Serum Albumin Binding and Esterase Activity: Mechanistic Interactions with Organophosphates

Abstract
:1. Introduction
2. Methods
2.1. Preparation of 3D-models of Ligands and Proteins
2.2. Molecular Docking Procedure and Dissociation Constant Estimation
2.3. Molecular Dynamic Simulation
3. Results
3.1. Esterase and Pseudoesterase Activity of Albumin: Theoretical Considerations on Kinetic Schemes and Constants
3.2. Albumin and Organophosphates: Molecular Modeling Studies
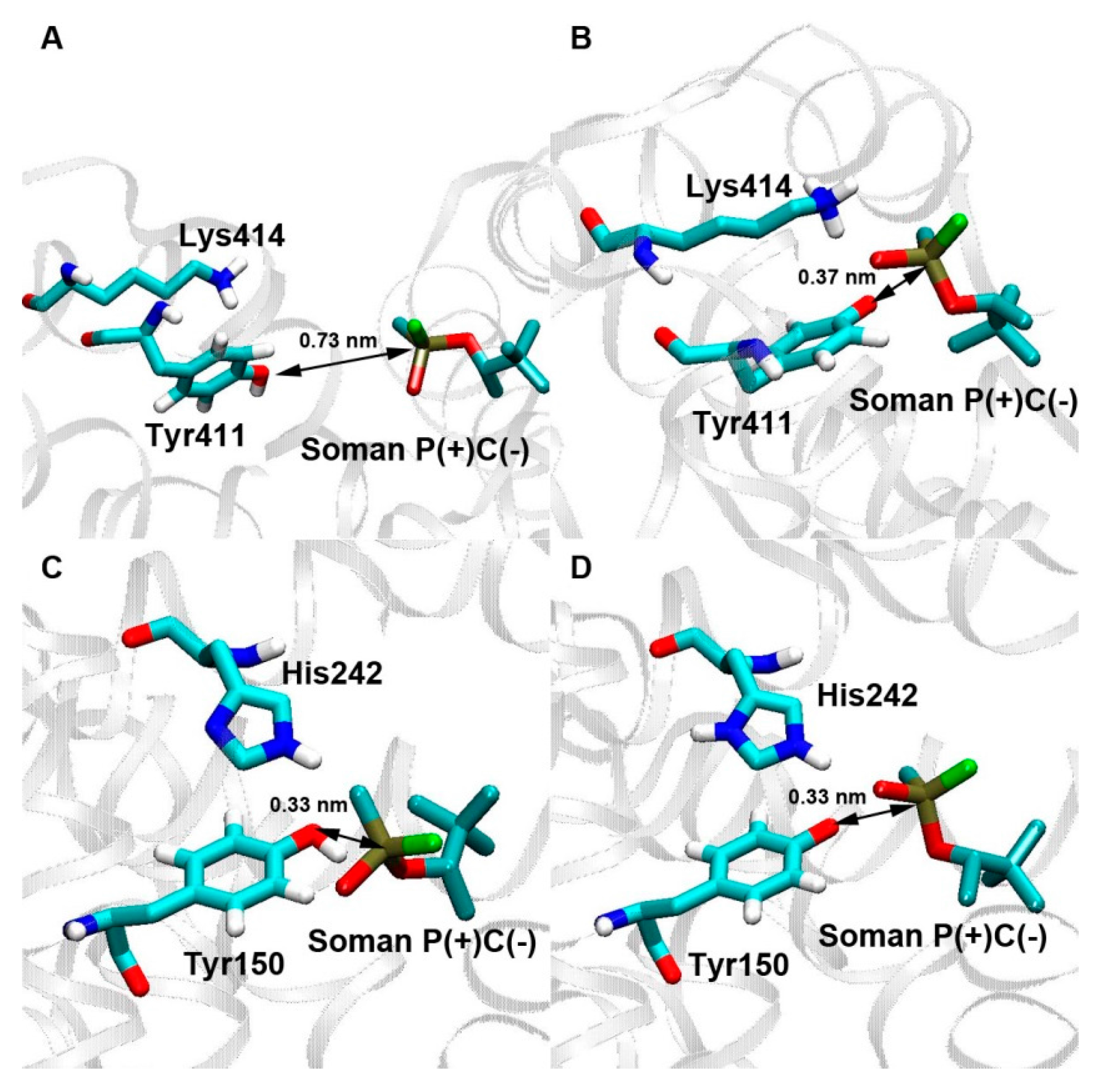
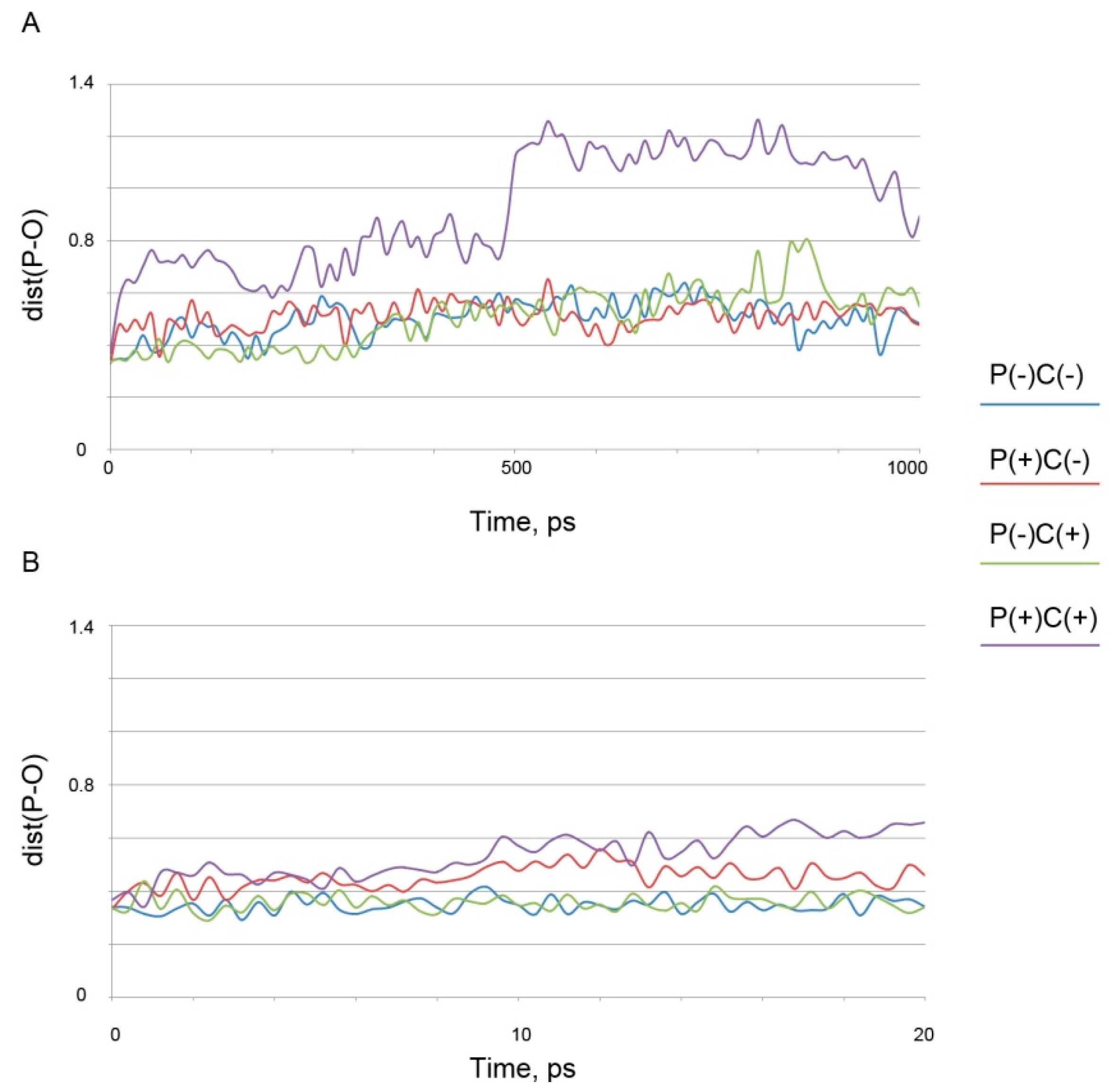
3.2.1. Studies on Interaction of Soman with Albumin
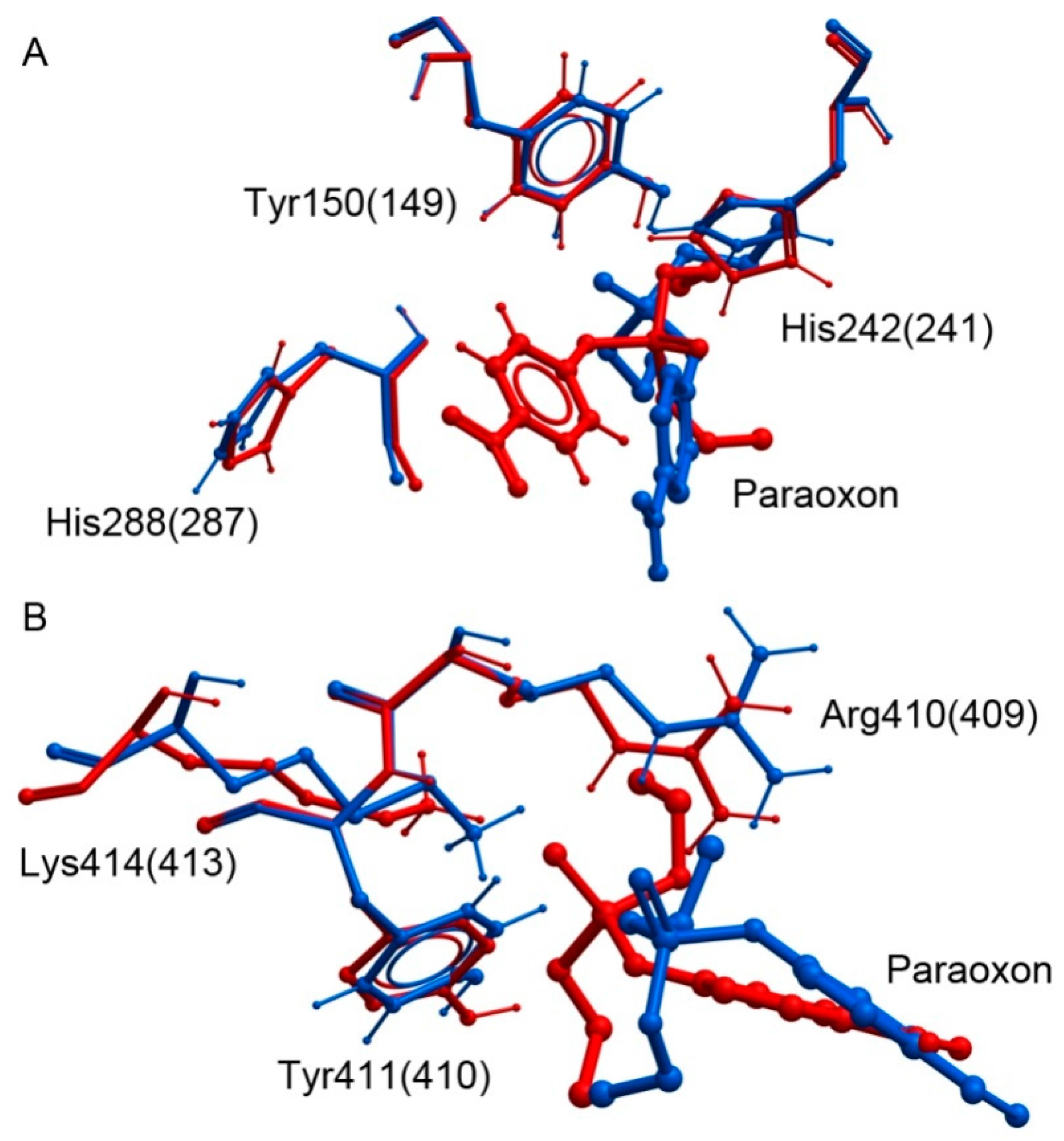
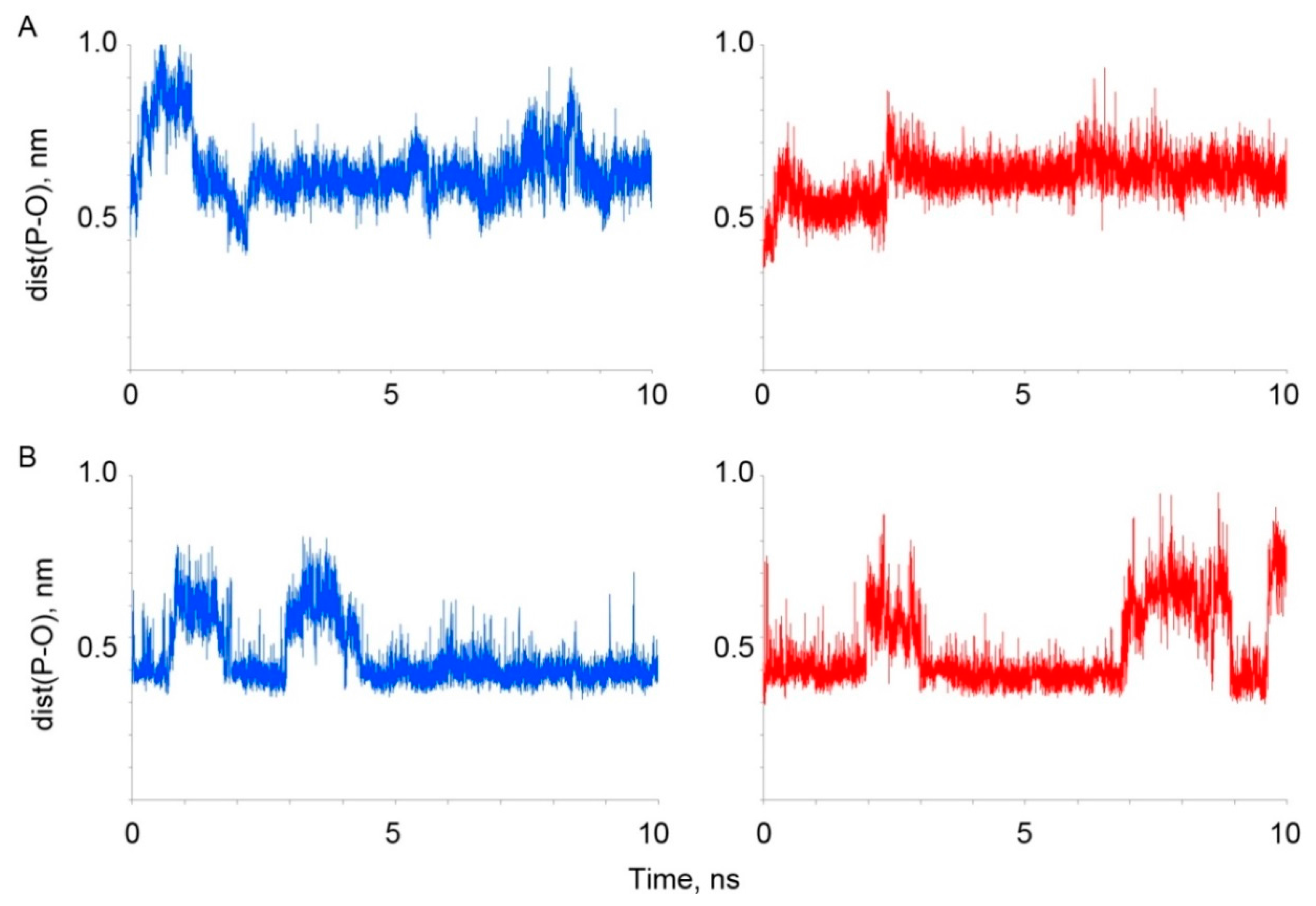
3.2.2. Comparative Analysis In Silico of Paraoxon Binding with Human and Bovine Serum Albumin
3.3. Substrate Specificity of Albumin and Potential for its Assignment to Enzyme Nomenclature
4. Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Macwilliam, J.A. Remarks on a new test for albumin and other proteids. Br. Med. J. 1891, 1, 837–840. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, J.F. A preliminary note on the fractional precipitation of the globulin and albumin of normal horse’s serum and diphtheric antitoxic serum, and the antitoxic strength of the precipitates. J. Exp. Med. 1899, 4, 649–654. [Google Scholar] [CrossRef] [PubMed]
- He, X.M.; Carter, D.C. Atomic structure and chemistry of human serum albumin. Nature 1992, 358, 209–215. [Google Scholar] [CrossRef]
- Sugio, S.; Kashima, A.; Mochizuki, S.; Noda, M.; Kobayashi, K. Crystal structure of human serum albumin at 2.5 Å resolution. Protein Eng. 1999, 12, 439–446. [Google Scholar] [CrossRef]
- Majorek, K.A.; Porebski, P.J.; Dayal, A.; Zimmerman, M.D.; Jablonska, K.; Stewart, A.J.; Chruszcz, M.; Minor, W. Structural and immunologic characterization of bovine, horse, and rabbit serum albumins. Mol. Immunol. 2012, 52, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Peters, T., Jr. All about albumin. In Biochemistry, Genetics, and Medical Applications; Academic Press Ltd.: London, UK, 1996. [Google Scholar]
- Bujacz, A. Structures of bovine, equine and leporine serum albumin. Acta Crystallogr. Sect. D 2012, 68, 1278–1289. [Google Scholar] [CrossRef] [PubMed]
- Ascenzi, P.; Bocedi, A.; Notari, S.; Fanali, G.; Fesce, R.; Fasano, M. Allosteric modulation of drug binding to human serum albumin. Mini Rev. Med. Chem. 2006, 6, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Ascenzi, P.; Fasano, M. Allostery in a monomeric protein: The case of human serum albumin. Biophys. Chem. 2010, 148, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Ghuman, J.; Zunszain, P.A.; Petitpas, I.; Bhattacharya, A.A.; Otagiri, M.; Curry, S. Structural basis of the drug-binding specificity of human serum albumin. J. Mol. Biol. 2005, 353, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Sand, K.M.; Bern, M.; Nilsen, J.; Noordzij, H.T.; Sandlie, I.; Andersen, J.T. Unraveling the Interaction between FcRn and Albumin: Opportunities for Design of Albumin-Based Therapeutics. Front. Immunol. 2015, 5, e00682. [Google Scholar] [CrossRef] [PubMed]
- Fasano, M.; Curry, S.; Terreno, E.; Galliano, M.; Fanali, G.; Narciso, P.; Notari, S.; Ascenzi, P. The extraordinary ligand binding properties of human serum albumin. IUBMB Life 2005, 57, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Casida, J.E.; Augustinsson, K.B. Reaction of plasma albumin with I-naphthyl N-methylcarbamate and certain other esters. Biochim. Biophys. Acta 1959, 36, 411–426. [Google Scholar] [CrossRef]
- Tildon, J.T.; Ogilvie, J.W. The esterase activity of bovine mercaptalbumin. The reaction of the protein with p-nitrophenyl acetate. J. Biol. Chem. 1972, 247, 1265–12671. [Google Scholar]
- Means, G.E.; Bender, M.L. Acetylation of human serum albumin by p-nitrophenyl acetate. Biochemistry 1975, 14, 4989–4994. [Google Scholar] [CrossRef] [PubMed]
- Tove, S.B. The esterolytic activity of serum albumin. Biochim. Biophys. Acta 1962, 5, 230–235. [Google Scholar] [CrossRef]
- Rainsford, K.D.; Ford, N.L.; Brooks, P.M.; Watson, H.M. Plasma aspirin esterases in normal individuals, patients with alcoholic liver disease and rheumatoid arthritis: Characterization and the importance of the enzymic components. Eur. J. Clin. Investig. 1980, 10, 413–420. [Google Scholar] [CrossRef]
- Dubois-Presle, N.; Lapicque, F.; Maurice, M.H.; Fournel-Gigleux, S.; Magdalou, J.; Abiteboul, M.; Siest, G.; Netter, P. Stereoselective esterase activity of human serum albumin toward ketoprofen glucuronide. Mol. Pharmacol. 1995, 47, 647–653. [Google Scholar] [PubMed]
- Kwon, C.H.; Maddison, K.; LoCastro, L.; Borch, R.F. Accelerated decomposition of 4-hydroxycyclophosphamide by human serum albumin. Cancer Res. 1987, 47, 1505–1508. [Google Scholar] [PubMed]
- Salvi, A.; Carrupt, P.; Mayer, J.M.; Testa, B. Esterase-like activity of human serum albumin toward prodrug esters of nicotinic acid. Drug Metab. Dispos. 1997, 25, 395–398. [Google Scholar] [PubMed]
- De Vriese, C.; Hacquebard, M.; Gregoire, F.; Carpentier, Y.; Delporte, C. Ghrelin interacts with human plasma lipoproteins. Endocrinology 2007, 148, 2355–2362. [Google Scholar] [CrossRef] [PubMed]
- Manoharan, I.; Boopathy, R. Diisopropylfluorophosphate-sensitive aryl acylamidase activity of fatty acid free human serum albumin. Arch. Biochem. Biophys. 2006, 452, 186–188. [Google Scholar] [CrossRef] [PubMed]
- Masson, P.; Froment, M.T.; Darvesh, S.; Schopfer, L.M.; Lockridge, O. Aryl acylamidase activity of human serum albumin with o-nitrotrifluoroacetanilide as the substrate. J. Enzym. Inhib. Med. Chem. 2007, 22, 463–469. [Google Scholar] [CrossRef]
- Sogorb, M.A.; Diaz Alejo, N.; Escudero, M.A.; Vilanova, E. Phosphotriesterase activity identified in purified serum albumins. Arch. Toxicol. 1998, 72, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Lockridge, O.; Xue, W.; Gaydess, A.; Grigoryan, H.; Ding, S.J.; Schopfer, L.M.; Hinrichs, S.H.; Masson, P. Pseudo-esterase activity of human albumin: Slow turnover on tyrosine 411 and stable acetylation of 82 residues including 59 lysines. J. Biol. Chem. 2008, 283, 22582–22590. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Nachon, F.; Froment, M.T.; Verdier, L.; Debouzy, J.C.; Brasme, B.; Gillon, E.; Schopfer, L.M.; Lockridge, O.; Masson, P. Binding and hydrolysis of soman by human serum albumin. Chem. Res. Toxicol. 2008, 21, 421–431. [Google Scholar] [CrossRef] [PubMed]
- John, H.; Breyer, F.; Thumfart, J.O.; Höchstetter, H.; Thiermann, H. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) for detection and identification of albumin phosphylation by organophosphorus pesticides and G- and V-type nerve agents. Anal. Bioanal. Chem. 2010, 398, 2677–2691. [Google Scholar] [CrossRef] [PubMed]
- Radilov, A.; Rembovskiy, V.; Rybalchenko, I.; Savelieva, E.; Podolskaya, E.; Babakov, V.; Ermolaeva, E.; Dulov, S.; Kuznetsov, S.; Mindukshev, I.; et al. Russian VX. In Handbook of the Toxicology of Chemical Warfare Agents; Gupta, R.C., Ed.; Academic Press/Elsevier: Amsterdam, the Netherlands, 2009; pp. 69–91. [Google Scholar]
- Hurst, R.; Bao, Y.; Ridley, S.; Williamson, G. Phospholipid hydroperoxide cysteine peroxidase activity of human serum albumin. Biochem. J. 1999, 338, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Cha, M.K.; Kim, I.H. Activation of thiol-dependent antioxidant activity of human serum albumin by alkaline pH is due to the B-like conformational change. Arch. Biochem. Biophys. 2000, 380, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Cha, M.K.; Kim, I.H. Disulfide between Cys392 and Cys438 of human serum albumin is redox-active, which is responsible for the thioredoxin-supported lipid peroxidase activity. Arch. Biochem. Biophys. 2006, 445, 19–25. [Google Scholar] [CrossRef]
- Fletcher, R.; Reeves, C.M. Function minimization by conjugate gradients. Comput. J. 1964, 7, 148–154. [Google Scholar] [CrossRef]
- HyperchemUsers Manual; Hypercube Inc.: Waterloo, ON, Canada, 1994.
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Bujacz, A.; Zielinski, K.; Sekula, B. Structural studies of bovine, equine, and leporine serum albumin complexes with naproxen. Proteins 2014, 82, 2199–2208. [Google Scholar] [CrossRef] [PubMed]
- Sekula, B.; Zielinski, K.; Bujacz, A. Crystallographic studies of the complexes of bovine and equine serum albumin with 3,5-diiodosalicylic acid. Int. J. Biol. Macromol. 2013, 60, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Grigera, J.R.; Straatsma, T.P. The missing term in effective pair potentials. J. Phys. Chem. 1987, 91, 6269–6271. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; di Nola, A.; van Gunsteren, W.F.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Bussi, G.; Zykova-Timan, T.; Parrinello, M. Isothermal-isobaric molecular dynamics using stochastic velocity rescaling. J. Chem. Phys. 2009, 130, 74101–74110. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.; Tymoczko, J.; Stryer, L. Biochemistry; W.H. Freeman and Company: New York, NY, USA; Basingstoke, UK, 2006. [Google Scholar]
- Dixon, M.; Webb, E.C. Enzymes, 3rd ed.; Ademic Press: New York, NY, USA, 1979. [Google Scholar]
- Junge, W.; Krisch, K. Current problems on the structure and classification of mammalian liver carboxylesterases (EC 3.1.1.1). Mol. Cell. Biochem. 1973, 1, 41–52. [Google Scholar] [CrossRef]
- Ascenzi, P.; Gioia, M.; Fanali, G.; Coletta, M.; Fasano, M. Pseudo-enzymatic hydrolysis of 4-nitrophenyl acetate by human serum albumin: pH-dependence of rates of individual steps. Biochem. Biophys. Res. Commun. 2012, 424, 451–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakurai, Y.; Ma, S.F.; Watanabe, H.; Yamaotsu, N.; Hirono, S.; Kurono, Y.; Kragh-Hansen, U.; Otagiri, M. Esterase-like activity of serum albumin: Characterization of its structural chemistry using p-nitrophenyl esters as substrates. Pharm. Res. 2004, 21, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Zdrazilová, P.; Stepánková, S.; Vránová, M.; Komers, K.; Komersová, A.; Cegan, A. Kinetics of total enzymatic hydrolysis of acetylcholine and acetylthiocholine. Z. Naturforsch. C 2006, 61, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Michaelis, L.; Menten, M. Die Kinetik der Invertinwirkung. Biochem. Z. 1913, 49, 333–369. [Google Scholar]
- Zaidi, N.; Ajmal, M.R.; Rabbani, G.; Ahmad, E.; Khan, R.H. A comprehensive insight into binding of hippuric acid to human serum albumin: A study to uncover its impaired elimination through hemodialysis. PLoS ONE 2013, 8, e71422. [Google Scholar] [CrossRef] [PubMed]
- Walker, C.H.; Mackness, M.I. Esterases: Problems of identification and classification. Biochem. Pharmacol. 1983, 32, 3265–3269. [Google Scholar] [CrossRef]
- Briggs, G.E.; Haldane, J.B. A Note on the Kinetics of Enzyme Action. Biochem. J. 1925, 19, 338–339. [Google Scholar] [CrossRef] [PubMed]
- Van Slyke, D.D.; Cullen, G.E. The Mode of Action of Urease and of Enzymes in General. J. Biol. Chem. 1914, 19, 141–180. [Google Scholar]
- Morrison, J.F. Kinetics of the reversible inhibition of enzyme-catalysed reactions by tight-binding inhibitors. Biochim. Biophys. Acta 1969, 185, 269–286. [Google Scholar] [CrossRef]
- Flach, E.H.; Schnell, S. Use and abuse of the quasi-steady-state approximation. Syst. Biol. (Stevenage) 2006, 153, 187–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuzmic, P. Application of the Van Slyke-Cullen irreversible mechanism in the analysis of enzymatic progress curves. Anal. Biochem. 2009, 394, 287–289. [Google Scholar] [CrossRef] [PubMed]
- Aldridge, W.N. Serum esterases. I. Two types of esterase (A and B) hydrolysingp-nitrophenyl acetate, propionate and butyrate, and a method for their determination. Biochem. J. 1953, 53, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Aldridge, W.N. Serum esterases. II. An enzyme hydrolysing diethyl p-nitrophenyl phosphate (E600) and its identity with the A-esterase of mammalian sera. Biochem. J. 1953, 53, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Suji, G.; Sivakami, S. Malondialdehyde, a lipid-derived aldehyde alters the reactivity of Cys34 and the esterase activity of serum albumin. Toxicol. In Vitro 2008, 22, 618–624. [Google Scholar] [CrossRef] [PubMed]
- Ascenzi, P.; Fasano, M. Pseudo-enzymatic hydrolysis of 4-nitrophenyl myristate by human serum albumin. Biochem. Biophys. Res. Commun. 2012, 422, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.K.; Kregar, I.; Turk, V.; Rupley, J.A. Lysozyme-catalyzed reaction of the N-acetylglucosaminehexasaccharide. Dependence of rate on pH. J. Biol. Chem. 1973, 248, 4786–4792. [Google Scholar] [PubMed]
- Li, B.; Sedlacek, M.; Manoharan, I.; Boopathy, R.; Duysen, E.G.; Masson, P.; Lockridge, O. Butyrylcholinesterase, paraoxonase, and albumin esterase, but not carboxylesterase, are present in human plasma. Biochem. Pharmacol. 2005, 70, 1673–1684. [Google Scholar] [CrossRef] [PubMed]
- Benschop, H.P.; de Jong, L.P. Toxicokinetics of nerve agents. In Chemical Warfare Agents: Toxicity at Low Levels; Somani, S.M., Romano, J.A., Jr., Eds.; CRC Press: Boca Raton, FL, USA, 2001; pp. 25–82. [Google Scholar]
- Sogorb, M.A.; Vilanova, E. Enzymes involved in the detoxification of organophosphorus, carbamate and pyrethroid insecticides through hydrolysis. Toxicol. Lett. 2002, 128, 215–228. [Google Scholar] [CrossRef]
- Sogorb, M.A.; García-Argüelles, S.; Carrera, V.; Vilanova, E. Serum albumin is as efficient as paraxonase in the detoxication of paraoxon at toxicologically relevant concentrations. Chem. Res. Toxicol. 2008, 21, 1524–1529. [Google Scholar] [CrossRef] [PubMed]
- Sogorb, M.A.; Vilanova, E. Serum albumins and detoxication of anti-cholinesterase agents. Chem. Biol. Interact. 2010, 187, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Kua, J.; Zhang, Y.; McCammon, A. Studying enzyme binding specificity in acetylcholinesterase using a combined molecular dynamics and multiple docking approach. J. Am. Chem. Soc. 2002, 124, 8260–8267. [Google Scholar] [CrossRef] [PubMed]
- Zhan, C.G.; Zheng, F.; Landry, D.W. Fundamental reaction mechanism for cocaine hydrolysis in human butyrylcholinesterase. J. Am. Chem. Soc. 2003, 125, 2462–2474. [Google Scholar] [CrossRef] [PubMed]
- Thrige, D.G.; Buur, J.R.; Jørgensen, F.S. Substrate binding and catalytic mechanism in phospholipase C from Bacillus cereus: A molecular mechanics and molecular dynamics study. Biopolymers 1997, 42, 319–336. [Google Scholar] [CrossRef]
- Moralev, S.N.; Rozengart, E.V. Comparative Enzymology of Cholinesterases; International University Line: La Jolla, CA, USA, 2007. [Google Scholar]
- Benschop, H.P.; Konings, C.A.; van Genderen, J.; de Jong, L.P. Isolation, in vitro activity, and acute toxicity in mice of the four stereoisomers of soman. Fundam. Appl. Toxicol. 1984, 4, 84–95. [Google Scholar] [CrossRef]
- Sant’Anna, C.M.R.; Viana, A.D.; do Nascimento, N.M. A semiempirical study of acetylcholine hydrolysis catalyzed by Drosophila melanogaster acetylcholinesterase. Bioorg. Chem. 2006, 34, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Ekici, O.D.; Paetzel, M.; Dalbey, R.E. Unconventional serine proteases: Variations on the catalytic Ser/His/Asp triad configuration. Protein Sci. 2008, 17, 2023–2037. [Google Scholar] [CrossRef] [PubMed]
- Read, R.W.; Riches, J.R.; Stevens, J.A.; Stubbs, S.J.; Black, R.M. Biomarkers of organophosphorus nerve agent exposure: Comparison of phosphylated butyrylcholinesterase and phosphylated albumin after oxime therapy. Arch. Toxicol. 2010, 84, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Sogorb, M.A.; Alvarez-Escalante, C.; Carrera, V.; Vilanova, E. An in vitro approach for demonstrating the critical role of serum albumin in the detoxication of the carbamate carbaryl at in vivo toxicologically relevant concentrations. Arch. Toxicol. 2007, 81, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Zhang, A.; Liu, W. Binding of phenthoate to bovine serum albumin and reduced inhibition on acetylcholinesterase. Pestic. Biochem. Physiol. 2007, 88, 176–180. [Google Scholar] [CrossRef]
- Huang, B.X.; Kim, H.Y.; Dass, C. Probing three-dimensional structure of bovine serum albumin by chemical cross-linking and mass spectrometry. J. Am. Soc. Mass Spectrom. 2004, 15, 1237–1247. [Google Scholar] [CrossRef] [PubMed]
- Kohita, H.; Matsushita, Y.; Moriguchi, I. Binding of carprofen to human and bovine serum albumins. Chem. Pharm. Bull. (Tokyo) 1994, 42, 937–940. [Google Scholar] [CrossRef] [PubMed]
- Antoni, G.; Casagli, M.C.; Bigio, M.; Borri, G.; Neri, P. Different interactions of human and bovine serum albumin with Cibacron Blue and Blue Dextran. Ital. J. Biochem. 1982, 31, 100–106. [Google Scholar] [PubMed]
- Brown, N.A.; Müller, W.E. Binding of coumarin anticoagulants to human and bovine serum albumin. Circular dichroism studies. Pharmacology 1978, 17, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, F.; Bordbar, A.K.; Divsalar, A.; Mohammadi, K.; Saboury, A.A. Analysis of binding interaction of curcumin and diacetylcurcumin with human and bovine serum albumin using fluorescence and circular dichroism spectroscopy. Protein J. 2009, 28, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Fonda, M.L.; Trauss, C.; Guempel, U.M. The binding of pyridoxal 5'-phosphate to human serum albumin. Arch. Biochem. Biophys. 1991, 288, 79–86. [Google Scholar] [CrossRef]
- Silva, D.; Cortez, C.M.; Cunha-Bastos, J.; Louro, S.R. Methyl parathion interaction with human and bovine serum albumin. Toxicol. Lett. 2004, 147, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z. Interaction of ergosterol with bovine serum albumin and human serum albumin by spectroscopic analysis. Mol. Biol. Rep. 2012, 39, 9493–9508. [Google Scholar] [CrossRef] [PubMed]
- Belinskaia, D.A.; Shmurak, V.I.; Prokof’eva, D.S.; Goncharov, N.V. Serum albumin: Search for new sites with esterase activity according to molecular modeling data. Bioorg. Khimiia 2014, 40, 541–549. [Google Scholar] [PubMed]
- Belinskaya, D.A.; Taborskaya, K.I.; Goncharov, N.V. Modulation of the sites of albumin interaction with paraoxon by fatty acids: Analysis using molecular modeling methods. Bioorg. Khimiia 2017, 43, 347–356. [Google Scholar]
- Belinskaya, D.A.; Shmurak, V.I.; Prokofieva, D.S.; Goncharov, N.V. Investigation of soman binding with albumin by a molecular modeling method. Toksikol. Vestnik (Toxicol. Bull.) 2012, 116, 13–19. [Google Scholar]
- Goncharov, N.V.; Belinskaia, D.A.; Razygraev, A.V.; Ukolov, A.I. On the Enzymatic Activity of Albumin. Bioorg. Khimiia 2015, 41, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Williams, N.H.; Harrison, J.M.; Read, R.W.; Black, R.M. Phosphylated tyrosine in albumin as a biomarker of exposure to organophosphorus nerve agents. Arch. Toxicol. 2007, 81, 627–639. [Google Scholar] [CrossRef] [PubMed]
- Kragh-Hansen, U. Molecular and practical aspects of the enzymatic properties of human serum albumin and of albumin-ligand complexes. Biochim. Biophys. Acta 2013, 1830, 5535–5544. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, A.O.; Jacobsen, J. Reactivity of the thiol group in human and bovine albumin at pH 3–9, as measured by exchange with 2,2′-dithiodipyridine. Eur. J. Biochem. 1980, 106, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.P.; Phillips, M.; McPherson, R.A.; Hensley, P. Serum albumin and the metabolism of disulfiram. Biochem. Pharmacol. 1986, 35, 3341–3347. [Google Scholar] [CrossRef]
- Cha, M.K.; Kim, I.H. Glutathione-linked thiol peroxidase activity of human serum albumin: A possible antioxidant role of serum albumin in blood plasma. Biochem. Biophys. Res. Commun. 1996, 222, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Kim, I.H. Thioredoxin-linked lipid hydroperoxide peroxidase activity of human serum albumin in the presence of palmitoyl coenzyme A. Free Radic. Biol. Med. 2001, 30, 327–333. [Google Scholar] [CrossRef]
- Roche, M.; Rondeau, P.; Singh, N.R.; Tarnus, E.; Bourdon, E. The antioxidant properties of serum albumin. FEBS Lett. 2008, 582, 1783–1787. [Google Scholar] [CrossRef] [PubMed]
- Iwao, Y.; Ishima, Y.; Yamada, J.; Noguchi, T.; Kragh-Hansen, U.; Mera, K.; Honda, D.; Suenaga, A.; Maruyama, T.; Otagiri, M. Quantitative evaluation of the role of cysteine and methionine residues in the antioxidant activity of human serum albumin using recombinant mutants. IUBMB Life 2012, 64, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Kato, R.; Akiyama, M.; Kawakami, H.; Komatsu, T. Superoxide dismutase activity of the naturally occurring human serum albumin-copper complex without hydroxyl radical formation. Chem. Asian J. 2014, 1, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Gryzunov, Y.A.; Arroyo, A.; Vigne, J.-L.; Zhao, Q.; Tyurin, V.A.; Hubel, C.A.; Gandley, R.E.; Vladimirov, Y.A.; Taylor, R.N.; Kagan, V.E. Binding of fatty acids facilitates oxidation of cysteine-34 and converts copper-albumin complexes from antioxidants to prooxidants. Arch. Biochem. Biophys. 2003, 413, 53–66. [Google Scholar] [CrossRef]
- Blanco, R.A.; Ziegler, T.R.; Carlson, B.A.; Cheng, P.-Y.; Park, Y.; Cotsonis, G.A.; Accardi, C.J.; Jones, D.P. Diurnal variation in glutathione and cysteine redox states in human plasma. Am. J. Clin. Nutr. 2007, 86, 1016–1023. [Google Scholar] [PubMed]
- Ursini, F.; Maiorino, M.; Roveri, A. Phospholipid hydroperoxide glutathione peroxidase (PHGPx): More than an antioxidant enzyme? Biomed. Environ. Sci. 1997, 10, 327–332. [Google Scholar] [PubMed]
- Imai, H.; Nakagawa, Y. Biological significance of phospholipid hydroperoxide glutathione peroxidase (PHGPx, GPx4) in mammalian cells. Free Radic. Biol. Med. 2003, 34, 145–169. [Google Scholar] [CrossRef]
- Yoo, M.H.; Gu, X.; Xu, X.M.; Kim, J.Y.; Carlson, B.A.; Patterson, A.D.; Cai, H.; Gladyshev, V.N.; Hatfield, D.L. Delineating the role of glutathione peroxidase 4 in protecting cells against lipid hydroperoxide damage and in Alzheimer’s disease. Antioxid. Redox Signal. 2010, 12, 819–827. [Google Scholar] [CrossRef] [PubMed]
- BRENDA. The Comprehensive Enzyme Information System. Available online: http://www.brenda-enzymes.org/ (accessed on 30 May 2017).
- Fischer, K.; Kettunen, J.; Würtz, P.; Haller, T.; Havulinna, A.S.; Kangas, A.J.; Soininen, P.; Esko, T.; Tammesoo, M.L.; Mägi, R.; et al. Biomarker profiling by nuclear magnetic resonance spectroscopy for the prediction of all-cause mortality: An observational study of 17,345 persons. PLoS Med. 2014, 11, e1001606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georges, H.; Presle, N.; Buronfosse, T.; Fournel-Gigleux, S.; Netter, P.; Magdalou, J.; Lapicque, F. In Vitro stereoselective degradation of carprofen glucuronide by human serum albumin. Characterization of sites and reactive amino acids. Chirality 2000, 12, 53–62. [Google Scholar] [CrossRef]
- Williams, A.M.; Dickinson, R.G. Studies on the reactivity of acyl glucuronides—VI. Modulation of reversible and covalent interaction of diflunisal acyl glucuronide and its isomers with human plasma protein in vitro. Biochem. Pharmacol. 1994, 47, 457–467. [Google Scholar] [CrossRef]
- Drmanovic, Z.; Voyatzi, S.; Kouretas, D.; Sahpazidou, D.; Papageorgiou, A.; Antonoglou, O. Albumin possesses intrinsic enolase activity towards dihydrotestosterone which can differentiate benign from malignant breast tumors. Anticancer Res. 1999, 19, 4113–4124. [Google Scholar] [PubMed]
- Matsushita, S.; Isima, Y.; Chuang, V.T.G.; Watanabe, H.; Tanase, S.; Maruyama, T.; Otagiri, M. Functional analysis of recombinant human serum albumin domains for pharmaceutical applications. Pharm. Res. 2004, 21, 1924–1932. [Google Scholar] [CrossRef] [PubMed]
- Gresner, P.; Dolník, M.; Waczulíková, I.; Bryszewska, M.; Sikurová, L.; Watala, C. Increased blood plasma hydrolysis of acetylsalicylic acid in type 2 diabetic patients: A role of plasma esterases. Biochim. Biophys. Acta 2006, 1760, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Liyasova, M.S.; Schopfer, L.M.; Lockridge, O. Reaction of human albumin with aspirin in vitro: Mass spectrometric identification of acetylated lysines 199, 402, 519, and 545. Biochem. Pharmacol. 2010, 79, 784–791. [Google Scholar] [CrossRef] [PubMed]
- Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (NC-IUBMB). The Enzyme List, Class 3—Hydrolases. Generated from the ExplorEnz database. September 2010. Available online: http://www.enzyme-database.org/downloads/ec3.pdf (accessed on 30 May 2017).
- Schomburg, D.; Schomburg, I. Springer Handbook of Enzymes. EC Number Index; Springer-Verlag: Heidelberg/Berlin, Germany; New York, NY, USA, 2013. [Google Scholar]
- Kurdyukov, I.D.; Shmurak, V.I.; Nadeev, A.D.; Voitenko, N.G.; Prokofieva, D.S.; Goncharov, N.V. “Esterase status” of an organism at exposure by toxic substances and pharmaceuticals. Toksikol. Vestnik (Toxicol. Bull.) 2012, 6, 6–13. [Google Scholar]
- Devonshire, A.L.; Moores, G.D. A carboxylesterase with broad substrate specificity causes organ-ophosphorus, carbamate and pyrethroid resistance in peach-potato aphids (myzuspersicae). Pestic. Biochem. Physiol. 1982, 18, 235–246. [Google Scholar] [CrossRef]
- Augustinsson, K.B.; Heimburger, G. Enzymatic hydrolysis of organophosphorus compounds. I. Occurrence of enzymes hydrolysing dimethyl-amido-ethoxy-phosphoryl cyanide (Tabun). Acta Chem. Scand. 1954, 8, 753–761. [Google Scholar] [CrossRef]
- Augustinsson, K.B.; Heimburger, G. Enzymatic hydrolysis of organophosphorus compounds. II. Analysis of reaction products in experiments with tabun and some properties of blood plasma tabunase. Acta Chem. Scand. 1954, 8, 762–767. [Google Scholar] [CrossRef]
- Augustinsson, K.B.; Heimburger, G. Enzymatic hydrolysis of organophosphorus compounds. IV. Specificity studies. Acta Chem. Scand. 1954, 8, 1533–1541. [Google Scholar] [CrossRef]
- Cohen, J.A.; Warringa, M.G.P.J. Purification and properties of dialkylfluorophosphatase. Biochim. Biophys. Acta 1957, 26, 29–39. [Google Scholar] [CrossRef]
- Mounter, L.A. The Enzymes; Boyer, P.D., Lardy, H., Myrback, K., Eds.; Academic Press: New York, NY, USA, 1960; pp. 541–550. [Google Scholar]
- Reiner, E.; Aldridge, W.N.; Hoskin, C.G. Enzymes Hydrolysing Organophosphorus Compounds; Ellis Horwood Ltd.: Chichester, UK, 1989. [Google Scholar]
- Davies, H.G.; Richter, R.J.; Keifer, M.; Broomfield, C.A.; Sowalla, J.; Furlong, C.E. The effect of the human serum paraoxonase polymorphism is reversed with diazoxon, soman and sarin. Nat. Genet. 1996, 14, 334–336. [Google Scholar] [CrossRef] [PubMed]
- Aviram, M.; Vaya, J. Paraoxonase 1 activities, regulation, and interactions with atherosclerotic lesion. Curr. Opin. Lipidol. 2013, 24, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Gan, K.N.; Smolen, A.; Eckerson, H.W.; La Du, B.N. Purification of human serum paraoxonase/arylesterase. Evidence for one esterase catalyzing both activities. Drug Metab. Dispos. 1991, 19, 100–106. [Google Scholar] [PubMed]
- Jakubowski, H. Calcium-dependent human serum homocysteine thiolactone hydrolase. A protective mechanism against protein N-homocysteinylation. J. Biol. Chem. 2000, 275, 3957–3962. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H.; Ambrosius, W.T.; Pratt, J.H. Genetic determinants of homocysteine thiolactonase activity in humans: Implications for atherosclerosis. FEBS Lett. 2001, 491, 35–39. [Google Scholar] [CrossRef]
- Furlong, C.E.; Richter, R.J.; Chaplineand, C.J.; Crabb, W. Purification of rabbit and human serum paraoxonase. Biochemistry 1991, 30, 10133–10140. [Google Scholar] [CrossRef] [PubMed]
- Hassett, C.; Richter, R.J.; Humbert, R.; Chapline, C.; Crabb, J.W.; Omiencinski, C.J.; Furlong, C.E. Characterization of cDNA clones encoding rabbit and human serum paraoxonase: The mature protein retains its signal sequence. Biochemistry 1991, 30, 10141–10149. [Google Scholar] [CrossRef] [PubMed]
- Khersonsky, O.; Tawfik, D.S. The histidine 115-histidine 134 dyad mediates the lactonase activity of mammalian serum paraoxonases. J. Biol. Chem. 2006, 281, 7649–7656. [Google Scholar] [CrossRef] [PubMed]
- Acay, A.; Erdenen, F.; Altunoglu, E.; Erman, H.; Muderrisoglu, C.; Korkmaz, G.G.; Gelisgen, R.; Tabak, O.; Uzun, H. Evaluation of serum paraoxonase and arylesterase activities in subjects with asthma and chronic obstructive lung disease. Clin. Lab. 2013, 59, 1331–1337. [Google Scholar] [CrossRef] [PubMed]
- Atli, M. Hydroperoxide levels in adult football players after three days football tournament. Afr. Health Sci. 2013, 13, 565–570. [Google Scholar] [PubMed]
- Kati, C.; Karadas, S.; Aslan, M.; Gonullu, H.; Duran, L.; Demir, H. Serum paraoxonase and arylesterase activities and oxidative stress levels in patients with SSRI intoxication. J. Membr. Biol. 2014, 247, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Walker, C.H. The classification of esterases which hydrolyse organophosphates: Recent developments. Chem. Biol. Interact. 1993, 87, 17–24. [Google Scholar] [CrossRef]
- Sogorb, M.A.; Carrera, V.; Vilanova, E. Hydrolysis of carbaryl by human serum albumin. Arch. Toxicol. 2004, 78, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Pen, J.; Beintema, J.J. Nomenclature of esterases. Biochem. J. 1986, 240, 933. [Google Scholar] [CrossRef] [PubMed]
- McDonald, A.G.; Tipton, K.F. Fifty-five years of enzyme classification: advances and difficulties. FEBS J. 2014, 281, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Snow, T.A.; Kemper, R.A.; Jepson, G.W. Binding of perfluorooctanoic acid to rat and human plasma proteins. Chem. Res. Toxicol. 2003, 16, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Goncharov, N.V.; Terpilovsky, M.A.; Shmurak, V.I.; Belinskaia, D.A.; Avdonin, P.V. Comparative analysis of esterase and paraoxonase activity of different types of albumin. [Article in Russian]. Zh Evol. Biokhim Fiziol. 2017, 53, 241–250. [Google Scholar]
- Aubry, A.F.; Markoglou, N.; McGann, A. Comparison of drug binding interactions on human, rat and rabbit serum albumin using high-performance displacement chromatography. Comp. Biochem. Physiol. C Pharmacol. Toxicol. Endocrinol. 1995, 112, 257–266. [Google Scholar] [CrossRef]
- Frandsen, P.C.; Brodersen, R. Bilirubin/rat serum albumin interaction. Acta Chem. Scand. B 1986, 40, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Vyas, V.K.; Ukawala, R.D.; Ghate, M.; Chintha, C. Homology Modeling: A Fast Tool for Drug Discovery: Current Perspectives. Indian J. Pharm. Sci. 2012, 74, 1–17. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not apply. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
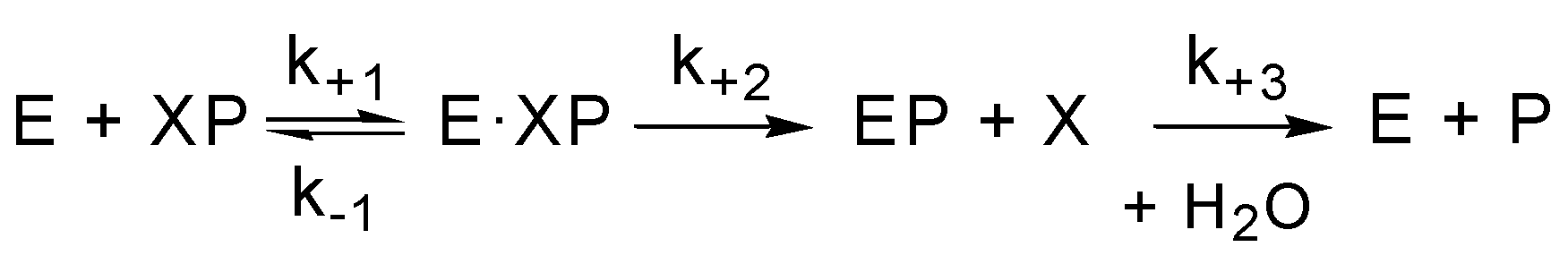{kind=link}
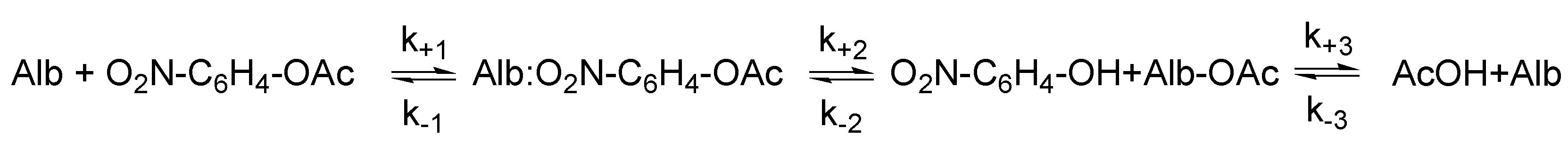{kind=link}
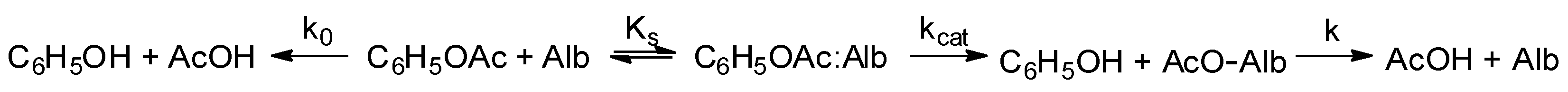{kind=link}
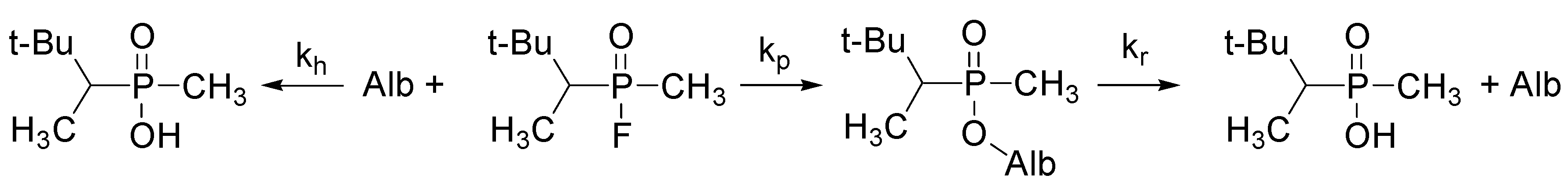{kind=link}
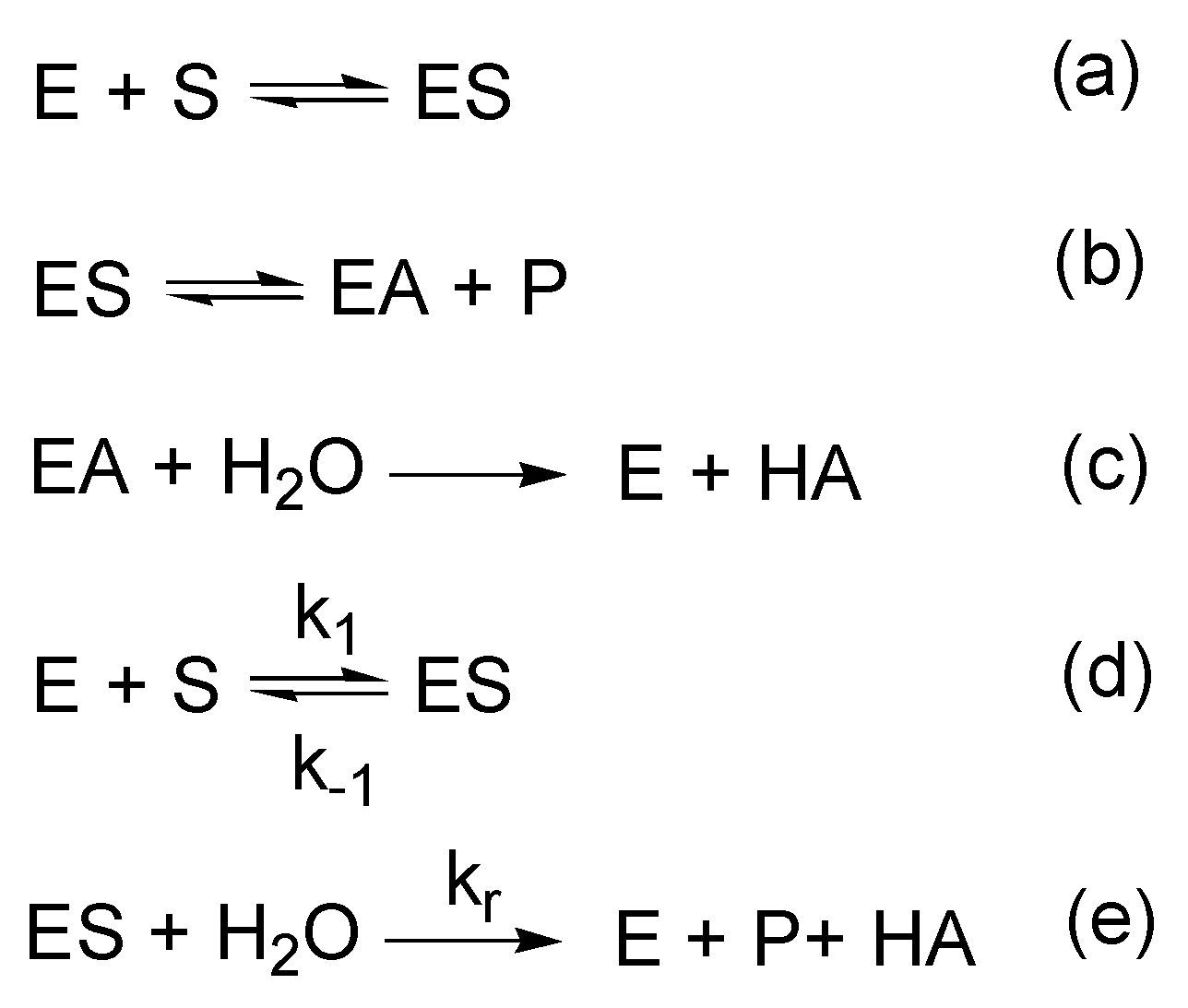{kind=link}
| Binding Site | Kd, μM | Dist (P-O(N)), nm | Amino Acid Closest to the Ligand | Distance to the Closest Amino Acid, nm | Acetylation by p-Nitrophenyl Acetate [25] |
|---|---|---|---|---|---|
| Tyr411 | 601 ± 26 | 0.72 | - | - | + |
| Tyr150 | 280 ± 3 | 0.36 | - | - | - |
| Ser192 | 581 ± 10 | 1.06 | Ser193 | 0.38 | + |
| Ser193 | 518 ± 14 | 0.38 | - | - | - |
| Thr243 | 290 ± 6 | 1.28 | Tyr150 | 0.35 | - |
| Ser287 | 295 ± 15 | 0.77 | Tyr150 | 0.36 | + |
| Lys402 | 737 ± 27 | 0.37 | - | - | + |
| Ser454 | 572 ± 17 | 1.14 | Tyr411 | 0.71 | + |
| Thr540 | 759 ± 21 | 0.87 | Lys402 | 0.37 | + |
| Binding Site | Binding Parameters | Stereoisomers of Soman | |||
|---|---|---|---|---|---|
| P(−)C(−) | P(+)C(−) | P(−)C(+) | P(+)C(+) | ||
| Ser193 | Kd, μM | 518 ± 14 | 598 ± 5 | 787 ± 27 | 633 ± 7 |
| dist(P-O(N)), nm | 0.38 | 0.41 | 0.37 | 0.40 | |
| Lys402 | Kd, μM | 737 ± 27 | 902 ± 45 | 763 ± 25 | 1031 ± 70 |
| dist(P-O(N)), nm | 0.37 | 0.43 | 0.37 | 0.55 | |
| Binding Site HSA(BSA) | Binding Parameters | HSA | BSA |
|---|---|---|---|
| Tyr150 (149) | Kd, μM | 4 ± 0.3 | 9 ± 0.7 |
| dist(P-O), nm | 0.36 | 0.37 | |
| Tyr411 (410) | Kd, μM | 87 ± 5 | 13 ± 1 |
| dist(P-O), nm | 0.37 | 0.35 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goncharov, N.V.; Belinskaia, D.A.; Shmurak, V.I.; Terpilowski, M.A.; Jenkins, R.O.; Avdonin, P.V. Serum Albumin Binding and Esterase Activity: Mechanistic Interactions with Organophosphates. Molecules 2017, 22, 1201. https://doi.org/10.3390/molecules22071201
Goncharov NV, Belinskaia DA, Shmurak VI, Terpilowski MA, Jenkins RO, Avdonin PV. Serum Albumin Binding and Esterase Activity: Mechanistic Interactions with Organophosphates. Molecules. 2017; 22(7):1201. https://doi.org/10.3390/molecules22071201
Chicago/Turabian StyleGoncharov, Nikolay V., Daria A. Belinskaia, Vladimir I. Shmurak, Maxim A. Terpilowski, Richard O. Jenkins, and Pavel V. Avdonin. 2017. "Serum Albumin Binding and Esterase Activity: Mechanistic Interactions with Organophosphates" Molecules 22, no. 7: 1201. https://doi.org/10.3390/molecules22071201