
Biases in Viral Metagenomics-Based Detection, Cataloguing and Quantification of Bacteriophage Genomes in Human Faeces, a Review

 , , ,
, , , 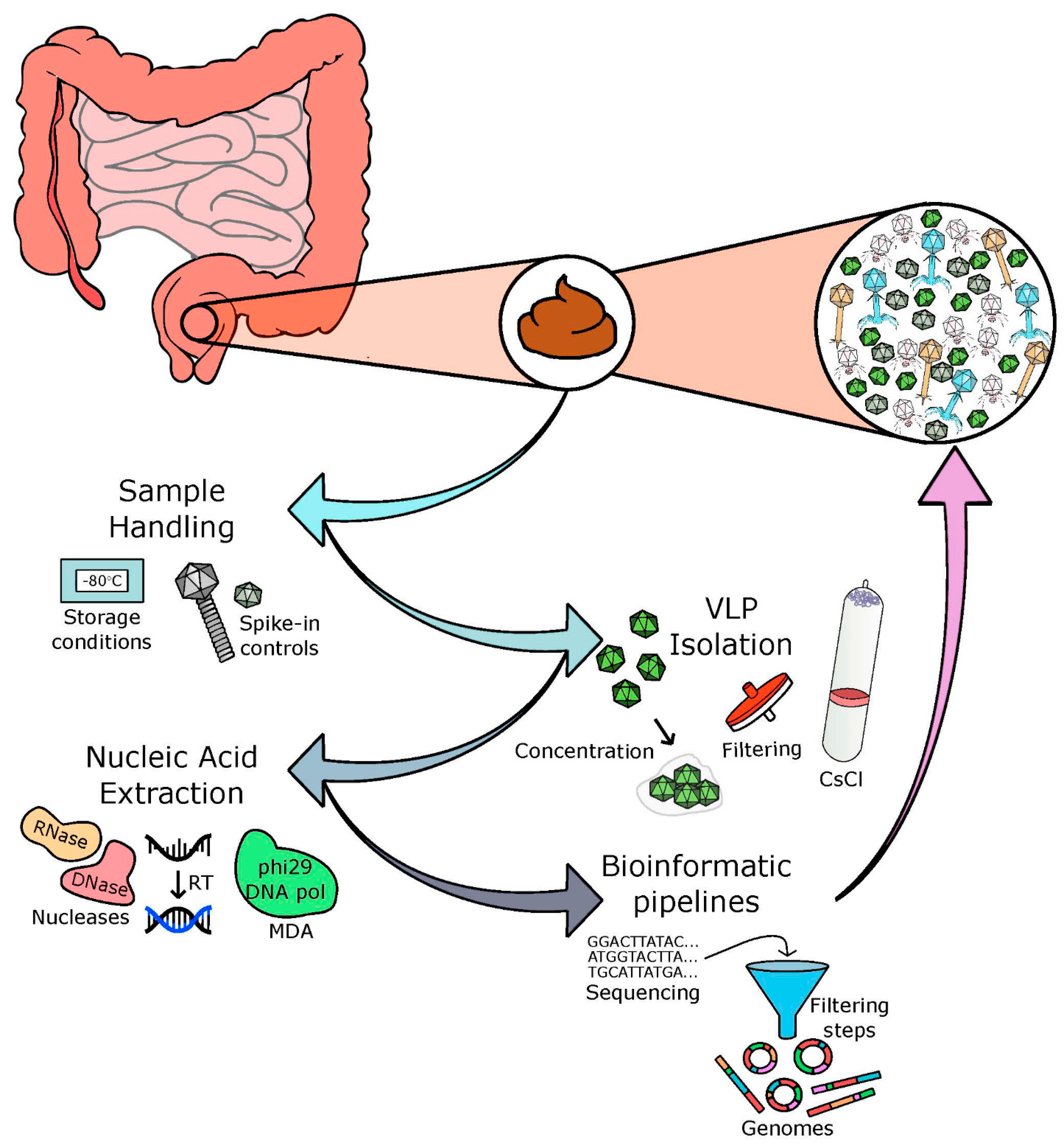{kind=link}
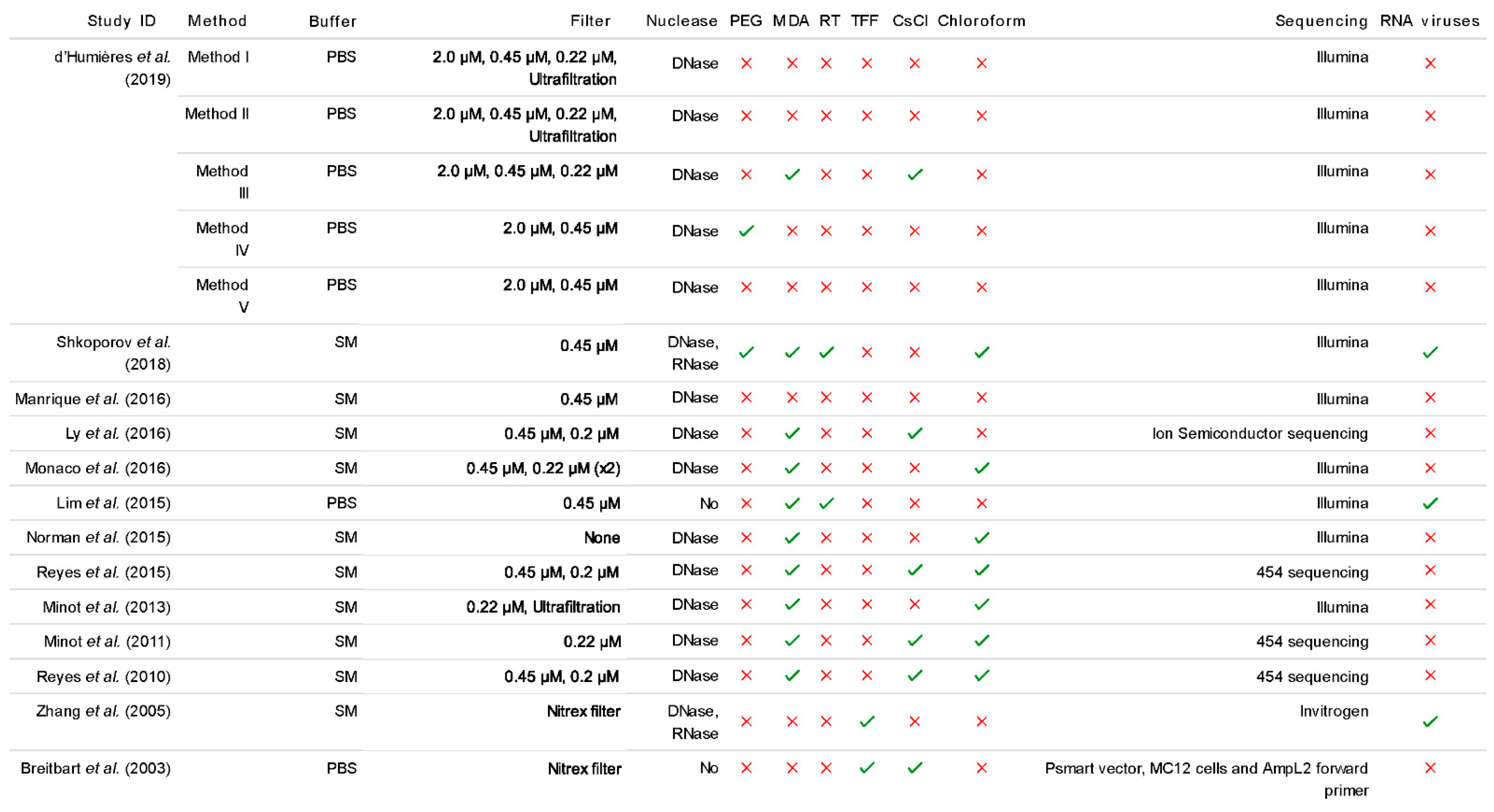{kind=link}
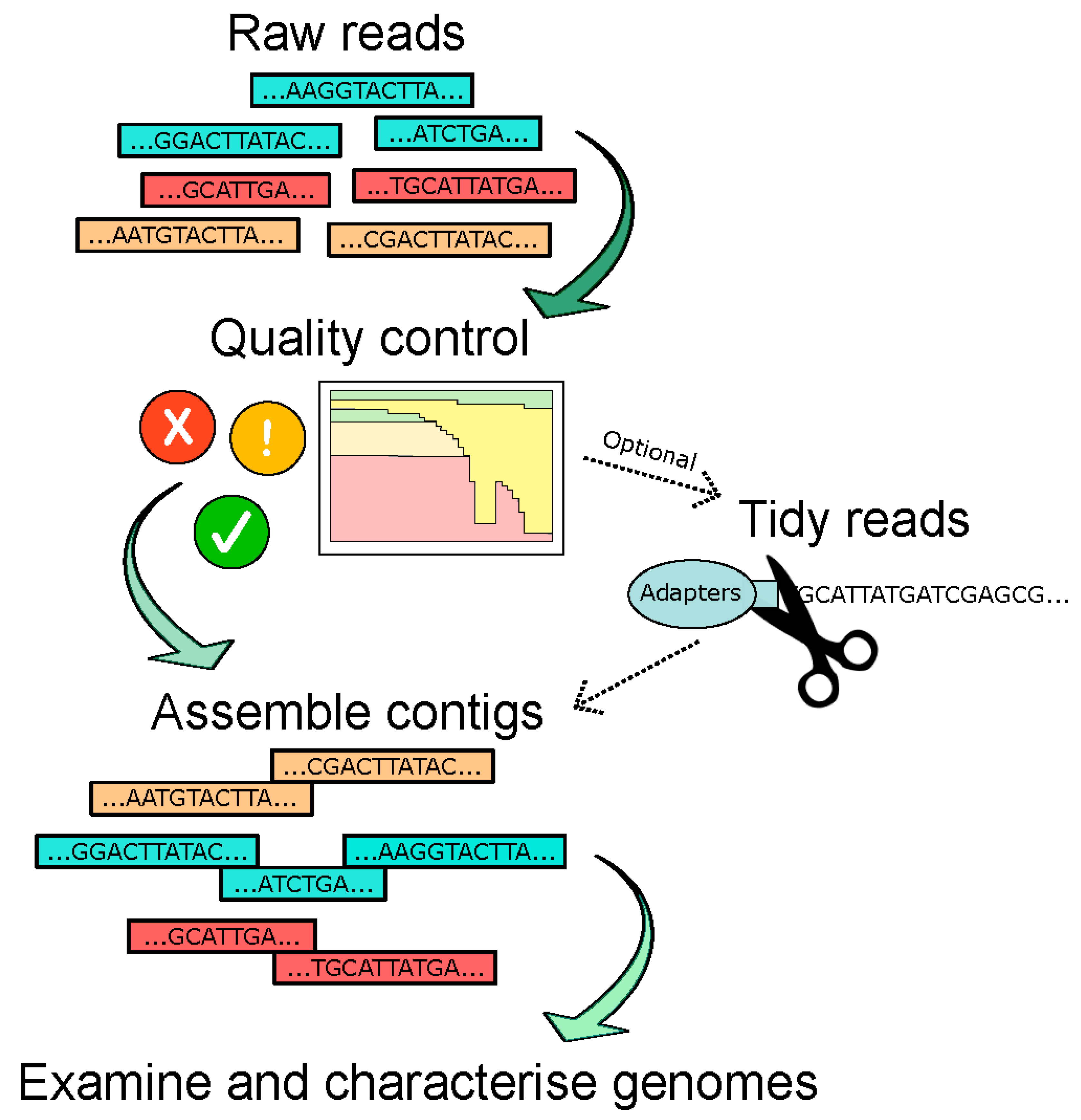{kind=link}
Abstract
:1. Introduction
2. Sample Handling
3. VLP Isolation
4. Nucleic Acid Extraction and Library Preparation
5. Bioinformatic Pipelines
6. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shkoporov, A.N.; Hill, C. Bacteriophages of the Human Gut: The “Known Unknown” of the Microbiome. Cell Host Microbe 2019, 25, 195–209. [Google Scholar] [CrossRef] [Green Version]
- Breitbart, M.; Hewson, I.; Felts, B.; Mahaffy, J.M.; Nulton, J.; Salamon, P.; Rohwer, F. Metagenomic Analyses of an Uncultured Viral Community from Human Feces. J. Bacteriol. 2003, 185, 6220–6223. [Google Scholar] [CrossRef] [Green Version]
- Hsu, B.B.; Gibson, T.E.; Yeliseyev, V.; Liu, Q.; Lyon, L.; Bry, L.; Silver, P.A.; Gerber, G.K. Dynamic Modulation of the Gut Microbiota and Metabolome by Bacteriophages in a Mouse Model. Cell Host Microbe 2019, 25, 803–814.e5. [Google Scholar] [CrossRef] [Green Version]
- Mirzaei, M.K.; Khan, A.A.; Ghosh, P.; Taranu, Z.E.; Taguer, M.; Ru, J.; Chowdhury, R.; Kabir, M.; Deng, L.; Mondal, D.; et al. Bacteriophages Isolated from Stunted Children Can Regulate Gut Bacterial Communities in an Age-Specific Manner. Cell Host Microbe 2020, 27, 199–212.e5. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Enault, F.; Hurwitz, B.L.; Sullivan, M.B. VirSorter: Mining viral signal from microbial genomic data. PeerJ 2015, 3, e985. [Google Scholar] [CrossRef] [PubMed]
- Feargalr/Demovir. Available online: https://github.com/feargalr/Demovir (accessed on 15 February 2021).
- Shkoporov, A.N.; Ryan, F.J.; Draper, L.A.; Forde, A.; Stockdale, S.R.; Daly, K.M.; McDonnell, S.A.; Nolan, J.A.; Sutton, T.D.; Dalmasso, M.; et al. Reproducible protocols for metagenomic analysis of human faecal phageomes. Microbiome 2018, 6, 1–17. [Google Scholar] [CrossRef] [PubMed]
- D’Humières, C.; Touchon, M.; Dion, S.; Cury, J.; Ghozlane, A.; Garcia-Garcera, M.; Bouchier, C.; Ma, L.; Denamur, E.; Rocha, E. A simple, reproducible and cost-effective procedure to analyse gut phageome: From phage isolation to bioinformatic approach. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Conceição-Neto, N.; Zeller, M.; Lefrère, H.; De Bruyn, P.; Beller, L.; Deboutte, W.; Yinda, C.K.; Lavigne, R.; Maes, P.; Van Ranst, M.; et al. Modular approach to customise sample preparation procedures for viral metagenomics: A reproducible protocol for virome analysis. Sci. Rep. 2015, 5, 16532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnamurthy, S.R.; Wang, D. Origins and challenges of viral dark matter. Virus Res. 2017, 239, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Hallam, S.J.; Woyke, T.; Sullivan, M.B. Viral dark matter and virus–host interactions resolved from publicly available microbial genomes. eLife 2015, 4, e08490. [Google Scholar] [CrossRef] [PubMed]
- Ogilvie, L.A.; Jones, B.V. The human gut virome: A multifaceted majority. Front. Microbiol. 2015, 6, 918. [Google Scholar] [CrossRef] [Green Version]
- Guerin, E.; Hill, C. Shining Light on Human Gut Bacteriophages. Front. Cell. Infect. Microbiol. 2020, 10, 481. [Google Scholar] [CrossRef]
- Sutton, T.D.S.; Hill, C. Gut Bacteriophage: Current Understanding and Challenges. Front. Endocrinol. 2019, 10, 784. [Google Scholar] [CrossRef] [PubMed]
- Forster, S.C.; Kumar, N.; Anonye, B.O.; Almeida, A.; Viciani, E.; Stares, M.D.; Dunn, M.; Mkandawire, T.T.; Zhu, A.; Shao, Y.; et al. A human gut bacterial genome and culture collection for improved metagenomic analyses. Nat. Biotechnol. 2019, 37, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Duhaime, M.B.; Deng, L.; Poulos, B.T.; Sullivan, M.B. Towards quantitative metagenomics of wild viruses and other ultra-low concentration DNA samples: A rigorous assessment and optimization of the linker amplification method. Environ. Microbiol. 2012, 14, 2526–2537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chibani-Chennoufi, S.; Bruttin, A.; Dillmann, M.-L.; Brüssow, H. Phage-Host Interaction: An Ecological Perspective. J. Bacteriol. 2004, 186, 3677–3686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Łoś, M.; Węgrzyn, G. Pseudolysogeny. Adv. Appl. Microbiol. 2012, 82, 339–349. [Google Scholar]
- Etank, M.; Bryant, D.A. Nutrient requirements and growth physiology of the photoheterotrophic Acidobacterium, Chloracidobacterium thermophilum. Front. Microbiol. 2015, 6, 226. [Google Scholar] [CrossRef] [Green Version]
- Edwards, R.A.; Vega, A.A.; Norman, H.M.; Ohaeri, M.; Levi, K.; Dinsdale, E.A.; Cinek, O.; Aziz, R.K.; McNair, K.; Barr, J.J.; et al. Global phylogeography and ancient evolution of the widespread human gut virus crAssphage. Nat. Microbiol. 2019, 4, 1727–1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerin, E.; Shkoporov, A.; Stockdale, S.R.; Clooney, A.G.; Ryan, F.J.; Sutton, T.D.; Draper, L.A.; Gonzalez-Tortuero, E.; Ross, R.P.; Hill, C. Biology and Taxonomy of crAss-like Bacteriophages, the Most Abundant Virus in the Human Gut. Cell Host Microbe 2018, 24, 653–664.e6. [Google Scholar] [CrossRef] [Green Version]
- Dutilh, B.E.; Cassman, N.; McNair, K.; Sanchez, S.E.; Silva, G.G.Z.; Boling, L.; Barr, J.J.; Speth, D.R.; Seguritan, V.; Aziz, R.K.; et al. A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nat. Commun. 2014, 5, 4498. [Google Scholar] [CrossRef] [Green Version]
- Shkoporov, A.N.; Khokhlova, E.V.; Fitzgerald, C.B.; Stockdale, S.R.; Draper, L.A.; Ross, R.P.; Hill, C. ΦCrAss001 represents the most abundant bacteriophage family in the human gut and infects Bacteroides intestinalis. Nat. Commun. 2018, 9, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Roux, S.; Adriaenssens, E.M.; Dutilh, B.E.; Koonin, E.V.; Kropinski, A.M.; Krupovic, M.; Kuhn, J.H.; Lavigne, R.; Brister, J.R.; Varsani, A.; et al. Minimum Information about an Uncultivated Virus Genome (MIUViG). Nat. Biotechnol. 2019, 37, 29–37. [Google Scholar] [CrossRef]
- Thurber, R.V.; Haynes, M.; Breitbart, M.; Wegley, L.; Rohwer, F. Laboratory procedures to generate viral metagenomes. Nat. Protoc. 2009, 4, 470–483. [Google Scholar] [CrossRef] [PubMed]
- Minot, S.; Sinha, R.; Chen, J.; Li, H.; Keilbaugh, S.A.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. The human gut virome: Inter-individual variation and dynamic response to diet. Genome Res. 2011, 21, 1616–1625. [Google Scholar] [CrossRef] [Green Version]
- Reyes, A.; Haynes, M.; Hanson, N.; Angly, F.E.; Heath, A.C.; Rohwer, F.; Gordon, J.I. Viruses in the faecal microbiota of monozygotic twins and their mothers. Nat. Cell Biol. 2010, 466, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Breitbart, M.; Lee, W.H.; Run, J.-Q.; Wei, C.L.; Soh, S.W.L.; Hibberd, M.L.; Liu, E.T.; Rohwer, F.; Ruan, Y. RNA Viral Community in Human Feces: Prevalence of Plant Pathogenic Viruses. PLoS Biol. 2005, 4, e3. [Google Scholar] [CrossRef] [Green Version]
- Callanan, J.; Stockdale, S.R.; Shkoporov, A.; Draper, L.A.; Ross, R.P.; Hill, C. Expansion of known ssRNA phage genomes: From tens to over a thousand. Sci. Adv. 2020, 6, eaay5981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, M.; Lin, X.-D.; Tian, J.-H.; Chen, L.-J.; Chen, X.; Li, C.-X.; Qin, X.-C.; Li, J.; Cao, J.-P.; Eden, J.-S.; et al. Redefining the invertebrate RNA virosphere. Nat. Cell Biol. 2016, 540, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, S.R.; Janowski, A.B.; Zhao, G.; Barouch, D.; Wang, D. Hyperexpansion of RNA Bacteriophage Diversity. PLoS Biol. 2016, 14, e1002409. [Google Scholar] [CrossRef]
- Vestergaard, G.; Aramayo, R.; Basta, T.; Häring, M.; Peng, X.; Brügger, K.; Chen, L.; Rachel, R.; Boisset, N.; Garrett, R.A.; et al. Structure of the Acidianus Filamentous Virus 3 and Comparative Genomics of Related Archaeal Lipothrixviruses. J. Virol. 2007, 82, 371–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, C.; Chipman, P.R.; Battisti, A.J.; Bowman, V.D.; Renesto, P.; Raoult, D.; Rossmann, M.G. Cryo-electron Microscopy of the Giant Mimivirus. J. Mol. Biol. 2005, 353, 493–496. [Google Scholar] [CrossRef]
- Gorzelak, M.A.; Gill, S.K.; Tasnim, N.; Ahmadi-Vand, Z.; Jay, M.; Gibson, D.L. Methods for Improving Human Gut Microbiome Data by Reducing Variability through Sample Processing and Storage of Stool. PLoS ONE 2015, 10, e0134802. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.H. The stability of bacterial viruses in solutions of salts. J. Gen. Physiol. 1949, 32, 579–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDaniel, L.D.; Young, E.; Delaney, J.; Ruhnau, F.; Ritchie, K.B.; Paul, J.H. High Frequency of Horizontal Gene Transfer in the Oceans. Science 2010, 330, 50. [Google Scholar] [CrossRef] [Green Version]
- Bushman, F. Lateral DNA Transfer; Cold Spring Harbor Laboratory Press: Cold Spring Harbour, NY, USA, 2002; ISBN 978-0-87969-603-0. [Google Scholar]
- Norman, J.M.; Handley, S.A.; Baldridge, M.T.; Droit, L.; Liu, C.Y.; Keller, B.C.; Kambal, A.; Monaco, C.L.; Zhao, G.; Fleshner, P.; et al. Disease-Specific Alterations in the Enteric Virome in Inflammatory Bowel Disease. Cell 2015, 160, 447–460. [Google Scholar] [CrossRef] [Green Version]
- Links, M.G.; Dumonceaux, T.J.; Hemmingsen, S.M.; Hill, J.E. The Chaperonin-60 Universal Target Is a Barcode for Bacteria That Enables De Novo Assembly of Metagenomic Sequence Data. PLoS ONE 2012, 7, e49755. [Google Scholar] [CrossRef] [Green Version]
- Hill, J.E.; Penny, S.L.; Crowell, K.G.; Goh, S.H.; Hemmingsen, S.M. cpnDB: A Chaperonin Sequence Database. Genome Res. 2004, 14, 1669–1675. [Google Scholar] [CrossRef] [Green Version]
- Hoyles, L.; McCartney, A.L.; Neve, H.; Gibson, G.R.; Sanderson, J.D.; Heller, K.J.; van Sinderen, D. Characterization of virus-like particles associated with the human faecal and caecal microbiota. Res. Microbiol. 2014, 165, 803–812. [Google Scholar] [CrossRef] [Green Version]
- Czajkowski, R.; Ozymko, Z.; Lojkowska, E. Application of zinc chloride precipitation method for rapid isolation and concentration of infectious Pectobacterium spp. and Dickeya spp. lytic bacteriophages from surface water and plant and soil extracts. Folia Microbiol. 2015, 61, 29–33. [Google Scholar] [CrossRef] [Green Version]
- Casey, A.; Jordan, K.; Neve, H.; Coffey, A.; McAuliffe, O. A tail of two phages: Genomic and functional analysis of Listeria monocytogenes phages vB_LmoS_188 and vB_LmoS_293 reveal the receptor-binding proteins involved in host specificity. Front. Microbiol. 2015, 6, 1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fauquet, C.; Mayo, M.A.; Maniloff, J.; Desselberger, U.; Ball, L.A. Virus Taxonomy - Eighth Report of the International Committee on the Taxonomy of Viruses. Viruses 2005, 83, 988–992. [Google Scholar]
- Kleiner, M.; Hooper, L.V.; Duerkop, B.A. Evaluation of methods to purify virus-like particles for metagenomic sequencing of intestinal viromes. BMC Genom. 2015, 16, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, T.; Gilbert, J.; Meyer, F. Metagenomics - a guide from sampling to data analysis. Microb. Inform. Exp. 2012, 2, 3. [Google Scholar] [CrossRef] [Green Version]
- Shaw, K.J.; Thain, L.; Docker, P.T.; Dyer, C.E.; Greenman, J.; Greenway, G.M.; Haswell, S.J. The use of carrier RNA to enhance DNA extraction from microfluidic-based silica monoliths. Anal. Chim. Acta 2009, 652, 231–233. [Google Scholar] [CrossRef]
- Garmaeva, S.; Sinha, T.; Kurilshikov, A.; Fu, J.; Wijmenga, C.; Zhernakova, A. Studying the gut virome in the metagenomic era: Challenges and perspectives. BMC Biol. 2019, 17, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, E.M.; Farkas, K.; Harrison, C.; Jones, D.L.; Allison, H.E.; McCarthy, A.J. Viromic Analysis of Wastewater Input to a River Catchment Reveals a Diverse Assemblage of RNA Viruses. mSystems 2018, 3, e00025-18. [Google Scholar] [CrossRef] [Green Version]
- Acheson, N.H.; Tamm, I. Ribonuclease Sensitivity of Semliki Forest Virus Nucleocapsids. J. Virol. 1970, 5, 714–717. [Google Scholar] [CrossRef] [Green Version]
- Džunková, M.; Garcia-Garcerà, M.; Martínez-Priego, L.; D’Auria, G.; Calafell, F.; Moya, A. Direct Sequencing from the Minimal Number of DNA Molecules Needed to Fill a 454 Picotiterplate. PLoS ONE 2014, 9, e97379. [Google Scholar] [CrossRef]
- Yilmaz, S.; Allgaier, M.; Hugenholtz, P. Multiple displacement amplification compromises quantitative analysis of metagenomes. Nat. Methods 2010, 7, 943–944. [Google Scholar] [CrossRef]
- Dean, F.B.; Hosono, S.; Fang, L.; Wu, X.; Faruqi, A.F.; Bray-Ward, P.; Sun, Z.; Zong, Q.; Du, Y.; Du, J.; et al. Comprehensive human genome amplification using multiple displacement amplification. Proc. Natl. Acad. Sci. USA 2002, 99, 5261–5266. [Google Scholar] [CrossRef] [Green Version]
- Minot, S.; Bryson, A.; Chehoud, C.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. Rapid evolution of the human gut virome. Proc. Natl. Acad. Sci. USA 2013, 110, 12450–12455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waller, A.S.; Yamada, T.; Kristensen, D.M.; Kultima, J.R.; Sunagawa, S.; Koonin, E.V.; Bork, P. Classification and quantification of bacteriophage taxa in human gut metagenomes. ISME J. 2014, 8, 1391–1402. [Google Scholar] [CrossRef]
- McCann, A.; Ryan, F.J.; Stockdale, S.R.; Dalmasso, M.; Blake, T.; Ryan, C.A.; Stanton, C.; Mills, S.; Ross, P.R.; Hill, C. Viromes of one year old infants reveal the impact of birth mode on microbiome diversity. PeerJ 2018, 6, e4694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roux, S.; Solonenko, N.E.; Dang, V.T.; Poulos, B.T.; Schwenck, S.M.; Goldsmith, D.B.; Coleman, M.L.; Breitbart, M.; Sullivan, M.B. Towards quantitative viromics for both double-stranded and single-stranded DNA viruses. PeerJ 2016, 4, e2777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, E.S.; Zhou, Y.; Zhao, G.; Bauer, I.K.; Droit, L.; Ndao, I.M.; Warner, B.B.; Tarr, P.I.; Wang, D.; Holtz, L.R. Early life dynamics of the human gut virome and bacterial microbiome in infants. Nat. Med. 2015, 21, 1228–1234. [Google Scholar] [CrossRef]
- Gregory, A.C.; Zablocki, O.; Zayed, A.A.; Howell, A.; Bolduc, B.; Sullivan, M.B. The Gut Virome Database Reveals Age-Dependent Patterns of Virome Diversity in the Human Gut. Cell Host Microbe 2020, 28, 724–740.e8. [Google Scholar] [CrossRef] [PubMed]
- Manrique, P.; Bolduc, B.; Walk, S.T.; Van Der Oost, J.; De Vos, W.M.; Young, M.J. Healthy human gut phageome. Proc. Natl. Acad. Sci. USA 2016, 113, 10400–10405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohlander, S.K.; Espinosa, R.; Le Beau, M.M.; Rowley, J.D.; Diaz, M.O. A method for the rapid sequence-independent amplification of microdissected chromosomal material. Genome 1992, 13, 1322–1324. [Google Scholar] [CrossRef]
- Krishnamurthy, S.R.; Wang, D. Extensive conservation of prokaryotic ribosomal binding sites in known and novel picobirnaviruses. Virology 2018, 516, 108–114. [Google Scholar] [CrossRef]
- Kapusinszky, B.; Minor, P.; Delwart, E. Nearly Constant Shedding of Diverse Enteric Viruses by Two Healthy Infants. J. Clin. Microbiol. 2012, 50, 3427–3434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ly, M.; Jones, M.B.; Abeles, S.R.; Santiago-Rodriguez, T.M.; Gao, J.; Chan, I.C.; Ghose, C.; Pride, D.T. Transmission of viruses via our microbiomes. Microbiome 2016, 4, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Monaco, C.L.; Gootenberg, D.B.; Zhao, G.; Handley, S.A.; Ghebremichael, M.S.; Lim, E.S.; Lankowski, A.; Baldridge, M.T.; Wilen, C.B.; Flagg, M.; et al. Altered Virome and Bacterial Microbiome in Human Immunodeficiency Virus-Associated Acquired Immunodeficiency Syndrome. Cell Host Microbe 2016, 19, 311–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes, A.; Blanton, L.V.; Gordon, J.I.; Cao, S.; Zhao, G.; Manary, M.J.; Trehan, I.; Smith, M.I.; Wang, D.; Virgin, H.W.; et al. Gut DNA viromes of Malawian twins discordant for severe acute malnutrition. Proc. Natl. Acad. Sci. USA 2015, 112, 11941–11946. [Google Scholar] [CrossRef] [Green Version]
- Starr, E.P.; Nuccio, E.E.; Pett-Ridge, J.; Banfield, J.F.; Firestone, M.K. Metatranscriptomic reconstruction reveals RNA viruses with the potential to shape carbon cycling in soil. Proc. Natl. Acad. Sci. USA 2019, 116, 25900–25908. [Google Scholar] [CrossRef] [Green Version]
- Hölzer, M.; Marz, M. Software Dedicated to Virus Sequence Analysis “Bioinformatics Goes Viral”. Adv. Appl. Microbiol. 2017, 99, 233–257. [Google Scholar] [CrossRef]
- Castromejia, J.L.; Muhammed, M.K.; Kot, W.; Neve, H.; Franz, C.M.A.P.; Hansen, L.H.; Vogensen, F.K.; Nielsen, D.S. Optimizing protocols for extraction of bacteriophages prior to metagenomic analyses of phage communities in the human gut. Microbiome 2015, 3, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Nooij, S.; Schmitz, D.; Vennema, H.; Kroneman, A.; Koopmans, M.P.G. Overview of Virus Metagenomic Classification Methods and Their Biological Applications. Front. Microbiol. 2018, 9, 749. [Google Scholar] [CrossRef]
- Lorenzi, H.A.; Hoover, J.; Inman, J.; Safford, T.; Murphy, S.; Kagan, L.; Williamson, S.J. TheViral MetaGenome Annotation Pipeline (VMGAP):an automated tool for the functional annotation of viral Metagenomic shotgun sequencing data. Stand. Genom. Sci. 2011, 4, 418–429. [Google Scholar] [CrossRef] [Green Version]
- Wommack, K.E.; Bhavsar, J.; Polson, S.W.; Chen, J.; Dumas, M.; Srinivasiah, S.; Furman, M.; Jamindar, S.; Nasko, D.J. VIROME: A standard operating procedure for analysis of viral metagenome sequences. Stand. Genom. Sci. 2012, 6, 427–439. [Google Scholar] [CrossRef] [Green Version]
- Roux, S.; Tournayre, J.; Mahul, A.; Debroas, D.; Enault, F. Metavir 2: New tools for viral metagenome comparison and assembled virome analysis. BMC Bioinform. 2014, 15, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angly, F.; Rodriguez-Brito, B.; Bangor, D.; McNairnie, P.; Breitbart, M.; Salamon, P.; Felts, B.; Nulton, J.; Mahaffy, J.; Rohwer, F. PHACCS, an online tool for estimating the structure and diversity of uncultured viral communities using metagenomic information. BMC Bioinform. 2005, 6, 41. [Google Scholar] [CrossRef] [Green Version]
- Lorenz, J.G.; Jackson, W.E.; Beck, J.C.; Hanner, R. The problems and promise of DNA barcodes for species diagnosis of primate biomaterials. Philos. Trans. R. Soc. B Biol. Sci. 2005, 360, 1869–1877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, D.; Priyadarshini, P.; Vrati, S. Unraveling the Web of Viroinformatics: Computational Tools and Databases in Virus Research. J. Virol. 2015, 89, 1489–1501. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Song, K.; Deng, C.; Ahlgren, N.A.; Fuhrman, J.A.; Li, Y.; Xie, X.; Poplin, R.; Sun, F. Identifying viruses from metagenomic data using deep learning. Quant. Biol. 2020, 8, 64–77. [Google Scholar] [CrossRef] [Green Version]
- Alawi, M.; Burkhardt, L.; Indenbirken, D.; Reumann, K.; Christopeit, M.; Kröger, N.; Lütgehetmann, M.; Aepfelbacher, M.; Fischer, N.; Grundhoff, A. DAMIAN: An open source bioinformatics tool for fast, systematic and cohort based analysis of microorganisms in diagnostic samples. Sci. Rep. 2019, 9, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Salzberg, S.L.; Phillippy, A.M.; Zimin, A.; Puiu, D.; Magoc, T.; Koren, S.; Treangen, T.J.; Schatz, M.C.; Delcher, A.L.; Roberts, M.; et al. GAGE: A critical evaluation of genome assemblies and assembly algorithms. Genome Res. 2012, 22, 557–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, I.; Ponsero, A.J.; Bomhoff, M.; Youens-Clark, K.; Hartman, J.H.; Hurwitz, B.L. Libra: Scalablek-mer–based tool for massive all-vs-all metagenome comparisons. GigaScience 2018, 8, 8. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Ahlgren, N.A.; Lu, Y.Y.; Fuhrman, J.A.; Sun, F. VirFinder: A novel k-mer based tool for identifying viral sequences from assembled metagenomic data. Microbiome 2017, 5, 1–20. [Google Scholar] [CrossRef]
- Viral Genomes. Available online: https://www.ncbi.nlm.nih.gov/genome/viruses/ (accessed on 28 January 2021).
- Goodacre, N.; Aljanahi, A.; Nandakumar, S.; Mikailov, M.; Khan, A.S. A Reference Viral Database (RVDB) To Enhance Bioinformatics Analysis of High-Throughput Sequencing for Novel Virus Detection. mSphere 2018, 3, e00069-18. [Google Scholar] [CrossRef] [Green Version]
- Pickett, B.E.; Sadat, E.L.; Zhang, Y.; Noronha, J.M.; Squires, R.B.; Hunt, V.; Liu, M.; Kumar, S.; Zaremba, S.; Gu, Z.; et al. ViPR: An open bioinformatics database and analysis resource for virology research. Nucleic Acids Res. 2011, 40, D593–D598. [Google Scholar] [CrossRef] [PubMed]
- NCBI Resources Genbank. Available online: http://www.ncbi.nlm.nih.gov/genbank/ (accessed on 19 September 2012).
- Grazziotin, A.L.; Koonin, E.V.; Kristensen, D.M. Prokaryotic Virus Orthologous Groups (pVOGs): A resource for comparative genomics and protein family annotation. Nucleic Acids Res. 2017, 45, D491–D498. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Callanan, J.; Stockdale, S.R.; Shkoporov, A.; Draper, L.A.; Ross, R.P.; Hill, C. Biases in Viral Metagenomics-Based Detection, Cataloguing and Quantification of Bacteriophage Genomes in Human Faeces, a Review. Microorganisms 2021, 9, 524. https://doi.org/10.3390/microorganisms9030524
Callanan J, Stockdale SR, Shkoporov A, Draper LA, Ross RP, Hill C. Biases in Viral Metagenomics-Based Detection, Cataloguing and Quantification of Bacteriophage Genomes in Human Faeces, a Review. Microorganisms. 2021; 9(3):524. https://doi.org/10.3390/microorganisms9030524
Chicago/Turabian StyleCallanan, Julie, Stephen R. Stockdale, Andrey Shkoporov, Lorraine A. Draper, R. Paul Ross, and Colin Hill. 2021. "Biases in Viral Metagenomics-Based Detection, Cataloguing and Quantification of Bacteriophage Genomes in Human Faeces, a Review" Microorganisms 9, no. 3: 524. https://doi.org/10.3390/microorganisms9030524