them for such long time scales [24,106].
The use of advanced mathematical techniques can play a significant role in the upscaling of β-ARS models to the whole-organ level. The recent work by Meyer et al., applying model reduction techniques by quasi-steady-state approximations, was able to simplify the β-ARS cascade of the Soltis and Saucerman model into a minimal version of only two ordinary differential equations while retaining most of the relevant features of the original model [50]. Another approach, already used in the study by Sanchez-Alonso et al. [75], is the direct implementation of the β-ARS effects in the model. The changes in the magnitude of the ionic currents that were observed in the Heijman model after PKA phosphorylation were applied to the electrophysiological model used for the 3D simulations. The computational cost is lower, as there is no coupling with the β-ARS model, and thus the number of equations that need to be solved is reduced. However, such an approach is suboptimal, as part of the changes induced by β-ARS (such as shifts in activation/inactivation curves, or the influence on gate activation kinetics) is effectively lost. A better solution could be the full implementation of the coupled model (electrophysiology + β-ARS), with a 3D model initialisation based on the state variables of the cellular model after the appropriate number of beats (depending on the level of β-adrenergic phosphorylation of interest). This initialisation approach, widely exploited in other 3D electrophysiological and electromechanical studies [107,108], could allow the simulation of different stages of the β-ARS cascade in whole 3D models.
6. Conclusions
Over the last two decades, great advances have been made towards the mechanistic modelling and multiscale simulation of the cardiac β-ARS response. While significant progress has already been accomplished, more complete and comprehensive descriptions of this physiologically crucial signalling process are expected to further contribute to an improved understanding of its role in healthy and pathological conditions. In particular, the integration of these β-ARS models into whole-heart computational models, linked with tissue electrophysiology and mechanics, is envisioned as the next crucial step in the realisation of their full potential to bring novel insights into the modulation of human cardiac function. These perspectives will qualify digital twin technologies with enhanced capabilities for arrhythmic risk stratification under physical exercise or emotional stress, as well as for the safety pharmacology and personalisation of therapies, such as beta-blockers, one of the most recurred treatments in patients of heart failure, atrial fibrillation, survivors of myocardial infarction, or at risk of suffering major adverse cardiac events.
Author Contributions
Conceptualization, R.D. and A.B.-O.; methodology, R.D.; software, R.D.; validation, R.D.; formal analysis, R.D. and A.B.-O.; investigation, R.D.; resources, A.B.-O.; data curation, R.D.; writing—original draft preparation, R.D.; writing—review and editing, R.D. and A.B.-O. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by a British Heart Foundation (BHF) Intermediate Basic Science Fellowship to A.B.-O. (FS/17/22/32644). The authors acknowledge additional support from an Infrastructure for Impact Award from the National Centre for the Replacement, Refinement and Reduction of Animals in Research (NC/P001076/1), and the Oxford BHF Centre of Research Excellence (RE/13/1/30181), PRACE for awarding us access to Piz Daint at the Swiss National Supercomputing Centre, Switzerland (ICEI-PRACE grants icp005 and icp013), and the use of the University of Oxford Advanced Research Computing (ARC) facility (http://dx.doi.org/10.5281/zenodo.22558) (accessed on 4 July 2021).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The codes used in this work can be found at: https://github.com/JQXGong/Ohara-beta-adrenergic (Gong et al. [56]) and https://github.com/saucermanlab/model-archive (Soltis and Saucerman [29]) (accessed on 4 July 2021).
Acknowledgments
We thank David Sampedro-Puente and Esther Pueyo, University of Zaragoza, Spain, for their help in the integration of the Xie et al. and O’Hara-Rudy models.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Lands, A.M.; Arnold, A.; McAuliff, J.P.; Luduena, F.P.; Brown, T.G. Differentiation of receptor systems activated by sympathomimetic amines. Nature 1967, 214, 597–598. [Google Scholar] [CrossRef] [PubMed]
- Lands, A.M.; Luduena, F.P.; Buzzo, H.J. Differentiation of receptors responsive to isoproterenol. Life Sci. 1967, 6, 2241–2249. [Google Scholar] [CrossRef]
- Coppini, R.; Ferrantini, C.; Yao, L.; Fan, P.; Del Lungo, M.; Stillitano, F.; Sartiani, L.; Tosi, B.; Suffredini, S.; Tesi, C.; et al. Late sodium current inhibition reverses electromechanical dysfunction in human hypertrophic cardiomyopathy. Circulation 2013, 127, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.R.; Chen, L.; Rao, M.; Chen, K.; Song, J.P.; Hu, S.S. A modified method for isolation of human cardiomyocytes to model cardiac diseases. J. Transl. Med. 2018, 16, 288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grandi, E.; Ripplinger, C.M. Antiarrhythmic mechanisms of beta blocker therapy. Pharmacol. Res. 2019, 146, 104274. [Google Scholar] [CrossRef] [PubMed]
- Pogwizd, S.M.; Schlotthauer, K.; Li, L.; Yuan, W.; Bers, D.M. Arrhythmogenesis and Contractile Dysfunction in Heart Failure. Circ. Res. 2001, 88, 1159–1167. [Google Scholar] [CrossRef] [Green Version]
- Desantiago, J.; Ai, X.; Islam, M.; Acuna, G.; Ziolo, M.T.; Bers, D.M.; Pogwizd, S.M. Arrhythmogenic effects of β2-adrenergic stimulation in the failing heart are attributable to enhanced sarcoplasmic reticulum Ca load. Circ. Res. 2008, 102, 1389–1397. [Google Scholar] [CrossRef] [Green Version]
- Myles, R.C.; Wang, L.; Kang, C.; Bers, D.M.; Ripplinger, C.M. Local β-adrenergic stimulation overcomes source-sink mismatch to generate focal arrhythmia. Circ. Res. 2012, 110, 1454–1464. [Google Scholar] [CrossRef] [Green Version]
- Ferrantini, C.; Pioner, J.M.; Mazzoni, L.; Gentile, F.; Tosi, B.; Rossi, A.; Belardinelli, L.; Tesi, C.; Palandri, C.; Matucci, R.; et al. Late sodium current inhibitors to treat exercise-induced obstruction in hypertrophic cardiomyopathy: An in vitro study in human myocardium. Br. J. Pharmacol. 2018, 175, 2635–2652. [Google Scholar] [CrossRef] [Green Version]
- Goldenberg, I.; Thottathil, P.; Lopes, C.M.; Moss, A.J.; McNitt, S.; Jin, O.U.; Robinson, J.L.; Zareba, W.; Ackerman, M.J.; Kaufman, E.S.; et al. Trigger-specific ion-channel mechanisms, risk factors, and response to therapy in type 1 long QT syndrome. Heart Rhythm 2012, 9, 49–56. [Google Scholar] [CrossRef]
- Uchi, J.; Rice, J.J.; Ruwald, M.H.; Parks, X.X.; Ronzier, E.; Moss, A.J.; Zareba, W.; Lopes, C.M. Impaired IKs channel activation by Ca2+-dependent PKC shows correlation with emotion/arousal-triggered events in LQT1. J. Mol. Cell. Cardiol. 2015, 79, 203–211. [Google Scholar] [CrossRef] [Green Version]
- Danielsen, T.K.; Manotheepan, R.; Sadredini, M.; Leren, I.S.; Edwards, A.G.; Vincent, K.P.; Lehnart, S.E.; Sejersted, O.M.; Sjaastad, I.; Haugaa, K.H.; et al. Arrhythmia initiation in catecholaminergic polymorphic ventricular tachycardia type 1 depends on both heart rate and sympathetic stimulation. PLoS ONE 2018, 13, e0207100. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.; Ko, K.S.; Rhee, B.D.; Han, J.; Kim, N. Different effects of prolonged β-adrenergic stimulation on heart and cerebral artery. Integr. Med. Res. 2014, 3, 204–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelhardt, S.; Hein, L.; Wiesmann, F.; Lohse, M.J. Progressive hypertrophy and heart failure in β1-adrenergic receptor transgenic mice. Proc. Natl. Acad. Sci. USA 1999, 96, 7059–7064. [Google Scholar] [CrossRef] [Green Version]
- Ostrom, R.S.; Naugle, J.E.; Hase, M.; Gregorian, C.; Swaney, J.S.; Insel, P.A.; Brunton, L.L.; Meszaros, J.G. Angiotensin II enhances adenylyl cyclase signaling via Ca2+/calmodulin: Gq-Ga cross-talk regulates collagen production in cardiac fibroblasts. J. Biol. Chem. 2003, 278, 24461–24468. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.M.; Antoons, G. Arrhythmogenic Mechanisms in Heart Failure: Linking β-Adrenergic Stimulation, Stretch, and Calcium. Front. Physiol. 2018, 9, 1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beuckelmann, D.J.; Erdmann, E. Ca2+-currents and intracellular [Ca2+]i-transients in single ventricular myocytes isolated from terminally failing human myocardium. In Cellular and Molecular Alterations in the Failing Human Heart; Steinkopff: Heidelberg, Germany, 1992; pp. 235–243. [Google Scholar] [CrossRef]
- Harding, S.E.; Jones, S.M.; O’Gara, P.; Vescovo, G.; Poole-Wilson, P.A. Reduced β-agonist sensitivity in single atrial cells from failing human hearts. Am. J. Physiol. Heart Circ. Physiol. 1990, 259. [Google Scholar] [CrossRef]
- Leosco, D.; Rengo, G.; Iaccarino, G.; Filippelli, A.; Lymperopoulos, A.; Zincarelli, C.; Fortunato, F.; Golino, L.; Marchese, M.; Esposito, G.; et al. Exercise training and β-blocker treatment ameliorate age-dependent impairment of β-adrenergic receptor signaling and enhance cardiac responsiveness to adrenergic stimulation. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, 1596–1603. [Google Scholar] [CrossRef] [Green Version]
- Lucia, C.D.; Eguchi, A.; Koch, W.J. New Insights in Cardiac β-Adrenergic Signaling During Heart Failure and Aging. Front. Pharmacol. 2018, 9, 904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozlik-Feldmann, R.; Kramer, H.H.; Wicht, H.; Feldmann, R.; Netz, H.; Reinhardt, D. Distribution of Myocardial β-Adrenoceptor Subtypes and Coupling to the Adenylate Cyclase in Children With Congenital Heart Disease and Implications for Treatment. J. Clin. Pharmacol. 1993, 33, 588–595. [Google Scholar] [CrossRef]
- Lazzerini, P.E.; Boutjdir, M.; Capecchi, P.L. COVID-19, Arrhythmic Risk, and Inflammation. Circulation 2020, 142, 7–9. [Google Scholar] [CrossRef] [Green Version]
- Sutanto, H.; Heijman, J. Beta-Adrenergic Receptor Stimulation Modulates the Cellular Proarrhythmic Effects of Chloroquine and Azithromycin. Front. Physiol. 2020, 11, 587709. [Google Scholar] [CrossRef]
- Corral-Acero, J.; Margara, F.; Marciniak, M.; Rodero, C.; Loncaric, F.; Feng, Y.; Gilbert, A.; Fernandes, J.F.; Bukhari, H.A.; Wajdan, A.; et al. The “Digital Twin” to enable the vision of precision cardiology. Eur. Heart J. 2020, 41, 4556–4564. [Google Scholar] [CrossRef]
- Woo, A.Y.H.; Xiao, R.P. β-Adrenergic receptor subtype signaling in heart: From bench to bedside. Acta Pharmacol. Sin. 2012, 33, 335–341. [Google Scholar] [CrossRef] [Green Version]
- Bers, D.M. Calcium cycling and signaling in cardiac myocytes. Annu. Rev. Physiol. 2008, 70, 23–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volders, P.G.A.; Stengl, M.; Van Opstal, J.M.; Gerlach, U.; Spätjens, R.L.H.M.G.; Beekman, J.D.M.; Sipido, K.R.; Vos, M.A. Probing the contribution of IKs to canine ventricular repolarization: Key role for β-adrenergic receptor stimulation. Circulation 2003, 107, 2753–2760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baba, S.; Dun, W.; Boyden, P.A. Can PKA activators rescue Na+ channel function in epicardial border zone cells that survive in the infarcted canine heart? Cardiovasc. Res. 2004, 64, 260–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soltis, A.R.; Saucerman, J.J. Synergy between CaMKII substrates and β-adrenergic signaling in regulation of cardiac myocyte Ca2+ handling. Biophys. J. 2010, 99, 2038–2047. [Google Scholar] [CrossRef] [Green Version]
- Negroni, J.A.; Morotti, S.; Lascano, E.C.; Gomes, A.V.; Grandi, E.; Puglisi, J.L.; Bers, D.M. Β-Adrenergic Effects on Cardiac Myofilaments and Contraction in an Integrated Rabbit Ventricular Myocyte Model. J. Mol. Cell. Cardiol. 2015, 81, 162–175. [Google Scholar] [CrossRef] [Green Version]
- Despa, S.; Bossuyt, J.; Han, F.; Ginsburg, K.S.; Jia, L.G.; Kutchai, H.; Tucker, A.L.; Bers, D.M. Phospholemman-phosphorylation mediates the β-adrenergic effects on Na/K pump function in cardiac myocytes. Circ. Res. 2005, 97, 252–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginsburg, K.S.; Bers, D.M. Modulation of excitation-contraction coupling by isoproterenol in cardiomyocytes with controlled SR Ca2+ load and Ca2+ current trigger. J. Physiol. 2004, 556, 463–480. [Google Scholar] [CrossRef]
- Stelzer, J.E.; Patel, J.R.; Walker, J.W.; Moss, R.L. Differential roles of cardiac myosin-binding protein C and cardiac troponin I in the myofibrillar force responses to protein kinase A phosphorylation. Circ. Res. 2007, 101, 503–511. [Google Scholar] [CrossRef] [Green Version]
- Krüger, M.; Linke, W.A. Protein kinase-A phosphorylates titin in human heart muscle and reduces myofibrillar passive tension. J. Muscle Res. Cell Motil. 2006, 27, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Rudy, Y. Early afterdepolarizations in cardiac myocytes: Mechanism and rate dependence. Biophys. J. 1995, 68, 949–964. [Google Scholar] [CrossRef] [Green Version]
- Saucerman, J.J.; Brunton, L.L.; Michailova, A.P.; McCulloch, A.D. Modeling β-Adrenergic Control of Cardiac Myocyte Contractility in Silico. J. Biol. Chem. 2003, 278, 47997–48003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenstein, J.L.; Tanskanen, A.J.; Winslow, R.L. Modeling the actions of β-adrenergic signaling on excitation- contraction coupling processes. Ann. N. Y. Acad. Sci. 2004, 1015, 16–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iancu, R.V.; Jones, S.W.; Harvey, R.D. Compartmentation of cAMP signaling in cardiac myocytes: A computational study. Biophys. J. 2007, 92, 3317–3331. [Google Scholar] [CrossRef] [Green Version]
- Heijman, J.; Volders, P.G.A.; Westra, R.L.; Rudy, Y. Local control of β-adrenergic stimulation: Effects on ventricular myocyte electrophysiology and Ca2+-transient. J. Mol. Cell. Cardiol. 2011, 50, 863–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bondarenko, V.E. A Compartmentalized Mathematical Model of the β1-Adrenergic Signaling System in Mouse Ventricular Myocytes. PLoS ONE 2014, 9, e89113. [Google Scholar] [CrossRef] [Green Version]
- Khalilimeybodi, A.; Daneshmehr, A.; Sharif-Kashani, B. Investigating β-adrenergic-induced cardiac hypertrophy through computational approach: Classical and non-classical pathways. J. Physiol. Sci. 2018, 68, 503–520. [Google Scholar] [CrossRef]
- Tomek, J.; Bueno-Orovio, A.; Passini, E.; Zhou, X.; Minchole, A.; Britton, O.; Bartolucci, C.; Severi, S.; Shrier, A.; Virag, L.; et al. Development, calibration, and validation of a novel human ventricular myocyte model in health, disease, and drug block. eLife 2019, 8, e48890. [Google Scholar] [CrossRef]
- Tanskanen, A.J.; Greenstein, J.L.; O’Rourke, B.; Winslow, R.L. The role of stochastic and modal gating of cardiac L-type Ca2+ channels on early after-depolarizations. Biophys. J. 2005, 88, 85–95. [Google Scholar] [CrossRef] [Green Version]
- Saucerman, J.J.; McCulloch, A.D. Mechanistic systems models of cell signaling networks: A case study of myocyte adrenergic regulation. Prog. Biophys. Mol. Biol. 2004, 85, 261–278. [Google Scholar] [CrossRef]
- Zaccolo, M.; Pozzan, T. Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes. Science 2002, 295, 1711–1715. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, V.; Pak, E.; Robinson, R.B.; Steinberg, S.F. β2-Adrenergic receptor actions in neonatal and adult rat ventricular myocytes. Circ. Res. 1995, 76, 40–52. [Google Scholar] [CrossRef]
- Vittone, L.; Mundiña-Weilenmann, C.; Said, M.; Mattiazzi, A. Mechanisms involved in the acidosis enhancement of the isoproterenol-induced phosphorylation of phospholamban in the intact heart. J. Biol. Chem. 1998, 273, 9804–9811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, R.P.; Lakatta, E.G. β1-Adrenoceptor stimulation and β2-adrenoceptor stimulation differ in their effects on contraction, cytosolic Ca2+, and Ca2+ current in single rat ventricular cells. Circ. Res. 1993, 73, 286–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.H.; Saucerman, J.J. Phospholemman is a negative feed-forward regulator of Ca2+ in β-adrenergic signaling, accelerating β-adrenergic inotropy. J. Mol. Cell. Cardiol. 2012, 52, 1048–1055. [Google Scholar] [CrossRef] [Green Version]
- Meyer, E.E.; Clancy, C.E.; Lewis, T.J. Dynamics of adrenergic signaling in cardiac myocytes and implications for pharmacological treatment. J. Theor. Biol. 2021, 519, 110619. [Google Scholar] [CrossRef]
- Warrier, S.; Belevych, A.E.; Ruse, M.; Eckert, R.L.; Zaccolo, M.; Pozzan, T.; Harvey, R.D. β-adrenergic- and muscarinic receptor-induced changes in cAMP activity in adult cardiac myocytes detected with FRET-based biosensor. Am. J. Physiol. Cell Physiol. 2005, 289, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Hohl, C.M.; Li, Q. Compartmentation of cAMP in adult canine ventricular myocytes: Relation to single-cell free Ca2+ transients. Circ. Res. 1991, 69, 1369–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagykaldi, Z.; Kem, D.; Lazzara, R.; Szabo, B. Canine ventricular myocyte β2-adrenoceptors are not functionally coupled to L-type calcium current. J. Cardiovasc. Electrophysiol. 1999, 10, 1240–1251. [Google Scholar] [CrossRef]
- Johnson, D.M.; Heijman, J.; Pollard, C.E.; Valentin, J.P.; Crijns, H.J.G.M.; Abi-Gerges, N.; Volders, P.G.A. IKs restricts excessive beat-to-beat variability of repolarization during beta-adrenergic receptor stimulation. J. Mol. Cell. Cardiol. 2010, 48, 122–130. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, T.; Rudy, Y. Arrhythmia formation in subclinical (“silent”) long QT syndrome requires multiple insults: Quantitative mechanistic study using the KCNQ1 mutation Q357R as example. Heart Rhythm 2012, 9, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Gong, J.Q.X.; Susilo, M.E.; Sher, A.; Musante, C.J.; Sobie, E.A. Quantitative analysis of variability in an integrated model of human ventricular electrophysiology and β-adrenergic signaling. J. Mol. Cell. Cardiol. 2020, 143, 96–106. [Google Scholar] [CrossRef]
- Robertson, S.P.; Johnson, J.D.; Holroyde, M.J.; Kraniass, E.G.; Potter, J.D.; Solaro, R.J. The Effect of Troponin I Phosphorylation on the Ca2+-binding of Bovine Cardiac Troponin * Properties of the Ca2+-regulatory Site. J. Biol. Chem. 1982, 257, 260–263. [Google Scholar] [CrossRef]
- Xie, Y.; Grandi, E.; Puglisi, J.L.; Sato, D.; Bers, D.M. β-adrenergic stimulation activates early afterdepolarizations transiently via kinetic mismatch of PKA targets. J. Mol. Cell. Cardiol. 2013, 58, 153–161. [Google Scholar] [CrossRef] [Green Version]
- O’Hara, T.; Virág, L.; Varró, A.; Rudy, Y. Simulation of the Undiseased Human Cardiac Ventricular Action Potential: Model Formulation and Experimental Validation. PLoS Comput. Biol. 2011, 7, e1002061. [Google Scholar] [CrossRef] [Green Version]
- Pueyo, E.; Orini, M.; Rodríguez, J.F.; Taggart, P. Interactive effect of beta-adrenergic stimulation and mechanical stretch on low-frequency oscillations of ventricular action potential duration in humans. J. Mol. Cell. Cardiol. 2016, 97, 93–105. [Google Scholar] [CrossRef] [Green Version]
- Dai, L.; Zang, Y.; Zheng, D.; Luo, R.; Xia, L. Modelling the integrated regulation role of β-adrenergic signaling and CaMKII in human myocyte electrophysiological properties. Comput. Cardiol. 2013, 40, 389–392. [Google Scholar]
- Kuzumoto, M.; Takeuchi, A.; Nakai, H.; Oka, C.; Noma, A.; Matsuoka, S. Simulation analysis of intracellular Na+ and Cl− homeostasis during β1-adrenergic stimulation of cardiac myocyte. Prog. Biophys. Mol. Biol. 2008, 96, 171–186. [Google Scholar] [CrossRef]
- Surdo, N.C.; Berrera, M.; Koschinski, A.; Brescia, M.; Machado, M.R.; Carr, C.; Wright, P.; Gorelik, J.; Morotti, S.; Grandi, E.; et al. FRET biosensor uncovers cAMP nano-domains at β-adrenergic targets that dictate precise tuning of cardiac contractility. Nat. Commun. 2017, 8, 15031. [Google Scholar] [CrossRef]
- Lyon, A.; Dupuis, L.J.; Arts, T.; Crijns, H.J.G.M.; Prinzen, F.W.; Delhaas, T.; Heijman, J.; Lumens, J. Differentiating the effects of β-adrenergic stimulation and stretch on calcium and force dynamics using a novel electromechanical cardiomyocyte model. Am. J. Physiol. Heart Circ. Physiol. 2020, 319, H519–H530. [Google Scholar] [CrossRef]
- Dupuis, L.J.; Lumens, J.; Arts, T.; Delhaas, T. High tension in sarcomeres hinders myocardial relaxation: A computational study. PLoS ONE 2018, 13, e0204642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moss, A.J.; Zareba, W. Long QT syndrome: Therapeutic considerations. In Cardiac Electrophysiology: From Cell to Bedside; Elsevier Inc.: Amsterdam, The Netherlands, 2004; pp. 660–667. ISBN 9780721603230. [Google Scholar]
- Terrenoire, C.; Clancy, C.E.; Cormier, J.W.; Sampson, K.J.; Kass, R.S. Autonomic control of cardiac action potentials: Role of potassium channel kinetics in response to sympathetic stimulation. Circ. Res. 2005, 96, e25–e34. [Google Scholar] [CrossRef] [Green Version]
- Kang, C.; Badiceanu, A.; Brennan, J.A.; Gloschat, C.; Qiao, Y.; Trayanova, N.A.; Efimov, I.R. β-adrenergic stimulation augments transmural dispersion of repolarization via modulation of delayed rectifier currents IKs and IKr in the human ventricle. Sci. Rep. 2017, 7, 15922. [Google Scholar] [CrossRef]
- Kilfoil, P.J.; Lotteau, S.; Zhang, R.; Yue, X.; Aynaszyan, S.; Solymani, R.E.; Cingolani, E.; Marbán, E.; Goldhaber, J.I. Distinct features of calcium handling and β-adrenergic sensitivity in heart failure with preserved versus reduced ejection fraction. J. Physiol. 2020, 598, 5091–5108. [Google Scholar] [CrossRef]
- Morotti, S.; Edwards, A.G.; Mcculloch, A.D.; Bers, D.M.; Grandi, E. A novel computational model of mouse myocyte electrophysiology to assess the synergy between Na+ loading and CaMKII. J. Physiol. 2014, 592, 1181–1197. [Google Scholar] [CrossRef] [PubMed]
- Passini, E.; Mincholé, A.; Coppini, R.; Cerbai, E.; Rodriguez, B.; Severi, S.; Bueno-Orovio, A. Mechanisms of pro-arrhythmic abnormalities in ventricular repolarisation and anti-arrhythmic therapies in human hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 2016, 96, 72–81. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Bueno-Orovio, A.; Orini, M.; Hanson, B.; Hayward, M.; Taggart, P.; Lambiase, P.D.; Burrage, K.; Rodriguez, B. In Vivo and in Silico Investigation into Mechanisms of Frequency Dependence of Repolarization Alternans in Human Ventricular Cardiomyocytes. Circ. Res. 2016, 118, 266–278. [Google Scholar] [CrossRef]
- Mora, M.T.; Gong, J.Q.X.; Sobie, E.A.; Trenor, B. The role of β-adrenergic system remodeling in human heart failure: A mechanistic investigation. J. Mol. Cell. Cardiol. 2021, 153, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Loucks, A.D.; O’Hara, T.; Trayanova, N.A. Degradation of T-Tubular Microdomains and Altered cAMP Compartmentation Lead to Emergence of Arrhythmogenic Triggers in Heart Failure Myocytes: An in silico Study. Front. Physiol. 2018, 9, 1737. [Google Scholar] [CrossRef]
- Sanchez-Alonso, J.L.; Loucks, A.; Schobesberger, S.; van Cromvoirt, A.M.; Poulet, C.; Chowdhury, R.A.; Trayanova, N.; Gorelik, J. Nanoscale regulation of L-type calcium channels differentiates between ischemic and dilated cardiomyopathies. EBioMedicine 2020, 57, 102845. [Google Scholar] [CrossRef]
- Iyer, V.; Hajjar, R.J.; Armoundas, A.A. Mechanisms of abnormal calcium homeostasis in mutations responsible for catecholaminergic polymorphic ventricular tachycardia. Circ. Res. 2007, 100, 22–31. [Google Scholar] [CrossRef] [Green Version]
- Tomek, J.; Rodriguez, B.; Bub, G.; Heijman, J. β-Adrenergic receptor stimulation inhibits proarrhythmic alternans in postinfarction border zone cardiomyocytes: A computational analysis. Am. J. Physiol. Heart. Circ. Physiol. 2017, 313, H338–H353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomek, J.; Hao, G.; Tomková, M.; Lewis, A.; Carr, C.; Paterson, D.J.; Rodriguez, B.; Bub, G.; Herring, N. β-Adrenergic Receptor Stimulation and Alternans in the Border Zone of a Healed Infarct: An ex vivo Study and Computational Investigation of Arrhythmogenesis. Front. Physiol. 2019, 10, 350. [Google Scholar] [CrossRef] [Green Version]
- Tsumoto, K.; Ashihara, T.; Naito, N.; Shimamoto, T.; Amano, A.; Kurata, Y.; Kurachi, Y. Specific decreasing of Na+ channel expression on the lateral membrane of cardiomyocytes causes fatal arrhythmias in Brugada syndrome. Sci. Rep. 2020, 10, 19964. [Google Scholar] [CrossRef]
- Sampedro-Puente, D.A.; Fernandez-Bes, J.; Szentandrássy, N.; Nánási, P.; Taggart, P.; Pueyo, E. Time Course of Low-Frequency Oscillatory Behavior in Human Ventricular Repolarization Following Enhanced Sympathetic Activity and Relation to Arrhythmogenesis. Front. Physiol. 2020, 10, 1547. [Google Scholar] [CrossRef] [Green Version]
- Brodde, O.E.; Bruck, H.; Leineweber, K.; Seyfarth, T. Presence, distribution and physiological function of adrenergic and muscarinic receptor subtypes in the human heart. Basic Res. Cardiol. 2001, 96, 528–538. [Google Scholar] [CrossRef] [PubMed]
- McCabe, K.J.; Rangamani, P. Computational modeling approaches to cAMP/PKA signaling in cardiomyocytes. J. Mol. Cell. Cardiol. 2021, 154, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Judina, A.; Gorelik, J.; Wright, P.T. Studying signal compartmentation in adult cardiomyocytes. Biochem. Soc. Trans. 2020, 48, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Schobesberger, S.; Wright, P.; Tokar, S.; Bhargava, A.; Mansfield, C.; Glukhov, A.V.; Poulet, C.; Buzuk, A.; Monszpart, A.; Sikkel, M.; et al. T-tubule remodelling disturbs localized β2-adrenergic signalling in rat ventricular myocytes during the progression of heart failure. Cardiovasc. Res. 2017, 113, 770–782. [Google Scholar] [CrossRef]
- Ruzsnavszky, F.; Hegyi, B.; Kistamás, K.; Váczi, K.; Horváth, B.; Szentandrássy, N.; Bányász, T.; Nánási, P.P.; Magyar, J. Asynchronous activation of calcium and potassium currents by isoproterenol in canine ventricular myocytes. Naunyn. Schmiedebergs. Arch. Pharmacol. 2014, 387, 457–467. [Google Scholar] [CrossRef]
- Liu, G.X.; Choi, B.R.; Ziv, O.; Li, W.; de Lange, E.; Qu, Z.; Koren, G. Differential conditions for early after-depolarizations and triggered activity in cardiomyocytes derived from transgenic LQT1 and LQT2 rabbits. J. Physiol. 2012, 590, 1171–1180. [Google Scholar] [CrossRef]
- Bers, D.M.; Zaccolo, M. Whole-Cell cAMP and PKA Activity are Epiphenomena, Nanodomain Signaling Matters. Physiology 2019, 34, 240–249. [Google Scholar] [CrossRef]
- Shugg, T.; Hudmon, A.; Overholser, B.R. Neurohormonal Regulation of IKs in Heart Failure: Implications for Ventricular Arrhythmogenesis and Sudden Cardiac Death. J. Am. Heart Assoc. 2020, 9, e016900. [Google Scholar] [CrossRef] [PubMed]
- Morales, D.; Hermosilla, T.; Varela, D. Calcium-dependent inactivation controls cardiac L-type Ca2+ currents under β-adrenergic stimulation. J. Gen. Physiol. 2018, 151, 786–797. [Google Scholar] [CrossRef] [Green Version]
- Treinys, R.; Kanaporis, G.; Fischmeister, R.; Jurevičius, J. Metabolic inhibition induces transient increase of L-type Ca2+ current in human and rat cardiac myocytes. Int. J. Mol. Sci. 2019, 20, 1501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, A.G.; Louch, W.E. Species-Dependent Mechanisms of Cardiac Arrhythmia: A Cellular Focus. Clin. Med. Insights Cardiol. 2017, 11. [Google Scholar] [CrossRef] [PubMed]
- Varshneya, M.; Devenyi, R.A.; Sobie, E.A. Slow Delayed Rectifier Current Protects Ventricular Myocytes From Arrhythmic Dynamics Across Multiple Species. Circ. Arrhythm. Electrophysiol. 2018, 11, e006558. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Morotti, S.; Tapa, S.; Stuart, S.D.F.; Jiang, Y.; Bers, D.M.; Wang, Z.; Myles, R.C.; Brack, K.E.; Andr, G.; et al. Different paths, same destination: Divergent action potential responses produce conserved cardiac fight-or-flight response in mouse and rabbit hearts. J. Physiol. 2019, 15, 3867–3883. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Shen, Y.T.; Kalthof, B.; Iwase, M.; Sato, N.; Uechi, M.; Vatner, S.F.; Vatner, D.E. Identification and functional role of β-adrenergic receptor subtypes in primate and rodent: In vivo versus isolated myocytes. J. Mol. Cell. Cardiol. 1996, 28, 1307–1317. [Google Scholar] [CrossRef]
- Chen, P.S.; Chen, L.S.; Fishbein, M.C.; Lin, S.F.; Nattel, S. Role of the autonomic nervous system in atrial fibrillation: Pathophysiology and therapy. Circ. Res. 2014, 114, 1500–1515. [Google Scholar] [CrossRef] [Green Version]
- Hindricks, G.; Potpara, T.; Dagres, N.; Bax, J.J.; Boriani, G.; Dan, G.A.; Fauchier, L.; Kalman, J.M.; Lane, D.A.; Lettino, M.; et al. 2020 ESC Guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European Association for Cardio-Thoracic Surgery (EACTS). Eur. Heart J. 2021, 42, 373–498. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, S.; Wang, W.; Wang, K.; Shen, W. A Mathematical Model of the Mouse Atrial Myocyte With Inter-Atrial Electrophysiological Heterogeneity. Front. Physiol. 2020, 11, 972. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, W.; Wang, K.; Shen, W.; Zhang, H. Alternans in Mouse Atrial Cardiomyocytes: A Computational Study on the Influence of Cell-Cell Coupling and β-Adrenergic Stimulation. IEEE Access 2020, 8, 84806–84820. [Google Scholar] [CrossRef]
- Kneller, J.; Zou, R.; Vigmond, E.J.; Wang, Z.; Leon, L.J.; Nattel, S. Cholinergic Atrial Fibrillation in a Computer Model of a Two-Dimensional Sheet of Canine Atrial Cells With Realistic Ionic Properties. Circ. Res. 2002, 90, e73–e87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matene, E.; Vinet, A.; Jacquemet, V. Dynamics of atrial arrhythmias modulated by time-dependent acetylcholine concentration: A simulation study. Europace 2014, 16, iv11–iv20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayer, J.D.; Boukens, B.J.; Krul, S.P.J.; Roney, C.H.; Driessen, A.H.G.; Berger, W.R.; van den Berg, N.W.E.; Verkerk, A.O.; Vigmond, E.J.; Coronel, R.; et al. Acetylcholine Delays Atrial Activation to Facilitate Atrial Fibrillation. Front. Physiol. 2019, 10, 1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawano, H.; Okada, R.; Yano, K. Histological study on the distribution of autonomic nerves in the human heart. Heart Vessels 2003, 18, 32–39. [Google Scholar] [CrossRef]
- Di Bona, A.; Vita, V.; Costantini, I.; Zaglia, T. Towards a clearer view of sympathetic innervation of cardiac and skeletal muscles. Prog. Biophys. Mol. Biol. 2020, 154, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Ajijola, O.A.; Lux, R.L.; Khahera, A.; Kwon, O.; Aliotta, E.; Ennis, D.B.; Fishbein, M.C.; Ardell, J.L.; Shivkumar, K. Sympathetic modulation of electrical activation in normal and infracted myocardium: Implications for arrhythmogenesis. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H608–H621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osanlouy, M.; Bandrowski, A.; de Bono, B.; Brooks, D.; Cassarà, A.M.; Christie, R.; Ebrahimi, N.; Gillespie, T.; Grethe, J.S.; Guercio, L.A.; et al. The SPARC DRC: Building a Resource for the Autonomic Nervous System Community. Front. Physiol. 2021, 12, 693735. [Google Scholar] [CrossRef] [PubMed]
- Niederer, S.A.; Lumens, J.; Trayanova, N.A. Computational models in cardiology. Nat. Rev. Cardiol. 2019, 16, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.J.; Santiago, A.; Zhou, X.; Wang, L.; Margara, F.; Levrero-Florencio, F.; Das, A.; Kelly, C.; Dallarmellina, E.; Vazquez, M.; et al. Human biventricular electromechanical simulations on the progression of electrocardiographic and mechanical abnormalities in post-myocardial infarction. Europace 2021, 23, I143–I152. [Google Scholar] [CrossRef] [PubMed]
- Levrero-Florencio, F.; Margara, F.; Zacur, E.; Bueno-Orovio, A.; Wang, Z.J.; Santiago, A.; Aguado-Sierra, J.; Houzeaux, G.; Grau, V.; Kay, D.; et al. Sensitivity analysis of a strongly-coupled human-based electromechanical cardiac model: Effect of mechanical parameters on physiologically relevant biomarkers. Comput. Methods Appl. Mech. Eng. 2020, 361, 112762. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic representation of the ionic currents and subcellular targets considered in modelling studies of β-ARS and the subsequent PKA phosphorylation cascade. The currents marked with an encircled P represent the different PKA targets. Adapted from [42] under the Creative Commons Attribution (CC BY 4.0) license.
Figure 1.
Schematic representation of the ionic currents and subcellular targets considered in modelling studies of β-ARS and the subsequent PKA phosphorylation cascade. The currents marked with an encircled P represent the different PKA targets. Adapted from [42] under the Creative Commons Attribution (CC BY 4.0) license.

Figure 2.
(A) Phosphorylation curves of the different PKA targets in the Xie model [58] (left) and in the Gong model [56] (right). (B) Evolution of action potential duration (APD) in each case. Phosphorylation curves and APD were calculated using the O’Hara-Rudy model [59] of a human endocardial myocyte, paced at 1 Hz, with simulated ISO administration after 50 beats.
Figure 2.
(A) Phosphorylation curves of the different PKA targets in the Xie model [58] (left) and in the Gong model [56] (right). (B) Evolution of action potential duration (APD) in each case. Phosphorylation curves and APD were calculated using the O’Hara-Rudy model [59] of a human endocardial myocyte, paced at 1 Hz, with simulated ISO administration after 50 beats.

Figure 3.
Action potential (AP), intracellular calcium transient (Ca2+), and different ionic currents and intracellular calcium fluxes obtained using the O’Hara-Rudy model without β-ARS (red, continuous lines) [59] and after the simulation of the β-ARS response using the Gong [56] (yellow, dashed line) and the Xie [58] (blue, dotted lines) models. Results are presented for the last beat under the pacing protocol described in Figure 2. The ionic currents and intracellular calcium fluxes shown are: L-type calcium current (ICaL); fast sodium current (INa); late sodium current (INaL); sodium–calcium exchanger (INaCa); sodium–potassium pump (INaK); rapid delayed rectifier potassium current (IKr); slow delayed rectifier potassium current (IKs); transient outward potassium current (Ito); calcium uptake via the SERCA pump (Jup) and ryanodine receptor calcium release (Jrel).
Figure 3.
Action potential (AP), intracellular calcium transient (Ca2+), and different ionic currents and intracellular calcium fluxes obtained using the O’Hara-Rudy model without β-ARS (red, continuous lines) [59] and after the simulation of the β-ARS response using the Gong [56] (yellow, dashed line) and the Xie [58] (blue, dotted lines) models. Results are presented for the last beat under the pacing protocol described in Figure 2. The ionic currents and intracellular calcium fluxes shown are: L-type calcium current (ICaL); fast sodium current (INa); late sodium current (INaL); sodium–calcium exchanger (INaCa); sodium–potassium pump (INaK); rapid delayed rectifier potassium current (IKr); slow delayed rectifier potassium current (IKs); transient outward potassium current (Ito); calcium uptake via the SERCA pump (Jup) and ryanodine receptor calcium release (Jrel).

Figure 4.
Connectivity diagrams of autonomic nervous systems for individual species, available through the SPARC portal (https://sparc.science/maps) (accessed on 4 July 2021). Reproduced from [105] under Creative Commons Attribution (CC BY 4.0) license.
Figure 4.
Connectivity diagrams of autonomic nervous systems for individual species, available through the SPARC portal (https://sparc.science/maps) (accessed on 4 July 2021). Reproduced from [105] under Creative Commons Attribution (CC BY 4.0) license.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Doste, R.; Bueno-Orovio, A. Multiscale Modelling of β-Adrenergic Stimulation in Cardiac Electromechanical Function. Mathematics 2021, 9, 1785. https://doi.org/10.3390/math9151785
AMA Style
Doste R, Bueno-Orovio A. Multiscale Modelling of β-Adrenergic Stimulation in Cardiac Electromechanical Function. Mathematics. 2021; 9(15):1785. https://doi.org/10.3390/math9151785
Chicago/Turabian StyleDoste, Ruben, and Alfonso Bueno-Orovio. 2021. "Multiscale Modelling of β-Adrenergic Stimulation in Cardiac Electromechanical Function" Mathematics 9, no. 15: 1785. https://doi.org/10.3390/math9151785
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.