
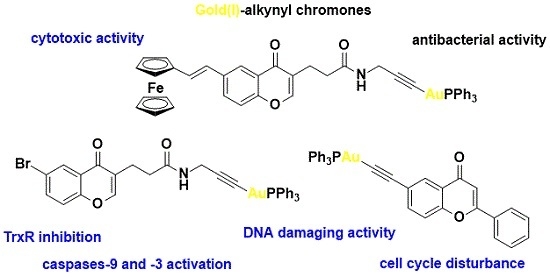
Anticancer and Antibacterial Activity Studies of Gold(I)-Alkynyl Chromones

Abstract
:
1. Introduction
2. Results and Discussion
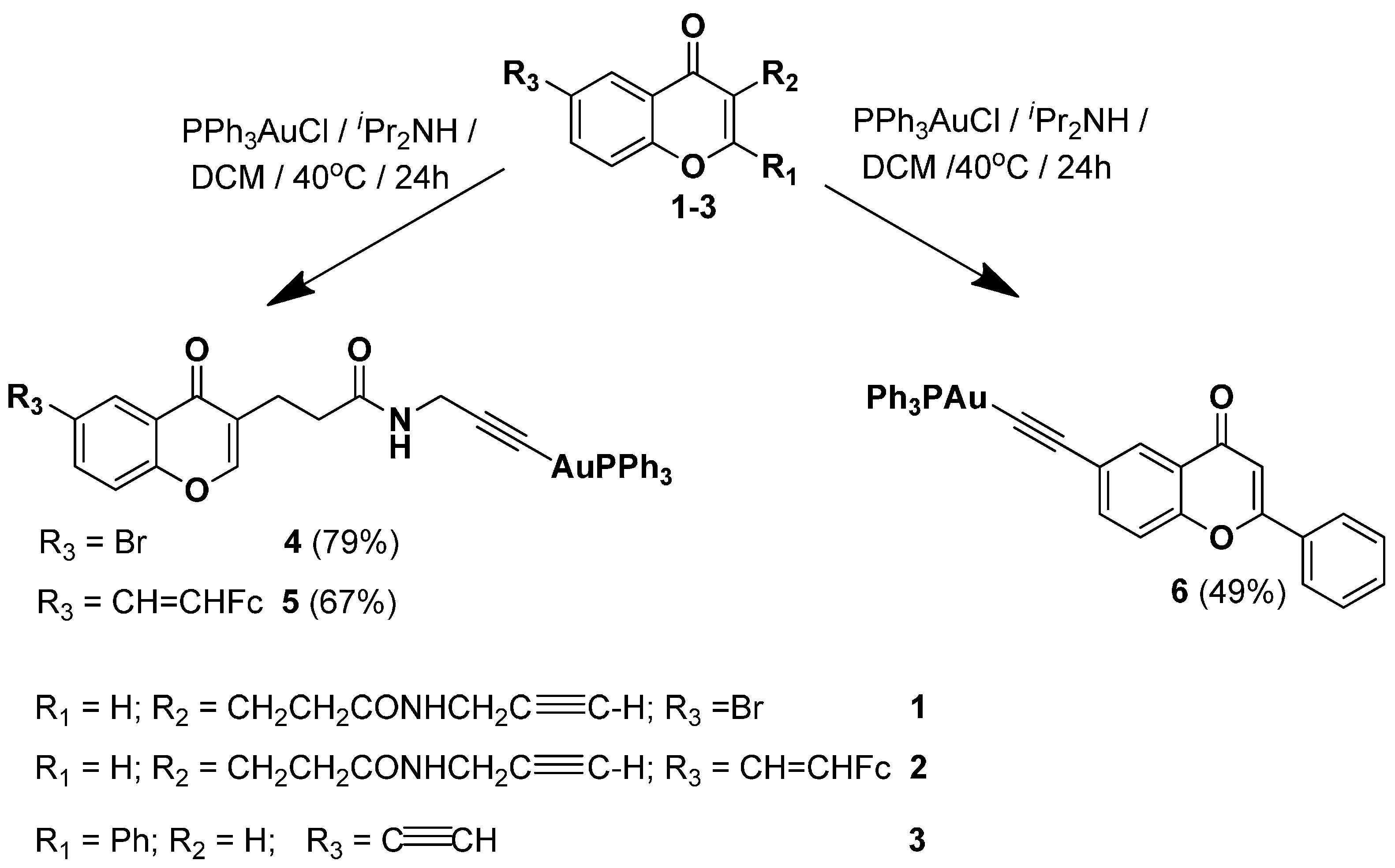
2.1. Synthesis of Complexes 4–6

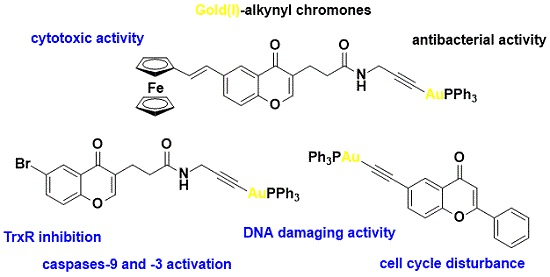
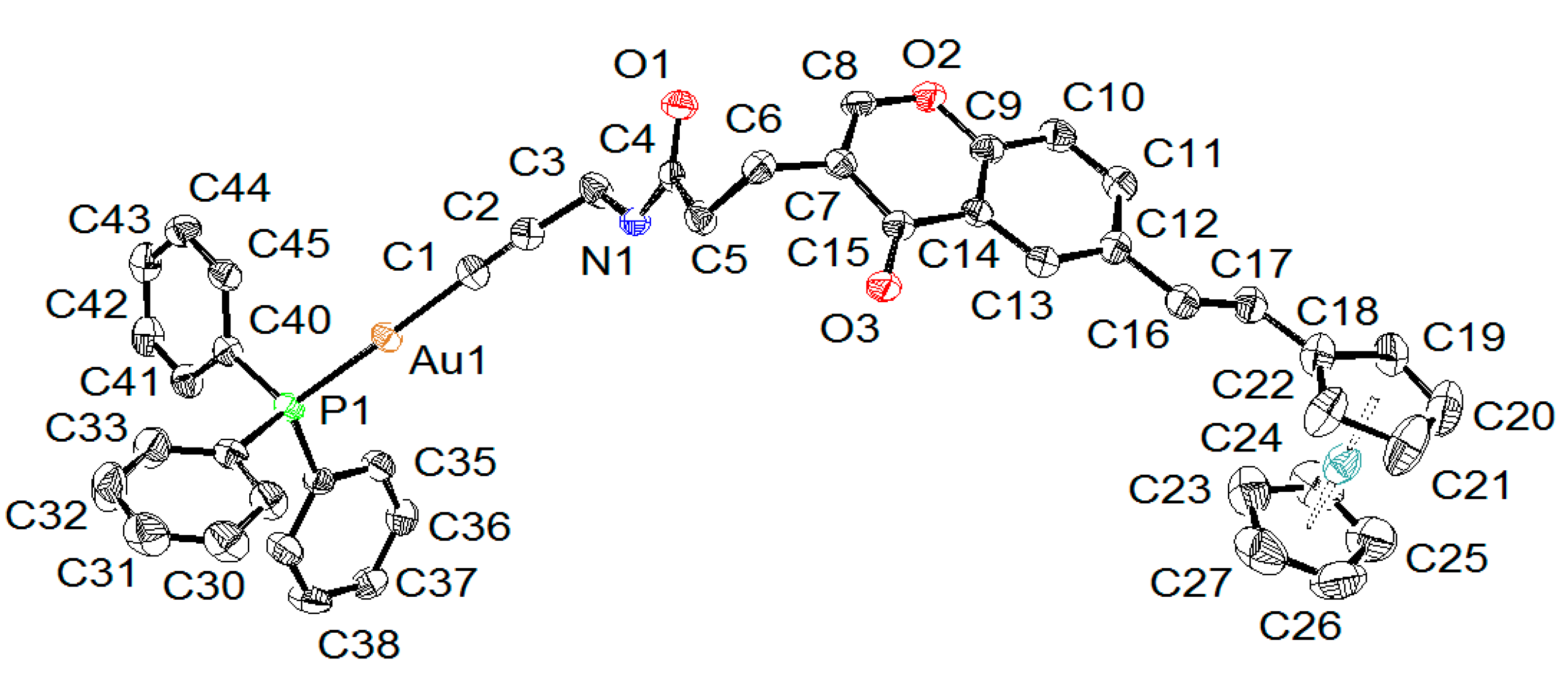
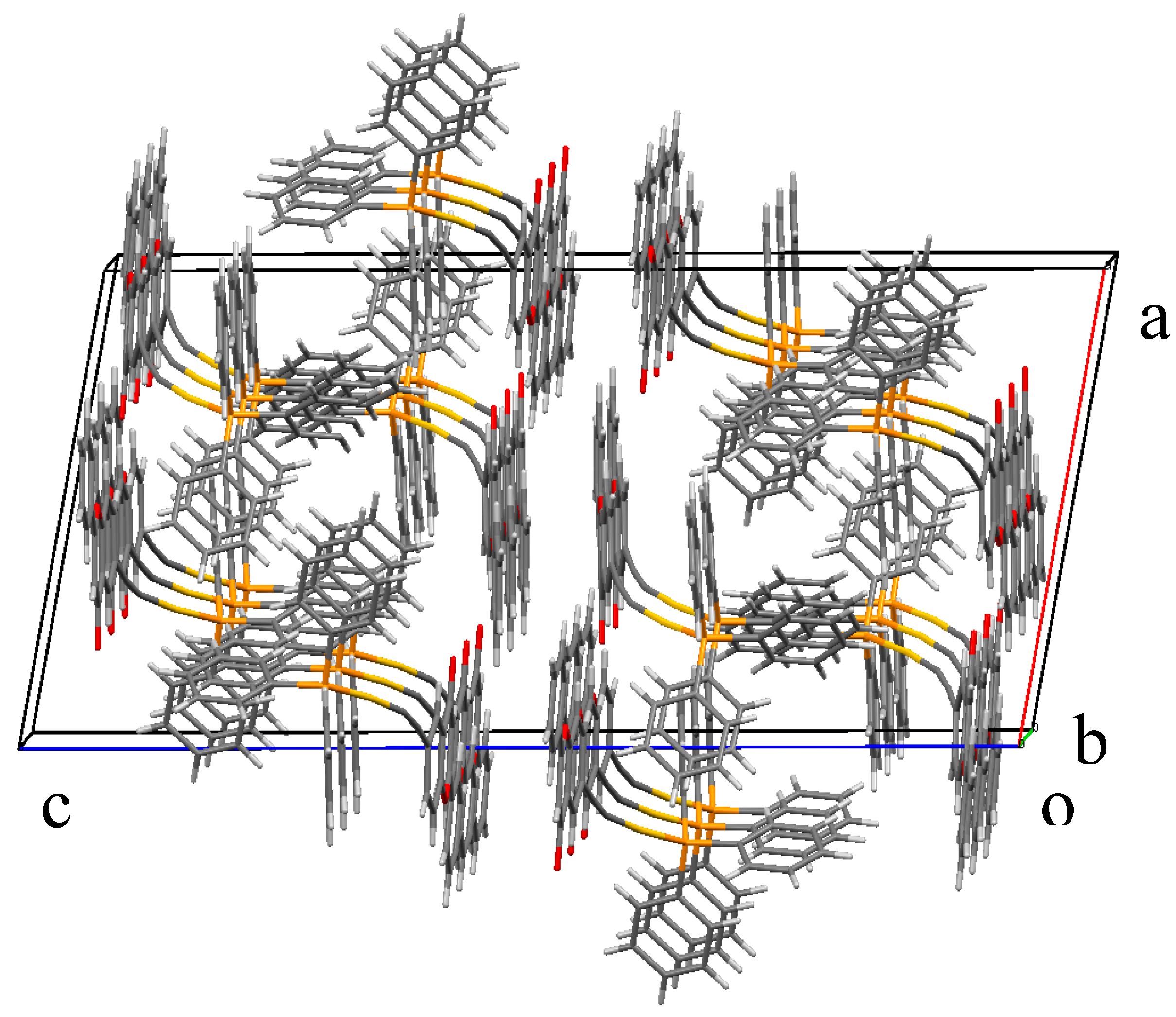
2.2. X-ray Diffraction Study of Compounds



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 5 | 6A | 6B |
|---|---|---|---|
| Interatomic Distances (Å) | |||
| Au-C | 1.996(4) | 1.989(5) | 1.988(4) |
| Au-P | 2.2787(10) | 2.2739(12) | 2.2720(11) |
| C≡C | 1.205(6) | 1.214(6) | 1.199(6) |
| Angles (°) | |||
| P-Au-C | 178.13(14) | 174.00(12) | 172.46(12) |
| Au-C≡C | 176.5(4) | 164.3(4) | 165.9(4) |

2.3. Biological Results
2.3.1. Antiproliferative Effect toward Human Cancer Cells
| Cell Line | IC50 (µmol/L) | |||
|---|---|---|---|---|
| 4 | 5 | 6 | Auranofin | |
| HepG2 | 10.0 ± 1.3 | >120 | 19.5 ± 1.9 | 50.0 ± 2.7 |
| MCF-7 | 5.5 ± 0.7 | 11.0 ± 0.8 | 13.0 ± 2.3 | 13.1 ± 0.9 |
| MDA-MB-231 | 9.7 ± 2.5 | 49.7 ± 1,7 | 10.0 ± 3.4 | 3.0 ± 0.6 |
| CCRF-CEM | 3.5 ± 1.1 | 3.8 ± 0.9 | 4.2 ± 0.9 | 6.0 ± 1.2 |
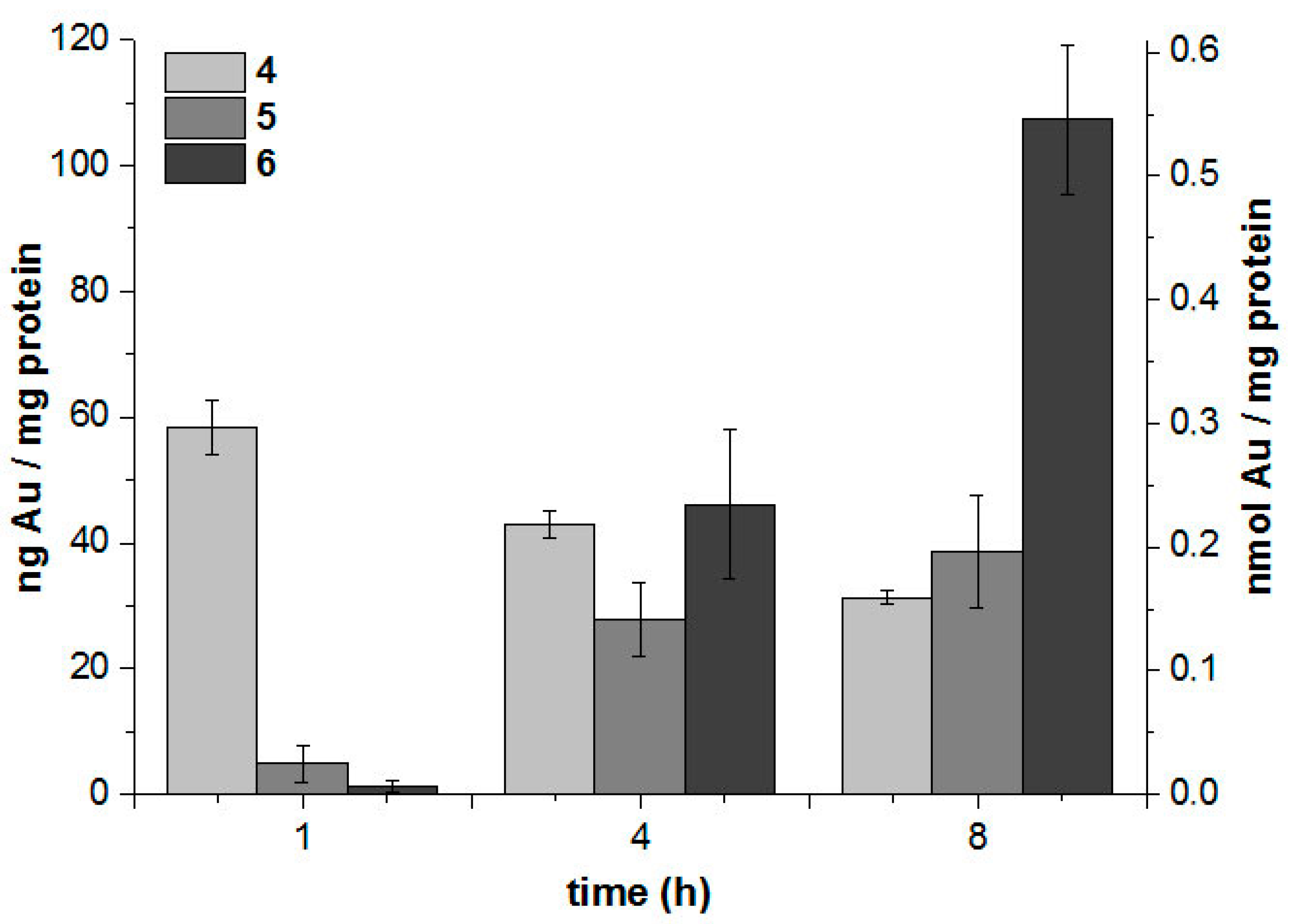
2.3.2. Cellular Uptake with High-Resolution Continuum-Source Atomic Absorption Spectroscopy

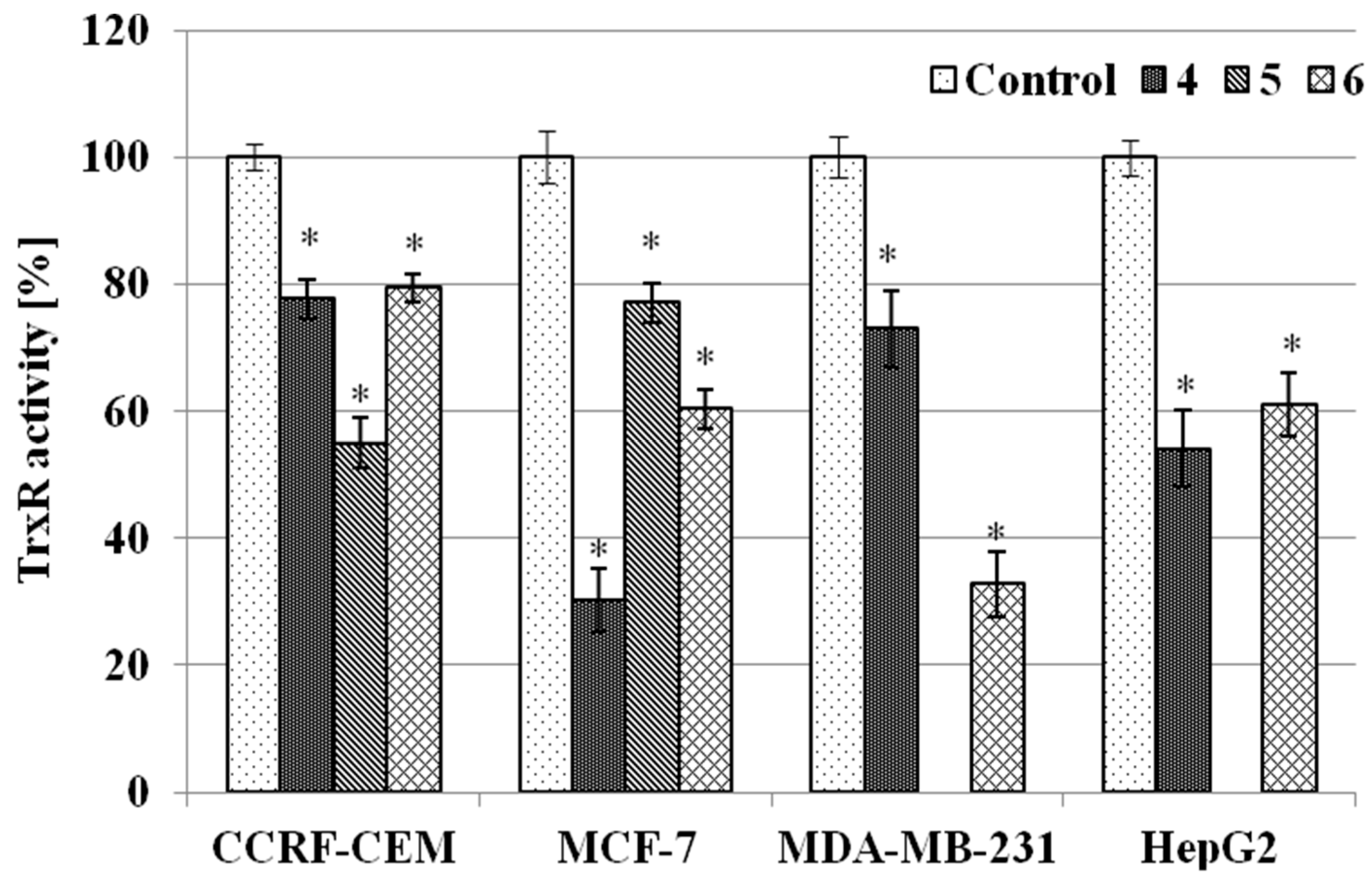
2.3.3. TrxR Inhibition Assay

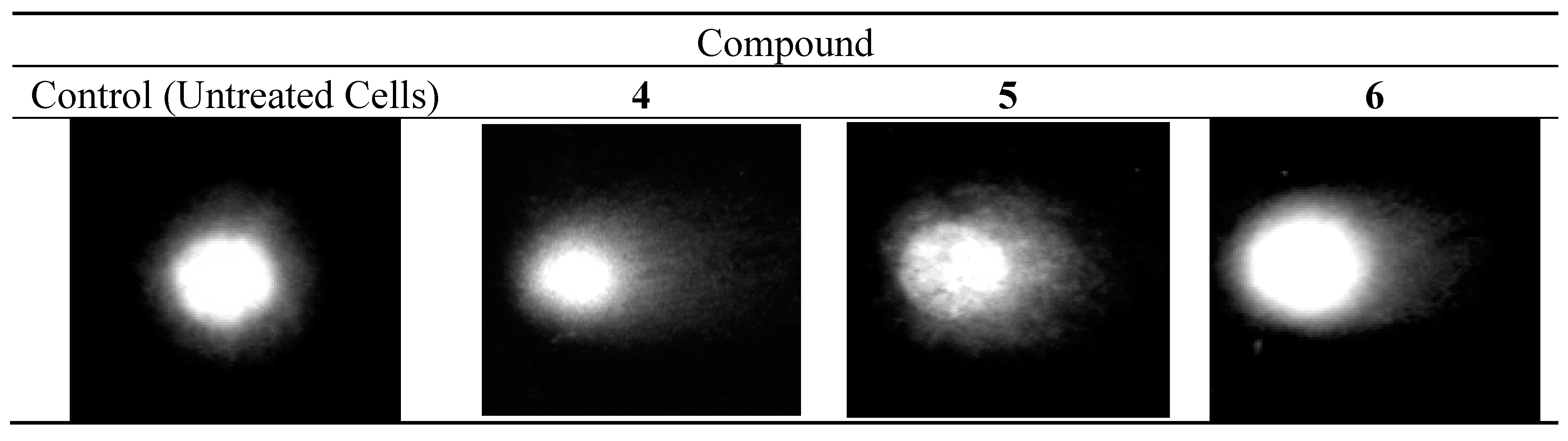
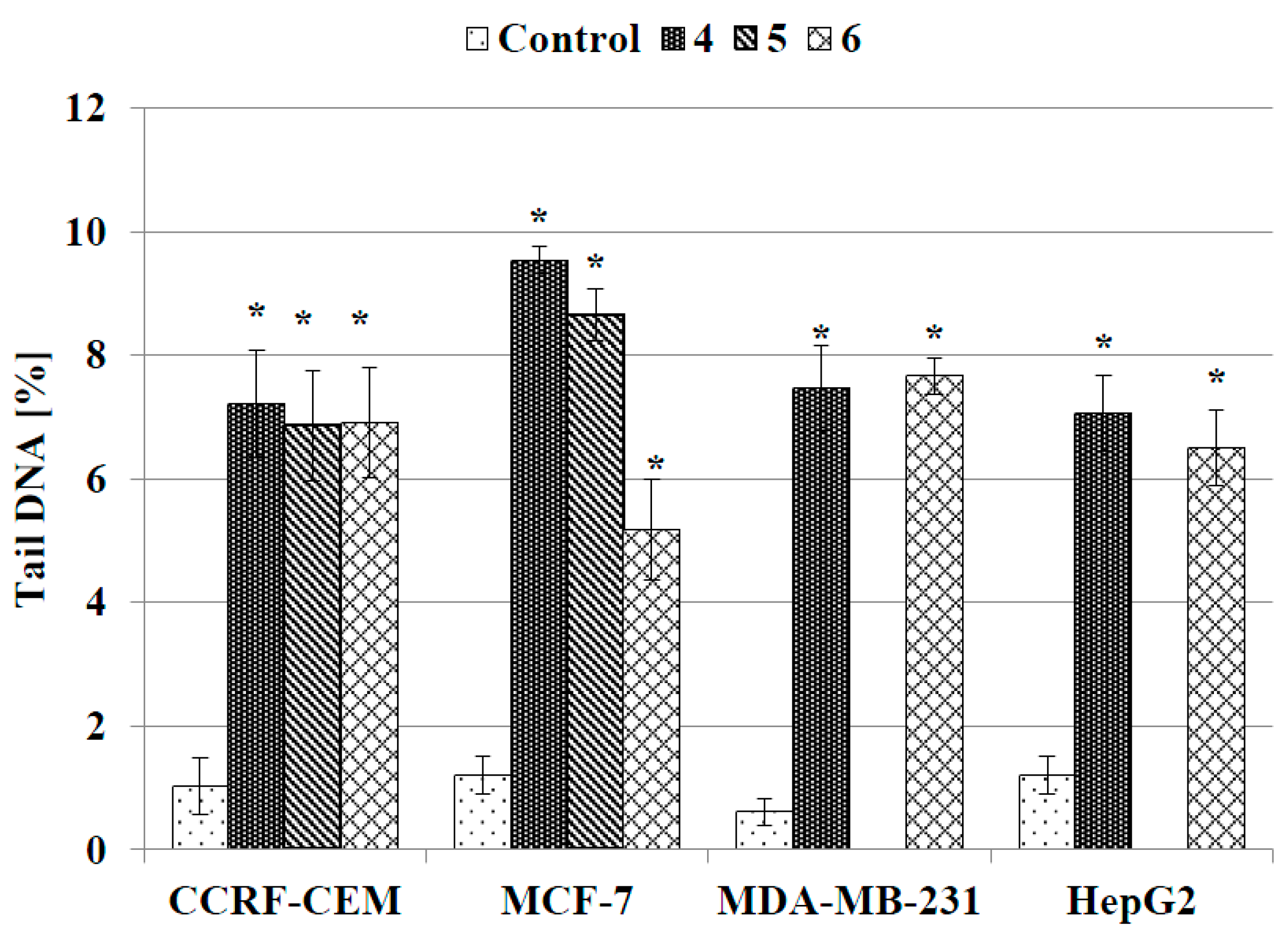
2.3.4. DNA Comet Assay


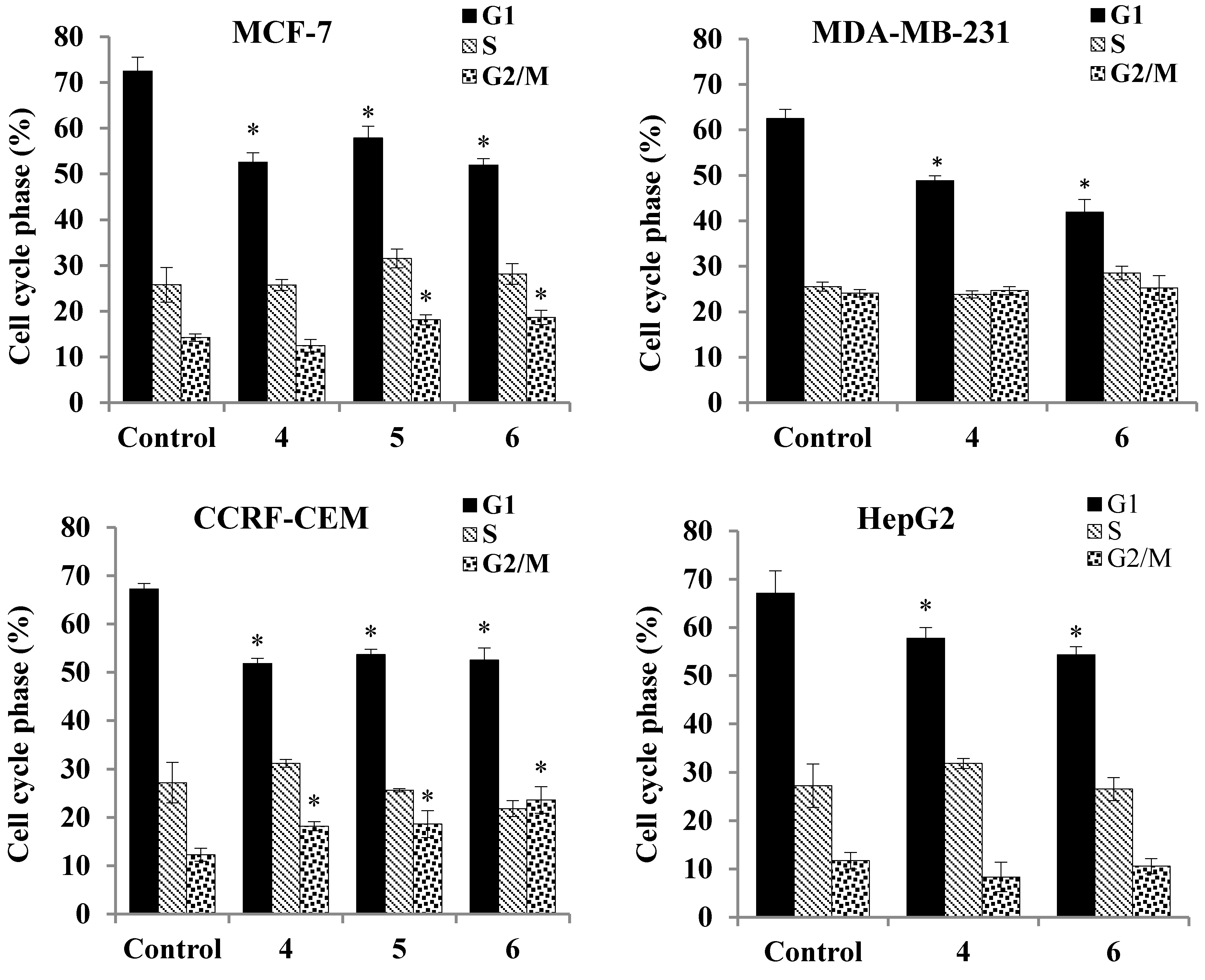
2.3.5. Cell Cycle Analysis

| Cell Line | Cell Cycle Analysis Of DNA Histograms (Sub-G1 Fraction) | |||
|---|---|---|---|---|
| Control (Untreated Cell) | Compound | |||
| 4 | 5 | 6 | ||
| HepG2 | 1.95 ± 0.23 | 15.31 ± 1.64 * | - | 12.46 ± 1 * |
| MDA-MB-231 | 2.39 ± 0.64 | 13.32 ± 1.39 * | - | 5.0 ± 2.51 |
| CCRF-CEM | 2.81 ± 0.51 | 13.33 ± 1.37 * | 6.18 ± 1.1 | 9.64 * ± 1.12 |
| MCF-7 | 2.87 ± 0.9 | 18.58 ± 2.27 * | 9.42 ± 1 * | 10.48 ± 0.6 * |
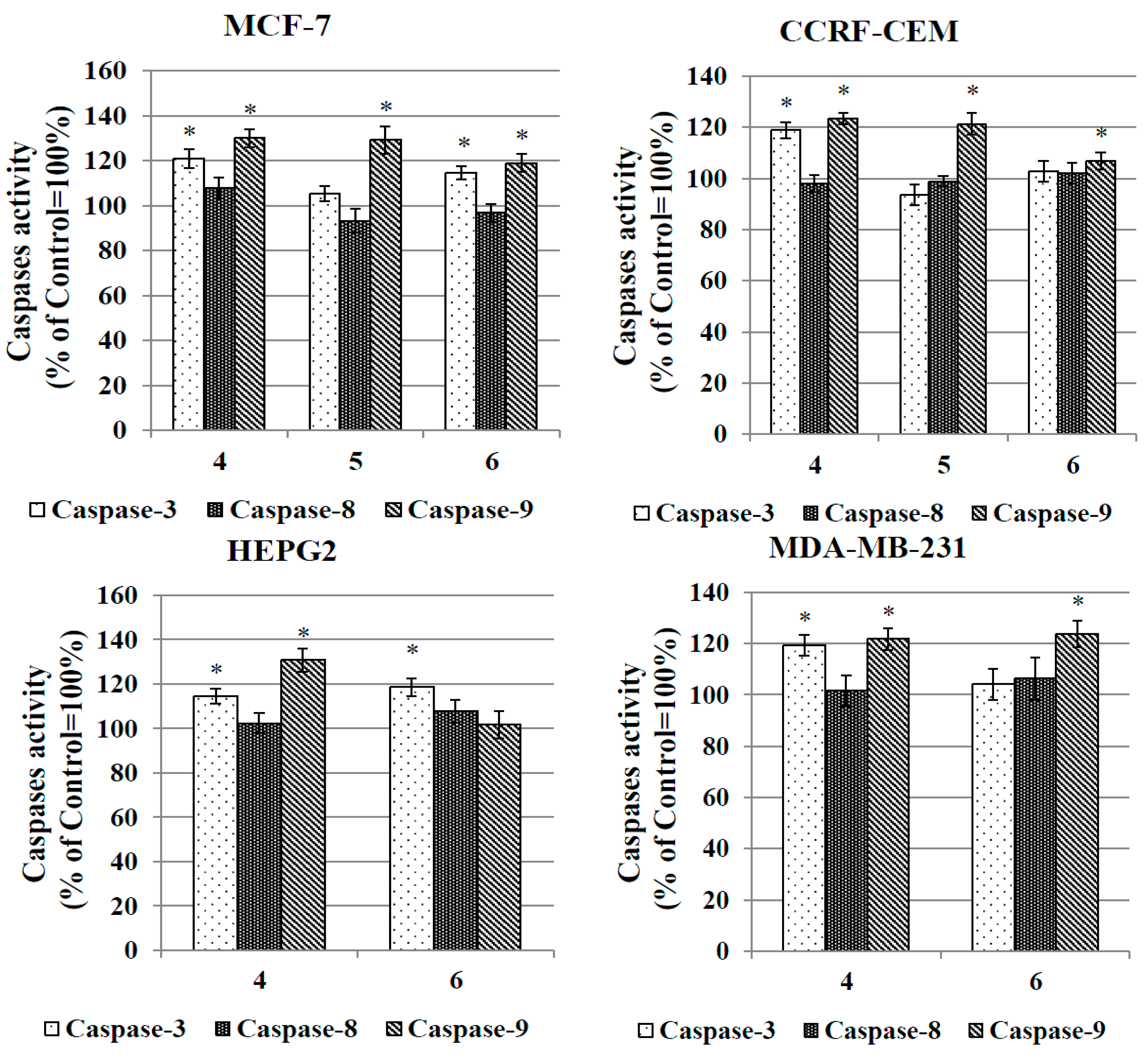
2.3.6. Caspase Activation Assay

2.3.7. Evaluation of Antibacterial and Hemolytic Activity
| Microorganism | MIC (μg/mL) | |||
|---|---|---|---|---|
| 4 | 5 | 6 | Au(PPh3)Cl | |
| S. aureus subsp. aureus ATCC® 29213™ (MSSA) | 4 | 4 | 32 | 2 |
| S. aureus subsp. aureus ATCC® 43300 (MRSA) | 2 | 2 | 2 | 1 |
| E. coli ATCC® 25922 | >256 | >256 | >256 | 16 |
| E. coli ATCC®BAA-198 | >256 | >256 | >256 | 32 |
| Clinical isolates: S. aureus | ||||
| (MSSA) 26/11 | 4 | 4 | 32 | 2 |
| (MSSA) 30/11 | 4 | 4 | 32 | 2 |
| (MRSA) 7/10 | 4 | 4 | 16 | 2 |
| (MRSA) 41/12 | 4 | 4 | 32 | 2 |
3. Experimental Section
3.1. General Information
3.2. Chemistry
3.2.1. Synthesis of 4
3.2.2. Synthesis of 5
3.2.3. Synthesis of 6
3.3. X-ray Crystallography
3.4. Biology
3.4.1. Cell Lines
3.4.2. Cell Culture
3.4.3. Evaluation of Cytotoxic Activity (MTT Assay)
3.4.4. Thioredoxin Reductase Inhibition Assay
3.4.5. DNA Comet Assay
3.4.6. Cell Cycle Analysis
3.4.7. Caspases Activation Assay
Caspase-8 and Caspase-9 Activities
Caspase-3 Activity
3.4.8. Statistical Analysis
3.4.9. Cellular Uptake and AAS Measurements
Antibacterial Activity
Hemolysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Rosenberg, B.; Van, C.L.; Krigas, T. Inhibition of Cell Division in Escherichia coli by Electrolysis Products from a Platinum Electrode. Nature 1965, 205, 698–699. [Google Scholar] [CrossRef] [PubMed]
- Cisplatin: Chemistry and Biochemistry of a Leading Anticancer Drug; Lippert, B. (Ed.) Wiley: New York, NY, USA, 1999.
- Berners-Price, S.J. Activating Platinum Anticancer Complexes with Visible Light. Angew. Chem. Int. Ed. 2011, 50, 804–805. [Google Scholar] [CrossRef] [PubMed]
- Allardyce, C.S.; Dyson, P.J.; Ellis, D.J.; Heath, S.I. [Ru(η6-p-cymene)Cl2(pta)] (pta = 1,3,5-triaza-7-phosphatricyclo-[3.3.1.1]decane): A water soluble compound that exhibits pH dependent DNA binding providing selectivity for diseased cells. Chem. Commun. 2001, 15, 1396–1397. [Google Scholar] [CrossRef]
- Pacor, S.; Zorzet, S.; Cocchietto, M.; Bacac, M.; Vadori, M.; Turrin, C.; Gava, B.; Castellarin, A.; Sava, G. Intratumoral NAMI-A Treatment Triggers Metastasis Reduction, Which Correlates to CD44 Regulation and Tumor Infiltrating Lymphocyte Recruitment. J. Pharmacol. Exp. Ther. 2004, 310, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Hartinger, C.G.; Jakupec, M.A.; Zorbas-Seifried, S.; Groessl, M.; Egger, A.; Berger, W.; Zorbas, H.; Dyson, P.J.; Keppler, B.K. KP1019, A New Redox-Active Anticancer Agent—Preclinical Development and Results of a Clinical Phase I Study in Tumor Patients. Chem. Biodivers. 2008, 5, 2140–2155. [Google Scholar] [CrossRef] [PubMed]
- Debreczeni, J.E.; Bullock, A.N.; Atilla, G.E.; Williams, D.S.; Bregman, H.; Knapp, S.; Meggers, E. Ruthenium Half-Sandwich Complexes Bound to Protein Kinase Pim-1. Angew. Chem. Int. Ed. 2006, 45, 1580–1585. [Google Scholar] [CrossRef] [PubMed]
- Vessières, A.; Top, S.; Beck, W.; Hillard, E.; Jaouen, G. Metal complex SERMs (selective oestrogen receptor modulators). The influence of different metal units on breast cancer cell antiproliferative effects. Dalton Trans. 2006, 4, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Citta, A.; Folda, A.; Bindoli, A.; Pigeon, P.; Top, S.; Vessières, A.; Salmain, M.; Jaouen, G.; Rigobello, M.P. Evidence for Targeting Thioredoxin Reductases with Ferrocenyl Quinone Methides. A Possible Molecular Basis for the Antiproliferative Effect of Hydroxyferrocifens on Cancer Cells. J. Med. Chem. 2014, 57, 8849–8859. [Google Scholar] [CrossRef] [PubMed]
- Rubbiani, R.; Wahrig, B.; Ott, I. Historical and biochemical aspects of a seventeenth century gold-based aurum vitae recipe. J. Biol. Inorg. Chem. 2014, 19, 961–965. [Google Scholar] [CrossRef] [PubMed]
- Koch, R. Ueber bacteriologische Forschung. Dtsch. Med. Wochenstr. 1890, 16, 756–757. [Google Scholar]
- Forestier, J. Rheumatoid arthritis and its treatment by gold salts: The results of six years’ experience. J. Lab. Clin. Med. 1935, 20, 827–840. [Google Scholar] [CrossRef]
- Simon, T.M.; Kunishima, D.H.; Vibert, G.J.; Lorber, A. Screening Trial with the Coordinated Gold Compound Auranofin Using Mouse Lymphocytic Leukemia P388. Cancer Res. 1981, 41, 94–97. [Google Scholar] [PubMed]
- Mirabelli, C.K.; Johnson, R.K.; Sung, C.M.; Faucette, L.; Muirhead, K.; Crooke, S.T. Evaluation of the in Vivo Antitumor Activity and in Vitro Cytotoxic Properties of Auranofin, a Coordinated Gold Compound, in Murine Tumor Models. Cancer Res. 1985, 45, 32–39. [Google Scholar] [PubMed]
- Ott, I. On the medicinal chemistry of gold complexes as anticancer drugs. Coord. Chem. Rev. 2009, 253, 1670–1681. [Google Scholar] [CrossRef]
- Liu, W.; Gust, R. Metal N-heterocyclic carbene complexes as potential antitumor metallodrugs. Chem. Soc. Rev. 2013, 42, 755–773. [Google Scholar] [CrossRef] [PubMed]
- Hackenberg, F.; Tacke, M. Benzyl-substituted metallocarbene antibiotics and anticancer drugs. Dalton Trans. 2014, 43, 8144–8153. [Google Scholar] [CrossRef] [PubMed]
- Cisnetti, F.; Gautier, A. Metal/N-Heterocyclic Carbene Complexes: Opportunities for the Development of Anticancer Metallodrugs. Angew. Chem. Int. Ed. 2013, 52, 11976–11978. [Google Scholar] [CrossRef] [PubMed]
- Glišić, B.D.; Djuran, M.I. Gold complexes as antimicrobial agents: An overview of different biological activities in relation to the oxidation state of the gold ion and the ligand structure. Dalton Trans. 2014, 43, 5950–5969. [Google Scholar] [CrossRef]
- Tacke, M. Benzyl-substituted carbene–metal complexes: Potential for novel antibiotics and anticancer drugs? J. Organomet. Chem. 2015, 782, 17–21. [Google Scholar] [CrossRef]
- Bagowski, C.P.; You, Y.; Scheffler, H.; Vlecken, D.H.; Schmitz, D.J.; Ott, I. Naphthalimide gold(I) phosphine complexes as anticancer metallodrugs. Dalton Trans. 2009, 48, 10799–10805. [Google Scholar] [CrossRef] [PubMed]
- Ott, I.; Qian, X.; Xu, Y.; Vlecken, D.H.W.; Marques, I.J.; Kubutat, D.; Will, J.; Sheldrick, W.S.; Jesse, P.; Prokop, A.; et al. A Gold(I) Phosphine Complex Containing a Naphthalimide Ligand Functions as a TrxR Inhibiting Antiproliferative Agent and Angiogenesis Inhibitor. J. Med. Chem. 2009, 52, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Wang, V.-H.; Shih, W.-C.; Chang, H.C.; Kuo, Y.-Y.; Hung, W.-C.; Ong, T.-G.; Li, W.-S. Preparation and Characterization of Amino-Linked Heterocyclic Carbene Palladium, Gold, and Silver Complexes and Their Use as Anticancer Agents That Act by Triggering Apoptotic Cell Death. J. Med. Chem. 2011, 54, 5245–5249. [Google Scholar] [CrossRef] [PubMed]
- Rubbiani, R.; Salassa, L.; de Almeida, A.; Casini, A.; Ott, I. Cytotoxic Gold(I) N-heterocyclic Carbene Complexes with Phosphane Ligands as Potent Enzyme Inhibitors. ChemMedChem. 2014, 9, 1205–1210. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, B.; de Almeida, A.; van der Burgt, E.P.M.; Picquet, M.; Citta, A.; Folda, A.; Rigobello, M.P.; le Gendre, P.; Bodio, E.; Casini, A.; et al. New Gold(I) Organometallic Compounds with Biological Activity in Cancer Cells. Eur. J. Inorg. Chem. 2014, 27, 4532–4536. [Google Scholar] [CrossRef]
- Arcau, J.; Andermark, V.; Rodrigues, M.; Giannicchi, I.; Pérez-Garcia, L.; Ott, I.; Rodríguez, L. Synthesis and Biological Activity of Gold(I) N-Heterocyclic Carbene Complexes with Long Aliphatic Side Chains. Eur. J. Inorg. Chem. 2014, 35, 6117–6125. [Google Scholar] [CrossRef]
- Hickey, J.L.; Ruhayel, R.A.; Barnard, P.J.; Baker, M.V.; Berners-Price, S.J.; Filipovska, A. Mitochondria-Targeted Chemotherapeutics: The Rational Design of Gold(I) N-Heterocyclic Carbene Complexes That Are Selectively Toxic to Cancer Cells and Target Protein Selenols in Preference to Thiols. J. Am. Chem. Soc. 2008, 130, 12570–12571. [Google Scholar] [CrossRef] [PubMed]
- Wetzel, C.; Kunz, P.C.; Kassack, M.U.; Hamacher, A.; Böhler, P.; Watjen, W.; Ott, I.; Rubbiani, R.; Spingler, B. Gold(I) complexes of water-soluble diphos-type ligands: Synthesis, anticancer activity, apoptosis and thioredoxin reductase inhibition. Dalton Trans. 2011, 40, 9212–9220. [Google Scholar] [CrossRef] [PubMed]
- Balasingham, R.G.; Williams, C.F.; Mottram, H.J.; Coogan, M.P.; Pope, S.J.A. Gold(I) Complexes Derived from Alkynyloxy-Substituted Anthraquinones: Syntheses, Luminescence, Preliminary Cytotoxicity, and Cell Imaging Studies. Organometallics 2012, 31, 5835–5843. [Google Scholar] [CrossRef]
- Meyer, A.; Gutiérrez, A.; Ott, I.; Rodríguez, L. Phosphine-bridged dinuclear gold(I) alkynyl complexes: Thioredoxin reductase inhibition and cytotoxicity. Inorg. Chim. Acta. 2013, 398, 72–76. [Google Scholar] [CrossRef]
- Chui, C.-H.; Wong, R.S.-M.; Gambari, R.; Cheng, G.Y.-M.; Yuen, M.C.-W.; Chan, K.-W.; Tong, S.-W.; Tong, F.-Y.; Lau, F.-Y.; Lai, P.B.-S.; et al. Antitumor activity of diethynylfluorene derivatives of gold(I). Bioorg. Med. Chem. 2009, 17, 7872–7877. [Google Scholar] [CrossRef] [PubMed]
- Schuh, E.; Valiahdi, S.M.; Jakupec, M.A.; Keppler, B.K.; Chiba, P.; Mohr, F. Synthesis and biological studies of some gold(I) complexes containing functionalised alkynes. Dalton Trans. 2009, 48, 10841–10845. [Google Scholar] [CrossRef] [PubMed]
- Vergara, E.; Cerrada, E.; Casini, A.; Zava, O.; Laguna, M.; Dyson, P.J. Antiproliferative Activity of Gold(I) Alkyne Complexes Containing Water-Soluble Phosphane Ligands. Organometallics 2010, 29, 2596–2603. [Google Scholar] [CrossRef]
- Meyer, A.; Bagowski, C.P.; Kokoschka, M.; Stefanopoulou, M.; Alborzinia, H.; Can, S.; Vlecken, D.H.; Sheldrick, W.S.; Wölfl, S.; Ott, I.; et al. On the Biological Properties of Alkynyl Phosphine Gold(I) Complexes. Angew. Chem. Int. Ed. 2012, 51, 8895–8899. [Google Scholar] [CrossRef] [PubMed]
- Stockland, R.A.; Kohler, M.C.; Guzei, I.A.; Kastner, M.E.; Bawiec, J.A.; Labaree, D.C.; Hochberg, R.B. Organometallic Complexes Containing 17-Ethynyl-17β-hydroxyandrost-4-en-3-one and Related Ethynyl Steroids. Organometallics 2006, 25, 2475–2485. [Google Scholar] [CrossRef]
- Rana, B.K.; Nandy, A.; Bertolasi, V.; Bielawski, C.W.; Saha, K.D.; Dinda, J. Novel Gold(I)- and Gold(III)-N-Heterocyclic Carbene Complexes: Synthesis and Evaluation of Their Anticancer Properties. Organometallics 2014, 33, 2544–2548. [Google Scholar] [CrossRef]
- Rubbiani, R.; Zehnder, T.N.; Mari, C.; Blacque, O.; Venkatesan, K.; Gasser, G. Anticancer Profile of a Series of Gold(III) (2-phenyl)pyridine Complexes. ChemMedChem 2014, 9, 2781–2790. [Google Scholar] [CrossRef] [PubMed]
- Zou, T.; Lum, C.T.; Chui, S.-Y.; Che, C.-M. Gold(III) Complexes Containing N-Heterocyclic Carbene Ligands: Thiol “Switch-on” Fluorescent Probes and Anti-Cancer Agents. Angew. Chem. Int. Ed. 2013, 52, 2930–2933. [Google Scholar] [CrossRef] [PubMed]
- Barnard, P.J.; Berners-Price, S.J. Targeting the mitochondrial cell death pathway with gold compounds. Coord. Chem. Rev. 2007, 251, 1889–1902. [Google Scholar] [CrossRef]
- Holenya, P.; Can, S.; Rubbiani, R.; Alborzinia, H.; Jünger, A.; Cheng, X.; Ott, I.; Wölfl, S. Detailed analysis of pro-apoptotic signaling and metabolic adaptation triggered by a N-heterocyclic carbine-gold(I) complex. Metallomics 2014, 6, 1591–1601. [Google Scholar] [CrossRef] [PubMed]
- Pratesi, A.; Gabbiani, C.; Michelucci, E.; Ginanneschi, M.; Papini, A.M.; Rubbiani, R.; Ott, I.; Messori, L. Insights on the mechanism of thioredoxin reductase inhibition by Gold N-heterocyclic carbene compounds using the synthetic linear Selenocysteine containing C-terminal peptide hTrxR(488-499): An ESI-MS investigation. J. Inorg. Biochem. 2014, 136, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Can, P.; Alborzinia, H.; Rubbiani, R.; Ott, I.; Wölfl, S. A TrxR inhibiting gold(I) NHC complex induces apoptosis through ASK1-p38-MAPK signaling in pancreatic cancer cells. Mol. Cancer 2014, 13, 221–236. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, A.; Matos, M.J.; Garrido, J.; Uriarte, E.; Borges, F. Chromone: A Valid Scaffold in Medicinal Chemistry. Chem. Rev. 2014, 9, 4960–4992. [Google Scholar] [CrossRef] [PubMed]
- Keri, R.S.; Budagumpi, S.; Pai, R.K.; Balakrishna, R.G. Chromones as a privileged scaffold in drug discovery: A review. Eur. J. Med. Chem. 2014, 78, 340–374. [Google Scholar] [CrossRef] [PubMed]
- Kurzwernhart, A.; Kandioller, W.; Bartel, C.; Bächler, S.; Trondl, R.; Mühlgassner, G.; Jakupiec, M.A.; Arion, V.B.; Marko, D.; Keppler, B.K.; et al. Targeting the DNA-topoisomerase complex in a double-strike approach with a topoisomerase inhibiting moiety and covalent DNA binder. Chem. Commun. 2012, 48, 4839–4841. [Google Scholar] [CrossRef] [PubMed]
- Kurzwernhart, A.; Kandioller, W.; Bächler, S.; Bartel, C.; Martic, S.; Buczkowska, M.; Mühlgassner, G.; Jakupiec, M.A.; Kraatz, H.-B.; Bednarski, P.J.; et al. Structure–Activity Relationships of Targeted RuII(η6-p-Cymene) Anticancer Complexes with Flavonol-Derived Ligands. J. Med. Chem. 2012, 55, 10512–10522. [Google Scholar] [CrossRef] [PubMed]
- Monserrat, J.-P.; Tiwari, K.N.; Quentin, L.; Pigeon, P.; Jaouen, G.; Vessières, A.; Chabot, G.G.; Hillard, E.A. Ferrocenyl flavonoid-induced morphological modifications of endothelial cells and cytotoxicity against B16 murine melanoma cells. J. Organomet. Chem. 2013, 734, 78–85. [Google Scholar] [CrossRef]
- Monserrat, J.-P.; Al-Safi, R.I.; Tiwari, K.N.; Quentin, L.; Chabot, G.G.; Vessières, A.; Jaouen, G.; Neamati, N.; Hillard, E.A. Ferrocenyl chalcone difluoridoborates inhibit HIV-1 integrase and display low activity towards cancer and endothelial cells. Bioorg. Med. Chem. Lett. 2011, 20, 6195–6197. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, K.; Koceva-Chyła, A.; Szczupak, Ł.; Hikisz, P.; Bernasińska, J.; Rajnisz, A.; Solecka, J.; Therrien, B. Ferrocenylvinyl-flavones: Synthesis, structure, anticancer and antibacterial activity studies. J. Organomet. Chem. 2013, 741, 153–161. [Google Scholar] [CrossRef]
- Kowalski, K.; Hikisz, P.; Szczupak, Ł.; Therrien, B.; Koceva-Chyła, A. Ferrocenyl and dicobalt hexacarbonyl chromones—New organometallics inducing oxidative stress and arresting human cancer cells in G2/M phase. Eur. J. Med. Chem. 2014, 81, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, K.; Szczupak, Ł.; Oehninger, L.; Ott, I.; Hikisz, P.; Koceva-Chyła, A.; Therrien, B. Ferrocenyl derivatives of pterocarpene and coumestan: Synthesis, structure and anticancer activity studies. J. Organomet. Chem. 2014, 772, 49–59. [Google Scholar] [CrossRef]
- Kowalski, K.; Szczupak, Ł.; Bernaś, T.; Czerwieniec, R. Luminescent rhenium(I)chromone bioconjugate: Synthesis, photophysical properties, and confocal luminescence microscopy investigation. J. Organomet. Chem. 2015, 782, 124–130. [Google Scholar] [CrossRef]
- Patonay, T.; Pazurik, I.; Ábrahám, A. C-Alkynylation of Chromones by Sonogashira Reaction. Aust. J. Chem. 2013, 66, 646–654. [Google Scholar] [CrossRef]
- Pomestchenko, I.E.; Polyansky, D.E.; Castellano, F.N. Influence of a Gold(I)-Acetylide Subunit on the Photophysics of Re(Phen)(CO)3Cl. Inorg. Chem. 2005, 44, 3412–3421. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, Y.; Moriuchi, T.; Hirao, T. Organogold(I)-uracil conjugates: Synthesis and structural characterization. J. Organomet. Chem. 2015, 782, 77–81. [Google Scholar] [CrossRef]
- Collins, A.R. The comet assay for DNA damage and repair. Mol. Biotechnol. 2004, 26, 249–261. [Google Scholar] [CrossRef]
- Ormerod, M.G.; Collins, K.L.; Rodriguez-Tarduchy, G.; Robertson, D. Apoptosis in interleukin-3-dependent haemopoietic cells: Quantification by two flow cytometric methods. J. Immunol. Methods 1992, 153, 57–65. [Google Scholar] [CrossRef]
- Darzynkiewicz, Z.; Bruno, S.; Del Bino, G.; Gorczyca, W.; Hotz, M.A.; Lassota, P.; Traganos, F. Features of apoptotic cells measured by flow cytometry. Cytometry 1992, 13, 795–808. [Google Scholar] [CrossRef] [PubMed]
- Hotz, M.A.; Gong, J.; Traganos, F.; Darzynkiewicz, Z. Flow cytometric detection of apoptosis: Comparison of the assays of in situ DNA degradation and chromatin changes. Cytometry 1994, 15, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Koceva-Chyła, A.; Jędrzejczak, M.; Skierski, J.; Kania, K.; Jóźwiak, Z. Mechanisms of induction of apoptosis by anthraquinone anticancer drugs aclarubicin and mitoxantrone in comparison with doxorubicin: Relation to drug cytotoxicity and caspase-3 activation. Apoptosis 2005, 10, 1497–1514. [Google Scholar] [CrossRef] [PubMed]
- Kruidering, M.; Evan, G.I. Caspase-8 in Apoptosis: The Beginning of “The End”? IUBMB Life 2000, 50, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Alenzi, F.Q.; Lotfy, M.; Wyse, R. Swords of Cell Death: Caspase Activation and Regulation. Asian Pac. J. Cancer Prev. 2010, 11, 271–280. [Google Scholar] [PubMed]
- Juo, P.; Kuo, C.J.; Yuan, J.; Blenis, J. Essential requirement for caspase-8/FLICE in the initiation of the Fas-induced apoptotic cascade. Curr. Biol. 1998, 8, 1001–1008. [Google Scholar] [CrossRef]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase Functions in Cell Death and Disease. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Wolf, B.B.; Schuler, M.; Echeverri, F.; Green, D.R. Caspase-3 Is the Primary Activator of Apoptotic DNA Fragmentation via DNA Fragmentation Factor-45/Inhibitor of Caspase-activated DNase Inactivation. J. Biol. Chem. 1999, 274, 30651–30656. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. A. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. ORTEP-3 for Windows—A version of ORTEP-III with a Graphical User Interface (GUI). J. Appl. Cryst. 1997, 30, 565. [Google Scholar] [CrossRef]
- Bruno, I.J.; Cole, J.C.; Edgington, P.R.; Kessler, M.; Macrae, C.F.; McCabe, P.; Pearson, J.; Taylor, R. New software for searching the Cambridge Structural Database and visualizing crystal structures. Acta Cryst. B 2002, 58, 389–397. [Google Scholar] [CrossRef]
- Rubbiani, R.; Can, S.; Kitanovic, I.; Alborzinia, H.; Stefanopoulou, M.; Kokoschka, M.; Mönchgesang, S.; Sheldrick, W.S.; Wölfl, S.; Ott, I.; et al. Comparative in Vitro Evaluation of N-Heterocyclic Carbene Gold(I) Complexes of the Benzimidazolylidene Type. J. Med. Chem. 2011, 54, 8646–8657. [Google Scholar] [CrossRef] [PubMed]
- Approved Standard-Eighth Edition. M07-A8Method for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically. CLSI: 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898, USA, 2009; ISBN 1-56238-689-1.
- Knopik-Skrocka, A.; Bielawski, J. Differences in amphotericin-B-induced hemolysis between human erythrocytes from male and female donors. Biol. Lett. 2005, 42, 49–60. [Google Scholar]
- Sample Availability: Samples of the compounds 4–6 are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hikisz, P.; Szczupak, Ł.; Koceva-Chyła, A.; Guśpiel, A.; Oehninger, L.; Ott, I.; Therrien, B.; Solecka, J.; Kowalski, K. Anticancer and Antibacterial Activity Studies of Gold(I)-Alkynyl Chromones. Molecules 2015, 20, 19699-19718. https://doi.org/10.3390/molecules201119647
Hikisz P, Szczupak Ł, Koceva-Chyła A, Guśpiel A, Oehninger L, Ott I, Therrien B, Solecka J, Kowalski K. Anticancer and Antibacterial Activity Studies of Gold(I)-Alkynyl Chromones. Molecules. 2015; 20(11):19699-19718. https://doi.org/10.3390/molecules201119647
Chicago/Turabian StyleHikisz, Paweł, Łukasz Szczupak, Aneta Koceva-Chyła, Adam Guśpiel, Luciano Oehninger, Ingo Ott, Bruno Therrien, Jolanta Solecka, and Konrad Kowalski. 2015. "Anticancer and Antibacterial Activity Studies of Gold(I)-Alkynyl Chromones" Molecules 20, no. 11: 19699-19718. https://doi.org/10.3390/molecules201119647