Immersion Bioprinting of Tumor Organoids in Multi-Well Plates for Increasing Chemotherapy Screening Throughput

 ,
,  , , ,
, , ,
Abstract
: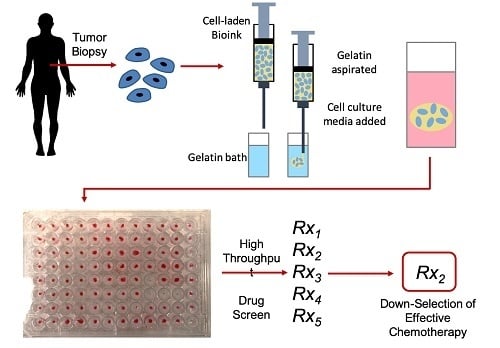
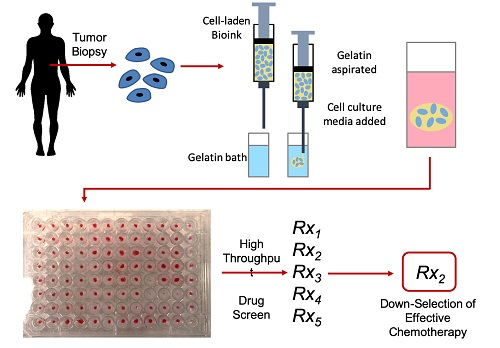{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Hydrogel Bioink Formulations and Preparation
2.2. Immersion Bioprinting Evaluation
2.3. Cell Line Culture
2.4. Cell Line Organoid Immersion Bioprinting
2.5. Viability and Proliferation Analysis
2.6. Patient-Derived Tumor Biospecimen Processing
2.7. Patient-Derived Tumor Organoid Chemotherapy Screening
3. Results and Discussion
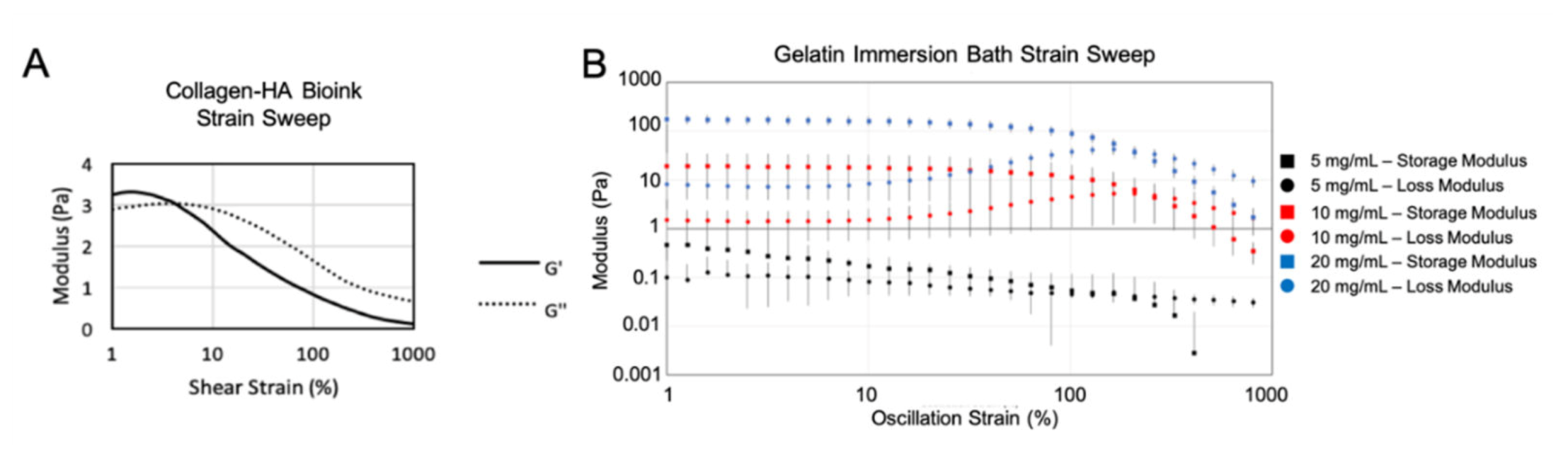
3.1. Hydrogel Bioink Formulation Immersion Bioprinting Evaluation
3.2. Cell Line Tumor Organoid Bioprinting and Viability/Proliferation Analysis
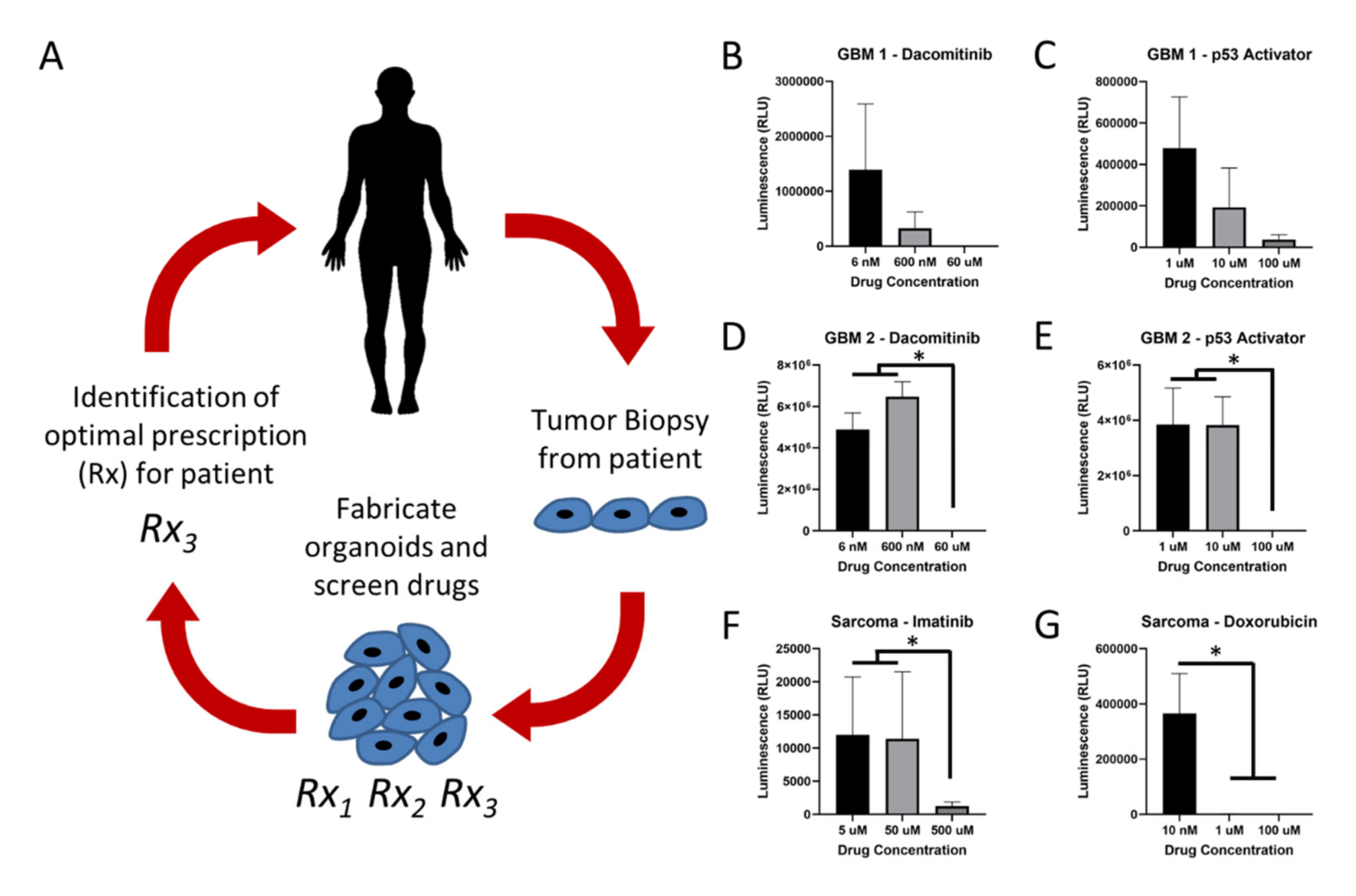
3.3. Patient-Derived Tumor Organoid Bioprinting and Chemotherapy Screening
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Mak, I.W.; Evaniew, N.; Ghert, M. Lost in translation: Animal models and clinical trials in cancer treatment. Am. J. Transl. Res. 2014, 6, 114–118. [Google Scholar]
- Pound, P.; Ritskes-Hoitinga, M. Is it possible to overcome issues of external validity in preclinical animal research? Why most animal models are bound to fail. J. Transl. Med. 2018, 16, 304. [Google Scholar] [CrossRef] [Green Version]
- Turner, N.C.; Reis-Filho, J.S. Genetic heterogeneity and cancer drug resistance. Lancet Oncol. 2012, 13, e178–e185. [Google Scholar] [CrossRef]
- Tran, N.H.; Cavalcante, L.L.; Lubner, S.J.; Mulkerin, D.L.; LoConte, N.K.; Clipson, L.; Matkowskyj, K.A.; Deming, D.A. Precision medicine in colorectal cancer: The molecular profile alters treatment strategies. Ther. Adv. Med Oncol. 2015, 7, 252–262. [Google Scholar] [CrossRef] [Green Version]
- Hayes, D.F.; Schott, A.F. Personalized Medicine: Genomics Trials in Oncology. Trans. Am. Clin. Climatol. Assoc. 2015, 126, 133–143. [Google Scholar]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef]
- Gorre, M.E.; Mohammed, M.; Ellwood, K.; Hsu, N.; Paquette, R.; Rao, P.N.; Sawyers, C.L. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 2001, 293, 876–880. [Google Scholar] [CrossRef] [Green Version]
- Ahasic, A.M.; Christiani, D.C. Personalized Critical Care Medicine: How Far Away Are We? Semin. Respir. Crit. Care Med. 2015, 36, 809–822. [Google Scholar]
- Sohal, D.P.; Rini, B.I.; Khorana, A.A.; Dreicer, R.; Abraham, J.; Procop, G.W.; Saunthararajah, Y.; Pennell, N.A.; Stevenson, J.P.; Pelley, R.; et al. Prospective Clinical Study of Precision Oncology in Solid Tumors. J. Natl. Cancer Inst. 2015, 183, djv332. [Google Scholar] [CrossRef]
- Kunz-Schughart, L.A.; Freyer, J.P.; Hofstaedter, F.; Ebner, R. The use of 3-D cultures for high-throughput screening: The multicellular spheroid model. J. Biomol. Screen 2004, 9, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Ho, W.J.; Pham, E.A.; Kim, J.W.; Ng, C.W.; Kim, J.H.; Kamei, D.T.; Wu, B.M. Incorporation of multicellular spheroids into 3-D polymeric scaffolds provides an improved tumor model for screening anticancer drugs. Cancer Sci. 2010, 101, 2637–2643. [Google Scholar] [CrossRef]
- Drewitz, M.; Helbling, M.; Fried, N.; Bieri, M.; Moritz, W.; Lichtenberg, J.; Kelm, J.M. Towards automated production and drug sensitivity testing using scaffold-free spherical tumor microtissues. Biotechnol. J. 2011, 6, 1488–1496. [Google Scholar] [CrossRef]
- Skardal, A.; Devarasetty, M.; Rodman, C.; Atala, A.; Soker, S. Liver-Tumor Hybrid Organoids for Modeling Tumor Growth and Drug Response In Vitro. Ann. Biomed. Eng. 2015, 43, 2361–2373. [Google Scholar] [CrossRef] [Green Version]
- Skardal, A.; Devarasetty, M.; Forsythe, S.; Atala, A.; Soker, S. A reductionist metastasis-on-a-chip platform for in vitro tumor progression modeling and drug screening. Biotechnol. Bioeng. 2016, 113, 2020–2032. [Google Scholar] [CrossRef] [Green Version]
- Huh, D.; Hamilton, G.A.; Ingber, D.E. From 3D cell culture to organs-on-chips. Trends Cell Biol. 2011, 21, 745–754. [Google Scholar] [CrossRef] [Green Version]
- Ghaemmaghami, A.M.; Hancock, M.J.; Harrington, H.; Kaji, H.A. Khademhosseini, Biomimetic tissues on a chip for drug discovery. Drug Discov. Today 2012, 17, 173–181. [Google Scholar] [CrossRef] [Green Version]
- Skardal, A.; Murphy, S.V.; Devarasetty, M.; Mead, I.; Kang, H.W.; Seol, Y.J.; Zhang, Y.S.; Shin, S.R.; Zhao, L.; Aleman, J.; et al. Multi-tissue interactions in an integrated three-tissue organ-on-a-chip platform. Sci. Rep. 2017, 7, 8837. [Google Scholar] [CrossRef]
- Skardal, A.; Shupe, T.; Atala, A. Organoid-on-a-chip and body-on-a-chip systems for drug screening and disease modeling. Drug Discov. Today 2016, 21, 1399–1411. [Google Scholar] [CrossRef]
- Skardal, A.; Devarasetty, M.; Soker, S.; Hall, A.R. In situ patterned micro 3-D liver constructs for parallel toxicology testing in a fluidic device. Biofabrication 2015, 7, 031001. [Google Scholar] [CrossRef] [Green Version]
- Mazzocchi, A.R.; Rajan, S.A.P.; Votanopoulos, K.I.; Hall, A.R.; Skardal, A. In vitro patient-derived 3D mesothelioma tumor organoids facilitate patient-centric therapeutic screening. Sci. Rep. 2018, 8, 2886. [Google Scholar] [CrossRef] [Green Version]
- Mazzocchi, A.; Devarasetty, M.; Herberg, S.; Petty, W.J.; Marini, F.; Miller, L.D.; Kucera, G.L.; Dukes, D.K.; Ruiz, J.; Skardal, A.; et al. Pleural Effusion Aspirate for Use in 3D Lung Cancer Modeling and Chemotherapy Screening, ACS Biomater. Sci. Eng. 2019, 5, 1937–1943. [Google Scholar]
- Votanopoulos, K.I.; Mazzocchi, A.; Sivakumar, H.; Forsythe, S.; Aleman, J.; Levine, E.A.; Skardal, A. Appendiceal Cancer Patient-Specific Tumor Organoid Model for Predicting Chemotherapy Efficacy Prior to Initiation of Treatment: A Feasibility Study. Ann. Surg. Oncol. 2019, 26, 139–147. [Google Scholar] [CrossRef]
- Forsythe, S.; Mehta, N.; Devarasetty, M.; Sivakumar, H.; Gmeiner, W.; Soker, S.; Votanopoulos, K.; Skardal, A. Development of a Colorectal Cancer 3D Micro-tumor Construct Platform From Cell Lines and Patient Tumor Biospecimens for Standard-of-Care and Experimental Drug Screening. Ann. Biomed. Eng. 2020, 48, 940–952. [Google Scholar] [CrossRef]
- Kang, H.W.; Lee, S.J.; Ko, I.K.; Kengla, C.; Yoo, J.J.; Atala, A. A 3D bioprinting system to produce human-scale tissue constructs with structural integrity. Nat. Biotechnol. 2016, 34, 312–319. [Google Scholar] [CrossRef]
- Mazzocchi, A.R.; Soker, S.; Skardal, A. Biofabrication Technologies for Developing In Vitro Tumor Models. In Tumor Organoids; Soker, S., Skardal, A., Eds.; Springer Nature: Berlin, Germany, 2017. [Google Scholar]
- Skardal, A.; Atala, A. Biomaterials for integration with 3-d bioprinting. Ann. Biomed. Eng. 2015, 43, 730–746. [Google Scholar] [CrossRef]
- Skardal, A.; Devarasetty, M.; Kang, H.W.; Mead, I.; Bishop, C.; Shupe, T.; Lee, S.J.; Jackson, J.; Yoo, J.; Soker, S.; et al. A hydrogel bioink toolkit for mimicking native tissue biochemical and mechanical properties in bioprinted tissue constructs. Acta Biomater. 2015, 25, 24–34. [Google Scholar] [CrossRef]
- Skardal, A.; Devarasetty, M.; Kang, H.W.; Seol, Y.J.; Forsythe, S.D.; Bishop, C.; Shupe, T.; Soker, S.; Atala, A. Bioprinting Cellularized Constructs Using a Tissue-specific Hydrogel Bioink. J. Vis. Exp. 2016, 110, e53606. [Google Scholar] [CrossRef] [Green Version]
- Skardal, A.; Zhang, J.; McCoard, L.; Oottamasathien, S.; Prestwich, G.D. Dynamically crosslinked gold nanoparticle-hyaluronan hydrogels. Adv. Mater. 2010, 22, 4736–4740. [Google Scholar] [CrossRef]
- Skardal, A.; Zhang, J.; McCoard, L.; Xu, X.; Oottamasathien, S.; Prestwich, G.D. Photocrosslinkable hyaluronan-gelatin hydrogels for two-step bioprinting. Tissue Eng. Part A 2010, 16, 2675–2685. [Google Scholar] [CrossRef] [Green Version]
- Skardal, A.; Zhang, J.; Prestwich, G.D. Bioprinting vessel-like constructs using hyaluronan hydrogels crosslinked with tetrahedral polyethylene glycol tetracrylates. Biomaterials 2010, 31, 6173–6181. [Google Scholar] [CrossRef]
- Clark, C.C.; Aleman, J.; Mutkus, L.; Skardal, A. A mechanically robust thixotropic collagen and hyaluronic acid bioink supplemented with gelatin nanoparticles. Bioprinting 2019, 16, e00058. [Google Scholar] [CrossRef]
- Mazzocchi, A.; Devarasetty, M.; Huntwork, R.; Soker, S.; Skardal, A. Optimization of collagen type I-hyaluronan hybrid bioink for 3D bioprinted liver microenvironments. Biofabrication 2018, 11, 015003. [Google Scholar] [CrossRef]
- Skardal, A. Perspective: “Universal” bioink technology for advancing extrusion bioprinting-based biomanufacturing. Bioprinting 2018, 10, e00026. [Google Scholar] [CrossRef]
- Antoshin, A.A.; Churbanov, S.N.; Minaev, N.V.; Zhang, Y.; Shpichka, A.I.; Timashev, P.S. LIFT-bioprinting, is it worth it? Bioprinting 2019, 15, e00052. [Google Scholar] [CrossRef]
- Hinton, T.J.; Jallerat, Q.; Palchesko, R.N.; Park, J.H.; Grodzicki, M.S.; Shue, H.J.; Ramadan, M.H.; Hudson, A.R.; Feinberg, A.W. Three-dimensional printing of complex biological structures by freeform reversible embedding of suspended hydrogels. Sci. Adv. 2015, 1, e1500758. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.; Hudson, A.R.; Shiwarski, D.J.; Tashman, J.W.; Hinton, T.J.; Yerneni, S.; Bliley, J.M.; Campbell, P.G.; Feinberg, A.W. 3D bioprinting of collagen to rebuild components of the human heart. Science 2019, 365, 482–487. [Google Scholar] [CrossRef]
- Skardal, A.; Smith, L.; Bharadwaj, S.; Atala, A.; Soker, S.; Zhang, Y. Tissue specific synthetic ECM hydrogels for 3-D in vitro maintenance of hepatocyte function. Biomaterials 2012, 33, 4565–4575. [Google Scholar] [CrossRef] [Green Version]
- Devarasetty, M.; Skardal, A.; Cowdrick, K.; Marini, F.; Soker, S. Bioengineered Submucosal Organoids for In Vitro Modeling of Colorectal Cancer. Tissue Eng. Part A 2017, 23, 1026–1041. [Google Scholar] [CrossRef]
- Gettler, B.C.; Zakhari, J.S.; Gandhi, P.S.; Williams, S.K. Formation of Adipose Stromal Vascular Fraction Cell-Laden Spheroids Using a Three-Dimensional Bioprinter and Superhydrophobic Surfaces. Tissue Eng. Part C Methods 2017, 23, 516–524. [Google Scholar] [CrossRef]
- Pang, Y.; Yang, J.; Hui, Z.; Grottkau, B.E. Robotic Patterning a Superhydrophobic Surface for Collective Cell Migration Screening. Tissue Eng. Part C Methods 2018, 24, 205–213. [Google Scholar] [CrossRef]
- Devarasetty, M.; Mazzocchi, A.R.; Skardal, A. Applications of Bioengineered 3D Tissue and Tumor Organoids in Drug Development and Precision Medicine: Current and Future. BioDrugs 2018, 32, 53–68. [Google Scholar] [CrossRef]
- Mazzocchi, A.; Soker, S.; Skardal, A. 3D bioprinting for high-throughput screening: Drug screening, disease modeling, and precision medicine applications. Appl. Phys. Rev. 2019, 6, 011302. [Google Scholar] [CrossRef]
- Engelman, J.A.; Zejnullahu, K.; Gale, C.M.; Lifshits, E.; Gonzales, A.J.; Shimamura, T.; Zhao, F.; Vincent, P.W.; Naumov, G.N.; Bradner, J.E. PF00299804, an irreversible pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res. 2007, 67, 11924–11932. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Zhou, L.; Hong, B.; van den Heuvel, A.P.; Prabhu, V.V.; Warfel, N.A.; Kline, C.L.; Dicker, D.T.; Kopelovich, L.; El-Deiry, W.S. Small-Molecule NSC59984 Restores p53 Pathway Signaling and Antitumor Effects against Colorectal Cancer via p73 Activation and Degradation of Mutant p53. Cancer Res. 2015, 78, 3842–3852. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.C.; Zhang, J.X.; Rashid, A.; Yeung, S.C.; Szklaruk, J.; Hess, K.; Xie, K.; Ellis, L.; Abbruzzese, J.L.; Ajani, J.A. Clinical and in vitro studies of imatinib in advanced carcinoid tumors. Clin. Cancer Res. 2007, 13, 234–240. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Kalen, A.L.; Smith, B.J.; Cullen, J.J.; Oberley, L.W. Enhancing the antitumor activity of adriamycin and ionizing radiation. Cancer Res. 2009, 69, 4294–4300. [Google Scholar] [CrossRef] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maloney, E.; Clark, C.; Sivakumar, H.; Yoo, K.; Aleman, J.; Rajan, S.A.P.; Forsythe, S.; Mazzocchi, A.; Laxton, A.W.; Tatter, S.B.; et al. Immersion Bioprinting of Tumor Organoids in Multi-Well Plates for Increasing Chemotherapy Screening Throughput. Micromachines 2020, 11, 208. https://doi.org/10.3390/mi11020208
Maloney E, Clark C, Sivakumar H, Yoo K, Aleman J, Rajan SAP, Forsythe S, Mazzocchi A, Laxton AW, Tatter SB, et al. Immersion Bioprinting of Tumor Organoids in Multi-Well Plates for Increasing Chemotherapy Screening Throughput. Micromachines. 2020; 11(2):208. https://doi.org/10.3390/mi11020208
Chicago/Turabian StyleMaloney, Erin, Casey Clark, Hemamylammal Sivakumar, KyungMin Yoo, Julio Aleman, Shiny A. P. Rajan, Steven Forsythe, Andrea Mazzocchi, Adrian W. Laxton, Stephen B. Tatter, and et al. 2020. "Immersion Bioprinting of Tumor Organoids in Multi-Well Plates for Increasing Chemotherapy Screening Throughput" Micromachines 11, no. 2: 208. https://doi.org/10.3390/mi11020208