
Design, Synthesis, and Biological Evaluation of Proteolysis Targeting Chimeras (PROTACs) for the Dual Degradation of IGF-1R and Src

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Design and Synthesis
2.2. CPR3 (12a) and CPR4 (12b), Synthesized PROTAC Compounds, Inhibited Cancer Cell Proliferation
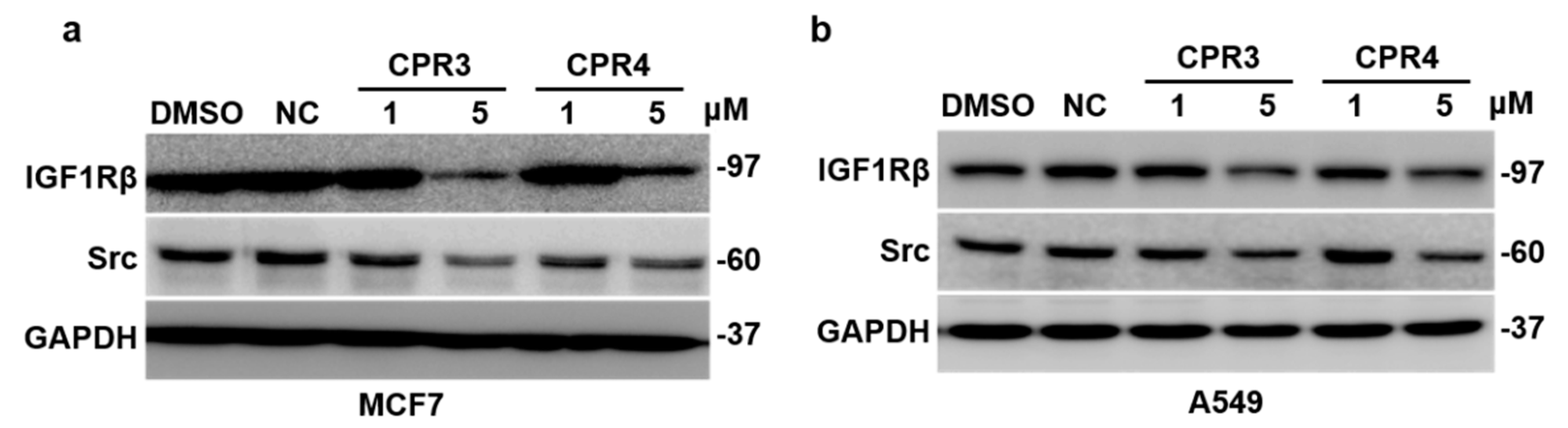
2.3. CPR3 and CPR4 Degraded Both Src and IGF-1R Proteins
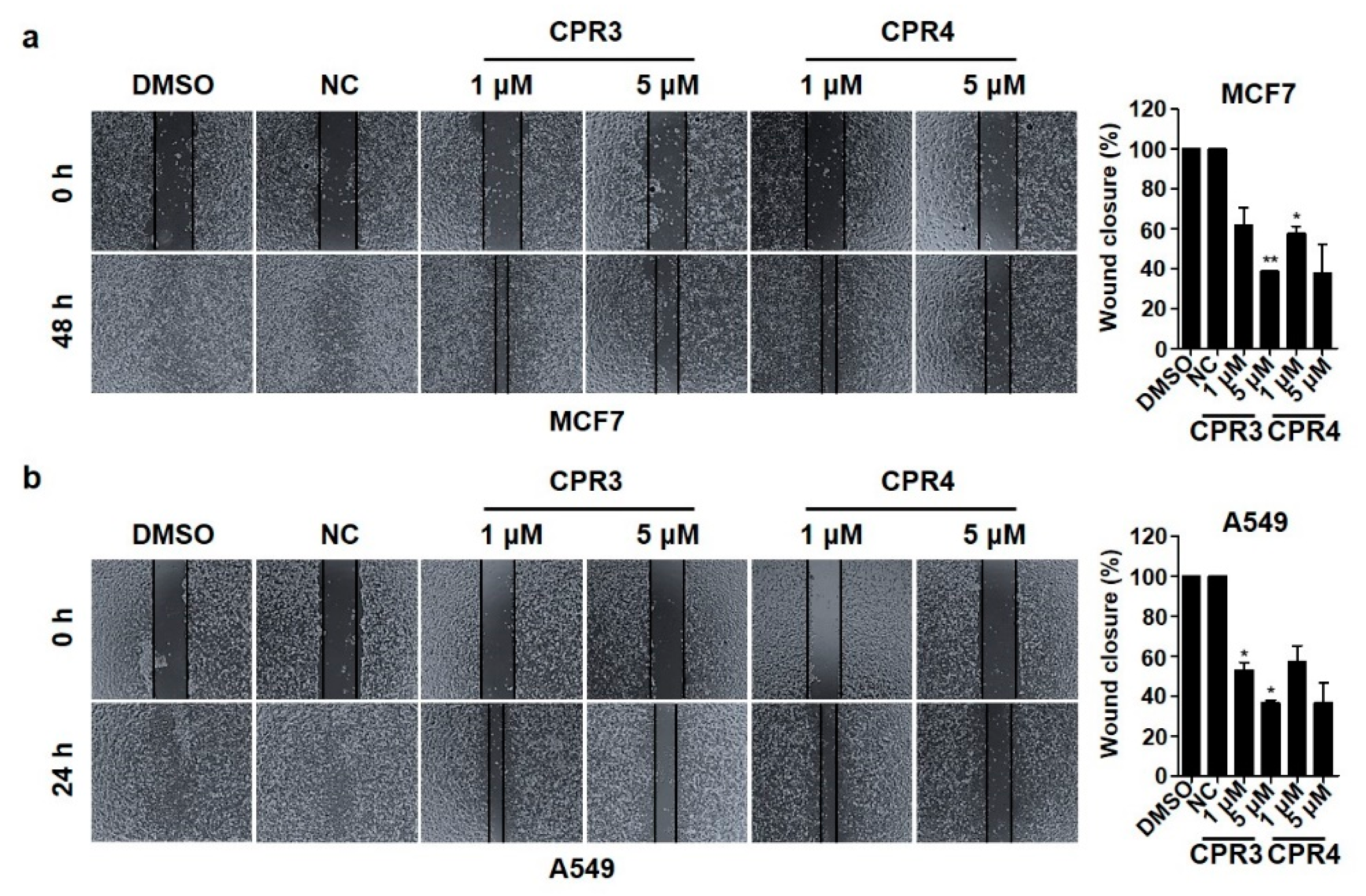
2.4. Invasion and Migration Ability Were Suppressed by CPR3 and CPR4 Treatment in Both MCF7 and A549 Cells
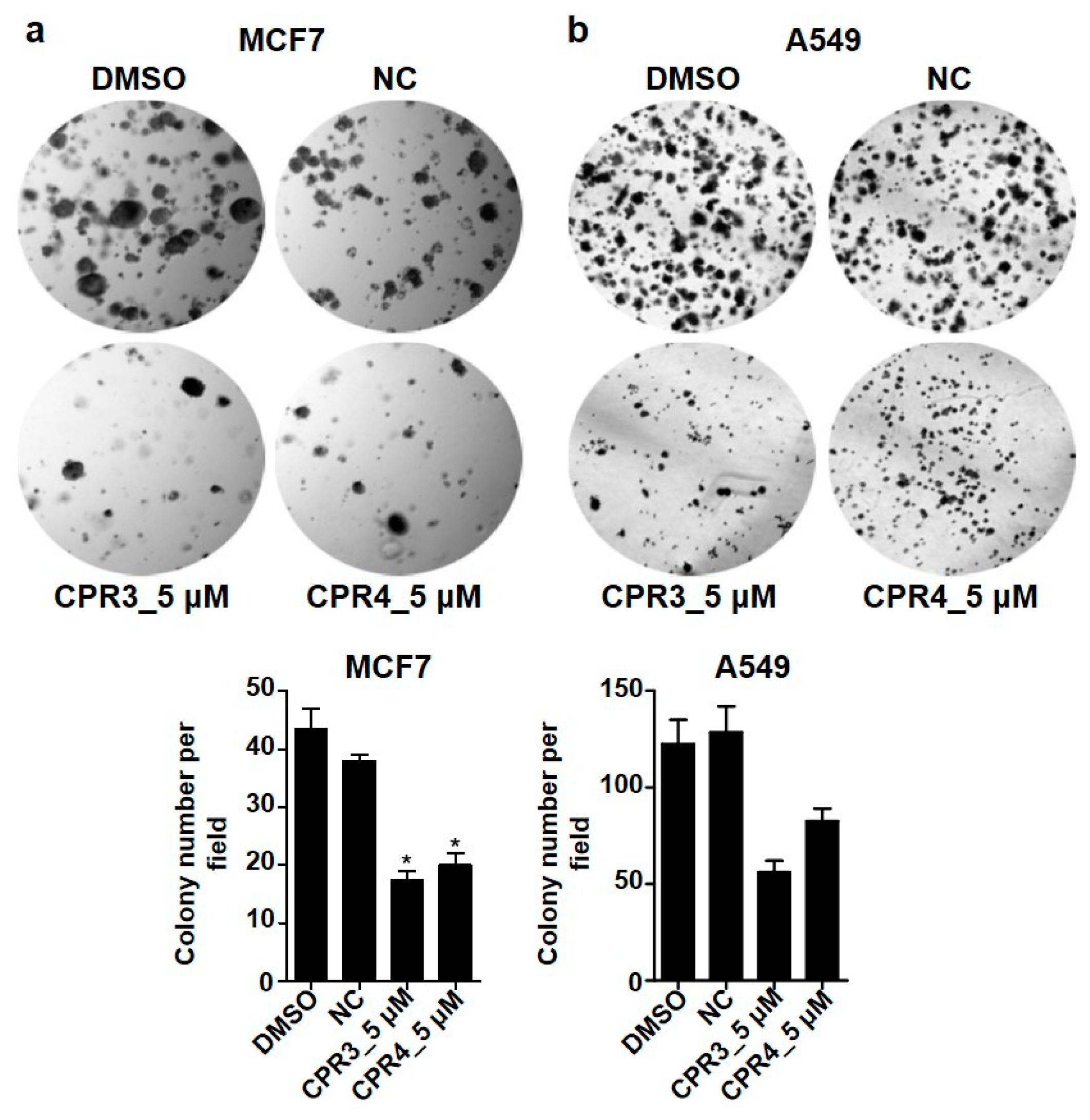
2.5. PROTAC Compounds Inhibited the Cell Growth of Both MCF7 and A549 Cells in the Soft Agar Colony Formation Assay
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.1.1. General Information
4.1.2. Synthesis of 10a–c, 12a–d, 16a–b, 18a–b and 21a–b
4.2. Anticancer Activity
4.2.1. MTT Assay
4.2.2. Western Blot Assay
4.2.3. Wound Healing and Invasion Assay
4.2.4. Soft Agar Colony Formation Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Salami, J.; Crews, C.M. Waste disposal-An attractive strategy for cancer therapy. Science 2017, 355, 1163–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winter, G.E.; Buckley, D.L.; Paulk, J.; Roberts, J.M.; Souza, A.; Dhe-Paganon, S.; Bradner, J.E. Drug Development. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381. [Google Scholar] [CrossRef] [Green Version]
- Mullard, A. First targeted protein degrader hits the clinic. Nat. Rev. Drug Discov. 2019. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, P.P.; Hamann, L.G. Development of targeted protein degradation therapeutics. Nat. Chem. Biol. 2019, 15, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Toure, M.; Crews, C.M. Small-Molecule PROTACS: New Approaches to Protein Degradation. Angew. Chem. Int. Ed. Engl. 2016, 55, 1966–1973. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.C.; Crews, C.M. Induced protein degradation: An emerging drug discovery paradigm. Nat. Rev. Drug Discov. 2017, 16, 101–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef] [Green Version]
- Schneekloth, A.R.; Pucheault, M.; Tae, H.S.; Crews, C.M. Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorg. Med. Chem. Lett. 2008, 18, 5904–5908. [Google Scholar] [CrossRef] [Green Version]
- Itoh, Y.; Ishikawa, M.; Naito, M.; Hashimoto, Y. Protein knockdown using methyl bestatin-ligand hybrid molecules: Design and synthesis of inducers of ubiquitination-mediated degradation of cellular retinoic acid-binding proteins. J. Am. Chem. Soc. 2010, 132, 5820–5826. [Google Scholar] [CrossRef]
- Zhou, B.; Hu, J.; Xu, F.; Chen, Z.; Bai, L.; Fernandez-Salas, E.; Lin, M.; Liu, L.; Yang, C.Y.; Zhao, Y.; et al. Discovery of a Small-Molecule Degrader of Bromodomain and Extra-Terminal (BET) Proteins with Picomolar Cellular Potencies and Capable of Achieving Tumor Regression. J. Med. Chem. 2018, 61, 462–481. [Google Scholar] [CrossRef]
- Zengerle, M.; Chan, K.H.; Ciulli, A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol. 2015, 10, 1770–1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, T.T.; Gao, N.; Li, Q.Q.; Chen, P.G.; Yang, X.F.; Chen, Y.X.; Zhao, Y.F.; Li, Y.M. Specific Knockdown of Endogenous Tau Protein by Peptide-Directed Ubiquitin-Proteasome Degradation. Cell Chem. Biol. 2016, 23, 453–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazo, J.S.; Sharlow, E.R. Drugging Undruggable Molecular Cancer Targets. Annu. Rev. Pharm. Toxicol. 2016, 56, 23–40. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J.S.; Whittle, M.C.; Nakamura, K.; Abell, A.N.; Midland, A.A.; Zawistowski, J.S.; Johnson, N.L.; Granger, D.A.; Jordan, N.V.; Darr, D.B.; et al. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell 2012, 149, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Visakorpi, T.; Hyytinen, E.; Koivisto, P.; Tanner, M.; Keinänen, R.; Palmberg, C.; Palotie, A.; Tammela, T.; Isola, J.; Kallioniemi, O.P. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat. Genet. 1995, 9, 401–406. [Google Scholar] [CrossRef]
- Hon, W.C.; Wilson, M.I.; Harlos, K.; Claridge, T.D.; Schofield, C.J.; Pugh, C.W.; Maxwell, P.H.; Ratcliffe, P.J.; Stuart, D.I.; Jones, E.Y. Structural basis for the recognition of hydroxyproline in HIF-1 alpha by pVHL. Nature 2002, 417, 975–978. [Google Scholar] [CrossRef]
- Schneekloth, J.S., Jr.; Fonseca, F.N.; Koldobskiy, M.; Mandal, A.; Deshaies, R.; Sakamoto, K.; Crews, C.M. Chemical genetic control of protein levels: Selective in vivo targeted degradation. J. Am. Chem. Soc. 2004, 126, 3748–3754. [Google Scholar] [CrossRef] [Green Version]
- Gadd, M.S.; Testa, A.; Lucas, X.; Chan, K.H.; Chen, W.Z.; Lamont, D.J.; Zengerle, M.; Ciulli, A. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat. Chem. Biol. 2017, 13, 514. [Google Scholar] [CrossRef]
- Ito, T.; Ando, H.; Suzuki, T.; Ogura, T.; Hotta, K.; Imamura, Y.; Yamaguchi, Y.; Handa, H. Identification of a primary target of thalidomide teratogenicity. Science 2010, 327, 1345–1350. [Google Scholar] [CrossRef] [Green Version]
- Raina, K.; Lu, J.; Qian, Y.M.; Altieri, M.; Gordon, D.; Rossi, A.M.K.; Wang, J.; Chen, X.; Dong, H.Q.; Siu, K.; et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 7124–7129. [Google Scholar] [CrossRef] [Green Version]
- Bondeson, D.P.; Mares, A.; Smith, I.E.D.; Ko, E.; Campos, S.; Miah, A.H.; Mulholland, K.E.; Routly, N.; Buckley, D.L.; Gustafson, J.L.; et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 2015, 11, 611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, A.C.; Toure, M.; Hellerschmied, D.; Salami, J.; Jaime-Figueroa, S.; Ko, E.; Hines, J.; Crews, C.M. Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew. Chem. Int. Ed. Engl. 2016, 55, 807–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olson, C.M.; Jiang, B.S.; Erb, M.A.; Liang, Y.K.; Doctor, Z.M.; Zhang, Z.N.; Zhang, T.H.; Kwiatkowski, N.; Boukhali, M.; Green, J.L.; et al. Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation. Nat. Chem. Biol. 2018, 14, 163. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Wang, E.S.; Donovan, K.A.; Liang, Y.; Fischer, E.S.; Zhang, T.; Gray, N.S. Development of Dual and Selective Degraders of Cyclin-Dependent Kinases 4 and 6. Angew. Chem. Int. Ed. Engl. 2019, 58, 6321–6326. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Yang, Z.; Gao, H.; Yang, H.; Zhu, S.; An, Z.; Wang, J.; Li, Q.; Chandarlapaty, S.; Deng, H.; et al. Potent and Preferential Degradation of CDK6 via Proteolysis Targeting Chimera Degraders. J. Med. Chem. 2019, 62, 7575–7582. [Google Scholar] [CrossRef]
- Smith, B.E.; Wang, S.L.; Jaime-Figueroa, S.; Harbin, A.; Wang, J.; Hamman, B.D.; Crews, C.M. Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase. Nat. Commun. 2019, 10, 131. [Google Scholar] [CrossRef] [Green Version]
- Bondeson, D.P.; Smith, B.E.; Burslem, G.M.; Buhimschi, A.D.; Hines, J.; Jaime-Figueroa, S.; Wang, J.; Hamman, B.D.; Ishchenko, A.; Crews, C.M. Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead. Cell Chem. Biol. 2018, 25, 78–87. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Pourpak, A.; Morris, S.W. Inhibition of the Insulin-like Growth Factor-1 Receptor (IGF1R) Tyrosine Kinase as a Novel Cancer Therapy Approach. J. Med. Chem. 2009, 52, 4981–5004. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Li, W.; Craddock, B.P.; Foreman, K.W.; Mulvihill, M.J.; Ji, Q.S.; Miller, W.T.; Hubbard, S.R. Small-molecule inhibition and activation-loop trans-phosphorylation of the IGF1 receptor. EMBO J. 2008, 27, 1985–1994. [Google Scholar] [CrossRef] [Green Version]
- Pollak, M.N.; Schernhammer, E.S.; Hankinson, S.E. Insulin-like growth factors and neoplasia. Nat. Rev. Cancer 2004, 4, 505–518. [Google Scholar] [CrossRef]
- Min, H.Y.; Yun, H.J.; Lee, J.S.; Lee, H.J.; Cho, J.; Jang, H.J.; Park, S.H.; Liu, D.; Oh, S.H.; Lee, J.J.; et al. Targeting the insulin-like growth factor receptor and Src signaling network for the treatment of non-small cell lung cancer. Mol. Cancer 2015, 14, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dziadziuszko, R.; Camidge, D.R.; Hirsch, F.R. The insulin-like growth factor pathway in lung cancer. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2008, 3, 815–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeatman, T.J. A renaissance for SRC. Nat. Rev. Cancer 2004, 4, 470–480. [Google Scholar] [CrossRef]
- Dayyani, F.; Parikh, N.U.; Varkaris, A.S.; Song, J.H.; Moorthy, S.; Chatterji, T.; Maity, S.N.; Wolfe, A.R.; Carboni, J.M.; Gottardis, M.M.; et al. Combined Inhibition of IGF-1R/IR and Src family kinases enhances antitumor effects in prostate cancer by decreasing activated survival pathways. PLoS ONE 2012, 7, e51189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, D.H.; Lee, H.J.; Min, H.Y.; Choi, S.P.; Lee, M.S.; Lee, J.W.; Johnson, F.M.; Mehta, K.; Lippman, S.M.; Glisson, B.S.; et al. Combating resistance to anti-IGFR antibody by targeting the integrin beta3-Src pathway. J. Natl. Cancer Inst. 2013, 105, 1558–1570. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.J.; Pham, P.C.; Hyun, S.Y.; Baek, B.; Kim, B.; Kim, Y.; Min, H.Y.; Lee, J.; Lee, H.Y. Development of a 4-aminopyrazolo[3,4-d]pyrimidine-based dual IGF1R/Src inhibitor as a novel anticancer agent with minimal toxicity. Mol. Cancer 2018, 17, 50. [Google Scholar] [CrossRef]
- Kwarcinski, F.E.; Steffey, M.E.; Fox, C.C.; Soellner, M.B. Discovery of Bivalent Kinase Inhibitors via Enzyme-Templated Fragment Elaboration. ACS Med. Chem. Lett. 2015, 6, 898–901. [Google Scholar] [CrossRef] [Green Version]
- Kwarcinski, F.E.; Fox, C.C.; Steffey, M.E.; Soellner, M.B. Irreversible inhibitors of c-Src kinase that target a nonconserved cysteine. ACS Chem. Biol. 2012, 7, 1910–1917. [Google Scholar] [CrossRef] [Green Version]
- Tandon, R.; Senthil, V.; Nithya, D.; Pamidiboina, V.; Kumar, A.; Malik, S.; Chaira, T.; Diwan, M.; Gupta, P.; Venkataramanan, R.; et al. RBx10080307, a dual EGFR/IGF-1R inhibitor for anticancer therapy. Eur. J. Pharmacol. 2013, 711, 19–26. [Google Scholar] [CrossRef]
- Tandon, R.; Kapoor, S.; Vali, S.; Senthil, V.; Nithya, D.; Venkataramanan, R.; Sharma, A.; Talwadkar, A.; Ray, A.; Bhatnagar, P.K.; et al. Dual epidermal growth factor receptor (EGFR)/insulin-like growth factor-1 receptor (IGF-1R) inhibitor: A novel approach for overcoming resistance in anticancer treatment. Eur. J. Pharmacol. 2011, 667, 56–65. [Google Scholar] [CrossRef]
- Buchanan, J.L.; Newcomb, J.R.; Carney, D.P.; Chaffee, S.C.; Chai, L.; Cupples, R.; Epstein, L.F.; Gallant, P.; Gu, Y.; Harmange, J.C.; et al. Discovery of 2,4-bis-arylamino-1,3-pyrimidines as insulin-like growth factor-1 receptor (IGF-1R) inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 2394–2399. [Google Scholar] [CrossRef] [PubMed]
- Schenone, S.; Radi, M.; Musumeci, F.; Brullo, C.; Botta, M. Biologically driven synthesis of pyrazolo[3,4-d]pyrimidines as protein kinase inhibitors: An old scaffold as a new tool for medicinal chemistry and chemical biology studies. Chem. Rev. 2014, 114, 7189–7238. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Ahmad, I.; Chhikara, B.S.; Tiwari, R.; Mandal, D.; Parang, K. Synthesis of 3-phenylpyrazolopyrimidine-1,2,3-triazole conjugates and evaluation of their Src kinase inhibitory and anticancer activities. Bioorg. Med. Chem. Lett. 2011, 21, 1342–1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abellan-Flos, M.; Tanc, M.; Supuran, C.T.; Vincent, S.P. Exploring carbonic anhydrase inhibition with multimeric coumarins displayed on a fullerene scaffold. Org. Biomol. Chem. 2015, 13, 7445–7451. [Google Scholar] [CrossRef]
- Song, Z.; Jin, Y.; Ge, Y.; Wang, C.; Zhang, J.; Tang, Z.; Peng, J.; Liu, K.; Li, Y.; Ma, X. Synthesis and biological evaluation of azole-diphenylpyrimidine derivatives (AzDPPYs) as potent T790M mutant form of epidermal growth factor receptor inhibitors. Bioorg. Med. Chem. 2016, 24, 5505–5512. [Google Scholar] [CrossRef]
- Lu, J.; Qian, Y.; Altieri, M.; Dong, H.; Wang, J.; Raina, K.; Hines, J.; Winkler, J.D.; Crew, A.P.; Coleman, K.; et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol. 2015, 22, 755–763. [Google Scholar] [CrossRef] [Green Version]
- Degorce, S.L.; Boyd, S.; Curwen, J.O.; Ducray, R.; Halsall, C.T.; Jones, C.D.; Lach, F.; Lenz, E.M.; Pass, M.; Pass, S.; et al. Discovery of a Potent, Selective, Orally Bioavailable, and Efficacious Novel 2-(Pyrazol-4-ylamino)-pyrimidine Inhibitor of the Insulin-like Growth Factor-1 Receptor (IGF-1R). J. Med. Chem. 2016, 59, 4859–4866. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, G.; Mao, J.; Xie, M.; Zhao, M.; Guo, X.; Liang, S.; Li, H.; Li, X.; Wang, R. IGF-1R Inhibition Suppresses Cell Proliferation and Increases Radiosensitivity in Nasopharyngeal Carcinoma Cells. Mediat. Inflamm. 2019, 5497467. [Google Scholar] [CrossRef] [Green Version]
- Cox, O.T.; O’Shea, S.; Tresse, E.; Bustamante-Garrido, M.; Kiran-Deevi, R.; O’Connor, R. IGF-1 Receptor and Adhesion Signaling: An Important Axis in Determining Cancer Cell Phenotype and Therapy Resistance. Front. Endocrinol. 2015, 6, 106. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Wang, Y.; Lin, X.; Sun, G.; Parang, K. Synthesis and evaluation of 3-phenylpyrazolo[3,4-d]pyrimidine-peptide conjugates as Src kinase inhibitors. ChemMedChem 2007, 2, 1346–1360. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manda, S.; Lee, N.K.; Oh, D.-C.; Lee, J. Design, Synthesis, and Biological Evaluation of Proteolysis Targeting Chimeras (PROTACs) for the Dual Degradation of IGF-1R and Src. Molecules 2020, 25, 1948. https://doi.org/10.3390/molecules25081948
Manda S, Lee NK, Oh D-C, Lee J. Design, Synthesis, and Biological Evaluation of Proteolysis Targeting Chimeras (PROTACs) for the Dual Degradation of IGF-1R and Src. Molecules. 2020; 25(8):1948. https://doi.org/10.3390/molecules25081948
Chicago/Turabian StyleManda, Sudhakar, Na Keum Lee, Dong-Chan Oh, and Jeeyeon Lee. 2020. "Design, Synthesis, and Biological Evaluation of Proteolysis Targeting Chimeras (PROTACs) for the Dual Degradation of IGF-1R and Src" Molecules 25, no. 8: 1948. https://doi.org/10.3390/molecules25081948