Interface Characterization Between Polyethylene/ Silica in Engineered Cementitious Composites by Molecular Dynamics Simulation

Abstract
:1. Introduction
2. Details of the Simulation
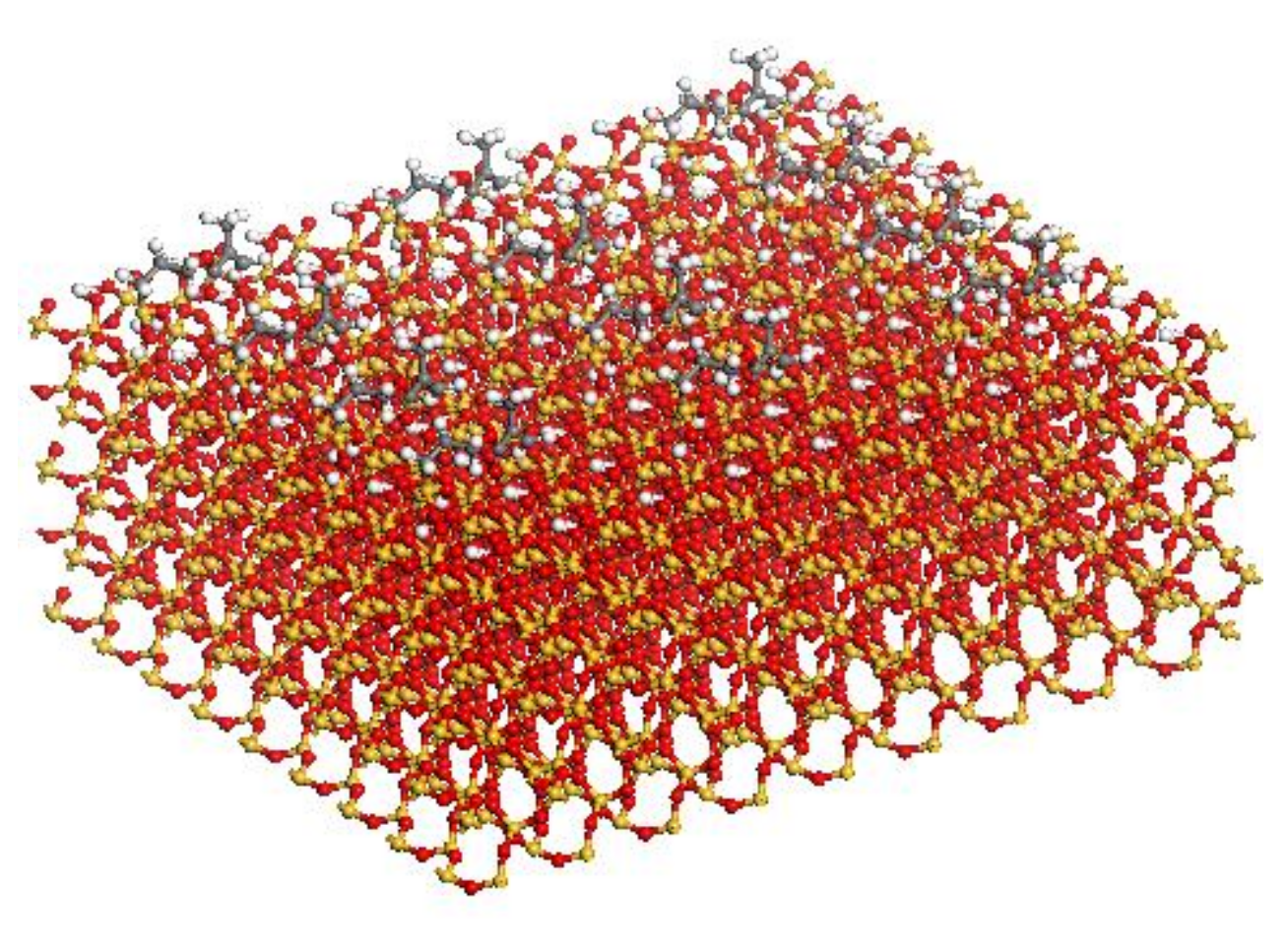
2.1. MD Models
2.2. Simulation of the Tensile Deformation of the Interfaces
3. Results and Discussion
3.1. Thermodynamic Work of Adhesion
3.2. Tensile Elongation of the Interface
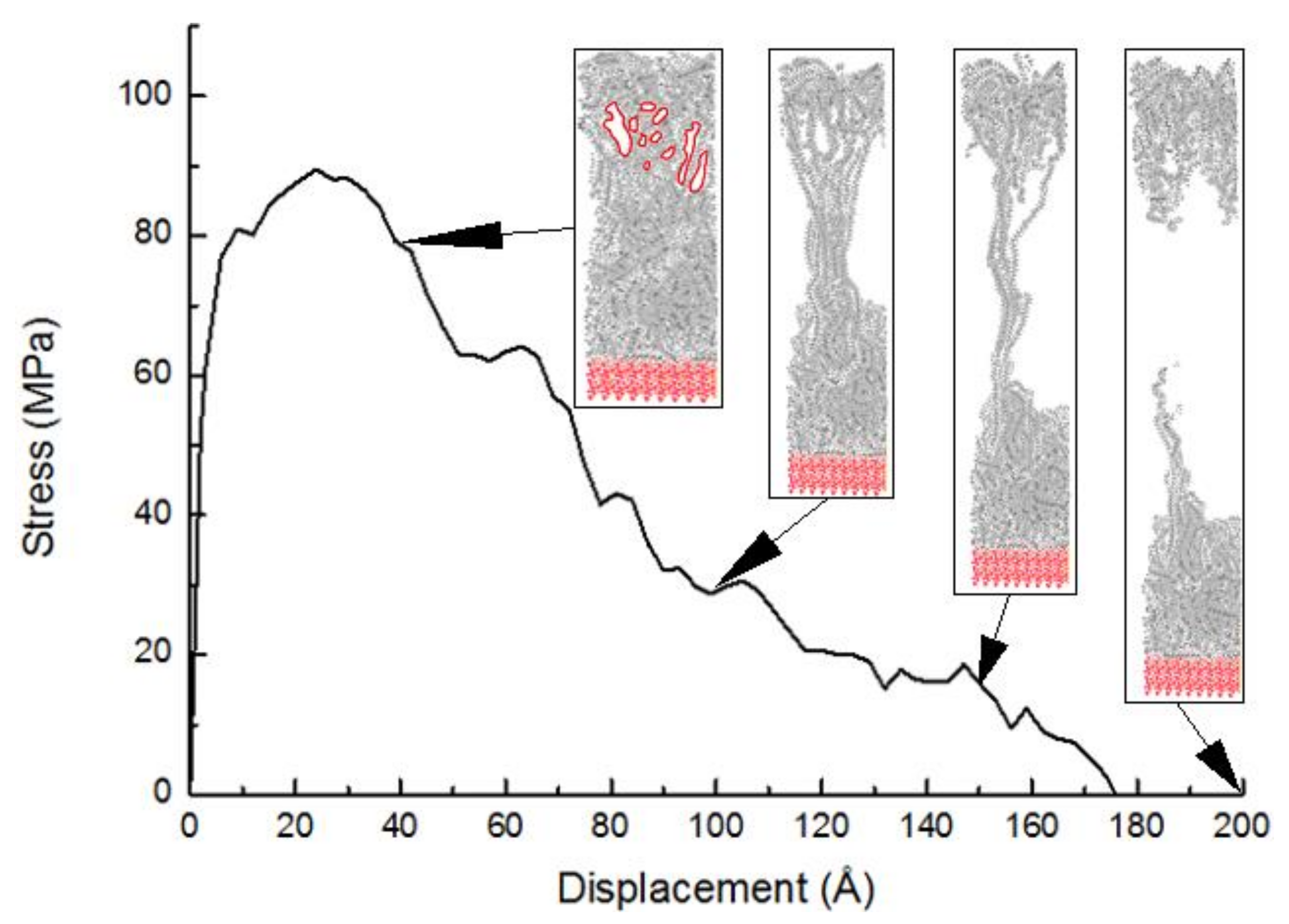
3.2.1. Stress-Displacement Behavior
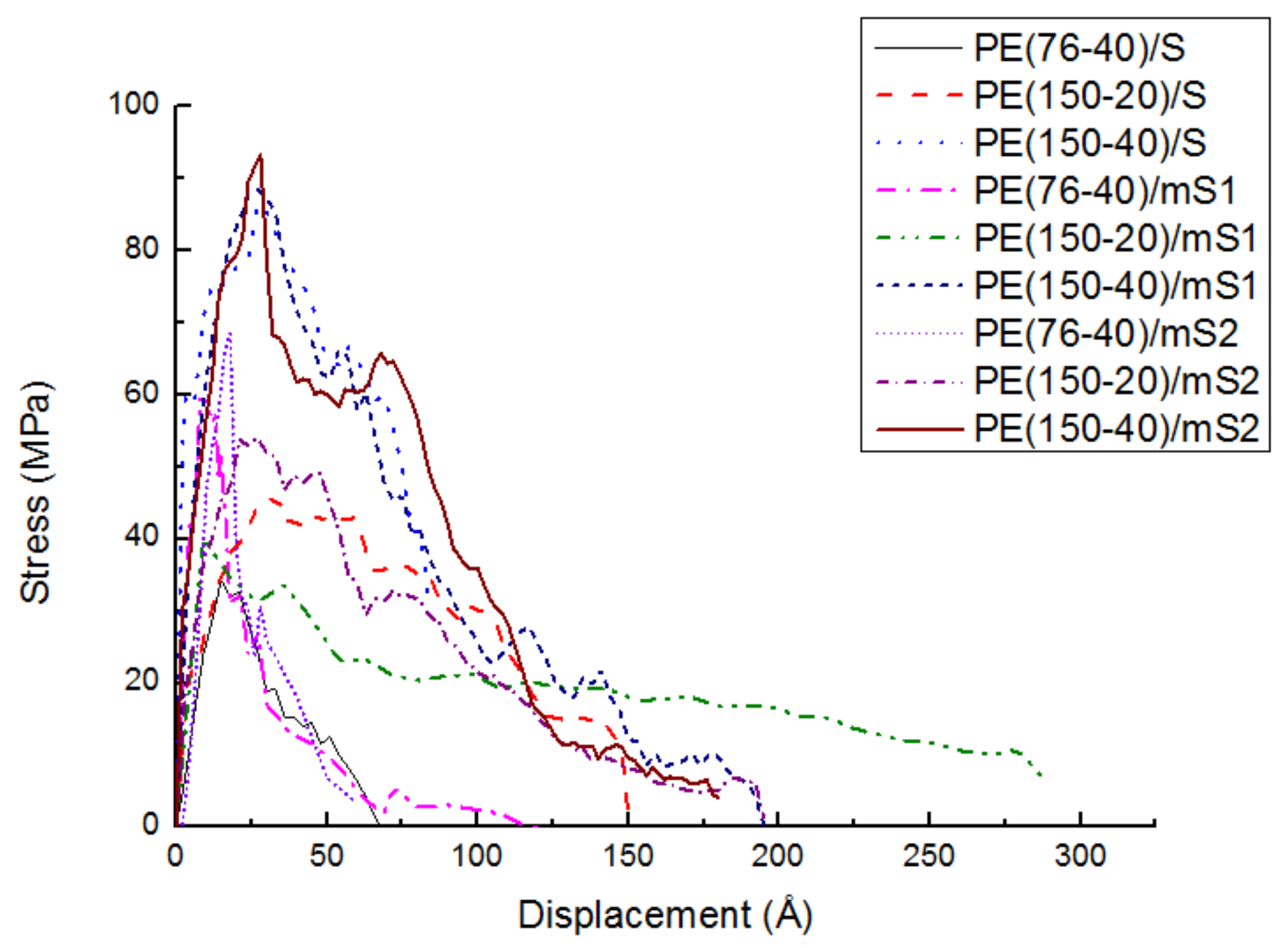
3.2.2. Effect of Chain Length, Number of Chains and Coupling Agents
3.2.3. Density Profile Analysis
3.2.4. Potential Energy Evolutions
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Li, V.; Lim, Y.; Chan, Y. Feasibility study of a passive smart self-healing cementitious composite. Compos. Part B Eng. 1998, 29, 819–827. [Google Scholar] [CrossRef] [Green Version]
- Yu, K.; Dai, J.; Lu, Z.; Poon, C. Rate-dependent tensile properties of ultra-high performance engineered cementitious composites (UHP-ECC). Cem. Concr. Compos. 2018, 93, 218–234. [Google Scholar] [CrossRef]
- Yu, K.; Yu, J.; Dai, J.; Lu, Z.; Shah, S. Development of ultra-high performance engineered cementitious composites using polyethylene (PE) fibers. Constr. Build. Mater. 2018, 158, 217–227. [Google Scholar] [CrossRef]
- Mihashi, H.; Nishiwaki, T. Development of engineered self-healing and self-repairing concrete-state-of-the-art report. J. Adv. Concr. Technol. 2012, 10, 170–184. [Google Scholar] [CrossRef]
- Zhou, S.; Zhu, H.H.; Ju, J.W.; Yan, Z.G.; Chen, Q. Modeling microcapsule-enabled self-healing cementitious composite materials using discrete element method. Int. J. Damage Mech. 2017, 26, 340–357. [Google Scholar] [CrossRef]
- Van Tittelboom, K.; De Belie, N. Self-healing in cementitious materials—A review. Materials 2013, 6, 2182–2217. [Google Scholar] [CrossRef] [PubMed]
- Sidiq, A.; Gravina, R.; Giustozzi, F. Is concrete healing really efficient? A review. Constr. Build. Mater. 2019, 205, 257–273. [Google Scholar] [CrossRef]
- Büyüköztürk, O.; Buehler, M.; Lau, D.; Tuakta, C. Structural solution using molecular dynamics: Fundamentals and a case study of epoxy-silica interface. Int. J. Solids Struct. 2011, 48, 2131–2140. [Google Scholar] [CrossRef] [Green Version]
- Nikkhah, S.J.; Moghbeli, M.R.; Hashemianzadeh, S.M. Dynamic study of deformation and adhesion of an amorphous polyethylene/graphene interface: A simulation study. Macromol. Theor. Simul. 2016, 25, 533–549. [Google Scholar] [CrossRef]
- Tam, L.; Zhou, A.; Yu, Z.; Qiu, Q.; Lau, D. Understanding the effect of temperature on the interfacial behavior of CFRP-wood composite via molecular dynamics simulations. Compos. Part B Eng. 2017, 109, 227–237. [Google Scholar] [CrossRef]
- Chen, Q.; Zhu, H.H.; Yan, Z.G.; Ju, J.W.; Jiang, Z.W.; Wang, Y.Q. A multiphase micromechanical model for hybrid fiber reinforced concrete considering the aggregate and ITZ effects. Constr. Build. Mater. 2016, 114, 839–850. [Google Scholar] [CrossRef]
- Liu, S.; Liu, C.; Deng, L. Machine learning approaches for protein–protein interaction hot spot prediction: Progress and comparative assessment. Molecules 2018, 23, 2535. [Google Scholar] [CrossRef]
- Elghobashi-Meinhardt, N. Exploring peptide–solvent interactions: A computational study. Molecules 2018, 23, 2355. [Google Scholar] [CrossRef] [PubMed]
- Krawczyk, J. Solid wettability modification via adsorption of antimicrobial sucrose fatty acid esters and some other sugar-based surfactants. Molecules 2018, 23, 1597. [Google Scholar] [CrossRef] [PubMed]
- Piquer, L.R.; Sánchez, R.R.; Sañudo, E.C.; Echeverría, J. Understanding the molecule-electrode interface for molecular spintronic devices: A computational and experimental study. Molecules 2018, 23, 1441. [Google Scholar] [CrossRef]
- Nnaji, N.; Nwaji, N.; Mack, J.; Nyokong, T. Corrosion resistance of aluminum against acid activation: Impact of benzothiazole-substituted gallium phthalocyanine. Molecules 2019, 24, 207. [Google Scholar] [CrossRef] [PubMed]
- Park, C.; Jung, J.; Yun, G. Thermomechanical properties of mineralized nitrogen-doped carbon nanotube/polymer nanocomposites by molecular dynamics simulations. Compos. Part B Eng. 2019, 161, 639–650. [Google Scholar] [CrossRef]
- Zhuang, X.Y.; Zhou, S. Molecular dynamics study of an amorphous polyethylene/silica interface with shear tests. Materials 2018, 11, 929. [Google Scholar] [CrossRef]
- Zhang, H.P.; Lu, X.; Leng, Y.; Fang, L.; Qu, S.; Feng, B.; Weng, J.; Wang, J. Molecular dynamics simulations on the interaction between polymers and hydroxyapatite with and without coupling agents. Acta biomater. 2009, 5, 1169–1181. [Google Scholar] [CrossRef]
- Chen, Y.; Chia, J.Y.H.; Su, Z.C.; Tay, T.E.; Tan, V.B.C. Mechanical characterization of interfaces in epoxy-clay nanocomposites by molecular simulations. Polymer 2013, 54, 766–773. [Google Scholar] [CrossRef]
- Yi, P.; Locker, C.R.; Rutledge, G.C. Molecular dynamics simulation of homogeneous crystal nucleation in polyethylene. Macromolecules 2013, 46, 4723–4733. [Google Scholar] [CrossRef]
- Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Mayo, S.L.; Olafson, B.D.; Goddard, W.A. DREIDING: A generic force field for molecular simulations. J. Phys. Chem. 1990, 94, 8897–8909. [Google Scholar] [CrossRef]
- Vu-Bac, N.; Bessa, M.A.; Rabczuk, T.; Liu, W.K. A multiscale model for the quasi-static thermo-plastic behavior of highly cross-linked glassy polymers. Macromolecules 2015, 48, 6713–6723. [Google Scholar] [CrossRef]
- Paek, E.; Hwang, G.S. A computational analysis of graphene adhesion on amorphous silica. J. Appl. Phys. 2013, 113, 164901. [Google Scholar] [CrossRef]
- Viani, L.; Risko, C.; Toney, M.F.; Breiby, D.W.; Brédas, J.L. Substrate-induced variations of molecular packing, dynamics, and intermolecular electronic couplings in pentacene monolayers on the amorphous silica dielectric. ACS Nano 2014, 8, 690–700. [Google Scholar] [CrossRef]
- Nosé, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 2002, 100, 191–198. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef]
- Awasthi, A.P.; Lagoudas, D.C.; Hammerand, D.C. Modeling of graphene-polymer interfacial mechanical behavior using molecular dynamics. Modelling Simul. Mater. Sci. Eng. 2008, 17, 015002. [Google Scholar] [CrossRef]
- Han, J.; Gee, R.H.; Boyd, R.H. Glass transition temperatures of polymers from molecular dynamics simulations. Macromolecules 1994, 27, 7781–7784. [Google Scholar] [CrossRef]
- Pothan, L.A.; Thomas, S.; Groeninckx, G. The role of fibre/matrix interactions on the dynamic mechanical properties of chemically modified banana fibre/polyester composites. Compos. Part A Appl. Sci. Manufac. 2006, 37, 1260–1269. [Google Scholar] [CrossRef]
- Xie, Y.; Hill, C.A.; Xiao, Z.; Militz, H.; Mai, C. Silane coupling agents used for natural fiber/polymer composites: A review. Compos. Part A Appl. Sci. Manufac. 2010, 41, 806–819. [Google Scholar] [CrossRef]
- Miller, J.D.; Ishida, H.; Maurer, F.H.J. Dynamic-mechanical properties of interfacially modified glass sphere filled polyethylene. Rheol. Acta. 1988, 27, 397–404. [Google Scholar] [CrossRef]
- McFarren, G.A.; Sanderson, T.F.; Schappell, F.G. Azidosilane polymer-filler coupling agent. Polym. Eng. Sci. 1977, 17, 46–49. [Google Scholar] [CrossRef]
- Nikkhah, S.J.; Moghbeli, M.R.; Hashemianzadeh, S.M. A molecular simulation study on the adhesion behavior of a functionalized polyethylene-functionalized graphene interface. Phys. Chem. Chem. Phys. 2015, 17, 27414–27427. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhuang, X.; Muthu, J.; Mabrouki, T.; Fontaine, M.; Gong, Y.; Rabczuk, T. Load transfer of graphene/carbon nanotube/polyethylene hybrid nanocomposite by molecular dynamics simulation. Compos. Part B Eng. 2014, 63, 27–33. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Work of Adhesion [mJ m−2] |
|---|---|
| PE(76-40)/S | 93.14 |
| PE(76-40)/mS1 | 3205.253 |
| PE(150-20)/S | 163.3887 |
| PE(150-20)/mS1 | 3235.154 |
| PE(150-40)/S | 335.7157 |
| PE(150-40)/mS1 | 3217.675 |
| System | Adhesion Strength (MPa) | Distance (Å) |
|---|---|---|
| PE(76-40)/S | 34.2 | 15 |
| PE(76-40)/mS1 | 59.4 | 8 |
| PE(76-40)/mS2 | 68.6 | 18 |
| PE(150-20)/S | 42.8 | 26 |
| PE(150-20)/mS1 | 43 | 9 |
| PE(150-20)/mS2 | 54.0 | 27 |
| PE(150-40)/S | 86.3 | 27 |
| PE(150-40)/mS1 | 88.5 | 27 |
| PE(150-40)/mS2 | 93.2 | 28 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, S.; Vu-Bac, N.; Arash, B.; Zhu, H.; Zhuang, X. Interface Characterization Between Polyethylene/ Silica in Engineered Cementitious Composites by Molecular Dynamics Simulation. Molecules 2019, 24, 1497. https://doi.org/10.3390/molecules24081497
Zhou S, Vu-Bac N, Arash B, Zhu H, Zhuang X. Interface Characterization Between Polyethylene/ Silica in Engineered Cementitious Composites by Molecular Dynamics Simulation. Molecules. 2019; 24(8):1497. https://doi.org/10.3390/molecules24081497
Chicago/Turabian StyleZhou, Shuai, Nam Vu-Bac, Behrouz Arash, Hehua Zhu, and Xiaoying Zhuang. 2019. "Interface Characterization Between Polyethylene/ Silica in Engineered Cementitious Composites by Molecular Dynamics Simulation" Molecules 24, no. 8: 1497. https://doi.org/10.3390/molecules24081497