
Synthesis and Chemical Properties of 3-Phosphono-coumarins and 1,2-Benzoxaphosphorins as Precursors for Bioactive Compounds


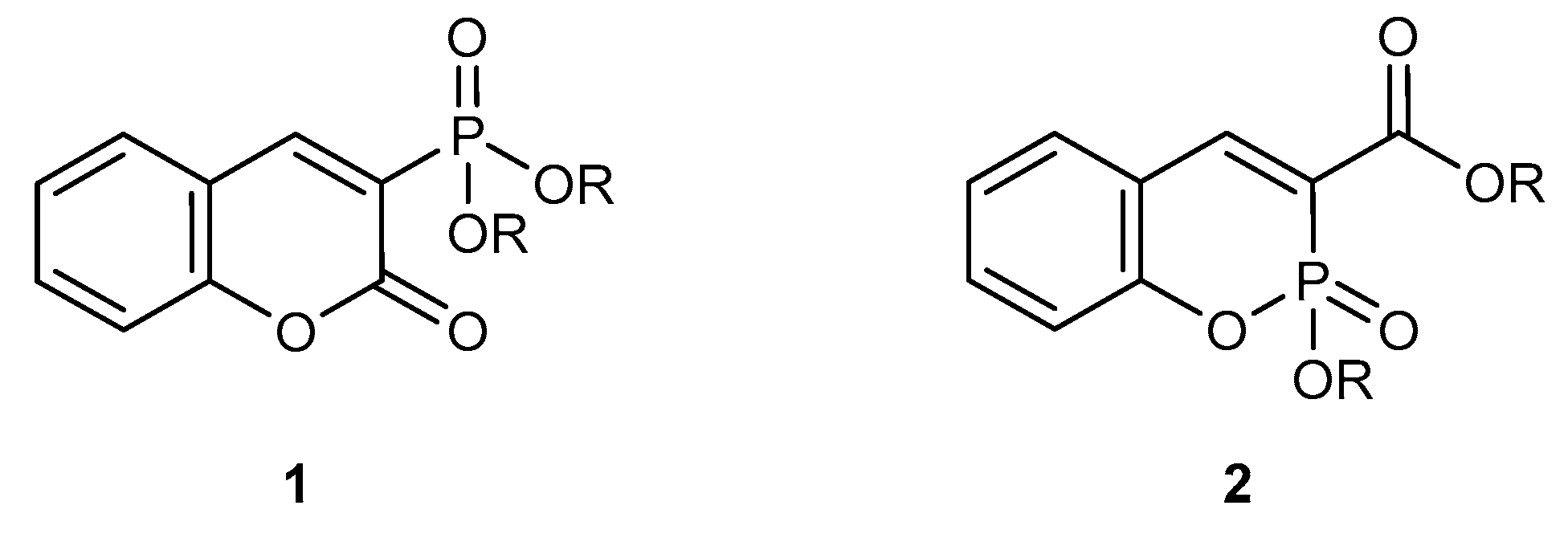
Abstract
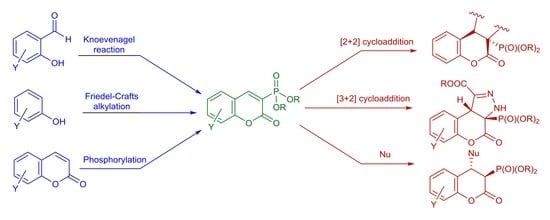
:
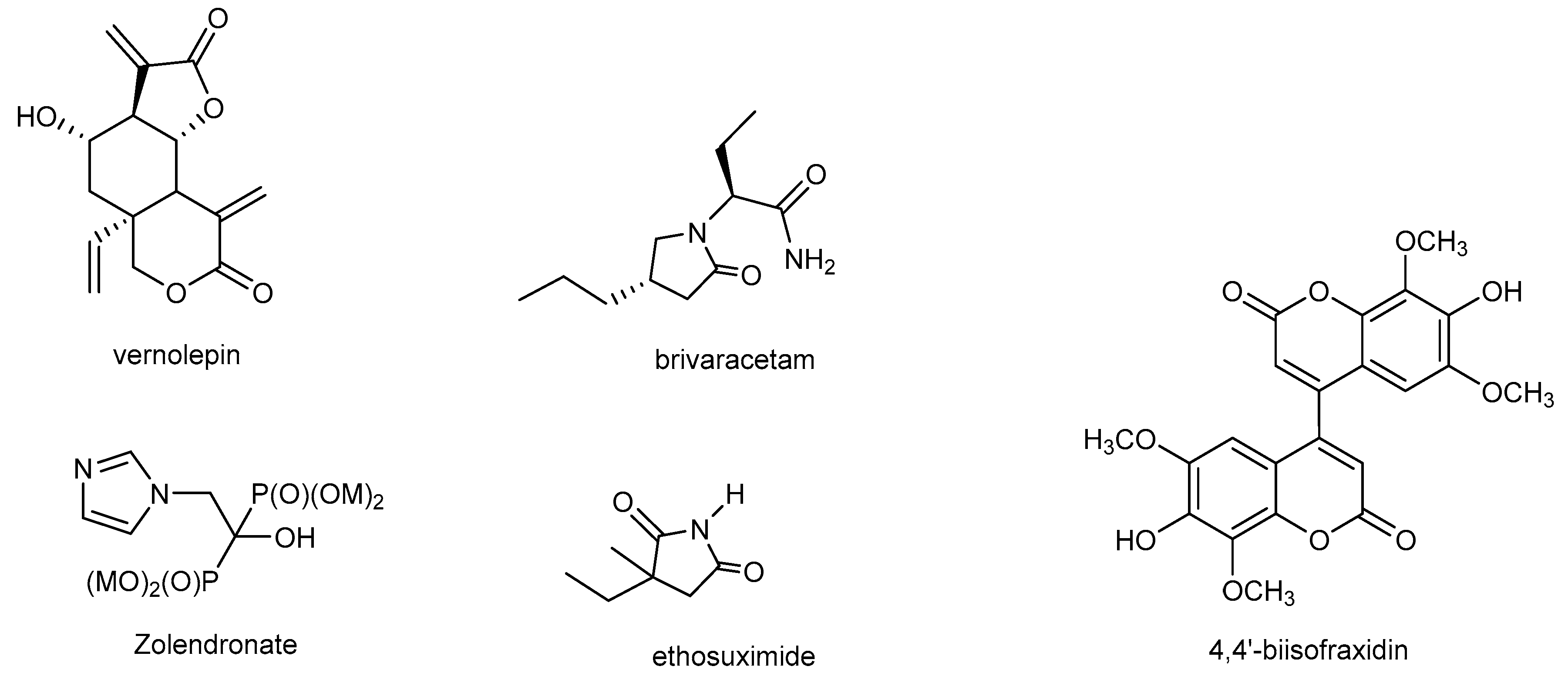
1. Introduction
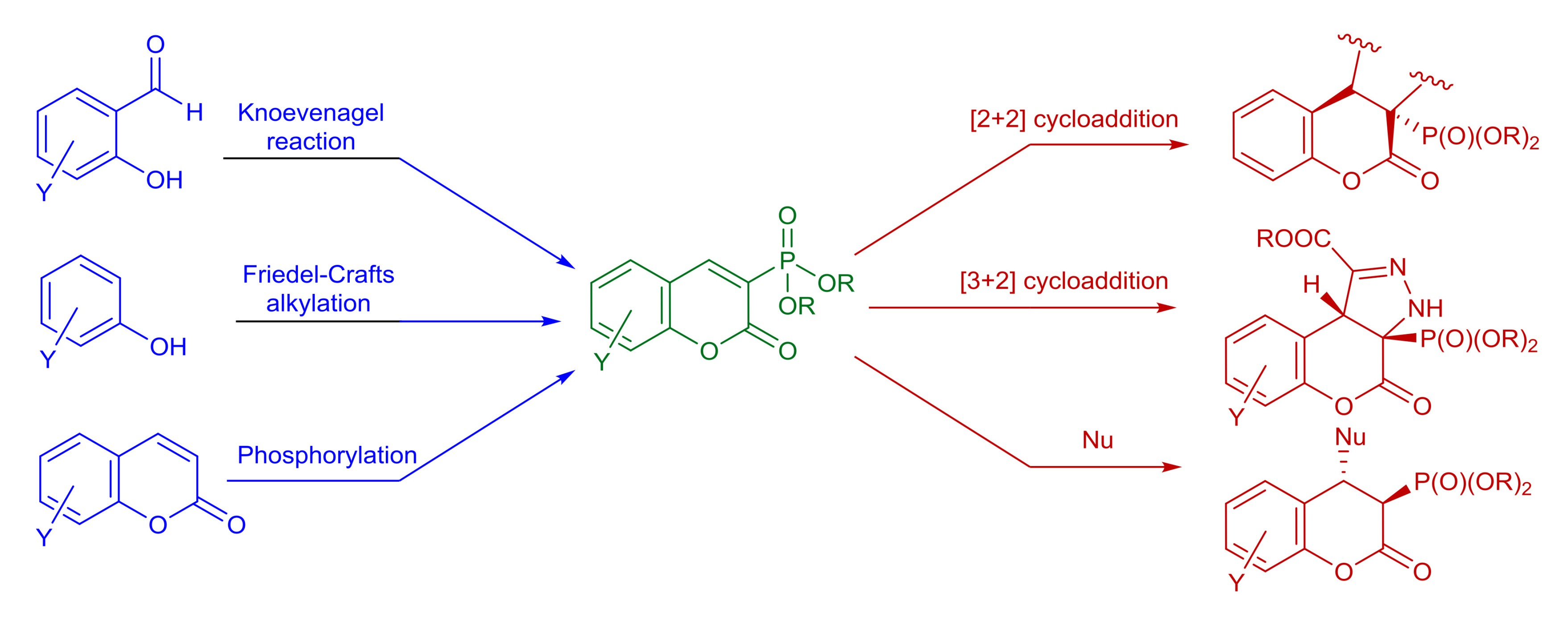
2. Synthesis and Some Reactions of Dialkyl 2-oxo-2H-1-benzopyran-3-phosphonates 1
2.1. Synthesis of Dialkyl 2-oxo-2H-1-benzopyran-3-phosphonates 1
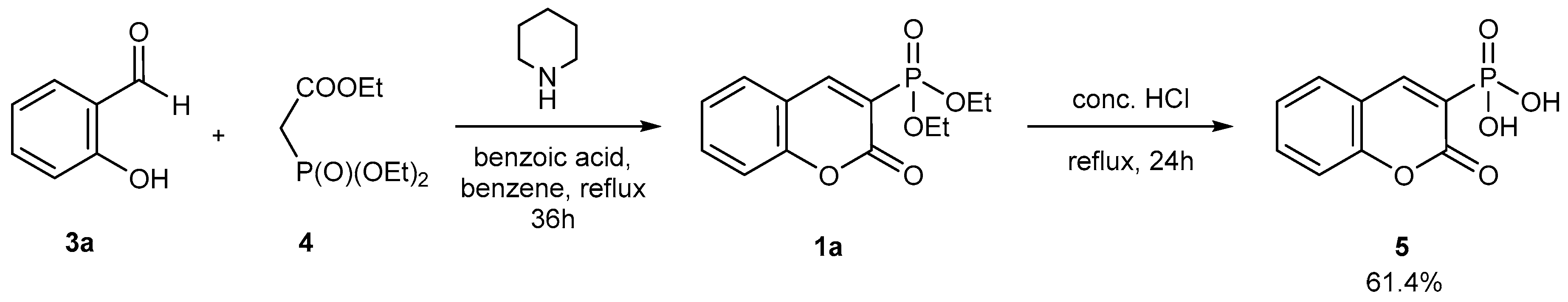
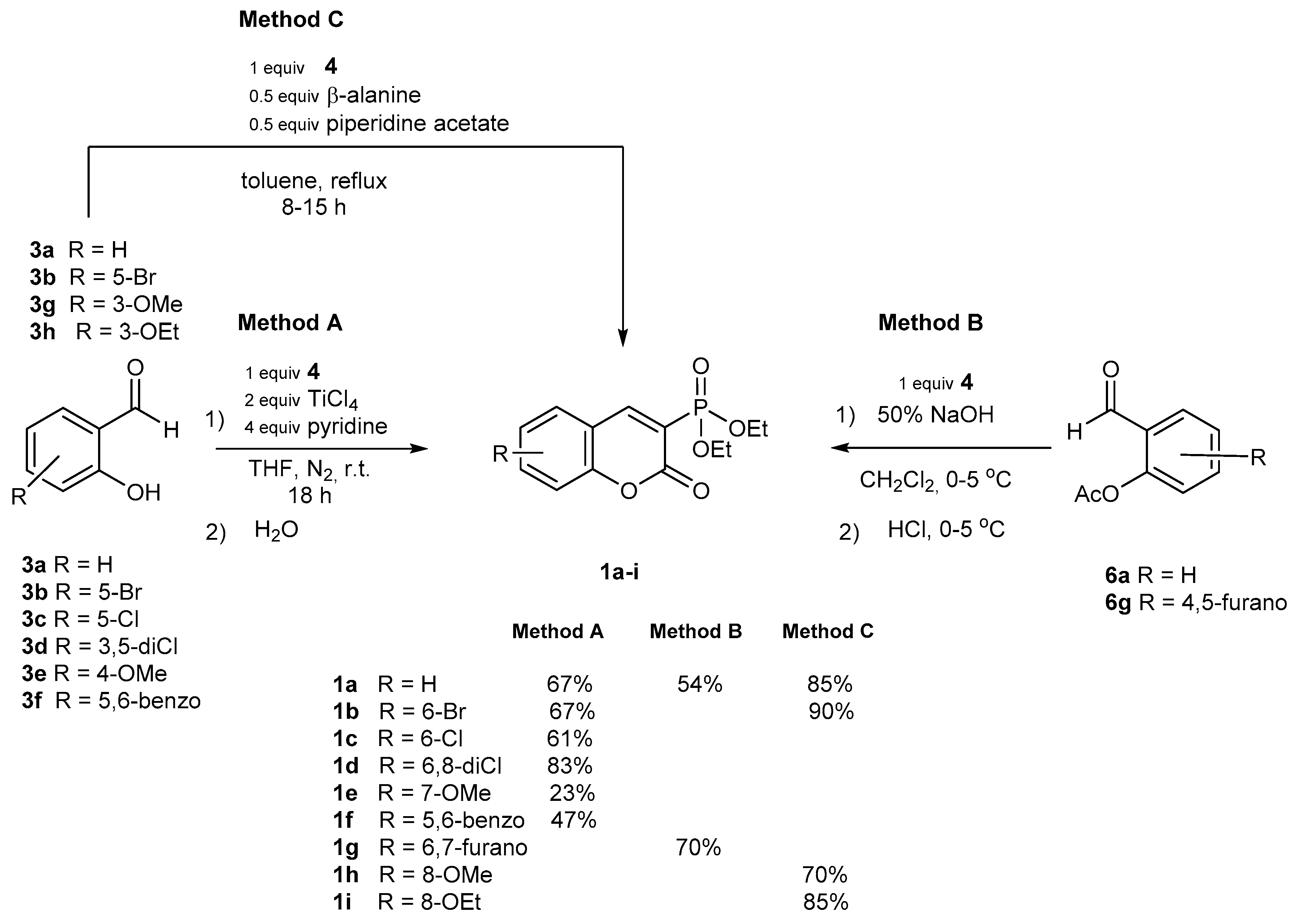
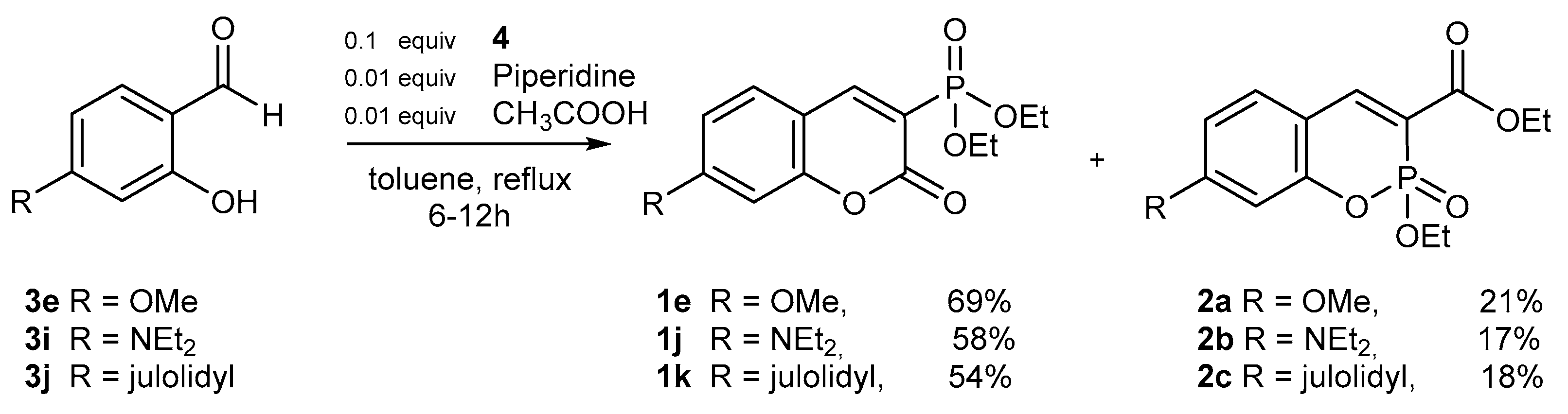
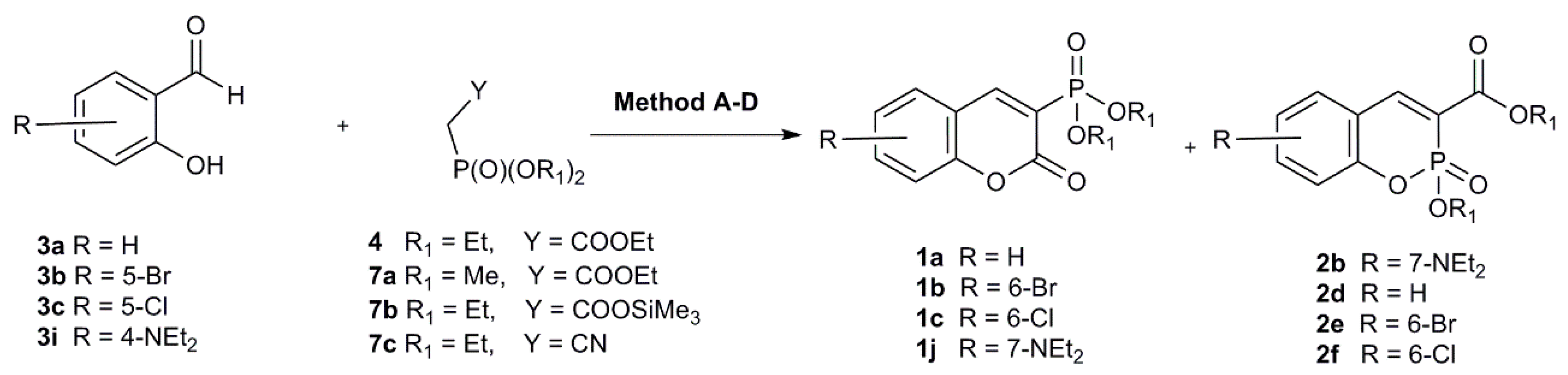
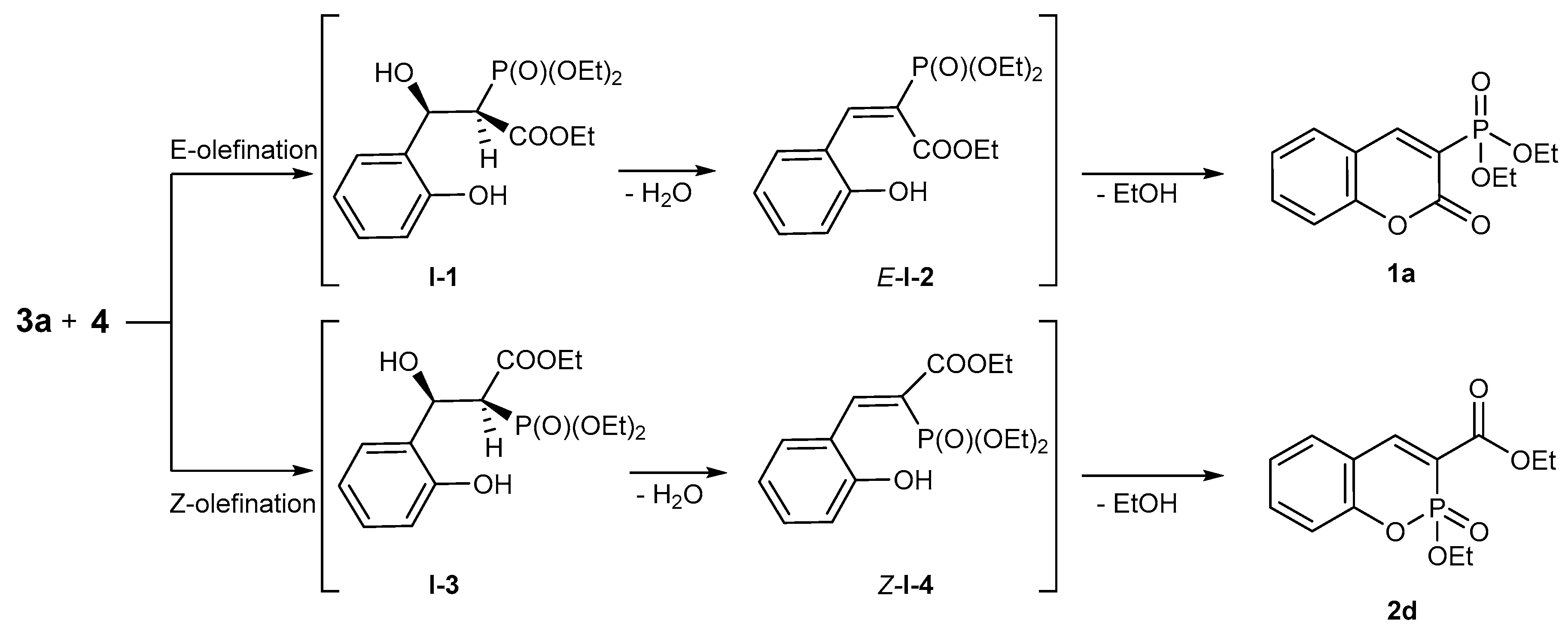
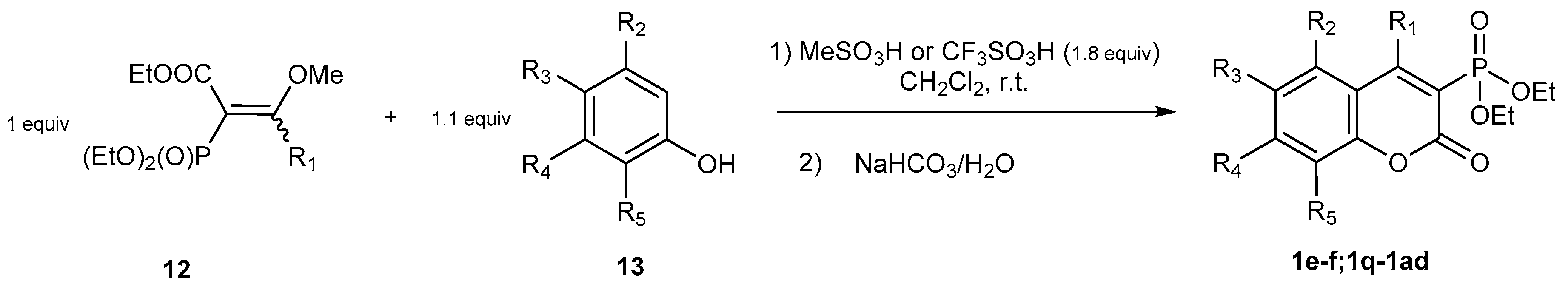
2.1.1. Synthetic Protocols Applying Knoevenagel Reaction
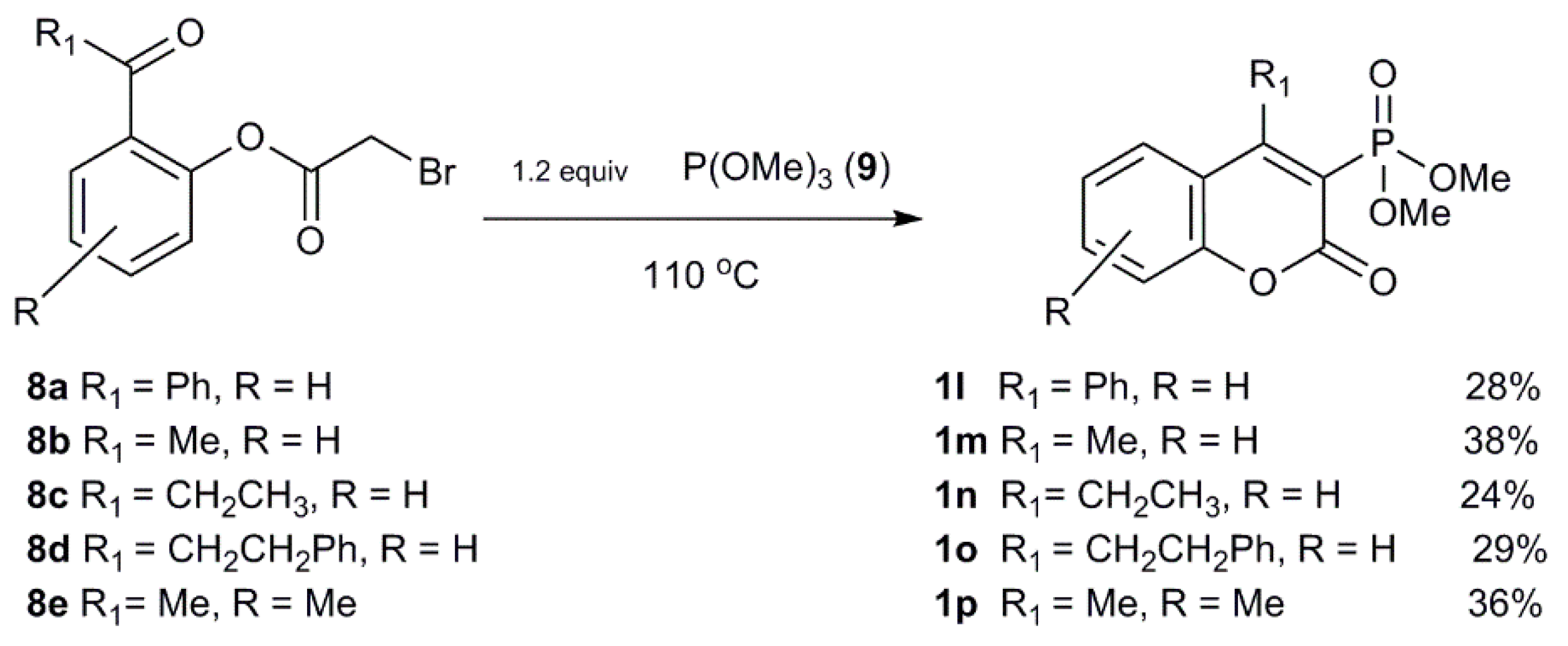
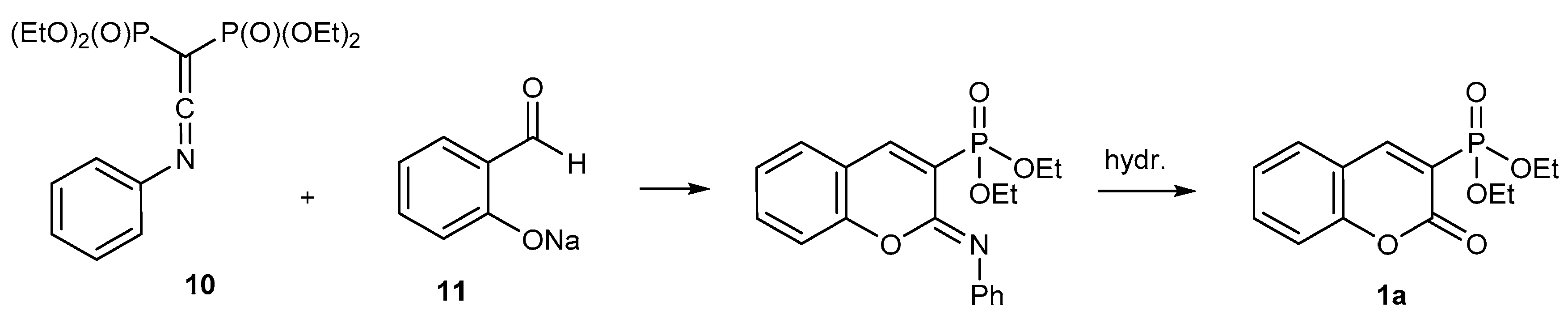
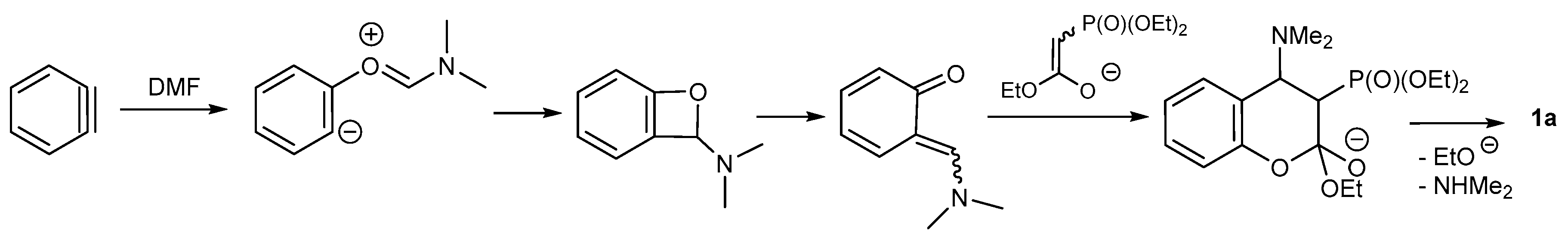
2.1.2. Synthetic Procedure Including Phosphoryl Ketenimines
2.1.3. Synthetic Procedure Including Vinylphosphonates—Friedel-Crafts Alkylation of Phenols
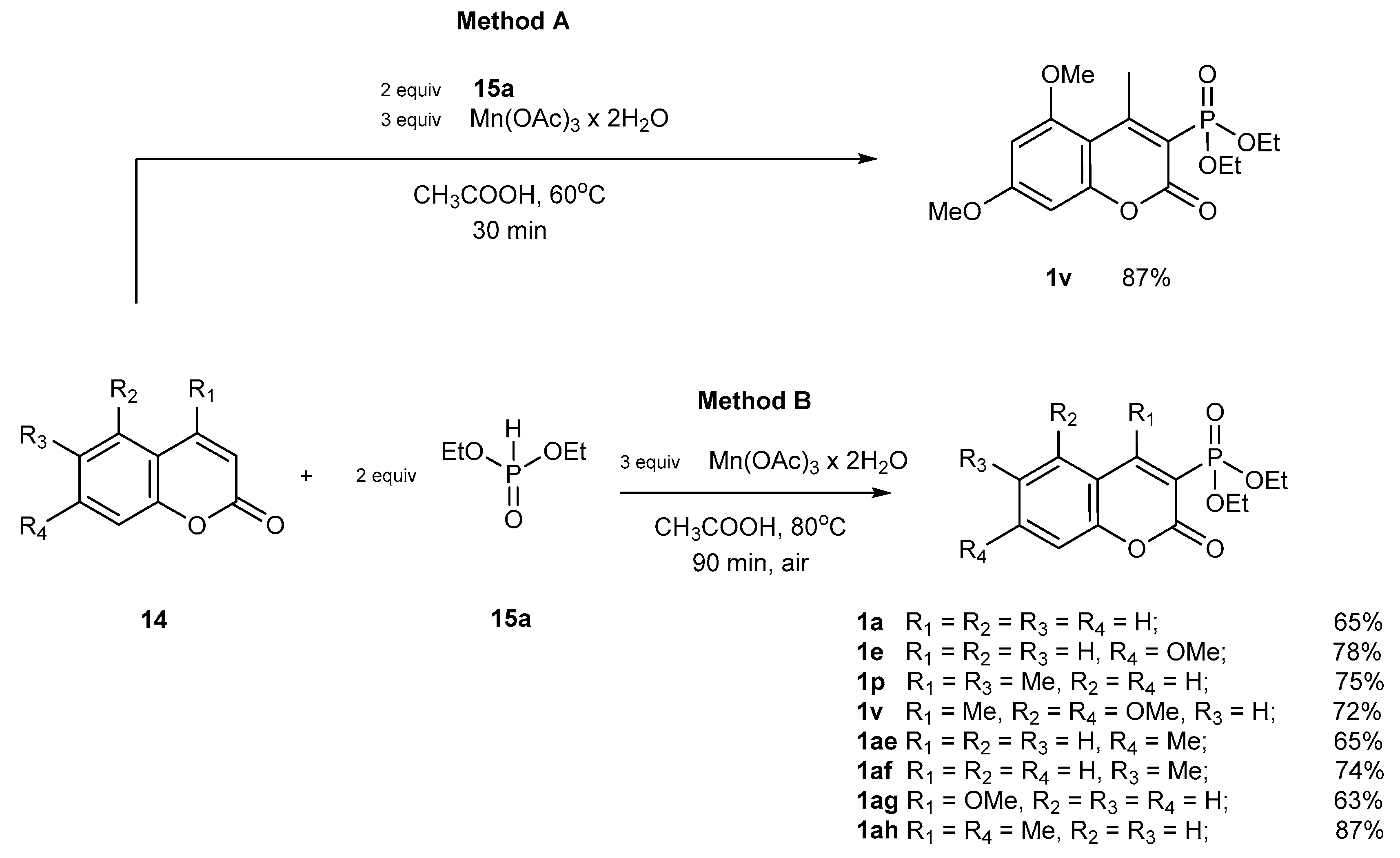
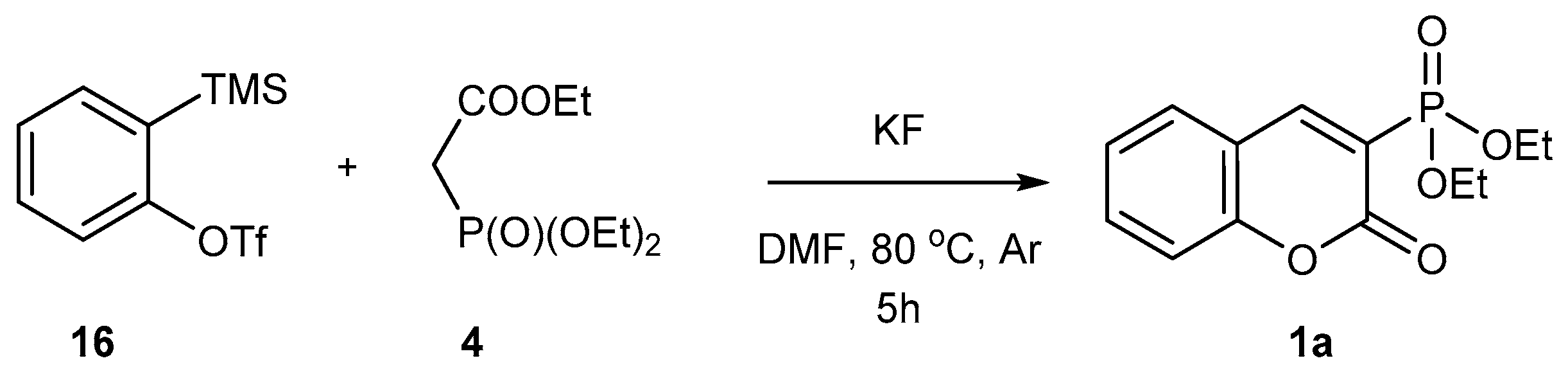
2.1.4. Phosphorylation of Coumarins
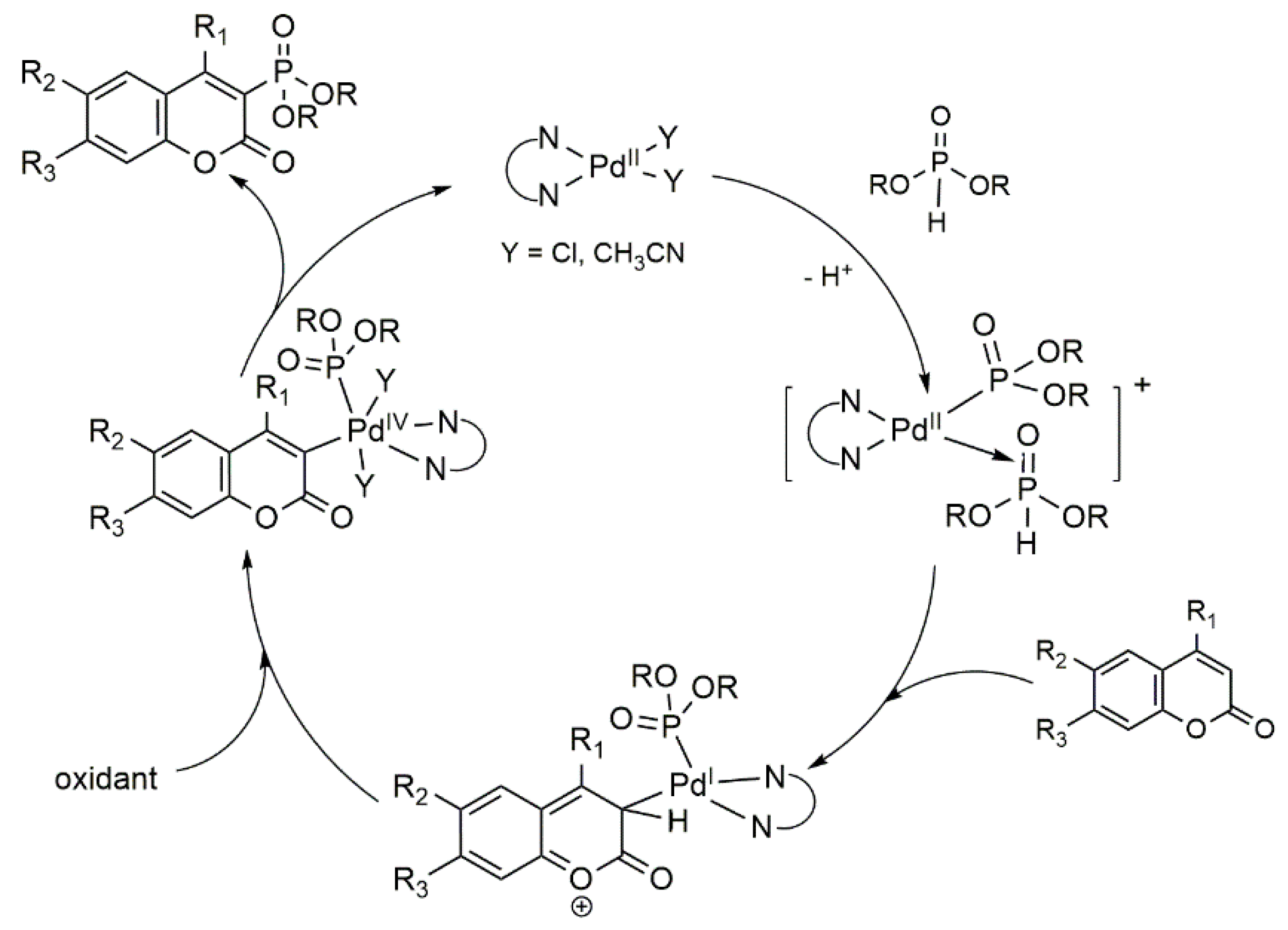
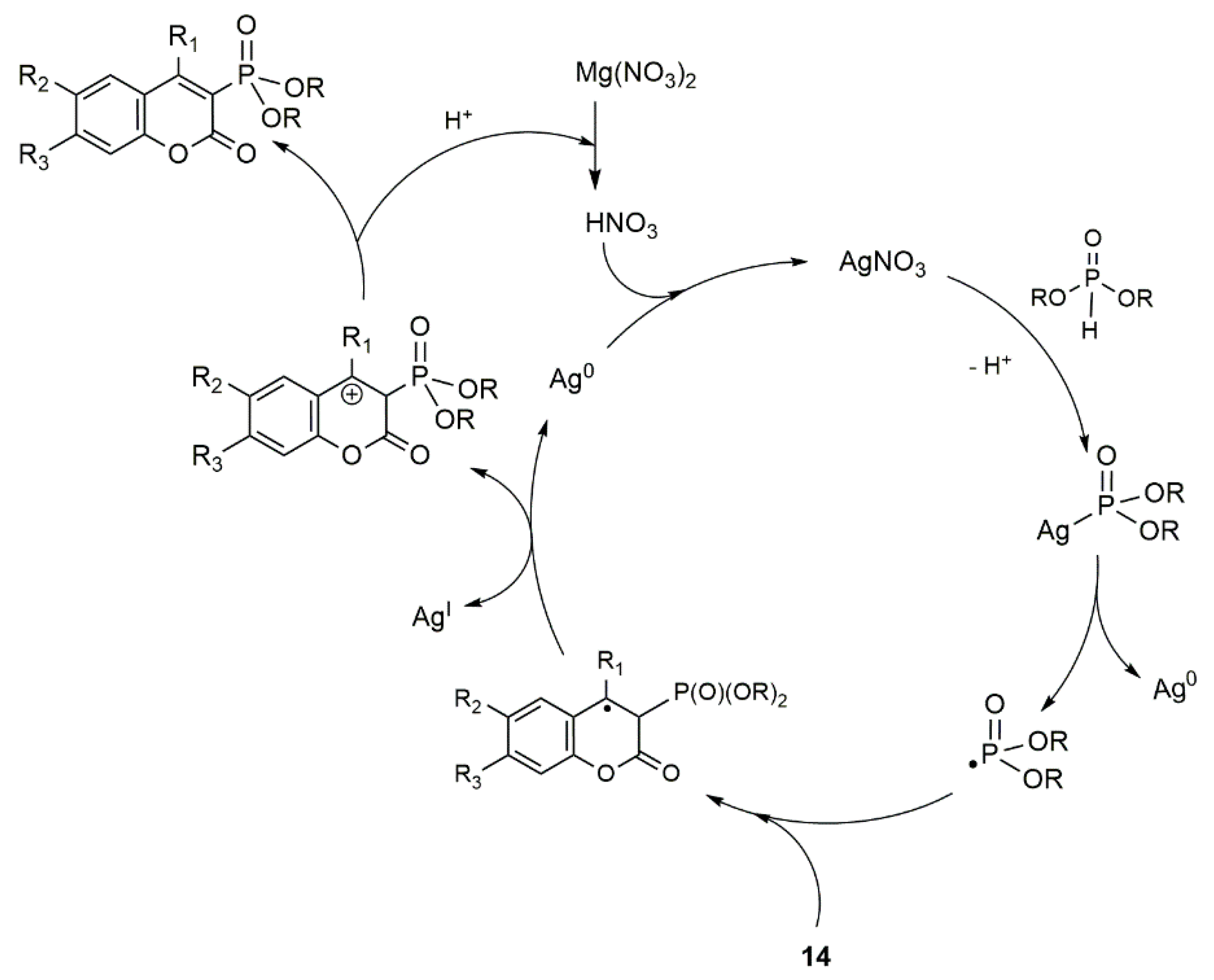
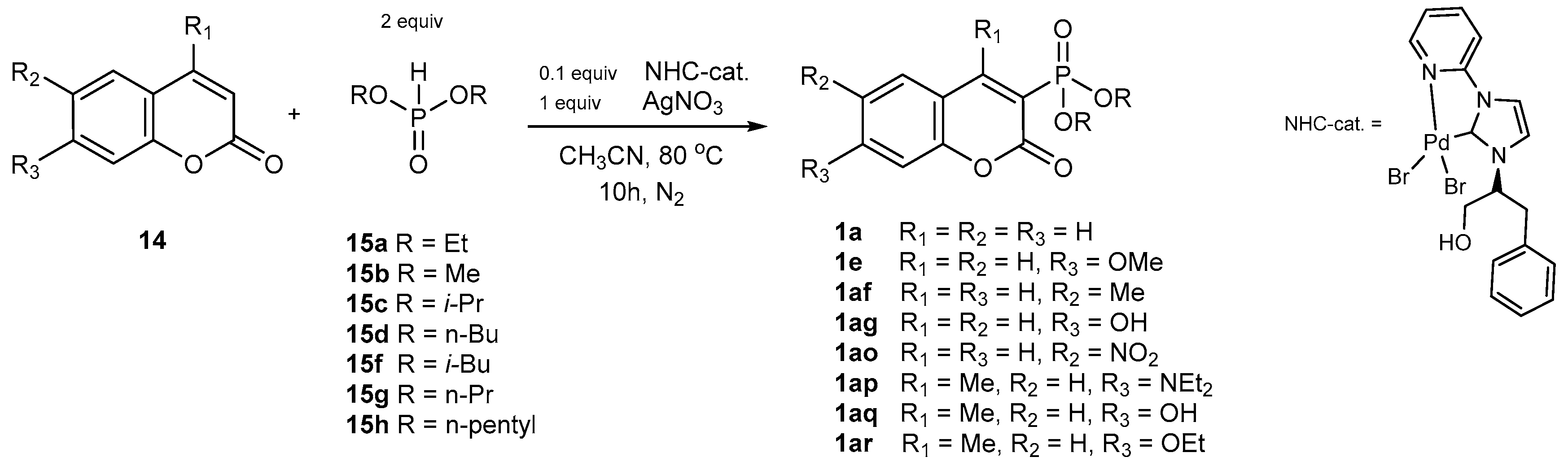
Catalytic Phosphorylation
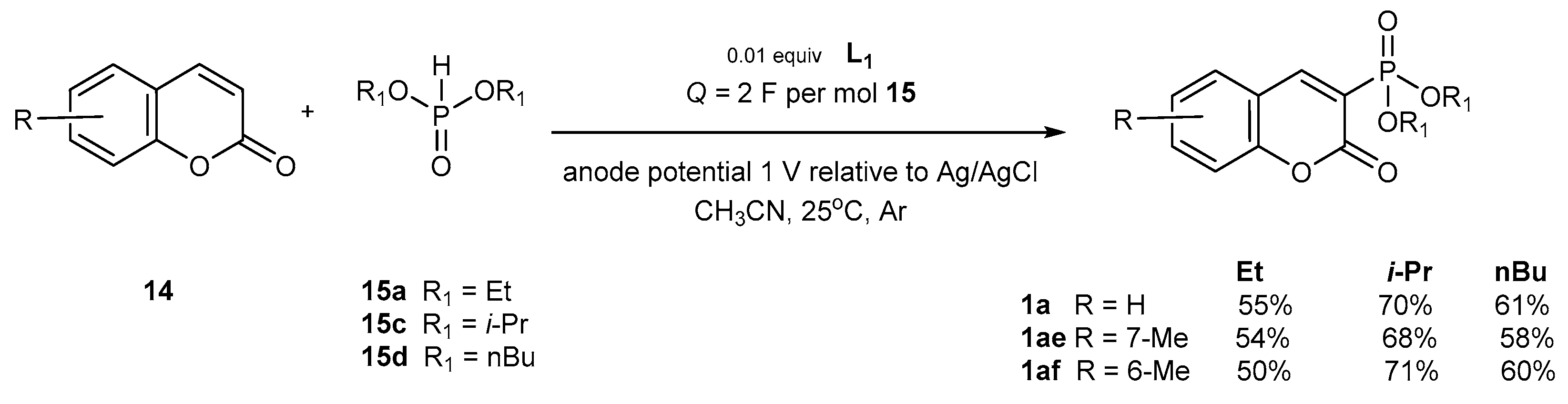
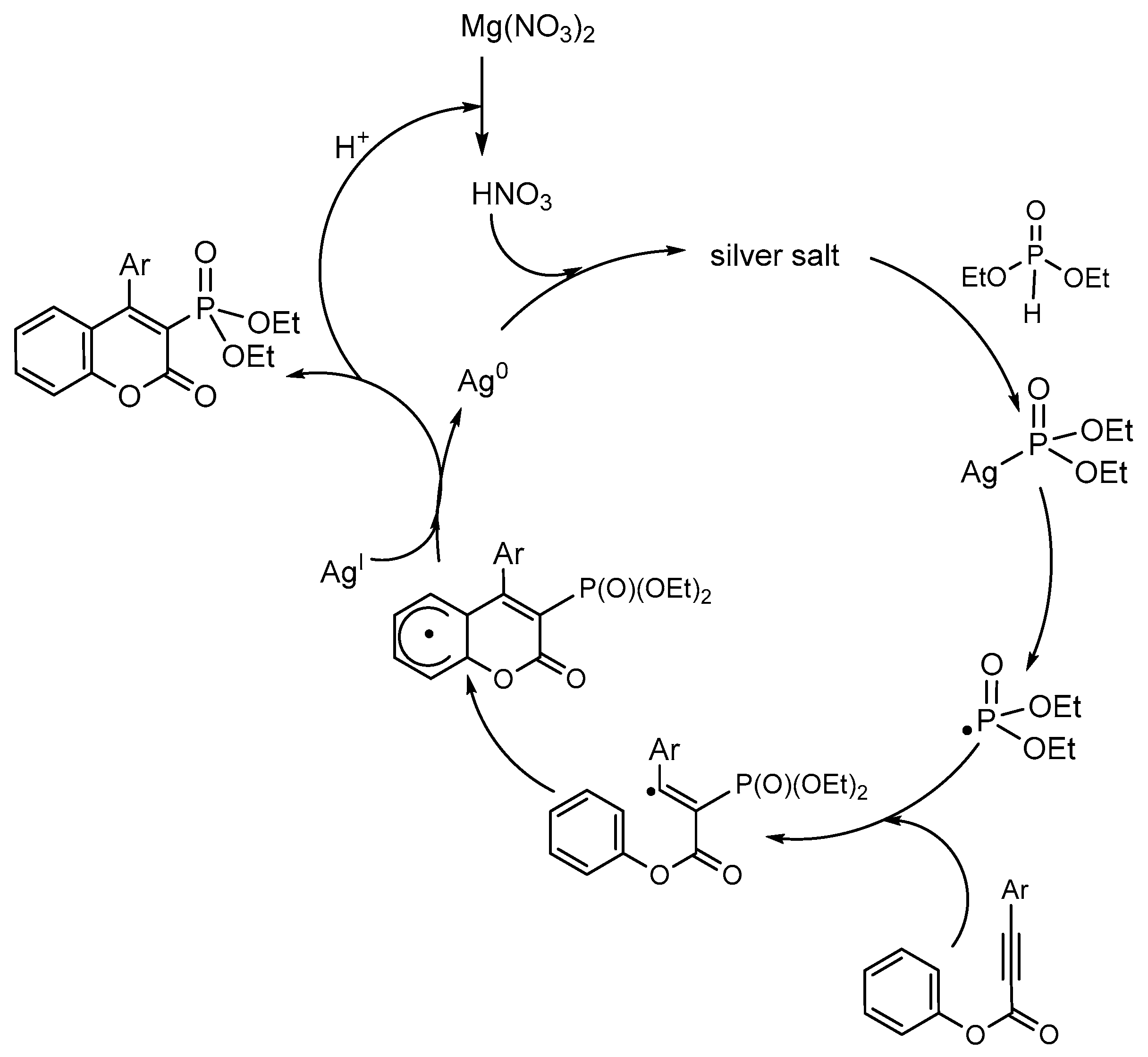
Electrochemical Phosphorylation
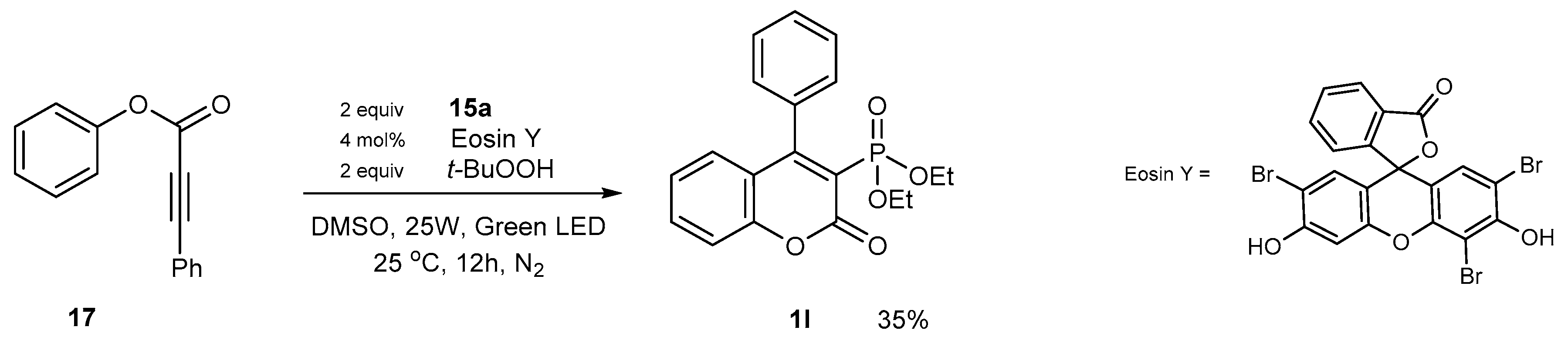
2.1.5. Synthetic Protocols Involving Coupling Reactions
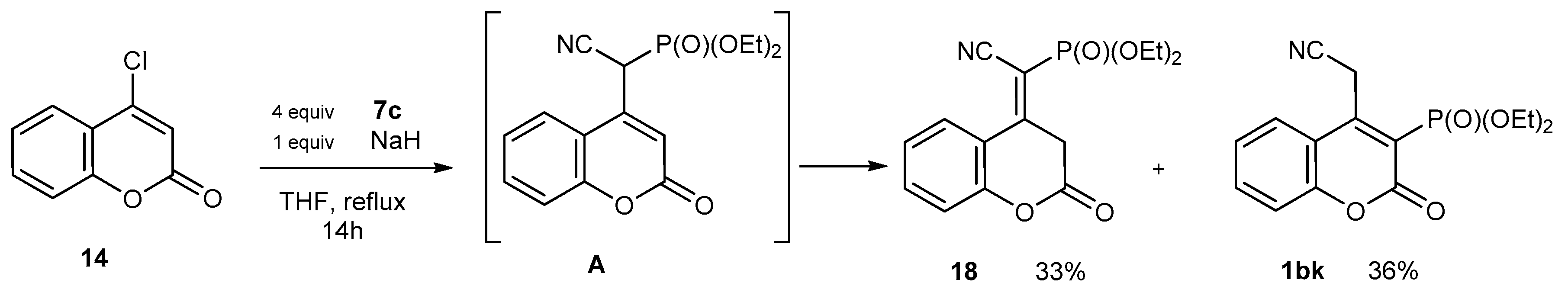
2.1.6. Rearrangement Reactions
2.2. Reaction of Dialkyl 2-oxo-2H-1-benzopyran-3-phosphonates 1
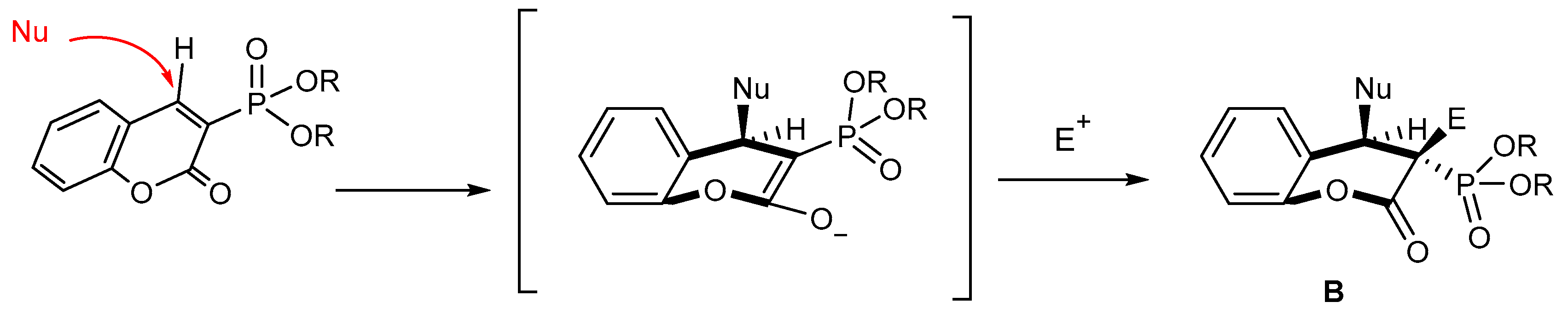
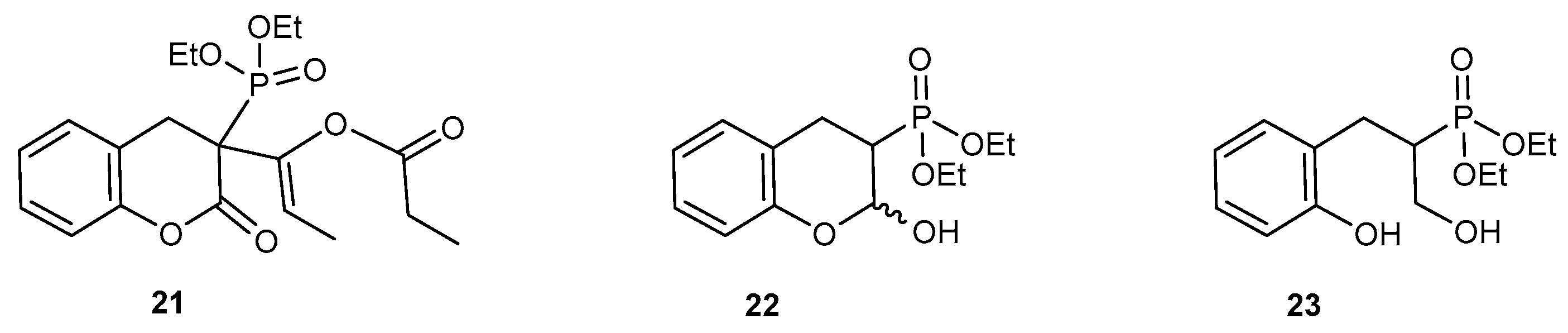
2.2.1. Reactions with Nucleophilic Reagents
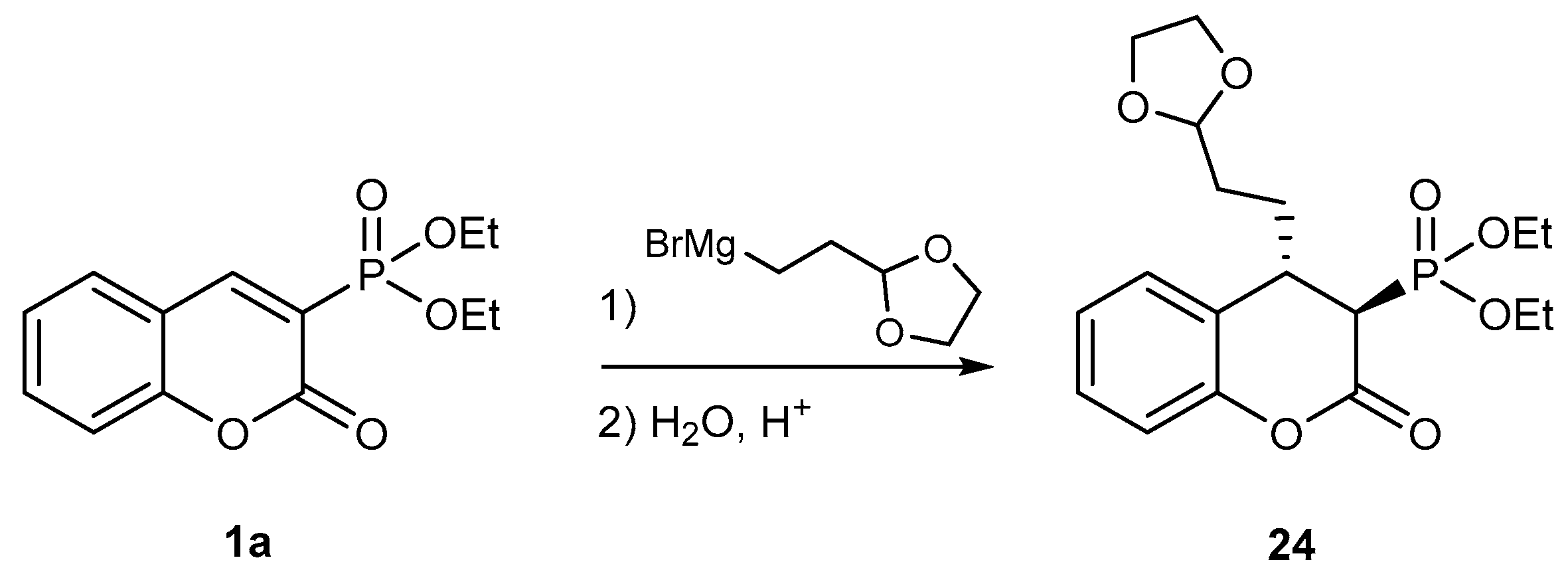
2.2.2. Reactions with Hydrides
2.2.3. Reactions with Organometallic Reagents
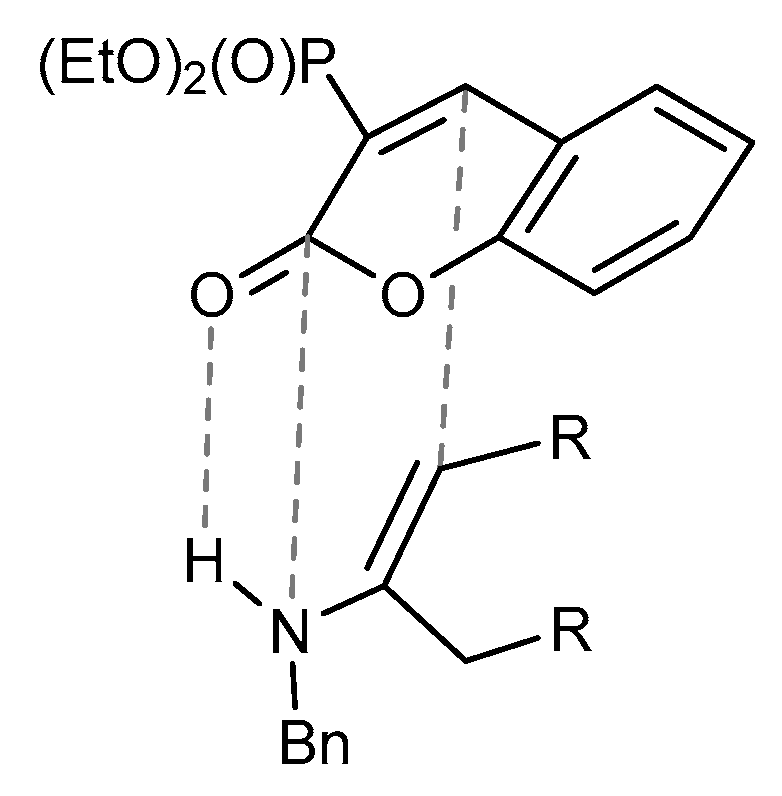
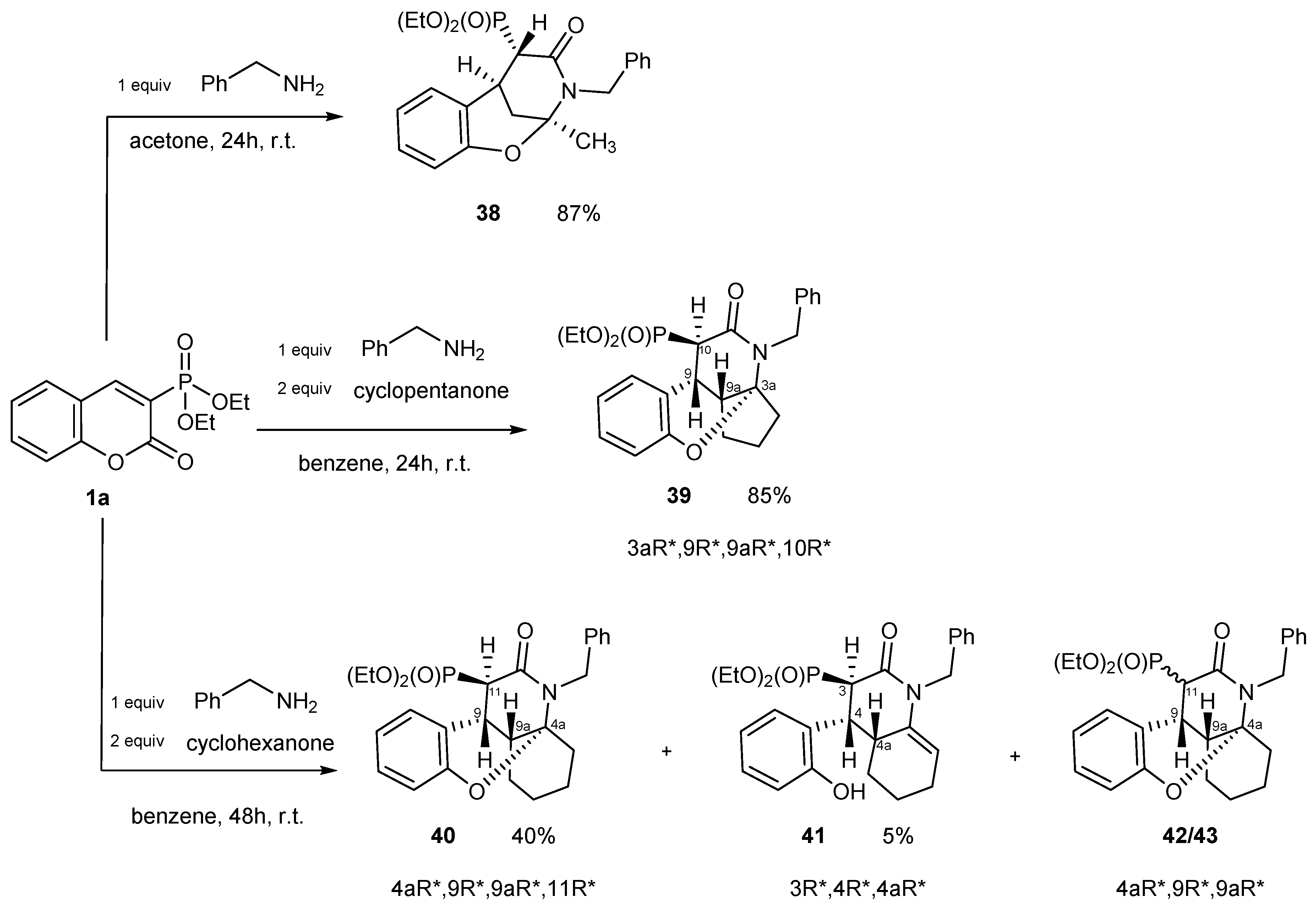
2.2.4. Three-Component Reactions of Diethyl 2-oxo-2H-1-benzopyran-3-phosphonate (1a) with Compounds Bearing Carbonyl and Amino Groups
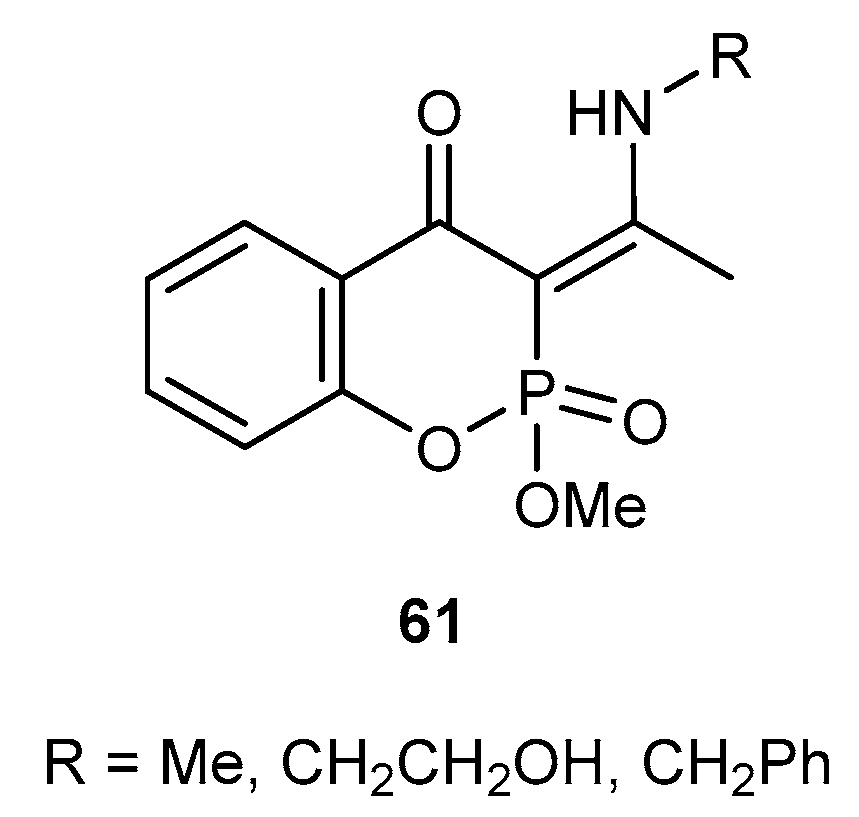
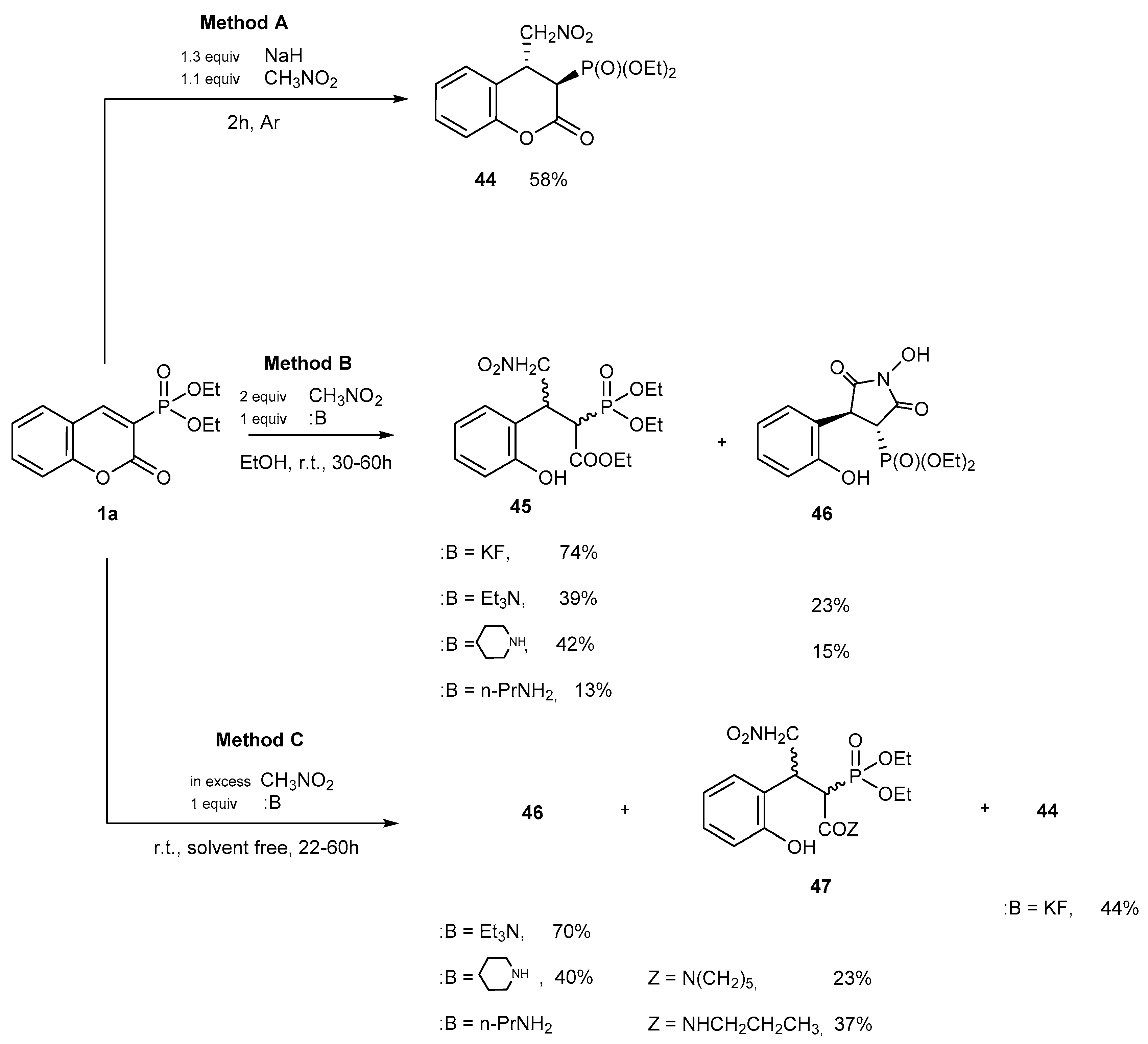
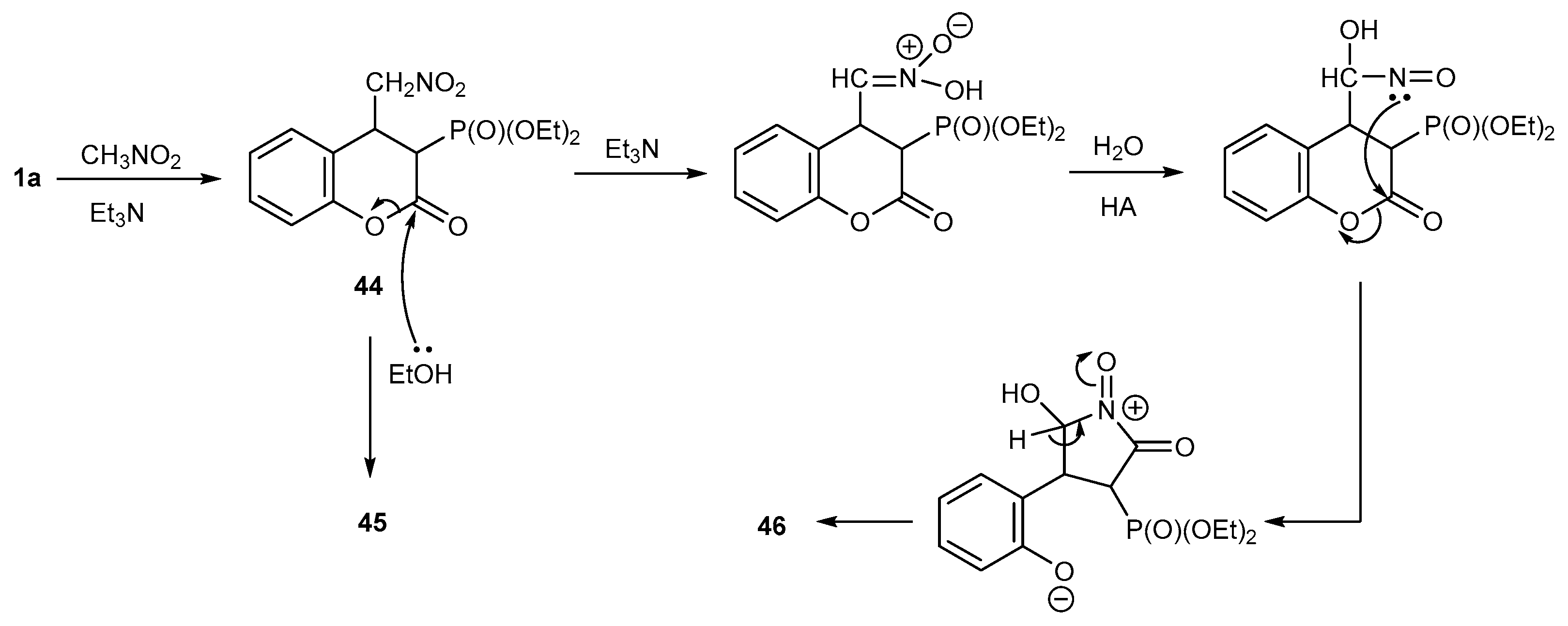
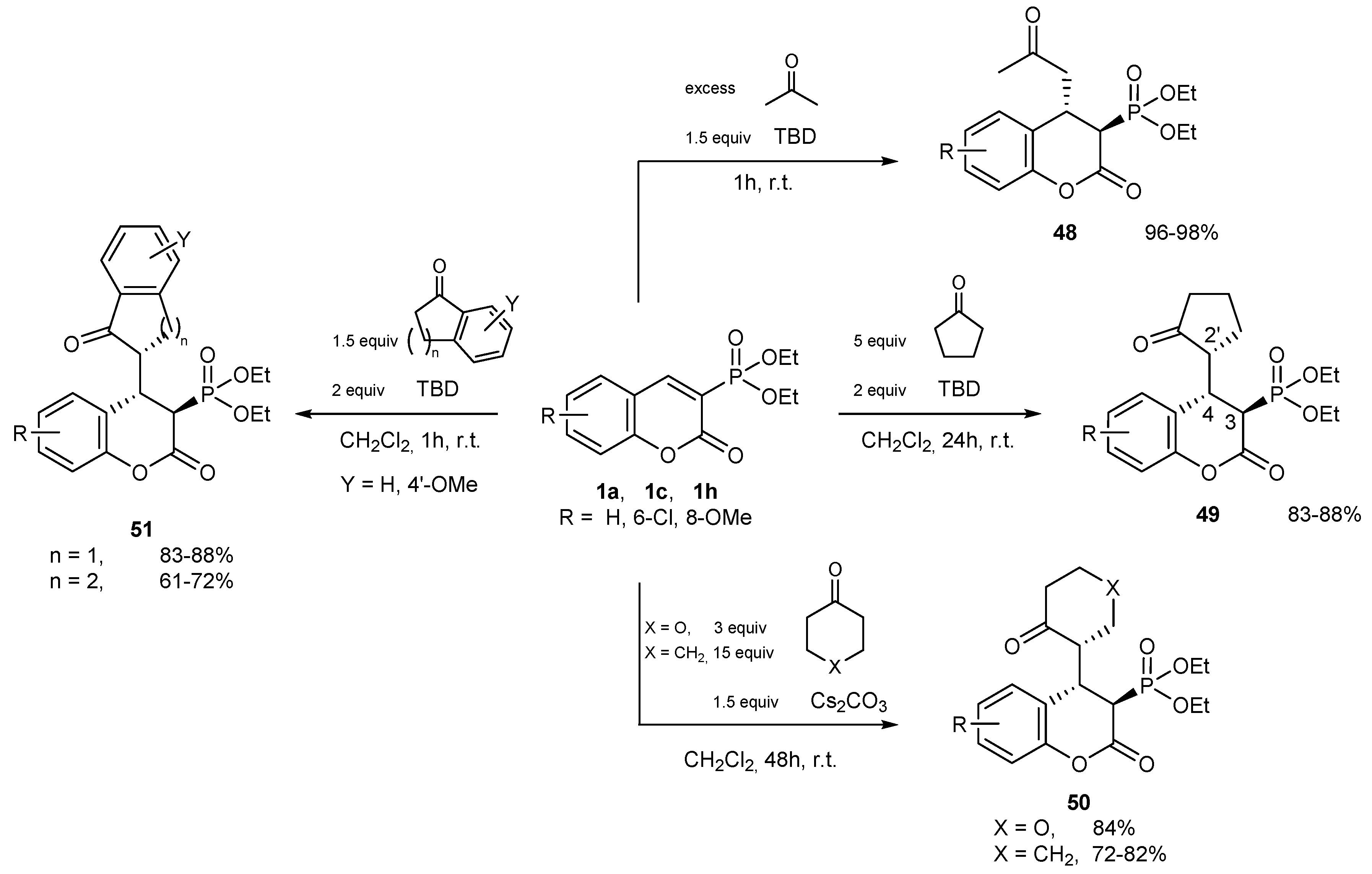
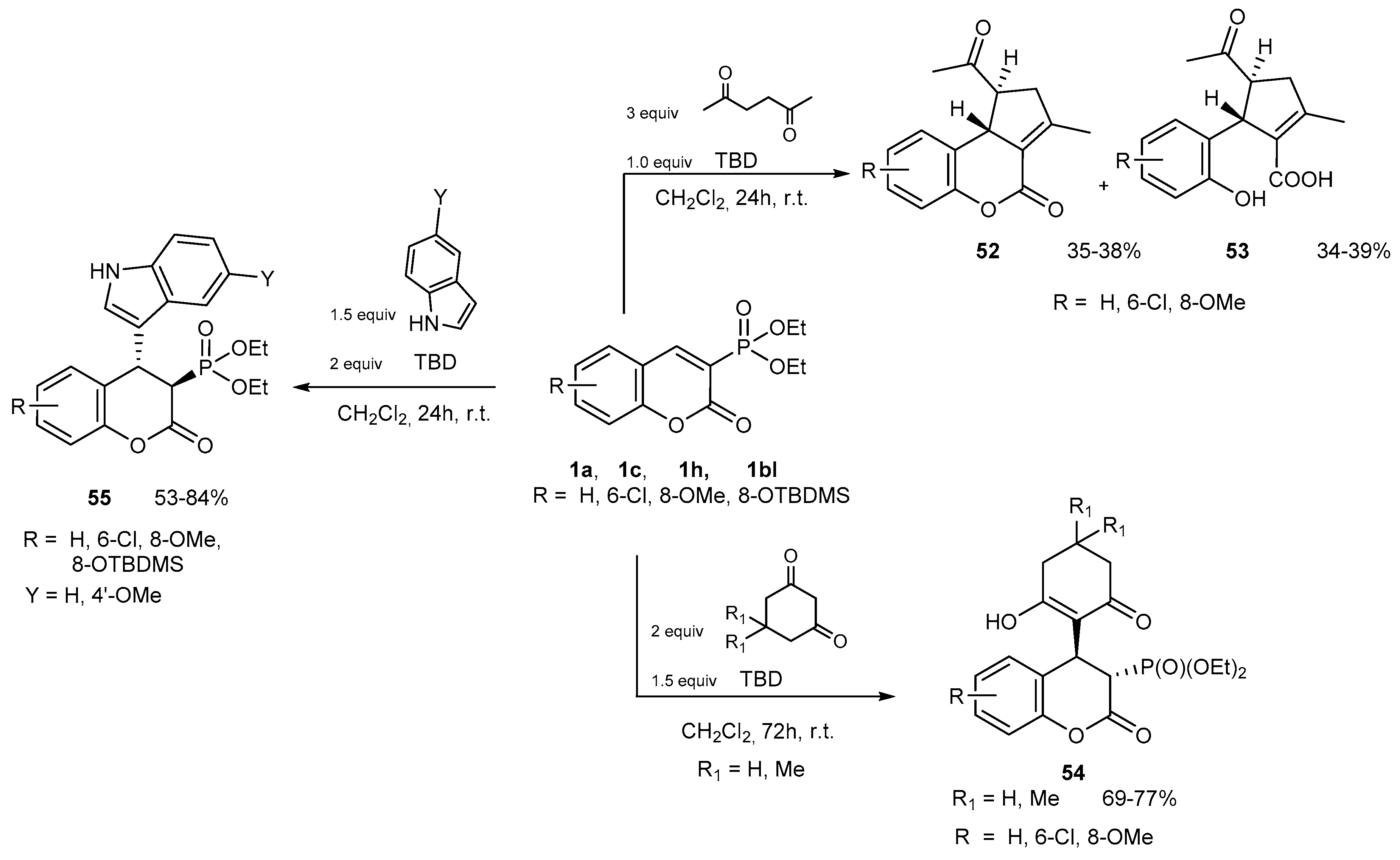
Reactions with CH-acidic Compounds
3. Synthesis and Some Reactions of Alkyl 1,2-benzoxaphosphorin-3-carboxylates 2
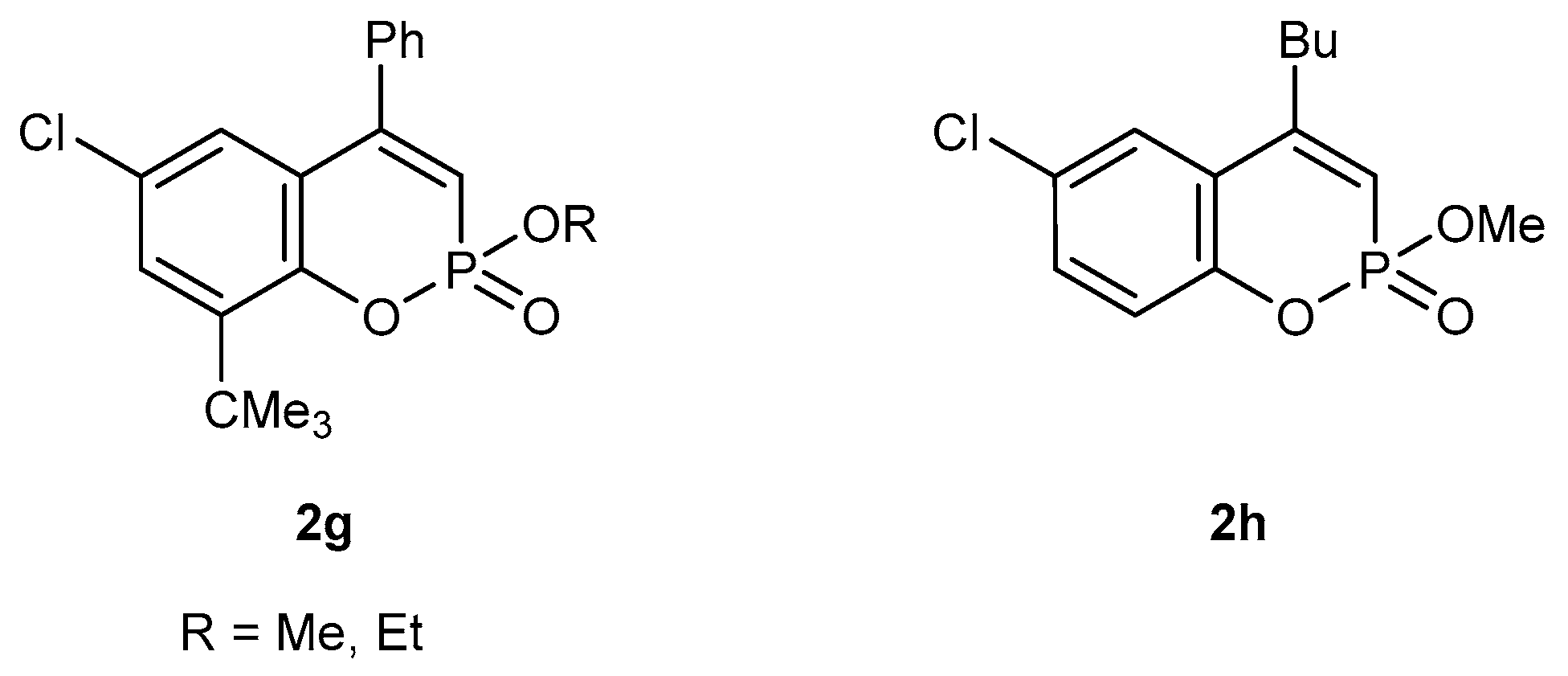
3.1. Synthesis of Substituted Alkyl 1,2-benzoxaphosphorin-3-carboxylates 2
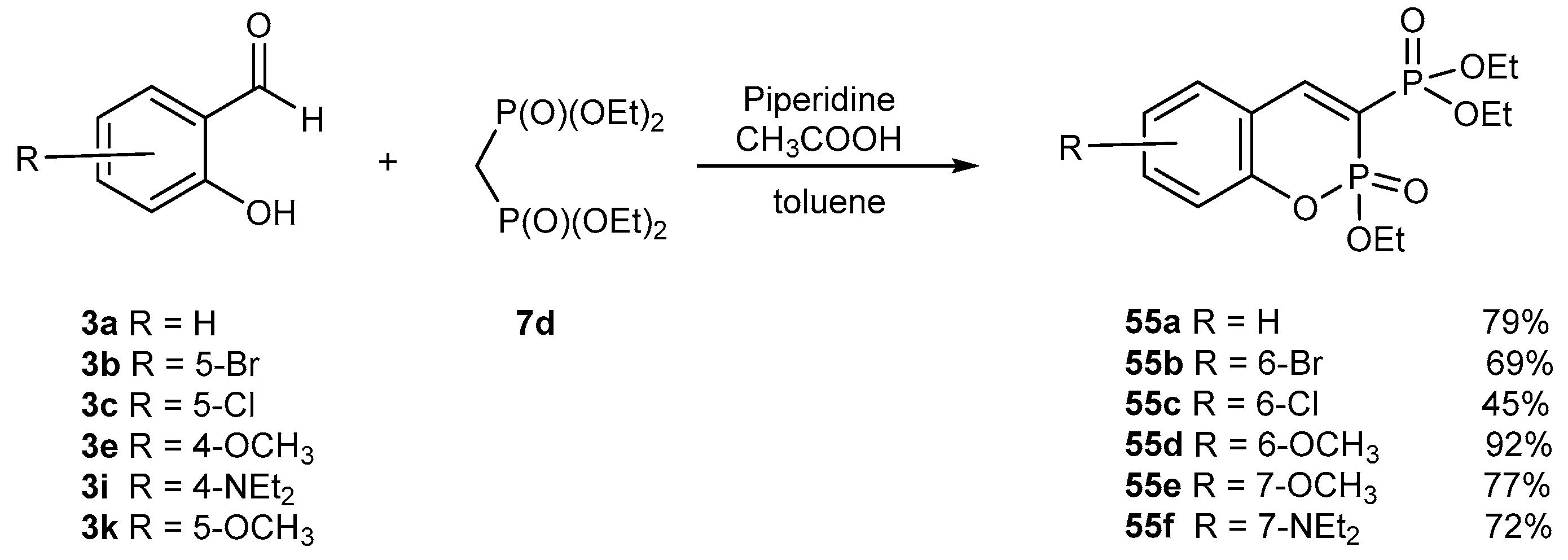
3.1.1. Synthetic Protocols Involving Knoevenagel Condensation Reaction
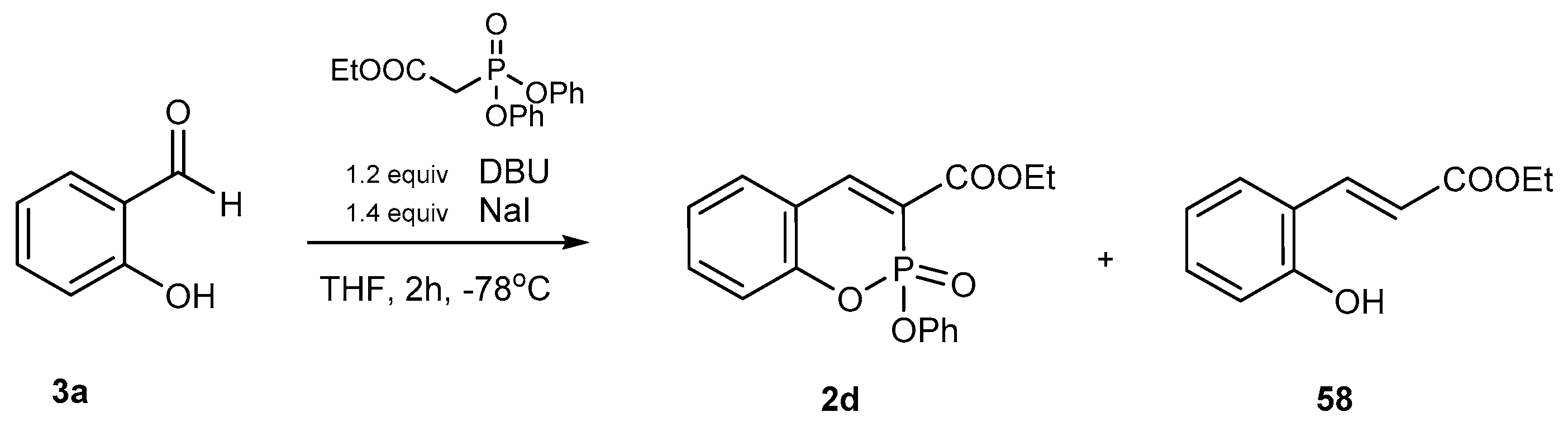
3.1.2. Synthetic Protocols Including Intermolecular Horner-Wadsworth-Emmons Reaction
3.1.3. Synthetic Protocols on the Reaction of Oxaphospholes with Terminal Acetylenes
3.1.4. Synthetic Protocols on A Reaction of Dialkyl Benzylphosphonates with Methyl Salicylate
3.1.5. Synthetic Protocols on for Reactions of 2-Ethoxyvinylphosphonic Dichloride with Substituted Phenols
3.1.6. Synthetic Protocols for Gold-Catalyzed Hydroarylation of Aryl Alkynylphosphonates
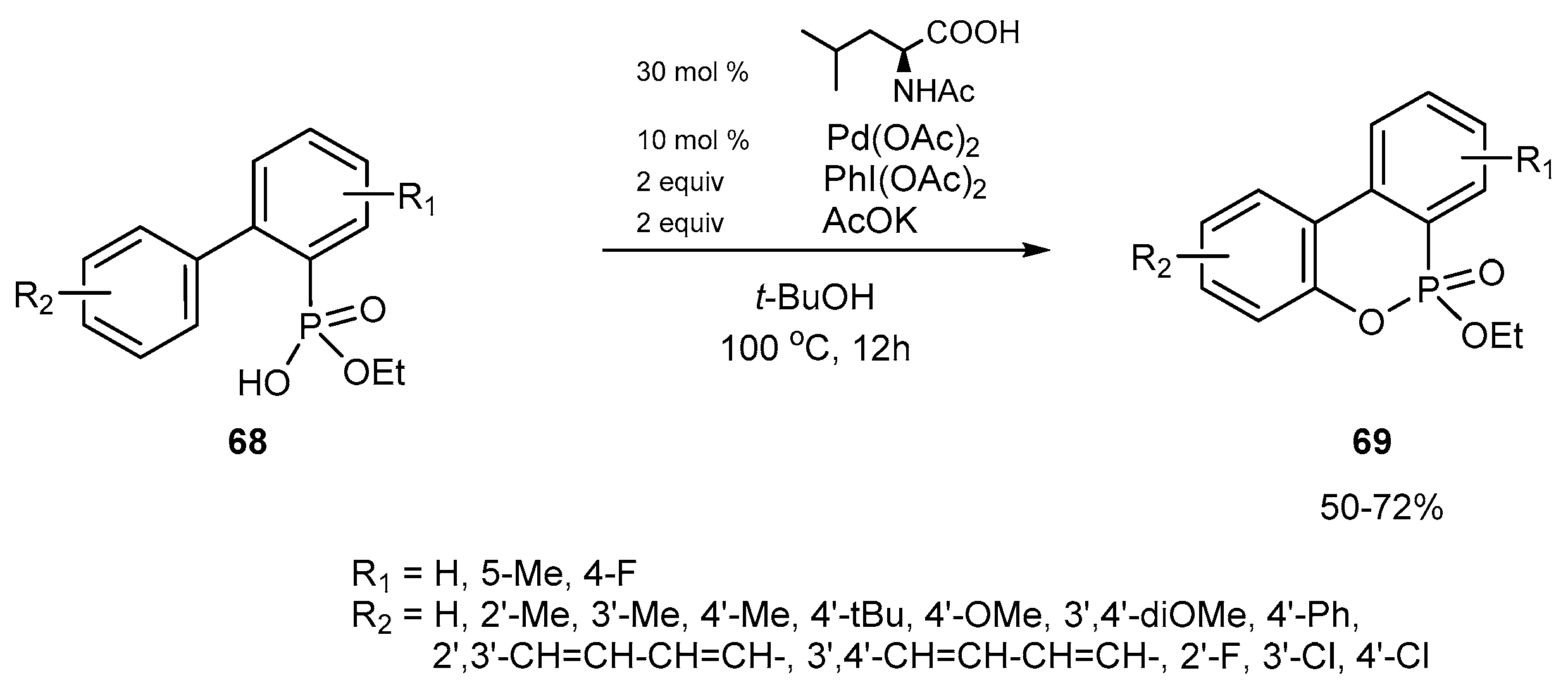
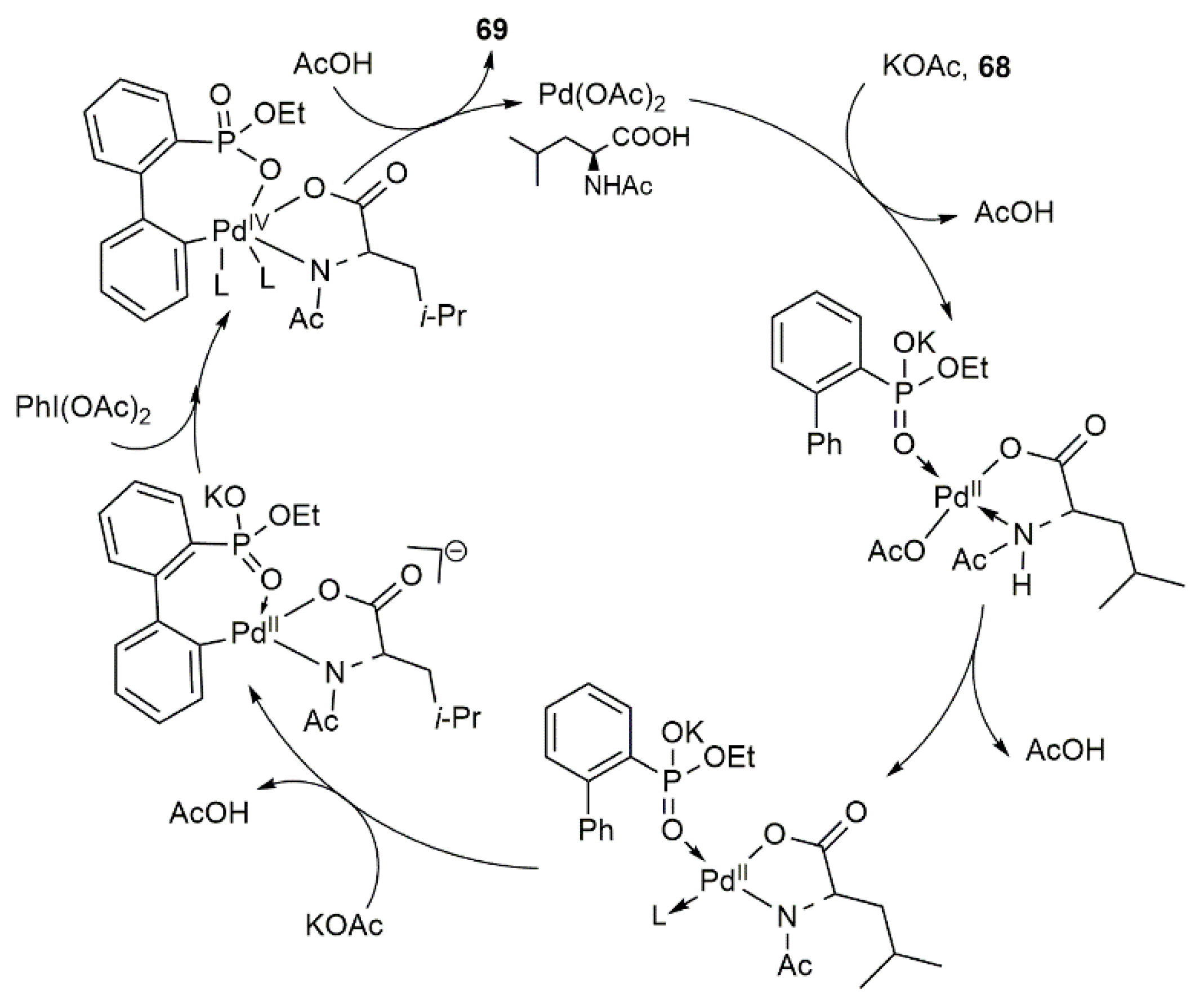
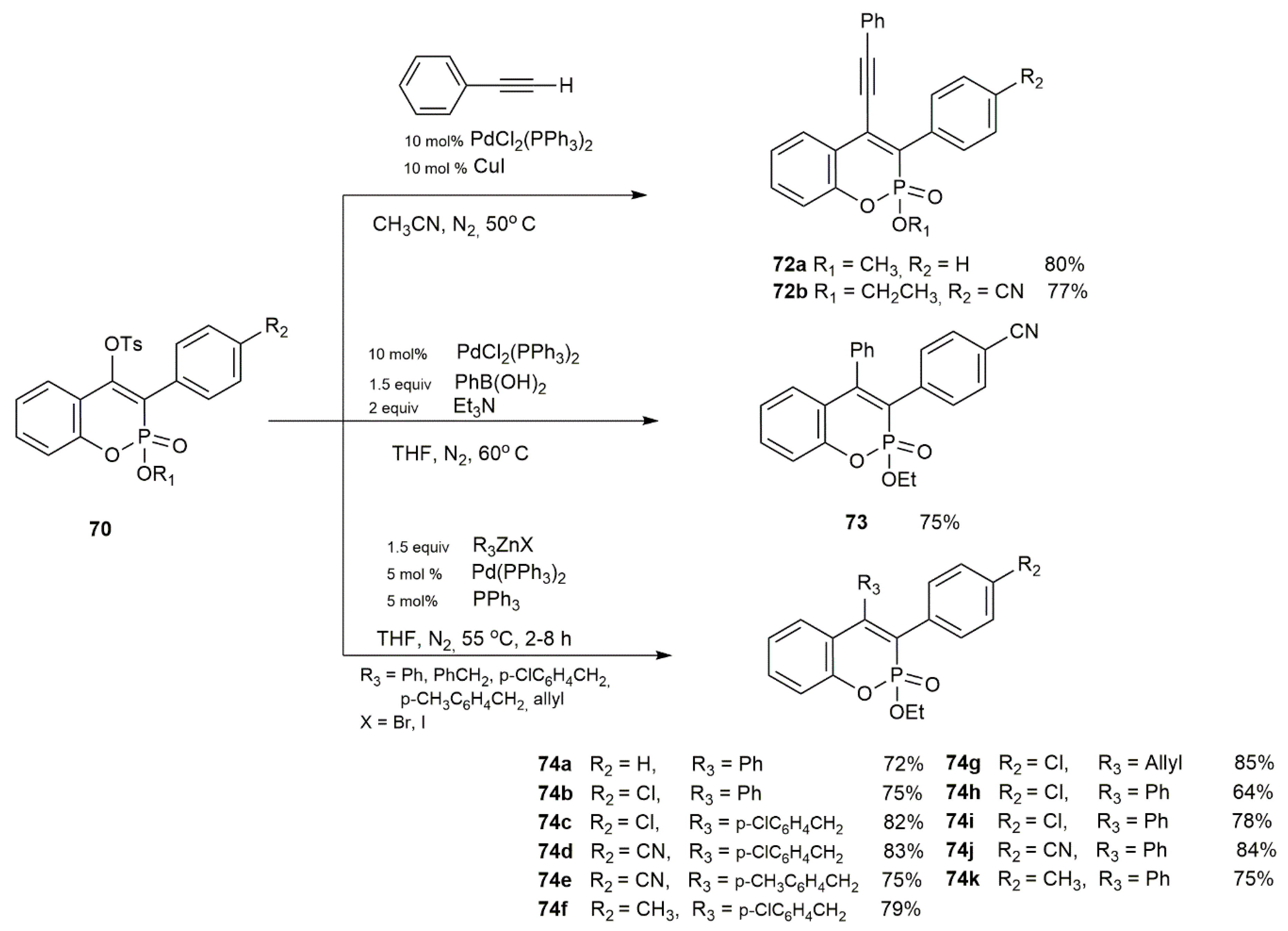
3.1.7. Synthetic Protocols for Pd-Catalyzed Intramolecular Cross-Coupling Reactions of Ethyl 2-(aryl)arylphosphonates
3.2. Chemical Reactions of Alkyl 1,2-benzoxaphosphorin-3-carboxylates 2
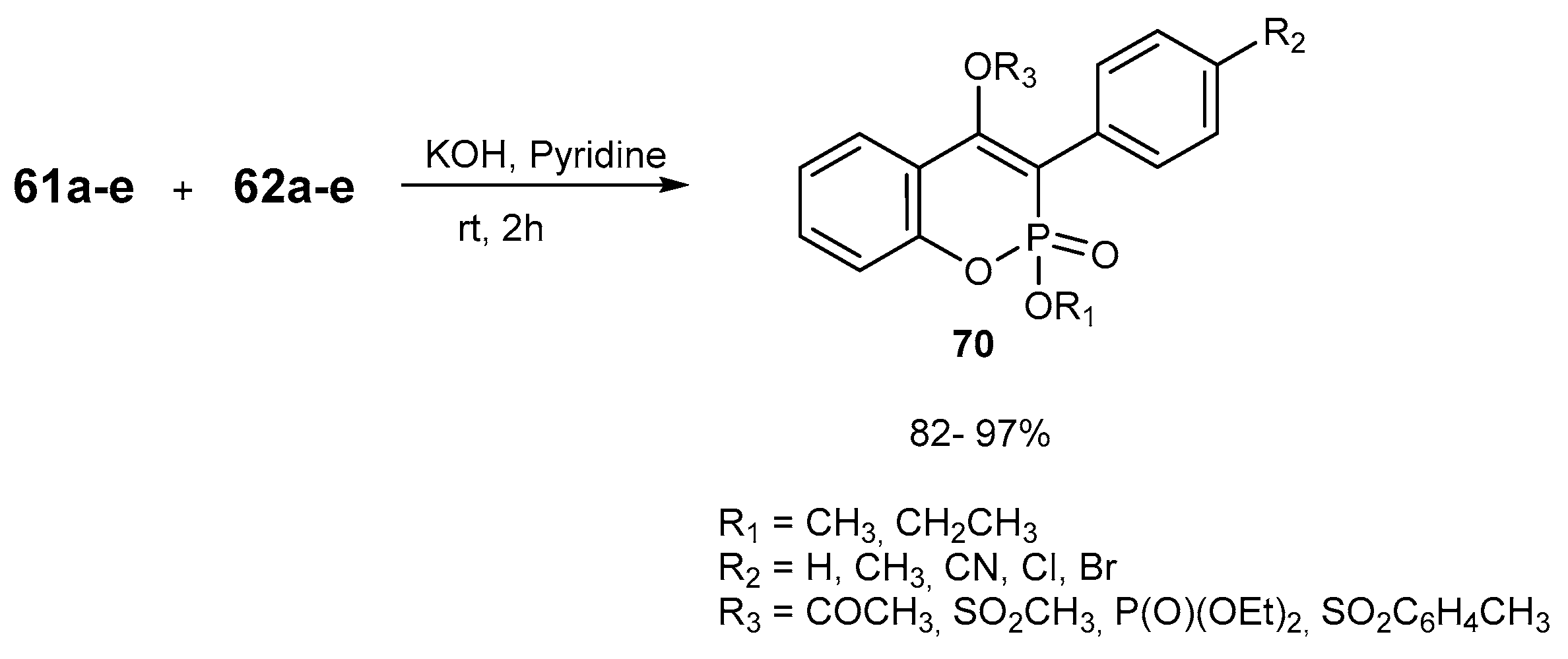
3.2.1. Reactions Resulting in the Formation of 4-O-substituted 1,2-benzoxaphosphorines
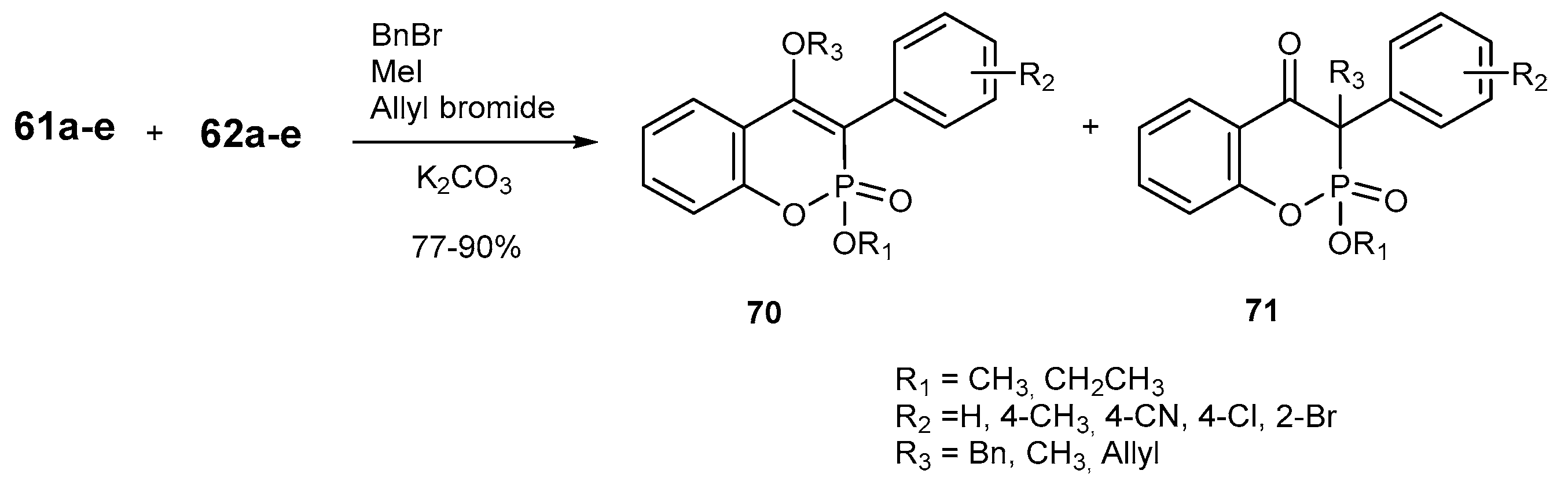
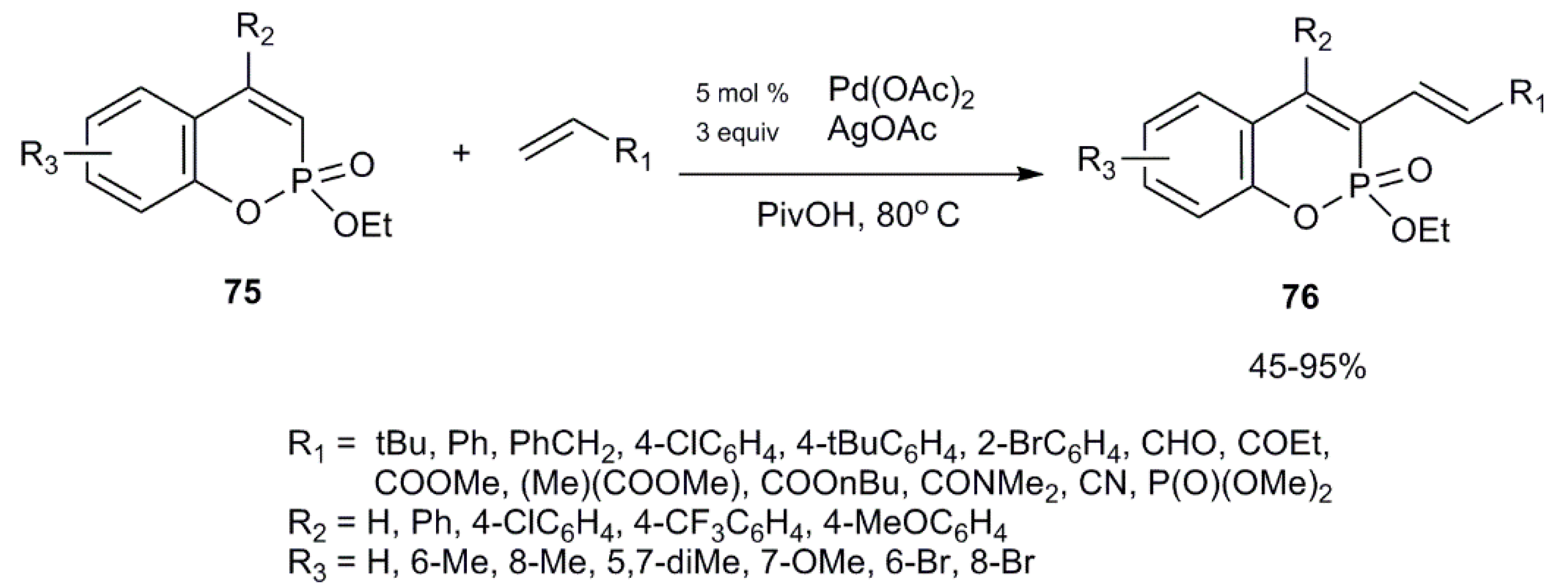
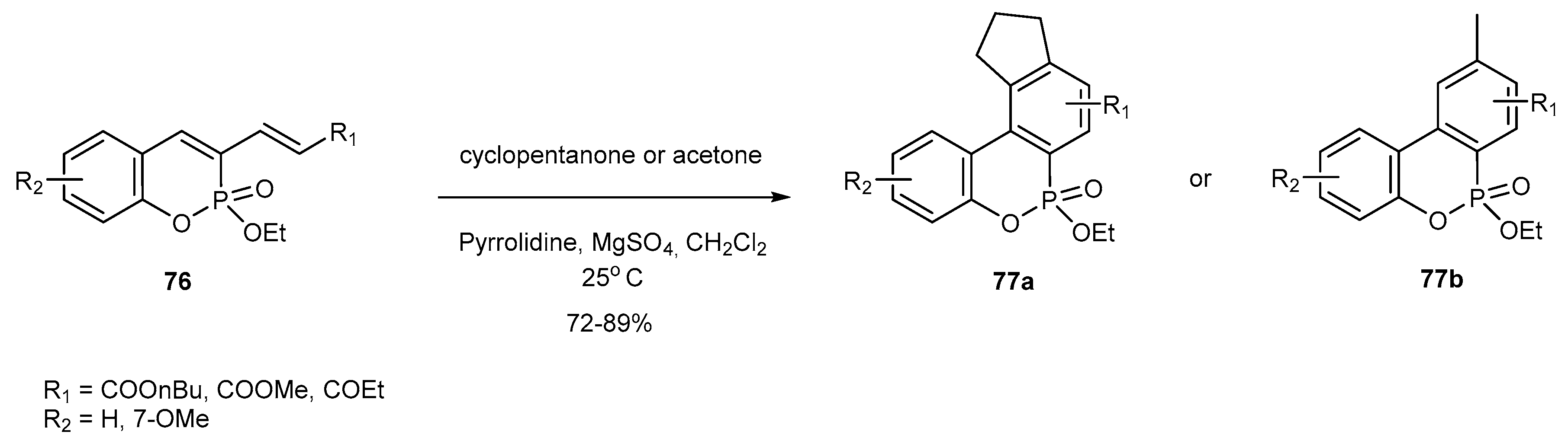
3.2.2. Participation in Coupling Reactions
4. Reactions of [2+2] and [3+2] Cycloadditions of Dialkyl 2-oxo-2H-1-benzopyran-3-phosphonates and Alkyl 1,2-benzoxaphosphorin-3-carboxylates
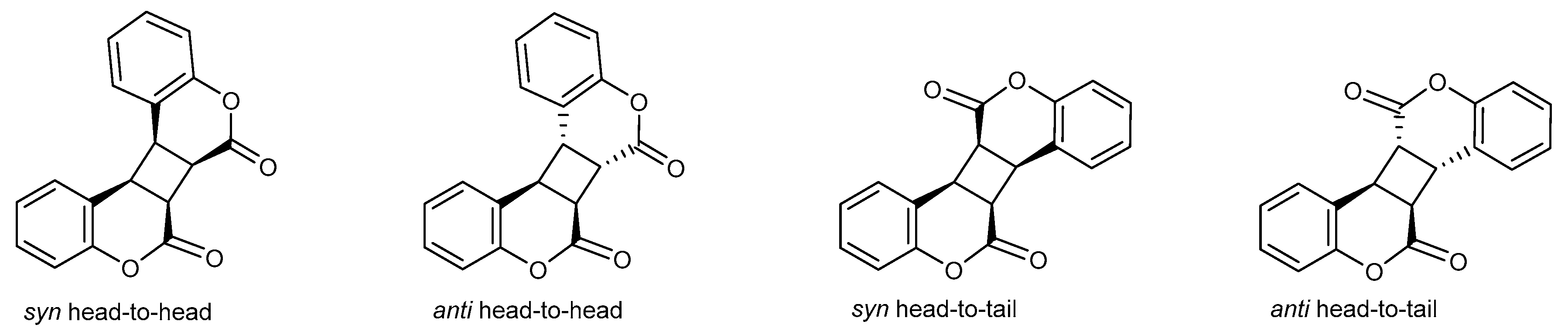
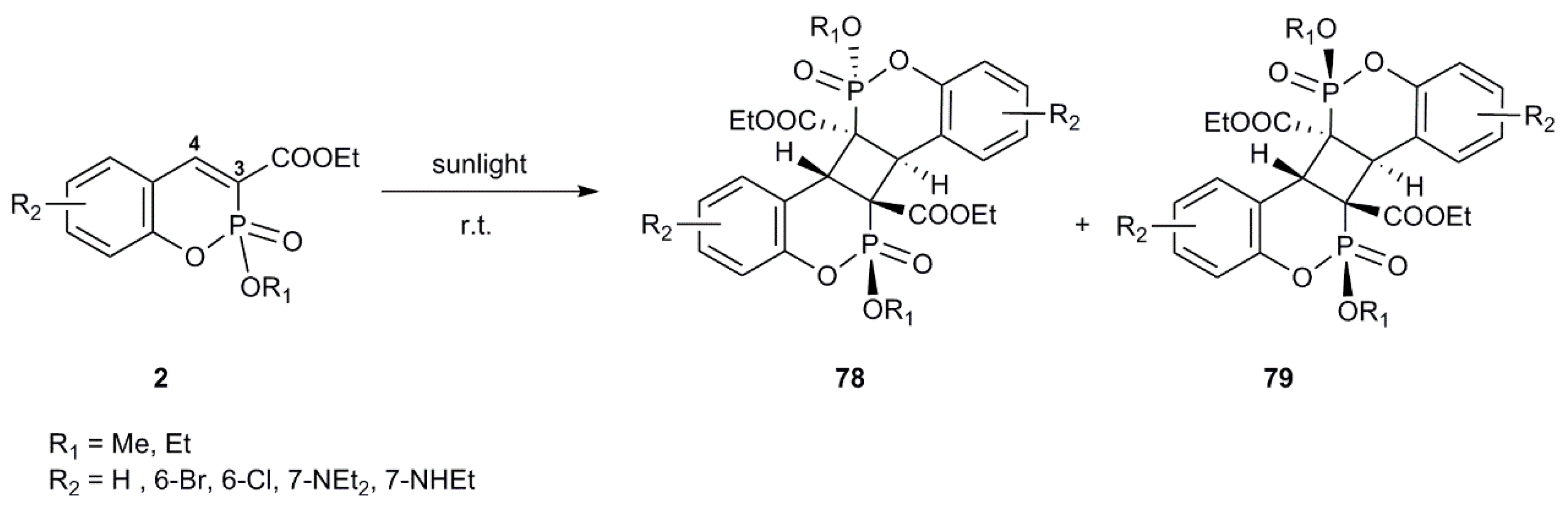
4.1. [2+2] Cycloaddition Reactions
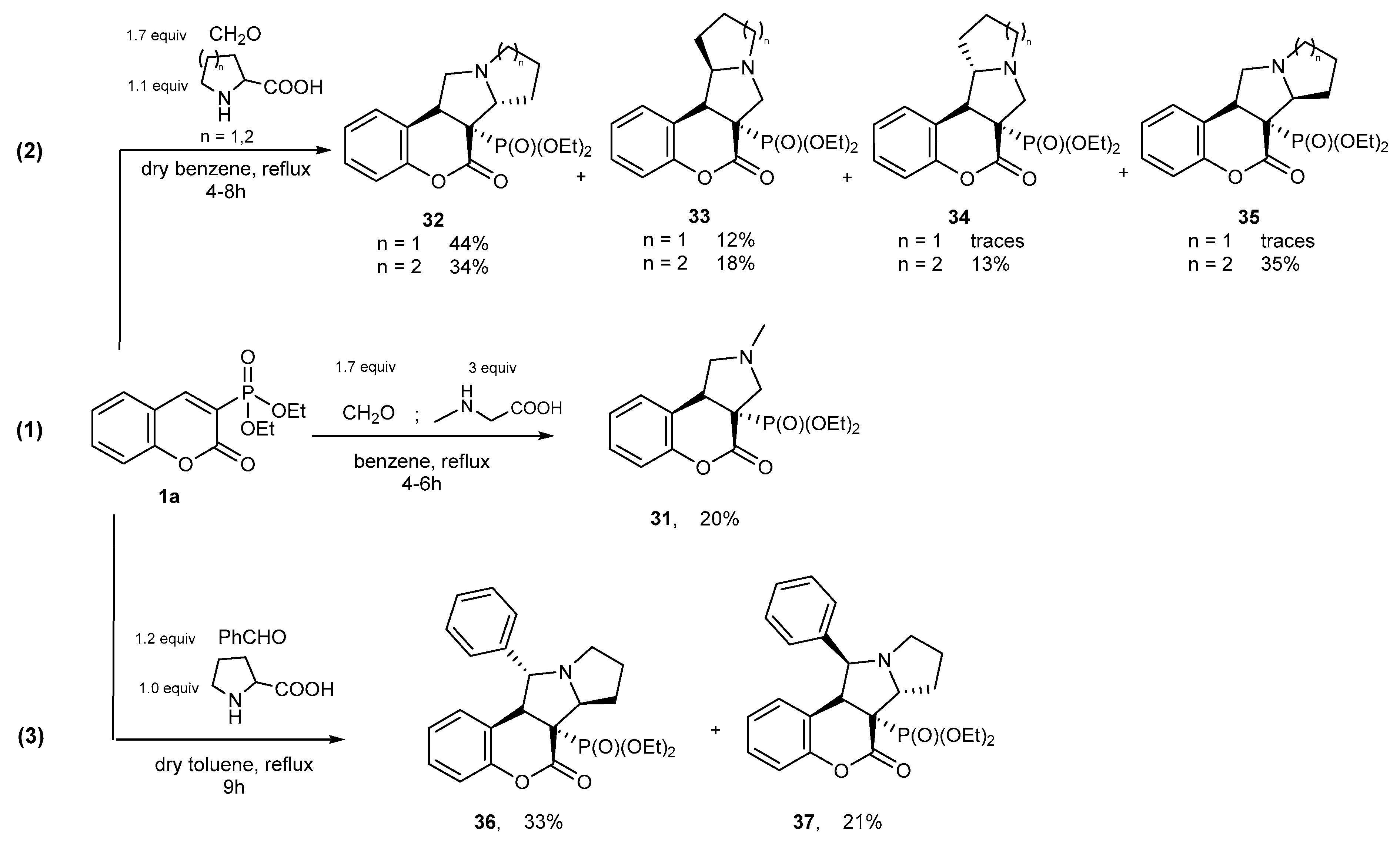
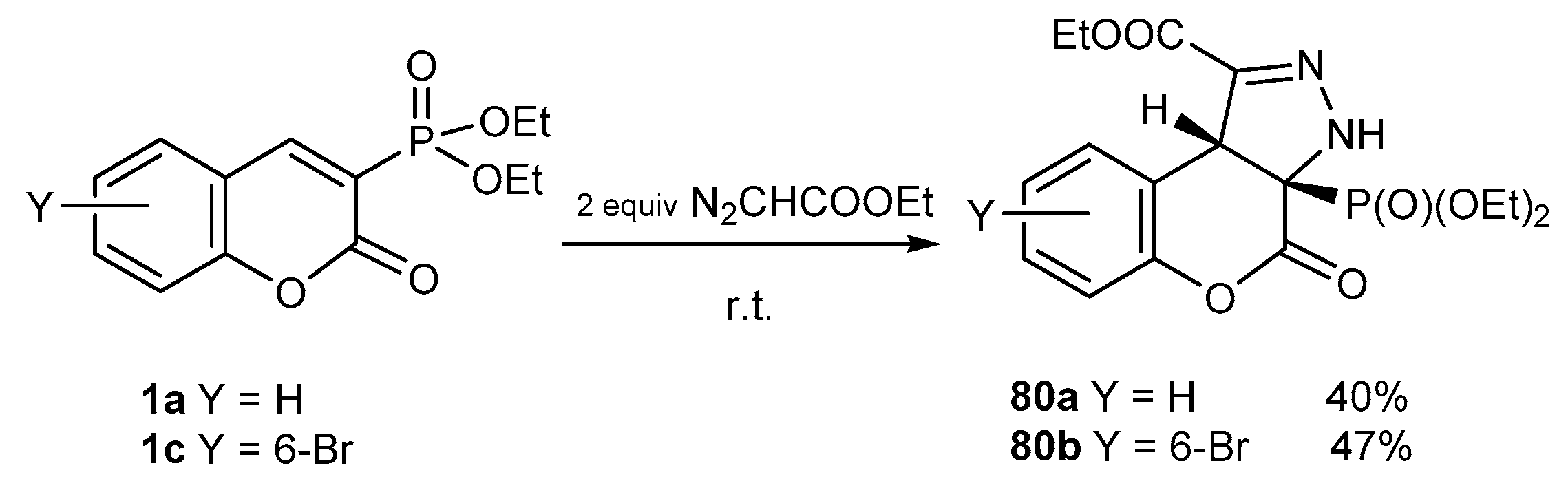
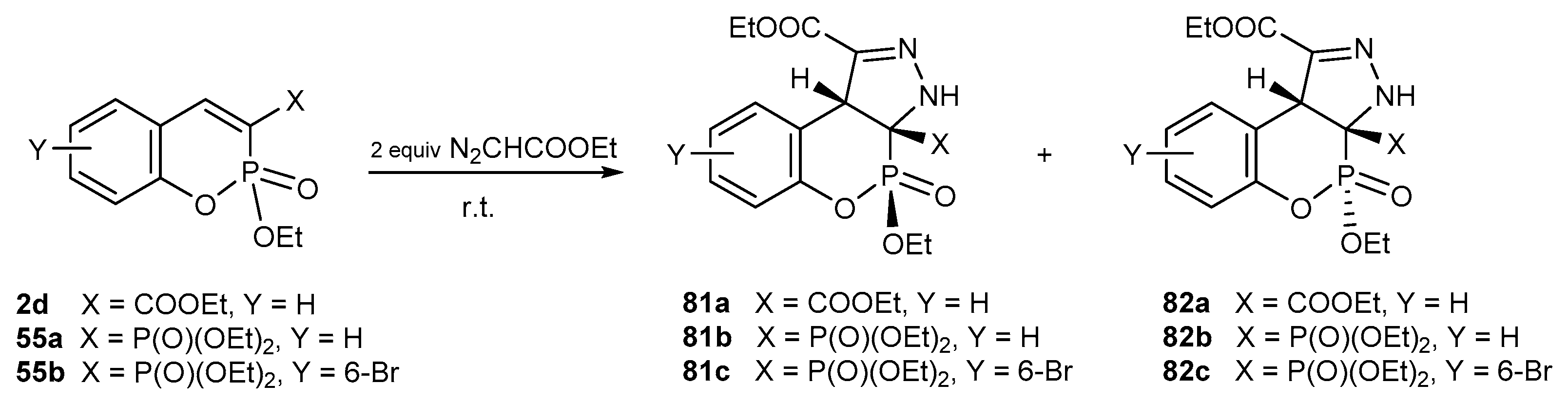
4.2. [3+2] Cycloaddition Reactions
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Borges, F.; Roleira, F.; Milhazes, N.; Santana, L.; Uriarte, E. Simple coumarins and analogues in medicinal chemistry: Occurrence, synthesis and biological activity. Curr. Med. Chem. 2005, 12, 887–916. [Google Scholar] [CrossRef] [PubMed]
- Riveiro, M.E.; De Kimpe, N.; Moglioni, A.; Vázquez, R.; Monczor, F.; Shayo, C.; Davio, C. Coumarins: Old compounds with novel promising therapeutic perspectives. Curr. Med. Chem. 2010, 17, 1325–1338. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Cho, M.; Karlsberg, K.; Zhou, D.; Yuan, Y.-C. Biochemical and Biological Characterization of a Novel Anti-aromatase Coumarin Derivative. J. Biol. Chem. 2004, 279, 48071–48078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, M.V.; Kulkarni, G.M.; Lin, C.-H.; Sun, C.-M. Recent Advances in Coumarins and 1-Azacoumarins as Versatile Biodynamic Agents. Curr. Med. Chem. 2006, 13, 2795–2818. [Google Scholar] [CrossRef] [PubMed]
- Al-Majedy, Y.; Kadhum, A.A.; Ibraheem, H.; Al-Amiery, A.; Moneim, A.A.; Mohamad, A.B. A Systematic Review on Pharmacological Activities of 4-Methylumbelliferon. Sys. Rev. Pharm. 2018, 9, 49–54. [Google Scholar] [CrossRef]
- Kitson, R.; Millemaggi, A.; Taylor, R. The Renaissance of a-Methylene-g-butyrolactones: New Synthetic Approaches. Angew. Chem. Int. Ed. 2009, 48, 9426–9451. [Google Scholar] [CrossRef]
- Zhang, S.; Won, Y.-K.; Ong, C.-N.; Shen, H.-M. Anti-Cancer Potential of Sesquiterpene Lactones: Bioactivity and Molecular Mechanisms. Curr. Med. Chem. Anti-Cancer Agents 2005, 5, 239–249. [Google Scholar] [CrossRef]
- Janecka, A.; Wyrębska, A.; Gach, K.; Fichna, J.; Janecki, T. Natural and synthetic α-methylenelactones and α-methylenelactams with anticancer potential. Drug Discov. Today 2012, 7, 561–572. [Google Scholar] [CrossRef]
- Albrecht, L.; Albrecht, A.; Janecki, T. α-Alkylidene-γ- and δ-lactones and lactams. In Natural Lactones and Lactams: Synthesis, Occurrence and Biological Activity; Janecki, T., Ed.; Wiley-VCH: Weinheim, Germany, 2014; pp. 147–192. ISBN 9783527666911. [Google Scholar]
- Perucca, E. The new generation of antiepileptic drugs: Advantages and disadvantages. Br. J. Clin. Pharmacol. 1996, 42, 531–543. [Google Scholar] [CrossRef]
- Wong, M.; Defina, J.; Andrews, P. Conformational Analysis of Clinically Active Anticonvulsant Drugs. J. Med. Chem. 1986, 29, 562–572. [Google Scholar] [CrossRef]
- Tasso, S.; Bruno-Blanch, L.; Moon, S.; Estiú, G. Pharmacophore searching and QSAR analysis in the design of anticonvulsant drugs. J. Mol. Struct. (Theochem) 2000, 504, 229–240. [Google Scholar] [CrossRef]
- Hussain, H.; Hussain, J.; Al-Harrasi, A.; Krohn, K. The chemistry and biology of biscoumarins. Tetrahedron 2012, 68, 2553–2578. [Google Scholar] [CrossRef]
- Budzisz, E. Synthesis, reactions and biological activity of phosphorus-containing derivatives of chromone and coumarin. Phosphorus Sulfur Silicon 2004, 179, 2131–2147. [Google Scholar] [CrossRef]
- Robinson, C.N.; Addison, J.F. Condensation of Triethyl Phosphonoacetate with Aromatic Aldehydes. J. Org. Chem. 1966, 31, 4325–4326. [Google Scholar] [CrossRef]
- Singh, R.K.; Rogers, M.D. An efficient synthesis of diethyl coumarin-3-phosphonates. J. Heterocycl. Chem. 1985, 22, 1713–1714. [Google Scholar] [CrossRef]
- Chen, C.H.; Fox, J.L.; Lippert, E.K. Synthesis of phosphacoumarins and phosphonocoumarins. Two new classes of fluorescent dyes. J. Heterocycl. Chem. 1987, 24, 931–932. [Google Scholar] [CrossRef]
- Bouyssou, P.; Chenault, J. Phosphonates and phosphine oxides as reagents in a one-pot synthesis of coumarins. Tetrahedron Lett. 1991, 32, 5341–5344. [Google Scholar] [CrossRef]
- Falsone, G.; Cateni, F.; De Nardo, M.; Darai, M. Synthesis of 3-Alkylcoumarins and 3-Alkyl-α,β-unsaturated δ-Lactones from 3-Diethylphosphonocoumarins, 3-Diethylphosphonolactones and Aldehydes. Zeitschrift für Naturforschung B 1993, 48, 1391–1397. [Google Scholar] [CrossRef]
- Bojilova, A.; Nikolova, R.; Ivanov, C.; Rodios, N.; Terzis, A.; Raptopoulou, C. A comparative study of the interaction of salicylaldehydes with phosphonoacetates under Knoevenagel reaction conditions. Synthesis of 1,2-benzoxaphosphorines and their dimers. Tetrahedron 1996, 52, 12597–12612. [Google Scholar] [CrossRef]
- Kostka, K.; Pastuszko, S.; Kotynski, A.; Budzisz, E. 4-Derivatives Coumarin-3-Phosphonic Acids and Esters. Phosphorus Sulfur Silicon 1998, 134, 199–209. [Google Scholar] [CrossRef]
- Bestmann, H.; Lehnen, H. Synthese und reaktionen des N-phenyl-bis(diethylphosphonato)-ketenimins. Tetrahedron Lett. 1991, 32, 4279–4282. [Google Scholar] [CrossRef]
- Janecki, T.; Albrecht, A.; Koszuk, J.; Mondranka, J.; Slowak, D. A simple and effective synthesis of activated vinylphosphonates from 3-methoxy-2-diethoxyphosphorylacrylate. Tetrahedron Lett. 2010, 51, 2274–2276. [Google Scholar] [CrossRef]
- Mondranka, J.; Albrecht, A.; Janecki, T. A Convenient Entry to 3-Methylidenechroman-2-ones and 2-Methylidenedihydrobenzochromen-3-ones. Synlett 2010, 19, 2867–2870. [Google Scholar] [CrossRef]
- Mondranka, J.; Albrecht, A.; Jakubowski, R.; Krawczyk, H.; Rozalski, M.; Krajewska, U.; Janecka, A.; Wyrebska, A.; Rozalska, B.; Janecki, T. Synthesis and biological evaluation of α-methylidene-δ-lactones with 3,4-dihydrocoumarin skeleton. Biorg. Med. Chem. 2012, 20, 5017–5026. [Google Scholar] [CrossRef]
- Jakubowski, R.; Romorska, D.; Dlugosz, A.; Janecka, A.; Krajewska, U.; Rozalski, M.; Mirowski, M.; Bartosik, T.; Janecki, T. Synthesis of 4,4-Disubstituted 3-Methylidenechroman-2-ones as Potent Anticancer Agents. Chem. Med. Chem. 2017, 12, 599–605. [Google Scholar] [CrossRef]
- Pomorska, D.; Gach-Janczak, K.; Jakubowski, R.; Janecki, T.; Szymanski, J.; Janecka, A. Evaluation of anticancer properties of a new α-methylene-δ-lactone DL-249 on two cancer cell lines. Open Life Sci. 2017, 12, 178–179. [Google Scholar] [CrossRef]
- Pan, X.; Zou, J.; Zhang, G.; Zhang, W. Manganese(III)-mediated direct phosphonation of arylalkenes and arylalkynes. Chem. Commun. 2010, 46, 1721–1723. [Google Scholar] [CrossRef]
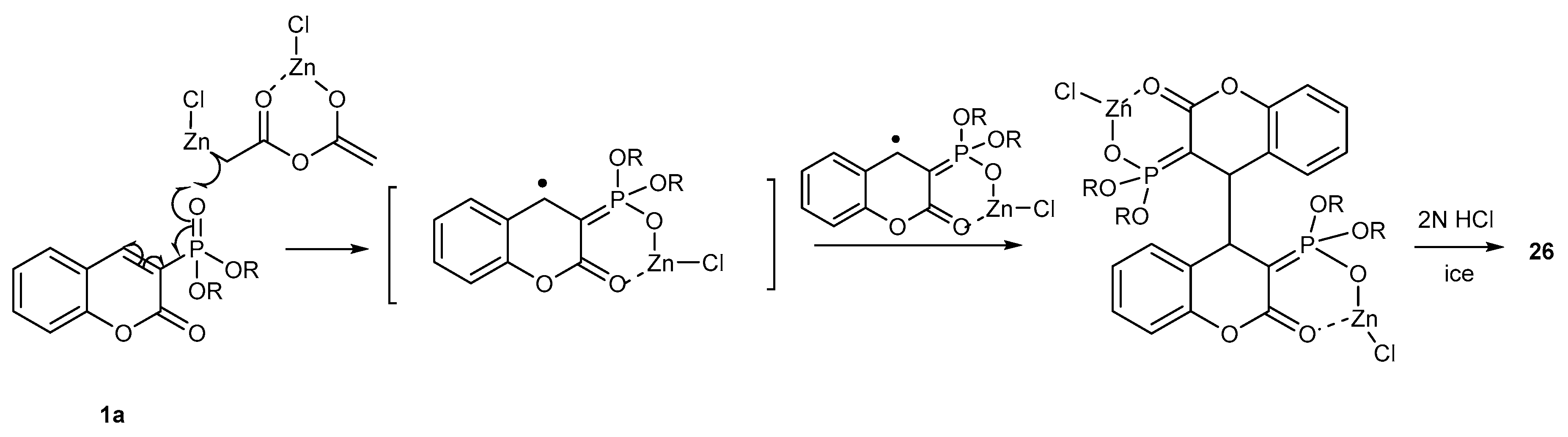
- Zhou, P.; Jiang, Y.; Zou, J.; Zhang, W. Manganese(III) Acetate Mediated Free-Radical Phosphonylation of Flavones and Coumarins. Synthesis 2012, 44, 1043–1050. [Google Scholar] [CrossRef]
- Mi, X.; Huang, M.; Zhang, J.; Wang, C.; Wu, Y. Regioselective Palladium-Catalyzed Phosphonation of Coumarins with Dialkyl H-Phosphonates via C–H Functionalization. Org. Lett. 2013, 15, 6266–6269. [Google Scholar] [CrossRef]
- Yuan, J.; Li, Y.; Yang, L.; Mai, W.; Mao, P.; Xiao, Y.; Qu, L. Silver-catalyzed direct Csp2-H radical phosphorylation of coumarins with H-phosphites. Tetrahedron 2015, 71, 8178–8186. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, X.; Yuan, J.; Xiao, Y.; Mao, P. Catalytic activity of chelating N-heterocyclic carbene palladium complexes towards selective phosphorylation of coumarins. J. Organomet. Chem. 2016, 818, 179–184. [Google Scholar] [CrossRef]
- Strekalova, S.; Khrizanforov, M.; Gryaznova, T.; Khrizanforova, V.; Budnikova, Y.H. Electrochemical phosphorylation of coumarins catalyzed by transition metal complexes (Ni—Mn, Co—Mn). Russ. Chem. Bull. 2016, 65, 1295–1298. [Google Scholar] [CrossRef]
- Grinenko, V.; Khrizanforov, M.; Strekalova, S.; Khrizanforova, V.; Kholin, K.; Gryaznova, T.; Budnikova, Y.H. Electrooxidative phosphorylation of coumarins by bimetallic catalytic systems Ni(II)/Mn(II) or Co(II)/Mn(II). Phosphorus Sulfur Silicon Relat. Elem. 2016, 191, 1660–1661. [Google Scholar] [CrossRef]
- Khrizanforov, M.; Strekalova, S.; Kholin, K.; Khrizanforova, V.; Kadirov, M.; Gryaznova, T.; Budnikova, Y.H. Novel approach to metal-induced oxidative phosphorylation of aromatic compounds. Catal. Today 2017, 279, 133–141. [Google Scholar] [CrossRef]
- Yoshida, H.; Ito, Y.; Ohshita, J. Three-component coupling using arynes and DMF: Straightforward access to coumarins via ortho-quinone methides. Chem. Commun. 2011, 47, 8512–8514. [Google Scholar] [CrossRef]
- Mi, X.; Wang, C.; Huang, M.; Zhang, J.; Wu, Y.; Wu, Y. Silver-Catalyzed Synthesis of 3-Phosphorated Coumarins via Radical Cyclization of Alkynoates and Dialkyl H-Phosphonates. Org. Lett. 2014, 12, 3356–3359. [Google Scholar] [CrossRef]
- Liu, D.; Chen, J.; Wang, X.; Xu, P. Metal-Free, Visible-Light-Promoted Synthesis of 3-Phosphorylated Coumarins via Radical C−P/C−C Bond Formation. ASC Commun. 2017, 359, 2773–2777. [Google Scholar] [CrossRef]
- Abdou, W.; Shaddy, A. Application of Dialkyl Cyanomethylphosphonates in Synthesis of Biologically Active Phosphono-Substituted Heterocycles and Vinylphosphonates. Synth. Commun. 2006, 31, 1953–1964. [Google Scholar] [CrossRef]
- Alexieva, V.; Karanov, E.; Nikolova, R.; Bojilova, A. Plant Growth Regulating Activity of Some Phosphorus Derivatives of Coumarin. Bulg. J. Plant Physiol. 1995, 21, 45–51. [Google Scholar]
- Budzisz, E.; Brzezcnska, E.; Krajewska, U.; Rozatski, M. Cytotoxic effects, alkylating properties and molecular modelling of coumarin derivatives and their phosphonic analogues. Eur. J. Med. Chem. 2003, 38, 597–603. [Google Scholar] [CrossRef]
- Bojilova, A.; Ivanov, C. Two new routes to esters of 2-oxochroman-4-acetic acid. Synthesis 1976, 4, 267–268. [Google Scholar] [CrossRef]
- Ivanov, C.; Bojilova, A. Umwandlung von 2-oxo-2H-1-benzopyran-3-carbonsaureestern in 2-oxo-4-chromanessigsaureester—eine neue Umlagerung. Chem. Ber. 1978, 111, 3755–3763. [Google Scholar] [CrossRef]
- Bojilova, A.; Rodios, N.; Nikolova, R.; Ivanov, C. Reactions of 3-acyl-substituted-2H-1-benzopyran-2-ones with acid anhydrides, II. Liebigs Ann. Chem. 1991, 12, 1279–1284. [Google Scholar] [CrossRef]
- Bojilova, A.; Rodios, N.; Nikolova, R.; Ivanov, C. Potassium fluoride promoted reaction of 3-acylsubstituted 2H-1-benzopyran-2-ones with acid anhydrides. An improved method for the synthesis of 4-(2-oxoalkyl)-2H-chroman-2-ones. Part III. Synth. Commun. 1992, 22, 741–754. [Google Scholar] [CrossRef]
- Bojilova, A.; Ivanov, C. On the Reaction of 3-Phenylcoumarin with Organomagnesium Compounds. Synthesis 1974, 708–709. [Google Scholar] [CrossRef]
- Kasantsev, A.V.; Butyaikin, V.V.; Otrashchenkov, E.A.; Muldakhmetov, Z.M. Conjugated addition of lithium and magnesium derivatives of o-carboranes to 3-carbethoxycoumarin. Russ. Chem. Bull. 1995, 44, 1976–1977. [Google Scholar] [CrossRef]
- Nikolova, R.D.; Bojilova, A.; Rodios, N.A. A new and efficient method for conjugate addition of trialkylphosphites to 3-acylsubstituted coumarins. Tetrahedron 2004, 60, 10335–10342. [Google Scholar] [CrossRef]
- Desyatkln, V.G.; Beletskaya, I.P. Asymmetric Friedel–Crafts/Michael Reaction of Indoles and Pyrroles with Coumarin-3-carbonylates. Synthesis 2017, 49, 4327–4334. [Google Scholar] [CrossRef]
- Petkova, N.I.; Nikolova, R.D.; Kostov, K.L.; Mineva, T.; Vayssilov, G.N. Theoretical and Experimental Local Reactivity Parameters of 3-Substituted Coumarin Derivatives. J. Phys. Chem. A 2014, 118, 11062–11073. [Google Scholar] [CrossRef]
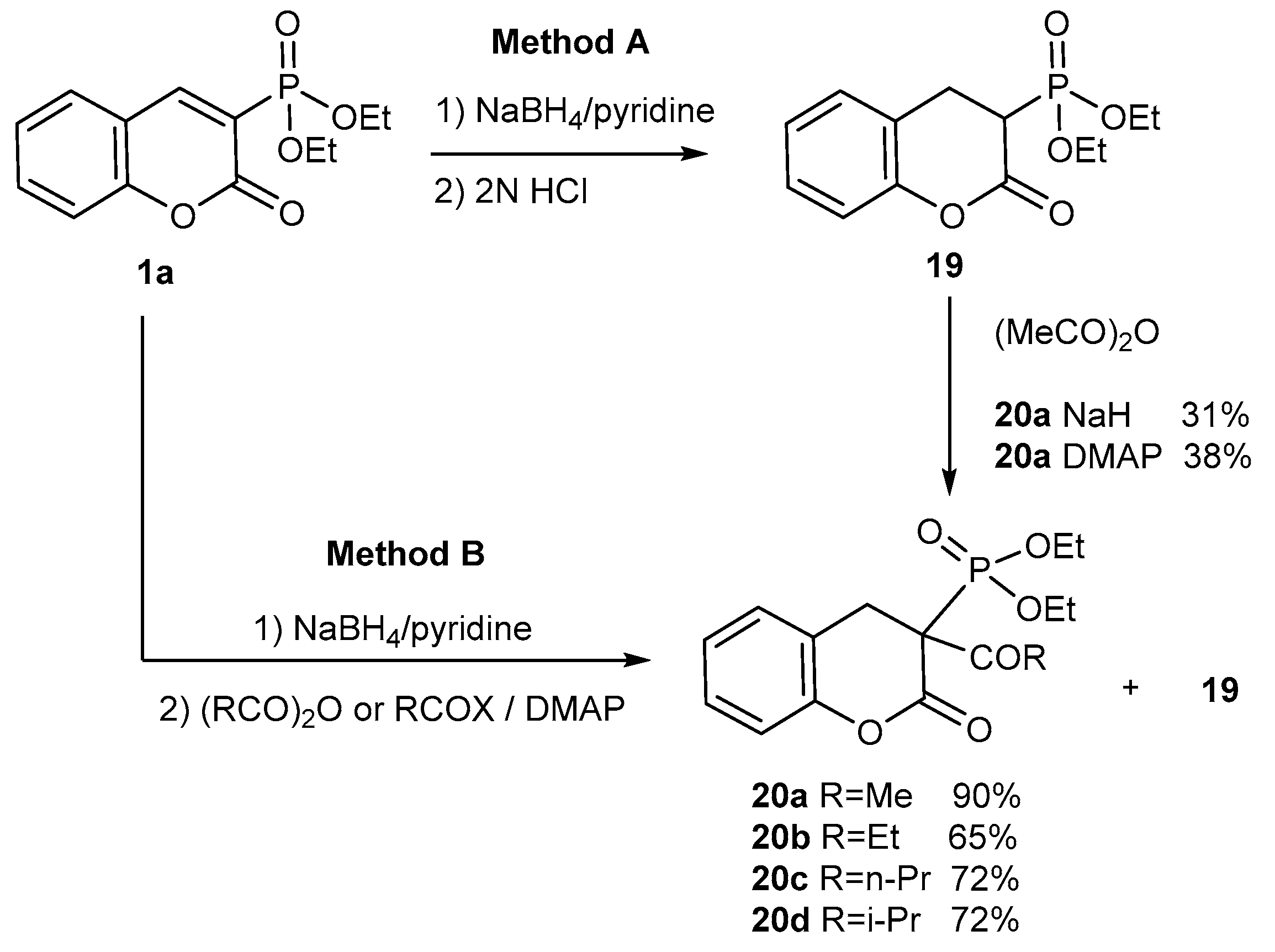
- Petkova, N.I.; Nikolova, R.D.; Bojilova, A.G.; Rodios, N.A.; Raptopoulou, C.P. Hydrogenation/Regioselective C-Acylation Reaction of Diethyl Coumarin-3-phosphonate with NaBH4/Acid Anhydrides: A New One-Pot Tandem Reaction. Synth. Commun. 2006, 36, 509–524. [Google Scholar] [CrossRef]
- Janecki, T.; Wąsek, T. A Novel Route to Substituted 3-Methylidenechroman-2-ones and 3-Methylchromen-2-ones. Tetrahedron 2004, 60, 1049–1055. [Google Scholar] [CrossRef]
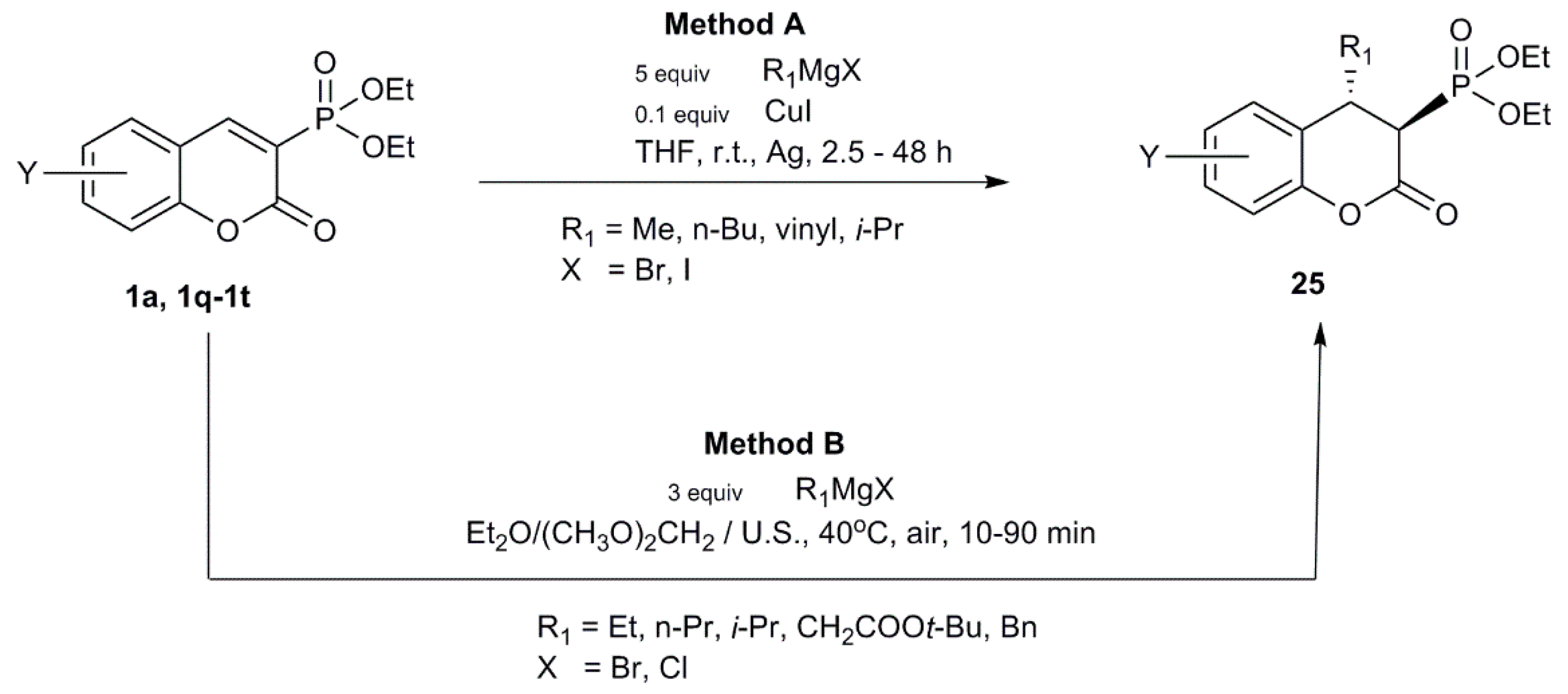
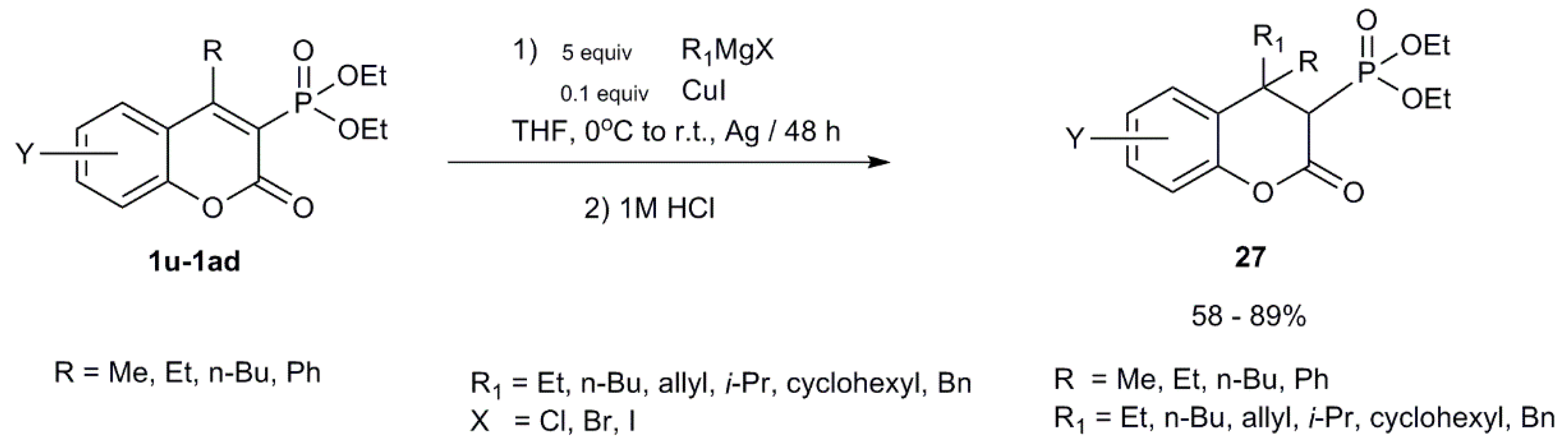
- Koleva, A.I.; Petkova, N.I.; Nikolova, R.D. Ultrasound-Assisted Conjugate Addition of Organometallic Reagents to 3-Diethylphosphonocoumarin. Synlett 2016, 27, 2676–2680. [Google Scholar] [CrossRef] [Green Version]
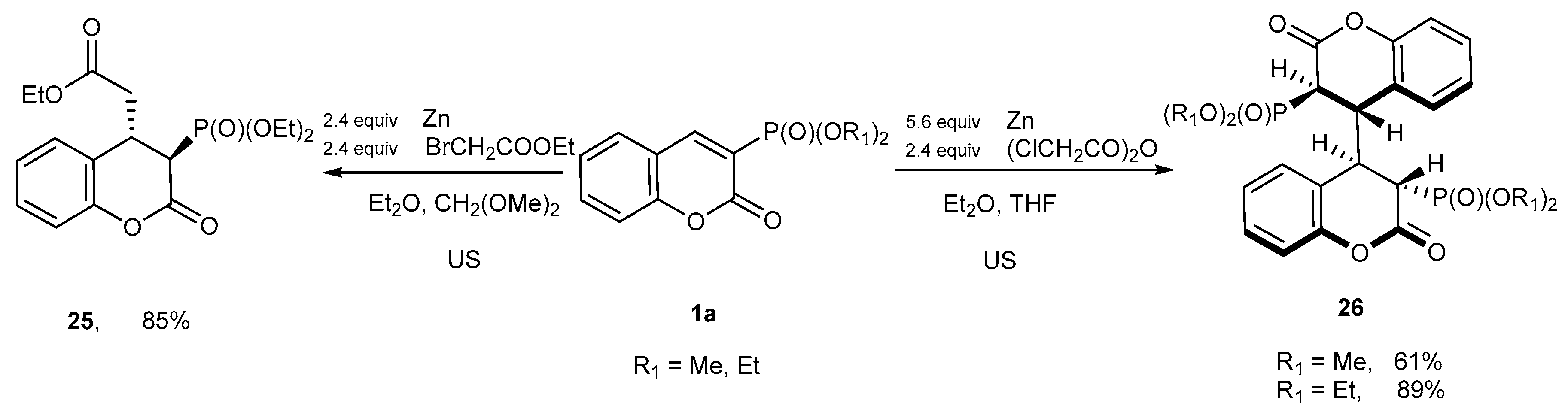
- Koleva, A.I.; Petkova, N.I.; Nikolova, R.D. Ultrasound-Assisted Metal-Mediated Method for the Formation of Tetrahydro-3,3′-Disubstituted Biscoumarins. Molecules 2018, 23, 2810. [Google Scholar] [CrossRef]
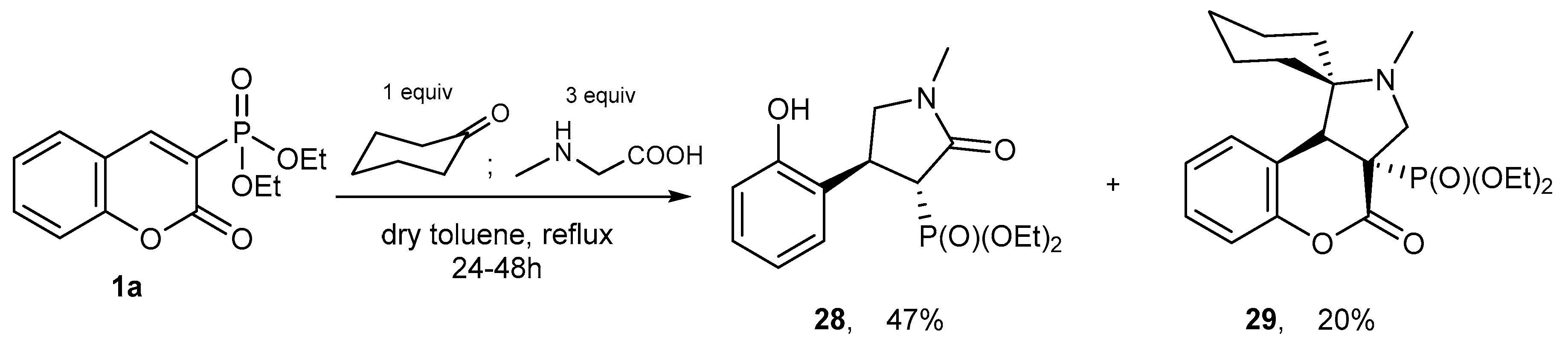
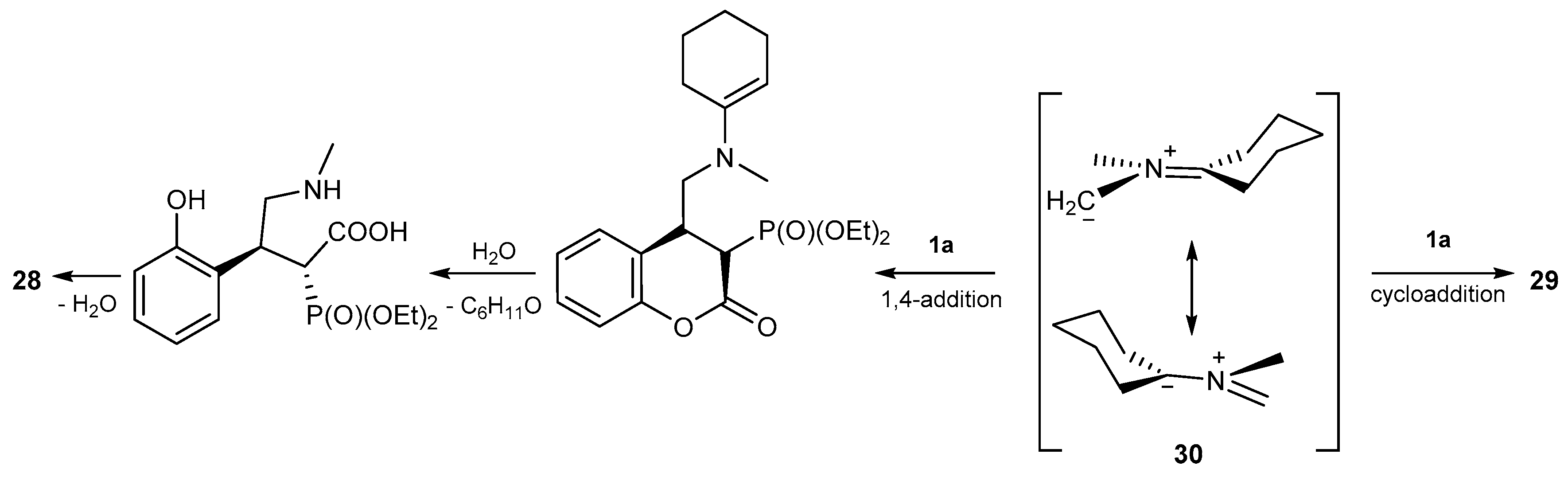
- Moshkin, V.S.; Sosnovskikh, V.Y.; Röschenthaler, G. Nucleophilic properties of a nonstabilized azomethine ylide derived from sarcosine and cyclohexanone. A novel domino reaction leading to substituted 4-aryl-2-pyrrolidones. Tetrahedron Lett. 2012, 53, 3568–3572. [Google Scholar] [CrossRef]
- Moshkin, V.S.; Sosnovskikh, V.Y.; Röschenthaler, G. Synthesis of benzopyranopyrrolidines via 1,3-dipolar cycloaddition of nonstabilized azomethine ylides with 3-substituted coumarins. Tetrahedron 2013, 69, 5884–5892. [Google Scholar] [CrossRef] [Green Version]
- Deredas, D.; Albrecht, Ł.; Maniukiewicz, W.; Wojciechowski, J.; Wolf, W.M.; Paluch, P.; Janecki, T.; Rόżalski, M.; Krajewska, U.; Janecka, A.; Krawczyk, H. Three-component reaction of 3-(diethoxyphosphoryl)coumarin, enolizable ketones and primary amines: Simple, stereoselective synthesis of benzo[1,3]oxazocine skeletons. RSC Adv. 2013, 3, 6821–6832. [Google Scholar] [CrossRef]
- Ilieva, E.D.; Petkova, N.I.; Nikolova, R.D. Ring Opening Reactions of 3-Phosphonocoumarin under Michael Reaction Conditions. Phosphorus Sulfur Silicon 2012, 187, 39–50. [Google Scholar] [CrossRef]
- Deredas, D.; Huben, K.; Maniukiewicz, W.; Krawczyk, H. Highly syn-diastereoselective Michael addition of enolizable ketones to 3-(diethoxyphosphoryl)coumarin. Tetrahedron 2014, 70, 8925–8929. [Google Scholar] [CrossRef]
- Deredas, D.; Huben, K.; Janecka, A.; Długosz, A.; Pomorska, D.K.; Mirowski, M.; Krajewska, U.; Janecki, T.; Rόżalski, M.; Krawczyk, H. Synthesis and anticancer properties of 3-methylene-4-(2-oxoalkyl)-3,4-dihydrocoumarins. MedChemComm 2016, 7, 1745–1758. [Google Scholar] [CrossRef]
- Deredas, D.; Huben, K.; Maniukiewicz, W.; Krawczyk, H. Tandem Conjugate Addition–Intramolecular Horner–Wadsworth–Emmons Olefination Approach to the Synthesis of Cyclopentene[c]chroman-2-ones and Cyclopent-1-enecarboxylates. Synlett 2014, 25, 280–282. [Google Scholar] [CrossRef]
- Deredas, D.; Łągiewka, B.; Maniukiewicz, W. A facile synthesis of novel 9-substituted 2,3,4,9-tetrahydro-1H-xanthen-1-ones. Tetrahedron Lett. 2019, 60, 538–540. [Google Scholar] [CrossRef]
- Deredas, D.; Krawczyk, H.; Huben, K. An efficient synthesis of 3-diethoxyphosphoryl-4-(1H-indol-3-yl)-3,4-dihydrocoumarins: A convenient approach to 3-methylene-4-(indol-3-yl)-3,4-dihydrocoumarins. Arkivoc 2018, 120–133. [Google Scholar] [CrossRef]
- Petkova, N.; Bojilova, A.; Nikolova, R.; Ivanov, C.; Rodios, N.; Kopf, J. Synthesis of Heterocyclic Methylenebisphosphonates by 1,3-Dipolar Cycloaddition of Ethyl Diazoacetate to 1,2-Benzoxaphosphorin-3-phosphonates. Tetrahedron 2009, 65, 1639–1647. [Google Scholar] [CrossRef]
- Takeuchi, Y.; Ueda, N.; Uesugi, K.; Abe, H.; Nishioka, H.; Harayama, T. Convenient Synthesis of a Simple Coumarin from Salicylaldehyde and Wittig Reagent. IV1a-c: Improved Synthetic Method of Substituted Coumarins. Heterocycles 2003, 59, 217–224. [Google Scholar] [CrossRef]
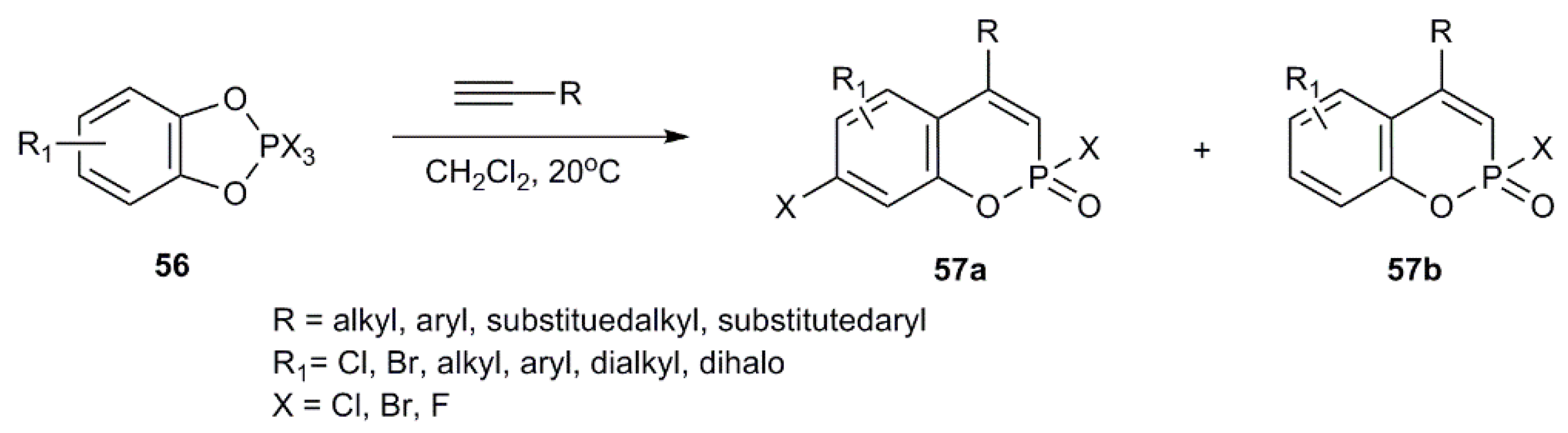
- Mironov, V.F.; Bogdanov, A.V.; Nemtarev, A.V.; Shtyrlina, A.A.; Varaksina, E.N.; Cherkasov, V.K.; Dobrynin, A.B.; Krivolapov, D.B.; Musin, R.Z.; Litvinov, I.A.; et al. Reactions of 3,5-di(tert-butyl)-1,2-benzoquinone with terminal acetylenes in the presence of phosphorus trichloride. ipso-Substitution of the tert-butyl group. Russ. Chem. Bull. 2007, 56, 1900–1910. [Google Scholar] [CrossRef]
- Nemtarev, A.V.; Mironov, V.F.; Varaksina, E.N.; Gubaidullin, A.T.; Krivolapov, D.B.; Musin, R.Z.; Litvinov, I.A. Reaction of arylenedioxytrihalophosphoranes with acetylenes 12. Alkylacetylenes in the reaction with 2,2,2-trihalobenzo-1,3,2-dioxaphospholes. Russ. Chem. Bull. 2014, 63, 149–177. [Google Scholar] [CrossRef]
- Mironov, V.F.; Nemtarev, A.V. Reactions of Arylenedioxytrihalophosphoranes with Acetylenes: XV.1 Reaction of 2,2,2-Tribromo-4,6-di-tert-butylbenzo-1,3,2λ5-dioxaphospholedioxaphosphole with Pent-1-yne. Rev. J. Chem. 2011, 1, 27–55. [Google Scholar] [CrossRef]
- Aniskin, A.S.; Nemtarev, A.V.; Baranov, D.S.; Mironov, V.F.; Vasilevskii, S.F. Effect of Electronic Nature of Substituents in Arylacetylene on the Rate of Reaction with 2,2,2-Trichloroareno-1,3,2-dioxaphosphols. Russ. J. Gen. Chem. 2013, 83, 1010–1012. [Google Scholar] [CrossRef]
- Nemtarev, A.V.; Mironov, V.F.; Aniskin, A.S.; Baranov, D.S.; Mironova, E.V.; Krivolapov, D.B.; Musin, R.Z.; Vasilevskii, S.F.; Druzhkov, N.O.; Cherkasov, V.K. Reaction of arylenedioxytrihalophosphoranes with acetylenes 11. Electronic effect of the substituent in arylacetylene on the reaction rate. Russ. Chem. Bull. 2013, 62, 55–70. [Google Scholar] [CrossRef]
- Mironov, V.F.; Nemtarev, A.V.; Varaksina, E.N.; Shtyrlina, A.A.; Gubaidullin, A.T.; Litvinov, I.A.; Dobrynin, A.B. Reactions of phenylenedioxytrihalophosphoranes with arylacetylenes: XIII. Reaction of 5-tert-butyl-2,2,2-trihalo-1,3,2λ5-benzodioxaphospholes with acetylenes. Russ. J. Org. Chem. 2014, 50, 864–887. [Google Scholar] [CrossRef]
- Nemtarev, A.V.; Mironov, V.F.; Fayzullin, R.R.; Litvinov, I.A.; Musin, R.Z. Reactions of Arylenedioxytrihalophosphoranes with Acetylenes: XV.1 Reaction of 2,2,2-Tribromo-4,6-di-tert-butylbenzo-1,3,2λ5-dioxaphospholedioxaphosphole with Pent-1-yne. Russ. J. Gen. Chem. 2018, 88, 2290–2295. [Google Scholar] [CrossRef]
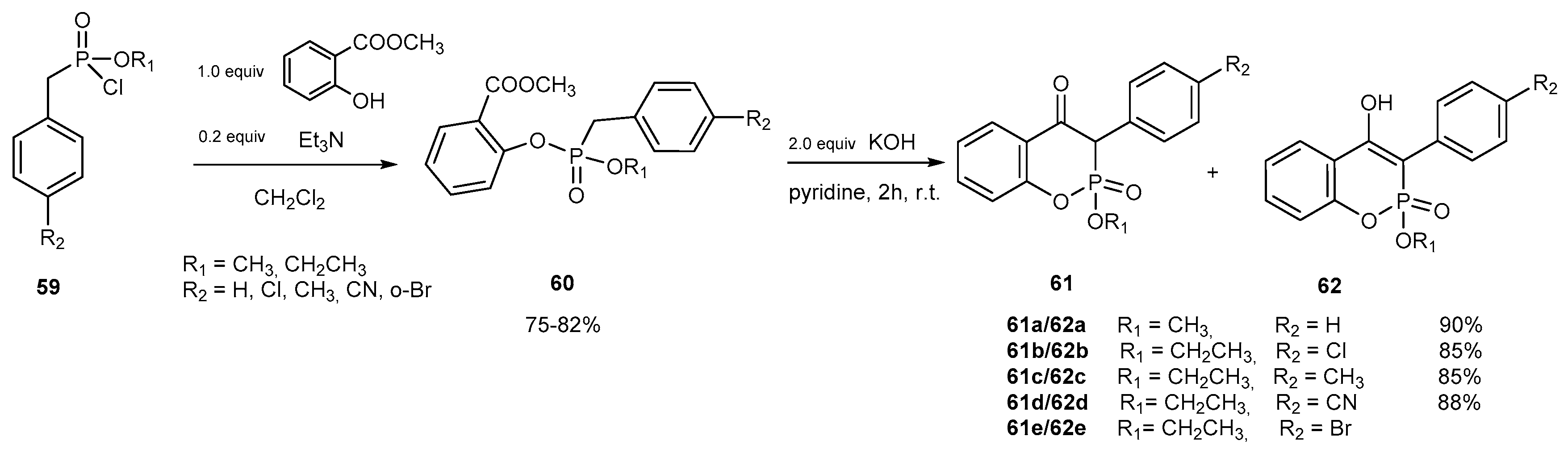
- Li, X.; Zhang, D.; Pang, H.; Shen, F.; Fu, H.; Jiang, Y.; Zhao, Y. Synthesis of a Diverse Series of Phosphacoumarins with Biological Activity. Org. Lett. 2005, 7, 4919–4922. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, Y.; Fu, H.; Jiang, Y.; Zhao, Y. Alkylation reactions of phosphachroman-2,4-diones and 4-hydroxy phosphacoumarins. Bioorg Chem. 2006, 34, 105–113. [Google Scholar] [CrossRef] [PubMed]
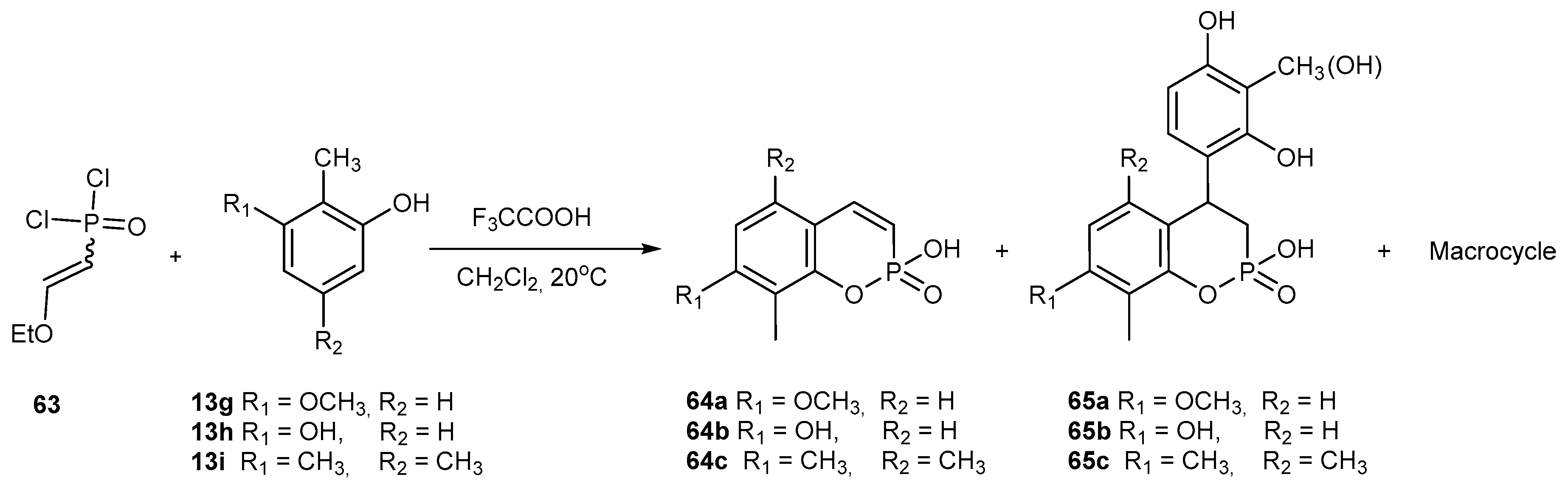
- Burilov, A.R.; Knyazeva, I.R.; Kasymova, E.M.; Sadykova, Y.M.; Pudovik, M.A.; Habicher, W.D.; Sinyashin, O.G. New P-Containing Linear and Macrocyclic Polyphenols. Phosphorus Sulfur Silicon 2011, 186, 884–887. [Google Scholar] [CrossRef]
- Sadykova, Y.M.; Dalmatova, N.V.; Burilov, A.R.; Pudovik, M.A.; Dobrynin, A.B. Reaction of 3-methoxy-2-methylphenol with 2-ethoxyvinylphosphonic dichloride. Russ. Chem. Bull. 2011, 60, 2078–2080. [Google Scholar] [CrossRef]
- Sadykova, Y.M.; Sadikova, L.M.; Badrtdinova, A.R.; Dobrynin, A.B.; Burilov, A.R.; Pudovik, M.A. Condensation of 2-Ethoxyvinylphosphonic Acid Dichloroanhydride with 2,3,5-Trimethylphenol. Novel Method for Preparation of Phosphacoumarins. Phosphorus Sulfur Silicon 2015, 190, 2267–2272. [Google Scholar] [CrossRef]
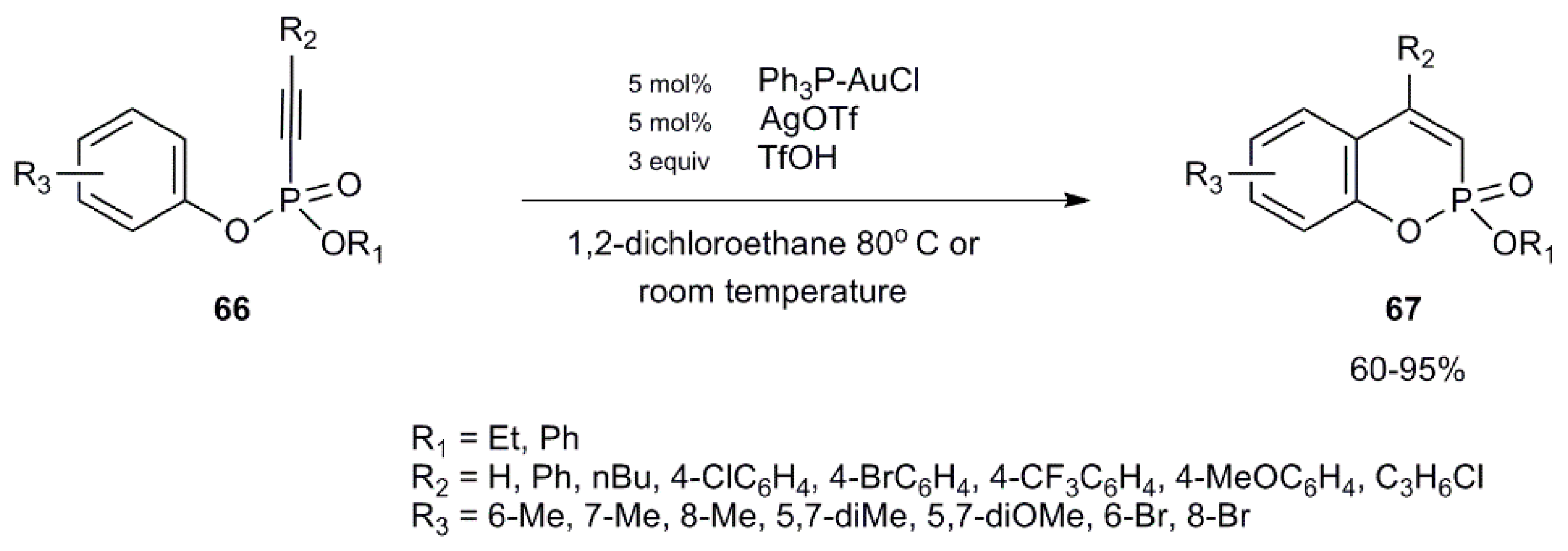
- Kim, C.-E.; Ryu, T.; Kim, S.; Lee, K.; Lee, C.-H.; Lee, P.H. Gold-Catalyzed Hydroarylation of Aryl Alkynylphosphonates for the Synthesis of Phosphacoumarins. Adv. Synth. Catal. 2013, 355, 2873–2883. [Google Scholar] [CrossRef]
- Shin, S.; Kang, D.; Jeon, W.H.; Lee, P.H. Synthesis of ethoxy dibenzooxaphosphorin oxides through palladium-catalyzed C(sp2)–H activation/C–O formation. Beilstein J. Org. Chem. 2014, 10, 1220–1227. [Google Scholar] [CrossRef]
- Li, X.; Shen, F.; Fu, H.; Jiang, Y.; Zhao, Y. Synthesis of 4-Substituted Phosphacoumarins via Cross-Coupling of 4-Tosylphosphacoumarins with Organozinc Reagents. Synlett 2006, 4, 630–632. [Google Scholar] [CrossRef]
- Kim, C.-E.; Son, J.-Y.; Shin, S.; Seo, B.; Lee, P.H. Alkenylation of Phosphacoumarins via Aerobic Oxidative Heck Reactions and Their Synthetic Application to Fluorescent Benzophosphacoumarins. Org. Lett. 2015, 17, 908–911. [Google Scholar] [CrossRef]
- Nikolova, R.; Vayssilov, G.N.; Rodios, N.; Bojilova, A. Regio- and Stereoselective [2+2] Photodimerization of 3-Substituted 2-Alkoxy-2-oxo-2H-1,2-benzoxaphosphorines. Molecules 2002, 7, 420–432. [Google Scholar] [CrossRef]































































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
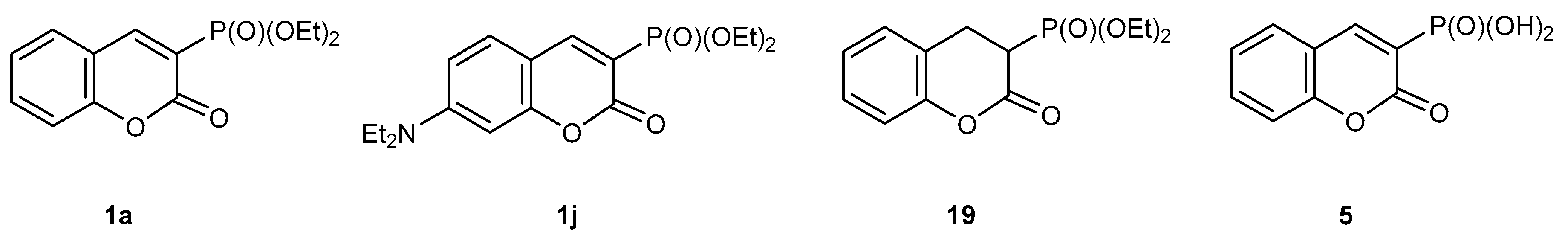{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
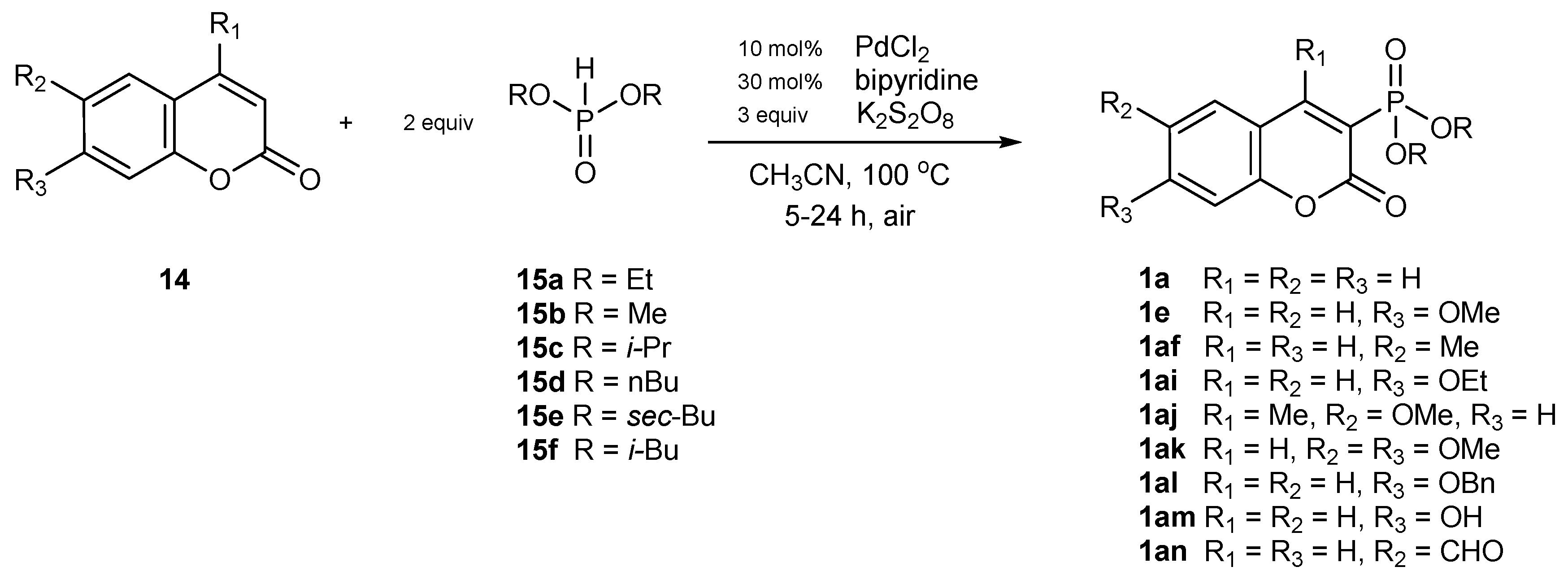{kind=link}
{kind=link}
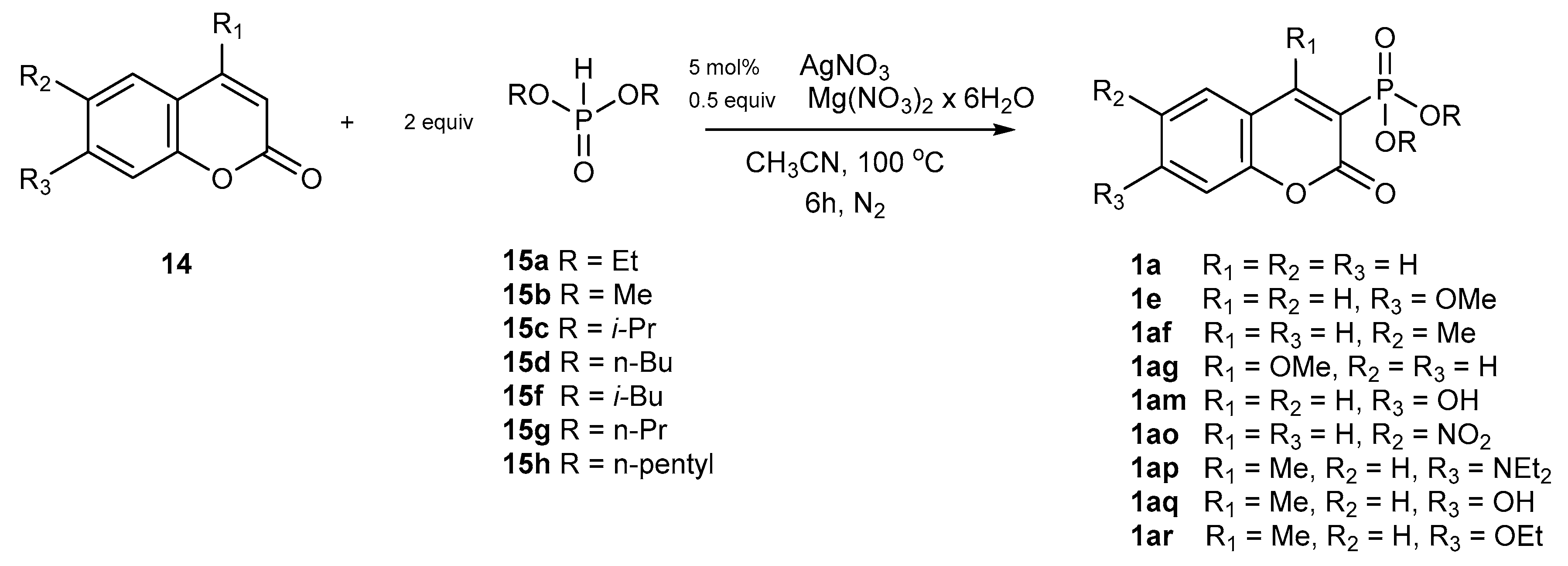{kind=link}
{kind=link}
{kind=link}
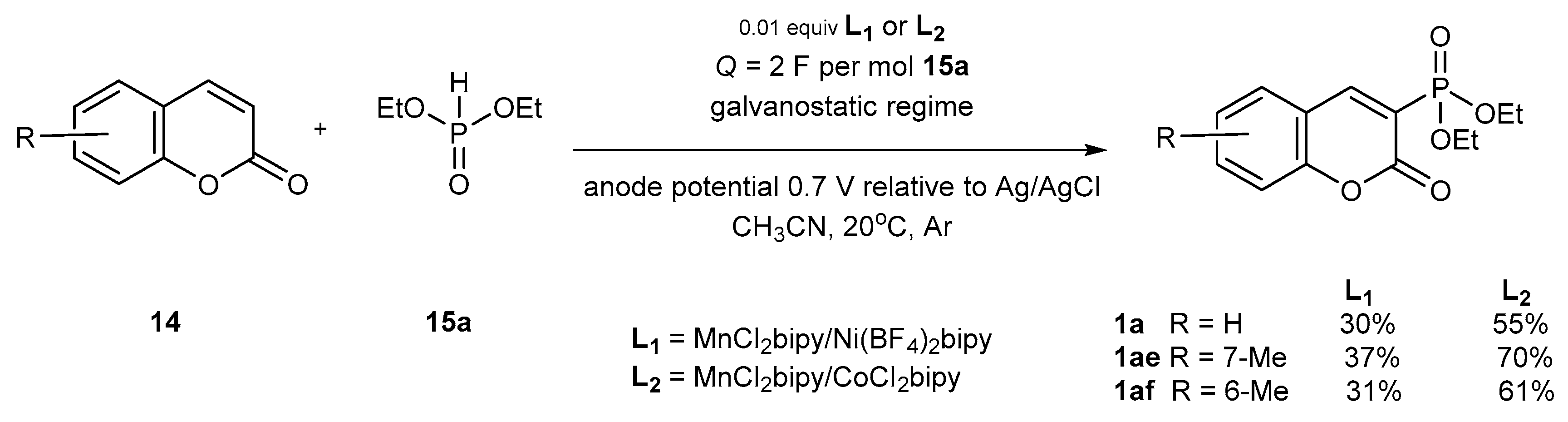{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| R1 | X | R | Method | Overall Yield | 1 a | 2 a |
|---|---|---|---|---|---|---|
| Me | COOEt | H | A | 51 | 33 | 11 |
| A1 | 56 | 27 | 21 | |||
| B | 20 | 20 | 0 | |||
| Et | COOEt | H | A | 87 | 70 | 9 |
| A1 | 98 | 84 | 14 | |||
| B | 50 | 50 | 0 | |||
| C | 74 | 66 | 8 | |||
| Et | COOSi(Me)3 | H | A | 59 | 47 | 8 |
| C | 28 | 20 | 2 | |||
| D | 73 | 57 | 9 | |||
| Et | CN | H | A | 69 | 45 | 24 |
| A1 | 52 | 17 | 35 | |||
| B | 49 | 49 | 0 | |||
| C | 55 | 47 | 8 | |||
| D | 60 | 42 | 18 | |||
| Et | COOEt | 6-Br | A | 98 | 77 | 19 |
| B | 81 | 81 | 0 | |||
| C | 50 | 40 | 4 | |||
| Et | COOEt | 6-Cl | A | 78 | 65 | 9 |
| B | 71 | 71 | 0 | |||
| C | 67 | 41 | 20 | |||
| Et | COOEt | 7-NEt2 | A | 81 | 64 | 17 |
| B | 3 | 3 | 0 | |||
| C | 86 | 71 | 15 |
| 1 | R1 | R2 | R3 | R4 | R5 | Reaction Time [days] | Yields | Ref |
|---|---|---|---|---|---|---|---|---|
| 1q | H | OMe | H | OMe | H | 6d | 88% | [23,24,25] |
| 1e | H | H | H | OMe | H | 10d | 81% | [24,25] |
| 1r | H | H | -CH2OCH2- | H | 60d | 73% | [24,25] | |
| 1f | H | -CH=CH-CH=CH- | H | H | 10d | 88% | [24,25] | |
| 1s | H | -CH=C(OH)-CH=CH- | H | H | 18d | 95% | [24,25] | |
| 1t | H | H | H | -CH=CH-HC=CH- | 14d | 9% | [24] | |
| 1u | Me | H | H | OMe | H | 3d | 81% | [26,27] |
| 1v | Me | OMe | H | OMe | H | 3d | 69% | [26] |
| 1w | Me | H | H | -CH=CH-HC=CH- | 3d | 60% | [26] | |
| 1x | Et | H | H | OMe | H | 3d | 59% | [26] |
| 1y | Et | OMe | H | OMe | H | 3d | 47% | [26] |
| 1z | Et | H | H | -CH=CH-HC=CH- | 3d | 26% | [26] | |
| 1aa | n-Bu | H | H | OMe | H | 3d | 38% | [26] |
| 1ab | n-Bu | OMe | H | OMe | H | 3d | 31% | [26] |
| 1ac | Ph | H | H | OMe | H | 3d | 85% | [26] |
| 1ad | Ph | OMe | H | OMe | H | 3d | 70% | [26] |
| Substituent R1 | Reflux | US | ||
|---|---|---|---|---|
| Reaction Time [min] | Yield [%] | Reaction Time [min] | Yield [%] | |
| Et | 50 | 64 | 10 | 89 |
| n-Pr | 60 | 77 | 30 | 94 |
| i-Pr | 80 | 69 | 50 | 74 |
| PhCH2 | 125 | 38 | 90 | 53 |
| CH2COOt-Bu | 75 | 78 | 30 | 95 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koleva, A.I.; Petkova-Yankova, N.I.; Nikolova, R.D. Synthesis and Chemical Properties of 3-Phosphono-coumarins and 1,2-Benzoxaphosphorins as Precursors for Bioactive Compounds. Molecules 2019, 24, 2030. https://doi.org/10.3390/molecules24112030
Koleva AI, Petkova-Yankova NI, Nikolova RD. Synthesis and Chemical Properties of 3-Phosphono-coumarins and 1,2-Benzoxaphosphorins as Precursors for Bioactive Compounds. Molecules. 2019; 24(11):2030. https://doi.org/10.3390/molecules24112030
Chicago/Turabian StyleKoleva, Ana I., Nevena I. Petkova-Yankova, and Rositca D. Nikolova. 2019. "Synthesis and Chemical Properties of 3-Phosphono-coumarins and 1,2-Benzoxaphosphorins as Precursors for Bioactive Compounds" Molecules 24, no. 11: 2030. https://doi.org/10.3390/molecules24112030