Role of Lipid-Based and Polymer-Based Non-Viral Vectors in Nucleic Acid Delivery for Next-Generation Gene Therapy

 and
and
Abstract
:1. Introduction
2. Lipid-Based Vectors
2.1. Messenger RNA (mRNA) Delivery by Lipid Based Vectors
2.2. siRNA Delivery by Lipid Based Vectors
2.3. Antisense Oligonucleotide Delivery by Lipid Based Vectors
3. Cationic Polymers
3.1. mRNA Delivery by Cationic Polymeric Vectors
3.2. siRNA Delivery by Cationic Polymeric Vectors
3.3. Antisense Oligonucleotide Delivery by Cationic Polymeric Vectors
4. Conjugate Delivery Systems
5. Genome Editing
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kay, M.A. State-of-the-art gene-based therapies: The road ahead. Nat. Rev. Genet. 2011, 12, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Quijano, E.; Bahal, R.; Ricciardi, A.; Saltzman, W.M.; Glazer, P.M. Therapeutic peptide nucleic acids: Principles, limitations, and opportunities. Yale J. Biol. Med. 2017, 90, 583–598. [Google Scholar] [PubMed]
- Houa, K.K.; Panb, H.; Schlesingerc, P.H.; Wicklineb, S.A. A Role for Peptides in Overcoming Endosomal Entrapment in siRNA Delivery—A Focus on Melittin. Biotechnol. Adv. 2015, 33, 931–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borgatti, M.; Romanelli, M.; Saviano, M.; Pedone, C.; Lampronti, I.; Breda, L.; Nastruzzi, C.; Bianchi, N.; Mischiati, C.; Gambari, R. Resistance of decoy PNA-DNA chimeras to enzymatic degradation in cellular extracts and serum. Oncol. Res. 2003, 13, 279–287. [Google Scholar] [CrossRef]
- Yin, H.; Kanasty, R.L.; Eltoukhy, A.A.; Vegas, A.J.; Dorkin, J.R.; Anderson, D.G. Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 2014, 15, 541–555. [Google Scholar] [CrossRef]
- Shima, M.S.; Wang, X.; Ragan, R.; Kwon, Y.J. Dynamics of Nucleic Acid/Cationic Polymer Complexation and Disassembly under Biologically Simulated Conditions Using In Situ Atomic Force Microscopy. Bone 2010, 73, 845–856. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Mahato, R.I. Lipid and polymeric carrier-mediated nucleic acid delivery. Expert Opin. Drug Deliv. 2010, 7, 1209–1226. [Google Scholar] [CrossRef] [Green Version]
- Longmire, M.L.; Choyke, P.L.; Kobayashi, H. Clearance Properties of Nano-sized Particles and Molecules as Imagin Agents: Consideration and Caveats. Nanomedicine 2012, 3, 703–717. [Google Scholar] [CrossRef] [Green Version]
- Fang, J.; Nakamura, H.; Maeda, H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv. Drug Deliv. Rev. 2011, 63, 136–151. [Google Scholar] [CrossRef]
- Torchilin, V.P. Recent advances with liposomes as pharmaceutical carriers. Nat. Rev. Drug Discov. 2005, 4, 145–160. [Google Scholar] [CrossRef]
- Felgner, P.L.; Gadek, T.R.; Holm, M.; Roman, R.; Chan, H.W.; Wenz, M.; Northrop, J.P.; Ringold, G.M.; Danielsen, M. Lipofection: A highly efficient, lipid-mediated DNA-transfection procedure. Proc. Natl. Acad. Sci. USA 1987, 84, 7413–7417. [Google Scholar] [CrossRef] [Green Version]
- Koltover, I.; Salditt, T.; Rädler, J.O.; Safinya, C.R. An inverted hexagonal phase of cationic liposome-DNA complexes related to DNA release and delivery. Science 1998, 281, 78–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, H.; Hui, K.M. Synthesis of a novel series of cationic lipids that can act as efficient gene delivery vehicles through systematic heterocyclic substitution of cholesterol derivatives. Gene Ther. 2001, 8, 855–863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, H.; Zhang, S.; Wang, B.; Cui, S.; Yan, J. Toxicity of cationic lipids and cationic polymers in gene delivery. J. Control. Release 2006, 114, 100–109. [Google Scholar] [CrossRef]
- Hattori, Y.; Suzuki, S.; Kawakami, S.; Yamashita, F.; Hashida, M. The role of dioleoylphosphatidylethanolamine (DOPE) in targeted gene delivery with mannosylated cationic liposomes via intravenous route. J. Control. Release 2005, 108, 484–495. [Google Scholar] [CrossRef]
- Dabkowska, A.P.; Barlow, D.J.; Hughes, A.V.; Campbell, R.A.; Quinn, P.J.; Lawrence, M.J. The effect of neutral helper lipids on the structure of cationic lipid monolayers. J. R. Soc. Interface 2012, 9, 548–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semple, S.C.; Chonn, A.; Cullis, P.R. Influence of cholesterol on the association of plasma proteins with liposomes. Biochemistry 1996, 35, 2521–2525. [Google Scholar] [CrossRef] [PubMed]
- Thewalt, J.L.; Bloom, M. Phosphatidylcholine: Cholesterol phase diagrams. Biophys. J. 1992, 63, 1176–1181. [Google Scholar] [CrossRef] [Green Version]
- Barenholz, Y. Doxil®—The first FDA-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef]
- Jokerst, J.; Lobovkina, T.; Zare, R.N.; Gambhir, S.S. Nanoparticle PEGylation for imaging and therapy. Nanomedicine 2011, 6, 715–728. [Google Scholar] [CrossRef] [Green Version]
- Bao, Y.; Jin, Y.; Chivukula, P.; Zhang, J.; Liu, Y.; Liu, J.; Clamme, J.P.; Mahato, R.I.; Ng, D.; Ying, W.; et al. Effect of PEGylation on biodistribution and gene silencing of siRNA/lipid nanoparticle complexes. Pharm. Res. 2013, 30, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Lee, R.J. The role of helper lipids in lipid nanoparticles (LNPs) designed for oligonucleotide delivery. Adv. Drug Deliv. Rev. 2016, 99, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Filion, M.C.; Phillips, N.C. Toxicity and immunomodulatory activity of liposomal vectors formulated with cationic lipids toward immune effector cells. Biochim. Biophys. Acta Biomembr. 1997, 1329, 345–356. [Google Scholar] [CrossRef] [Green Version]
- Knudsen, K.B.; Northeved, H.; Pramod Kumar, E.K.; Permin, A.; Gjetting, T.; Andresen, T.L.; Larsen, S.; Wegener, K.M.; Lykkesfeldt, J.; Jantzen, K.; et al. In vivo toxicity of cationic micelles and liposomes. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 467–477. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, J.J.; Palmer, L.; Ossanlou, M.; Maclachlan, I.; Graham, R.W.; Zhang, Y.P.; Hope, M.J.; Scherrer, P.; Cullis, P.R. Stabilized plasmid-lipid particles: Construction and characterization. Gene Ther. 1999, 6, 271–281. [Google Scholar] [CrossRef] [Green Version]
- Ambegia, E.; Ansell, S.; Cullis, P.; Heyes, J.; Palmer, L.; MacLachlan, I. Stabilized plasmid-lipid particles containing PEG-diacylglycerols exhibit extended circulation lifetimes and tumor selective gene expression. Biochim. Biophys. Acta Biomembr. 2005, 1669, 155–163. [Google Scholar] [CrossRef] [Green Version]
- Cullis, P.R. Stabilized plasmid-lipid particles for systemic gene therapy. Cell. Mol. Biol. Lett. 2002, 7, 226. [Google Scholar] [CrossRef]
- Batzri, S.; Korn, E.D. Single bilayer liposomes prepared without sonication. BBA Biomembr. 1973, 298, 1015–1019. [Google Scholar] [CrossRef]
- Evers, M.J.W.; Kulkarni, J.A.; van der Meel, R.; Cullis, P.R.; Vader, P.; Schiffelers, R.M. State-of-the-Art Design and Rapid-Mixing Production Techniques of Lipid Nanoparticles for Nucleic Acid Delivery. Small Methods 2018, 2, 1700375. [Google Scholar] [CrossRef]
- Jayaraman, M.; Ansell, S.M.; Mui, B.L.; Tam, Y.K.; Chen, J.; Du, X.; Butler, D.; Eltepu, L.; Matsuda, S.; Narayanannair, J.K.; et al. Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angew. Chem. Int. Ed. 2012, 51, 8529–8533. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, K.A.; Dorkin, J.R.; Vegas, A.J.; Chang, P.H.; Veiseh, O.; Matthews, J.; Fenton, O.S.; Zhang, Y.; Olejnik, K.T.; Yesilyurt, V.; et al. Degradable lipid nanoparticles with predictable in vivo siRNA delivery activity. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Hafez, I.M.; Maurer, N.; Cullis, P.R. On the mechanism whereby cationic lipids promote intracellular delivery of polynucleic acids. Gene Ther. 2001, 8, 1188–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jirikowski, G.F.; Sanna, P.P.; Maciejewski-lenoir, D.; Bloom, F.E.; Jirikowski, G.F.; Sanna, P.P.; Maciejewski-lenoir, D.; Bloom, F.E. Reversal of Diabetes Insipidus in Brattleboro Rats: Intrahypothalamic Injection of Vasopressin mRNA. Science 1992, 255, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Pardia, N.; Tuyishimea, S.; Muramatsua, H.; Karikoa, K.; Muib, B.L.; Tamb, Y.K.; Maddenb, T.D.; Hopeb, M.J.; Weissmana, D. Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J. Control. Release 2015, 217, 345–351. [Google Scholar] [CrossRef] [Green Version]
- Kauffman, K.J.; Dorkin, J.R.; Yang, J.H.; Heartlein, M.W.; Derosa, F.; Mir, F.F.; Fenton, O.S.; Anderson, D.G. Optimization of Lipid Nanoparticle Formulations for mRNA Delivery in Vivo with Fractional Factorial and Definitive Screening Designs. Nano Lett. 2015, 15, 7300–7306. [Google Scholar] [CrossRef]
- Nabhan, J.F.; Wood, K.M.; Rao, V.P.; Morin, J.; Bhamidipaty, S.; Labranche, T.P.; Gooch, R.L.; Bozal, F.; Bulawa, C.E.; Guild, B.C. Intrathecal delivery of frataxin mRNA encapsulated in lipid nanoparticles to dorsal root ganglia as a potential therapeutic for Friedreich’s ataxia. Sci. Rep. 2016, 6, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Sabnis, S.; Kumarasinghe, E.S.; Salerno, T.; Mihai, C.; Ketova, T.; Senn, J.J.; Lynn, A.; Bulychev, A.; McFadyen, I.; Chan, J.; et al. A Novel Amino Lipid Series for mRNA Delivery: Improved Endosomal Escape and Sustained Pharmacology and Safety in Non-human Primates. Mol. Ther. 2018, 26, 1509–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbink, P.; Larocca, R.A.; De La Barrera, R.A.; Bricault, C.A.; Moseley, E.T.; Boyd, M.; Kirilova, M.; Li, Z.; Ng’ang’a, D.; Nanayakkara, O.; et al. Protective Efficacy of Multiple Vaccine Platforms Against Zika Virus Challenge in Rhesus Monkeys. Science 2016, 353, 1129–1132. [Google Scholar] [CrossRef] [Green Version]
- Dowd, K.A.; Ko, S.Y.; Morabito, K.M.; Yang, E.S.; Pelc, R.S.; DeMaso, C.R.; Castilho, L.R.; Abbink, P.; Boyd, M.; Nityanandam, R.; et al. Rapid development of a DNA vaccine for Zika virus. Science 2016, 354, 237–240. [Google Scholar] [CrossRef] [Green Version]
- Muthumani, K.; Griffin, B.D.; Agarwal, S.; Kudchodkar, S.B.; Reuschel, E.L.; Choi, H.; Kraynyak, K.A.; Duperret, E.K.; Keaton, A.A.; Chung, C.; et al. In vivo protection against ZIKV infection and pathogenesis through passive antibody transfer and active immunisation with a prMEnv DNA vaccine. NPJ Vaccines 2016, 1, 1–11. [Google Scholar] [CrossRef]
- Ciaramella, G.; Shresta, S.; Pierson, T.C.; Salazar, V.; Dowd, K.A.; Richner, J.M.; Tang, W.W.; Himansu, S.; Butler, S.L.; Diamond, M.S.; et al. Modified mRNA Vaccines Protect against Zika Virus Infection. Cell 2017, 169, 176. [Google Scholar] [CrossRef] [Green Version]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, M.K.; Xu, S.; Fire, A. RNA as a target of double-stranded RNA-mediated genetic interference in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 1998, 95, 15502–15507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar] [CrossRef]
- Tomari, Y.; Zamore, P.D. Perspective: Machines for RNAi. Genes Dev. 2005, 19, 517–529. [Google Scholar] [CrossRef] [Green Version]
- Hammond, S.M.; Bernstein, E.; Beach, D.; Hannon, G.J. An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature 2000, 404, 293–296. [Google Scholar] [CrossRef]
- Bernstein, E.; Caudy, A.A.; Hammond, S.M.; Hannon, G.J. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 2001, 409, 363–366. [Google Scholar] [CrossRef]
- Ando, Y.; Coelho, T.; Berk, J.L.; Cruz, M.W.; Ericzon, B.G.; Ikeda, S.I.; Lewis, W.D.; Obici, L.; Planté-Bordeneuve, V.; Rapezzi, C.; et al. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J. Rare Dis. 2013, 8, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gertz, M.A.; Benson, M.D.; Dyck, P.J.; Grogan, M.; Coelho, T.; Cruz, M.; Berk, J.L.; Plante-Bordeneuve, V.; Schmidt, H.H.J.; Merlini, G. Diagnosis, Prognosis, and Therapy of Transthyretin Amyloidosis. J. Am. Coll. Cardiol. 2015, 66, 2451–2466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef]
- Zhang, X.; Goel, V.; Robbie, G.J. Pharmacokinetics of Patisiran, the First Approved RNA Interference Therapy in Patients With Hereditary Transthyretin-Mediated Amyloidosis. J. Clin. Pharmacol. 2019, 60, 573–585. [Google Scholar] [CrossRef] [Green Version]
- Suhr, O.B.; Coelho, T.; Buades, J.; Pouget, J.; Conceicao, I.; Berk, J.; Schmidt, H.; Waddington-Cruz, M.; Campistol, J.M.; Bettencourt, B.R.; et al. Efficacy and safety of patisiran for familial amyloidotic polyneuropathy: A phase II multi-dose study. Orphanet J. Rare Dis. 2015, 10, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ball, R.L.; Hajj, K.A.; Vizelman, J.; Bajaj, P.; Whitehead, K.A. Lipid Nanoparticle Formulations for Enhanced Co-delivery of siRNA and mRNA. Nano Lett. 2018, 18, 3814–3822. [Google Scholar] [CrossRef] [PubMed]
- Joseph, L.; Robinson, J.G. Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) Inhibition and the Future of Lipid Lowering Therapy. Prog. Cardiovasc. Dis. 2015, 58, 19–31. [Google Scholar] [CrossRef]
- Cohen, J.C.; Boerwinkle, E.; Mosley, T.H.; Hobbs, H.H. Sequence Variations in PCSK9, Low LDL, and Protection against Coronary Heart Disease. N. Engl. J. Med. 2006, 354, 1264–1272. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Hyodo, K.; Suzuki, T.; Tanaka, Y.; Kikuchi, H.; Ishihara, H. Biodegradable lipid nanoparticles induce a prolonged RNA interference-mediated protein knockdown and show rapid hepatic clearance in mice and nonhuman primates. Int. J. Pharm. 2017, 519, 34–43. [Google Scholar] [CrossRef]
- Zamecnik, P.C.; Stephenson, M.L. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 280–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juliano, R.L. The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 2016, 44, 6518–6548. [Google Scholar] [CrossRef] [PubMed]
- Geary, R.S.; Norris, D.; Yu, R.; Bennett, C.F. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv. Drug Deliv. Rev. 2015, 87, 46–51. [Google Scholar] [CrossRef] [Green Version]
- Crooke, S.T.; Wang, S.; Vickers, T.A.; Shen, W.; Liang, X.H. Cellular uptake and trafficking of antisense oligonucleotides. Nat. Biotechnol. 2017, 35, 230–237. [Google Scholar] [CrossRef]
- Glazier, D.A.; Liao, J.; Roberts, B.; Li, X.; Yang, K.; Stevens, C.M.; Tang, W. Chemical Synthesis and Biological Application of Modified Oligonucleotides. Bioconjug. Chem. 2020, 31, 1213–1233. [Google Scholar] [CrossRef] [PubMed]
- Mathew, V.; Wang, A.K. Inotersen: New promise for the treatment of hereditary transthyretin amyloidosis. Drug Des. Devel. Ther. 2019, 13, 1515–1525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagedorn, P.H.; Persson, R.; Funder, E.D.; Albæk, N.; Diemer, S.L.; Hansen, D.J.; Møller, M.R.; Papargyri, N.; Christiansen, H.; Hansen, B.R.; et al. Locked nucleic acid: Modality, diversity, and drug discovery. Drug Discov. Today 2018, 23, 101–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Summerton, J.; Weller, D. Morpholino antisense oligomers: Design, preparation, and properties. Antisense Nucleic Acid Drug Dev. 1997, 7, 157–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, P.E. Peptide nucleic acid (PNA). A structural DNA mimic. Mater. Res. Soc. Symp. Proc. 1994, 330, 3–6. [Google Scholar] [CrossRef]
- Shen, X.; Corey, D.R. Chemistry, mechanism and clinical status of antisense oligonucleotides and duplex RNAs. Nucleic Acids Res. 2018, 46, 1584–1600. [Google Scholar] [CrossRef]
- Dominski, Z.; Kole, R. Restoration of correct splicing in thalassemic pre-mRNA by antisense oligonucleotides. Proc. Natl. Acad. Sci. USA 1993, 90, 8673–8677. [Google Scholar] [CrossRef] [Green Version]
- Havens, M.A.; Hastings, M.L. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res. 2016, 44, 6549–6563. [Google Scholar] [CrossRef]
- Knudsen, H.; Nielsen, P.E. Antisense properties of duplex- and triplex-forming PNAs. Nucleic Acids Res. 1996, 24, 494–500. [Google Scholar] [CrossRef] [Green Version]
- Dean, D.A. Peptide nucleic acids: Versatile tools for gene therapy strategies. Adv. Drug Deliv. Rev. 2000, 44, 81–95. [Google Scholar] [CrossRef] [Green Version]
- Summerton, J. Morpholino antisense oligomers: The case for an RNase H-independent structural type. Biochim. Biophys. Acta 1999, 1489, 141–158. [Google Scholar] [CrossRef]
- Yang, L.; Ma, F.; Liu, F.; Chen, J.; Zhao, X.; Xu, Q. Efficient Delivery of Antisense Oligonucleotides Using Bioreducible Lipid Nanoparticles In Vitro and In Vivo. Mol. Ther. Nucleic Acids 2020, 19, 1357–1367. [Google Scholar] [CrossRef]
- Gleave, M.E.; Monia, B.P. Antisense therapy for cancer. Nat. Rev. Cancer 2005, 5, 468–479. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Liu, Q.; Li, H.; Kang, C.; Liu, Y.; Guo, T.; Shang, K.; Yan, C.; Cheng, G.; Lee, R.J. Lipid Nanoparticles Loaded with an Antisense Oligonucleotide Gapmer Against Bcl-2 for Treatment of Lung Cancer. Pharm. Res. 2017, 34, 310–320. [Google Scholar] [CrossRef]
- Boussif, O.; Lezoualc’h, F.; Zanta, M.A.; Mergny, M.D.; Scherman, D.; Demeneixt, B.; Behr, J.–P. A Versatile Vector for Gene and Oligonucleotide Transfer into Cells in Culture and in vivo: Polyethylenimine. Proc. Natl. Acad. Sci. USA 1995, 92, 7297–7301. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.Y.; Wu, C.H. Receptor-mediated in vitro gene transformation by a soluble DNA carrier system. J. Biol. Chem. 1987, 262, 4429–4432. [Google Scholar]
- Behr, J.P. The proton sponge: A trick to enter cells the viruses did not exploit. Chimia 1997, 51, 34–36. [Google Scholar]
- Vermeulen, L.M.P.; Brans, T.; Samal, S.K.; Dubruel, P.; Demeester, J.; De Smedt, S.C.; Remaut, K.; Braeckmans, K. Endosomal Size and Membrane Leakiness Influence Proton Sponge-Based Rupture of Endosomal Vesicles. ACS Nano 2018, 12, 2332–2345. [Google Scholar] [CrossRef] [Green Version]
- Wojnilowicz, M.; Glab, A.; Bertucci, A.; Caruso, F.; Cavalieri, F. Super-resolution Imaging of Proton Sponge-Triggered Rupture of Endosomes and Cytosolic Release of Small Interfering RNA. ACS Nano 2019, 13, 187–202. [Google Scholar] [CrossRef]
- Godbey, W.T.; Wu, K.K.; Mikos, A.G. Size matters: Molecular weight affects the efficiency of poly(ethylenimine) as a gene delivery vehicle. J. Biomed. Mater. Res. 1999, 45, 268–275. [Google Scholar] [CrossRef]
- Fischer, D.; Bieber, T.; Li, Y.; Elsässer, H.P.; Kissel, T. A novel non-viral vector for DNA delivery based on low molecular weight, branched polyethylenimine: Effect of molecular weight on transfection efficiency and cytotoxicity. Pharm. Res. 1999, 16, 1273–1279. [Google Scholar] [CrossRef]
- Gao, X.; Yao, L.; Song, Q.; Zhu, L.; Xia, Z.; Xia, H.; Jiang, X.; Chen, J.; Chen, H. The association of autophagy with polyethylenimine-induced cytotoxity in nephritic and hepatic cell lines. Biomaterials 2011, 32, 8613–8625. [Google Scholar] [CrossRef]
- Green, J.J.; Langer, R.; Anderson, D.G. A combinatorial polymer library approach yields insight into nonviral gene delivery. Acc. Chem. Res. 2008, 41, 749–759. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Liu, J.; Cheng, C.J.; Patel, T.R.; Weller, C.E.; Piepmeier, J.M.; Jiang, Z.; Saltzman, W.M. Biodegradable poly(amine-co-ester) terpolymers for targeted gene delivery. Nat. Mater. 2012, 11, 82–90. [Google Scholar] [CrossRef] [Green Version]
- Kauffman, A.C.; Piotrowski-Daspit, A.S.; Nakazawa, K.H.; Jiang, Y.; Datye, A.; Saltzman, W.M. Tunability of Biodegradable Poly(amine-co-ester) Polymers for Customized Nucleic Acid Delivery and Other Biomedical Applications. Biomacromolecules 2018, 19, 3861–3873. [Google Scholar] [CrossRef]
- Obata, Y.; Saito, S.; Takeda, N.; Takeoka, S. Plasmid DNA-encapsulating liposomes: Effect of a spacer between the cationic head group and hydrophobic moieties of the lipids on gene expression efficiency. Biochim. Biophys. Acta Biomembr. 2009, 1788, 1148–1158. [Google Scholar] [CrossRef] [Green Version]
- Piest, M.; Engbersen, J.F.J. Effects of charge density and hydrophobicity of poly(amido amine)s for non-viral gene delivery. J. Control. Release 2010, 148, 83–90. [Google Scholar] [CrossRef]
- Patel, A.K.; Kaczmarek, J.C.; Bose, S.; Kauffman, K.J.; Mir, F.; Heartlein, M.W.; DeRosa, F.; Langer, R.; Anderson, D.G. Inhaled Nanoformulated mRNA Polyplexes for Protein Production in Lung Epithelium. Adv. Mater. 2019, 31. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Gaudin, A.; Zhang, J.; Agarwal, T.; Song, E.; Kauffman, A.C.; Tietjen, G.T.; Wang, Y.; Jiang, Z.; Cheng, C.J.; et al. A “top-down” approach to actuate poly(amine-co-ester) terpolymers for potent and safe mRNA delivery. Biomaterials 2018, 176, 122–130. [Google Scholar] [CrossRef]
- Jiang, Y.; Lu, Q.; Wang, Y.; Xu, E.; Ho, A.; Singh, P.; Wang, Y.; Jiang, Z.; Yang, F.; Tietjen, G.T.; et al. Quantitating Endosomal Escape of a Library of Polymers for mRNA Delivery. Nano Lett. 2020, 20, 1117–1123. [Google Scholar] [CrossRef]
- Cui, J.; Qin, L.; Zhang, J.; Abrahimi, P.; Li, H.; Li, G.; Tietjen, G.T.; Tellides, G.; Pober, J.S.; Mark Saltzman, W. Ex vivo pretreatment of human vessels with siRNA nanoparticles provides protein silencing in endothelial cells. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Cui, J.; Piotrowski-Daspit, A.S.; Zhang, J.; Shao, M.; Bracaglia, L.G.; Utsumi, T.; Seo, Y.E.; DiRito, J.; Song, E.; Wu, C.; et al. Poly(amine-co-ester) nanoparticles for effective Nogo-B knockdown in the liver. J. Control. Release 2019, 304, 259–267. [Google Scholar] [CrossRef]
- Munier, S.; Messai, I.; Delair, T.; Verrier, B.; Ataman-Önal, Y. Cationic PLA nanoparticles for DNA delivery: Comparison of three surface polycations for DNA binding, protection and transfection properties. Colloids Surf. B Biointerfaces 2005, 43, 163–173. [Google Scholar] [CrossRef]
- Morin, G.B. The human telomere terminal transferase enzyme is a ribonucleoprotein that synthesizes TTAGGG repeats. Cell 1989, 59, 521–529. [Google Scholar] [CrossRef]
- Holt, S.E.; Shay, J.W.; Wright, W.E. Refining the telomere-telomerase hypothesis of aging and cancer. Nat. Biotechnol. 1996, 14, 836–839. [Google Scholar] [CrossRef]
- Pitts, A.E.; Corey, D.R. Inhibition of human telomerase by 2′-O-methyl-RNA. Proc. Natl. Acad. Sci. USA 1998, 95, 11549–11554. [Google Scholar] [CrossRef] [Green Version]
- Nafee, N.; Taetz, S.; Schneider, M.; Schaefer, U.F.; Lehr, C.M. Chitosan-coated PLGA nanoparticles for DNA/RNA delivery: Effect of the formulation parameters on complexation and transfection of antisense oligonucleotides. Nanomed. Nanotechnol. Biol. Med. 2007, 3, 173–183. [Google Scholar] [CrossRef]
- Feinbaum, R.; Ambros, V.; Lee, R. The C. elegans Heterochronic Gene lin-4 Encodes Small RNAs with Antisense Complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar]
- Friedman, R.C.; Farh, K.K.H.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [Green Version]
- Catalanotto, C.; Cogoni, C.; Zardo, G. MicroRNA in control of gene expression: An overview of nuclear functions. Int. J. Mol. Sci. 2016, 17, 1712. [Google Scholar] [CrossRef] [Green Version]
- Baumann, V.; Winkler, J. MiRNA-based therapies: Strategies and delivery platforms for oligonucleotide and non-oligonucleotide agents. Future Med. Chem. 2014, 6, 1967–1984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.; Croce, C.M. The role of microRNAs in human cancer. Signal Transduct. Target. Ther. 2016, 1. [Google Scholar] [CrossRef] [Green Version]
- Seo, Y.E.; Suh, H.W.; Bahal, R.; Josowitz, A.; Zhang, J.; Song, E.; Cui, J.; Noorbakhsh, S.; Jackson, C.; Bu, T.; et al. Nanoparticle-mediated intratumoral inhibition of miR-21 for improved survival in glioblastoma. Biomaterials 2019, 201, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Shmueli, R.B.; Anderson, D.G.; Green, J.J. Electrostatic surface modifications to improve gene delivery. Expert Opin. Drug Deliv. 2010, 7, 535–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.S.; Green, J.J.; Love, K.T.; Sunshine, J.; Langer, R.; Anderson, D.G. Gold, poly(β-amino ester) nanoparticles for small interfering RNA delivery. Nano Lett. 2009, 9, 2402–2406. [Google Scholar] [CrossRef] [Green Version]
- Runcie, K.; Budman, D.R.; John, V.; Seetharamu, N. Bi-specific and tri-specific antibodies- the next big thing in solid tumor therapeutics. Mol. Med. 2018, 24, 1–15. [Google Scholar] [CrossRef]
- Tsuchikama, K.; An, Z. Antibody-drug conjugates: Recent advances in conjugation and linker chemistries. Protein Cell 2018, 9, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Kaplon, H.; Muralidharan, M.; Schneider, Z.; Reichert, J.M. Antibodies to watch in 2020. MAbs 2020, 12, 1703531. [Google Scholar] [CrossRef] [Green Version]
- Johnston, M.C.; Scott, C.J. Antibody conjugated nanoparticles as a novel form of antibody drug conjugate chemotherapy. Drug Discov. Today Technol. 2018, 30, 63–69. [Google Scholar] [CrossRef]
- Okamoto, A.; Asai, T.; Hirai, Y.; Shimizu, K.; Koide, H.; Minamino, T.; Oku, N. Systemic Administration of siRNA with Anti-HB-EGF Antibody-Modified Lipid Nanoparticles for the Treatment of Triple-Negative Breast Cancer. Mol. Pharm. 2018, 15, 1495–1504. [Google Scholar] [CrossRef]
- Soutschek, J.; Akinc, A.; Bramlage, B.; Charisse, K.; Constien, R.; Donoghue, M.; Elbashir, S.; Gelck, A.; Hadwiger, P.; Harborth, J.; et al. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature 2004, 432, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Ambardekar, V.V.; Han, H.; Varney, M.L.; Vinogradov, S.V.; Singh, R.K.; Vetro, J.A. The modification of siRNA with 3′ cholesterol to increase nuclease protection and suppression of native mRNA by select siRNA polyplexes. Biomaterials 2011, 32, 1404–1411. [Google Scholar] [CrossRef] [Green Version]
- Alnylam. Alnylam® Development Pipeline of Investigational RNAi Therapeutics. Available online: https://www.alnylam.com/alnylam-rnai-pipeline/2020 (accessed on 20 May 2020).
- Wooddell, C.I.; Rozema, D.B.; Hossbach, M.; John, M.; Hamilton, H.L.; Chu, Q.; Hegge, J.O.; Klein, J.J.; Wakefield, D.H.; Oropeza, C.E.; et al. Hepatocyte-targeted RNAi therapeutics for the treatment of chronic hepatitis B virus infection. Mol. Ther. 2013, 21, 973–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wooddell, C.I.; Yuen, M.F.; Chan, H.L.Y.; Gish, R.G.; Locarnini, S.A.; Chavez, D.; Ferrari, C.; Given, B.D.; Hamilton, J.; Kanner, S.B.; et al. Rnai-based treatment of chronically infected patients and chimpanzees reveals that integrated hepatitis b virus DNA is a source of hbsag. Sci. Transl. Med. 2017, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Nantz, M.H.; Zern, M.A. Targeting hepatocytes for drug and gene delivery: Emerging novel approaches and applications. Front. Biosci. 2002, 7, d717–d725. [Google Scholar]
- de Paula Brandão, P.R.; Titze-de-Almeida, S.S.; Titze-de-Almeida, R. Leading RNA Interference Therapeutics Part 2: Silencing Delta-Aminolevulinic Acid Synthase 1, with a Focus on Givosiran. Mol. Diagnosis Ther. 2020, 24, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.; Taubel, J.; Bush, J.; Borodovsky, A.; Kawahata, N.; Mclean, H.; Powell, C.; Chaturvedi, P.; Warner, G.; Garg, P.; et al. A Subcutaneously Administered Investigational RNAi Therapeutic (ALN-CC5) Targeting Complement C5 for Treatment of PNH and Complement-Mediated Diseases: Interim Phase 1 Study Results. Blood 2015, 128, 2413. [Google Scholar] [CrossRef]
- Hill, A.; Valls, A.G.; Griffin, M.; Munir, T.; Borodovsky, A.; Kawahata, N.; Mclean, H.; Shi, K.; Partisano, A.M.; Kim, J.; et al. A subcutaneously administered investigational RNAi therapeutic (ALN-CC5) targeting complement C5 for treatment of PNH and complement-mediated diseases: Preliminary phase 1/2 study results in patients with PNH. Blood 2016, 128, 3891. [Google Scholar] [CrossRef]
- Liebow, A.; Li, X.; Racie, T.; Hettinger, J.; Bettencourt, B.R.; Najafian, N.; Haslett, P.; Fitzgerald, K.; Holmes, R.P.; Erbe, D.; et al. An investigational RNAi therapeutic targeting glycolate oxidase reduces oxalate production in models of primary hyperoxaluria. J. Am. Soc. Nephrol. 2017, 28, 494–503. [Google Scholar] [CrossRef] [Green Version]
- Judge, D.P.; Kristen, A.V.; Grogan, M.; Maurer, M.S.; Falk, R.H.; Hanna, M.; Gillmore, J.; Garg, P.; Vaishnaw, A.K.; Harrop, J.; et al. Phase 3 Multicenter Study of Revusiran in Patients with Hereditary Transthyretin-Mediated (hATTR) Amyloidosis with Cardiomyopathy (ENDEAVOUR). Cardiovasc. Drugs Ther. 2020, 34, 357–370. [Google Scholar] [CrossRef] [Green Version]
- Pasi, K.J.; Georgiev, P.; Mant, T.; Lissitchkov, T.; Creagh, M.D.; Bevan, D.; Austin, S.; Hay, C.R.; Hegemann, I.; Kazmi, R.; et al. Fitusiran, an Investigational RNAi Therapeutic Targeting Antithrombin for the Treatment of Hemophilia: Updated Results from a Phase 1 and Phase 1/2 Extension Study in Patients with Inhibitors. Blood 2016, 128, 1397. [Google Scholar] [CrossRef]
- Pipe, S.; Ragni, M.V.; Négrier, C.; Yu, Q.; Bajwa, N.; Caminis, J.; Mei, B.; Andersson, S.R. Fitusiran, an RNAi Therapeutic Targeting Antithrombin to Restore Hemostatic Balance in Patients with Hemophilia a or B with or without Inhibitors: Management of Acute Bleeding Events. Blood 2019, 134, 1138. [Google Scholar] [CrossRef]
- Fitzgerald, K.; White, S.; Borodovsky, A.; Bettencourt, B.R.; Strahs, A.; Clausen, V.; Wijngaard, P.; Horton, J.D.; Taubel, J.; Brooks, A.; et al. A highly durable RNAi therapeutic inhibitor of PCSK9. N. Engl. J. Med. 2017, 376, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Ray, K.K.; Landmesser, U.; Leiter, L.A.; Kallend, D.; Dufour, R.; Karakas, M.; Hall, T.; Troquay, R.P.T.; Turner, T.; Visseren, F.L.J.; et al. Inclisiran in patients at high cardiovascular risk with elevated LDL cholesterol. N. Engl. J. Med. 2017, 376, 1430–1440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, K.K.; Wright, R.S.; Kallend, D.; Koenig, W.; Leiter, L.A.; Raal, F.J.; Bisch, J.A.; Richardson, T.; Jaros, M.; Wijngaard, P.L.J.; et al. Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. N. Engl. J. Med. 2020, 382, 1507–1519. [Google Scholar] [CrossRef]
- Graham, M.J.; Lee, R.G.; Brandt, T.A.; Tai, L.J.; Fu, W.; Peralta, R.; Yu, R.; Hurh, E.; Paz, E.; McEvoy, B.W.; et al. Cardiovascular and metabolic effects of ANGPTL3 antisense oligonucleotides. N. Engl. J. Med. 2017, 377, 222–232. [Google Scholar] [CrossRef]
- Han, K.; Cremer, J.; Elston, R.; Oliver, S.; Baptiste-Brown, S.; Chen, S.; Gardiner, D.; Davies, M.; Saunders, J.; Hamatake, R.; et al. A Randomized, Double-Blind, Placebo-Controlled, First-Time-in-Human Study to Assess the Safety, Tolerability, and Pharmacokinetics of Single and Multiple Ascending Doses of GSK3389404 in Healthy Subjects. Clin. Pharmacol. Drug Dev. 2019, 8, 790–801. [Google Scholar] [CrossRef] [Green Version]
- McCaleb, M.; Hughes, S.; Greary, R.; Monia, B.; Grossman, T. Pharmacodynamic efficacy of IONIS-FB-LRX, a complement factor B antisense oligonucleotide, in a phase 1 clinical study. Mol. Immunol. 2018, 102, 187–188. [Google Scholar] [CrossRef]
- Ferrone, J.D.; Bhattacharjee, G.; Revenko, A.S.; Zanardi, T.A.; Warren, M.S.; Derosier, F.J.; Viney, N.J.; Pham, N.C.; Kaeser, G.E.; Baker, B.F.; et al. IONIS-PKK Rx a Novel Antisense Inhibitor of Prekallikrein and Bradykinin Production. Nucleic Acid Ther. 2019, 29, 82–91. [Google Scholar] [CrossRef] [Green Version]
- Rozema, D.B.; Lewis, D.L.; Wakefield, D.H.; Wong, S.C.; Klein, J.J.; Roesch, P.L.; Bertin, S.L.; Reppen, T.W.; Chu, Q.; Blokhin, A.V.; et al. Dynamic PolyConjugates for targeted in vivo delivery of siRNA to hepatocytes. Proc. Natl. Acad. Sci. USA 2007, 104, 12982–12987. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.C.; Klein, J.J.; Hamilton, H.L.; Chu, Q.; Frey, C.L.; Trubetskoy, V.S.; Hegge, J.; Wakefield, D.; Rozema, D.B.; Lewis, D.L. Co-injection of a targeted, reversibly masked endosomolytic polymer dramatically improves the efficacy of cholesterol-conjugated small interfering RNAs in vivo. Nucleic Acid Ther. 2012, 22, 399–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Yang, Y.; Hong, W.; Huang, M.; Wu, M.; Zhao, X. Applications of genome editing technology in the targeted therapy of human diseases: Mechanisms, advances and prospects. Signal Transduct. Target. Ther. 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Gaj, T.; Gersbach, C.A.; Barbas, C.F. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Driscoll, M.; Jeggo, P.A. The role of double-strand break repair—Insights from human genetics. Nat. Rev. Genet. 2006, 7, 45–54. [Google Scholar] [CrossRef]
- Carroll, D. Genome engineering with zinc-finger nucleases. Genetics 2011, 188, 773–782. [Google Scholar] [CrossRef] [Green Version]
- Joung, J.K.; Sander, J.D. TALENs: A widely applicable technology for targeted genome editing. Nat. Rev. Mol. Cell Biol. 2013, 14, 49–55. [Google Scholar] [CrossRef] [Green Version]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [Green Version]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Mahiny, A.J.; Dewerth, A.; Mays, L.E.; Alkhaled, M.; Mothes, B.; Malaeksefat, E.; Loretz, B.; Rottenberger, J.; Brosch, D.M.; Reautschnig, P.; et al. In vivo genome editing using nuclease-encoding mRNA corrects SP-B deficiency. Nat. Biotechnol. 2015, 33, 584–586. [Google Scholar] [CrossRef]
- Conway, A.; Mendel, M.; Kim, K.; McGovern, K.; Boyko, A.; Zhang, L.; Miller, J.C.; DeKelver, R.C.; Paschon, D.E.; Mui, B.L.; et al. Non-viral Delivery of Zinc Finger Nuclease mRNA Enables Highly Efficient In Vivo Genome Editing of Multiple Therapeutic Gene Targets. Mol. Ther. 2019, 27, 866–877. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Ji, W.; Hall, J.M.; Hu, Q.; Wang, C.; Beisel, C.L.; Gu, Z. Efficient Delivery of CRISPR-Cas9 for Genome Editing via Self-Assembled DNA Nanoclews. Angew. Chem. Int. Ed. Engl. 2015, 54, 12029–12033. [Google Scholar] [CrossRef]
- Jiang, C.; Mei, M.; Li, B.; Zhu, X.; Zu, W.; Tian, Y.; Wang, Q.; Guo, Y.; Dong, Y.; Tan, X. A non-viral CRISPR/Cas9 delivery system for therapeutically targeting HBV DNA and pcsk9 in vivo. Cell Res. 2017, 27, 440–443. [Google Scholar] [CrossRef] [PubMed]
- Jia, N.; Ye, Y.; Wang, Q.; Zhao, X.; Hu, H.; Chen, D.; Qiao, M. Preparation and evaluation of poly(L-histidine) based pH-sensitive micelles for intracellular delivery of doxorubicin against MCF-7/ADR cells. Asian J. Pharm. Sci. 2017, 12, 433–441. [Google Scholar] [CrossRef]
- Rezvantalab, S.; Drude, N.I.; Moraveji, M.K.; Güvener, N.; Koons, E.K.; Shi, Y.; Lammers, T.; Kiessling, F. PLGA-based nanoparticles in cancer treatment. Front. Pharmacol. 2018, 9, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, X.; Pan, W.; Su, T.; Zhang, M.; Dong, W.; Qi, X. Recent advances in natural polymer-based drug delivery systems. React. Funct. Polym. 2020, 148, 104501. [Google Scholar] [CrossRef]
- Doktorovová, S.; Kovačević, A.B.; Garcia, M.L.; Souto, E.B. Preclinical safety of solid lipid nanoparticles and nanostructured lipid carriers: Current evidence from in vitro and in vivo evaluation. Eur. J. Pharm. Biopharm. 2016, 108, 235–252. [Google Scholar] [CrossRef] [PubMed]







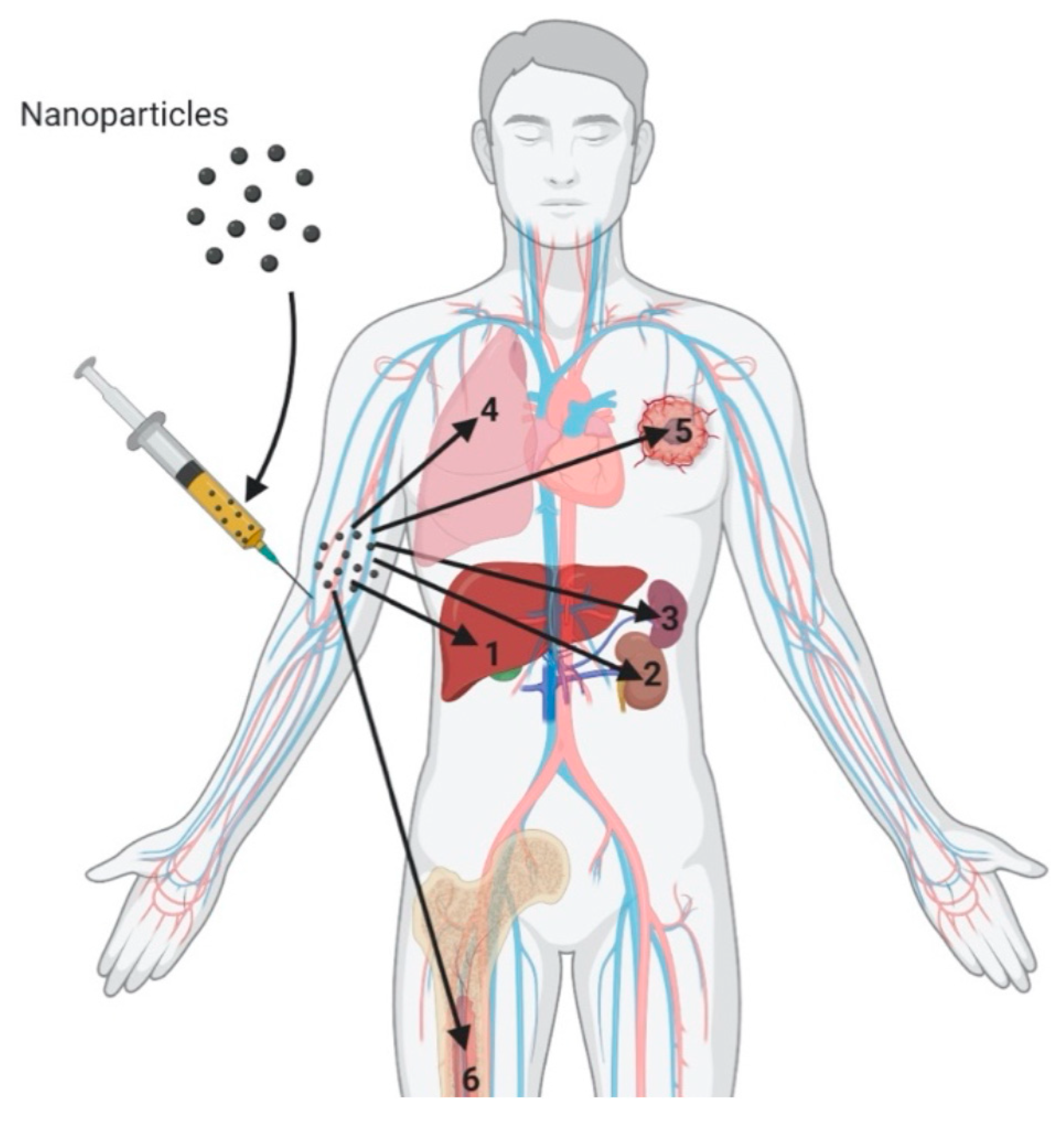{kind=link}
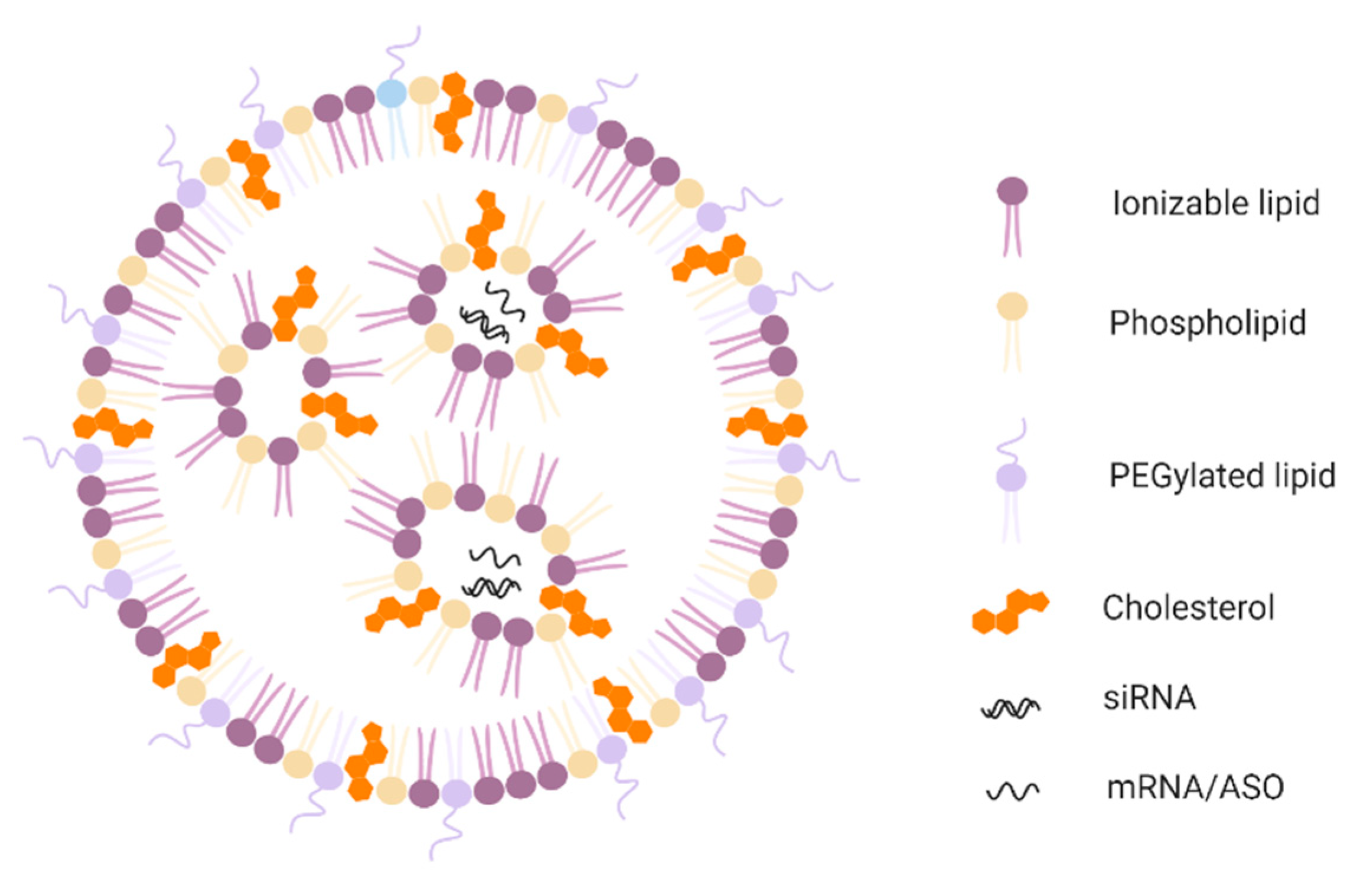{kind=link}
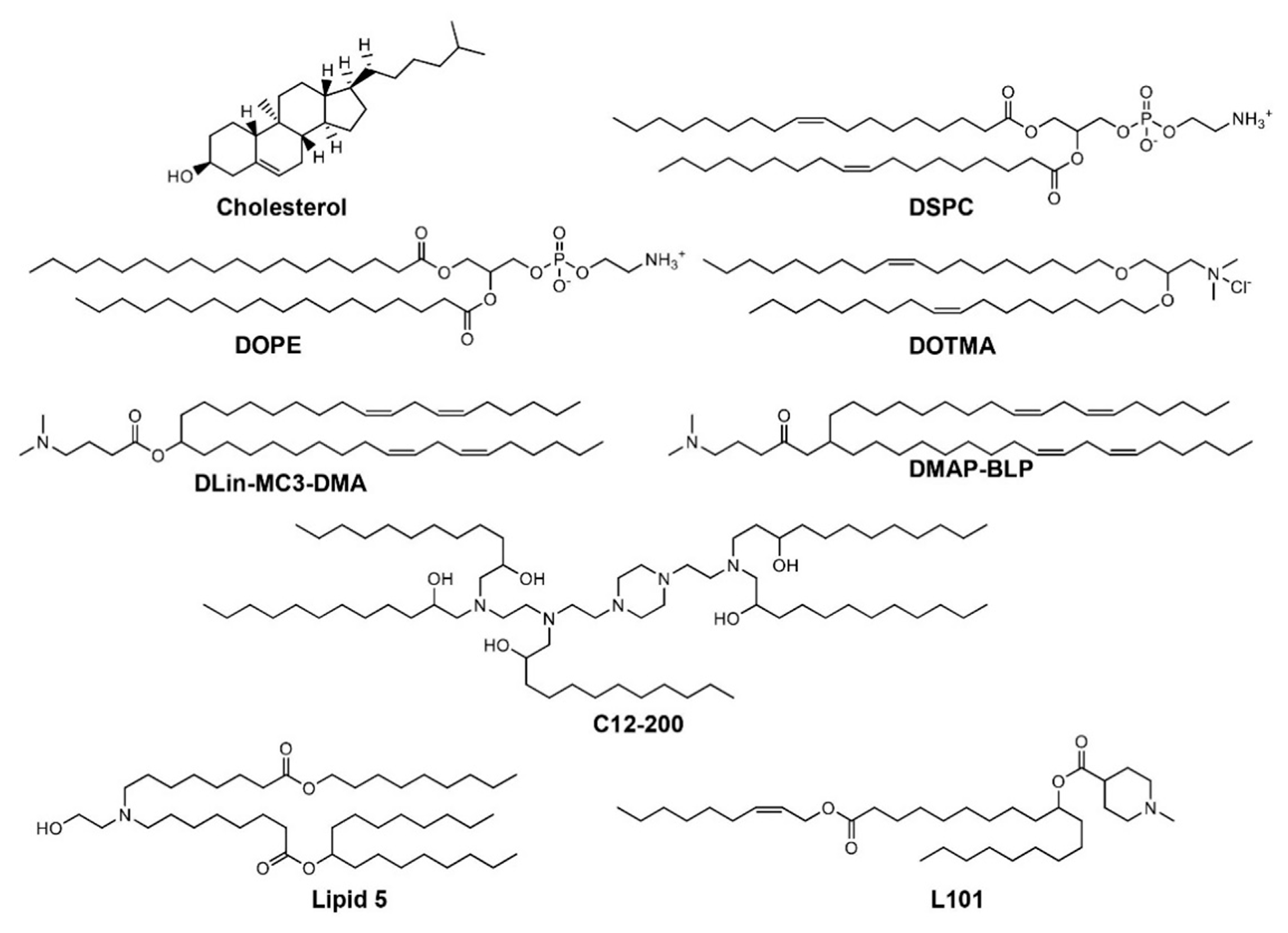{kind=link}
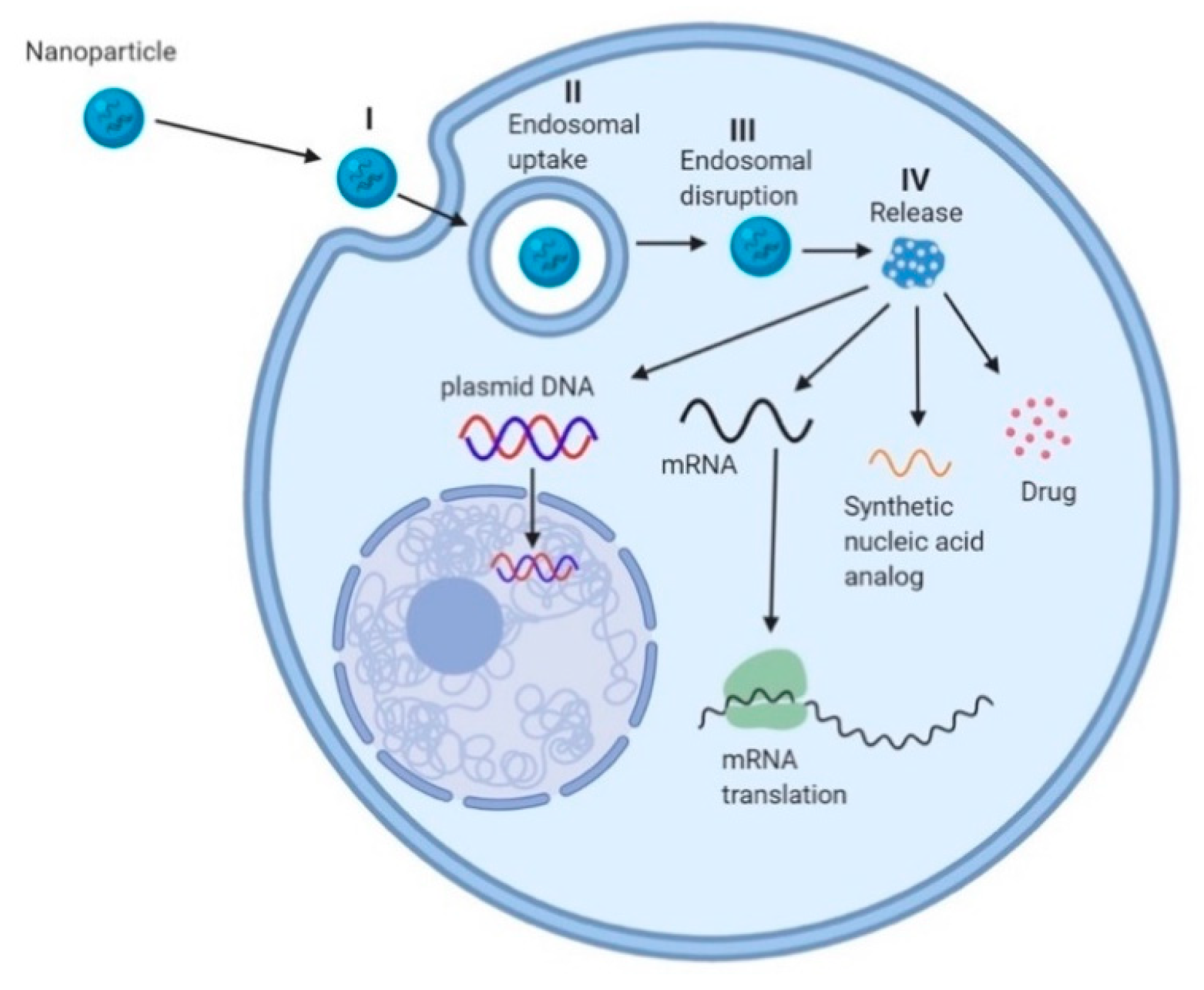{kind=link}
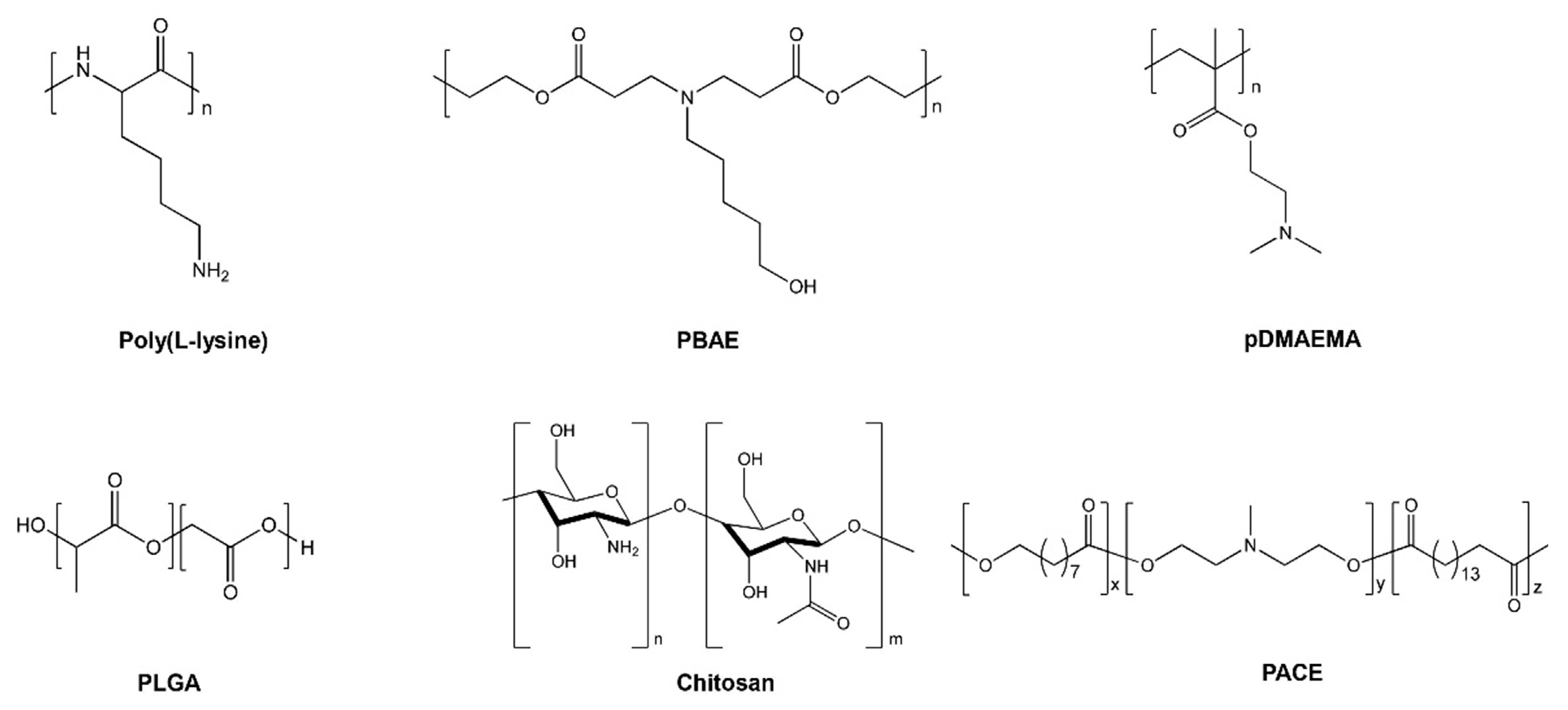{kind=link}
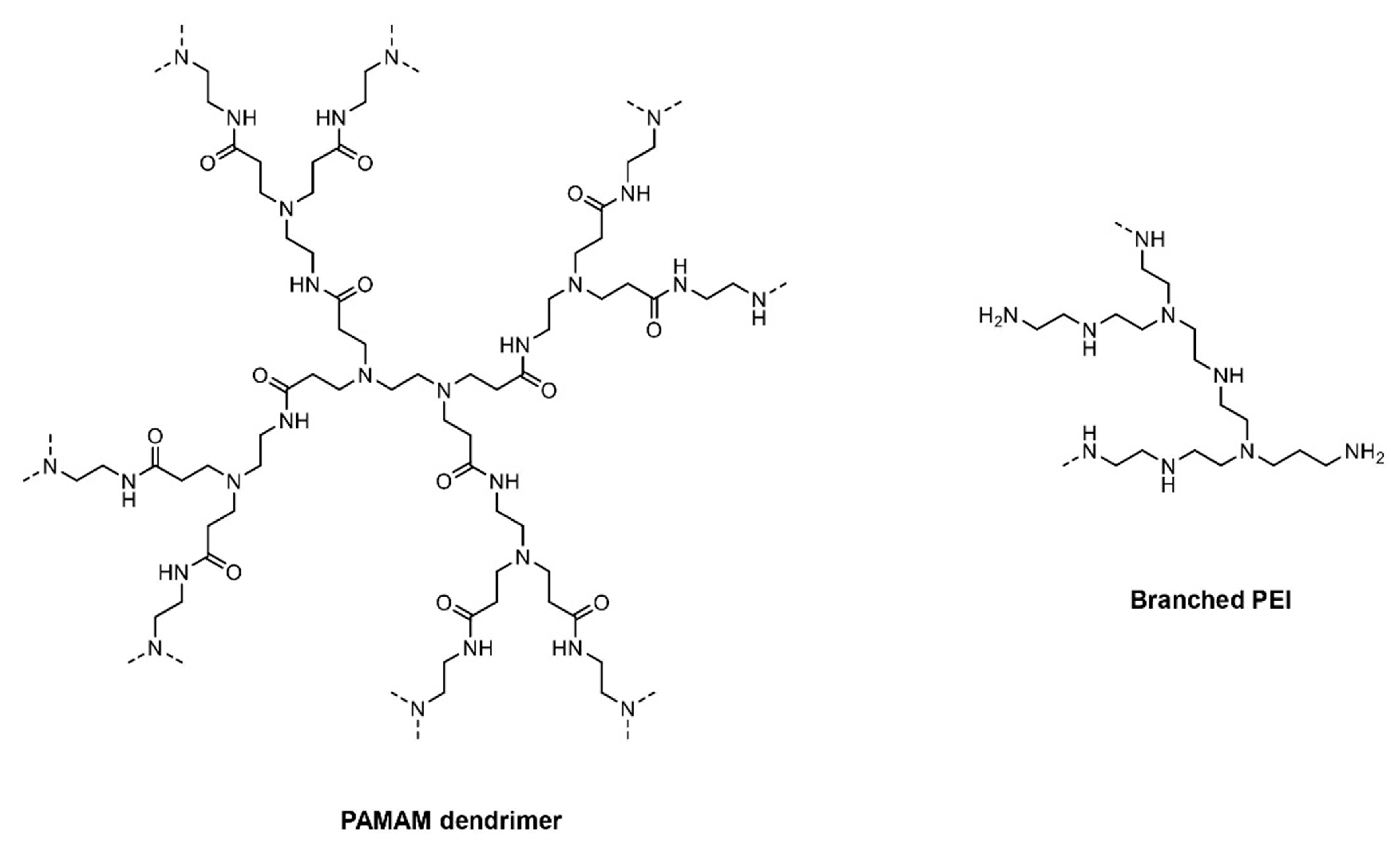{kind=link}
| SN | Nucleic Acid | Gene/Target | Vector | Disease/Condition | Route of Delivery | Animal/Cell Line Used | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | mRNA | EPO | LNPs | - | IV | C57BL/6 mice | [35] |
| 2 | mRNA | Luciferase | LNPs | - | IP, IM, SC, IV, ID and ITr | BALB/c mice | [34] |
| 3 | mRNA | FXN | LNPs | Friedreich’s ataxia | ICV | BALB/c mice | [36] |
| 4 | mRNA | Luciferase and hEPO | LNPs | - | IV | CD-1 mice, rats, cynomolgus monkeys | [37] |
| 5 | mRNA | pRM and E | LNPs | Zika | IM | AG129 mice | [41] |
| 6 | siRNA and mRNA | Factor VII and luciferase | LNPs | - | IV | C57BL/6 mice | [53] |
| 7 | siRNA | PCSK9 | LNPs | Elevated LDL-Cholesterol | IV | C57BL/6 mice and monkeys | [56] |
| 8 | mRNA | PCSK9 | LNPs | Elevated LDL-Cholesterol | IV | BALB/c mice | [72] |
| 9 | G3139-GAP | Bcl-2 | LNPs | Lung cancer | IV | BALB/c mice | [74] |
| 10 | mRNA | Luciferase | hPBAEs NPs | - | Inhalation | C57BL/6 mice | [88] |
| 11 | mRNA | EPO | PACE NPs | - | IV | BALB/c mice | [89] |
| 12 | siRNA | CIITA | PACE NPs | Tissue transplantation | Incubation | SCID/beige mice | [91] |
| 13 | siRNA | Nogo-B | PACE NPs | Hepatic fibrosis and alcoholic liver disease | Spleen | C57BL/6 mice | [92] |
| 14 | ASO | Human telomerase | Chitosan-coated PLGA NPs | Lung cancer | Cellular uptake | A549 cancer cell line | [97] |
| 15 | ASO | miR-21 | PACE NPs | Glioblastoma | CED | Fischer 344 rats | [103] |
| 16 | siRNA | PLK-1 | EGF antibody Anti-HB -LNPs | Triple negative breast cancer | IV | BALB/c mice | [110] |
| 17 | siRNA | ApoB | PBAVE NPs | - | IV | ICR mice | [132] |
| 18 | ZFN mRNA | SFTB | Chitosan-coated PLGA NPs | Lung disease | ITh | SP-B transgenic mice | [140] |
| 19 | ZFN mRNA | TTR and PCSK9 | LNPs | Elevated LDL-Cholesterol and amyloidosis | IV | CD-1 mice | [141] |
| 20 | Cas9 and sgRNA | EGFP | PEI coated DNA nanoclew | Osteosarcoma | IT | Nude mice | [144] |
| 21 | Cas9 and sgRNA | PCSK 9 and HBV | LLNs | Elevated LDL-Cholesterol and Hepatitis B | IV | C57BL/6 mice | [142] |
| SN | Nucleic Acid | Target | Vector | Disease | Route of Delivery | Clinical Trial | Status |
|---|---|---|---|---|---|---|---|
| 1 | mRNA | OX40L | LNPs | Solid tumors and lymphomas | IT | NCT03739931 | Active |
| 2 | mRNA | OX40L | LNPs | Solid tumors, lymphomas and ovarian cancer | IT | NCT03323398 | Active |
| 3 | mRNA | S-protein | LNPs | COVID-19 | IM | NCT04283461 | Active |
| 4 | mRNA | OTC | LNPs | OTC deficiency | IV | NCT03767270 | Withdrawn |
| 5 | mRNA | prM and E | LNPs | Zika | IM | NCT04064905 | Active |
| 6 | mRNA | Pentamer and T cell antigen | LNPs | CMV infection | IM | NCT03382405 | Active |
| 7 | siRNA | MYC | LNPs | Hematological and solid tumors | IV | NCT02110563 | Terminated |
| 8 | siRNA | MYC | LNPs | Hepatocellular carcinoma | IV | NCT02314052 | Terminated |
| 9 | siRNA | HSP47 | LNPs | Hepatic fibrosis | IV | NCT02227459 | Completed |
| 10 | siRNA | PLK1 | LNPs | Solid tumors | Hepatic IA | NCT01437007 | Completed |
| 11 | saRNA | CEBPA | Liposomal NPs | Hepatocellular carcinoma | IV | NCT02716012 | Active |
| 12 | siRNA | TGF-β1 and Cox-2 | NPs | Hypertrophic scar | ID | NCT02956317 | Unknown |
| 13 | siRNA | KRAS | PLGA matrix | Adeno-carcinoma | SI | NCT01676259 | Recruiting |
| 14 | siRNA | PKN3 | Liposomes | Pancreatic cancer | IV | NCT01808638 | Completed |
| 15 | siRNA | HBV antigen | LNPs | Hepatitis B | IV | NCT02631096 | Completed |
| 16 | siRNA | KSP and VEGF | LNPs | Solid tumors | IV | NCT01158079 | Completed |
| 17 | siRNA | PCSK9 | LNPs | Elevated LDL-Cholesterol | IV | NCT01437059 | Completed |
| 18 | siRNA | Bcl-2 | Gold NPs | GBM | IV | NCT03020017 | Completed |
| 19 | siRNA | RRM2 | Cyclodextrin polymer | Solid tumors | IV | NCT00689065 | Terminated |
| 20 | siRNA | GO | LNPs | Primary hyperoxaluria type 1 | IV | NCT02795325 | Terminated |
| 21 | ASO | Grb-2 | Liposomes | AML, ALL, MDS, CML | IV | NCT01159028 | Active |
| Advantages | Disadvantages | |
|---|---|---|
| Cationic polymeric vectors | • Control and sustained release kinetics | • Scale up and manufacturing is difficult |
| • Functional group conjugation is achievable for active targeting [104] | • High cationic charge favors endosomal uptake but offers cellular toxicity | |
| • Better stability for the encapsulation of negatively charged nucleic acid cargo | • Poor clinical translation | |
| • Offers a wide range of polymeric systems based on temperature, pH, light sensitive, hydrolysis and enzyme degradation | ||
| • Optimization of chemical and physical properties is highly achievable by use of different polymer chemistries | ||
| • Offers biodegradable polymer options such as PLGA [97,145] | ||
| • Offers several natural polymers such as chitosan [97,141], hyaluronic acid and collagen [146] | ||
| Lipid based vectors | • Easy scale up and manufacturing [28,29] | • Poor drug loading |
| • Good pharmacokinetic and safety profile [147] | • Requires extensive formulation work to optimize ideal concentration of lipid components | |
| • Excellent clinical translation | ||
| • Allows conjugated ligands to be designed |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wahane, A.; Waghmode, A.; Kapphahn, A.; Dhuri, K.; Gupta, A.; Bahal, R. Role of Lipid-Based and Polymer-Based Non-Viral Vectors in Nucleic Acid Delivery for Next-Generation Gene Therapy. Molecules 2020, 25, 2866. https://doi.org/10.3390/molecules25122866
Wahane A, Waghmode A, Kapphahn A, Dhuri K, Gupta A, Bahal R. Role of Lipid-Based and Polymer-Based Non-Viral Vectors in Nucleic Acid Delivery for Next-Generation Gene Therapy. Molecules. 2020; 25(12):2866. https://doi.org/10.3390/molecules25122866
Chicago/Turabian StyleWahane, Aniket, Akaash Waghmode, Alexander Kapphahn, Karishma Dhuri, Anisha Gupta, and Raman Bahal. 2020. "Role of Lipid-Based and Polymer-Based Non-Viral Vectors in Nucleic Acid Delivery for Next-Generation Gene Therapy" Molecules 25, no. 12: 2866. https://doi.org/10.3390/molecules25122866