NMR Spectroscopy for Metabolomics Research

 , , , ,
, , , ,  , , ,
, , , 
Abstract
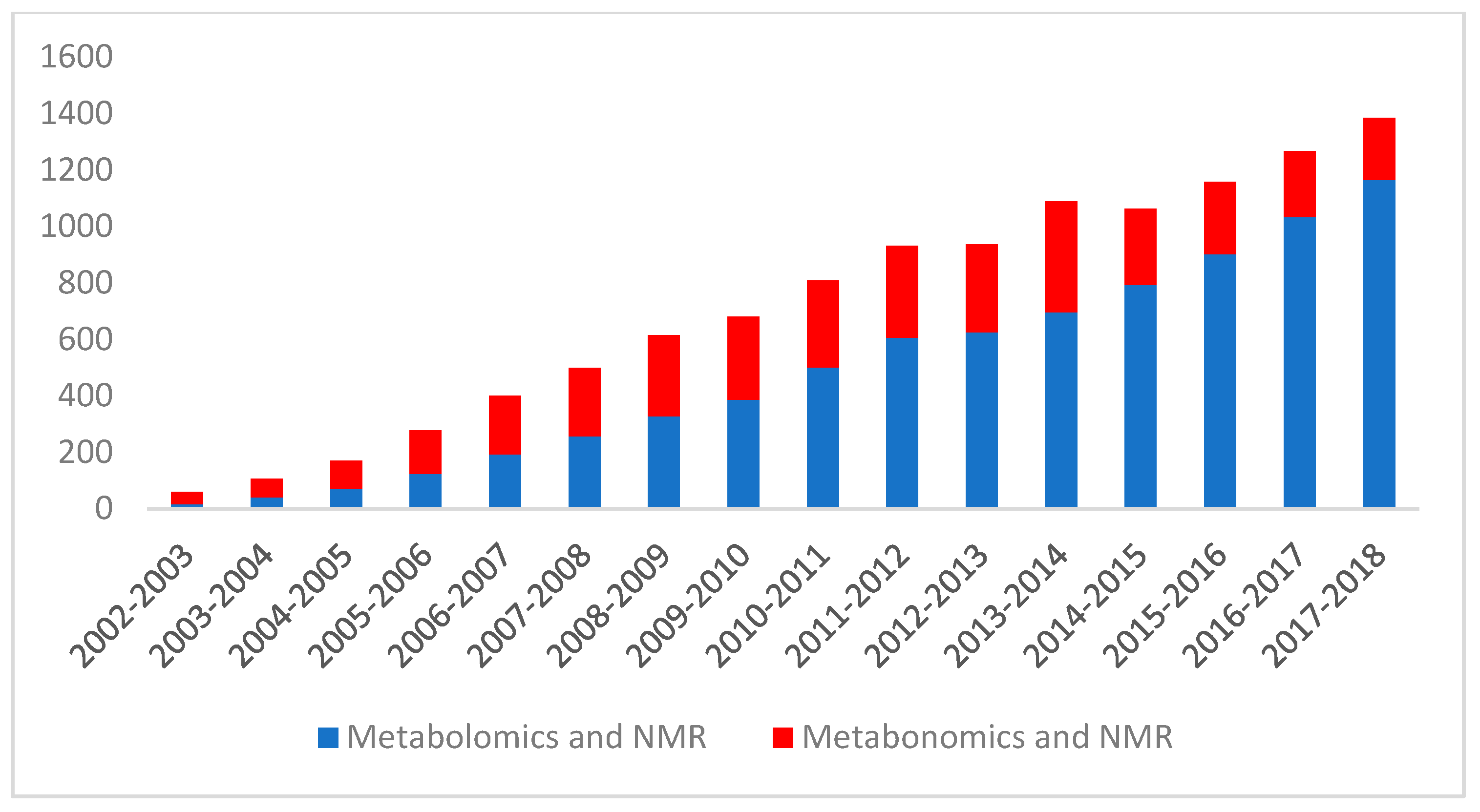
:1. Introduction
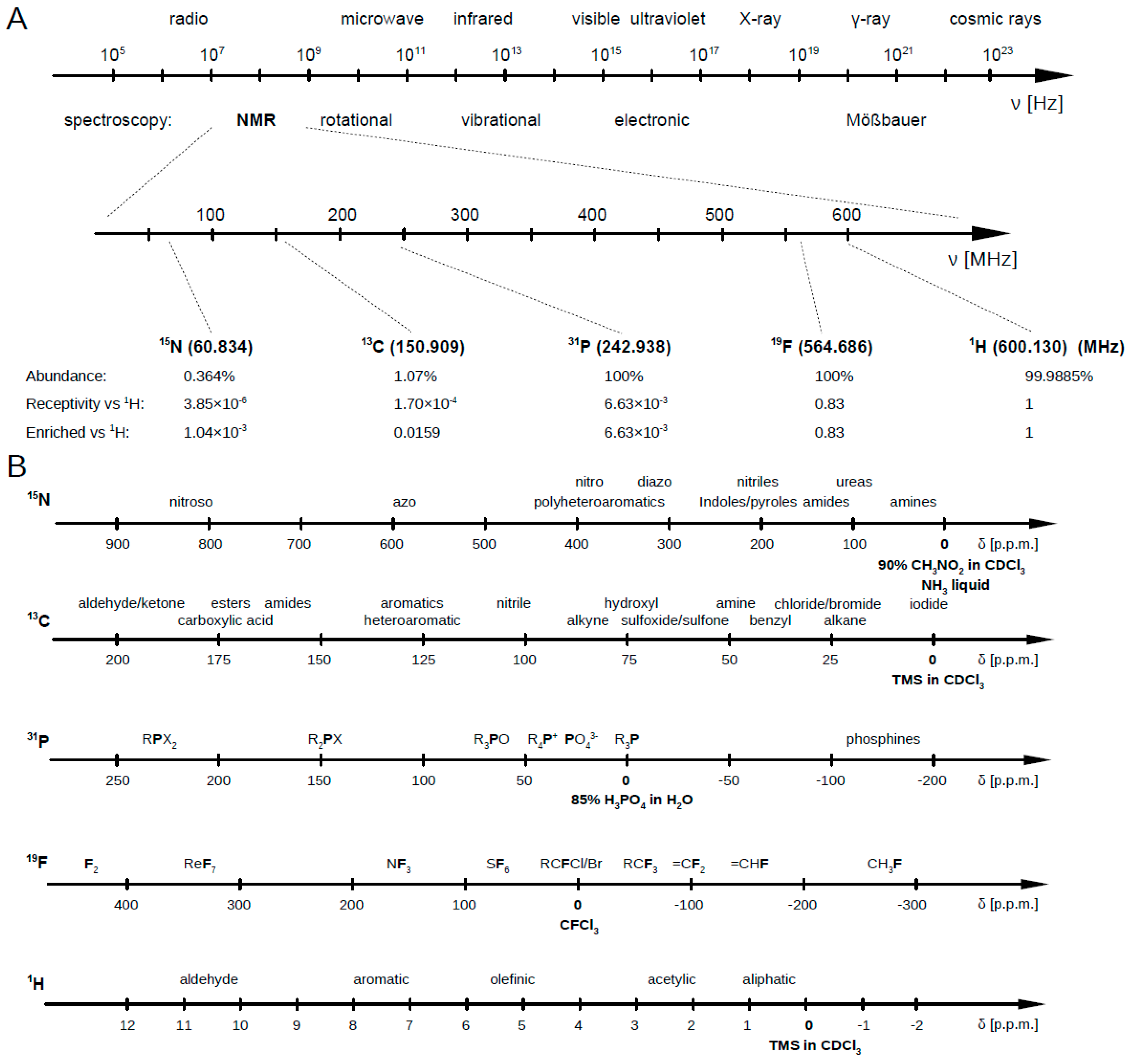
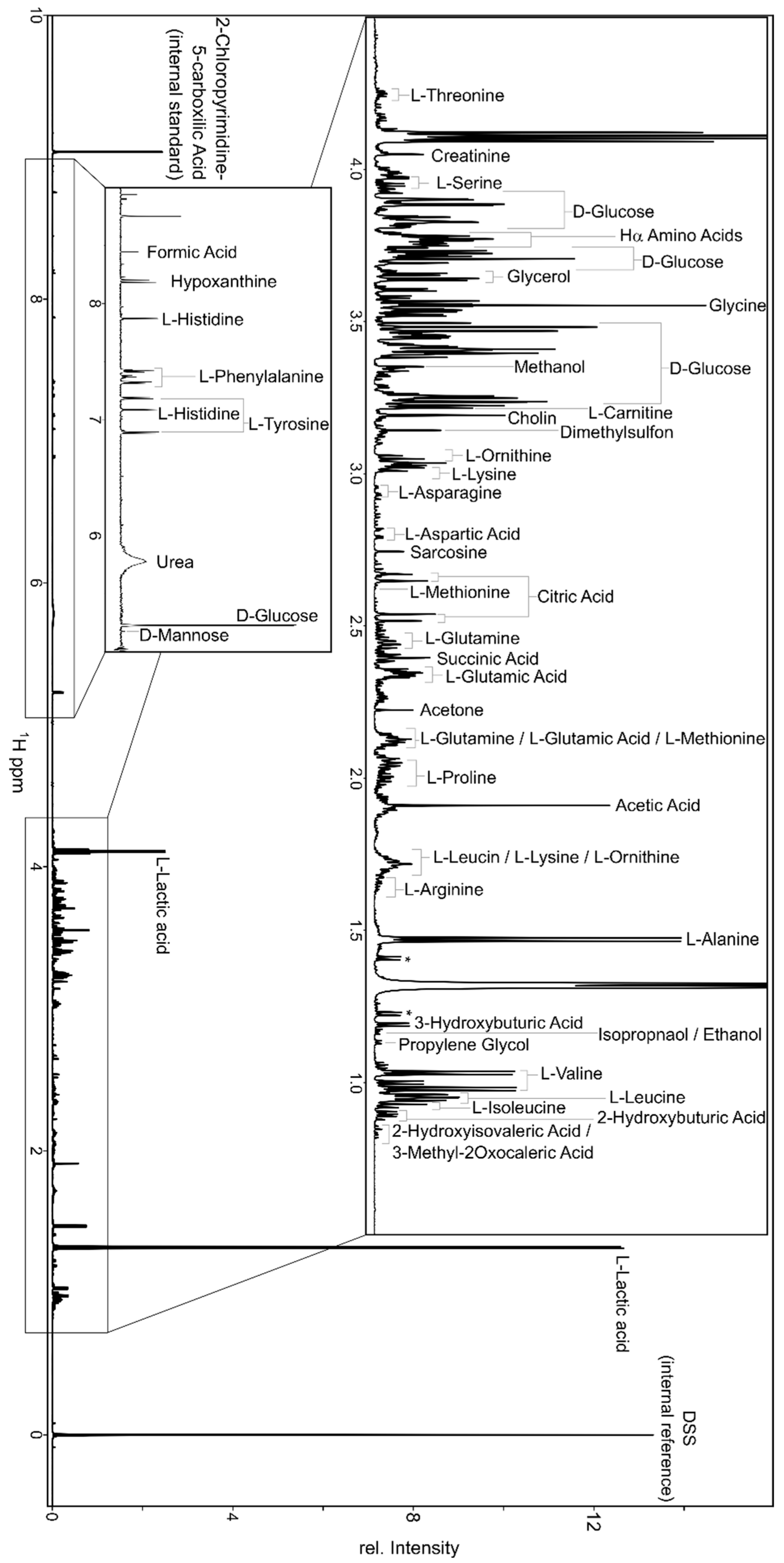
2. 1H NMR Spectroscopy for Metabolomics
2.1. 13C NMR Spectroscopy for Metabolomics
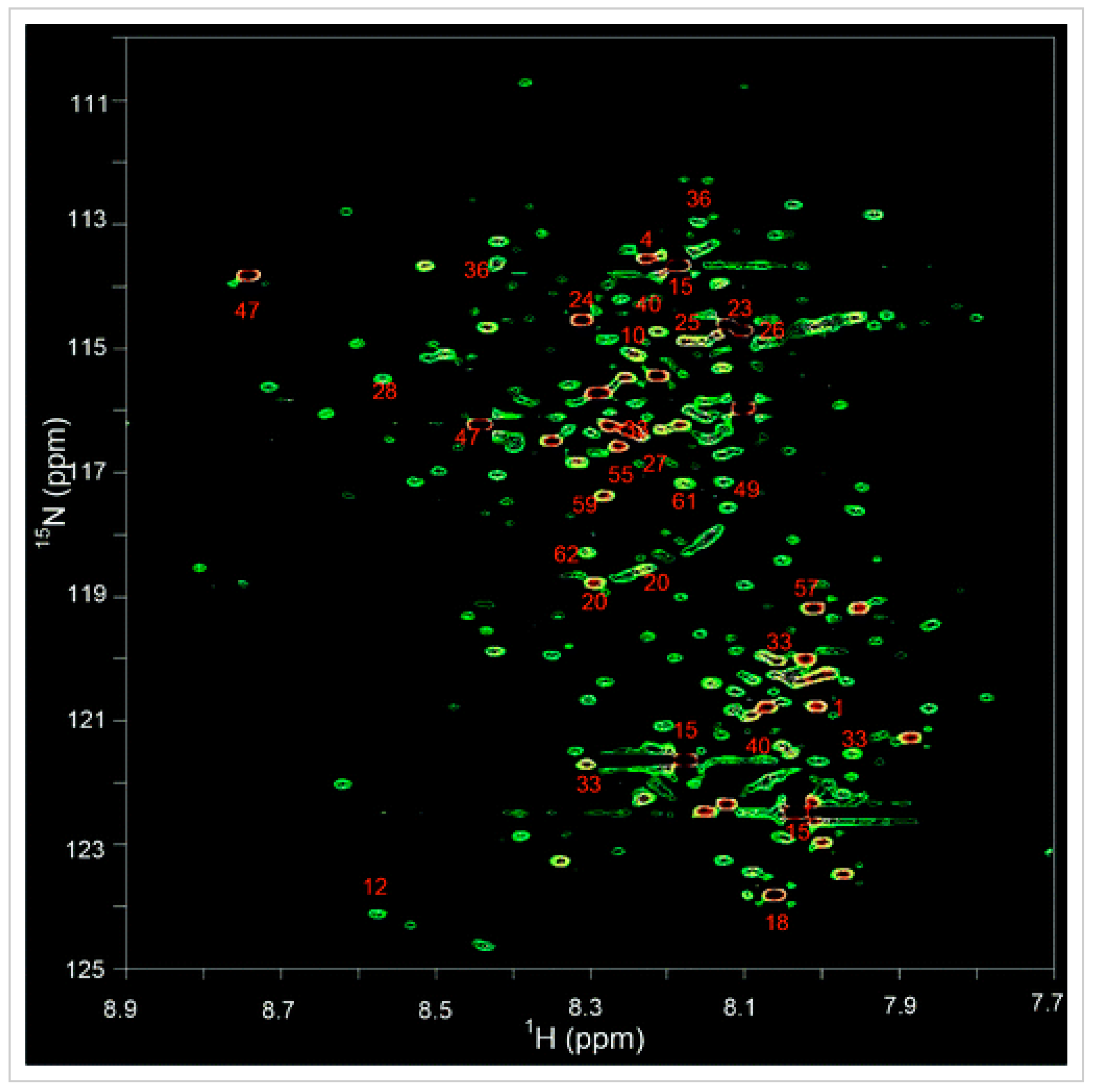
2.2. 15N NMR Spectroscopy for Metabolomics
2.3. 31P NMR Spectroscopy for Metabolomics
3. Two-Dimensional (2D) NMR Spectroscopy
3.1. Correlation Spectroscopy (COSY)
3.2. Total Correlation Spectroscopy (TOCSY)
3.3. 2D J-Resolved Spectroscopy (J-Res)
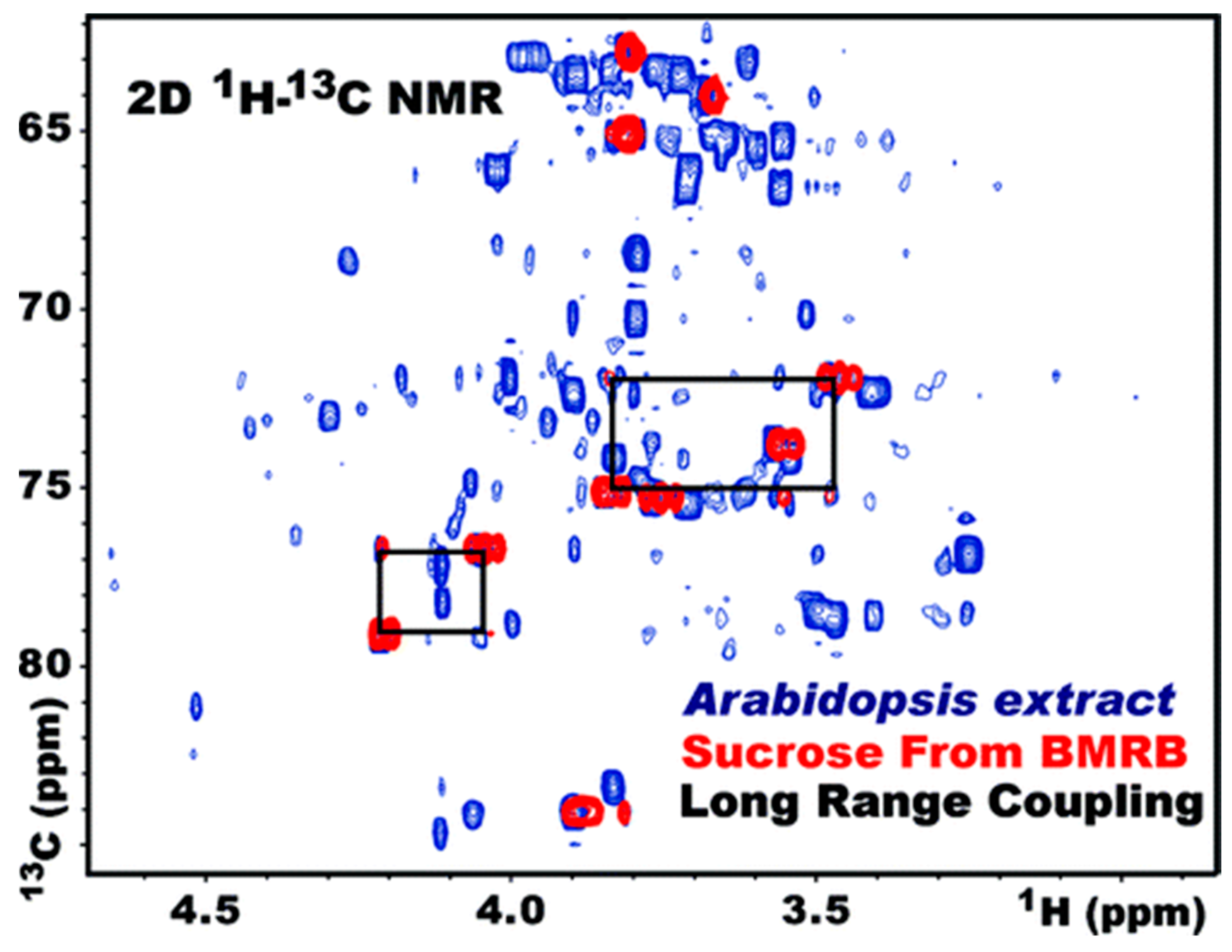
3.4. Heteronuclear Single Quantum Correlation Spectroscopy (HSQC)
3.5. Heteronuclear Multiple Bond Correlation (HMBC) Spectroscopy
4. NMR Databases and Software for Metabolite Identification
5. New NMR Methods in Metabolomics
5.1. High-Resolution Magic-Angle Spinning NMR Spectroscopy (HRMAS)
5.2. Hyperpolarization Methods
5.2.1. Dynamic Nuclear Polarization (DNP)
5.2.2. Applications of DNP in Metabolomics
5.2.3. Parahydrogen-Induced Polarization (PHIP) and Signal Amplification by Reversible Exchange (SABRE)
5.3. Fast NMR Methods
5.4. Pure-Shift NMR
5.5. LC-NMR and Other Hybrid NMR Approaches
6. New Developments with NMR Equipment
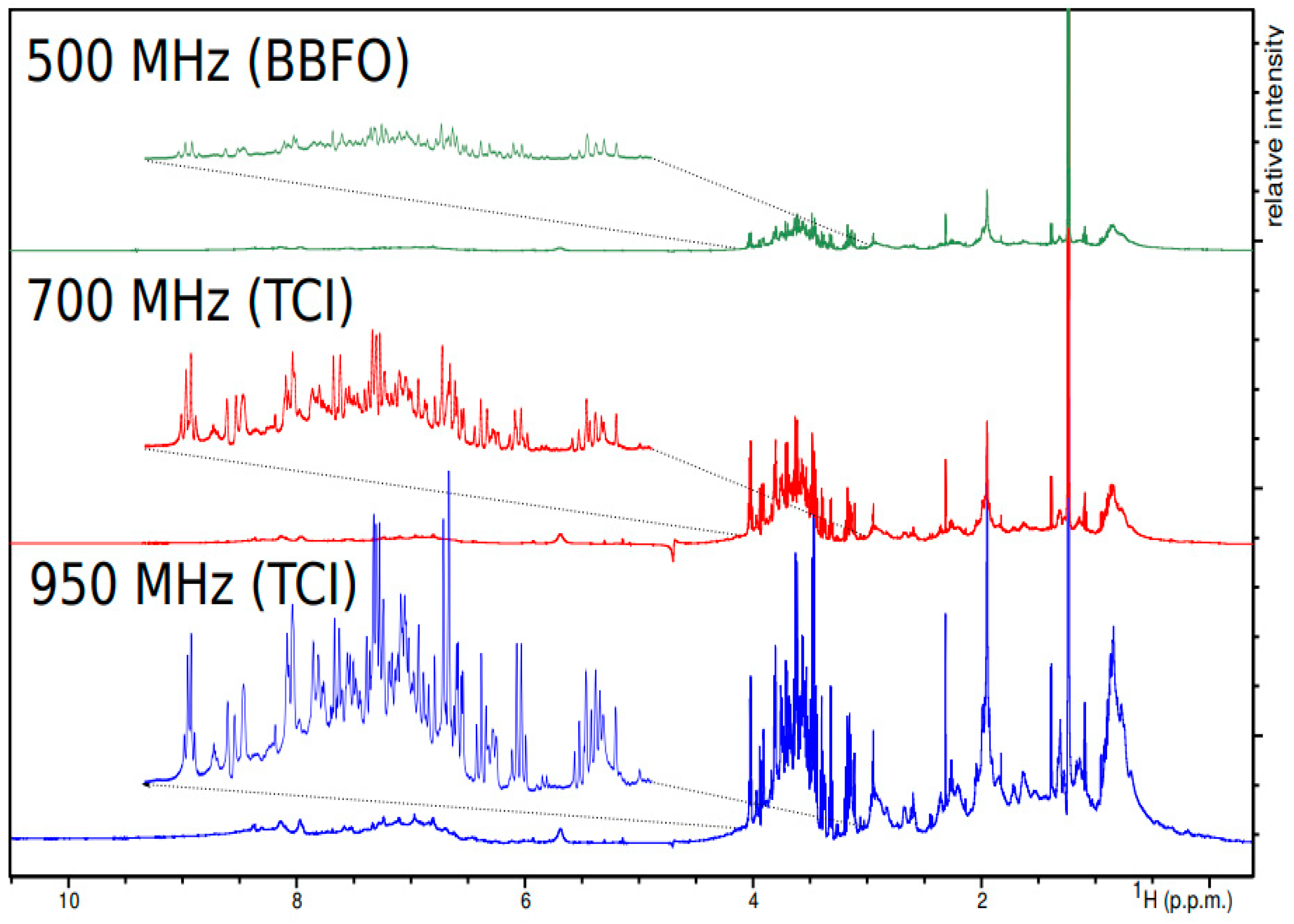
6.1. NMR Magnets
6.2. NMR Probes
7. Limitations of NMR in Metabolomics
8. Concluding Remarks and Future Prospects
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ciborowski, M.; Lipska, A.; Godzien, J.; Ferrarini, A.; Korsak, J.; Radziwon, P.; Tomasiak, M.; Barbas, C. Combination of LC-MS-and GC-MS-based Metabolomics to Study the Effect of Ozonated Autohemotherapy on Human Blood. J. Proteome Res. 2012, 11, 6231–6241. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.-L.; Zheng, P.; Liu, Z.; Xu, Y.; Mu, J.; Guo, J.; Huang, T.; Meng, H.-Q.; Xie, P. GC-MS based metabolomics identification of possible novel biomarkers for schizophrenia in peripheral blood mononuclear cells. Mol. Biosyst. 2014, 10, 2398–2406. [Google Scholar] [CrossRef] [PubMed]
- Raji, M.; Amad, M.; Emwas, A.H. Dehydrodimerization of pterostilbene during electrospray ionization mass spectrometry. Rapid Commun. Mass Spectrom. 2013, 27, 1260–1266. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Zhang, M.; Elmore, C.S.; Vishwanathan, K. An integrated strategy for in vivo metabolite profiling using high-resolution mass spectrometry based data processing techniques. Anal. Chim. Acta 2013, 780, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Tian, Y.; Li, G.; Li, Y.; Yin, X.; Peng, C.; Xu, F.; Zhang, Z. Discovery of safety biomarkers for realgar in rat urine using UFLC-IT-TOF/MS and H-1 NMR based metabolomics. Anal. Bioanal. Chem. 2013, 405, 4811–4822. [Google Scholar] [CrossRef] [PubMed]
- Vadla, N.C.; Davalagar, V.D.; Sripadi, P. Detection and characterization of N-alkyl diethanolamines and N-2-alkoxyethyl diethanolamines in milk by electrospray ionization mass spectrometry. Metabolomics 2013, 9, 623–630. [Google Scholar] [CrossRef]
- Allard, E.; Backstrom, D.; Danielsson, R.; Sjobberg, J.R.; Bergquist, J. Comparing Capillary Electrophoresis—Mass Spectrometry Fingerprints of Urine Samples Obtained after Intake of Coffee, Tea, or Water. Anal. Chem. 2008, 80, 8946–8955. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-H.; Chen, C.-H.; Lin, C.-J.; Chen, C.-C. Metabonomic Study with a High Performance Liquid Chromatography Coupling to a Triple Quadruple Mass Spectrometer to Identify Biomarkers from Urine of High-fat Fed and Streptozotocin Treated Rats. J. Food Drug Anal. 2009, 17, 28–35. [Google Scholar]
- Cho, S.-H.; Choi, M.H.; Kwon, O.S.; Lee, W.-Y.; Chung, B.C. Metabolic significance of bisphenol A-induced oxidative stress in rat urine measured by liquid chromatography-mass spectrometry. J. Appl. Toxicol. 2009, 29, 110–117. [Google Scholar] [CrossRef]
- Emwas, A.-H.M.; Al-Talla, Z.A.; Kharbatia, N.M. Sample collection and preparation of biofluids and extracts for gas chromatography-mass spectrometry. Methods Mol. Biol. 2015, 1277, 75–90. [Google Scholar]
- Emwas, A.-H.M.; Al-Talla, Z.A.; Yang, Y.; Kharbatia, N.M. Gas chromatography-mass spectrometry of biofluids and extracts. Methods Mol. Biol. 2015, 1277, 91–112. [Google Scholar] [PubMed]
- Al-Talla, Z.A.; Akrawi, S.H.; Emwas, A.H.M. Solid state NMR and bioequivalence comparison of the pharmacokinetic parameters of two formulations of clindamycin. Int. J. Clin. Pharmacol. Ther. 2011, 49, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Al-Talla, Z.A.; Akrawi, S.H.; Tolley, L.T.; Sioud, S.H.; Zaater, M.F.; Emwas, A.H. Bioequivalence assessment of two formulations of ibuprofen. Drug Des. Devel. 2011, 5, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, S.; Wang, M.; Shi, W.; Du, X.; Sun, C. The toxicity of 3-chloropropane-1,2-dipalmitate in Wistar rats and a metabonomics analysis of rat urine by ultra-performance liquid chromatography-mass spectrometry. Chem. Biol. Interact. 2013, 206, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Molz, P.; Ellwanger, J.H.; Iochims dos Santos, C.E.; Dias, J.F.; de Campos, D.; Corbellini, V.A.; Pra, D.; Lopes Putzke, M.T.; Rech Franke, S.I. A metabolomics approach to evaluate the effects of shiitake mushroom (Lentinula edodes) treatment in undernourished young rats. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 2014, 318, 194–197. [Google Scholar] [CrossRef]
- Szultka, M.; Krzeminski, R.; Walczak, J.; Jackowski, M.; Buszewski, B. Pharmacokinetic study of amoxicillin in human plasma by solid-phase microextraction followed by high-performance liquid chromatography-triple quadrupole mass spectrometry. Biomed. Chromatogr. 2014, 28, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Lan, K.; Zhang, Y.; Yang, J.; Xu, L. Simple quality assessment approach for herbal extracts using high performance liquid chromatography-UV based metabolomics platform. J. Chromatogr. A 2010, 1217, 1414–1418. [Google Scholar] [CrossRef]
- Liang, X.; Zhang, L.; Zhang, X.; Dai, W.; Li, H.; Hu, L.; Liu, H.; Su, J.; Zhang, W. Qualitative and quantitative analysis of traditional Chinese medicine Niu Huang Jie Du Pill using ultra performance liquid chromatography coupled with tunable UV detector and rapid resolution liquid chromatography coupled with time-of-flight tandem mass spectrometry. J. Pharm. Biomed. Anal. 2010, 51, 565–571. [Google Scholar]
- Zheng, S.; Yu, M.; Lu, X.; Huo, T.; Ge, L.; Yang, J.; Wu, C.; Li, F. Urinary metabonomic study on biochemical changes in chronic unpredictable mild stress model of depression. Clin. Chim. Acta 2010, 411, 204–209. [Google Scholar] [CrossRef]
- Kim, J.W.; Ryu, S.H.; Kim, S.; Lee, H.W.; Lim, M.-S.; Seong, S.J.; Kim, S.; Yoon, Y.-R.; Kim, K.-B. Pattern Recognition Analysis for Hepatotoxicity Induced by Acetaminophen Using Plasma and Urinary H-1 NMR-Based Metabolomics in Humans. Anal. Chem. 2013, 85, 11326–11334. [Google Scholar] [CrossRef]
- Wang, B.; Goodpaster, A.M.; Kennedy, M.A. Coefficient of variation, signal-to-noise ratio, and effects of normalization in validation of biomarkers from NMR-based metabonomics studies. Chemom. Intell. Lab. Syst. 2013, 128, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.-C.; Dai, Y.-Q.; Hui, R.-R.; Hua, J.; Chen, H.-J.; Luo, Q.-Y.; Li, J.-X. NMR-based metabonomic approach on the toxicological effects of a Cimicifuga triterpenoid. J. Appl. Toxicol. 2012, 32, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Calvani, R.; Miccheli, A.; Capuani, G.; Miccheli, A.T.; Puccetti, C.; Delfini, M.; Iaconelli, A.; Nanni, G.; Mingrone, G. Gut microbiome-derived metabolites characterize a peculiar obese urinary metabotype. Int. J. Obes. 2010, 34, 1095–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Huang, R.; Liu, L.; Peng, J.; Xiao, B.; Yang, J.; Miao, Z.; Huang, H. Metabonomics study of urine from Sprague-Dawley rats exposed to Huang-Yao-Zi using H-1 NMR spectroscopy. J. Pharm. Biomed. Anal. 2010, 52, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Gowda, G.A.N.; Ye, T.; Raftery, D. Advances in NMR-based biofluid analysis and metabolite profiling. Analyst 2010, 135, 1490–1498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wishart, D.S. Quantitative metabolomics using NMR. TrAC Trends Anal. Chem. 2008, 27, 228–237. [Google Scholar] [CrossRef]
- Zhang, S.; Gowda, G.A.N.; Asiago, V.; Shanaiah, N.; Barbas, C.; Raftery, D. Correlative and quantitative H-1 NMR-based metabolomics reveals specific metabolic pathway disturbances in diabetic rats. Anal. Biochem. 2008, 383, 76–84. [Google Scholar] [CrossRef]
- Emwas, A.-H.M. The strengths and weaknesses of NMR spectroscopy and mass spectrometry with particular focus on metabolomics research. Methods Mol. Biol. 2015, 1277, 161–193. [Google Scholar]
- Takis, P.G.; Ghini, V.; Tenori, L.; Turano, P.; Luchinat, C. Uniqueness of the NMR approach to metabolomics. TrAC Trends Anal. Chem. 2018. [Google Scholar] [CrossRef]
- Le Guennec, A.; Giraudeau, P.; Caldarelli, S. Evaluation of Fast 2D NMR for Metabolomics. Anal. Chem. 2014, 86, 5946–5954. [Google Scholar] [CrossRef]
- Blondel, C.; Khelalfa, F.; Reynaud, S.; Fauvelle, F.; Raveton, M. Effect of organochlorine pesticides exposure on the maize root metabolome assessed using high-resolution magic-angle spinning H-1 NMR spectroscopy. Environ. Pollut. 2016, 214, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Diserens, G.; Vermathen, M.; Precht, C.; Broskey, N.T.; Boesch, C.; Amati, F.; Dufour, J.F.; Vermathen, P. Separation of small metabolites and lipids in spectra from biopsies by diffusion-weighted HR-MAS NMR: A feasibility study. Analyst 2015, 140, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.S.; Coen, M.; Rhode, C.M.; Reily, M.D.; Robertson, D.G.; Holmes, E.; Lindon, J.C.; Nicholson, J.K. Chemical shift calibration of H-1 MAS NMR liver tissue spectra exemplified using a study of glycine protection of galactosamine toxicity. Magn. Reson. Chem. 2009, 47, S47–S53. [Google Scholar] [CrossRef] [PubMed]
- Jang, W.G.; Park, J.Y.; Lee, J.; Bang, E.; Kim, S.R.; Lee, E.K.; Yun, H.J.; Kang, C.M.; Hwang, G.S. Investigation of relative metabolic changes in the organs and plasma of rats exposed to X-ray radiation using HR-MAS H-1 NMR and solution H-1 NMR. NMR Biomed. 2016, 29, 507–518. [Google Scholar] [CrossRef]
- Calvo, N.; Beltran-Debon, R.; Rodriguez-Gallego, E.; Hernandez-Aguilera, A.; Guirro, M.; Marine-Casado, R.; Milla, L.; Alegret, J.M.; Sabench, F.; del Castillo, D.; et al. Liver fat deposition and mitochondrial dysfunction in morbid obesity: An approach combining metabolomics with liver imaging and histology. World J. Gastroenterol. 2015, 21, 7529–7544. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.Q.; Shou, J.X.; Li, X.Y.; Ma, L.; Zhu, X.H. Metabolic changes in acute cerebral infarction: Findings from proton magnetic resonance spectroscopic imaging. Exp. Ther. Med. 2014, 7, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.; Keshari, K.R.; Park, J.M. Cancer Metabolism and Tumor Heterogeneity: Imaging Perspectives Using MR Imaging and Spectroscopy. Contrast Media Mol. Imaging 2017, 2017, 6053879. [Google Scholar] [CrossRef]
- Simoes, R.V.; Martinez-Aranda, A.; Martin, B.; Cerdan, S.; Sierra, A.; Arus, C. Preliminary characterization of an experimental breast cancer cells brain metastasis mouse model by MRI/MRS. Magn. Reson. Mater. Phys. Biol. Med. 2008, 21, 237–249. [Google Scholar] [CrossRef]
- Yoon, H.; Yoon, D.; Yun, M.; Choi, J.S.; Park, V.Y.; Kim, E.K.; Jeong, J.; Koo, J.S.; Yoon, J.H.; Moon, H.J.; et al. Metabolomics of Breast Cancer Using High-Resolution Magic Angle Spinning Magnetic Resonance Spectroscopy: Correlations with 18F-FDG Positron Emission Tomography-Computed Tomography, Dynamic Contrast-Enhanced and Diffusion-Weighted Imaging MRI. PLoS ONE 2016, 11, e0159949. [Google Scholar] [CrossRef]
- Jeong, S.; Eskandari, R.; Park, S.M.; Alvarez, J.; Tee, S.S.; Weissleder, R.; Kharas, M.G.; Lee, H.; Keshari, K.R. Real-time quantitative analysis of metabolic flux in live cells using a hyperpolarized micromagnetic resonance spectrometer. Sci. Adv. 2017, 3, e1700341. [Google Scholar] [CrossRef]
- Motta, A.; Paris, D.; Melck, D. Monitoring Real-Time Metabolism of Living Cells by Fast Two-Dimensional NMR Spectroscopy. Anal. Chem. 2010, 82, 2405–2411. [Google Scholar] [CrossRef] [PubMed]
- Batool, F.; Emwas, A.H.; Gao, X.; Munawar, M.A.; Chotana, G.A. Synthesis and Suzuki Cross-Coupling Reactions of 2,6-Bis(trifluoromethyl) pyridine-4-boronic Acid Pinacol Ester. Synth. Stuttg. 2017, 49, 1327–1334. [Google Scholar]
- Elbaz, A.M.; Gani, A.; Hourani, N.; Emwas, A.H.; Sarathy, S.M.; Roberts, W.L. TG/DTG, FT-ICR Mass Spectrometry, and NMR Spectroscopy Study of Heavy Fuel Oil. Energy Fuels 2015, 29, 7825–7835. [Google Scholar] [CrossRef] [Green Version]
- Jameel, A.G.A.; Van Oudenhoven, V.; Emwas, A.H.; Sarathy, S.M. Predicting Octane Number Using Nuclear Magnetic Resonance Spectroscopy and Artificial Neural Networks. Energy Fuels 2018, 32, 6309–6329. [Google Scholar] [CrossRef] [Green Version]
- Rehman, Z.U.; Jeong, S.; Tabatabai, S.A.A.; Emwas, A.H.; Leiknes, T. Advanced characterization of dissolved organic matter released by bloom-forming marine algae. Desalin. Water Treat. 2017, 69, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Ye, T.; Mo, H.P.; Shanaiah, N.; Gowda, G.A.N.; Zhang, S.C.; Raftery, D. Chemoselective N-15 Tag for Sensitive and High-Resolution Nuclear Magnetic Resonance Profiling of the Carboxyl-Containing Metabolome. Anal. Chem. 2009, 81, 4882–4888. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, O.; van Zijl, P.; Cohen, J.S. Information from combined 1H and 31P NMR studies of cell extracts: Differences in metabolism between drug-sensitive and drug-resistant MCF-7 human breast cancer cells. Biochem. Biophys. Res. Commun. 1990, 169, 383–390. [Google Scholar] [CrossRef]
- Holmes, M.V.; Millwood, I.Y.; Kartsonaki, C.; Hill, M.R.; Bennett, D.A.; Boxall, R.; Guo, Y.; Xu, X.; Bian, Z.; Hu, R.Y.; et al. Lipids, Lipoproteins, and Metabolites and Risk of Myocardial Infarction and Stroke. J. Am. Coll. Cardiol. 2018, 71, 620–632. [Google Scholar] [CrossRef]
- Dona, A.C.; Kyriakides, M.; Scott, F.; Shephard, E.A.; Varshavi, D.; Veselkov, K.; Everett, J.R. A guide to the identification of metabolites in NMR-based metabonomics/metabolomics experiments. Comput. Struct. Biotechnol. J. 2016, 14, 135–153. [Google Scholar] [CrossRef] [Green Version]
- Emwas, A.-H.; Roy, R.; McKay, R.T.; Ryan, D.; Brennan, L.; Tenori, L.; Luchinat, C.; Gao, X.; Zeri, A.C.; Gowda, G.A.N.; et al. Recommendations and Standardization of Biomarker Quantification Using NMR-Based Metabolomics with Particular Focus on Urinary Analysis. J. Proteome Res. 2016, 15, 360–373. [Google Scholar] [CrossRef] [Green Version]
- Emwas, A.-H.M.S.; Reza, M.; Griffin, J.L.; Merzaban, J. NMR-based metabolomics in human disease diagnosis: Applications, limitations, and recommendations. Metabolomics 2013, 9, 1048–1072. [Google Scholar] [CrossRef]
- Maher, A.D.; Lindon, J.C.; Nicholson, J.K. H-1 NMR-based metabonomics for investigating diabetes. Future Med. Chem. 2009, 1, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Serkova, N.J.; Niemann, C.U. Pattern recognition and biomarker validation using quantitative H-1-NMR-based metabolomics. Expert Rev. Mol. Diagn. 2006, 6, 717–731. [Google Scholar] [CrossRef] [PubMed]
- Kostidis, S.; Addie, R.D.; Morreau, H.; Mayboroda, O.A.; Giera, M. Quantitative NMR analysis of intra-and extracellular metabolism of mammalian cells: A tutorial. Anal. Chim. Acta 2017, 980, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S. Emerging applications of metabolomics in drug discovery and precision medicine. Nat. Rev. Drug Discov. 2016, 15, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Markley, J.L.; Bruschweiler, R.; Edison, A.S.; Eghbalnia, H.R.; Powers, R.; Raftery, D.; Wishart, D.S. The future of NMR-based metabolomics. Curr. Opin. Biotechnol. 2017, 43, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Kruk, J.; Doskocz, M.; Jodlowska, E.; Zacharzewska, A.; Lakomiec, J.; Czaja, K.; Kujawski, J. NMR Techniques in Metabolomic Studies: A Quick Overview on Examples of Utilization. Appl. Magn. Reson. 2017, 48, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Emwas, A.H.; Saccenti, E.; Gao, X.; McKay, R.T.; dos Santos, V.; Roy, R.; Wishart, D.S. Recommended strategies for spectral processing and post-processing of 1D H-1-NMR data of biofluids with a particular focus on urine. Metabolomics 2018, 14, 31. [Google Scholar] [CrossRef] [PubMed]
- Brennan, L. NMR-based metabolomics: From sample preparation to applications in nutrition research. Prog. Nucl. Magn. Reson. Spectrosc. 2014, 83, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Dona, A.C.; Jiménez, B.; Schäfer, H.; Humpfer, E.; Spraul, M.; Lewis, M.R.; Pearce, J.T.M.; Holmes, E.; Lindon, J.C.; Nicholson, J.K. Precision High-Throughput Proton NMR Spectroscopy of Human Urine, Serum, and Plasma for Large-Scale Metabolic Phenotyping. Anal. Chem. 2014, 86, 9887–9894. [Google Scholar] [CrossRef] [PubMed]
- Bharti, S.K.; Roy, R. Quantitative H-1 NMR spectroscopy. TrAC Trends Anal. Chem. 2012, 35, 5–26. [Google Scholar] [CrossRef]
- Kikuchi, J.; Ito, K.; Date, Y. Environmental metabolomics with data science for investigating ecosystem homeostasis. Prog. Nucl. Magn. Reson. Spectrosc. 2018, 104, 56–88. [Google Scholar] [CrossRef] [PubMed]
- Holmes, E.; Nicholls, A.W.; Lindon, J.C.; Connor, S.C.; Connelly, J.C.; Haselden, J.N.; Damment, S.J.P.; Spraul, M.; Neidig, P.; Nicholson, J.K. Chemometric models for toxicity classification based on NMR spectra of biofluids. Chem. Res. Toxicol. 2000, 13, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Lindon, J.C.; Nicholson, J.K.; Holmes, E.; Everett, J.R. Metabonomics: Metabolic processes studied by NMR spectroscopy of biofluids. Concepts Magn. Reson. 2000, 12, 289–320. [Google Scholar] [CrossRef]
- Hao, J.; Astle, W.; De Iorio, M.; Ebbels, T.M.D. BATMAN-an R package for the automated quantification of metabolites from nuclear magnetic resonance spectra using a Bayesian model. Bioinformatics 2012, 28, 2088–2090. [Google Scholar] [CrossRef] [PubMed]
- Ravanbakhsh, S.; Liu, P.; Bjorndahl, T.C.; Mandal, R.; Grant, J.R.; Wilson, M.; Eisner, R.; Sinelnikov, I.; Hu, X.Y.; Luchinat, C.; et al. Accurate, Fully-Automated NMR Spectral Profiling for. PLoS ONE 2015, 10, e0124219. [Google Scholar] [CrossRef]
- Rohnisch, H.E.; Eriksson, J.; Mullner, E.; Agback, P.; Sandstrom, C.; Moazzami, A.A. AQuA: An Automated Quantification Algorithm for High-Throughput NMR-Based Metabolomics and Its Application in Human Plasma. Anal. Chem. 2018, 90, 2095–2102. [Google Scholar] [CrossRef]
- Canueto, D.; Gomez, J.; Salek, R.M.; Correig, X.; Canellas, N. rDolphin: A GUI R package for proficient automatic profiling of 1D H-1-NMR spectra of study datasets. Metabolomics 2018, 14, 24. [Google Scholar] [CrossRef]
- Cui, Q.; Lewis, I.A.; Hegeman, A.D.; Anderson, M.E.; Li, J.; Schulte, C.F.; Westler, W.M.; Eghbalnia, H.R.; Sussman, M.R.; Markley, J.L. Metabolite identification via the Madison Metabolomics Consortium Database. Nat. Biotechnol. 2008, 26, 162–164. [Google Scholar] [CrossRef]
- Tardivel, P.J.C.; Canlet, C.; Lefort, G.; Tremblay-Franco, M.; Debrauwer, L.; Concordet, D.; Servien, R. ASICS: An automatic method for identification and quantification of metabolites in complex 1D H-1 NMR spectra. Metabolomics 2017, 13, 109. [Google Scholar] [CrossRef]
- Karaman, I.; Ferreira, D.L.S.; Boulange, C.L.; Kaluarachchi, M.R.; Herrington, D.; Dona, A.C.; Castagne, R.; Moayyeri, A.; Lehne, B.; Loh, M.; et al. Workflow for Integrated Processing of Multicohort Untargeted H-1 NMR Metabolomics Data in Large-Scale Metabolic Epidemiology. J. Proteome Res. 2016, 15, 4188–4194. [Google Scholar] [CrossRef] [PubMed]
- Wurtz, P.; Kangas, A.J.; Soininen, P.; Lawlor, D.A.; Smith, G.D.; Ala-Korpela, M. Quantitative Serum Nuclear Magnetic Resonance Metabolomics in Large-Scale Epidemiology: A Primer on -Omic Technologies. Am. J. Epidemiol. 2017, 186, 1084–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tynkkynen, T.; Wang, Q.; Ekholm, J.; Anufrieva, O.; Ohukainen, P.; Vepsäläinen, J.; Männikkö, M.; Keinänen-Kiukaanniemi, S.; Holmes, M.V.; Goodwin, M.; et al. Proof of concept for quantitative urine NMR metabolomics pipeline for large-scale epidemiology and genetics. Int. J. Epidemiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Giraudeau, P.; Silvestre, V.; Akoka, S. Optimizing water suppression for quantitative NMR-based metabolomics: A tutorial review. Metabolomics 2015, 11, 1041–1055. [Google Scholar] [CrossRef]
- Gueron, M.; Plateau, P.; Decorps, M. Solvent Signal Suppression in NMR. Prog. Nucl. Magn. Reson. Spectrosc. 1991, 23, 135–209. [Google Scholar] [CrossRef]
- Zheng, G.; Price, W.S. Solvent signal suppression in NMR. Prog. Nucl. Magn. Reson. Spectrosc. 2010, 56, 267–288. [Google Scholar] [CrossRef]
- Hore, P.J. Solvent Suppression in Fourier-Transform Nuclear Magnetic-Resonance. J. Magn. Reson. 1983, 55, 283–300. [Google Scholar] [CrossRef]
- McKay, R.T. Recent Advances in Solvent Suppression for Solution NMR: A Practical Reference. In Annual Reports on Nmr Spectroscopy; Webb, G.A., Ed.; Elsevier: Amsterdam, The Netherlands, 2009; Volume 66, pp. 33–76. [Google Scholar]
- McKay, R.T. How the 1D-NOESY Suppresses Solvent Signal in Metabonomics NMR Spectroscopy: An Examination of the Pulse Sequence Components and Evolution. Concepts Magn. Reson. Part A 2011, 38, 197–220. [Google Scholar] [CrossRef]
- Lacy, P.; McKay, R.T.; Finkel, M.; Karnovsky, A.; Woehler, S.; Lewis, M.J.; Chang, D.; Stringer, K.A. Signal Intensities Derived from Different NMR Probes and Parameters Contribute to Variations in Quantification of Metabolites. PLoS ONE 2014, 9, e85732. [Google Scholar] [CrossRef]
- Sokolenko, S.; McKay, R.; Blondeel, E.J.M.; Lewis, M.J.; Chang, D.; George, B.; Aucoin, M.G. Understanding the variability of compound quantification from targeted profiling metabolomics of 1D-H-1-NMR spectra in synthetic mixtures and urine with additional insights on choice of pulse sequences and robotic sampling. Metabolomics 2013, 9, 887–903. [Google Scholar] [CrossRef]
- Mo, H.P.; Raftery, D. Pre-SAT180, a simple and effective method for residual water suppression. J. Magn. Reson. 2008, 190, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoult, D.I. Solvent Peak Saturation with Single-Phase and Quadrature Fourier Transformation. J. Magn. Reson. 1976, 21, 337–347. [Google Scholar] [CrossRef]
- Campbell, I.D.; Dobson, C.M.; Jeminet, G.; Williams, R.J.P. Pulsed NMR Methods for Observation and Assignment of Exchangeable Hydrogens—Application To Bacitracin. FEBS Lett. 1974, 49, 115–119. [Google Scholar] [CrossRef]
- Saude, E.J.; Sykes, B.D. Urine stability for metabolomic studies: Effects of preparation and storage. Metabolomics 2007, 3, 19–27. [Google Scholar] [CrossRef]
- Pinto, D.; Delaby, E.; Merico, D.; Barbosa, M.; Merikangas, A.; Klei, L.; Thiruvahindrapuram, B.; Xu, X.; Ziman, R.; Wang, Z.Z.; et al. Convergence of Genes and Cellular Pathways Dysregulated in Autism Spectrum Disorders. Am. J. Hum. Genet. 2014, 94, 677–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cloarec, O.; Dumas, M.E.; Craig, A.; Barton, R.H.; Trygg, J.; Hudson, J.; Blancher, C.; Gauguier, D.; Lindon, J.C.; Holmes, E.; et al. Statistical total correlation spectroscopy: An exploratory approach for latent biomarker identification from metabolic H-1 NMR data sets. Anal. Chem. 2005, 77, 1282–1289. [Google Scholar] [CrossRef] [PubMed]
- Fonville, J.M.; Maher, A.D.; Coen, M.; Holmes, E.; Lindon, J.C.; Nicholson, J.K. Evaluation of Full-Resolution J-Resolved H-1 NMR Projections of Biofluids for Metabonomics Information Retrieval and Biomarker Identification. Anal. Chem. 2010, 82, 1811–1821. [Google Scholar] [CrossRef]
- Abdul-Hamid, M.; Emwas, J.S.M.; Hacene, S. Theory and Applications of NMR-Based Metabolomics in Human Disease Diagnosis. In Applications of NMR Spectroscopy; Atta-ur-Rahman, M.I.C., Ed.; Bentham Science Publishers: Karachi, Pakistan, 2015; Volume 1, p. 38. [Google Scholar]
- Sandusky, P.; Appiah-Amponsah, E.; Raftery, D. Use of optimized 1D TOCSY NMR for improved quantitation and metabolomic analysis of biofluids. J. Biomol. NMR 2011, 49, 281–290. [Google Scholar] [CrossRef] [Green Version]
- Sandusky, P.; Raftery, D. Use of semiselective TOCSY and the Pearson Correlation for the metabonomic analysis of biofluid mixtures: Application to urine. Anal. Chem. 2005, 77, 7717–7723. [Google Scholar] [CrossRef]
- Doddrell, D.M.; Pegg, D.T.; Bendall, M.R. Distortionless Enhancement of NMR Signals by Polarization Transfer. J. Magn. Reson. 1982, 48, 323–327. [Google Scholar] [CrossRef]
- Merchak, N.; Silvestre, V.; Rouger, L.; Giraudeau, P.; Rizk, T.; Bejjani, J.; Akoka, S. Precise and rapid isotopomic analysis by H-1-C-13 2D NMR: Application to triacylglycerol matrices. Talanta 2016, 156, 239–244. [Google Scholar] [CrossRef]
- Wushensky, J.A.; Youngster, T.; Mendonca, C.M.; Aristilde, L. Flux Connections Between Gluconate Pathway, Glycolysis, and Pentose-Phosphate Pathway During Carbohydrate Metabolism in Bacillus megaterium QM B1551. Front. Microbiol. 2018, 9, 2789. [Google Scholar] [CrossRef] [PubMed]
- Buescher, J.M.; Antoniewicz, M.R.; Boros, L.G.; Burgess, S.C.; Brunengraber, H.; Clish, C.B.; DeBerardinis, R.J.; Feron, O.; Frezza, C.; Ghesquiere, B.; et al. A roadmap for interpreting C-13 metabolite labeling patterns from cells. Curr. Opin. Biotechnol. 2015, 34, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Chokkathukalam, A.; Kim, D.H.; Barrett, M.P.; Breitling, R.; Creek, D.J. Stable isotope- labeling studies in metabolomics: New insights into structure and dynamics of metabolic networks. Bioanalysis 2014, 6, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Pang, Q.Y.; Zhang, T.; Wang, Y.; Kong, W.W.; Guan, Q.J.; Yan, X.F.; Chen, S.X. Metabolomics of Early Stage Plant Cell-Microbe Interaction Using Stable Isotope Labeling. Front. Plant Sci. 2018, 9, 760. [Google Scholar] [CrossRef] [PubMed]
- Gowda, G.A.N.; Shanaiah, N.; Raftery, D. Isotope Enhanced Approaches in Metabolomics. In Isotope Labeling in Biomolecular NMR; Atreya, H.S., Ed.; Springer: Berlin/Heidelberg, Germany, 2012; Volume 992, pp. 147–164. [Google Scholar]
- Shanaiah, N.; Desilva, M.A.; Nagana Gowda, G.; Raftery, M.A.; Hainline, B.E.; Raftery, D. Class selection of amino acid metabolites in body fluids using chemical derivatization and their enhanced 13C NMR. Proc. Natl. Acad. Sci. USA 2007, 104, 11540–11544. [Google Scholar] [CrossRef] [PubMed]
- Keun, H.C.; Beckonert, O.; Griffin, J.L.; Richter, C.; Moskau, D.; Lindon, J.C.; Nicholson, J.K. Cryogenic probe 13C NMR spectroscopy of urine for metabonomic studies. Anal. Chem. 2002, 74, 4588–4593. [Google Scholar] [CrossRef] [PubMed]
- Keun, H.C.; Ebbels, T.M.D.; Antti, H.; Bollard, M.E.; Beckonert, O.; Schlotterbeck, G.; Senn, H.; Niederhauser, U.; Holmes, E.; Lindon, J.C. Analytical reproducibility in 1H NMR-based metabonomic urinalysis. Chem. Res. Toxicol. 2002, 15, 1380–1386. [Google Scholar] [CrossRef]
- Jang, C.; Chen, L.; Rabinowitz, J.D. Metabolomics and Isotope Tracing. Cell 2018, 173, 822–837. [Google Scholar] [CrossRef]
- Schatzlein, M.P.; Becker, J.; Schulze-Sunninghausen, D.; Pineda-Lucena, A.; Herance, J.R.; Luy, B. Rapid two-dimensional ALSOFAST-HSQC experiment for metabolomics and fluxomics studies: Application to a C-13-enriched cancer cell model treated with gold nanoparticles. Anal. Bioanal. Chem. 2018, 410, 2793–2804. [Google Scholar] [CrossRef]
- Heux, S.; Berges, C.; Millard, P.; Portais, J.C.; Letisse, F. Recent advances in high-throughput C-13-fluxomics. Curr. Opin. Biotechnol. 2017, 43, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Tayyari, F.; Gowda, G.A.N.; Gu, H.W.; Raftery, D. N-15-Cholamine-A Smart Isotope Tag for Combining NMR- and MS-Based Metabolite Profiling. Anal. Chem. 2013, 85, 8715–8721. [Google Scholar] [CrossRef] [PubMed]
- Korzhnev, D.M.; Religa, T.L.; Lundstrom, P.; Fersht, A.R.; Kay, L.E. The folding pathway of an FF domain: Characterization of an on-pathway intermediate state under folding conditions by N-15, C-13(alpha) and C-13-methyl relaxation dispersion and H-1/(2) H-exchange NMR Spectroscopy. J. Mol. Biol. 2007, 372, 497–512. [Google Scholar] [CrossRef] [PubMed]
- Arbogast, L.W.; Brinson, R.G.; Marino, J.P. Application of Natural Isotopic Abundance H-1-C-13- and H-1-N-15-Correlated Two-Dimensional NMR for Evaluation of the Structure of Protein Therapeutics. In Isotope Labeling of Biomolecules—Applications; Kelman, Z., Ed.; Academic Press: Cambridge, MA, USA, 2016; Volume 566, pp. 3–34. [Google Scholar]
- Dallmann, A.; Simon, B.; Duszczyk, M.M.; Kooshapur, H.; Pardi, A.; Bermel, W.; Sattler, M. Efficient Detection of Hydrogen Bonds in Dynamic Regions of RNA by Sensitivity-Optimized NMR Pulse Sequences. Angew. Chem. Int. Ed. 2013, 52, 10487–10490. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, C.; Schubert, M.; Duss, O.; Ravindranathan, S.; Allain, F.H.T. Structure determination and dynamics of protein-RNA complexes by NMR spectroscopy. Prog. Nucl. Magn. Reson. Spectrosc. 2011, 58, 1–61. [Google Scholar] [CrossRef] [PubMed]
- Zwahlen, C.; Legault, P.; Vincent, S.J.F.; Greenblatt, J.; Konrat, R.; Kay, L.E. Methods for measurement of intermolecular NOEs by multinuclear NMR spectroscopy: Application to a bacteriophage lambda N-peptide/boxB RNA complex. J. Am. Chem. Soc. 1997, 119, 6711–6721. [Google Scholar] [CrossRef]
- Hansen, D.F. Measurement of N-15 longitudinal relaxation rates in (NH4+)-N-15 spin systems to characterise rotational correlation times and chemical exchange. J. Magn. Reson. 2017, 279, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Berjanskii, M.V.; Wishart, D.S. A Simple Method to Measure Protein Side-Chain Mobility Using NMR Chemical Shifts. J. Am. Chem. Soc. 2013, 135, 14536–14539. [Google Scholar] [CrossRef]
- Mishima, M.; Hatanaka, M.; Yokoyama, S.; Ikegami, T.; Walchli, M.; Ito, Y.; Shirakawa, M. Intermolecular P-31-N-15 and P-31-H-1 scalar couplings across hydrogen bonds formed between a protein and a nucleotide. J. Am. Chem. Soc. 2000, 122, 5883–5884. [Google Scholar] [CrossRef]
- Pervushin, K.; Ono, A.; Fernandez, C.; Szyperski, T.; Kainosho, M.; Wuthrich, K. NMR scaler couplings across Watson-Crick base pair hydrogen bonds in DNA observed by transverse relaxation optimized spectroscopy. Proc. Natl. Acad. Sci. USA 1998, 95, 14147–14151. [Google Scholar] [CrossRef]
- Ruiz-Cabello, J.; Cohen, J.S. Phospholipid metabolites as indicators of cancer cell function. NMR Biomed. 1992, 5, 226–233. [Google Scholar] [CrossRef] [PubMed]
- DeSilva, M.A.; Shanaiah, N.; Gowda, G.A.N.; Rosa-Perez, K.; Hanson, B.A.; Raftery, D. Application of P-31 NMR spectroscopy and chemical derivatization for metabolite profiling of lipophilic compounds in human serum. Magn. Reson. Chem. 2009, 47, S74–S80. [Google Scholar] [CrossRef] [PubMed]
- Emwas, A.H.M.; Al-Talla, Z.A.; Guo, X.R.; Al-Ghamdi, S.; Al-Masri, H.T. Utilizing NMR and EPR spectroscopy to probe the role of copper in prion diseases. Magn. Reson. Chem. 2013, 51, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Mattar, S.M.; Emwas, A.H.; Calhoun, L.A. Spectroscopic studies of the intermediates in the conversion of 1,4,11,12-tetrahydro-9,10-anthraquinone to 9,10-anthraquinone by reaction with oxygen under basic conditions. J. Phys. Chem. A 2004, 108, 11545–11553. [Google Scholar] [CrossRef]
- Chu, S.; Maltsev, S.; Emwas, A.H.; Lorigan, G.A. Solid-state NMR paramagnetic relaxation enhancement immersion depth studies in phospholipid bilayers. J. Magn. Reson. 2010, 207, 89–94. [Google Scholar] [CrossRef] [Green Version]
- Sandusky, P.; Raftery, D. Use of selective TOCSY NMR experiments for quantifying minor components in complex mixtures: Application to the metabonomics of amino acids in honey. Anal. Chem. 2005, 77, 2455–2463. [Google Scholar] [CrossRef]
- Feraud, B.; Govaerts, B.; Verleysen, M.; de Tullio, P. Statistical treatment of 2D NMR COSY spectra in metabolomics: Data preparation, clustering-based evaluation of the Metabolomic Informative Content and comparison with H-1-NMR. Metabolomics 2015, 11, 1756–1768. [Google Scholar] [CrossRef]
- Kumar, A.; Ernst, R.R.; Wuthrich, K. A two-dimensional nuclear Overhauser enhancement (2D NOE) experiment for the elucidation of complete proton-proton cross-relaxation networks in biological macromolecules. Biochem. Biophys. Res. Commun. 1980, 95, 1–6. [Google Scholar] [CrossRef]
- Beckonert, O.; Keun, H.C.; Ebbels, T.M.D.; Bundy, J.; Holmes, E.; Lindon, J.C.; Nicholson, J.K. Metabolic profiling, metabolomic and metabonomic procedures for NMR spectroscopy of urine, plasma, serum and tissue extracts. Nat. Protoc. 2007, 2, 2692–2703. [Google Scholar] [CrossRef]
- Nicholson, J.K.; Foxall, P.J.D.; Spraul, M.; Farrant, R.D.; Lindon, J.C. 750 MHz 1H and 1H-13C NMR spectroscopy of human blood plasma. Anal. Chem. 1995, 67, 793–811. [Google Scholar] [CrossRef]
- Yuk, J.; McKelvie, J.R.; Simpson, M.J.; Spraul, M.; Simpson, A.J. Comparison of 1-D and 2-D NMR techniques for screening earthworm responses to sub-lethal endosulfan exposure. Environ. Chem. 2010, 7, 524–536. [Google Scholar] [CrossRef]
- Aguilar, J.A.; Adams, R.W.; Nilsson, M.; Morris, G.A. Suppressing exchange effects in diffusion-ordered NMR spectroscopy. J. Magn. Reson. 2014, 238, 16–19. [Google Scholar] [CrossRef] [PubMed]
- Mannina, L.; Sobolev, A.; Capitani, D.; Iaffaldano, N.; Rosato, M.; Ragni, P.; Reale, A.; Sorrentino, E.; D’Amico, I.; Coppola, R. NMR metabolic profiling of organic and aqueous sea bass extracts: Implications in the discrimination of wild and cultured sea bass. Talanta 2008, 77, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Jenner, J. Two Dimensional correlated spectroscopy (COSY). Magn. Reson. Med. 1989, 11, 316–330. [Google Scholar]
- Kono, H. (1)H and (13)C chemical shift assignment of the monomers that comprise carboxymethyl cellulose. Carbohydr. Polym. 2013, 97, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Hunt, C.T.; Boulanger, Y.; Fesik, S.W.; Armitage, I.M. NMR Analysis of the Structure and Metal Sequestering Properties of Metallothioneins. Environ. Health Perspect. 1984, 54, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Lown, J.W.; Hanstock, C.C. High-Field H-1-NMR Analysis of the 1-1 Intercalation Complex of the Antitumor Agent Mitoxantrone and the DNA Duplex D(Cpgpcpg) 2. J. Biomol. Struct. Dyn. 1985, 2, 1097–1106. [Google Scholar] [CrossRef]
- Macura, S.; Kumar, N.G.; Brown, L.R. Combined Use of Cosy and Double Quantum Two-Dimensional Nmr-Spectroscopy for Elucidation of Spin Systems in Polymyxin-B. Biochem. Biophys. Res. Commun. 1983, 117, 486–492. [Google Scholar] [CrossRef]
- Keifer, P.A. 90 degrees pulse width calibrations: How to read a pulse width array. Concepts Magn. Reson. 1999, 11, 165–180. [Google Scholar] [CrossRef]
- Kim, H.K.; Choi, Y.H.; Verpoorte, R. NMR-based metabolomic analysis of plants. Nat. Protoc. 2010, 5, 536–549. [Google Scholar] [CrossRef]
- Le Guennec, A.; Tea, I.; Antheaume, I.; Martineau, E.; Charrier, B.; Pathan, M.; Akoka, S.; Giraudeau, P. Fast Determination of Absolute Metabolite Concentrations by Spatially Encoded 2D NMR: Application to Breast Cancer Cell Extracts. Anal. Chem. 2012, 84, 10831–10837. [Google Scholar] [CrossRef] [PubMed]
- Sekiyama, Y.; Chikayama, E.; Kikuchi, J. Evaluation of a Semipolar Solvent System as a Step toward Heteronuclear Multidimensional NMR-Based Metabolomics for C-13-Labelled Bacteria, Plants, and Animals. Anal. Chem. 2011, 83, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Flores-Sanchez, I.J.; Choi, Y.H.; Verpoorte, R. Metabolite Analysis of Cannabis sativa L. by NMR Spectroscopy. In Functional Genomics: Methods and Protocols, 2nd ed.; Kaufmann, M., Klinger, C., Eds.; Springer: New York, NY, USA, 2012; Volume 815, pp. 363–375. [Google Scholar]
- Blasco, H.; Corcia, P.; Moreau, C.; Veau, S.; Fournier, C.; Vourc’h, P.; Emond, P.; Gordon, P.; Pradat, P.-F.; Praline, J.; et al. 1H-NMR-based metabolomic profiling of CSF in early amyotrophic lateral sclerosis. PLoS ONE 2010, 5, e13223. [Google Scholar] [CrossRef]
- Simon, O.Z. Applied NMR Spectroscopy for Chemists and Life Scientists; Wiley-VCH: Weinheim, Germany, 2013. [Google Scholar]
- Reynolds, W.F.; Enriquez, R.G. Choosing the best pulse sequences, acquisition parameters, postacquisition processing strategies, and probes for natural product structure elucidation by NMR spectroscopy. J. Nat. Prod. 2002, 65, 221–244. [Google Scholar] [CrossRef] [PubMed]
- Kupce, E.; Claridge, T.D.W. NOAH: NMR Supersequences for Small Molecule Analysis and Structure Elucidation. Angew. Chem. Int. Ed. 2017, 56, 11779–11783. [Google Scholar] [CrossRef] [PubMed]
- Bingol, K.; Bruschweiler-Li, L.; Li, D.-W.; Brüschweiler, R. Customized Metabolomics Database for the Analysis of NMR 1H–1H TOCSY and 13C–1H HSQC-TOCSY Spectra of Complex Mixtures. Anal. Chem. 2014, 86, 5494–5501. [Google Scholar] [CrossRef] [PubMed]
- Lucio-Gutiérrez, J.R.; Delgado-Montemayor, C.; Coello-Bonilla, J.; Waksman-Minsky, N.; Saucedo, A.L. Selective 1D-TOCSY and chemometrics to evaluate authenticity of Turnera diffusa and related botanical extracts. Phytochem. Lett. 2019, 30, 62–68. [Google Scholar] [CrossRef]
- Nagayama, K.; Wüthrich, K.; Bachmann, P.; Ernst, R.R. Two-dimensional J-resolved 1H n.m.r. spectroscopy for studies of biological macromolecules. Biochem. Biophys. Res. Commun. 1977, 78, 99–105. [Google Scholar] [CrossRef]
- Aue, W.; Karhan, J.; Ernst, R. Homonuclear broad band decoupling and two-dimensional J-resolved NMR spectroscopy. J. Chem. Phys. 1976, 64, 4226–4227. [Google Scholar] [CrossRef]
- Ludwig, C.; Viant, M.R. Two-dimensional J-resolved NMR spectroscopy: Review of a key methodology in the metabolomics toolbox. Phytochem. Anal. 2010, 21, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Foxall, P.J.D.; Parkinson, J.A.; Sadler, I.H.; Lindon, J.C.; Nicholson, J.K. Analysis of biological fluids using 600 MHz proton NMR spectroscopy: Application of homonuclear two-dimension J-resolved spectroscopy to urine and blood plasma for spectral simplification and assignment. J. Pharm. Biomed. Anal. 1993, 11, 21–31. [Google Scholar] [CrossRef]
- Yang, W.; Wang, Y.; Zhou, Q.; Tang, H. Analysis of human urine metabolites using SPE and NMR spectroscopy. Sci. China Ser. B Chem. 2008, 51, 218–225. [Google Scholar] [CrossRef] [Green Version]
- Lutz, N.W.; Maillet, S.; Nicoli, F.; Viout, P.; Cozzone, P.J. Further assignment of resonances in H-1 NMR spectra of cerebrospinal fluid (CSF). FEBS Lett. 1998, 425, 345–351. [Google Scholar] [CrossRef]
- Phalaraksh, C.; Lenz, E.M.; Lindon, J.C.; Nicholson, J.K.; Farrant, R.D.; Reynolds, S.E.; Wilson, I.D.; Osborn, D.; Weeks, J.M. NMR spectroscopic studies on the haemolymph of the tobacco hornworm, Manduca sexta: Assignment of H-1 and C-13 NMR spectra. Insect Biochem. Mol. Biol. 1999, 29, 795–805. [Google Scholar] [CrossRef]
- Kazimierczuk, K.; Zawadzka, A.; Kozminski, W. Narrow peaks and high dimensionalities: Exploiting the advantages of random sampling. J. Magn. Reson. 2009, 197, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Malmodin, D.; Billeter, M. Robust and versatile interpretation of spectra with coupled evolution periods using multi-way decomposition. Magn. Reson. Chem. 2006, 44, S185–S195. [Google Scholar] [CrossRef] [PubMed]
- Frydman, L.; Scherf, T.; Lupulescu, A. The acquisition of multidimensional NMR spectra within a single scan. Proc. Natl. Acad. Sci. USA 2002, 99, 15858–15862. [Google Scholar] [CrossRef] [Green Version]
- Shrot, Y.; Frydman, L. Single-scan NMR spectroscopy at arbitrary dimensions. J. Am. Chem. Soc. 2003, 125, 11385–11396. [Google Scholar] [CrossRef]
- Giraudeau, P.; Remaud, G.S.; Akoka, S. Evaluation of Ultrafast 2D NMR for Quantitative Analysis. Anal. Chem. 2009, 81, 479–484. [Google Scholar] [CrossRef]
- Viant, M.R. Improved methods for the acquisition and interpretation of NMR metabolomic data. Biochem. Biophys. Res. Commun. 2003, 310, 943–948. [Google Scholar] [CrossRef]
- Yi, Q.; Scalley-Kim, M.L.; Alm, E.J.; Baker, D. NMR characterization of residual structure in the denatured state of protein L. J. Mol. Biol. 2000, 299, 1341–1351. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-H.; Cha, E.-J.; Lim, J.-E.; Kwon, S.-H.; Kim, D.-H.; Cho, H.; Han, K.-H. Structural characterization of an intrinsically unfolded mini-HBX protein from hepatitis B virus. Mol. Cells 2012, 34, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Robertson, I.M.; Boyko, R.F.; Sykes, B.D. Visualizing the principal component of H-1, N-15-HSQC NMR spectral changes that reflect protein structural or functional properties: Application to troponin C. J. Biomol. Nmr 2011, 51, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.-K.; Parkinson, J.A.; Bella, J.; Wang, F.; Sadler, P.J. Penetrative DNA intercalation and G-base selectivity of an organometallic tetrahydroanthracene Ru-II anticancer complex. Chem. Sci. 2010, 1, 258–270. [Google Scholar] [CrossRef]
- Lewis, I.A.; Schommer, S.C.; Hodis, B.; Robb, K.A.; Tonelli, M.; Westler, W.M.; Sussman, M.R.; Markley, J.L. Method for determining molar concentrations of metabolites in complex solutions from two-dimensional 1H-13C NMR spectra. Anal. Chem. 2007, 79, 9385–9390. [Google Scholar] [CrossRef] [PubMed]
- Schanda, P.; Kupce, E.; Brutscher, B. SOFAST-HMQC experiments for recording two-dimensional heteronuclear correlation spectra of proteins within a few seconds. J. Biomol. NMR 2005, 33, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Bernini, P.; Bertini, I.; Luchinat, C.; Nepi, S.; Saccenti, E.; Schaefer, H.; Schuetz, B.; Spraul, M.; Tenori, L. Individual Human Phenotypes in Metabolic Space and Time. J. Proteome Res. 2009, 8, 4264–4271. [Google Scholar] [CrossRef]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Eisner, R.; Young, N.; Gautam, B.; Hau, D.D.; Psychogios, N.; Dong, E.; Bouatra, S.; et al. HMDB: A knowledgebase for the human metabolome. Nucleic Acids Res. 2009, 37, D603–D610. [Google Scholar] [CrossRef]
- Wishart, D.S.; Tzur, D.; Knox, C.; Eisner, R.; Guo, A.C.; Young, N.; Cheng, D.; Jewell, K.; Arndt, D.; Sawhney, S.; et al. HMDB: The human metabolome database. Nucleic Acids Res. 2007, 35, D521–D526. [Google Scholar] [CrossRef]
- Ulrich, E.L.; Akutsu, H.; Doreleijers, J.F.; Harano, Y.; Ioannidis, Y.E.; Lin, J.; Livny, M.; Mading, S.; Maziuk, D.; Miller, Z.; et al. BioMagResBank. Nucleic Acids Res. 2008, 36, D402–D408. [Google Scholar] [CrossRef]
- Kuhn, S.; Schlorer, N.E. Facilitating quality control for spectra assignments of small organic molecules: nmrshiftdb2-a free in-house NMR database with integrated LIMS for academic service laboratories. Magn. Reson. Chem. 2015, 53, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Jewison, T.; Guo, A.C.; Wilson, M.; Knox, C.; Liu, Y.F.; Djoumbou, Y.; Mandal, R.; Aziat, F.; Dong, E.; et al. HMDB 3.0-The Human Metabolome Database in 2013. Nucleic Acids Res. 2013, 41, D801–D807. [Google Scholar] [CrossRef] [PubMed]
- Farag, M.A.; Porzel, A.; Al-Hanimady, M.A.; Hegazy, M.E.F.; Meyer, A.; Mohamed, T.A.; Westphal, H.; Wessjohann, L.A. Soft Corals Biodiversity in the Egyptian Red Sea: A Comparative MS and NMR Metabolomics Approach of Wild and Aquarium Grown Species. J. Proteome Res. 2016, 15, 1274–1287. [Google Scholar] [CrossRef] [PubMed]
- Aru, V.; Lam, C.; Khakimov, B.; Hoefsloot, H.C.J.; Zwanenburg, G.; Lind, M.V.; Schafer, H.; van Duynhoven, J.; Jacobs, D.M.; Smilde, A.K.; et al. Quantification of lipoprotein profiles by nuclear magnetic resonance spectroscopy and multivariate data analysis. TrAC Trends Anal. Chem. 2017, 94, 210–219. [Google Scholar] [CrossRef]
- Takis, P.G.; Schafer, H.; Spraul, M.; Luchinat, C. Deconvoluting interrelationships between concentrations and chemical shifts in urine provides a powerful analysis tool. Nat. Commun. 2017, 8, 1662. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.R.; Lange, B.M. Open-Access Metabolomics Databases for Natural Product Research: Present Capabilities and Future Potential. Front. Bioeng. Biotechnol. 2015, 3, 10. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.G.; Bjorndahl, T.C.; Tang, P.; Wishart, D.S. MetaboMiner—Semi-automated identification of metabolites from 2D NMR spectra of complex biofluids. BMC Bioinform. 2008, 9, 507. [Google Scholar] [CrossRef] [PubMed]
- Alahmari, F.; Davaasuren, B.; Emwas, A.H.; Costa, P.; Rothenberger, A. Tris(ethylenediamine)nickel(II) thio-hydroxogermanate monohydrate: Synthesis, crystal structure, H-1 NMR, EPR, optical and magnetic properties. Inorg. Chim. Acta 2019, 488, 145–151. [Google Scholar] [CrossRef]
- Alahmari, F.; Dey, S.; Emwas, A.H.; Davaasuren, B.; Rothenberger, A. Layered copper thioaluminate K2Cu3AlS4: Synthesis, crystal structure, characterization and solid-state Al-27 and K-39 NMR studies. J. Alloy. Compd. 2019, 776, 1041–1047. [Google Scholar] [CrossRef]
- Schanda, P.; Ernst, M. Studying Dynamics by Magic-Angle Spinning Solid-State NMR Spectroscopy: Principles and Applications to Biomolecules. Prog. Nucl. Magn. Reson. Spectrosc. 2016, 96, 1–46. [Google Scholar] [CrossRef] [PubMed]
- Eddy, M.T.; Belenky, M.; Sivertsen, A.C.; Griffin, R.G.; Herzfeld, J. Selectively dispersed isotope labeling for protein structure determination by magic angle spinning NMR. J. Biomol. NMR 2013, 57, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Koito, Y.; Yamada, K.; Ando, S. Solid-state NMR and wide-angle X-ray diffraction study of hydrofluoroether/beta-cyclodextrin inclusion complex. J. Incl. Phenom. Macrocycl. Chem. 2013, 76, 143–150. [Google Scholar] [CrossRef]
- Bouhrara, M.; Ranga, C.; Fihri, A.; Shaikh, R.R.; Sarawade, P.; Emwas, A.-H.; Hedhili, M.N.; Polshettiwar, V. Nitridated Fibrous Silica (KCC-1) as a Sustainable Solid Base Nanocatalyst. ACS Sustain. Chem. Eng. 2013, 1, 1192–1199. [Google Scholar] [CrossRef]
- Jackson, M.D.; Moon, J.; Gotti, E.; Taylor, R.; Chae, S.R.; Kunz, M.; Emwas, A.-H.; Meral, C.; Guttmann, P.; Levitz, P.; et al. Material and Elastic Properties of Al-Tobermorite in Ancient Roman Seawater Concrete. J. Am. Ceram. Soc. 2013, 96, 2598–2606. [Google Scholar] [CrossRef]
- Pettinari, C.; Caruso, F.; Zaffaroni, N.; Villa, R.; Marchetti, F.; Pettinari, R.; Phillips, C.; Tanski, J.; Rossi, M. Synthesis, spectroscopy (IR, multinuclear NMR, ESI-MS), diffraction, density functional study and in vitro antiproliferative activity of pyrazole-beta-diketone dihalotin(IV) compounds on 5 melanoma cell lines. J. Inorg. Biochem. 2006, 100, 58–69. [Google Scholar] [CrossRef]
- Khan, M.T.; Busch, M.; Molina, V.G.; Emwas, A.-H.; Aubry, C.; Croue, J.-P. How different is the composition of the fouling layer of wastewater reuse and seawater desalination RO membranes? Water Res. 2014, 59, 271–282. [Google Scholar] [CrossRef]
- Bonhomme, C.; Gervais, C.; Laurencin, D. Recent NMR developments applied to organic-inorganic materials. Prog. Nucl. Magn. Reson. Spectrosc. 2014, 77, 1–48. [Google Scholar] [CrossRef] [PubMed]
- Sahloul, N.; Emwas, A.; Power, W.; Penlidis, A. Ethyl acrylate-hydroxyethyl acrylate and hydroxyethyl acrylate-methacrylic acid: Reactivity ratio estimation from cross-linked polymer using high resolution magic angle spinning spectroscopy. J. Macromol. Sci. Pure Appl. Chem. 2005, A42, 1369–1385. [Google Scholar] [CrossRef]
- Madhu, B.; Shaw, G.L.; Warren, A.Y.; Neal, D.E.; Griffiths, J.R. Response of Degarelix treatment in human prostate cancer monitored by HR-MAS H-1 NMR spectroscopy. Metabolomics 2016, 12, 120. [Google Scholar] [CrossRef] [PubMed]
- Morvan, D.; Demidem, A.; Papon, J.; De Latour, M.; Madelmont, J.C. Melanoma tumors acquire a new phospholipid metabolism phenotype under cystemustine as revealed by high-resolution magic angle spinning proton nuclear magnetic resonance spectroscopy of intact tumor samples. Cancer Res. 2002, 62, 1890–1897. [Google Scholar] [PubMed]
- Garrod, S.; Humpfer, E.; Spraul, M.; Connor, S.; Polley, S.; Connelly, J.; Lindon, J.; Nicholson, J.; Holmes, E. High-resolution magic angle spinning 1H NMR spectroscopic studies on intact rat renal cortex and medulla. Magn. Reson. Med. 1999, 41, 1108–1118. [Google Scholar] [CrossRef]
- Griffin, J.L.; Walker, L.A.; Garrod, S.; Holmes, E.; Shore, R.F.; Nicholson, J.K. NMR spectroscopy based metabonomic studies on the comparative biochemistry of the kidney and urine of the bank vole (Clethrionomys glareolus), wood mouse (Apodemus sylvaticus), white toothed shrew (Crocidura suaveolens) and the laboratory rat. Comp. Biochem. Physiol. Part. B Biochem. Mol. Biol. 2000, 127, 357–367. [Google Scholar] [CrossRef]
- Yang, J.; Xu, G.; Zheng, Y.; Kong, H.; Pang, T.; Lv, S.; Yang, Q. Diagnosis of liver cancer using HPLC-based metabonomics avoiding false-positive result from hepatitis and hepatocirrhosis diseases. J. Chromatogr. B 2004, 813, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Ratai, E.M.; Pilkenton, S.; Lentz, M.R.; Greco, J.B.; Fuller, R.A.; Kim, J.P.; He, J.; Cheng, L.L.; Gonzalez, R.G. Comparisons of brain metabolites observed by HRMAS 1H NMR of intact tissue and solution 1H NMR of tissue extracts in SIV-infected macaques. NMR Biomed. 2005, 18, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Griffin, J.L.; Troke, J.; Walker, L.A.; Shore, R.F.; Lindon, J.C.; Nicholson, J.K. The biochemical profile of rat testicular tissue as measured by magic angle spinning H-1 NMR spectroscopy. FEBS Lett. 2000, 486, 225–229. [Google Scholar] [CrossRef]
- Nishiyama, Y.; Endo, Y.; Nemoto, T.; Bouzier-Sore, A.K.; Wong, A. High-resolution NMR-based metabolic detection of microgram biopsies using a 1 mm HR mu MAS probe. Analyst 2015, 140, 8097–8100. [Google Scholar] [CrossRef] [PubMed]
- Lucas-Torres, C.; Huber, G.; Ichikawa, A.; Nishiyama, Y.; Wong, A. HR-μMAS NMR-Based Metabolomics: Localized Metabolic Profiling of a Garlic Clove with μg Tissues. Anal. Chem. 2018, 90, 13736–13743. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.; Lucas-Torres, C. Simultaneous metabolic mapping of different anatomies by 1H HR-MAS chemical shift imaging. Anal. Bioanal. Chem. 2019, 411, 1591–1599. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, L.; Zhang, H.; Deng, P.; Chen, J.; Zhou, B.; Hu, J.; Zou, J.; Lu, W.; Xiang, P.; et al. H-1 NMR-based metabolic profiling of human rectal cancer tissue. Mol. Cancer 2013, 12, 121. [Google Scholar] [CrossRef]
- Monleon, D.; Morales, J.M.; Gonzalez-Darder, J.; Talamantes, F.; Cortes, O.; Gil-Benso, R.; Loezin-Gines, C.; Cerda-Nicolas, M.; Celda, B. Benign and atypical meningioma metabolic signatures by high-resolution magic-angle spinning molecular profiling. J. Proteome Res. 2008, 7, 2882–2888. [Google Scholar] [CrossRef]
- Cacciatore, S.; Hu, X.Y.; Viertler, C.; Kap, M.; Bernhardt, G.A.; Mischinger, H.J.; Riegman, P.; Zatloukal, K.; Luchinat, C.; Turano, P. Effects of Intra- and Post-Operative Ischemia on the Metabolic Profile of Clinical Liver Tissue Specimens Monitored by NMR. J. Proteome Res. 2013, 12, 5723–5729. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.H.; Redwine, D.; Beshah, K.; Livazovic, S.; Canlas, C.G.; Guinov, A.; Emwas, A.H.M. Amide versus amine ratio in the discrimination layer of reverse osmosis membrane by solid state N-15 NMR and DNP NMR. J. Membr. Sci. 2019, 581, 243–251. [Google Scholar] [CrossRef]
- Zhai, W.X.; Feng, Y.L.; Liu, H.Q.; Rockenbauer, A.; Mance, D.; Li, S.Y.; Song, Y.G.; Baldus, M.; Liu, Y.P. Diastereoisomers of L-proline-linked trityl-nitroxide biradicals: Synthesis and effect of chiral configurations on exchange interactions. Chem. Sci. 2018, 9, 4381–4391. [Google Scholar] [CrossRef] [PubMed]
- Ardenkjaer-Larsen, J.H.; Fridlund, B.; Gram, A.; Hansson, G.; Hansson, L.; Lerche, M.H.; Servin, R.; Thaning, M.; Golman, K. Increase in signal-to-noise ratio of >10,000 times in liquid-state NMR. Proc. Natl. Acad. Sci. USA 2003, 100, 10158–10163. [Google Scholar] [CrossRef] [PubMed]
- Abragam, A.; Proctor, W.G. Une Nouvelle Methode de Polarisation Dynamique des Noyaux Atomiques Dans les Solides. C. R. Hebd. Seances Acad. Sci. 1958, 246, 2253–2256. [Google Scholar]
- Wenckebach, W.T. The Solid Effect. Appl. Magn. Reson. 2008, 34, 227. [Google Scholar] [CrossRef]
- Overhauser, A.W. Polarization of Nuclei in Metals. Phys. Rev. 1953, 92, 411–415. [Google Scholar] [CrossRef]
- Kessenikh, A.; Manenkov, A. Dynamic polarization of nuclei during saturation of nonuniformly broadened electron paramagnetic resonance lines. Sov. Phys.-Solid State 1963, 5, 835–837. [Google Scholar]
- Thurber, K.R.; Tycko, R. Theory for cross effect dynamic nuclear polarization under magic-angle spinning in solid state nuclear magnetic resonance: The importance of level crossings. J. Chem. Phys. 2012, 137, 084508. [Google Scholar] [CrossRef]
- Abragam, A.; Borghini, M. Chapter VIII Dynamic Polarization of Nuclear Targets. Prog. Low Temp. Phys. 1964, 4, 384–449. [Google Scholar]
- Abragam, A.; Goldman, M. Principles of dynamic nuclear polarisation. Rep. Prog. Phys. 1978, 41, 395. [Google Scholar] [CrossRef]
- Abragam, A. The Principles of Nuclear Magnetism; Oxford Science Publications: Oxford, UK, 1961. [Google Scholar]
- Emwas, A.H.; Saunders, M.; Ludwig, C.; Günther, U. Determinants for optimal enhancement in ex situ DNP experiments. Appl. Magn. Reson. 2008, 34, 483–494. [Google Scholar] [CrossRef]
- Bornet, A.; Maucourt, M.; Deborde, C.; Jacob, D.; Milani, J.; Vuichaud, B.; Ji, X.; Dumez, J.N.; Moing, A.; Bodenhausen, G.; et al. Highly Repeatable Dissolution Dynamic Nuclear Polarization for Heteronuclear NMR Metabolomics. Anal. Chem. 2016, 88, 6179–6183. [Google Scholar] [CrossRef] [PubMed]
- Kurhanewicz, J.; Vigneron, D.B.; Brindle, K.; Chekmenev, E.Y.; Comment, A.; Cunningham, C.H.; DeBerardinis, R.J.; Green, G.G.; Leach, M.O.; Rajan, S.S.; et al. Analysis of Cancer Metabolism by Imaging Hyperpolarized Nuclei: Prospects for Translation to Clinical Research. Neoplasia 2011, 13, 81–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lumata, L.; Yang, C.D.; Ragavan, M.; Carpenter, N.; DeBerardinis, R.J.; Merritt, M.E. Hyperpolarized C-13 Magnetic Resonance and Its Use in Metabolic Assessment of Cultured Cells and Perfused Organs. In Metabolic Analysis Using Stable Isotopes; Metallo, C.M., Ed.; Academic Press: New York, NY, USA, 2015; Volume 561, pp. 73–106. [Google Scholar]
- Christensen, C.E.; Karlsson, M.; Winther, J.R.; Jensen, P.R.; Lerche, M.H. Non-invasive In-cell Determination of Free Cytosolic NAD(+)/NADH Ratios Using Hyperpolarized Glucose Show Large Variations in Metabolic Phenotypes. J. Biol. Chem. 2014, 289, 2344–2352. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Liu, M.X.; Hilty, C. Parallelized Ligand Screening Using Dissolution Dynamic Nuclear Polarization. Anal. Chem. 2016, 88, 11178–11183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovtunov, K.V.; Pokochueva, E.V.; Salnikov, O.G.; Cousin, S.F.; Kurzbach, D.; Vuichoud, B.; Jannin, S.; Chekmenev, E.Y.; Goodson, B.M.; Barskiy, D.A.; et al. Hyperpolarized NMR Spectroscopy: D-DNP, PHIP, and SABRE Techniques. Chem. Asian J. 2018, 13, 1857–1871. [Google Scholar] [CrossRef]
- Kovtunov, K.V.; Zhivonitko, V.V.; Skovpin, I.V.; Barskiy, D.A.; Koptyug, I.V. Parahydrogen-Induced Polarization in Heterogeneous Catalytic Processes. In Hyperpolarization Methods in NMR Spectroscopy; Kuhn, L.T., Ed.; Springer: Berlin/Heidelberg, Germany, 2013; Volume 338, pp. 123–180. [Google Scholar]
- Kovtunov, K.V.; Truong, M.L.; Barskiy, D.A.; Salnikov, O.G.; Bukhtiyarov, V.I.; Coffey, A.M.; Waddell, K.W.; Koptyug, I.V.; Chekmenev, E.Y. Propane-d(6) Heterogeneously Hyperpolarized by Parahydrogen. J. Phys. Chem. C 2014, 118, 28234–28243. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.W.; Aguilar, J.A.; Atkinson, K.D.; Cowley, M.J.; Elliott, P.I.P.; Duckett, S.B.; Green, G.G.R.; Khazal, I.G.; Lopez-Serrano, J.; Williamson, D.C. Reversible Interactions with para-Hydrogen Enhance NMR Sensitivity by Polarization Transfer. Science 2009, 323, 1708–1711. [Google Scholar] [CrossRef] [Green Version]
- Adams, R.W.; Duckett, S.B.; Green, R.A.; Williamson, D.C.; Green, G.G.R. A theoretical basis for spontaneous polarization transfer in non-hydrogenative parahydrogen-induced polarization. J. Chem. Phys. 2009, 131, 194505. [Google Scholar] [CrossRef]
- Zacharias, N.M.; Chan, H.R.; Sailasuta, N.; Ross, B.D.; Bhattacharya, P. Real-time molecular imaging of tricarboxylic acid cycle metabolism in vivo by hyperpolarized 1-(13)C diethyl succinate. J. Am. Chem. Soc. 2012, 134, 934–943. [Google Scholar] [CrossRef] [PubMed]
- Dechent, J.F.; Buljubasich, L.; Schreiber, L.M.; Spiess, H.W.; Munnemann, K. Proton magnetic resonance imaging with para-hydrogen induced polarization. Phys. Chem. Chem. Phys. 2012, 14, 2346–2352. [Google Scholar] [CrossRef] [PubMed]
- Reineri, F.; Santelia, D.; Viale, A.; Cerutti, E.; Poggi, L.; Tichy, T.; Premkumar, S.S.D.; Gobetto, R.; Aime, S. Para-hydrogenated Glucose Derivatives as Potential C-13-Hyperpolarized Probes for Magnetic Resonance Imaging. J. Am. Chem. Soc. 2010, 132, 7186–7193. [Google Scholar] [CrossRef] [PubMed]
- Carravetta, M.; Levitt, M.H. Theory of long-lived nuclear spin states in solution nuclear magnetic resonance. I. Singlet states in low magnetic field. J. Chem. Phys. 2005, 122, 214505. [Google Scholar] [CrossRef] [PubMed]
- Barskiy, D.A.; Shchepin, R.V.; Coffey, A.M.; Theis, T.; Warren, W.S.; Goodson, B.M.; Chekmenev, E.Y. Over 20% N-15 Hyperpolarization in Under One Minute for Metronidazole, an Antibiotic and Hypoxia Probe. J. Am. Chem. Soc. 2016, 138, 8080–8083. [Google Scholar] [CrossRef] [PubMed]
- Warren, W.S.; Jenista, E.; Branca, R.T.; Chen, X. Increasing Hyperpolarized Spin Lifetimes Through True Singlet Eigenstates. Science 2009, 323, 1711–1714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharya, P.; Chekmenev, E.Y.; Perman, W.H.; Harris, K.C.; Lin, A.P.; Norton, V.A.; Tan, C.T.; Ross, B.D.; Weitekamp, D.P. Towards hyperpolarized (13)C-succinate imaging of brain cancer. J. Magn. Reson. 2007, 186, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Roth, M.; Kindervater, P.; Raich, H.P.; Bargon, J.; Spiess, H.W.; Munnemann, K. Continuous H-1 and C-13 Signal Enhancement in NMR Spectroscopy and MRI Using Parahydrogen and Hollow-Fiber Membranes. Angew. Chem. Int. Ed. 2010, 49, 8358–8362. [Google Scholar] [CrossRef] [PubMed]
- Marco-Rius, I.; Tayler, M.C.D.; Kettunen, M.I.; Larkin, T.J.; Timm, K.N.; Serrao, E.M.; Rodrigues, T.B.; Pileio, G.; Ardenkjaer-Larsen, J.H.; Levitt, M.H.; et al. Hyperpolarized singlet lifetimes of pyruvate in human blood and in the mouse. NMR Biomed. 2013, 26, 1696–1704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feliz, M.; García, J.; Aragón, E.; Pons, M. Fast 2D NMR Ligand Screening Using Hadamard Spectroscopy. J. Am. Chem. Soc. 2006, 128, 7146–7147. [Google Scholar] [CrossRef] [PubMed]
- Kupče, Ē.; Freeman, R. Fast multi-dimensional Hadamard spectroscopy. J. Magn. Reson. 2003, 163, 56–63. [Google Scholar] [CrossRef]
- Giraudeau, P.; Frydman, L. Ultrafast 2D NMR: An emerging tool in analytical spectroscopy. Annu. Rev. Anal. Chem. 2014, 7, 129–161. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, C.; Marin-Montesinos, I.; Saunders, M.G.; Emwas, A.H.; Pikramenou, Z.; Hammond, S.P.; Gunther, U.L. Application of ex situ dynamic nuclear polarization in studying small molecules. Phys. Chem Chem Phys. 2010, 12, 5868–5871. [Google Scholar] [CrossRef] [PubMed]
- Foroozandeh, M.; Adams, R.W.; Meharry, N.J.; Jeannerat, D.; Nilsson, M.; Morris, G.A. Ultrahigh-resolution NMR spectroscopy. Angew. Chem. 2014, 53, 6990–6992. [Google Scholar] [CrossRef] [PubMed]
- Foroozandeh, M.; Adams, R.W.; Nilsson, M.; Morris, G.A. Ultrahigh-Resolution Total Correlation NMR Spectroscopy. J. Am. Chem. Soc. 2014, 136, 11867–11869. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.A.; Aguilar, J.A.; Evans, R.; Haiber, S.; Nilsson, M. True Chemical Shift Correlation Maps: A TOCSY Experiment with Pure Shifts in Both Dimensions. J. Am. Chem. Soc. 2010, 132, 12770–12772. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, J.A.; Colbourne, A.A.; Cassani, J.; Nilsson, M.; Morris, G.A. Decoupling Two-Dimensional NMR Spectroscopy in Both Dimensions: Pure Shift NOESY and COSY. Angew. Chem. Int. Ed. 2012, 51, 6460–6463. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, J.A.; Faulkner, S.; Nilsson, M.; Morris, G.A. Pure Shift 1H NMR: A Resolution of the Resolution Problem? Angew. Chem. Int. Ed. 2010, 49, 3901–3903. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, J.A.; Nilsson, M.; Bodenhausen, G.; Morris, G.A. Spin echo NMR spectra without J modulation. Chem. Commun. 2012, 48, 811–813. [Google Scholar] [CrossRef]
- Zangger, K. Progress in Nuclear Magnetic Resonance Spectroscopy. Pure Shift NMR 2015, 86–87, 1–20. [Google Scholar]
- Timári, I.; Wang, C.; Hansen, A.L.; Costa dos Santos, G.; Yoon, S.O.; Bruschweiler-Li, L.; Brüschweiler, R. Real-Time Pure Shift HSQC NMR for Untargeted Metabolomics. Anal. Chem. 2019, 91, 2304–2311. [Google Scholar] [CrossRef] [PubMed]
- Zangger, K.; Sterk, H. Homonuclear Broadband-Decoupled NMR Spectra. J. Magn. Reson. 1997, 124, 486–489. [Google Scholar] [CrossRef]
- Aguilar, J.A.; Nilsson, M.; Morris, G.A. Simple Proton Spectra from Complex Spin Systems: Pure Shift NMR Spectroscopy Using BIRD. Angew. Chem. 2011, 123, 9890–9891. [Google Scholar] [CrossRef]
- Adams, R.W.; Byrne, L.; Kiraly, P.; Foroozandeh, M.; Paudel, L.; Nilsson, M.; Clayden, J.; Morris, G.A. Diastereomeric ratio determination by high sensitivity band-selective pure shift NMR spectroscopy. Chem. Commun. 2014, 50, 2512–2514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paudel, L.; Adams, R.W.; Király, P.; Aguilar, J.A.; Foroozandeh, M.; Cliff, M.J.; Nilsson, M.; Sándor, P.; Waltho, J.P.; Morris, G.A. Simultaneously Enhancing Spectral Resolution and Sensitivity in Heteronuclear Correlation NMR Spectroscopy. Angew. Chem. Int. Ed. 2013, 52, 11616–11619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lupulescu, A.; Olsen, G.L.; Frydman, L. Toward single-shot pure-shift solution 1H NMR by trains of BIRD-based homonuclear decoupling. J. Magn. Reson. 2012, 218, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Kiraly, P.; Adams, R.W.; Paudel, L.; Foroozandeh, M.; Aguilar, J.A.; Timári, I.; Cliff, M.J.; Nilsson, M.; Sándor, P.; Batta, G.; et al. Real-time pure shift 15N HSQC of proteins: A real improvement in resolution and sensitivity. J. Biomol. NMR 2015, 62, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.M.; Cabrera, R.; Maruenda, H. Ultra-Clean Pure Shift 1H-NMR applied to metabolomics profiling. Sci. Rep. 2019, 9, 6900. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Parihar, R.; Baishya, B. Identification of metabolites in coriander seeds (Coriandrum Sativum L.) aided by ultrahigh resolution total correlation NMR spectroscopy. Magn. Reson. Chem. 2019, 57, 304–316. [Google Scholar] [CrossRef]
- Simpson, A.J.; Tseng, L.H.; Simpson, M.J.; Spraul, M.; Braumann, U.; Kingery, W.L.; Kelleher, B.P.; Hayes, M.H.B. The application of LC-NMR and LC-SPE-NMR to compositional studies of natural organic matter. Analyst 2004, 129, 1216–1222. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, O.; Spraul, M. LC-NMR-MS in drug discovery. Drug Discov. Today 2003, 8, 624–631. [Google Scholar] [CrossRef]
- Appiah-Amponsah, E.; Owusu-Sarfo, K.; Gowda, G.A.N.; Ye, T.; Raftery, D. Combining Hydrophilic Interaction Chromatography (HILIC) and Isotope Tagging for Off-Line LC-NMR Applications in Metabolite Analysis. Metabolites 2013, 3, 575–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.J.; Capistrano, R.; Dhooghe, L.; Foubert, K.; Lemiere, F.; Maregesi, S.; Balde, A.; Apers, S.; Pieters, L. Herbal Medicines and Infectious Diseases: Characterization by LC-SPE-NMR of Some Medicinal Plant Extracts Used against Malaria. Planta Med. 2011, 77, 1139–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, F.; Fine, D.D.; Wherritt, D.J.; Lei, Z.T.; Sumner, L.W. PlantMAT: A Metabolomics Tool for Predicting the Specialized Metabolic Potential of a System and for Large-Scale Metabolite Identifications. Anal. Chem. 2016, 88, 11373–11383. [Google Scholar] [CrossRef] [PubMed]
- Grimes, J.H.; O’Connell, T.M. The application of micro-coil NMR probe technology to metabolomics of urine and serum. J. Biomol. NMR 2011, 49, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Tadanki, S.; Colon, R.D.; Moore, J.; Waddell, K.W. Double tuning a single input probe for heteronuclear NMR spectroscopy at low field. J. Magn. Reson. 2012, 223, 64–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haner, R.L.; Llanos, W.; Mueller, L. Small volume flow probe for automated direct-injection NMR analysis: Design and performance. J. Magn. Reson. 2000, 143, 69–78. [Google Scholar] [CrossRef]
- Bouatra, S.; Aziat, F.; Mandal, R.; Guo, A.C.; Wilson, M.R.; Knox, C.; Bjorndahl, T.C.; Krishnamurthy, R.; Saleem, F.; Liu, P.; et al. The human urine metabolome. PLoS ONE 2013, 8, e73076. [Google Scholar] [CrossRef]
- Psychogios, N.; Hau, D.D.; Peng, J.; Guo, A.C.; Mandal, R.; Bouatra, S.; Sinelnikov, I.; Krishnamurthy, R.; Eisner, R.; Gautam, B.; et al. The Human Serum Metabolome. PLoS ONE 2011, 6, e16957. [Google Scholar] [CrossRef]
- De Santana, A.P.; Jacomasso, T.; Riter, D.S.; Barison, A.; Iacomini, M.; Winnischofer, S.M.B.; Sassaki, G.L. NMR metabolic fingerprints of murine melanocyte and melanoma cell lines: Application to biomarker discovery. Sci. Rep. 2017, 7, 42324. [Google Scholar] [CrossRef]
- Zacharias, H.U.; Rehberg, T.; Mehrl, S.; Richtmann, D.; Wettig, T.; Oefner, P.J.; Spang, R.; Gronwald, W.; Altenbuchinger, M. Scale-Invariant Biomarker Discovery in Urine and Plasma Metabolite Fingerprints. J. Proteome Res. 2017, 16, 3596–3605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smolinska, A.; Blanchet, L.; Buydens, L.M.C.; Wijmenga, S.S. NMR and pattern recognition methods in metabolomics: From data acquisition to biomarker discovery: A review. Anal. Chim. Acta 2012, 750, 82–97. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.H.; Sun, H.; Qiu, S.; Wang, X.J. NMR-based metabolomics coupled with pattern recognition methods in biomarker discovery and disease diagnosis. Magn. Reson. Chem. 2013, 51, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Viant, M.R.; Rosenblum, E.S.; Tjeerdema, R.S. NMR-based metabolomics: A powerful approach for characterizing the effects of environmental stressors on organism health. Environ. Sci. Technol. 2003, 37, 4982–4989. [Google Scholar] [CrossRef] [PubMed]
- Cakir, T.; Hendriks, M.; Westerhuis, J.A.; Smilde, A.K. Metabolic network discovery through reverse engineering of metabolome data. Metabolomics 2009, 5, 318–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaub, J.; Reuss, M. In Vivo Dynamics of Glycolysis in Escherichia coli Shows Need for Growth-Rate Dependent Metabolome Analysis. Biotechnol. Prog. 2008, 24, 1402–1407. [Google Scholar] [CrossRef] [PubMed]
- Toya, Y.; Shimizu, H. Flux analysis and metabolomics for systematic metabolic engineering of microorganisms. Biotechnol. Adv. 2013, 31, 818–826. [Google Scholar] [CrossRef]
- Johnson, C.H.; Ivanisevic, J.; Siuzdak, G. Metabolomics: Beyond biomarkers and towards mechanisms. Nat. Rev. Mol. Cell Biol. 2016, 17, 451–459. [Google Scholar] [CrossRef]
- Corrias, G.; Balestrieri, A.; Politi, C.; Barberini, L.; Saba, L. Metabolomic and Imaging: A Literature Review. Curr. Med Imaging Rev. 2018, 14, 887–898. [Google Scholar] [CrossRef]
- Jagannathan, N.R.; Sharma, U. Breast Tissue Metabolism by Magnetic Resonance Spectroscopy. Metabolites 2017, 7, 25. [Google Scholar] [CrossRef]
- Pandey, R.; Caflisch, L.; Lodi, A.; Brenner, A.J.; Tiziani, S. Metabolomic signature of brain cancer. Mol. Carcinog. 2017, 56, 2355–2371. [Google Scholar] [CrossRef]
- Radjursoga, M.; Karlsson, G.B.; Lindqvist, H.M.; Pedersen, A.; Persson, C.; Pinto, R.C.; Ellegard, L.; Winkvist, A. Metabolic profiles from two different breakfast meals characterized by H-1 NMR-based metabolomics. Food Chem. 2017, 231, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Aru, V.; Khakimov, B.; Sorensen, K.M.; Engelsen, S.B. The foodome of bivalve molluscs: From hedonic eating to healthy diet. J. Food Compos. Anal. 2018, 69, 13–19. [Google Scholar] [CrossRef]
- Garcia-Garcia, A.B.; Lamichhane, S.; Castejon, D.; Cambero, M.I.; Bertram, H.C. H-1 HR-MAS NMR-based metabolomics analysis for dry-fermented sausage characterization. Food Chem. 2018, 240, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.S.; Zhong, X.; Li, S.S.; Zhang, G.R.; Liu, X. Metabolic characterization of natural and cultured Ophicordyceps sinensis from different origins by H-1 NMR spectroscopy. J. Pharm. Biomed. Anal. 2015, 115, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Sevillano, M.A.; Garcia-Barrera, T.; Gomez-Ariza, J.L. Environmental metabolomics: Biological markers for metal toxicity. Electrophoresis 2015, 36, 2348–2365. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Viant, M.R.; Tjeerdema, R.S. Metabolomics: Methodologies and applications in the environmental sciences. J. Pestic. Sci. 2006, 31, 245–251. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, M.; Meyer, K.A.; Jackson, T.M.; Schock, T.B.; Johnson, W.E.; Bearden, D.W. Application of NMR-based metabolomics for environmental assessment in the Great Lakes using zebra mussel (Dreissena polymorpha). Metabolomics 2015, 11, 1302–1315. [Google Scholar] [CrossRef]
- Dunn, W.B.; Goodacre, R.; Neyses, L.; Mamas, M. Integration of metabolomics in heart disease and diabetes research: Current achievements and future outlook. Bioanalysis 2011, 3, 2205–2222. [Google Scholar] [CrossRef]
- Guo, P.P.; Wei, D.D.; Wang, J.S.; Dong, G.; Zhang, Q.; Yang, M.H.; Kong, L.Y. Chronic toxicity of crude ricinine in rats assessed by H-1 NMR metabolomics analysis. RSC Adv. 2015, 5, 27018–27028. [Google Scholar] [CrossRef]
- Shi, J.; Cao, B.; Wang, X.W.; Aa, J.Y.; Duan, J.A.; Zhu, X.X.; Wang, G.J.; Liu, C.X. Metabolomics and its application to the evaluation of the efficacy and toxicity of traditional Chinese herb medicines. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2016, 1026, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Beger, R.D.; Sun, J.C.; Schnackenberg, L.K. Metabolomics approaches for discovering biomarkers of drug-induced hepatotoxicity and nephrotoxicity. Toxicol. Appl. Pharmacol. 2010, 243, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Yang, H.J.; Choi, M.J.; Kwon, H.N.; Kim, M.A.; Hong, S.S.; Park, S. Identification of Urinary Biomarkers Related to Cisplatin-Induced Acute Renal Toxicity Using NMR-Based Metabolomics. Biomol. Ther. 2011, 19, 38–44. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.J.; Choi, M.J.; Wen, H.; Kwon, H.N.; Jung, K.H.; Hong, S.W.; Kim, J.M.; Hong, S.S.; Park, S. An Effective Assessment of Simvastatin-Induced Toxicity with NMR-Based Metabonomics Approach. PLoS ONE 2011, 6, e16641. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Prakash, A.; Ruhela, R.K.; Medhi, B. Potential of metabolomics in preclinical and clinical drug development. Pharmacol. Rep. 2014, 66, 956–963. [Google Scholar] [CrossRef]
- Richardson, P.M.; Parrott, A.J.; Semenova, O.; Nordon, A.; Duckett, S.B.; Halse, M.E. SABRE hyperpolarization enables high-sensitivity H-1 and C-13 benchtop NMR spectroscopy. Analyst 2018, 143, 3442–3450. [Google Scholar] [CrossRef]
- Shchepin, R.V.; Barskiy, D.A.; Coffey, A.M.; Feldman, M.A.; Kovtunova, L.M.; Bukhtiyarov, V.I.; Kovtunov, K.V.; Goodson, B.M.; Koptyug, I.V.; Chekmenev, E.Y. Robust Imidazole-N-15(2) Synthesis for High-Resolution Low-Field (0.05 T) (15)NHyperpolarized NMR Spectroscopy. Chemistryselect 2017, 2, 4478–4483. [Google Scholar] [CrossRef]
- Schober, D.; Jacob, D.; Wilson, M.; Cruz, J.A.; Marcu, A.; Grant, J.R.; Moing, A.; Deborde, C.; de Figueiredo, L.F.; Haug, K.; et al. nmRML: A Community Supported Open Data Standard for the Description, Storage, and Exchange of NMR Data. Anal. Chem. 2018, 90, 649–656. [Google Scholar] [CrossRef]
- Sansone, S.A.; Rocca-Serra, P.; Field, D.; Maguire, E.; Taylor, C.; Hofmann, O.; Fang, H.; Neumann, S.; Tong, W.D.; Amaral-Zettler, L.; et al. Toward interoperable bioscience data. Nat. Genet. 2012, 44, 121–126. [Google Scholar] [CrossRef]
- Sud, M.; Fahy, E.; Cotter, D.; Azam, K.; Vadivelu, I.; Burant, C.; Edison, A.; Fiehn, O.; Higashi, R.; Nair, K.S.; et al. Metabolomics Workbench: An international repository for metabolomics data and metadata, metabolite standards, protocols, tutorials and training, and analysis tools. Nucleic Acids Res. 2016, 44, D463–D470. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NMR | Mass Spectrometry | |
|---|---|---|
| Reproducibility | High reproducibility is one of the fundamental advantages of NMR spectroscopy. | Compared to NMR spectroscopy, MS data are less reproducible. |
| Sensitivity | Intrinsically low but can be improved with multiple scans (time), higher magnet field strength, cryo-cooled and microprobes, and hyperpolarization methods. | High sensitivity is a major advantage of MS; metabolites with nanomolar concentrations can be readily detected |
| Selectivity | NMR is generally used for nonselective analysis. Peak overlaps from multiple detected metabolites pose major challenges. | MS is selective. However, in combination with chromatography (such as liquid and gas phase separation), it is a superior tool for targeted analysis. |
| Sample measurement | Enables relatively fast measurement using 1D 1H-NMR spectroscopy, where all metabolites at a detectable concentration level can be observed in one measurement. | Different ionization methods are required to maximize the number of detected metabolites. |
| Sample preparation | Involves minimal sample preparation, usually transferring the sample to an NMR tube and adding deuterated locking solvent. Can be automated. | More demanding; requires chromatography; requires sample derivatization for gas chromatography (GC)-MS. |
| Sample recovery | NMR is nondestructive and, hence, several analyses can be carried out on the same sample. Additionally, the sample can be recovered and stored for a long time. | MS is destructive technique; therefore, the sample cannot be recovered. However, it needs only a small amount of sample. |
| Quantitative analysis | NMR is inherently quantitative as the signal intensity is directly proportional to the metabolite concentrations and number of nuclei in the molecule. | The intensity of the MS line is often not correlated with metabolite concentrations as the ionization efficiency is also a determining factor. |
| Fluxomics Analysis | NMR permits both in vitro and in vivo metabolic flux analyses. Its inherently quantitative nature also enables precise quantification of precursors and products. Mapping of stable isotope locations and incorporating points in molecules is very easy via NMR. | MS can be used for fluxomics analysis; however, the destructive nature of MS-based methods means it is somewhat more limited than NMR-based fluxomics. In vivo fluxomics is not possible with MS, and isotope mapping is more difficult. |
| Tissue samples | Using high-resolution magic-angle sample spinning (HRMAS) NMR, it is possible to detect metabolites in tissue samples. | Although some MALDI-TOF approaches can be used to detect metabolites in tissue samples, these approaches are still far from being routine. |
| Number of detectable metabolites | Depending on spectral resolution, usually less than 200 metabolites can be unambiguously detected and identified in one measurement. | Using different MS techniques, it is possible to detect thousands of different metabolites and identify several hundred. |
| Targeted analysis | NMR spectroscopy can be used for both targeted and untargeted analyses, but it is not commonly used for targeted analyses. | Both GC-MS and liquid chromatography (LC)-MS are superior for targeted analyses |
| In vivo studies | Using magnetic resonance spectroscopy (MRS), in vivo investigation can be carried out most often using nuclei such as 1H and 31P. | Although desorption electrospray ionization (DESI) may be a useful way to analyze tissue samples during surgery, MS is not used for in vivo metabolomics studies. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Emwas, A.-H.; Roy, R.; McKay, R.T.; Tenori, L.; Saccenti, E.; Gowda, G.A.N.; Raftery, D.; Alahmari, F.; Jaremko, L.; Jaremko, M.; et al. NMR Spectroscopy for Metabolomics Research. Metabolites 2019, 9, 123. https://doi.org/10.3390/metabo9070123
Emwas A-H, Roy R, McKay RT, Tenori L, Saccenti E, Gowda GAN, Raftery D, Alahmari F, Jaremko L, Jaremko M, et al. NMR Spectroscopy for Metabolomics Research. Metabolites. 2019; 9(7):123. https://doi.org/10.3390/metabo9070123
Chicago/Turabian StyleEmwas, Abdul-Hamid, Raja Roy, Ryan T. McKay, Leonardo Tenori, Edoardo Saccenti, G. A. Nagana Gowda, Daniel Raftery, Fatimah Alahmari, Lukasz Jaremko, Mariusz Jaremko, and et al. 2019. "NMR Spectroscopy for Metabolomics Research" Metabolites 9, no. 7: 123. https://doi.org/10.3390/metabo9070123