
The Role of Natural Products in Drug Discovery and Development against Neglected Tropical Diseases

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Neglected Tropical Diseases: Definition, Classification and Epidemiology
1.2. Relative Public Health Impact
1.3. Challenges in Drug Discovery for NTDs and Challenges/Limitations of Current Therapies
1.4. Natural Products and Their Utility as Therapeutics: Advantages and Disadvantages
2. Human African Trypanosomiasis (Sleeping Sickness)
2.1. Background of the Disease
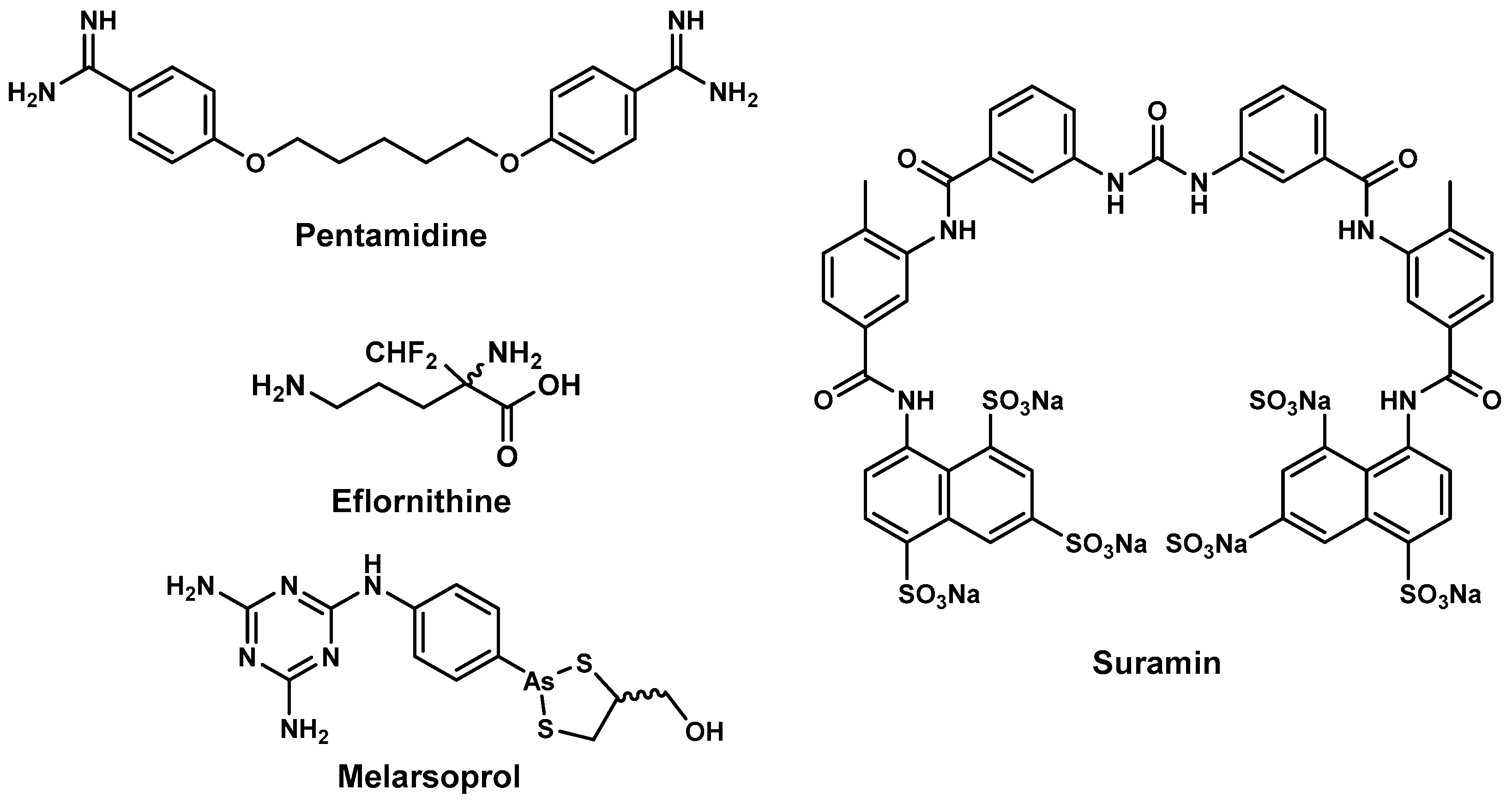
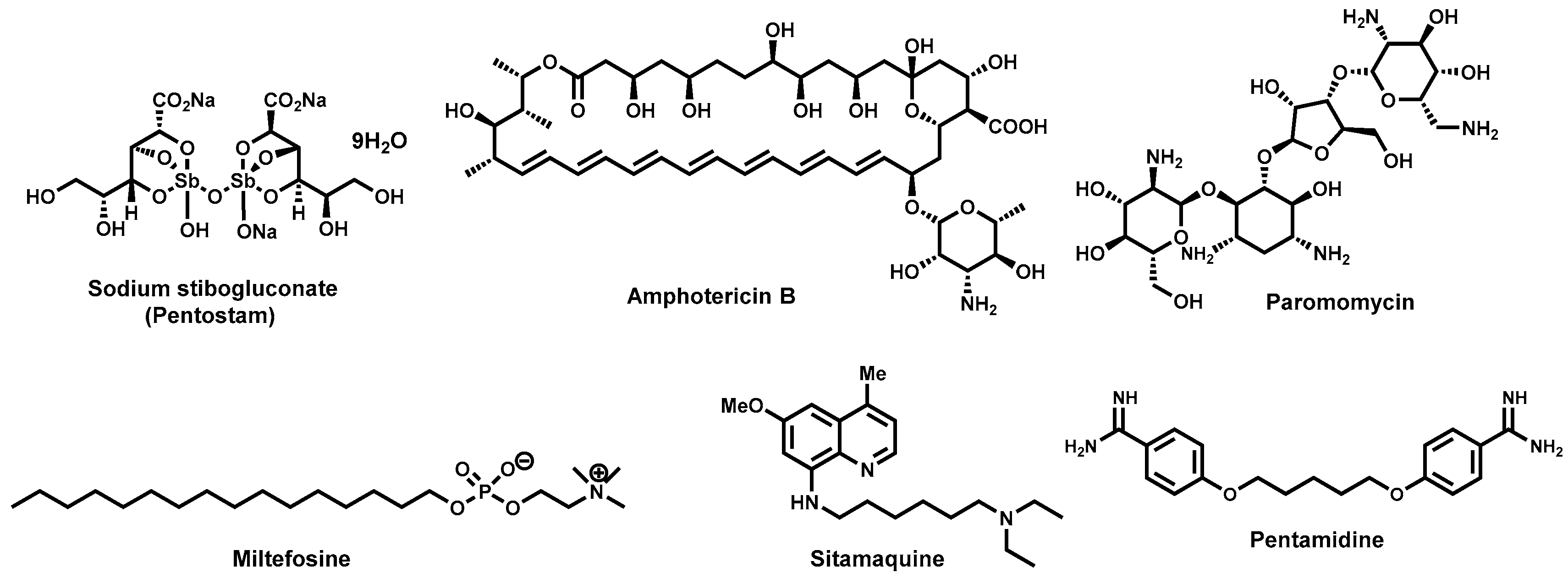
2.2. Antitrypanosomal Drugs and Current Shortfalls
2.3. Antitrypanosomal Natural Products
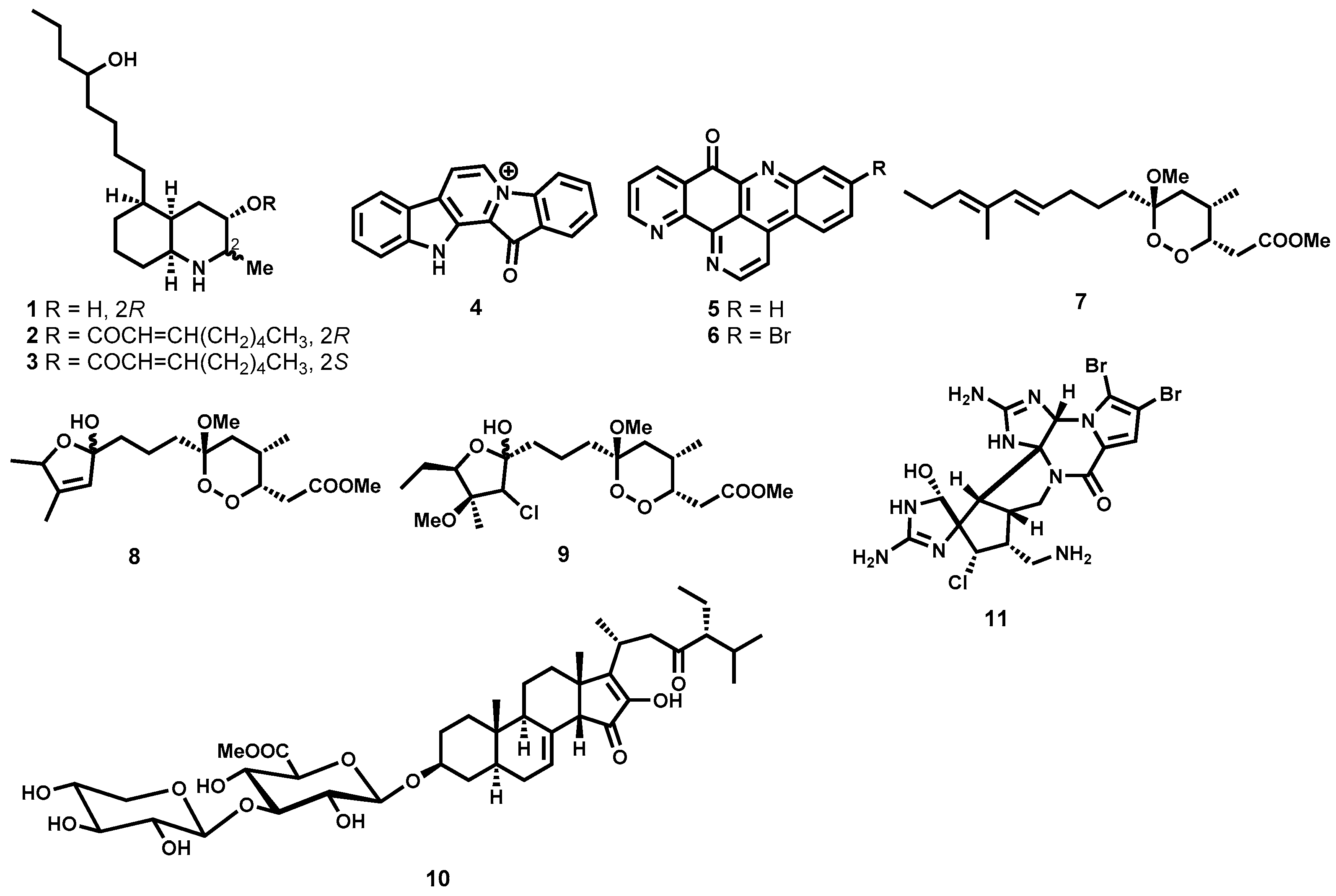
2.3.1. Antitrypanosomal Natural Products from Marine Sources
Alkaloids
Peroxides and Saponins
2.3.2. Antitrypanosomal Natural Products from Plant and Microbial Sources
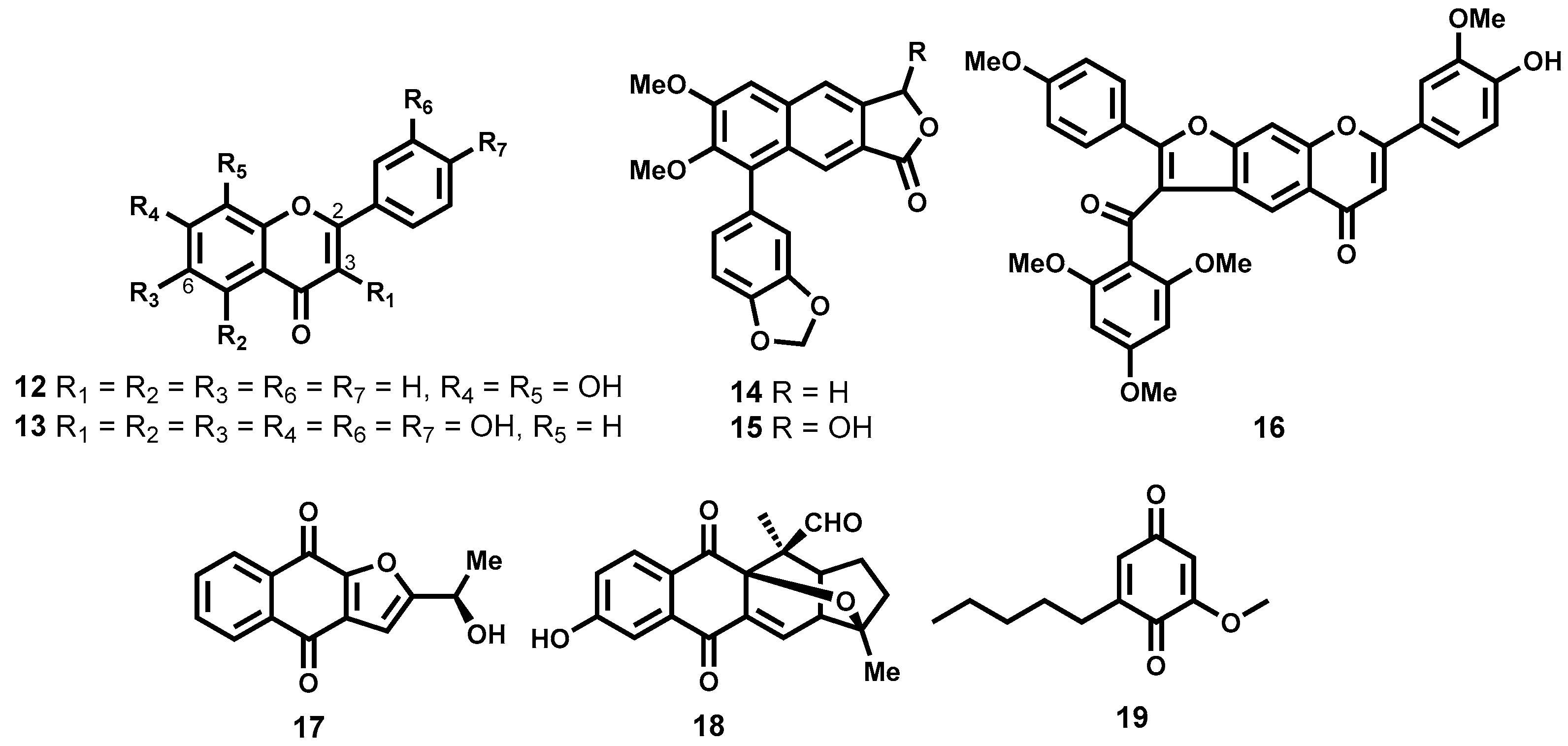
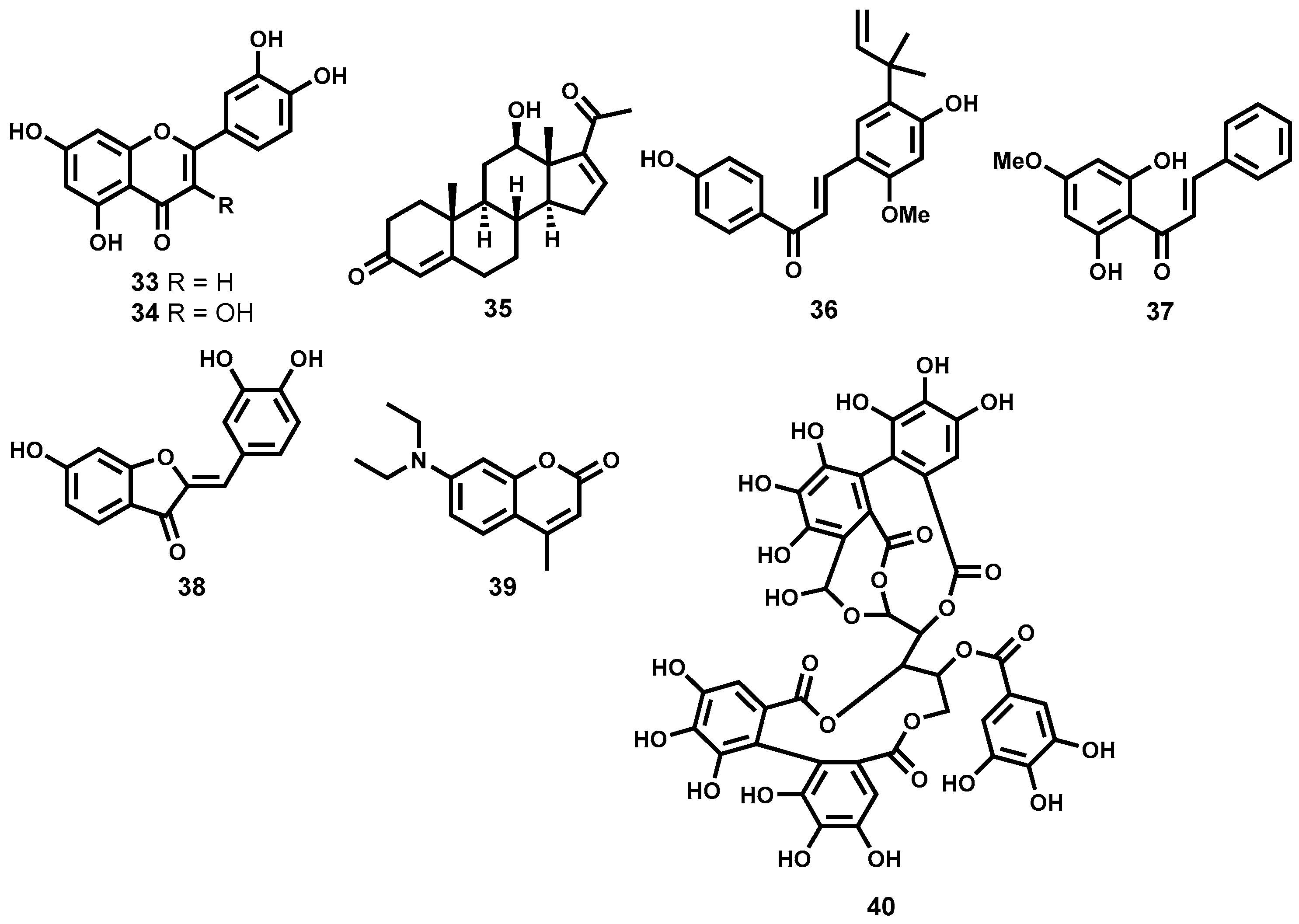
Phenolic Natural Products
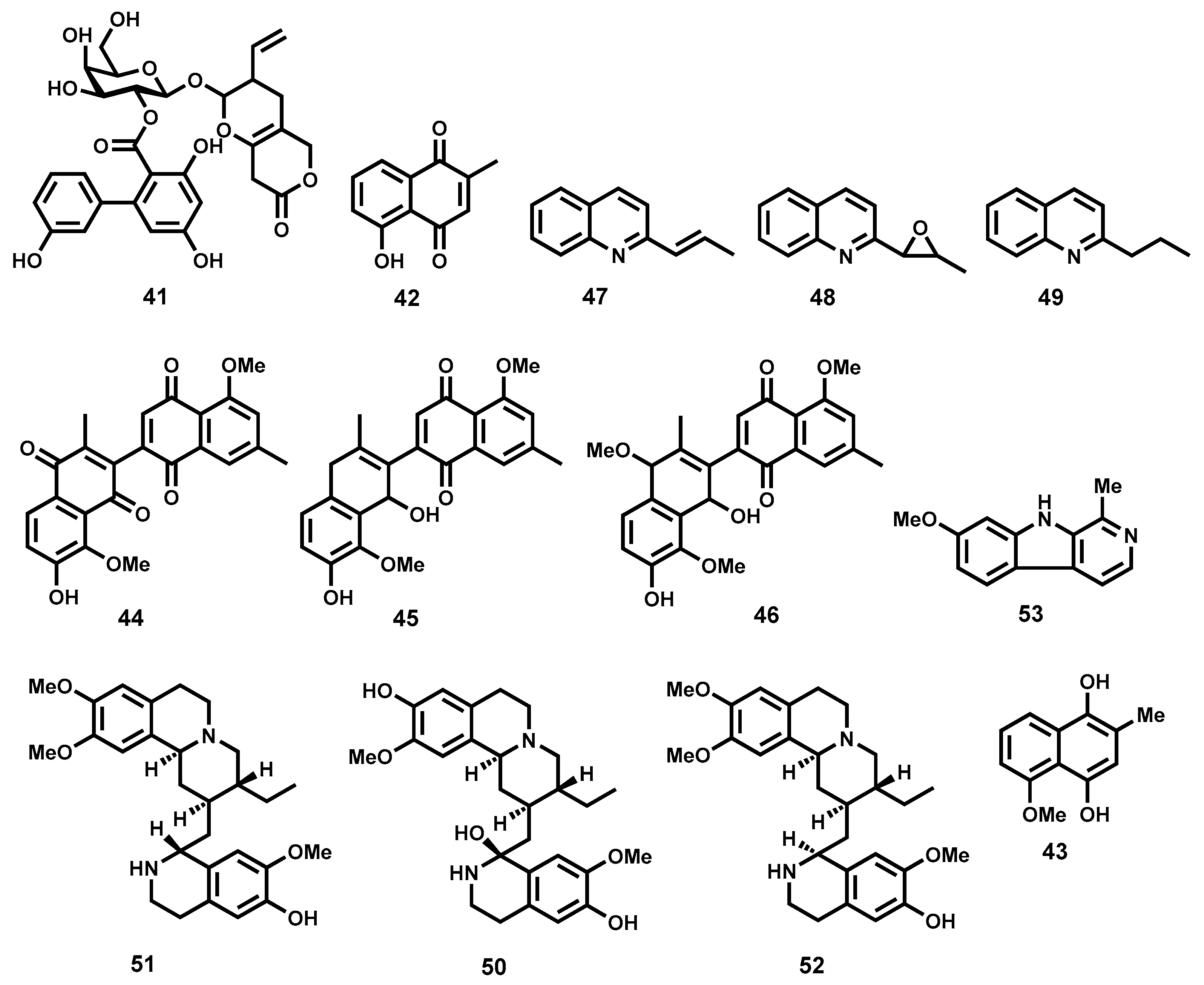
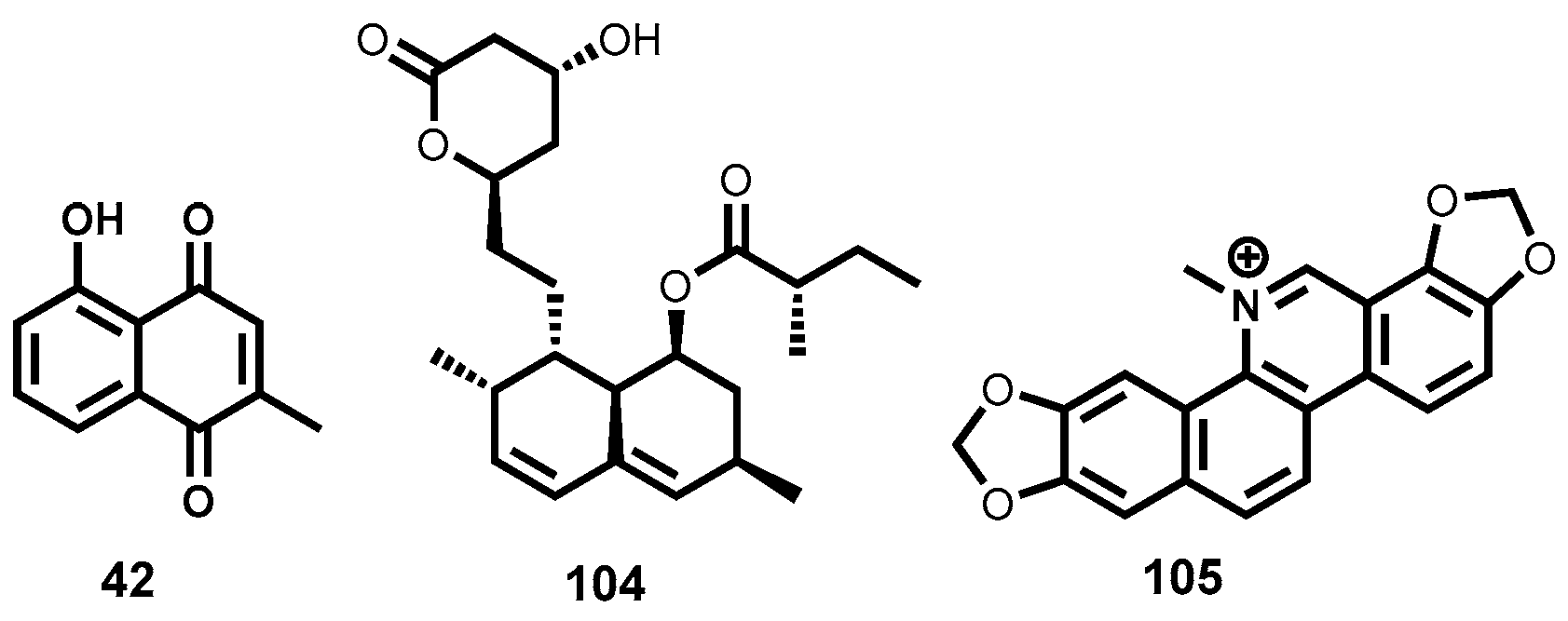
Quinones
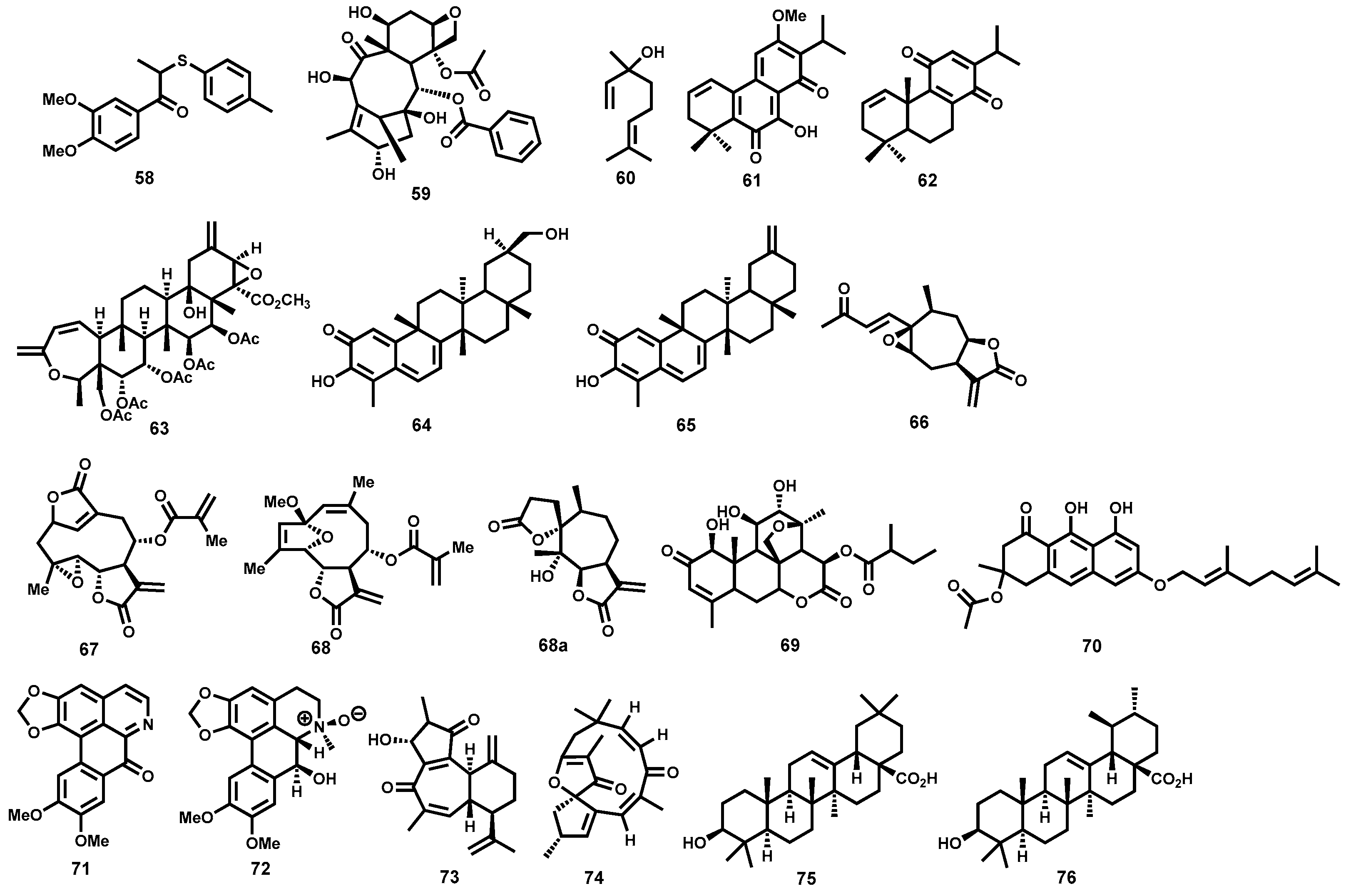
Terpenes and Other Metabolites
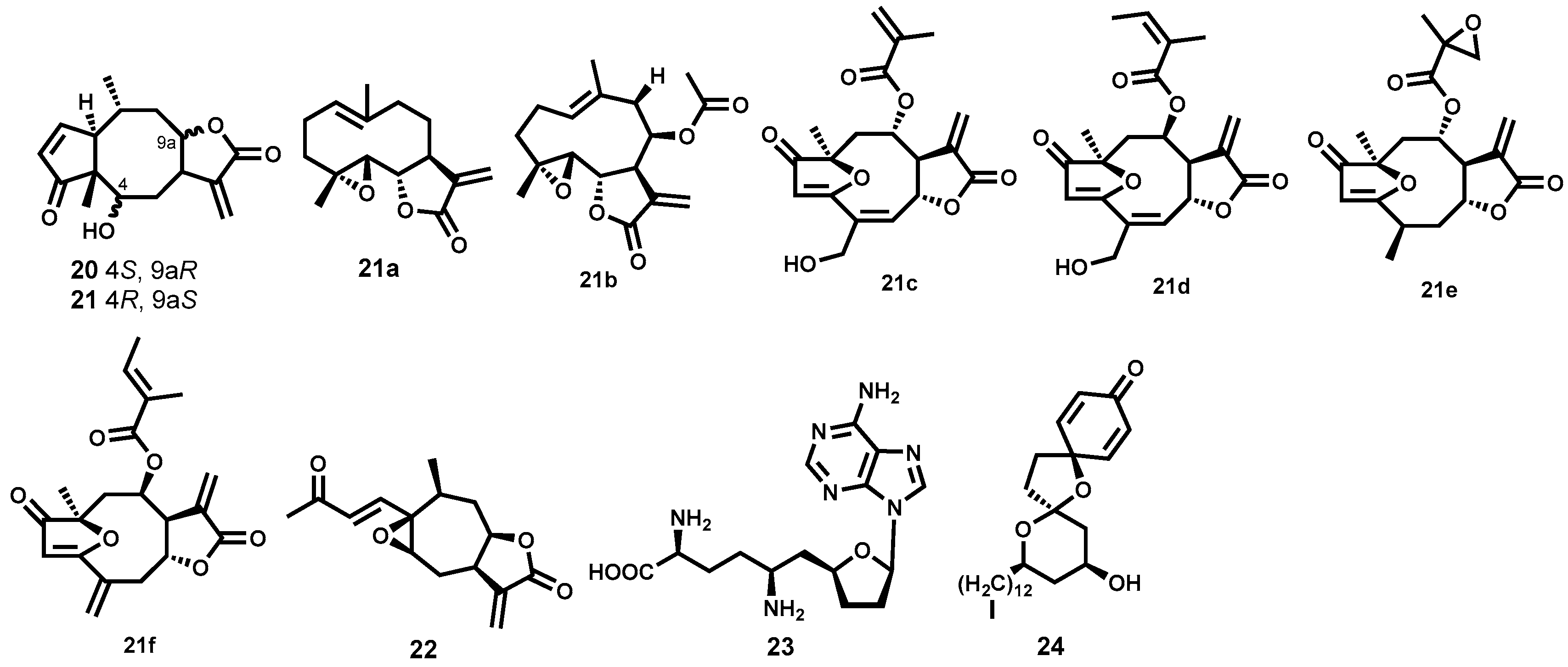
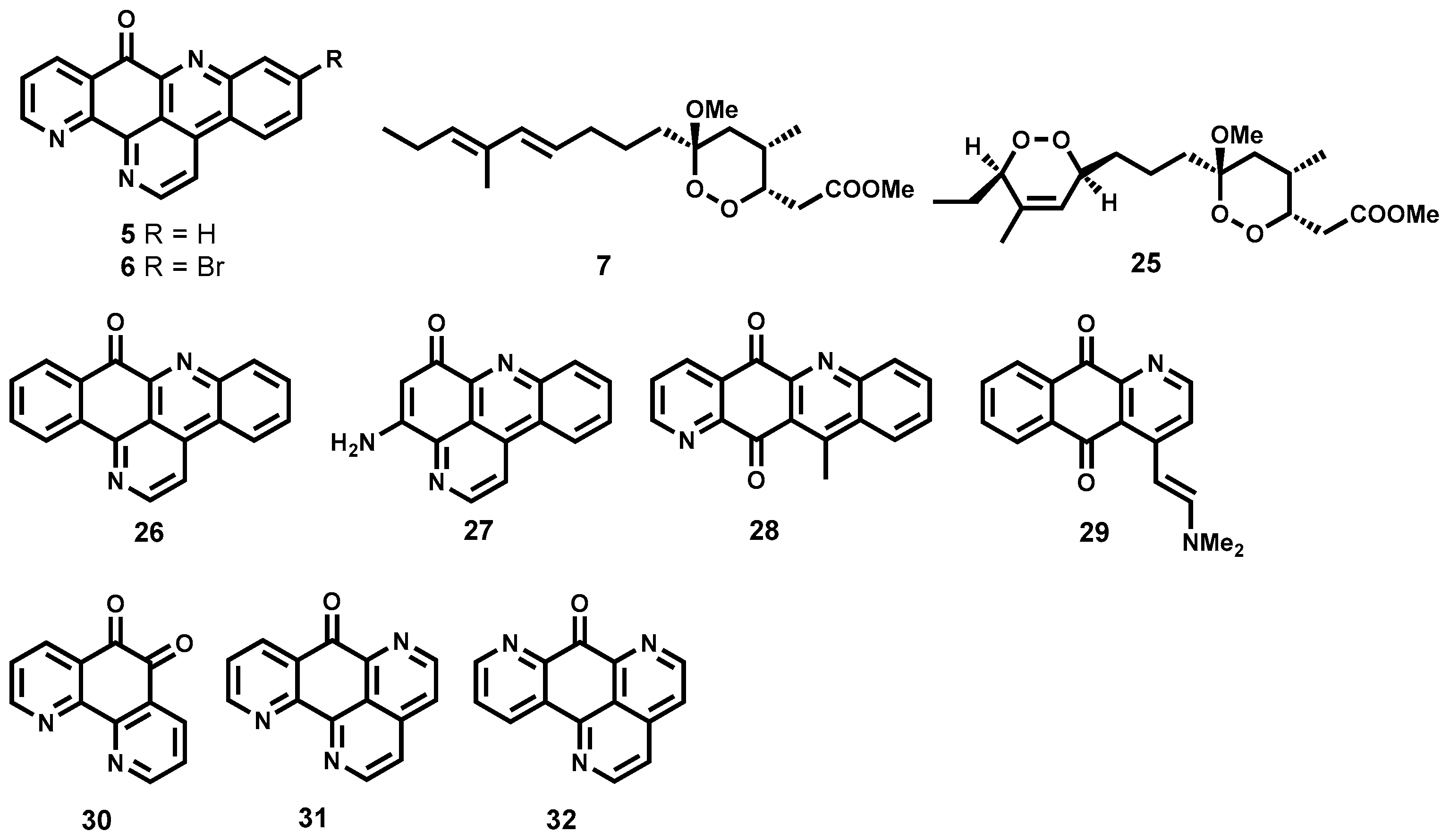
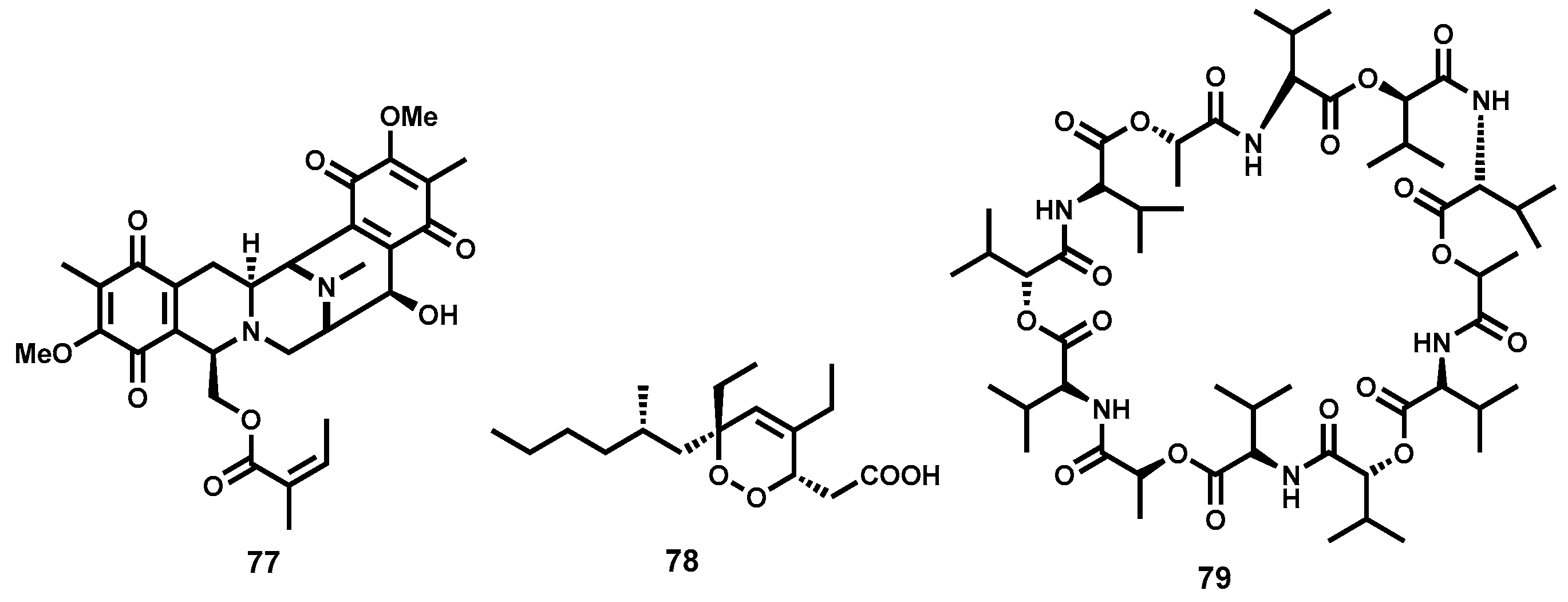
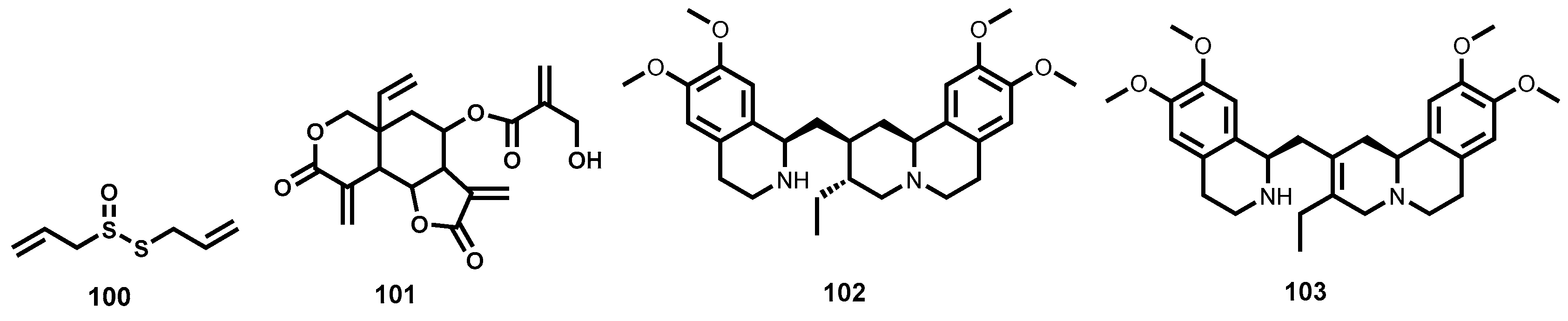
2.3.3. Semisynthetic Antitrypanosomal Natural Products
3. Leishmaniasis
3.1. Background of the Disease
3.2. Current Chemotherapy and Associated Challenges
3.3. Antileishmanial Natural Products
3.3.1. Antileishmanial Natural Products from Plant Sources
Flavonoids, Sterols, Chalcones, Coumarins, Tannins and Aurones
Iridoids, Quinones, and Quinoline Alkaloids
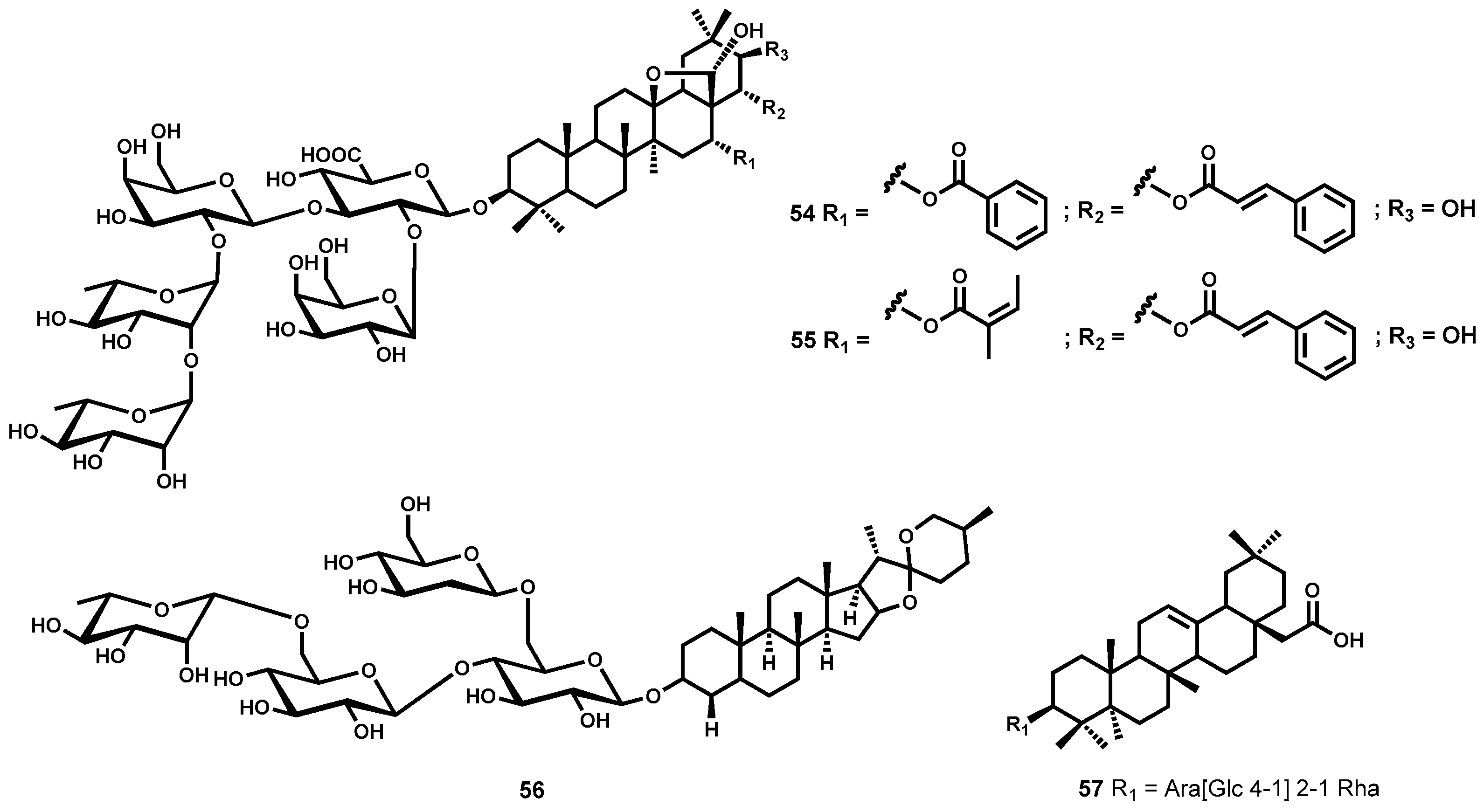
Saponins
Lignans, Taxoids, Terpenes, Anthranoids and Miscellaneous Antileishmanial Natural Products
3.3.2. Antileishmanial Natural Products from Marine Sources
3.3.3. Semi-Synthetic Antileishmanial Natural Products
4. Schistosomiasis
4.1. Background of the Disease
4.2. Intervention Strategies and Challenges
Parameters Assessed
4.3. Examples of Natural Product Leads or Drugs with Antischistosomal Activities
4.3.1. Epiisopilotulorine
4.3.2. Piplartine
4.3.3. Ginger
4.3.4. Phytol
4.3.5. Curcumin
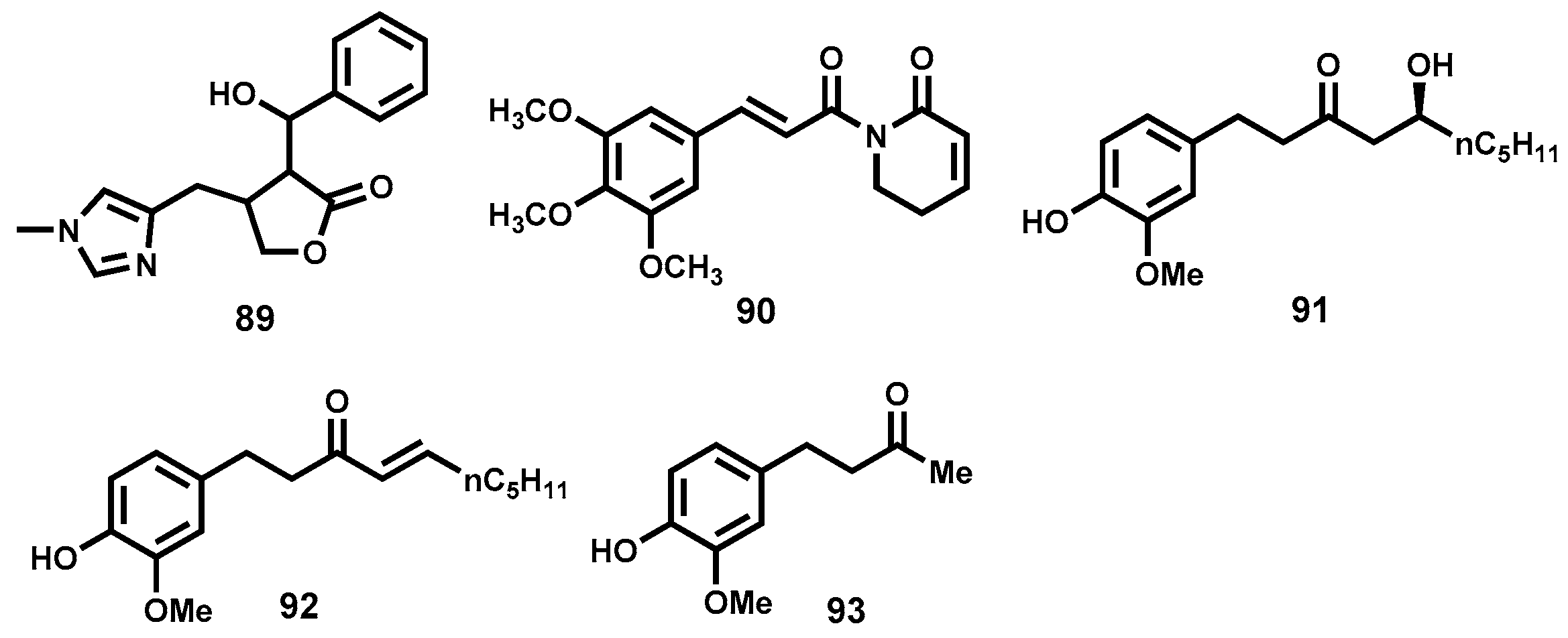
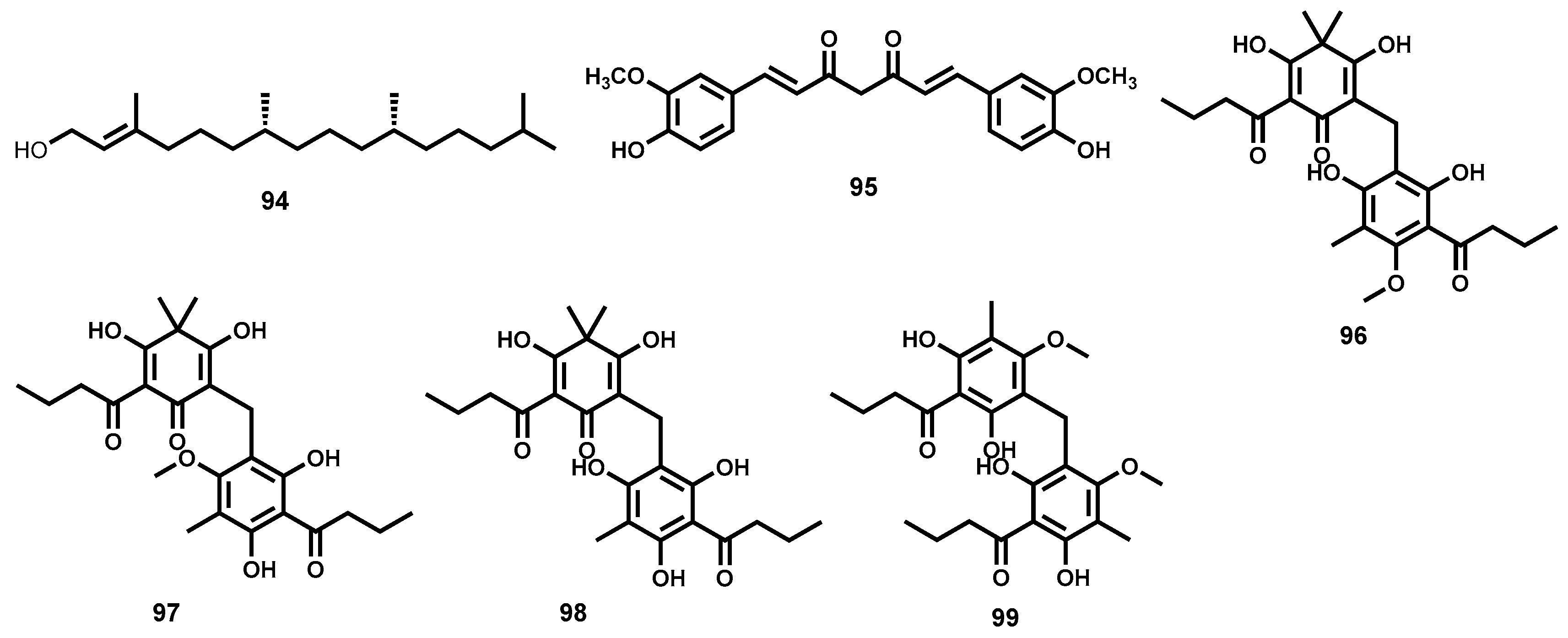
4.3.6. Phloroglucinols
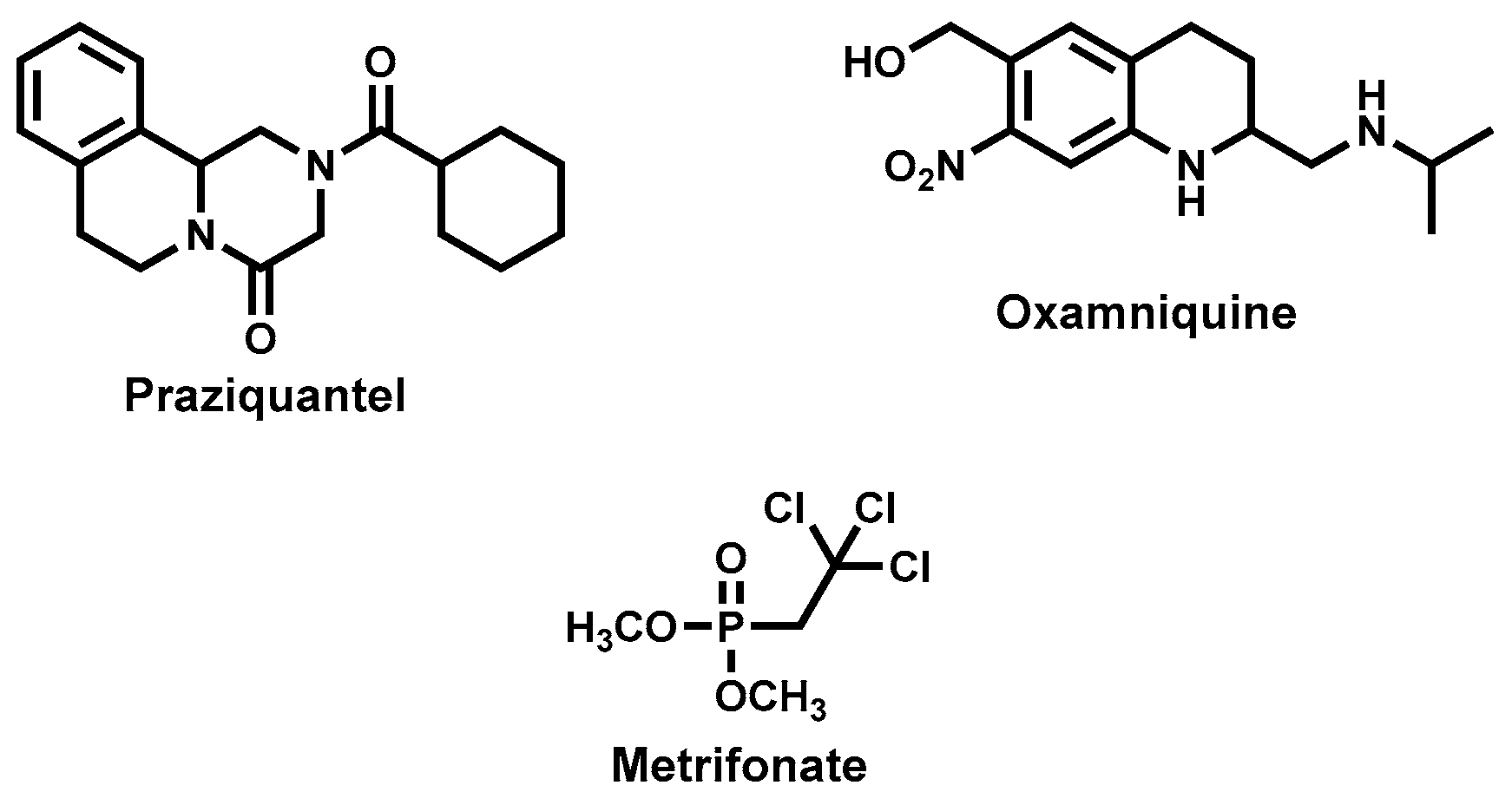
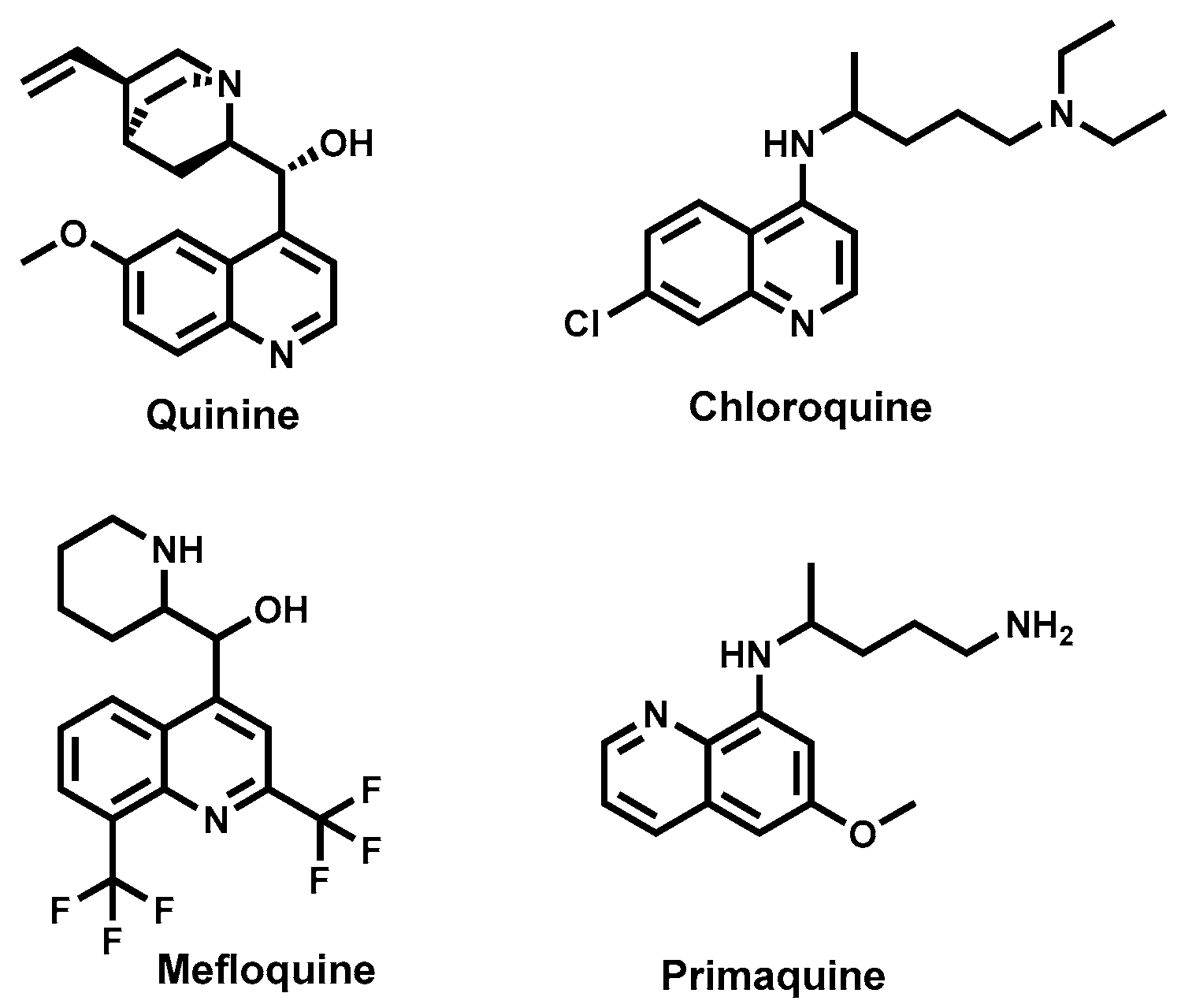
4.3.7. Cinchona Alkaloids—Quinine and Derivatives
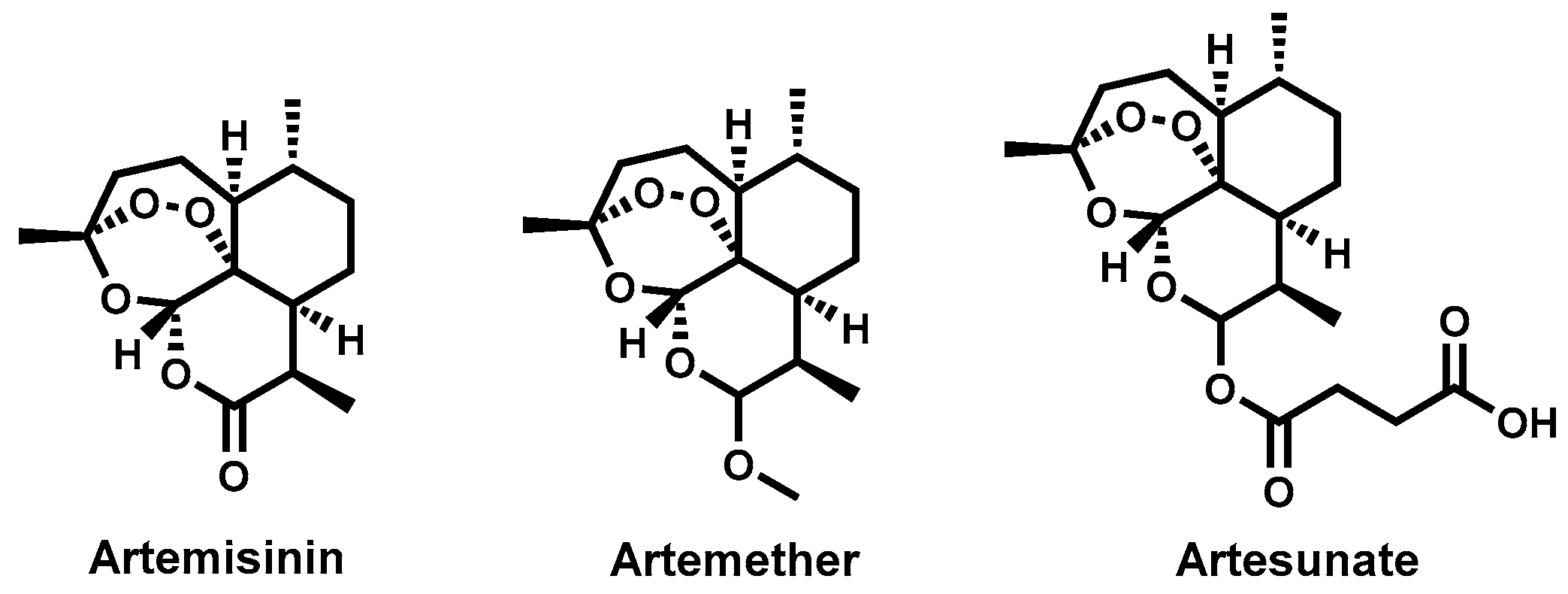
4.3.8. Artemisinins and Derivatives
4.3.9. Allicin
4.3.10. Vernonia amygdalina
4.3.11. Emetine
4.3.12. Other Natural Product Leads: Mevinolin (Lovastatin), Plumbagin and Sanguinarine
4.3.13. Summary
5. Lymphatic Filariasis
5.1. Background of the Disease
5.2. Naturally Derived Compounds for Lymphatic Filariasis
5.2.1. Plant Origin
5.2.2. Microbial Sources
5.2.3. Marine Sources
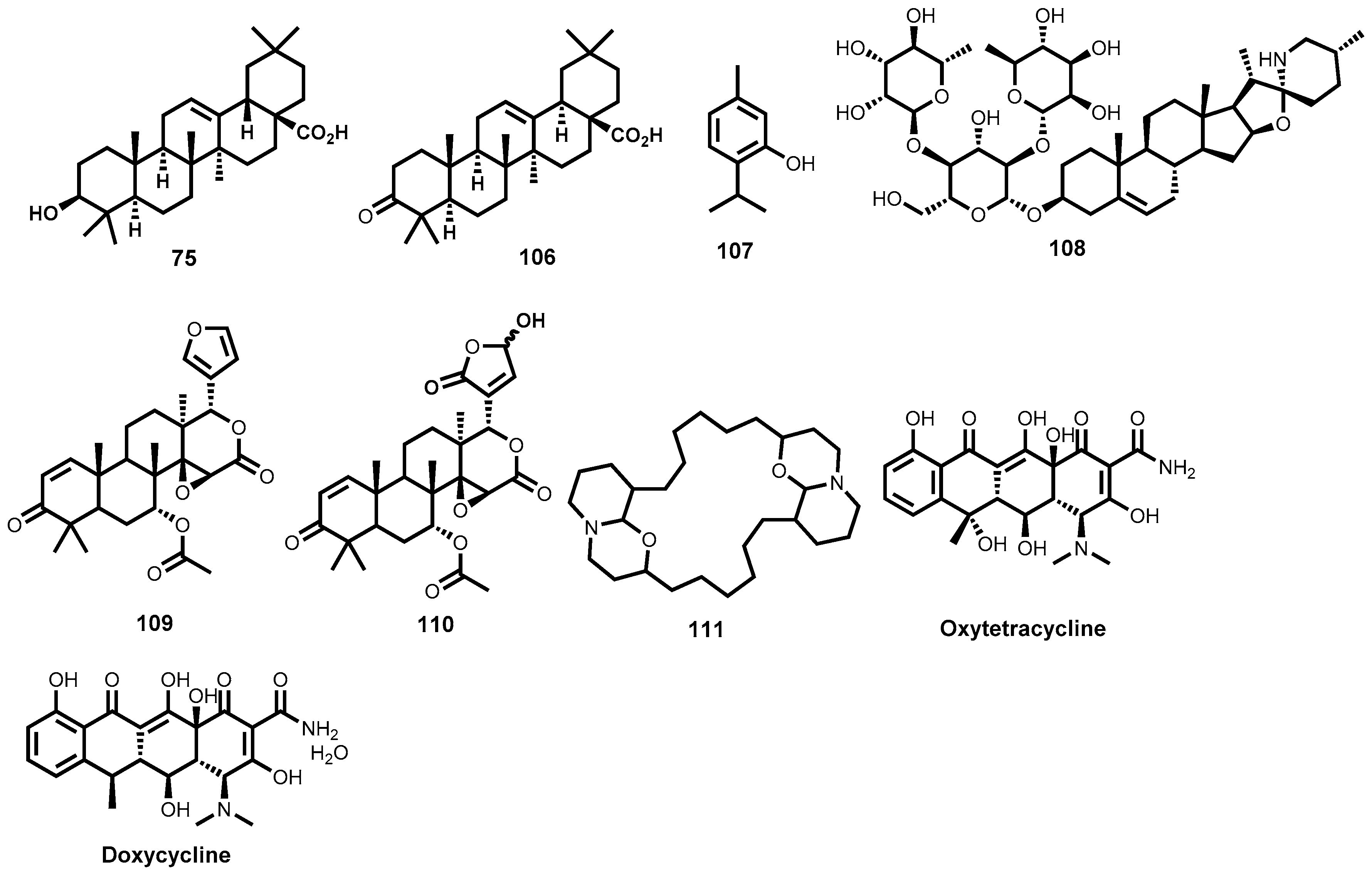
5.3. Semi-Synthetic/Total Synthetic Compounds Based on Natural Compound Templates
Pre-Clinical Compounds
6. Concluding Remarks
Conflicts of Interest
References
- Neglected Tropical Diseases. Available online: http://www.webcitation.org/6kb2aMCKW (accessed on 17 September 2016).
- Feasey, N.; Wansbrough-Jones, M.; Mabey, D.C.W.; Solomon, A.W. Neglected tropical diseases. Br. Med. Bull. 2010, 93, 179–200. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. World Health Report 2004: Changing History; World Health Organization: Geneva, Switzerland, 2004. [Google Scholar]
- Bern, C.; Maguire, J.H.; Alvar, J. Complexities of assessing the disease burden attributable to leishmaniasis. PLoS Negl. Trop. Dis. 2008, 2, e313. [Google Scholar] [CrossRef] [PubMed]
- Bethony, J.; Brooker, S.; Albonico, M.; Geiger, S.M.; Loukas, A.; Diemert, D.; Hotez, P.J. Soil-transmitted helminth infections: Ascariasis, trichuriasis, and hookworm. Lancet 2006, 367, 1521–1532. [Google Scholar] [CrossRef]
- Crompton, D.W.T.; Nesheim, M.C. Nutritional impact of intestinal helminthiasis during the human life cycle. Annu. Rev. Nutr. 2002, 22, 35–59. [Google Scholar] [CrossRef] [PubMed]
- Hotez, P.J.; Ferris, M.T. The antipoverty vaccines. Vaccine 2006, 24, 5787–5799. [Google Scholar] [CrossRef] [PubMed]
- Colley, D.G.; Bustinduy, A.L.; Secor, W.E.; King, C.H. Human schistosomiasis. Lancet 2014, 383, 2253–2264. [Google Scholar] [CrossRef]
- Perera, M.; Whitehead, M.; Molyneux, D.; Weerasooriya, M.; Gunatilleke, G. Neglected Patients with a Neglected Disease? A Qualitative Study of Lymphatic Filariasis. PLoS Negl. Trop. Dis. 2007, 1, e128. [Google Scholar] [CrossRef] [PubMed]
- Trachoma: Situation and Trends. Available online: http://www.webcitation.org/6kfb0UvfZ (accessed on 20 September 2016).
- NTD Overview: What are Neglected Tropical Diseases. Available online: http://www.webcitation.org/6kc3jm5cS (accessed on 18 September 2016).
- Hotez, P.J.; Kamath, A. Neglected tropical diseases in sub-Saharan Africa: Review of their prevalence, distribution, and disease burden. PLoS Negl. Trop. Dis. 2009, 3, e412. [Google Scholar] [CrossRef] [PubMed]
- Fenwick, A. The global burden of neglected tropical diseases. Public Health 2012, 126, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Pink, R.; Hudson, A.; Mouriès, M.-A.; Bendig, M. Opportunities and Challenges in Antiparasitic Drug Discovery. Nat. Rev. Drug Discov. 2005, 4, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.M. Natural Products as a Resource for New Drugs. Pharm. Res. 1996, 13, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Soejarto, D.D.; Farnsworth, N.R. Tropical Rain Forests: Potential Source of New Drugs? Perspect. Biol. Med. 1989, 32, 244–256. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Last 25 Years. J. Nat. Prod. 2007, 70, 461–477. [Google Scholar] [CrossRef] [PubMed]
- Tyler, V.E.; Brady, L.R.; Robbers, J.E. Pharmacognosy, 9th ed.; Lea & Febiger: Philadephia, PA, USA, 1998. [Google Scholar]
- Butler, M.S. Natural products to drugs: Natural product-derived compounds in clinical trials. Nat. Prod. Rep. 2008, 25, 475–516. [Google Scholar] [CrossRef] [PubMed]
- Chin, Y.-W.; Balunas, M.J.; Chai, H.B.; Kinghorn, A.D. Drug discovery from natural sources. AAPS J. 2006, 8, E239–E253. [Google Scholar] [CrossRef] [PubMed]
- Lam, K.S. New aspects of natural products in drug discovery. Trends Microbiol. 2007, 15, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, A. The impact of natural products upon modern drug discovery. Curr. Opin. Chem. Biol. 2008, 12, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.D.; Chu, M.; Oza, U.; Rajgarhia, V. The value of natural products to future pharmaceutical discovery. Nat. Prod. Rep. 2007, 24, 1225–1244. [Google Scholar] [CrossRef] [PubMed]
- McChesney, J.D.; Venkataraman, S.K.; Henri, J.T. Plant natural products: Back to the future or into extinction? Phytochemistry 2007, 68, 2015–2022. [Google Scholar] [CrossRef] [PubMed]
- Rishton, G.M. Natural Products as a Robust Source of New Drugs and Drug Leads: Past Successes and Present Day Issues. Am. J. Cardiol. 2008, 101, S43–S49. [Google Scholar] [CrossRef] [PubMed]
- Zofou, D.; Ntie-Kang, F.; Sippl, W.; Efange, S.M.N. Bioactive natural products derived from the Central African flora against neglected tropical diseases and HIV. Nat. Prod. Rep. 2013, 30, 1098–1120. [Google Scholar] [CrossRef] [PubMed]
- Ioset, J.-R. Natural Products for Neglected Diseases: A Review. Curr. Org. Chem. 2008, 12, 643–666. [Google Scholar] [CrossRef]
- Haridas, D.; Setzer, W.N. Phytomedicinal agents for treatment of schistosomiasis. In Drug Plants III; Govil, J.N., Singh, V.K., Eds.; Studium Press LLC: Houston, TX, USA, 2010; pp. 79–91. [Google Scholar]
- Ndjonka, D.; Rapado, L.; Silber, A.; Liebau, E.; Wrenger, C. Natural Products as a Source for Treating Neglected Parasitic Diseases. Int. J. Mol. Sci. 2013, 14, 3395–3439. [Google Scholar] [CrossRef] [PubMed]
- Neves, B.; Andrade, C.; Cravo, P. Natural Products as Leads in Schistosome Drug Discovery. Molecules 2015, 20, 1872–1903. [Google Scholar] [CrossRef] [PubMed]
- Ogungbe, I.V.; Singh, M.; Setzer, W.N. Antileishmanial natural products from plants. In Studies in Natural Products Chemistry; Rahman, A., Ed.; Elsevier: Oxford, UK, 2012; pp. 331–382. [Google Scholar]
- Hoet, S.; Opperdoes, F.; Brun, R.; Quetin-Leclercq, J. Natural products active against African trypanosomes: A step towards new drugs. Nat. Prod. Rep. 2004, 21, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Setzer, W.N.; Setzer, M.C. Antitrypanosomal agents from higher plants. In Biologically Active Natural Products for the 21st Century; Williams, L.A.D., Reese, P.B., Eds.; Research Signpost: Trivandrum, India, 2006; pp. 47–95. [Google Scholar]
- Schmidt, T.J.; Khalid, S.A.; Romanha, A.J.; Alves, T.M.; Biavatti, M.W.; Brun, R.; Da Costa, F.B.; de Castro, S.L.; Ferreira, V.F.; de Lacerda, M.V.G.; et al. The potential of secondary metabolites from plants as drugs or leads against protozoan neglected diseases—Part I. Curr. Med. Chem. 2012, 19, 2128–2175. [Google Scholar] [PubMed]
- Schmidt, T.J.; Khalid, S.A.; Romanha, A.J.; Alves, T.M.; Biavatti, M.W.; Brun, R.; Da Costa, F.B.; de Castro, S.L.; Ferreira, V.F.; de Lacerda, M.V.G.; et al. The potential of secondary metabolites from plants as drugs or leads against protozoan neglected diseases—Part II. Curr. Med. Chem. 2012, 19, 2176–2228. [Google Scholar] [CrossRef] [PubMed]
- Rocha, L.G.; Almeida, J.R.G.S.; Macêdo, R.O.; Barbosa-Filho, J.M. A review of natural products with antileishmanial activity. Phytomedicine 2005, 12, 514–535. [Google Scholar] [CrossRef] [PubMed]
- Kolodziej, H.; Kiderlen, A.F. Antileishmanial activity and immune modulatory effects of tannins and related compounds on Leishmania parasitised RAW 264.7 cells. Phytochemistry 2005, 66, 2056–2071. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.B.; Singh, R.K.; Srivastava, A.; Tripathi, V.J.; Tiwari, V.K. Fighting against Leishmaniasis: Search of alkaloids as future true potential anti-Leishmanial agents. Mini Rev. Med. Chem. 2009, 9, 107–123. [Google Scholar] [CrossRef] [PubMed]
- De Carvalho, P.B.; Ferreira, E.I. Leishmaniasis phytotherapy. Nature’s leadership against an ancient disease. Fitoterapia 2001, 72, 599–618. [Google Scholar] [CrossRef]
- Polonio, T.; Efferth, T. Leishmaniasis: Drug resistance and natural products (review). Int. J. Mol. Med. 2008, 22, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Mishra, B.B.; Bajpai, S.; Singh, R.K.; Tiwari, V.K. Natural product based leads to fight against leishmaniasis. Bioorg. Med. Chem. 2014, 22, 18–45. [Google Scholar] [CrossRef] [PubMed]
- Kayser, O.; Kiderlen, A.F.; Croft, S.L. Natural products as antiparasitic drugs. Parasitol. Res. 2003, 90, S55–S62. [Google Scholar] [CrossRef] [PubMed]
- Fournet, A.; Muñoz, V. Natural products as trypanocidal, antileishmanial and antimalarial drugs. Curr. Top. Med. Chem. 2002, 2, 1215–1237. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.M.; Werbovetz, K.A. Natural products from plants as drug candidates and lead compounds against leishmaniasis and trypanosomiasis. Curr. Med. Chem. 2006, 13, 2571–2598. [Google Scholar] [CrossRef] [PubMed]
- Akendengue, B.; Ngou-Milama, E.; Laurens, A.; Hocquemiller, R. Recent advances in the fight against leishmaniasis with natural products. Parasite 1999, 6, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Human African Trypanosomiasis. Available online: http://www.webcitation.org/6ki7VaGnQ (accessed on 22 September 2016).
- Trypanosomiasis, Human African (Sleeping Sickness). Available online: http://www.webcitation.org/6kidfPDjR (accessed on 22 September 2016).
- Docampo, R.; Moreno, S.N.J. Current chemotherapy of human African trypanosomiasis. Parasitol. Res. 2003, 90, S10–S13. [Google Scholar] [PubMed]
- International Federation of Pharmaceutical Manufacturers & Associations Health Partnerships for the Developing World. Sanofi-Aventis Sleeping Sickness Program. Available online: http://ifpma.org/index.php?id=287 (accessed on 9 July 2009).
- AG Bayer Bayer Sustainable Development Report 2004, Bayer AG, Leverkusen (2004). Available online: http://www.bayer.co.id/materials/File_Eng/publication_file_eng_229_8RXhl.pdf (accessed on 9 July 2009).
- Brun, R.; Blum, J.; Chappuis, F.; Burri, C. Human African trypanosomiasis. Lancet 2010, 375, 148–159. [Google Scholar] [CrossRef] [Green Version]
- World Health Organ (WHO). Epidemiology and Control of African Trypanosomiasis; World Health Organ Technical Report Series; World Health Organ: Geneva, Switzerland, 1986; p. 739. [Google Scholar]
- Gustafsson, L.L.; Beerman, B.; Aden Abdi, Y. Suramin. In Handbook of Drugs for Tropical Parasitic Infections, 1st ed.; Gustafsson, L.L., Beerman, B., Aden Abdi, Y., Eds.; Taylor & Francis: Basingstoke, UK, 1987; pp. 160–163. [Google Scholar]
- Jordan, A.M. Tsetse-flies (Glossinidae). In Medical Insects and Arachnids; Lane, R.P., Crosskey, R.W., Eds.; Chapman and Hall: London, UK, 1993; pp. 333–388. [Google Scholar]
- Chappuis, F.; Udayraj, N.; Stietenroth, K.; Meussen, A.; Bovier, P.A. Eflornithine Is Safer than Melarsoprol for the Treatment of Second-Stage Trypanosoma brucei gambiense Human African Trypanosomiasis. Clin. Infect. Dis. 2005, 41, 748–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burri, C.; Brun, R. Eflornithine for the treatment of human African trypanosomiasis. Parasitol. Res. 2003, 90, S49–S52. [Google Scholar] [PubMed]
- Simarro, P.P.; Jannin, J.; Cattand, P. Eliminating Human African Trypanosomiasis: Where Do We Stand and What Comes Next? PLoS Med. 2008, 5, e55. [Google Scholar] [CrossRef] [PubMed]
- Tagboto, S.; Townson, S. Antiparasitic properties of medicinal plants and other naturally occurring products. Adv. Parasitol. 2001, 50, 199–295. [Google Scholar] [PubMed]
- Wright, A.D.; Goclik, E.; König, G.M.; Kaminsky, R. Lepadins D−F: Antiplasmodial and Antitrypanosomal Decahydroquinoline Derivatives from the Tropical Marine Tunicate Didemnum sp. J. Med. Chem. 2002, 45, 3067–3072. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, G.; König, G.M.; Wright, A.D.; Kaminsky, R. A New Bioactive Sesterterpene and Antiplasmodial Alkaloids from the Marine Sponge Hyrtios cf. erecta. J. Nat. Prod. 2000, 63, 825–829. [Google Scholar] [CrossRef] [PubMed]
- Copp, B.R.; Kayser, O.; Brun, R.; Kiderlen, A.F. Antiparasitic activity of marine pyridoacridone alkaloids related to the ascididemins. Planta Med. 2003, 69, 527–531. [Google Scholar] [PubMed]
- Chianese, G.; Fattorusso, E.; Scala, F.; Teta, R.; Calcinai, B.; Bavestrello, G.; Dien, H.A.; Kaiser, M.; Tasdemir, D.; Taglialatela-Scafati, O. Manadoperoxides, a new class of potent antitrypanosomal agents of marine origin. Org. Biomol. Chem. 2012, 10, 7197–7207. [Google Scholar] [CrossRef] [PubMed]
- Regalado, E.L.; Tasdemir, D.; Kaiser, M.; Cachet, N.; Amade, P.; Thomas, O.P. Antiprotozoal Steroidal Saponins from the Marine Sponge Pandaros acanthifolium. J. Nat. Prod. 2010, 73, 1404–1410. [Google Scholar] [CrossRef] [PubMed]
- Scala, F.; Fattorusso, E.; Menna, M.; Taglialatela-Scafati, O.; Tierney, M.; Kaiser, M.; Tasdemir, D. Bromopyrrole Alkaloids as Lead Compounds against Protozoan Parasites. Mar. Drugs 2010, 8, 2162–2174. [Google Scholar] [CrossRef] [PubMed]
- Aiello, A.; D’Esposito, M.; Fattorusso, E.; Menna, M.; Müller, W.E.G.; Perović-Ottstadt, S.; Schröder, H.C. Novel bioactive bromopyrrole alkaloids from the Mediterranean sponge Axinella verrucosa. Bioorg. Med. Chem. 2006, 14, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Räz, B. Isolation and Evaluation of Antiparasitic Lead Compounds from African Medicinal Plants. Ph.D. Thesis, Universität Basel, Basel, Switzerland, 1998. [Google Scholar]
- Tarus, P.K.; Machocho, A.K.; Lang’at-Thoruwa, C.C.; Chhabra, S.C. Flavonoids from Tephrosia aequilata. Phytochemistry 2002, 60, 375–379. [Google Scholar] [CrossRef]
- Gertsch, J.; Thöni Tobler, R.; Brun, R.; Sticher, O.; Heilmann, J. Antifungal, antiprotozoal, cytotoxic and piscicidal properties of justicidin B and a new arylnaphthalide lignan from Phyllanthus piscatorum. Planta Med. 2003, 69, 420–424. [Google Scholar] [PubMed]
- Ramírez, I.; Carabot, A.; Meléndez, P.; Carmona, J.; Jimenez, M.; Patel, A.V.; Crabb, T.A.; Blunden, G.; Cary, P.D.; Croft, S.L.; et al. Cissampeloflavone, a chalcone-flavone dimer from Cissampelos pareira. Phytochemistry 2003, 64, 645–647. [Google Scholar]
- Moideen, S.V.K.; Houghton, P.J.; Rock, P.; Croft, S.L.; Aboagye-Nyame, F. Activity of Extracts and Naphthoquinones from Kigelia pinnata Against Trypanosoma brucei brucei and Trypanosoma brucei rhodesiense. Planta Med. 1999, 65, 536–540. [Google Scholar] [CrossRef] [PubMed]
- Gunatilaka, A.A.L.; Berger, J.M.; Evans, R.; Miller, J.S.; Wisse, J.H.; Neddermann, K.M.; Bursuker, I.; Kingston, D.G.I. Isolation, Synthesis, and Structure−Activity Relationships of Bioactive Benzoquinones from Miconia lepidota from the Suriname Rainforest 1. J. Nat. Prod. 2001, 64, 2–5. [Google Scholar] [CrossRef] [PubMed]
- Tasdemir, D.; Kaiser, M.; Brun, R.; Yardley, V.; Schmidt, T.J.; Tosun, F.; Ruedi, P. Antitrypanosomal and Antileishmanial Activities of Flavonoids and Their Analogues: In Vitro, In Vivo, Structure-Activity Relationship, and Quantitative Structure-Activity Relationship Studies. Antimicrob. Agents Chemother. 2006, 50, 1352–1364. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.J.; Brun, R.; Willuhn, G.; Khalid, S.A. Anti-trypanosomal activity of helenalin and some structurally related sesquiterpene lactones. Planta Med. 2002, 68, 750–751. [Google Scholar] [CrossRef] [PubMed]
- Julianti, T.; Hata, Y.; Zimmermann, S.; Kaiser, M.; Hamburger, M.; Adams, M. Antitrypanosomal sesquiterpene lactones from Saussurea costus. Fitoterapia 2011, 82, 955–959. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.J.; Da Costa, F.B.; Lopes, N.P.; Kaiser, M.; Brun, R. In Silico prediction and experimental evaluation of furanoheliangolide sesquiterpene lactones as potent agents against Trypanosoma brucei rhodesiense. Antimicrob. Agents Chemother. 2014, 58, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Schomburg, C.; Schuehly, W.; Da Costa, F.B.; Klempnauer, K.-H.; Schmidt, T.J. Natural sesquiterpene lactones as inhibitors of Myb-dependent gene expression: Structure–activity relationships. Eur. J. Med. Chem. 2013, 63, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, F.B.; Dias, D.A.; Callegari Lopes, J.L.; Vichnewski, W. Flavonoids and heliangolides from Lychnophora diamantinana. Phytochemistry 1993, 34, 261–263. [Google Scholar] [CrossRef]
- Arakawa, N.S.; Schorr, K.; Ambrósio, S.R.; Merfort, I.; Da Costa, F.B. Further Sesquiterpene Lactones from Viguiera robusta and the Potential Anti-Inflammatory Activity of a Heliangolide: Inhibition of Human Neutrophil Elastase Release. Z. Naturforschung C 2008, 63, 533–538. [Google Scholar] [CrossRef]
- Vichnewski, W.; Sarti, S.J.; Gilbert, B.; Herz, W. Goyazensolide, a schistosomicidal heliangolide from Eremanthus goyazensis. Phytochemistry 1976, 15, 191–193. [Google Scholar] [CrossRef]
- Freiburghaus, F.; Steck, A.; Pfander, H.; Brun, R. Bioassay-guided isolation of a diastereoisomer of kolavenol from Entada abyssinica active on Trypanosoma brucei rhodesiense. J. Ethnopharmacol. 1998, 61, 179–183. [Google Scholar] [CrossRef]
- Tchinda, A.T.; Tsopmo, A.; Tane, P.; Ayafor, J.F.; Connolly, J.D.; Sterner, O. Vernoguinosterol and vernoguinoside, trypanocidal stigmastane derivatives from Vernonia guineensis (Asteraceae). Phytochemistry 2002, 59, 371–374. [Google Scholar] [CrossRef]
- Del Rayo Camacho, M.; Phillipson, J.D.; Croft, S.L.; Marley, D.; Kirby, G.C.; Warhurst, D.C. Assessment of the Antiprotozoal Activity of Galphimia g lauca and the Isolation of New Nor-secofriedelanes and Nor-friedelanes. J. Nat. Prod. 2002, 65, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Del Rayo Camacho, M.; Phillipson, J.D.; Croft, S.L.; Kirby, G.C.; Warhurst, D.C.; Solis, P.N. Terpenoids from Guarea rhophalocarpa. Phytochemistry 2001, 56, 203–210. [Google Scholar] [CrossRef]
- Nour, A.M.M.; Khalid, S.A.; Kaiser, M.; Brun, R.; Abdallah, W.E.; Schmidt, T.J. The antiprotozoal activity of sixteen asteraceae species native to Sudan and bioactivity-guided isolation of xanthanolides from Xanthium brasilicum. Planta Med. 2009, 75, 1363–1368. [Google Scholar] [CrossRef] [PubMed]
- Kaminsky, R.; Schmid, C.; Brun, R. An in vitro selectivity index for evaluation of cytotoxicity of antitrypanosomal compounds. In Vitro Toxicol. 1996, 9, 315–324. [Google Scholar]
- Dube, D.K.; Mpimbaza, G.; Allison, A.C.; Lederer, E.; Rovis, L. Antitrypanosomal activity of sinefungin. Am. J. Trop. Med. Hyg. 1983, 32, 31–33. [Google Scholar] [PubMed]
- Zweygarth, E.; Röttcher, D. Efficacy of experimental trypanocidal compounds against a multiple drug-resistantTrypanosoma brucei brucei stock in mice. Parasitol. Res. 1989, 75, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Zweygarth, E.; Schillinger, D.; Kaufmann, W.; Rottcher, D. Evaluation of sinefungin for the treatment of Trypanosoma (Nannomonas) congolense infections in goats. Trop. Med. Parasitol. 1986, 37, 255–257. [Google Scholar] [PubMed]
- Heilmann, J.; Mayr, S.; Brun, R.; Rali, T.; Sticher, O. Antiprotozoal Activity and Cytotoxicity of Novel 1,7-Dioxadispiro[5.1.5.2]pentadeca-9,12-dien-11-one Derivatives from Amomum aculeatum. Helv. Chim. Acta 2000, 83, 2939–2945. [Google Scholar] [CrossRef]
- Heilmann, J.; Brun, R.; Mayr, S.; Rali, T.; Sticher, O. Minor cytotoxic and antibacterial compounds from the rhizomes of Amomum aculeatum. Phytochemistry 2001, 57, 1281–1285. [Google Scholar] [CrossRef]
- Chianese, G.; Scala, F.; Calcinai, B.; Cerrano, C.; Dien, H.; Kaiser, M.; Tasdemir, D.; Taglialatela-Scafati, O. Natural and Semisynthetic Analogues of Manadoperoxide B Reveal New Structural Requirements for Trypanocidal Activity. Mar. Drugs 2013, 11, 3297–3308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leishmaniasis. Available online: http://www.who.int/mediacentre/factsheets/fs375/en (accessed on 18 October 2016).
- Chappuis, F.; Sundar, S.; Hailu, A.; Ghalib, H.; Rijal, S.; Peeling, R.W.; Alvar, J.; Boelaert, M. Visceral leishmaniasis: what are the needs for diagnosis, treatment and control? Nat. Rev. Microbiol. 2007, 5, S7–S16. [Google Scholar] [CrossRef] [PubMed]
- Murray, H.W.; Berman, J.D.; Davies, C.R.; Saravia, N.G. Advances in leishmaniasis. Lancet 2005, 366, 1561–1577. [Google Scholar] [CrossRef]
- Amato, V.S.; Tuon, F.F.; Bacha, H.A.; Neto, V.A.; Nicodemo, A.C. Mucosal leishmaniasis. Acta Trop. 2008, 105, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sacks, D.; Noben-Trauth, N. The immunology of susceptibility and resistance to leishmania major in mice. Nat. Rev. Immunol. 2002, 2, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Von Stebut, E.; Udey, M.C. Requirements for Th1-dependent immunity against infection with Leishmania major. Microbes Infect. 2004, 6, 1102–1109. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Pandey, H.P.; Sundar, S. Visceral leishmaniasis (kala-azar): challenges ahead. Indian J. Med. Res. 2006, 123, 331–344. [Google Scholar] [PubMed]
- Maltezou, H.C. Drug Resistance in Visceral Leishmaniasis. J. Biomed. Biotechnol. 2010, 2010, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bern, C.; Adler-Moore, J.; Berenguer, J.; Boelaert, M.; den Boer, M.; Davidson, R.N.; Figueras, C.; Gradoni, L.; Kafetzis, D.A.; Ritmeijer, K.; et al. Reviews Of Anti-infective Agents: Liposomal Amphotericin B for the Treatment of Visceral Leishmaniasis. Clin. Infect. Dis. 2006, 43, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Izzedine, H.; Launay-Vacher, V.; Legrand, M.; Lieberherr, D.; Caumes, E.; Deray, G. ABT 378/r: A novel inhibitor of HIV-1 protease in haemodialysis. AIDS 2001, 15, 662–664. [Google Scholar] [CrossRef] [PubMed]
- Laniado-Laborín, R.; Cabrales-Vargas, M.N. Amphotericin B: Side effects and toxicity. Rev. Iberoam. Micol. 2009, 26, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Kumar, M.; Singh, R.K. Leishmaniasis: Current status of available drugs and new potential drug targets. Asian Pac. J. Trop. Med. 2012, 5, 485–497. [Google Scholar] [CrossRef]
- Sundar, S.; Chatterjee, M. Visceral leishmaniasis—Current therapeutic modalities. Indian J. Med. Res. 2006, 123, 345–352. [Google Scholar] [PubMed]
- Bhattacharya, S.K.; Sinha, P.K.; Sundar, S.; Thakur, C.P.; Jha, T.K.; Pandey, K.; Das, V.R.; Kumar, N.; Lal, C.; Verma, N.; et al. Phase 4 Trial of Miltefosine for the Treatment of Indian Visceral Leishmaniasis. J. Infect. Dis. 2007, 196, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Jha, T.K.; Sundar, S.; Thakur, C.P.; Bachmann, P.; Karbwang, J.; Fischer, C.; Voss, A.; Berman, J. Miltefosine, an Oral Agent, for the Treatment of Indian Visceral Leishmaniasis. N. Engl. J. Med. 1999, 341, 1795–1800. [Google Scholar] [CrossRef] [PubMed]
- Sundar, S.; Jha, T.K.; Thakur, C.P.; Bhattacharya, S.K.; Rai, M. Oral miltefosine for the treatment of Indian visceral leishmaniasis. Trans. R. Soc. Trop. Med. Hyg. 2006, 100, S26–S33. [Google Scholar] [CrossRef] [PubMed]
- Sundar, S.; Sinha, P.K.; Rai, M.; Verma, D.K.; Nawin, K.; Alam, S.; Chakravarty, J.; Vaillant, M.; Verma, N.; Pandey, K.; et al. Comparison of short-course multidrug treatment with standard therapy for visceral leishmaniasis in India: An open-label, non-inferiority, randomised controlled trial. Lancet 2011, 377, 477–486. [Google Scholar] [CrossRef]
- Thakur, C.P.; Kanyok, T.P.; Pandey, A.K.; Sinha, G.P.; Messick, C.; Olliaro, P. Treatment of visceral leishmaniasis with injectable paromomycin (aminosidine). An open-label randomized phase-II clinical study. Trans. R. Soc. Trop. Med. Hyg. 2000, 94, 432–433. [Google Scholar] [CrossRef]
- Thakur, B.B. Breakthrough in the management of visceral leishmaniasis. J. Assoc. Phys. India 2003, 51, 649–651. [Google Scholar]
- Jha, T.K.; Sundar, S.; Thakur, C.P.; Felton, J.M.; Sabin, A.J.; Horton, J. A phase II dose-ranging study of sitamaquine for the treatment of visceral leishmaniasis in India. Am. J. Trop. Med. Hyg. 2005, 73, 1005–1011. [Google Scholar] [PubMed]
- Wasunna, M.K.; Rashid, J.R.; Mbui, J.; Kirigi, G.; Kinoti, D.; Lodenyo, H.; Felton, J.M.; Sabin, A.J.; Albert, M.J.; Horton, J. A phase II dose-increasing study of sitamaquine for the treatment of visceral leishmaniasis in Kenya. Am. J. Trop. Med. Hyg. 2005, 73, 871–876. [Google Scholar] [PubMed]
- Das, V.N.; Ranjan, A.; Sinha, A.N.; Verma, N.; Lal, C.S.; Gupta, A.K.; Siddiqui, N.A.; Kar, S.K. A randomized clinical trial of low dosage combination of pentamidine and allopurinol in the treatment of antimony unresponsive cases of visceral leishmaniasis. J. Assoc. Phys. India 2001, 49, 609–613. [Google Scholar]
- Mittra, B.; Saha, A.; Chowdhury, A.R.; Pal, C.; Mandal, S.; Mukhopadhyay, S.; Bandyopadhyay, S.; Majumder, H.K. Luteolin, an abundant dietary component is a potent anti-leishmanial agent that acts by inducing topoisomerase II-mediated kinetoplast DNA cleavage leading to apoptosis. Mol. Med. 2000, 6, 527–541. [Google Scholar] [PubMed]
- Pan, L.; Lezama-Davila, C.M.; Isaac-Marquez, A.P.; Calomeni, E.P.; Fuchs, J.R.; Satoskar, A.R.; Kinghorn, A.D. Sterols with antileishmanial activity isolated from the roots of Pentalinon andrieuxii. Phytochemistry 2012, 82, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Christensen, S.B.; Blom, J.; Lemmich, E.; Nadelmann, L.; Fich, K.; Theander, T.G.; Kharazmi, A. Licochalcone A, a novel antiparasitic agent with potent activity against human pathogenic protozoan species of Leishmania. Antimicrob. Agents Chemother. 1993, 37, 2550–2556. [Google Scholar] [CrossRef] [PubMed]
- Zhai, L. The antileishmanial activity of novel oxygenated chalcones and their mechanism of action. J. Antimicrob. Chemother. 1999, 43, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Christensen, S.B.; Theander, T.G.; Kharazmi, A. Antileishmanial activity of licochalcone A in mice infected with Leishmania major and in hamsters infected with Leishmania donovani. Antimicrob. Agents Chemother. 1994, 38, 1339–1344. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhai, L.; Christensen, S.B.; Theander, T.G.; Kharazmi, A. Inhibition of Fumarate Reductase in Leishmania major and L. donovani by Chalcones. Antimicrob. Agents Chemother. 2001, 45, 2023–2029. [Google Scholar] [CrossRef] [PubMed]
- Torres-Santos, E.C.; Rodrigues, J.M.; Moreira, D.L.; Kaplan, M.A.; Rossi-Bergmann, B. Improvement of in vitro and in vivo antileishmanial activities of 2′,6′-dihydroxy-4′-methoxychalcone by entrapment in poly(d,l-lactide) nanoparticles. Antimicrob. Agents Chemother. 1999, 43, 1776–1778. [Google Scholar] [PubMed]
- Boeck, P.; Bandeira Falcão, C.A.; Leal, P.C.; Yunes, R.A.; Filho, V.C.; Torres-Santos, E.C.; Rossi-Bergmann, B. Synthesis of chalcone analogues with increased antileishmanial activity. Bioorg. Med. Chem. 2006, 14, 1538–1545. [Google Scholar] [CrossRef] [PubMed]
- Haudecoeur, R.; Boumendjel, A. Recent Advances in the Medicinal Chemistry of Aurones. Curr. Med. Chem. 2012, 19, 2861–2875. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, H.B.; Silva, M.; Ellena, J.; Rodrigues, B.D.G.; Almeida, A.L.C.; Vieira, P.C.; Oliva, G.; Thiemann, O.H. Aurapten, a coumarin with growth inhibition against Leishmania major promastigotes. Braz. J. Med. Biol. Res. 2004, 37, 1847–1852. [Google Scholar] [CrossRef] [PubMed]
- Oketch-Rabah, H.A.; Lemmich, E.; Dossaji, S.F.; Theander, T.G.; Olsen, C.E.; Cornett, C.; Kharazmi, A.; Christensen, S.B. Two New Antiprotozoal 5-Methylcoumarins from Vernonia brachycalyx. J. Nat. Prod. 1997, 60, 458–461. [Google Scholar] [CrossRef] [PubMed]
- Iranshahi, M.; Arfa, P.; Ramezani, M.; Jaafari, M.R.; Sadeghian, H.; Bassarello, C.; Piacente, S.; Pizza, C. Sesquiterpene coumarins from Ferula szowitsiana and in vitro antileishmanial activity of 7-prenyloxycoumarins against promastigotes. Phytochemistry 2007, 68, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Brenzan, M.A.; Santos, A.O.; Nakamura, C.V.; Filho, B.P.D.; Ueda-Nakamura, T.; Young, M.C.M.; Côrrea, A.G.; Júnior, J.A.; Morgado-Díaz, J.A.; Cortez, D.A.G. Effects of (−) mammea A/BB isolated from Calophyllum brasiliense leaves and derivatives on mitochondrial membrane of Leishmania amazonensis. Phytomedicine 2012, 19, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Haslam, E. Natural polyphenols (vegetable tannins) as drugs: Possible modes of action. J. Nat. Prod. 1996, 59, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Haslam, E.; Lilley, T.; Cai, Y.; Martin, R.; Mangnolato, D. Traditional Herbal Medicines—The Role of Polyphenols. Planta Med. 1989, 55, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kolodziej, H.; Kayser, O.; Kiderlen, A.F.; Ito, H.; Hatano, T.; Yoshida, T.; Foo, L.Y. Antileishmanial activity of hydrolyzable tannins and their modulatory effects on nitric oxide and tumour necrosis factor-alpha release in macrophages in vitro. Planta Med. 2001, 67, 825–832. [Google Scholar] [CrossRef] [PubMed]
- Gour, J.K.; Kumar, V.; Singh, N.; Bajpai, S.; Pandey, H.P.; Singh, R.K. Identification of Th1-responsive leishmanial excretory—Secretory antigens (LESAs). Exp. Parasitol. 2012, 132, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Majumder, H.K.; Chakravarty, A.K.; Mukhopadhyay, S.; Gil, R.R.; Cordell, G.A. Amarogentin, a Naturally Occurring Secoiridoid Glycoside and a Newly Recognized Inhibitor of Topoisomerase I from Leishmania donovani. J. Nat. Prod. 1996, 59, 27–29. [Google Scholar] [CrossRef] [PubMed]
- Medda, S. Evaluation of the in-vivo activity and toxicity of amarogentin, an antileishmanial agent, in both liposomal and niosomal forms. J. Antimicrob. Chemother. 1999, 44, 791–794. [Google Scholar] [CrossRef] [PubMed]
- Fournet, A.; Angelo, A.; Muñoz, V.; Roblot, F.; Hocquemiller, R.; Cavé, A. Biological and chemical studies of Pera benensis, a Bolivian plant used in folk medicine as a treatment of cutaneous leishmaniasis. J. Ethnopharmacol. 1992, 37, 159–164. [Google Scholar] [CrossRef]
- Hazra, B.; Sarkar, R.; Bhattacharyya, S.; Ghosh, P.K.; Chel, G.; Dinda, B. Synthesis of plumbagin derivatives and their inhibitory activities against Ehrlich ascites carcinomain vivo andLeishmania donovani Promastigotes in vitro. Phyther. Res. 2002, 16, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Fournet, A.; Barrios, A.A.; Muñoz, V.; Hocquemiller, R.; Cavé, A. Effect of natural naphthoquinones in BALB/c mice infected with Leishmania amazonensis and L. venezuelensis. Trop. Med. Parasitol. 1992, 43, 219–222. [Google Scholar] [PubMed]
- Fujii, N.; Yamashita, Y.; Arima, Y.; Nagashima, M.; Nakano, H. Induction of topoisomerase II-mediated DNA cleavage by the plant naphthoquinones plumbagin and shikonin. Antimicrob. Agents Chemother. 1992, 36, 2589–2594. [Google Scholar] [CrossRef]
- Mori-Yasumoto, K.; Izumoto, R.; Fuchino, H.; Ooi, T.; Agatsuma, Y.; Kusumi, T.; Satake, M.; Sekita, S. Leishmanicidal activities and cytotoxicities of bisnaphthoquinone analogues and naphthol derivatives from Burman Diospyros burmanica. Bioorg. Med. Chem. 2012, 20, 5215–5219. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Fuchino, H.; Satake, M.; Agatsuma, Y.; Sekita, S. In Vitro Screening of Leishmanicidal Activity in Myanmar Timber Extracts. Biol. Pharm. Bull. 2004, 27, 921–925. [Google Scholar] [CrossRef] [PubMed]
- Fournet, A.; Ferreira, M.E.; de Arias, A.R.; Ortiz, S.T.; Fuentes, S.; Nakayama, H.; Schinini, A.; Hocquemiller, R. In vivo efficacy of oral and intralesional administration of 2-substituted quinolines in experimental treatment of new world cutaneous leishmaniasis caused by Leishmania amazonensis. Antimicrob. Agents Chemother. 1996, 40, 2447–2451. [Google Scholar] [PubMed]
- Fournet, A.; Gantier, J.C.; Gautheret, A.; Leysalles, L.; Munos, M.H.; Mayrargue, J.; Moskowitz, H.; Cavé, A.; Hocquemiller, R. The activity of 2-substituted quinoline alkaloids in BALB/c mice infected with Leishmania donovani. J. Antimicrob. Chemother. 1994, 33, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, I.; Dunbar, D.C.; Khan, S.I.; Tekwani, B.L.; Bedir, E.; Takamatsu, S.; Ferreira, D.; Walker, L.A. Antiparasitic Alkaloids from Psychotria klugii. J. Nat. Prod. 2003, 66, 962–967. [Google Scholar] [CrossRef] [PubMed]
- Lala, S.; Pramanick, S.; Mukhopadhyay, S.; Bandyopadhyay, S.; Basu, M.K. Harmine: Evaluation of its Antileishmanial Properties in Various Vesicular Delivery Systems. J. Drug Target. 2004, 12, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Peng, W.; Chen, H.; Ma, Y.; Liu, X.; Hou, X.; Guan, H.; Xu, A. DNA binding properties of 9-substituted harmine derivatives. Biochem. Biophys. Res. Commun. 2005, 338, 1557–1563. [Google Scholar] [CrossRef] [PubMed]
- Germonprez, N.; Maes, L.; Van Puyvelde, L.; Van Tri, M.; Tuan, D.A.; de Kimpe, N. In Vitro and in Vivo Anti-Leishmanial Activity of Triterpenoid Saponins Isolated from Maesa b alansae and Some Chemical Derivatives. J. Med. Chem. 2005, 48, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Dutta, A.; Ghoshal, A.; Mandal, D.; Mondal, N.B.; Banerjee, S.; Sahu, N.P.; Mandal, C. Racemoside A, an anti-leishmanial, water-soluble, natural steroidal saponin, induces programmed cell death in Leishmania donovani. J. Med. Microbiol. 2007, 56, 1196–1204. [Google Scholar] [CrossRef] [PubMed]
- Delmas, F.; di Giorgio, C.; Elias, R.; Gasquet, M.; Azas, N.; Mshvildadze, V.; Dekanosidze, G.; Kemertelidze, E.; Timon-David, P. Antileishmanial activity of three saponins isolated from ivy, alpha-hederin, beta-hederin and hederacolchiside A1, as compared to their action on mammalian cells cultured in vitro. Planta Med. 2000, 66, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Di Giorgio, C.; Delmas, F.; Akhmedjanova, V.; Ollivier, E.; Bessonova, I.; Riad, E.; Timon-David, P. In vitro antileishmanial activity of diphyllin isolated from Haplophyllum bucharicum. Planta Med. 2005, 71, 366–369. [Google Scholar] [CrossRef] [PubMed]
- Georgopoulou, K.; Smirlis, D.; Bisti, S.; Xingi, E.; Skaltsounis, L.; Soteriadou, K. In vitro activity of 10-deacetylbaccatin III against Leishmania donovani promastigotes and intracellular amastigotes. Planta Med. 2007, 73, 1081–1088. [Google Scholar] [CrossRef] [PubMed]
- Rosa, M.d.S.S.; Mendonca-Filho, R.R.; Bizzo, H.R.; Rodrigues, I.d.A.; Soares, R.M.A.; Souto-Padron, T.; Alviano, C.S.; Lopes, A.H.C.S. Antileishmanial Activity of a Linalool-Rich Essential Oil from Croton cajucara. Antimicrob. Agents Chemother. 2003, 47, 1895–1901. [Google Scholar] [CrossRef]
- Tan, N.; Kaloga, M.; Radtke, O.A.; Kiderlen, A.F.; Öksüz, S.; Ulubelen, A.; Kolodziej, H. Abietane diterpenoids and triterpenoic acids from Salvia cilicica and their antileishmanial activities. Phytochemistry 2002, 61, 881–884. [Google Scholar] [CrossRef]
- Danelli, M.G.M.; Soares, D.C.; Abreu, H.S.; Peçanha, L.M.T.; Saraiva, E.M. Leishmanicidal effect of LLD-3 (1), a nor-triterpene isolated from Lophanthera lactescens. Phytochemistry 2009, 70, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Thiem, D.A.; Sneden, A.T.; Khan, S.I.; Tekwani, B.L. Bisnortriterpenes from Salacia m adagascariensis. J. Nat. Prod. 2005, 68, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Fuchino, H.; Koide, T.; Takahashi, M.; Sekita, S.; Satake, M. New sesquiterpene lactones from Elephantopus mollis and their leishmanicidal activities. Planta Med. 2001, 67, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Sulsen, V.P.; Frank, F.M.; Cazorla, S.I.; Anesini, C.A.; Malchiodi, E.L.; Freixa, B.; Vila, R.; Muschietti, L.V.; Martino, V.S. Trypanocidal and Leishmanicidal Activities of Sesquiterpene Lactones from Ambrosia tenuifolia Sprengel (Asteraceae). Antimicrob. Agents Chemother. 2008, 52, 2415–2419. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, I.; Bedir, E.; Khan, S.I.; Tekwani, B.L.; Khan, I.A.; Takamatsu, S.; Pelletier, J.; Walker, L.A. A New Antimalarial Quassinoid from Simaba o rinocensis. J. Nat. Prod. 2004, 67, 772–777. [Google Scholar] [CrossRef] [PubMed]
- Lenta, B.; Weniger, B.; Kaiser, M.; Vonthron-Sénécheau, C. Antileishmanial natural prenylated anthranoids. Planta Med. 2012, 78, PI290. [Google Scholar] [CrossRef]
- Da Silva, D.B.; Tulli, E.C.O.; Militão, G.C.G.; Costa-Lotufo, L.V.; Pessoa, C.; de Moraes, M.O.; Albuquerque, S.; de Siqueira, J.M. The antitumoral, trypanocidal and antileishmanial activities of extract and alkaloids isolated from Duguetia furfuracea. Phytomedicine 2009, 16, 1059–1063. [Google Scholar] [CrossRef] [PubMed]
- Schmeda-Hirschmann, G.; Razmilic, I.; Sauvain, M.; Moretti, C.; Muñoz, V.; Ruiz, E.; Balanza, E.; Fournet, A. Antiprotozoal activity of Jatrogrossidione from Jatropha grossidentata and Jatrophone from Jatropha isabellii. Phyther. Res. 1996, 10, 375–378. [Google Scholar] [CrossRef]
- Nakao, Y.; Shiroiwa, T.; Murayama, S.; Matsunaga, S.; Goto, Y.; Matsumoto, Y.; Fusetani, N. Identification of Renieramycin A as an Antileishmanial Substance in a Marine Sponge Neopetrosia sp. Mar. Drugs 2004, 2, 55–62. [Google Scholar] [CrossRef]
- Compagnone, R.S.; Piña, I.C.; Rangel, H.R.; Dagger, F.; Suárez, A.I.; Venkata Rami Reddy, M.; John Faulkner, D. Antileishmanial cyclic peroxides from the Palauan sponge Plakortis aff. angulospiculatus. Tetrahedron 1998, 54, 3057–3068. [Google Scholar] [CrossRef]
- Linington, R.G.; González, J.; Ureña, L.-D.; Romero, L.I.; Ortega-Barría, E.; Gerwick, W.H. Venturamides A and B: Antimalarial Constituents of the Panamanian Marine Cyanobacterium Oscillatoria sp. J. Nat. Prod. 2007, 70, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Pimentel-Elardo, S.M.; Kozytska, S.; Bugni, T.S.; Ireland, C.M.; Moll, H.; Hentschel, U. Anti-Parasitic Compounds from Streptomyces sp. Strains Isolated from Mediterranean Sponges. Mar. Drugs 2010, 8, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Vik, A.; Proszenyák, Á.; Vermeersch, M.; Cos, P.; Maes, L.; Gundersen, L.-L. Screening of Agelasine D and Analogs for Inhibitory Activity against Pathogenic Protozoa; Identification of Hits for Visceral Leishmaniasis and Chagas Disease. Molecules 2009, 14, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Chauhan, K.; Shivahare, R.; Vishwakarma, P.; Suthar, M.K.; Sharma, A.; Gupta, S.; Saxena, J.K.; Lal, J.; Chandra, P.; et al. Discovery of a New Class of Natural Product-Inspired Quinazolinone Hybrid as Potent Antileishmanial agents. J. Med. Chem. 2013, 56, 4374–4392. [Google Scholar] [CrossRef] [PubMed]
- Keiser, J. Antimalarials in the Treatment of Schistosomiasis. Curr. Pharm. Des. 2012, 18, 3531–3538. [Google Scholar] [CrossRef] [PubMed]
- Ingram-Sieber, K.; Cowan, N.; Panic, G.; Vargas, M.; Mansour, N.R.; Bickle, Q.D.; Wells, T.N.C.; Spangenberg, T.; Keiser, J. Orally Active Antischistosomal Early Leads Identified from the Open Access Malaria Box. PLoS Negl. Trop. Dis. 2014, 8, 30. [Google Scholar] [CrossRef] [PubMed]
- Lescano, S.Z.; Chieffi, P.P.; Canhassi, R.R.; Boulos, M.; Amato Neto, V. Antischistosomal activity of artemether in experimental Schistosomiasis mansoni. Rev. Saude Publica 2004, 38, 71–75. [Google Scholar] [PubMed]
- Njoroge, M.; Njuguna, N.M.; Mutai, P.; Ongarora, D.S.B.; Smith, P.W.; Chibale, K. Recent Approaches to Chemical Discovery and Development Against Malaria and the Neglected Tropical Diseases Human African Trypanosomiasis and Schistosomiasis. Chem. Rev. 2014, 114, 11138–11163. [Google Scholar] [CrossRef] [PubMed]
- Neves, B.J.; Andrade, C.H.; Cravo, P.V.L. Natural products as leads in schistosome drug discovery. Molecules 2015, 20, 1872–1903. [Google Scholar] [CrossRef] [PubMed]
- Utzinger, J.; Raso, G.; Brooker, S.; De Savigny, D.; Tanner, M.; Ornbjerg, N.; Singer, B.H.; N’goran, E.K. Schistosomiasis and neglected tropical diseases: Towards integrated and sustainable control and a word of caution. Parasitology 2009, 136, 1859–1874. [Google Scholar] [CrossRef] [PubMed]
- Schafer, T.; Hale, B. Gastrointestinal complications of schistosomiasis. Curr. Gastroenterol. Rep. 2001, 3, 293. [Google Scholar] [CrossRef] [PubMed]
- Mbabazi, P.S.; Andan, O.; Fitzgerald, D.W.; Chitsulo, L.; Engels, D.; Downs, J.A. Examining the relationship between urogenital schistosomiasis and HIV infection. PLoS Negl. Trop. Dis. 2011, 5. [Google Scholar] [CrossRef] [PubMed]
- Feldmeier, H.; Krantz, I.; Poggensee, G. Female Genital Schistosomiasis as a Risk-Factor for the Transmission of HIV. Int. J. STD AIDS 1994, 5, 368–372. [Google Scholar] [CrossRef]
- Kallestrup, P.; Zinyama, R.; Gomo, E.; Butterworth, A.E.; Mudenge, B.; van Dam, G.J.; Gerstoft, J.; Erikstrup, C.; Ullum, H. Schistosomiasis and HIV-1 infection in rural Zimbabwe: Effect of treatment of schistosomiasis on CD4 cell count and plasma HIV-1 RNA load. J. Infect. Dis. 2005, 192, 1956–1961. [Google Scholar] [CrossRef] [PubMed]
- Cioli, D.; Pica-Mattoccia, L.; Archer, S. Antischistosomal drugs: Past, present … and future? Pharmacol. Ther. 1995, 68, 35–85. [Google Scholar] [CrossRef]
- Utzinger, J.; Shuhua, X.; N’Goran, E.K.; Bergquist, R.; Tanner, M. The potential of artemether for the control of schistosomiasis. Int. J. Parasitol. 2001, 31, 1549–1562. [Google Scholar] [CrossRef]
- Xiao, S.H.; Catto, B.A.; Webster, L.T. Effects of praziquantel on different developmental stages of Schistosoma mansoni in vitro and in vivo. J. Infect. Dis. 1985, 151, 1130–1137. [Google Scholar] [CrossRef] [PubMed]
- Mostafa, O.M.S.; Eid, R.A.; Adly, M.A. Antischistosomal activity of ginger (Zingiber Officinale) against Schistosoma mansoni harbored in C57 mice. Parasitol. Res. 2011, 109, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Coles, G.C.; Bruce, J.I.; Kinoti, G.K.; Mutahi, W.T.; Dias, L.C.S.; Rocha, R.S.; Katz, N. The potential for drug resistance in schistosomiasis. Parasitol. Today 1987, 3, 349–350. [Google Scholar] [CrossRef]
- Danso-Appiah, A.; De Vlas, S.J. Interpreting low praziquantel cure rates of Schistosoma mansoni infections in Senegal. Trends Parasitol. 2002, 18, 125–129. [Google Scholar] [CrossRef]
- Faghiri, Z.; Camargo, S.M.R.; Huggel, K.; Forster, I.C.; Ndegwa, D.; Verrey, F.; Skelly, P.J. The tegument of the human parasitic worm Schistosoma mansoni as an excretory organ: The surface aquaporin SmAQP is a lactate transporter. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Hellemond, J.J.; Retra, K.; Brouwers, J.F.H.M.; van Balkom, B.W.M.; Yazdanbakhsh, M.; Shoemaker, C.B.; Tielens, A.G.M. Functions of the tegument of schistosomes: Clues from the proteome and lipidome. Int. J. Parasitol. 2006, 36, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, M.A.; de Oliveira, R.N.; Véras, L.M.C.; Lima, D.F.; Campelo, Y.D.M.; Campos, S.A.; Kuckelhaus, S.A.S.; Pinto, P.L.S.; Eaton, P.; Mafud, A.C.; et al. Anthelmintic Activity In Vivo of Epiisopiloturine against Juvenile and Adult Worms of Schistosoma mansoni. PLoS Negl. Trop. Dis. 2015, 9, e0003656. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Dutta, C.P. Alkaloids of Piper longum Linn. I. Structure and synthesis of piperlongumine and piperlonguminine. Tetrahedron 1967, 23, 1769–1781. [Google Scholar] [CrossRef]
- Bezerra, D.P.; Pessoa, C.; De Moraes, M.O.; Saker-Neto, N.; Silveira, E.R.; Costa-Lotufo, L.V. Overview of the therapeutic potential of piplartine (piperlongumine). Eur. J. Pharm. Sci. 2013, 48, 453–463. [Google Scholar] [CrossRef] [PubMed]
- De Moraes, J.; Nascimento, C.; Yamaguchi, L.F.; Kato, M.J.; Nakano, E. Schistosoma mansoni: In vitro schistosomicidal activity and tegumental alterations induced by piplartine on schistosomula. Exp. Parasitol. 2012, 132, 222–227. [Google Scholar] [CrossRef] [PubMed]
- De Moraes, J.; Nascimento, C.; Lopes, P.O.M.V.; Nakano, E.; Yamaguchi, L.F.; Kato, M.J.; Kawano, T. Schistosoma mansoni: In vitro schistosomicidal activity of piplartine. Exp. Parasitol. 2011, 127, 357–364. [Google Scholar] [CrossRef] [PubMed]
- De Moraes, J.; Keiser, J.; Ingram, K.; Nascimento, C.; Yamaguchi, L.F.; Bittencourt, C.R.; Bemquerer, M.P.; Leite, J.R.; Kato, M.J.; Nakano, E. In vitro synergistic interaction between amide piplartine and antimicrobial peptide dermaseptin against Schistosoma mansoni schistosomula and adult worms. Curr. Med. Chem. 2013, 20, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Bezerra, D.P.; Moura, D.J.; Rosa, R.M.; de Vasconcellos, M.C.; Silva, A.C.R.; de Moraes, M.O.; Silveira, E.R.; Lima, M.A.S.; Henriques, J.A.P.; Costa-Lotufo, L.V.; et al. Evaluation of the genotoxicity of piplartine, an alkamide of Piper tuberculatum, in yeast and mammalian V79 cells. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2008, 652, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Bezerra, D.P.; Militão, G.C.G.; de Castro, F.O.; Pessoa, C.; de Moraes, M.O.; Silveira, E.R.; Lima, M.A.S.; Elmiro, F.J.M.; Costa-Lotufo, L.V. Piplartine induces inhibition of leukemia cell proliferation triggering both apoptosis and necrosis pathways. Toxicol. In Vitro 2007, 21, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Raj, L.; Ide, T.; Gurkar, A.U.; Foley, M.; Schenone, M.; Li, X.; Tolliday, N.J.; Golub, T.R.; Carr, S.A.; Shamji, A.F.; et al. Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature 2011, 475, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Schaab, E.H.; Crotti, A.E.M.; Iamamoto, Y.; Kato, M.J.; Lotufo, L.V.C.; Lopes, N.P. Biomimetic oxidation of piperine and piplartine catalyzed by iron(III) and manganese(III) porphyrins. Biol. Pharm. Bull. 2010, 33, 912–916. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, L.; Bartlett, A.; Whitfield, P.J. In vitro and in vivo studies on the bioactivity of a ginger (Zingiber Officinale) extract towards adult schistosomes and their egg production. J. Helminthol. 2002, 76, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Adewunmi, C.O.; Oguntimein, B.O.; Furu, P. Molluscicidal and antischistosomal activities of Zingiber Officinale. Planta Med. 1990, 56, 374–376. [Google Scholar] [CrossRef] [PubMed]
- De Moraes, J.; de Oliveira, R.N.; Costa, J.P.; Junior, A.L.G.; de Sousa, D.P.; Freitas, R.M.; Allegretti, S.M.; Pinto, P.L.S. Phytol, a Diterpene Alcohol from Chlorophyll, as a Drug against Neglected Tropical Disease Schistosomiasis Mansoni. PLoS Negl. Trop. Dis. 2014, 8, 51. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, R.R.; Ghosh, S.K. Phytol-derived novel isoprenoid immunostimulants. Front. Immunol. 2012, 3, 49. [Google Scholar] [CrossRef] [PubMed]
- Vetter, W.; Schröder, M.; Lehnert, K. Differentiation of refined and virgin edible oils by means of the trans—And cis-phytol isomer distribution. J. Agric. Food Chem. 2012, 60, 6103–6107. [Google Scholar] [CrossRef] [PubMed]
- Hatcher, H.; Planalp, R.; Cho, J.; Torti, F.M.; Torti, S.V. Curcumin: From ancient medicine to current clinical trials. Cell. Mol. Life Sci. 2008, 65, 1631–1652. [Google Scholar] [CrossRef] [PubMed]
- Han, S.-S.; Keum, Y.-S.; Seo, H.-J.; Surh, Y.-J. Curcumin suppresses activation of NF-κB and AP-1 induced by phorbol ester in cultured human promyelocytic leukemia cells. J. Biochem. Mol. Biol. 2002, 35, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Magalhães, L.G.; Machado, C.B.; Morais, E.R.; Bueno De Carvalho Moreira, É.; Soares, C.S.; da Silva, S.H.; da Silva Filho, A.A.; Rodrigues, V. In vitro schistosomicidal activity of curcumin against Schistosoma mansoni adult worms. Parasitol. Res. 2009, 104, 1197–1201. [Google Scholar] [CrossRef] [PubMed]
- Na, M.; Jang, J.; Min, B.S.; Lee, S.J.; Lee, M.S.; Kim, B.Y.; Oh, W.K.; Ahn, J.S. Fatty acid synthase inhibitory activity of acylphloroglucinols isolated from Dryopteris crassirhizoma. Bioorg. Med. Chem. Lett. 2006, 16, 4738–4742. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-M.; Na, M.-K.; An, R.-B.; Min, B.-S.; Lee, H.-K. Antioxidant activity of two phloroglucinol derivatives from Dryopteris crassirhizoma. Biol. Pharm. Bull. 2003, 26, 1354–1356. [Google Scholar] [CrossRef] [PubMed]
- Kapadia, G.J.; Tokuda, H.; Konoshima, T.; Takasaki, M.; Takayasu, J.; Nishino, H. Anti-tumor promoting activity of Dryopteris phlorophenone derivatives. Cancer Lett. 1996, 105, 161–165. [Google Scholar] [CrossRef]
- Magalhães, L.G.; Kapadia, G.J.; da Silva Tonuci, L.R.; Caixeta, S.C.; Parreira, N.A.; Rodrigues, V.; da Silva Filho, A.A. In vitro schistosomicidal effects of some phloroglucinol derivatives from Dryopteris species against Schistosoma mansoni adult worms. Parasitol. Res. 2010, 106, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Kaur, K.; Jain, M.; Reddy, R.P.; Jain, R. Quinolines and structurally related heterocycles as antimalarials. Eur. J. Med. Chem. 2010, 45, 3245–3264. [Google Scholar] [CrossRef] [PubMed]
- Ridley, R.G. Medical need, scientific opportunity and the drive for antimalarial drugs. Nature 2002, 415, 686–693. [Google Scholar] [CrossRef] [PubMed]
- Keiser, J.; Chollet, J.; Xiao, S.H.; Mei, J.Y.; Jiao, P.Y.; Utzinger, J.; Tanner, M. Mefloquine—An aminoalcohol with promising antischistosomal properties in mice. PLoS Negl. Trop. Dis. 2009, 3, e305. [Google Scholar] [CrossRef] [PubMed]
- Ingram, K.; Ellis, W.; Keiser, J. Antischistosomal activities of mefloquine-related arylmethanols. Antimicrob. Agents Chemother. 2012, 56, 3207–3215. [Google Scholar] [CrossRef] [PubMed]
- Manneck, T.; Haggenmüller, Y.; Keiser, J. Morphological effects and tegumental alterations induced by mefloquine on schistosomula and adult flukes of Schistosoma mansoni. Parasitology 2010, 137, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Holtfreter, M.C.; Loebermann, M.; Klammt, S.; Sombetzki, M.; Bodammer, P.; Riebold, D.; Kinzelbach, R.; Reisinger, E.C. Schistosoma mansoni: Schistosomicidal effect of mefloquine and primaquine in vitro. Exp. Parasitol. 2011, 127, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Keiser, J.; Manneck, T.; Vargas, M. Interactions of mefloquine with praziquantel in the Schistosoma mansoni mouse model and in vitro. J. Antimicrob. Chemother. 2011, 66, 1791–1797. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.H.; Mei, J.Y.; Jiao, P.Y. Effect of mefloquine administered orally at single, multiple, or combined with artemether, artesunate, or praziquantel in treatment of mice infected with Schistosoma japonicum. Parasitol. Res. 2011, 108, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Kamchonwongpaisan, S.; Meshnick, S.R. The mode of action of the antimalarial artemisinin and its derivatives. Gen. Pharmacol. 1996, 27, 587–592. [Google Scholar] [CrossRef]
- Meshnick, S.R. Artemisinin: Mechanisms of action, resistance and toxicity. Int. J. Parasitol. 2002, 32, 1655–1660. [Google Scholar] [CrossRef]
- Klayman, D.L. Qinghaosu (artemisinin): An antimalarial drug from China. Science 1985, 228, 1049–1055. [Google Scholar] [CrossRef] [PubMed]
- Le, W.J.; You, J.Q.; Yang, Y.Q.; Mei, J.Y.; Guo, H.F.; Yang, H.Z.; Zhang, C.W. Studies on the efficacy of artemether in experimental schistosomiasis (author’s transl). Acta Pharm. Sin. 1982, 17, 187–193. [Google Scholar]
- Yin, J.W.; Yang, Y.Q.; Xiao, S.H.; Li, Y.; Jiang, H.J. [Comparative studies on histological and histochemical alterations of Schistosoma japonicum induced by arteether and artemether]. Zhongguo Yao Li Xue Bao 1991, 12, 478–480. [Google Scholar] [PubMed]
- Yue, W.J.; You, J.Q.; Mei, J.Y. [Effect of artemether on the tegumental surface antigen of Schistosoma japonicum]. Ji Sheng Chong Xue Yu Ji Sheng Chong Bing Za Zhi 1984, 2, 167–169. [Google Scholar] [PubMed]
- Wu, L.J.; Yang, H.Z.; Yang, Y.Q. Histological and histochemical changes of Schistosoma japonicum and host liver caused by artemether. Acta Pharm. Sin. 1983, 18, 7–14. [Google Scholar]
- Le, W.J.; You, J.Q.; Mei, J.Y. Chemotherapeutic effect of artesunate in experimental schistosomiasis. Acta Pharm. Sin. 1983, 18, 619–621. [Google Scholar]
- Xiao, S.H.; Yin, J.W.; Mei, J.Y.; You, J.Q.; Li, Y.; Jiang, H.J. Effect of arteether on Schistosoma japonicum. Acta Pharm. Sin. 1992, 27, 161–165. [Google Scholar]
- Abdel Aziz, S.S.; El-Badawy, N.M. Experimental trials of an artemisinin derivative in treatment of Schistosoma mansoni infected mice. J. Egypt. Soc. Parasitol. 2000, 30, 295–303. [Google Scholar] [PubMed]
- Shuhua, X.; Chollet, J.; Weiss, N.A.; Bergquist, R.N.; Tanner, M. Preventive effect of artemether in experimental animals infected with Schistosoma mansoni. Parasitol. Int. 2000, 49, 19–24. [Google Scholar] [CrossRef]
- Xiao, S.H.; You, J.Q.; Yang, Y.Q.; Wang, C.Z. Experimental studies on early treatment of schistosomal infection with artemether. Southeast Asian J. Trop. Med. Public Health 1995, 26, 306–318. [Google Scholar] [PubMed]
- Xiao, S.H.; You, J.Q.; Mei, J.Y.; Jiao, P.Y.; Guo, H.F.; Feng, Z. Preventive effect of artemether in rabbits infected with Schistosoma japonicum cercariae. Zhongguo Yao Li Xue Bao 1998, 19, 63–66. [Google Scholar] [PubMed]
- De Clercq, D.; Vercruysse, J.; Verlé, P.; Niasse, F.; Kongs, A.; Diop, M. Efficacy of artesunate against Schistosoma mansoni infections in Richard Toll, Senegal. Trans. R. Soc. Trop. Med. Hyg. 2000, 94, 90–91. [Google Scholar] [CrossRef]
- Utzinger, J.; N’Goran, E.K.; N’Dri, A.; Lengeler, C.; Xiao, S.; Tanner, M. Oral artemether for prevention of Schistosoma mansoni infection: Randomised controlled trial. Lancet 2000, 355, 1320–1325. [Google Scholar] [CrossRef]
- Xiao, S.H.; Booth, M.; Tanner, M. The prophylactic effects of artemether against Schistosoma japonicum infections. Parasitol. Today 2000, 16, 122–126. [Google Scholar] [CrossRef]
- Utzinger, J.; Chollet, J.; Jiqing, Y.; Jinyan, M.; Tanner, M.; Shuhua, X. Effect of combined treatment with praziquantel and artemether on Schistosoma japonicum and Schistosoma mansoni in experimentally infected animals. Acta Trop. 2001, 80, 9–18. [Google Scholar] [CrossRef]
- De Clercq, D.; Vercruysse, J.; Verlé, P.; Kongs, A.; Diop, M. Short communication: What is the effect of combining artesunate and praziquantel in the treatment of Schistosoma mansoni infections? Trop. Med. Int. Health 2000, 5, 744–746. [Google Scholar] [CrossRef]
- White, N.J.; Nosten, F.; Looareesuwan, S.; Watkins, W.M.; Marsh, K.; Snow, R.W.; Kokwaro, G.; Ouma, J.; Hien, T.T.; Molyneux, M.E.; et al. Averting a malaria disaster. Lancet 1999, 353, 1965–1967. [Google Scholar] [CrossRef]
- Meshnick, S.R.; Thomas, A.; Ranz, A.; Xu, C.M.; Pan, H.Z. Artemisinin (qinghaosu): The role of intracellular hemin in its mechanism of antimalarial action. Mol. Biochem. Parasitol. 1991, 49, 181–189. [Google Scholar] [CrossRef]
- Krungkrai, S.R.; Yuthavong, Y. The antimalarial action on Plasmodium falciparum of qinghaosu and artesunate in combination with agents which modulate oxidant stress. Trans. R. Soc. Trop. Med. Hyg. 1987, 81, 710–714. [Google Scholar] [CrossRef]
- Ingram, K.; Yaremenko, I.A.; Krylov, I.B.; Hofer, L.; Terentev, A.O.; Keiser, J. Identification of antischistosomal leads by evaluating bridged 1,2,4,5-tetraoxanes, alphaperoxides, and tricyclic monoperoxides. J. Med. Chem. 2012, 55, 8700–8711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantawy, M.M.; Ali, H.F.; Rizk, M.Z. Therapeutic Effects of Allium sativum and Allium cepa in Schistosoma mansoni experimental infection. Rev. Inst. Med. Trop. Sao Paulo 2011, 53, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Lima, C.M.B.L.; Freitas, F.I.D.S.; de Morais, L.C.S.L.; Cavalcanti, M.G.D.S.; Da Silva, L.F.; Padilha, R.J.R.; Barbosa, C.G.S.; dos Santos, F.A.B.; Alves, L.C.; Diniz, M.D.F.F.M. Ultrastructural study on the morphological changes to male worms of Schistosoma mansoni after in vitro exposure to allicin. Rev. Soc. Bras. Med. Trop. 2011, 44, 327–330. [Google Scholar] [CrossRef] [PubMed]
- Riad, N.H.A.; Fares, N.H.; Mostafa, O.M.S.; Mahmoud, Y. The Effect of Garlic on Murine Schistosomiasis Mansoni: A Histological and Ultrastructural Study on the Ileum. Res. J. Med. Med. Sci. 2008, 3, 188–201. [Google Scholar]
- Riad, N.H.A.; Fares, N.H.; Mostafa, O.M.S.; Mahmoud, Y. The Effect of Garlic on Some Parasitological Parameters and On Hepatic Tissue Reactions in Experimental Schistosomiasis Mansoni. J. Appl. Sci. Res. 2007, 3, 949–960. [Google Scholar]
- Koshimizu, K.; Ohigashi, H.; Huffman, M.A. Use of Vernonia amygdalina by wild chimpanzee: Possible roles of its bitter and related constituents. Physiol. Behav. 1994, 56, 1209–1216. [Google Scholar] [CrossRef]
- Jisaka, M.; Kawanaka, M.; Sugiyama, H.; Takegawa, K.; Huffman, M.A.; Ohigashi, H.; Koshimizu, K. Antischistosomal activities of sesquiterpene lactones and steroid glucosides from Vernonia amygdalina, possibly used by wild chimpanzees against parasite-related diseases. Biosci. Biotechnol. Biochem. 1992, 56, 845–846. [Google Scholar] [CrossRef] [PubMed]
- Wiegrebe, W.; Kramer, W.J.; Shamma, M. The emetine alkaloids. J. Nat. Prod. 1984, 47, 397–408. [Google Scholar] [CrossRef]
- Akinboye, E.S. Biological Activities of Emetine. Open Nat. Prod. J. 2011, 4, 8–15. [Google Scholar] [CrossRef]
- Grollman, A.P. Structural Basis for Inhibition of Protein Synthesis By Emetine and Cycloheximide Based on an Analogy between Ipecac Alkaloids and Glutarimide Antibiotics. Proc. Natl. Acad. Sci. USA 1966, 56, 1867–1874. [Google Scholar] [CrossRef] [PubMed]
- Grollman, A.P. Inhibitors of Protein Biosynthesis V. Effects of Emetine on Protein and Nucleic Acid Biosynthesis in HeLa Cells. J. Biol. Chem. 1968, 243, 4089–4094. [Google Scholar] [PubMed]
- Lietman, P.S. Mitochondrial protein synthesis: Inhibition by emetine hydrochloride. Mol. Pharmacol. 1971, 7, 122–128. [Google Scholar] [PubMed]
- Blanc, F.; Nosny, Y. [Treatment of schistosomiasis with injections of 2-dehydro-emetine]. Presse Med. 1968, 76, 1419–1420. [Google Scholar] [PubMed]
- Alonso, D.F.; Farina, H.G.; Skilton, G.; Gabri, M.R.; de Lorenzo, M.S.; Gomez, D.E. Reduction of mouse mammary tumor formation and metastasis by lovastatin, an inhibitor of the mevalonate pathway of cholesterol synthesis. Breast Cancer Res. Treat. 1998, 50, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; Brown, M.S. Regulation of the mevalonate pathway. Nature 1990, 343, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Z.; Foster, L.; Bennett, J.L. Antischistosomal action of mevinolin: Evidence that 3-hydroxy-methylglutaryl-coenzyme a reductase activity in Schistosoma mansoni is vital for parasite survival. Naunyn. Schmiedebergs. Arch. Pharmacol. 1990, 342, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.M.; Coultas, K.A. Identification of plumbagin and sanguinarine as effective chemotherapeutic agents for treatment of schistosomiasis. Int. J. Parasitol. Drugs Drug Resist. 2013, 3, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K.B.; Reddy, G.A.K.; Joy, J.M.; Rasheed, A.; Dalith, D. Natural antifilarial drugs: A review. Int. J. Pharmacol. Toxicol. 2011, 1, 1–10. [Google Scholar]
- Lymphatic Filariasis. Available online: http://www.webcitation.org/6mVkfD6pl (accessed on 4 December 2016).
- Bulman, C.A.; Bidlow, C.M.; Lustigman, S.; Cho-Ngwa, F.; Williams, D.; Rascón, A.A.; Tricoche, N.; Samje, M.; Bell, A.; Suzuki, B.; et al. Repurposing auranofin as a lead candidate for treatment of lymphatic filariasis and onchocerciasis. PLoS Negl. Trop. Dis. 2015, 9, e0003534. [Google Scholar] [CrossRef] [PubMed]
- Misra, N.; Sharma, M.; Raj, K.; Dangi, A.; Srivastava, S.; Misra-Bhattacharya, S. Chemical constituents and antifilarial activity of Lantana camara against human lymphatic filariid Brugia malayi and rodent filariid Acanthocheilonema viteae maintained in rodent hosts. Parasitol. Res. 2007, 100, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Mathew, N.; Misra-Bhattacharya, S.; Perumal, V.; Muthuswamy, K. Antifilarial lead molecules isolated from Trachyspermum ammi. Molecules 2008, 13, 2156–2168. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.; Wei, D.; Du, Y. An alternative total synthesis of solamargine. Sci. China Chem. 2012, 55, 1247–1251. [Google Scholar] [CrossRef]
- Misra, S.; Verma, M.; Mishra, S.K.; Srivastava, S.; Lakshmi, V.; Misra-Bhattacharya, S. Gedunin and photogedunin of Xylocarpus granatum possess antifilarial activity against human lymphatic filarial parasite Brugia malayi in experimental rodent host. Parasitol. Res. 2011, 109, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Petare, S.; Shinda, G.; Goswami, K.; Reddy, M.V.R. Novel drug designing rationale against B rugia malayi microfilariae using herbal extracts. Asian Pac. J. Trop. Dis. 2010, 846–850. [Google Scholar] [CrossRef]
- Srinivasan, L.; Mathew, N.; Muthuswamy, K. In vitro antifilarial activity of glutathione S-transferase inhibitors. Parasitol. Res. 2009, 105, 1179–1182. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, R.S.; Goswami, K.; Hande, S.; Bhoj, P. Evolution of anti-filarial therapeutics : An overview. J. Microbiol. Antimicrob. Agents 2015, 1, 16–22. [Google Scholar]
- Hoerauf, A.; Specht, S.; Büttner, M.; Pfarr, K.; Mand, S.; Fimmers, R.; Marfo-Debrekyei, Y.; Konadu, P.; Debrah, A.Y.; Bandi, C.; et al. Wolbachia endobacteria depletion by doxycycline as antifilarial therapy has macrofilaricidal activity in onchocerciasis: A randomized placebo-controlled study. Med. Microbiol. Immunol. 2008, 197, 295–311. [Google Scholar] [CrossRef] [PubMed]
- Lakshmi, V.; Srivastava, S.; Kumar Mishra, S.; Misra, S.; Verma, M.; Misra-Bhattacharya, S. In vitro and in vivo antifilarial potential of marine sponge, Haliclona exigua (Kirkpatrick), against human lymphatic filarial parasite Brugia malayi: Antifilarial activity of H. exigua. Parasitol. Res. 2009, 105, 1295–1301. [Google Scholar] [CrossRef] [PubMed]
- Gupta, J.; Misra, S.; Mishra, S.K.; Srivastava, S.; Srivastava, M.N.; Lakshmi, V.; Misra-Bhattacharya, S. Antifilarial activity of marine sponge Haliclona oculata against experimental Brugia malayi infection. Exp. Parasitol. 2012, 130, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Dhananjeyan, M.R.; Milev, Y.P.; Kron, M.A.; Nair, M.G. Synthesis and activity of substituted anthraquinones against a human filarial parasite, Brugia malayi. J. Med. Chem. 2005, 48, 2822–2830. [Google Scholar] [CrossRef] [PubMed]





















© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheuka, P.M.; Mayoka, G.; Mutai, P.; Chibale, K. The Role of Natural Products in Drug Discovery and Development against Neglected Tropical Diseases. Molecules 2017, 22, 58. https://doi.org/10.3390/molecules22010058
Cheuka PM, Mayoka G, Mutai P, Chibale K. The Role of Natural Products in Drug Discovery and Development against Neglected Tropical Diseases. Molecules. 2017; 22(1):58. https://doi.org/10.3390/molecules22010058
Chicago/Turabian StyleCheuka, Peter Mubanga, Godfrey Mayoka, Peggoty Mutai, and Kelly Chibale. 2017. "The Role of Natural Products in Drug Discovery and Development against Neglected Tropical Diseases" Molecules 22, no. 1: 58. https://doi.org/10.3390/molecules22010058