Photolysis of 5-Azido-3-Phenylisoxazole at Cryogenic Temperature: Formation and Direct Detection of a Nitrosoalkene


Abstract
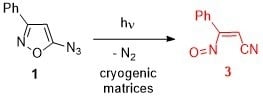
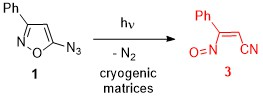
:
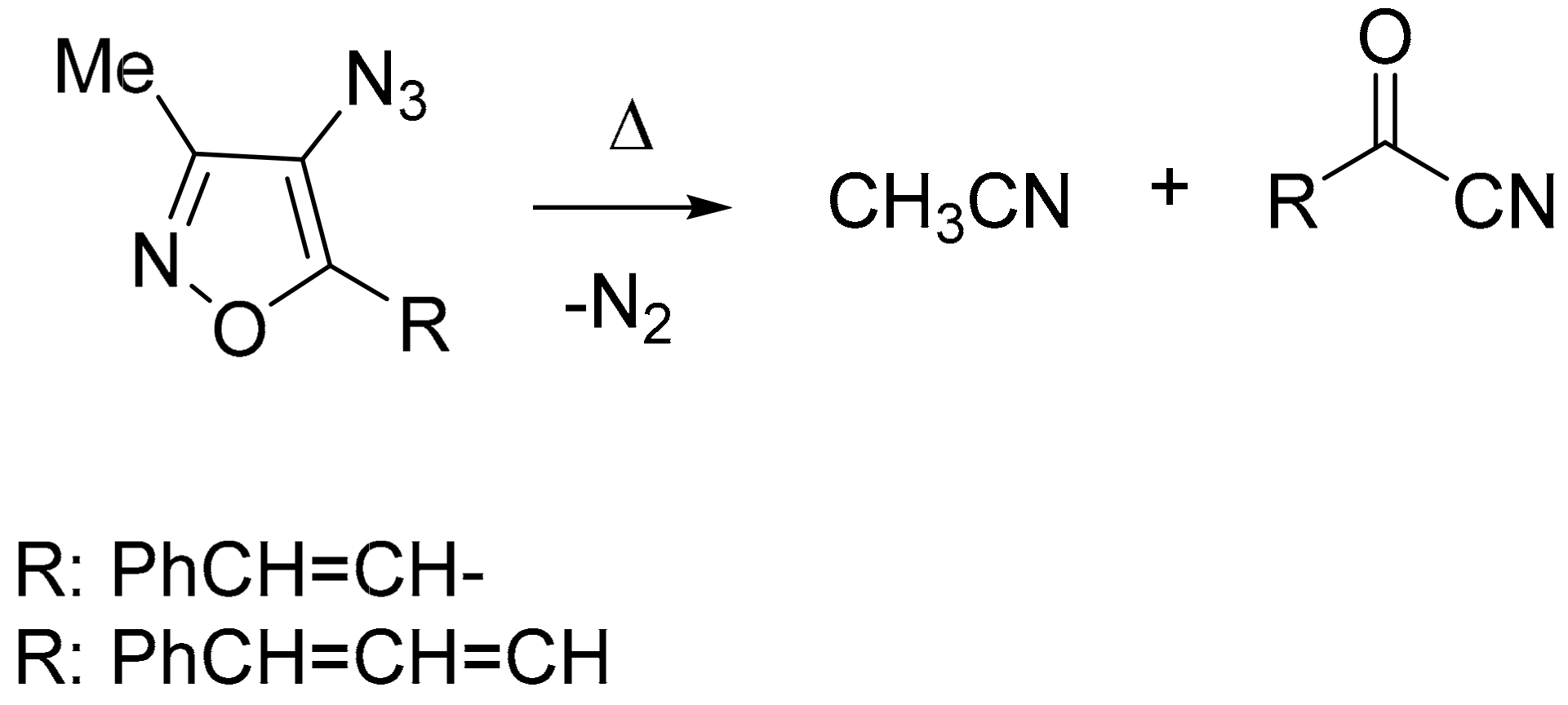
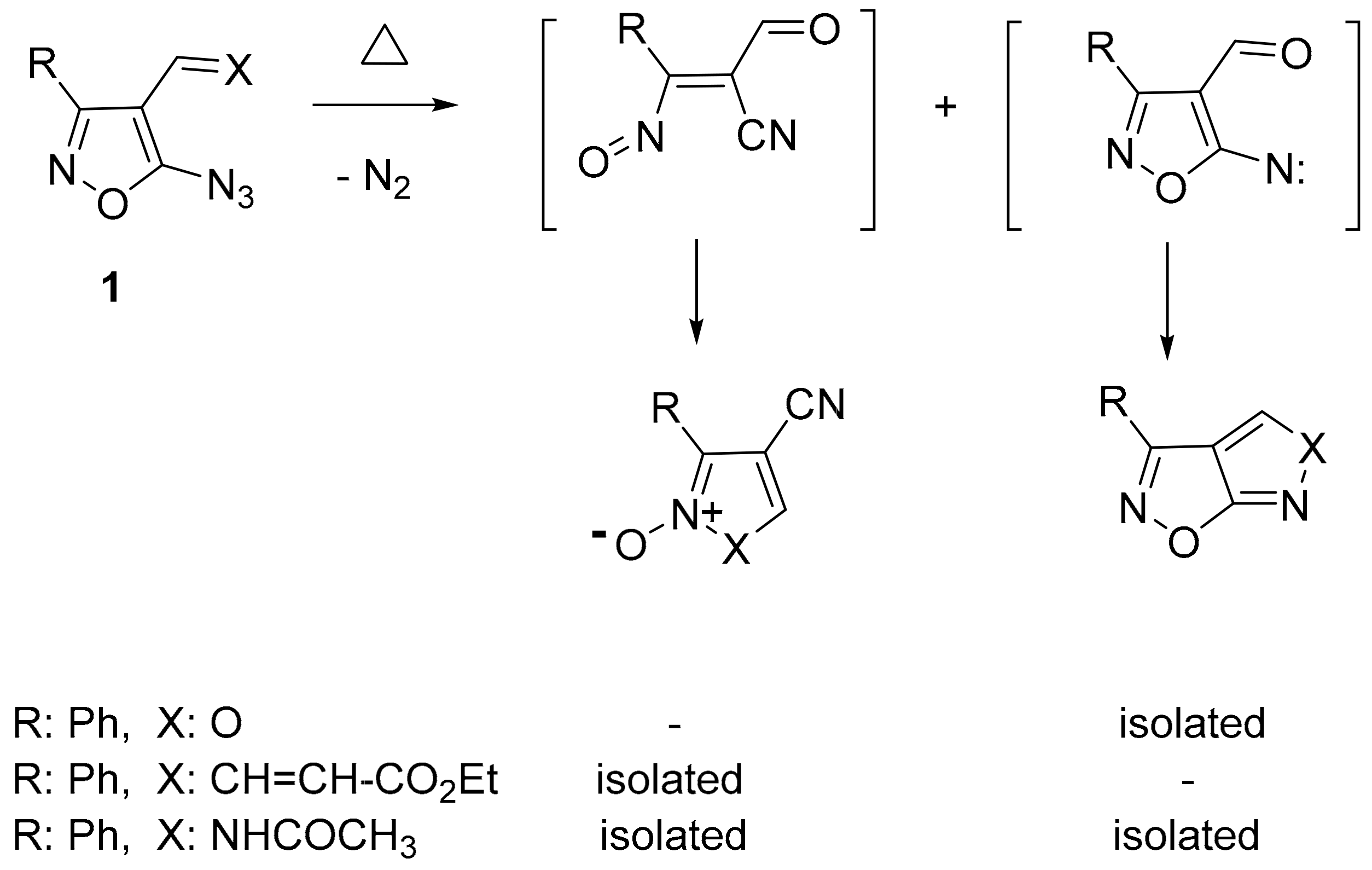
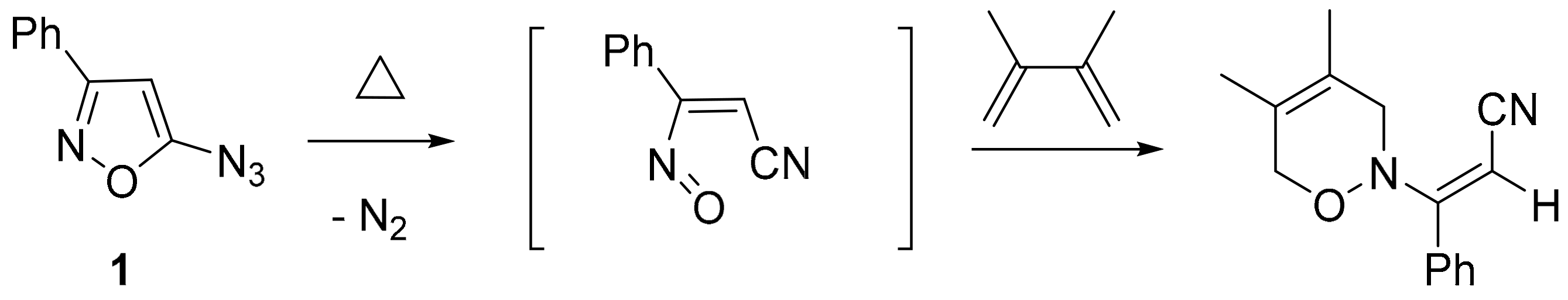
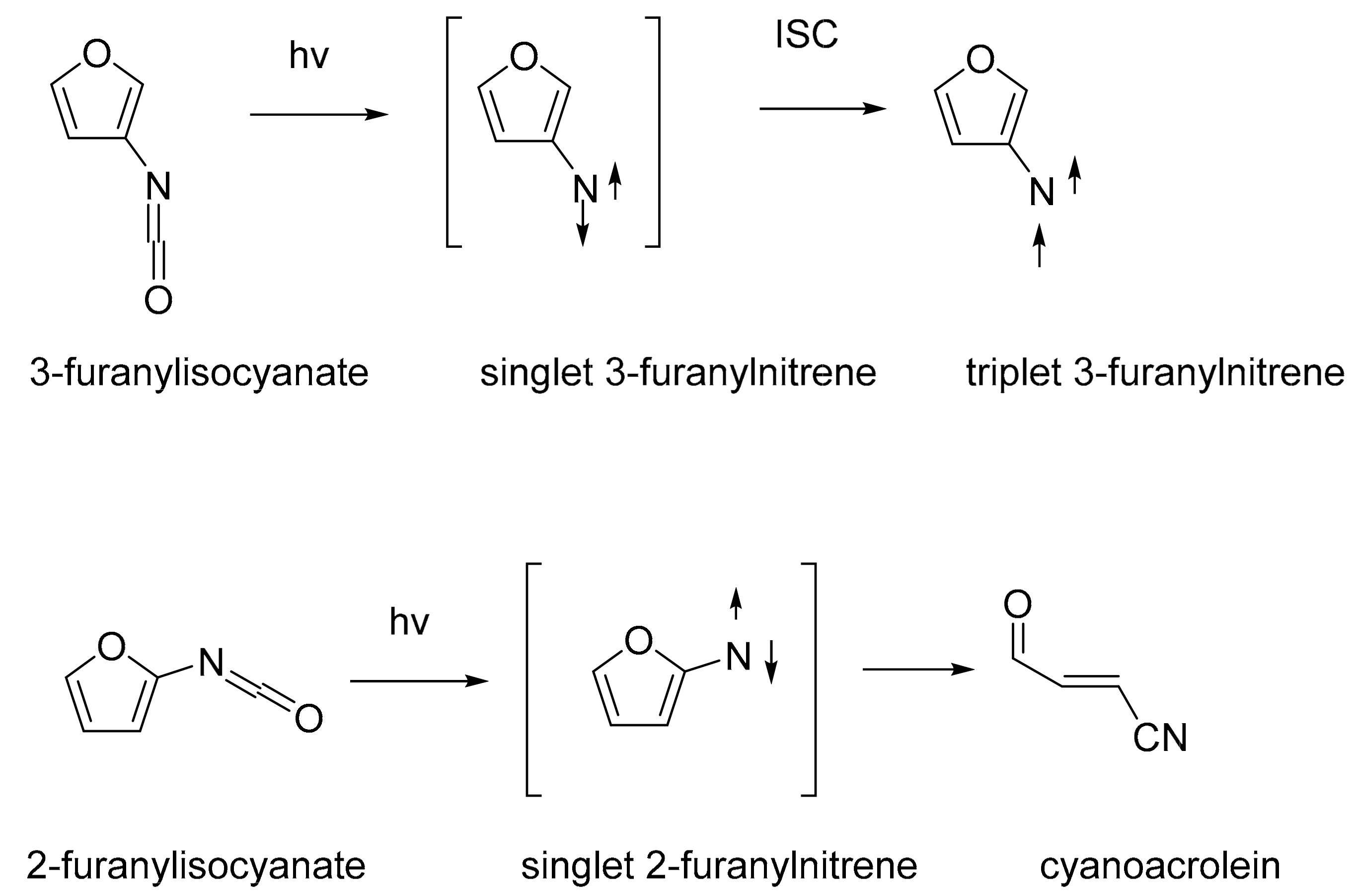
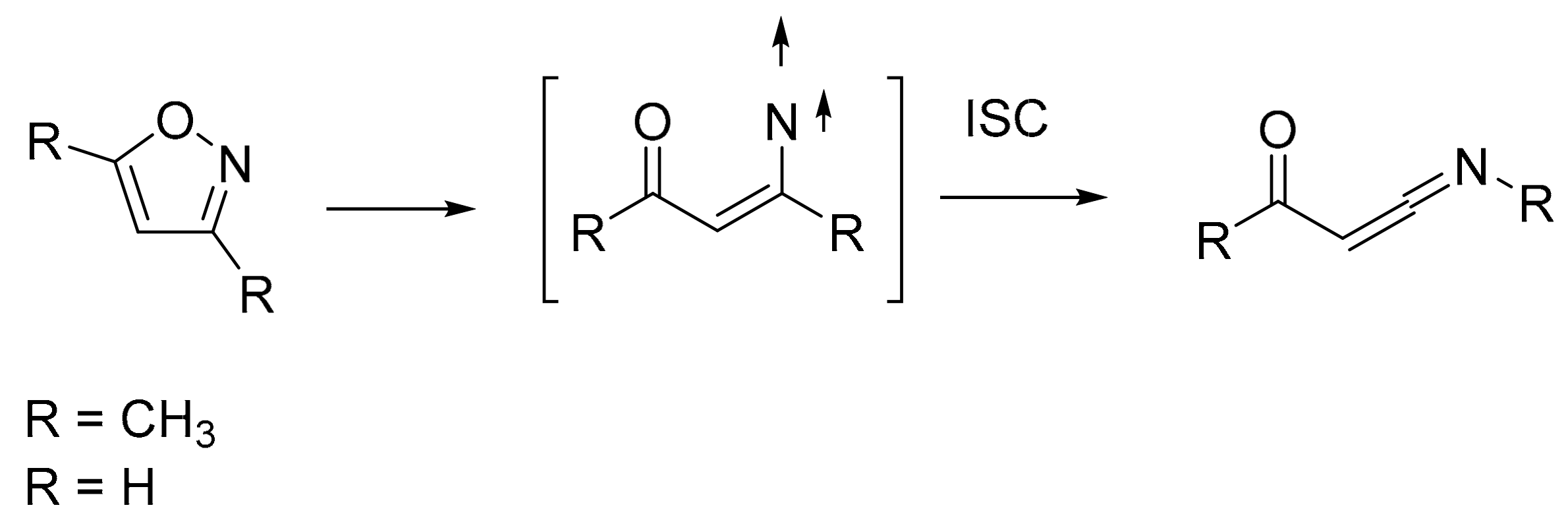
1. Introduction
2. Results
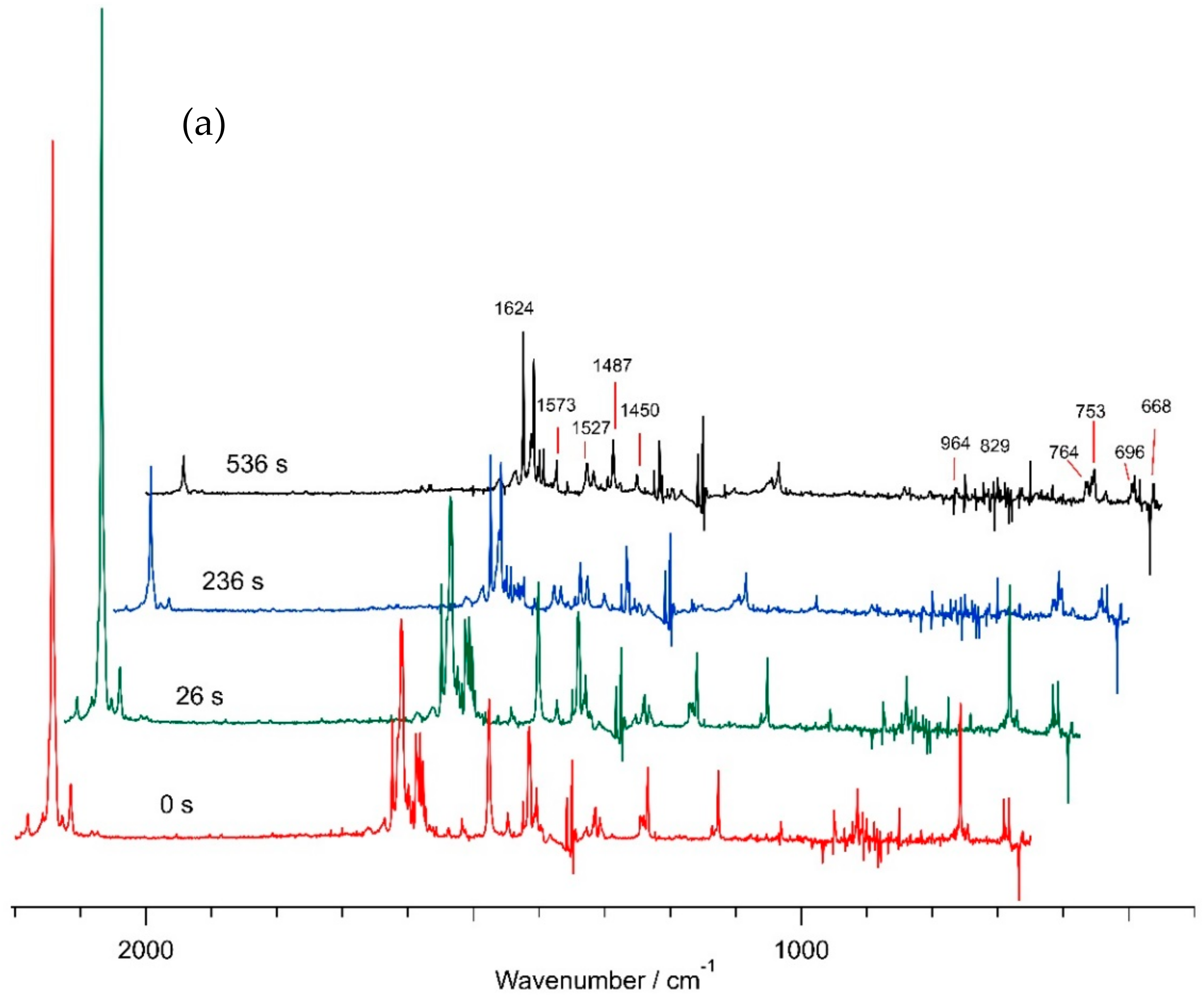
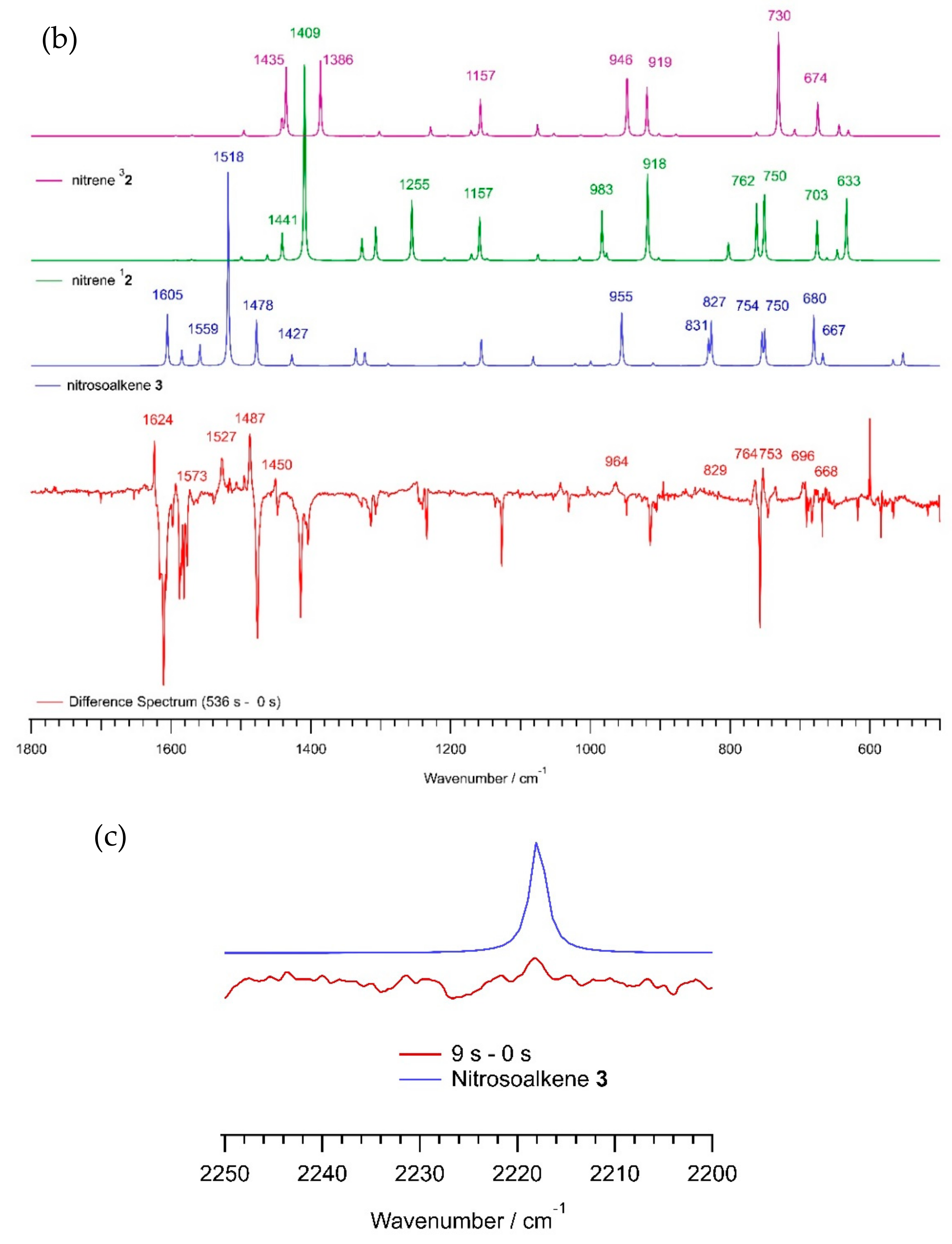
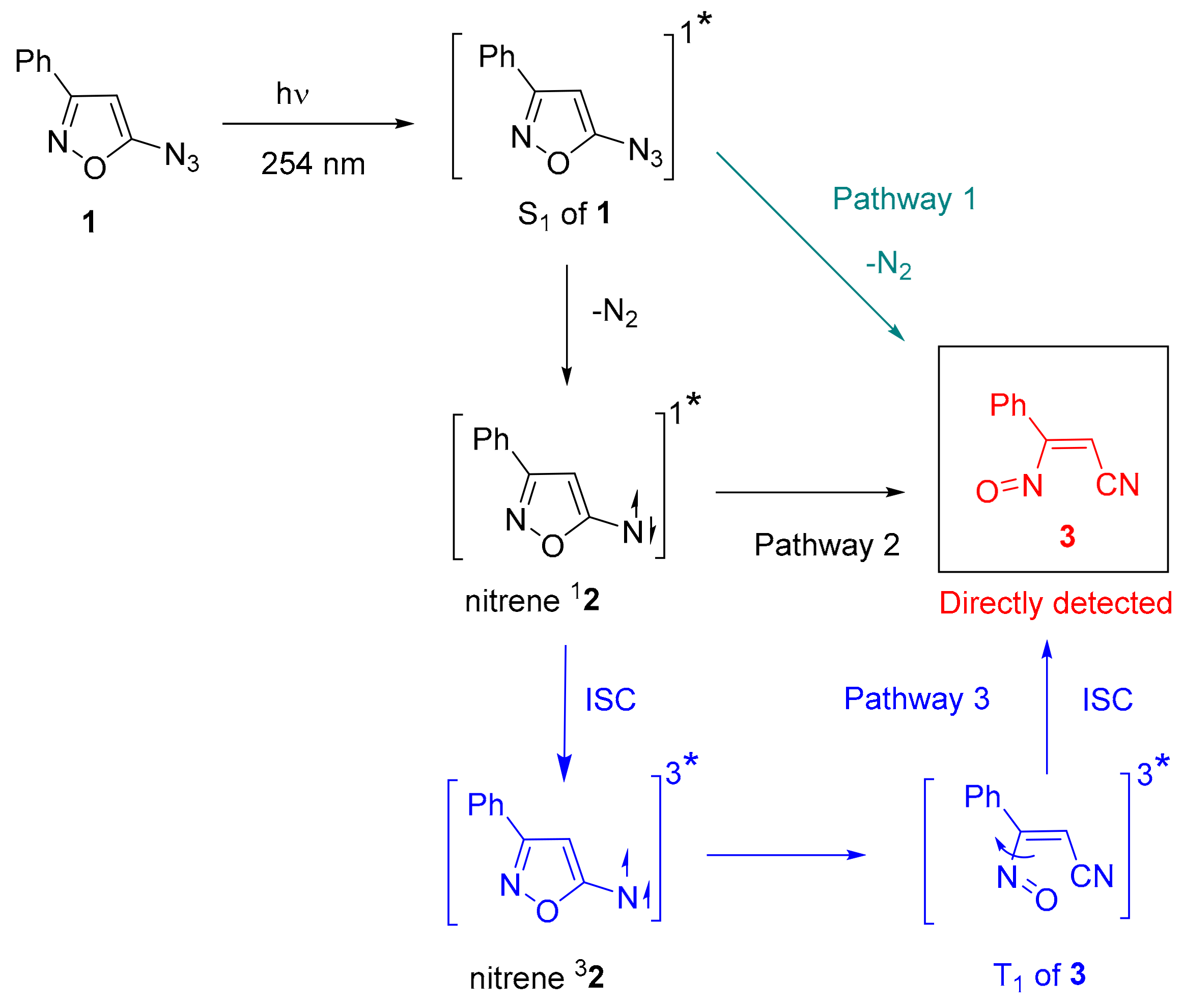
2.1. Matrix IR Spectroscopy
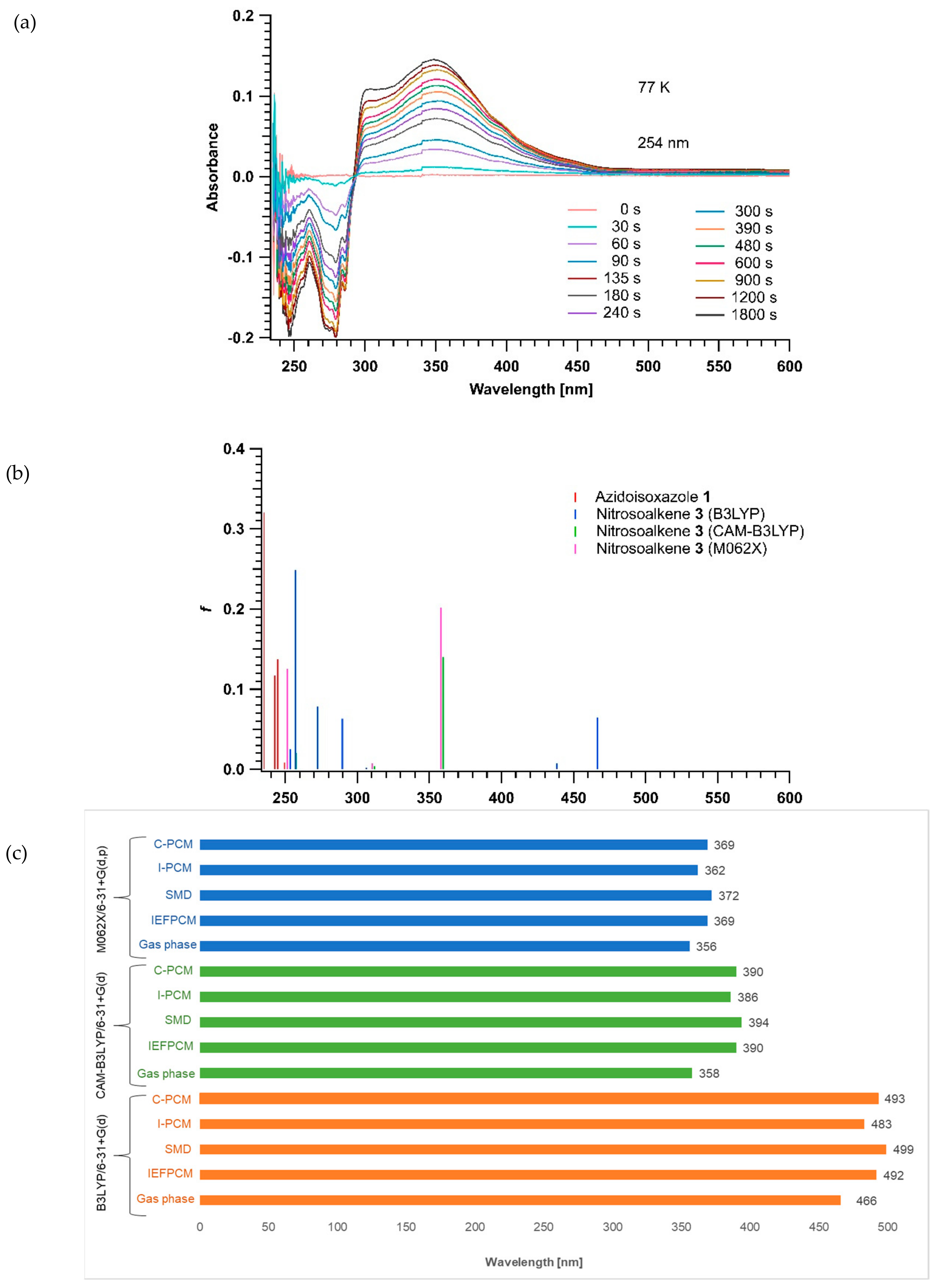
2.2. Glassy Matrix UV-Vis Absorption Spectroscopy
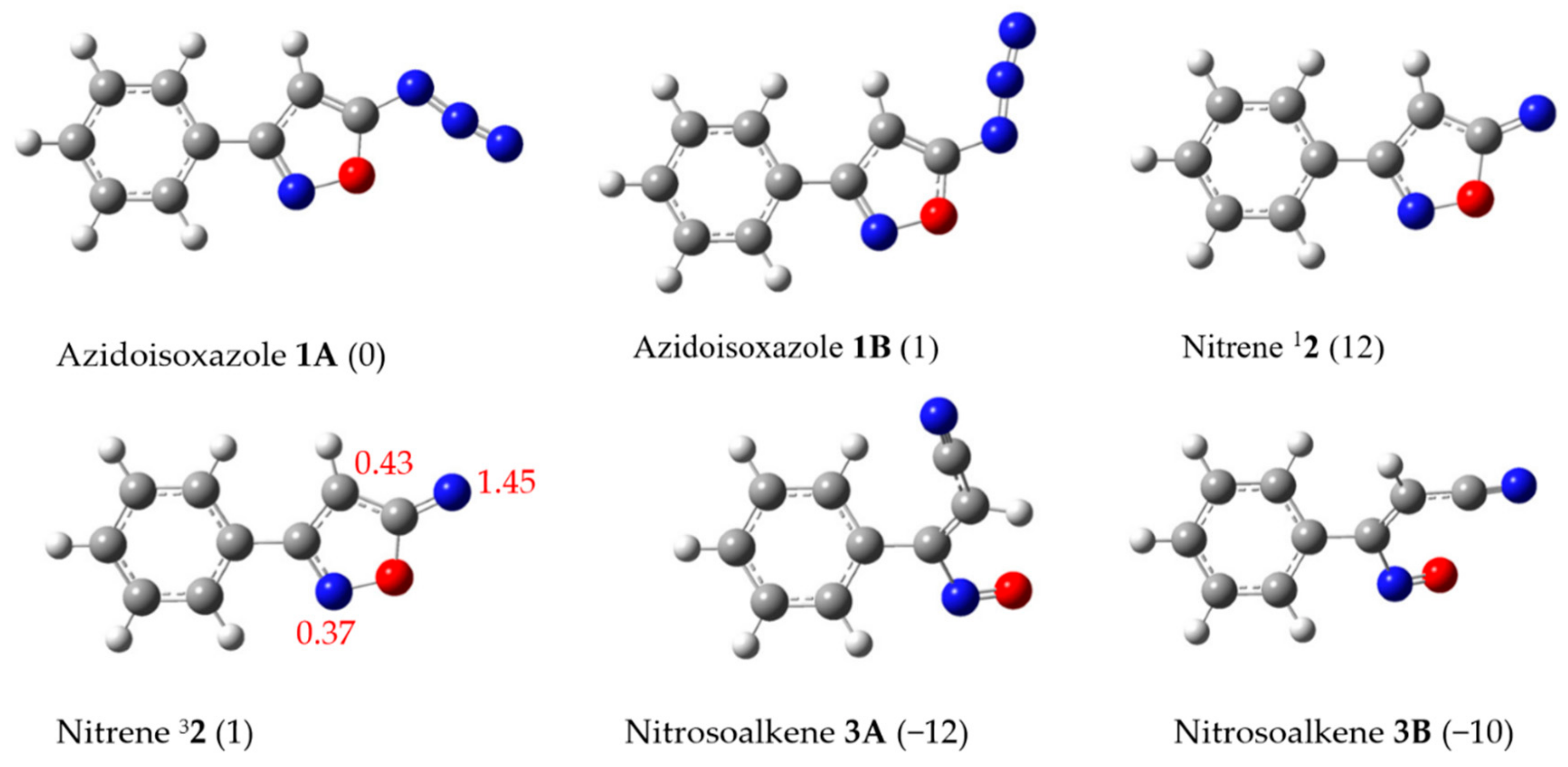
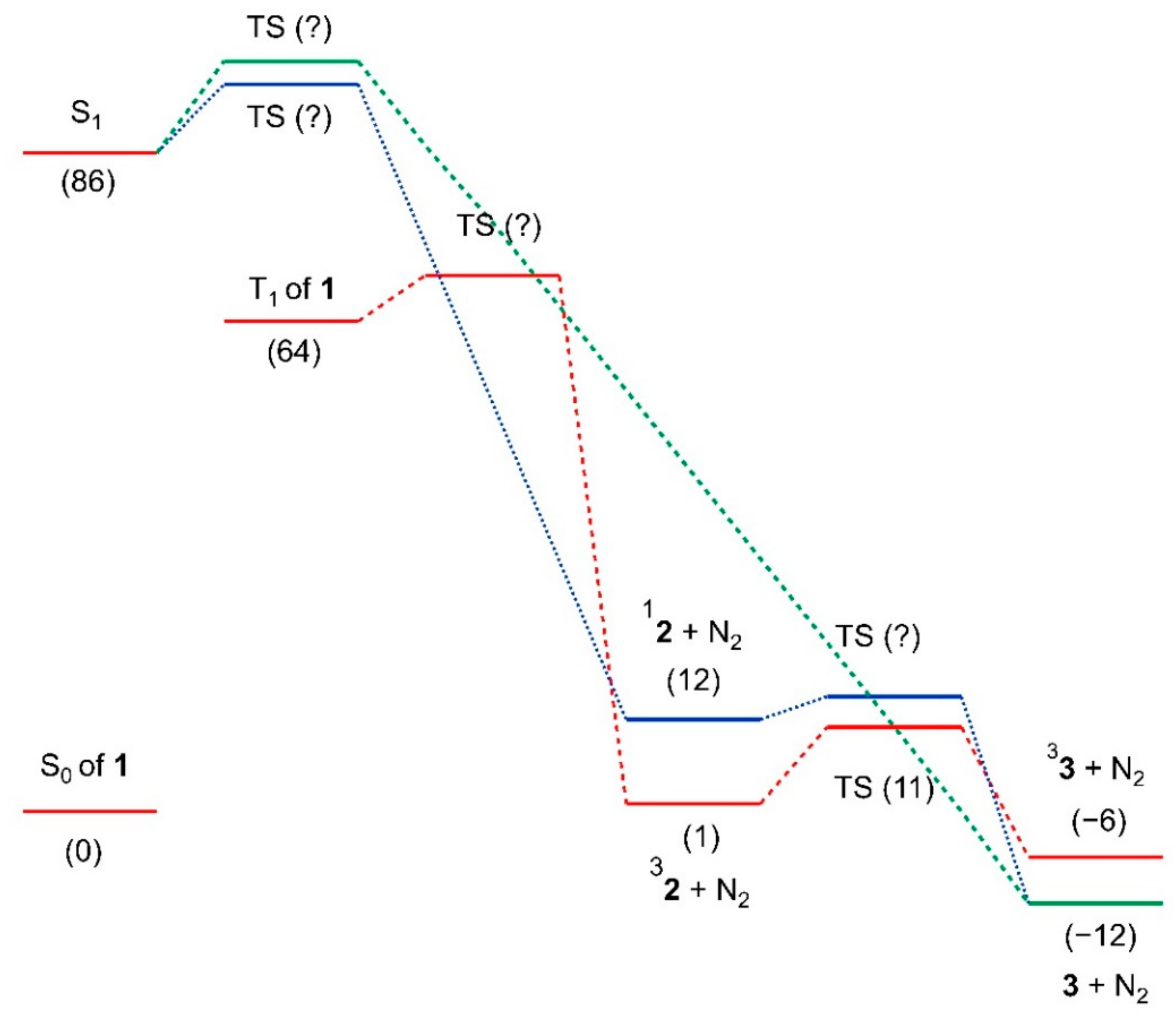
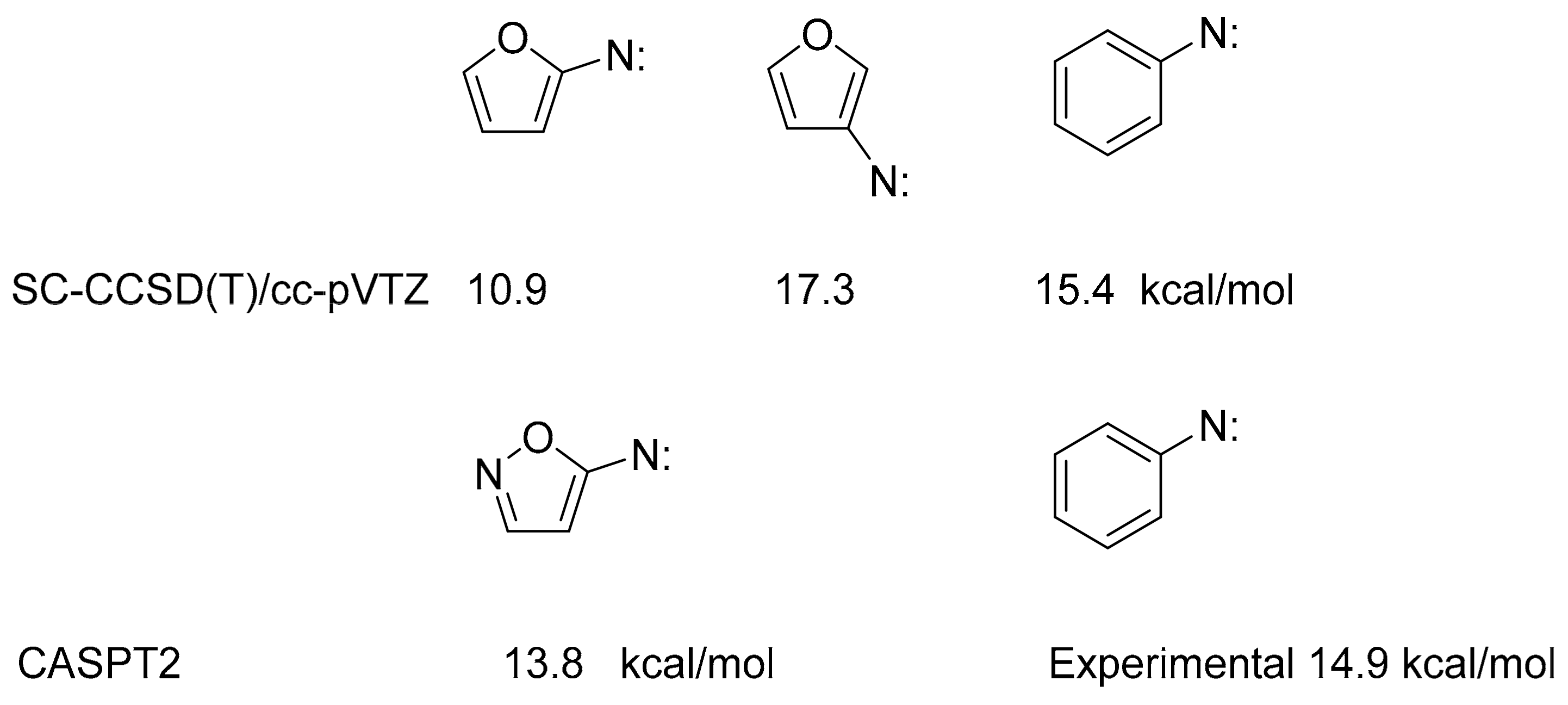
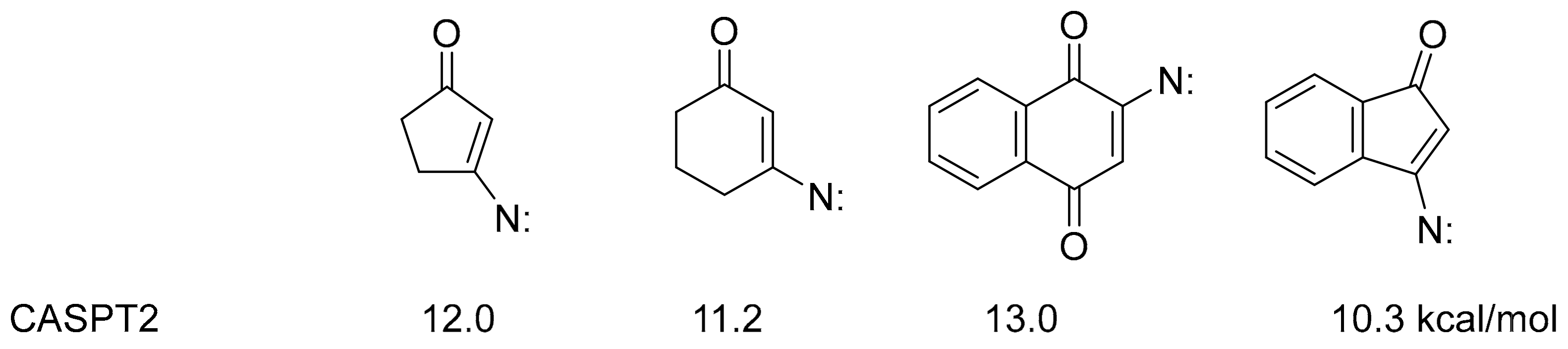
2.3. Quantum Chemical Calculations
3. Discussion
4. Materials and Methods
4.1. Preparation of Azidoisoxazole 1
4.2. Matrix IR Spectroscopy
4.3. Glassy Matrix UV-Vis Absorption Spectroscopy
4.4. Quantum Chemical Calculations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bräse, S.; Gil, C.; Knepper, K.; Zimmermann, V. Organic azides: An exploding diversity of a unique class of compounds. Angew. Chem. Int. Ed. 2005, 44, 5188–5240. [Google Scholar] [CrossRef] [PubMed]
- Kacprzak, K.; Skiera, I.; Piasecka, M.; Paryzek, Z. Alkaloids and isoprenoids modification by copper(I)-catalyzed Huisgen 1,3-dipolar cycloaddition (click chemistry): Toward new functions and molecular architectures. Chem. Rev. 2016, 116, 5689–5743. [Google Scholar] [CrossRef] [PubMed]
- Meldal, M.; Tornøe, C.W. Cu-catalyzed azide–alkyne cycloaddition. Chem. Rev. 2008, 108, 2952–3015. [Google Scholar] [CrossRef] [PubMed]
- Mandoli, A. Recent advances in recoverable systems for the copper-catalyzed azide-alkyne cycloaddition reaction (CuAAC). Molecules 2016, 21, 1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, D.M.; Yoon, T.P. Solar synthesis: Prospects in visible light photocatalysis. Science 2014, 343, 1239176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, U.; Thenna-Hewa, K.; Gudmundsdottir, A.D. Triplet vinylnitrenes. In Patai’s Chemistry of Functional Group; Rappoport, Z., Ed.; Wiley: Hoboken, NJ, USA, 2019; pp. 1–37. [Google Scholar]
- Fu, J.; Zanoni, G.; Anderson, E.A.; Bi, X. α-Substituted vinyl azides: An emerging functionalized alkene. Chem. Soc. Rev. 2017, 46, 7208–7228. [Google Scholar] [CrossRef]
- Hayashi, H.; Kaga, A.; Chiba, S. Application of vinyl azides in chemical synthesis: A recent update. J. Org. Chem. 2017, 82, 11981–11989. [Google Scholar] [CrossRef]
- L’Abbé, G.; Godts, F. Synthesis and thermal decomposition of 4-azidoisoxazoles. Bull. Soc. Chim. Belg. 1987, 96, 229–236. [Google Scholar] [CrossRef]
- Anderson, D.J.; Muchmore, C.R. Rearrangements of 5-azidoisoxazoles. J. Heterocycl. Chem. 1995, 32, 1189–1196. [Google Scholar] [CrossRef]
- Kumar, G.; Rajagopalan, K.; Swaminathan, S.; Balasubramanian, K.K. An interesting fragmentation during the thermolysis of azido isoxazoles. Tetrahedron Lett. 1979, 20, 4685–4686. [Google Scholar] [CrossRef]
- L’Abbé, G.; Dyall, L.; Meersman, K.; Dehaen, W. Rates and products from thermolysis of 5-azidoisoxazoles in solution. J. Chem. Soc. Perkin Trans. 2 1996, 2111–2118. [Google Scholar] [CrossRef]
- Foresman, J.B.; Frisch, Æ. Exploring Chemistry with Electronic Structure Methods; Gaussian, Inc.: Pittsburgh, PA, USA, 1996. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomasi, J.; Mennucci, B.; Cancès, E. The IEF version of the PCM solvation method: An overview of a new method addressed to study molecular solutes at the QM ab initio level. J. Mol. Struct.: THEOCHEM 1999, 464, 211–226. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Foresman, J.B.; Keith, T.A.; Wiberg, K.B.; Snoonian, J.; Frisch, M.J. Solvent effects. 5. Influence of cavity shape, truncation of electrostatics, and electron correlation on ab initio reaction field calculations. J. Phys. Chem. 1996, 100, 16098–16104. [Google Scholar] [CrossRef]
- Takano, Y.; Houk, K.N. Benchmarking the conductor-like polarizable continuum model (CPCM) for aqueous solvation free energies of neutral and ionic organic molecules. J. Chem. Theory Comput. 2005, 1, 70–77. [Google Scholar] [CrossRef]
- Cramer, C.J.; Truhlar, D.G. Implicit solvation models: Equilibria, structure, spectra, and dynamics. Chem. Rev. 1999, 99, 2161–2200. [Google Scholar] [CrossRef]
- Tirado-Rives, J.; Jorgensen, W.L. Performance of B3LYP density functional methods for a large set of organic molecules. J. Chem. Theory Comput. 2008, 4, 297–306. [Google Scholar] [CrossRef]
- Kruse, H.; Goerigk, L.; Grimme, S. Why the standard B3LYP/6-31G* model chemistry should not be used in DFT calculations of molecular thermochemistry: Understanding and correcting the problem. J. Org. Chem. 2012, 77, 10824–10834. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision, C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Travers, M.J.; Cowles, D.C.; Clifford, E.P.; Ellison, G.B. Photoelectron spectroscopy of the phenylnitrene anion. J. Am. Chem. Soc. 1992, 114, 8699–8701. [Google Scholar] [CrossRef]
- Wijeratne, N.R.; Da Fonte, M.; Ronemus, A.; Wyss, P.J.; Tahmassebi, D.; Wenthold, P.G. Photoelectron spectroscopy of chloro-substituted phenylnitrene anions. J. Phys. Chem. A 2009, 113, 9467–9473. [Google Scholar] [CrossRef]
- Hayes, J.C.; Sheridan, R.S. The IR spectrum of triplet phenylnitrene. On the origin of didehydroazepine in low temperature matrices. J. Am. Chem. Soc. 1990, 112, 5879–5881. [Google Scholar] [CrossRef]
- Leyva, E.; Platz, M.S.; Persy, G.; Wirz, J. Photochemistry of phenyl azide: The role of singlet and triplet phenylnitrene as transient intermediates. J. Am. Chem. Soc. 1986, 108, 3783–3790. [Google Scholar] [CrossRef]
- Wenthold, P.G. Spin-state dependent radical stabilization in nitrenes: The unusually small singlet–triplet splitting in 2-furanylnitrene. J. Org. Chem. 2012, 77, 208–214. [Google Scholar] [CrossRef]
- Feng, R.; Lu, Y.; Deng, G.; Xu, J.; Wu, Z.; Li, H.; Liu, Q.; Kadowaki, N.; Abe, M.; Zeng, X. Magnetically bistable nitrenes: Matrix isolation of furoylnitrenes in both singlet and triplet states and triplet 3-furylnitrene. J. Am. Chem. Soc. 2018, 140, 10–13. [Google Scholar] [CrossRef]
- Rajam, S.; Jadhav, A.V.; Li, Q.; Sarkar, S.K.; Singh, P.N.D.; Rohr, A.; Pace, T.C.S.; Li, R.; Krause, J.A.; Bohne, C.; et al. Triplet sensitized photolysis of a vinyl azide: Direct detection of a triplet vinyl azide and nitrene. J. Org. Chem. 2014, 79, 9325–9334. [Google Scholar] [CrossRef]
- Zhang, X.; Sarkar, S.K.; Weragoda, G.K.; Rajam, S.; Ault, B.S.; Gudmundsdottir, A.D. Comparison of the photochemistry of 3-methyl-2-phenyl-2H-azirine and 2-methyl-3-phenyl-2H-azirine. J. Org. Chem. 2014, 79, 653–663. [Google Scholar] [CrossRef]
- Lopes, S.; Nunes, C.M.; Gómez-Zavaglia, A.; Pinho e Melo, T.M.V.D.; Fausto, R. Structure and photochemical behavior of 3-azido-acrylophenones: A matrix isolation infrared spectroscopy study. Tetrahedron 2011, 67, 7794–7804. [Google Scholar] [CrossRef]
- Gamage, D.W.; Li, Q.; Ranaweera, R.A.A.U.; Sarkar, S.K.; Weragoda, G.K.; Carr, P.L.; Gudmundsdottir, A.D. Vinylnitrene formation from 3,5-diphenyl-isoxazole and 3-benzoyl-2-phenylazirine. J. Org. Chem. 2013, 78, 11349–11356. [Google Scholar] [CrossRef] [PubMed]
- Gatlin, D.M.; Karney, W.L.; Abe, M.; Ault, B.S.; Gudmundsdottir, A.D. Formation and reactivity of triplet vinylnitrenes as a function of ring size. J. Org. Chem. 2019, 84, 9215–9225. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.K.; Osisioma, O.; Karney, W.L.; Abe, M.; Gudmundsdottir, A.D. Using molecular architecture to control the reactivity of a triplet vinylnitrene. J. Am. Chem. Soc. 2016, 138, 14905–14914. [Google Scholar] [CrossRef]
- Sarkar, S.K.; Sawai, A.; Kanahara, K.; Wentrup, C.; Abe, M.; Gudmundsdottir, A.D. Direct detection of a triplet vinylnitrene, 1,4-naphthoquinone-2-ylnitrene, in solution and cryogenic matrices. J. Am. Chem. Soc. 2015, 137, 4207–4214. [Google Scholar] [CrossRef]
- Osisioma, O.; Chakraborty, M.; Ault, B.S.; Gudmundsdottir, A.D. Wavelength-dependent photochemistry of 2-azidovinylbenzene and 2-phenyl-2H-azirine. J. Mol. Struct. 2018, 1174, 94–101. [Google Scholar] [CrossRef]
- Ault, B.S. Infrared spectra of argon matrix-isolated alkali halide salt/water complexes. J. Am. Chem. Soc. 1978, 100, 2426–2433. [Google Scholar] [CrossRef]
- Stratmann, R.E.; Scuseria, G.E.; Frisch, M.J. An efficient implementation of time-dependent density-functional theory for the calculation of excitation energies of large molecules. J. Chem. Phys. 1998, 109, 8218–8224. [Google Scholar] [CrossRef]
- Bauernschmitt, R.; Ahlrichs, R. Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory. Chem. Phys. Lett. 1996, 256, 454–464. [Google Scholar] [CrossRef]
- Foresman, J.B.; Head-Gordon, M.; Pople, J.A.; Frisch, M.J. Toward a systematic molecular orbital theory for excited states. J. Phys. Chem. 1992, 96, 135–149. [Google Scholar] [CrossRef]
- Gonzalez, C.; Schlegel, H.B. An improved algorithm for reaction path following. J. Chem. Phys. 1989, 90, 2154–2161. [Google Scholar] [CrossRef]
- Gonzalez, C.; Schlegel, H.B. Reaction path following in mass-weighted internal coordinates. J. Phys. Chem. 1990, 94, 5523–5527. [Google Scholar] [CrossRef]
- Aquilante, F.; Autschbach, J.; Carlson, R.K.; Chibotaru, L.F.; Delcey, M.G.; De Vico, L.; Fernández Galván, I.; Ferré, N.; Frutos, L.M.; Gagliardi, L.; et al. MOLCAS 8: New capabilities for multiconfigurational quantum chemical calculations across the periodic table. J. Comput. Chem. 2016, 37, 506–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaftenaar, G.; Noordik, J.H. Molden: A pre- and post-processing program for molecular and electronic structures. J. Comput. Aided Mol. Des. 2000, 14, 123–134. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of azidoisoxazole 1 is available from the authors. |














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Singlet Energy |
|---|---|
| CASSCF(14,13)/cc-pVDZ | 13.9 |
| CASPT2(14,13)/cc-pVDZ | 15.2 |
| CASPT2(14,13)/ANO-S | 14.3 |
| CASPT2(14,13)/cc-pVTZ | 13.8 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banerjee, U.; Karney, W.L.; Ault, B.S.; Gudmundsdottir, A.D. Photolysis of 5-Azido-3-Phenylisoxazole at Cryogenic Temperature: Formation and Direct Detection of a Nitrosoalkene. Molecules 2020, 25, 543. https://doi.org/10.3390/molecules25030543
Banerjee U, Karney WL, Ault BS, Gudmundsdottir AD. Photolysis of 5-Azido-3-Phenylisoxazole at Cryogenic Temperature: Formation and Direct Detection of a Nitrosoalkene. Molecules. 2020; 25(3):543. https://doi.org/10.3390/molecules25030543
Chicago/Turabian StyleBanerjee, Upasana, William L. Karney, Bruce S. Ault, and Anna D. Gudmundsdottir. 2020. "Photolysis of 5-Azido-3-Phenylisoxazole at Cryogenic Temperature: Formation and Direct Detection of a Nitrosoalkene" Molecules 25, no. 3: 543. https://doi.org/10.3390/molecules25030543