
Determination of Chloramphenicol in Honey Using Salting-Out Assisted Liquid-Liquid Extraction Coupled with Liquid Chromatography-Tandem Mass Spectrometry and Validation According to 2002/657 European Commission Decision





Abstract
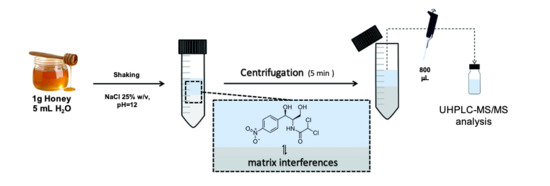
:
1. Introduction
2. Results and Discussion
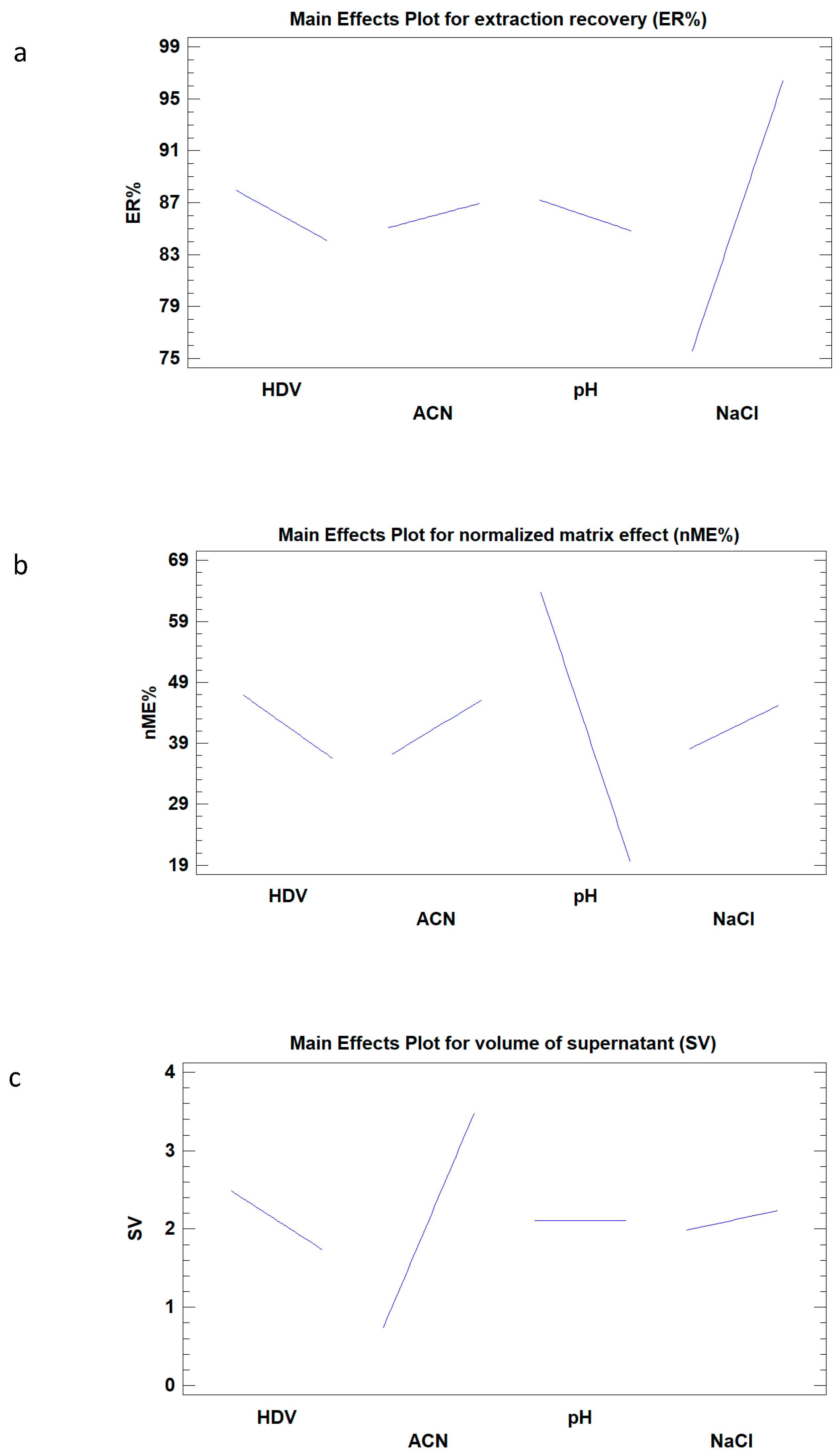
2.1. Optimization of SALLE by Experimental Design
2.2. Validation Protocol
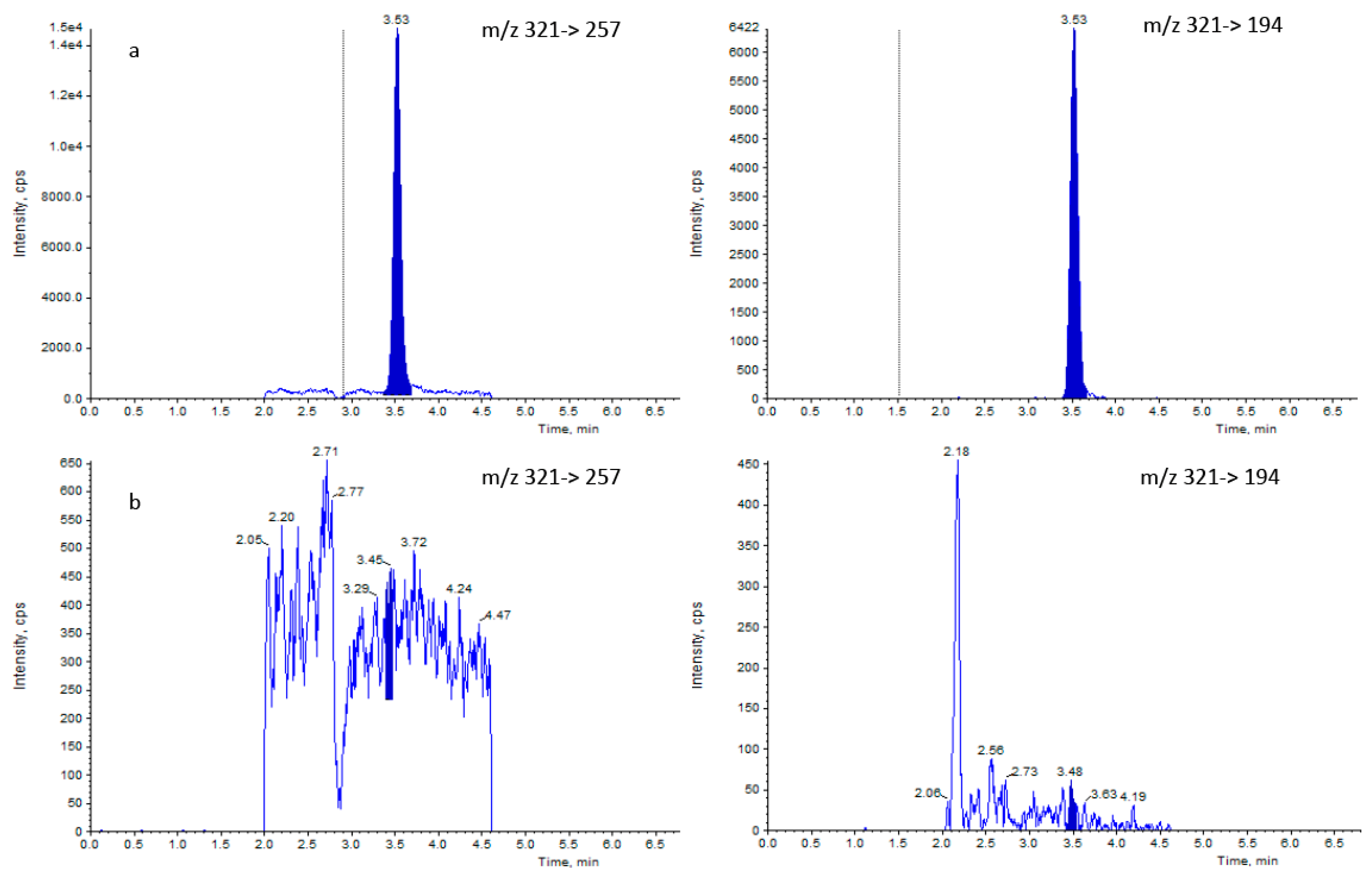
2.2.1. Liquid Chromatography and Mass Spectrometry Parameters
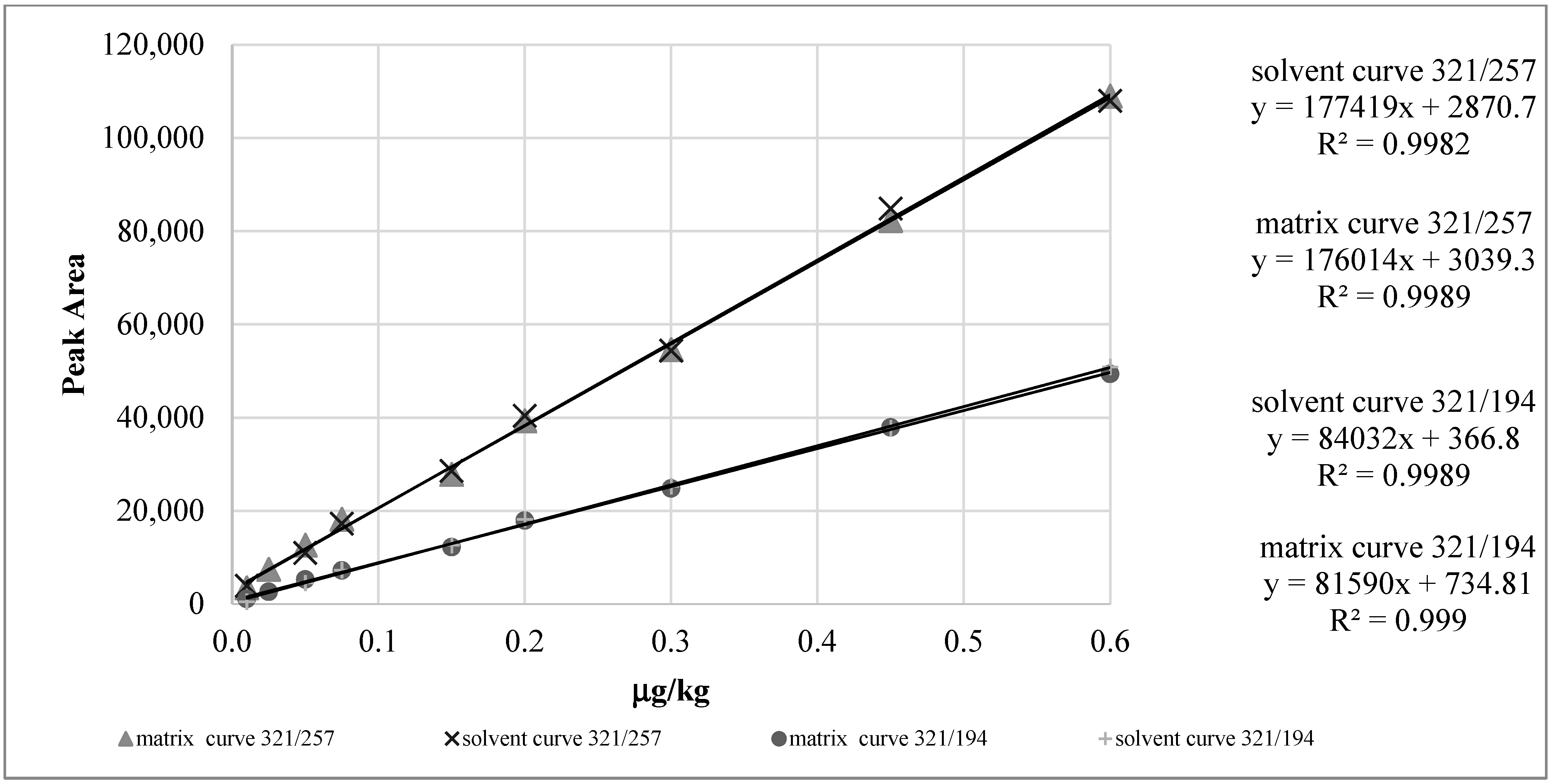
2.2.2. Calibration Curves and Linearity
2.2.3. Recovery, Repeatability (within-Laboratory Reproducibility) and Accuracy
2.2.4. Decision Limit (CCα) and Detection Capability (CCβ)
2.3. Real Samples Analysis
3. Material and Methods
3.1. Chemicals
3.2. Samples and Standard Solutions
3.3. Salting-Out Assisted Liquid-Liquid Extraction Procedure
3.4. UHPLC-MS/MS Analysis
3.5. Experimental Design
3.6. Matrix Effect Evaluation
3.7. Method Validation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Da Silva, P.M.; Gauche, C.; Gonzaga, L.V.; Costa, A.C.O.; Fett, R. Honey: Chemical composition, stability and authenticity. Food Chem. 2016, 196, 309–323. [Google Scholar] [CrossRef]
- Campone, L.; Piccinelli, A.L.; Pagano, I.; Carabetta, S.; Di Sanzo, R.; Russo, M.; Rastrelli, L. Determination of phenolic compounds in honey using dispersive liquid-liquid microextraction. J. Chromatogr. A 2014, 1334, 9–15. [Google Scholar] [CrossRef]
- Al-Waili, N.; Salom, K.; Al-Ghamdi, A.; Ansari, M.J. Antibiotic, pesticide, and microbial contaminants of honey: Human health hazards. Sci. World J. 2012, 2012, 930849. [Google Scholar] [CrossRef] [Green Version]
- Krongdang, S.; Evans, J.D.; Chen, Y.; Mookhploy, W.; Chantawannakul, P. Comparative susceptibility and immune responses of Asian and European honey bees to the American foulbrood pathogen, Paenibacillus larvae. Insect Sci. 2019, 26, 831–842. [Google Scholar] [CrossRef]
- Bargańska, Z.; Namieśnik, J.; Ślebioda, M. Determination of antibiotic residues in honey. TrAC Trends Anal. Chem. 2011, 30, 1035–1041. [Google Scholar] [CrossRef]
- Cundliffe, E.; McQuillen, K. Bacterial protein synthesis: The effects of antibiotics. J. Mol. Biol. 1967, 30, 137–146. [Google Scholar] [CrossRef]
- Watson, D.H. Pesticide, Veterinary and Other Residues in Food; Woodhead Publishing: Sawaton, UK, 2004. [Google Scholar]
- Commission Regulation. Commission Regulation (EU) No 37/2010 of 22 December 2009 on pharmacologically active substances and their classification regarding maximum residue limits in foodstuffs of animal origin. Off. J. Eur. Union 2010, 15, 1–72. [Google Scholar]
- The Commission of the European Communities. Commission Decision (2003/181/EC) of 13 March 2003 amending Decision 2002/657/EC as regards the setting of minimum required performance limits (MRPLs) for certain residues in food of animal origin. Off. J. Eur. Union 2003, 71, 17–18. [Google Scholar]
- Shen, H.Y.; Jiang, H.L. Screening, determination and confirmation of chloramphenicol in seafood, meat and honey using ELISA, HPLC-UVD, GC-ECD, GC-MS-EI-SIM and GCMS-NCI-SIM methods. Anal. Chim. Acta 2005, 535, 33–41. [Google Scholar] [CrossRef]
- Barreto, F.; Ribeiro, C.; Barcellos Hoff, R.; Dalla Costa, T. Determination of chloramphenicol, thiamphenicol, florfenicol and florfenicol amine in poultry, swine, bovine and fish by liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2016, 1449, 48–53. [Google Scholar] [CrossRef]
- Xie, X.; Wang, B.; Pang, M.; Zhao, X.; Xie, K.; Zhang, Y.; Wang, R.; Shi, H.; Zhang, G.; Zhang, T.; et al. Quantitative analysis of chloramphenicol, thiamphenicol, florfenicol and florfenicol amine in eggs via liquid chromatography-electrospray ionization tandem mass spectrometry. Food Chem. 2018, 269, 542–548. [Google Scholar] [CrossRef]
- European Commission. Commission Decision (2002/657/EC) of 12 August 2002 implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results. Off. J. Eur. Commun. 2002, 50, 8–36. [Google Scholar]
- Rodziewicz, L.; Zawadzka, I. Rapid determination of chloramphenicol residues in milk powder by liquid chromatography-elektrospray ionization tandem mass spectrometry. Talanta 2008, 75, 846–850. [Google Scholar] [CrossRef]
- Sheridan, R.; Policastro, B.; Thomas, S.; Rice, D. Analysis and occurrence of 14 sulfonamide antibacterials and chloramphenicol in honey by solid-phase extraction followed by LC/MS/MS analysis. J. Agric. Food Chem. 2008, 56, 3509–3516. [Google Scholar] [CrossRef]
- Pan, X.D.; Wu, P.G.; Jiang, W.; Ma, B.J. Determination of chloramphenicol, thiamphenicol, and florfenicol in fish muscle by matrix solid-phase dispersion extraction (MSPD) and ultra-high pressure liquid chromatography tandem mass spectrometry. Food Control 2015, 52, 34–38. [Google Scholar] [CrossRef]
- Samanidou, V.; Kehagia, M.; Kabir, A.; Furton, K.G. Matrix molecularly imprinted mesoporous sol-gel sorbent for efficient solid-phase extraction of chloramphenicol from milk. Anal. Chim. Acta 2016, 914, 62–74. [Google Scholar] [CrossRef]
- Campone, L.; Celano, R.; Piccinelli, A.L.; Pagano, I.; Cicero, N.; Di Sanzo, R.; Carabetta, S.; Russo, M.; Rastrelli, L. Ultrasound assisted dispersive liquid-liquid microextraction for fast and accurate analysis of chloramphenicol in honey. Food Res. Int. 2019, 115, 572–579. [Google Scholar] [CrossRef]
- Campone, L.; Piccinelli, A.L.; Celano, R.; Russo, M.; Valdés, A.; Ibáñez, C.; Rastrelli, L. A fully automated method for simultaneous determination of aflatoxins and ochratoxin A in dried fruits by pressurized liquid extraction and online solid-phase extraction cleanup coupled to ultra-high-pressure liquid chromatography-tandem mass spectrometry. Anal. Bioanal. Chem. 2015, 407, 2899–2911. [Google Scholar] [CrossRef] [PubMed]
- Campone, L.; Piccinelli, A.L.; Celano, R.; Russo, M.; Rastrelli, L. Rapid analysis of aflatoxin M1 in milk using dispersive liquid-liquid microextraction coupled with ultrahigh pressure liquid chromatography tandem mass spectrometry. Anal. Bioanal. Chem. 2013, 405, 8645–8652. [Google Scholar] [CrossRef]
- Campone, L.; Piccinelli, A.L.; Celano, R.; Rastrelli, L. PH-controlled dispersive liquid-liquid microextraction for the analysis of ionisable compounds in complex matrices: Case study of ochratoxin A in cereals. Anal. Chim. Acta 2012, 754, 61–66. [Google Scholar] [CrossRef]
- Campone, L.; Piccinelli, A.L.; Rastrelli, L. Dispersive liquid-liquid microextraction combined with high-performance liquid chromatography-tandem mass spectrometry for the identification and the accurate quantification by isotope dilution assay of Ochratoxin A in wine samples. Anal. Bioanal. Chem. 2011, 399, 1279–1286. [Google Scholar] [CrossRef]
- Valente, I.M.; Gonçalves, L.M.; Rodrigues, J.A. Another glimpse over the salting-out assisted liquid-liquid extraction in acetonitrile/water mixtures. J. Chromatogr. A 2013, 1308, 58–62. [Google Scholar] [CrossRef]
- Rice, N.M.; Irving, H.M.N.H.; Leonard, M.A. Nomenclature for liquid-liquid distribution (solvent extraction) (IUPAC Recommendations 1993). Pure Appl. Chem. 2007, 65, 2373–2396. [Google Scholar] [CrossRef]
- Teju, E.; Tadesse, B.; Megersa, N. Salting-out-assisted liquid–liquid extraction for the preconcentration and quantitative determination of eight herbicide residues simultaneously in different water samples with high-performance liquid chromatography. Sep. Sci. Technol. 2017, 1–11. [Google Scholar] [CrossRef]
- Campone, L.; Piccinelli, A.L.; Celano, R.; Pagano, I.; Russo, M.; Rastrelli, L. Rapid and automated analysis of aflatoxin M1 in milk and dairy products by online solid phase extraction coupled to ultra-high-pressure-liquid-chromatography tandem mass spectrometry. J. Chromatogr. A 2016, 1428, 212–219. [Google Scholar] [CrossRef]
- Vera Candioti, L.; De Zan, M.M.; Cámara, M.S.; Goicoechea, H.C. Experimental design and multiple response optimization. Using the desirability function in analytical methods development. Talanta 2014, 124, 123–138. [Google Scholar] [CrossRef]
- Antignac, J.P.; Le Bizec, B.; Monteau, F.; Andre, F. Validation of analytical methods based on mass spectrometric detection according to the ‘2002/657/EC’ European decision: Guideline and application. Anal. Chim. Acta 2003, 483, 325–334. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factor | p-Value SV (mL) | p-Value ER (%) | p-Valuen ME (%) |
|---|---|---|---|
| HDV (mL) | 0.0001 | 0.1900 | 0.0951 |
| ACN (mL) | 0.0001 | 0.5151 | 0.1476 |
| pH | 1.0000 | 0.4120 | 0.0003 |
| NaCl (%) | 0.0987 | 0.0001 | 0.2382 |
| Botanical Origin | Ion Ratio 194/257 | CVIR (%) |
|---|---|---|
| CAP STD (QC) | 42 | 2 |
| Acacia honey | 43 | 4 |
| Chestnut honey | 45 | 3 |
| Citrus honey | 41 | 12 |
| Levels (μg mL−1) | ||||||||
|---|---|---|---|---|---|---|---|---|
| 0.1 | 0.1 | 0.3 | 0.3 | 0.45 | 0.45 | 0.6 | 0.6 | |
| Transition (m/z) | 321→257 | 321→194 | 321→257 | 321→194 | 321→257 | 321→194 | 321→257 | 321→194 |
| Acacia | ||||||||
| Recovery (%) ± SD | 94 ± 4.7 | 98 ± 9.2 | 95 ±1.4 | 92 ± 3.6 | 100.3 ±1.19 | 100.1 ± 2.7 | 95 ±3.3 | 105.1 ± 7.1 |
| Precision (CV) | 5.03 | 9.63 | 1.45 | 3.33 | 1.15 | 2.33 | 3.55 | 6.73 |
| Chestnuts | ||||||||
| Recovery (%) ± SD | 97 ± 6.5 | 96 ± 5.36 | 98 ± 3.3 | 94 ± 2.36 | 100.8 ± 3.5 | 101 ± 2.7 | 100.2 ± 3.3 | 98.8 ± 3.2 |
| Precision (CV) | 6.75 | 5.67 | 3.38 | 2.37 | 3.45 | 2.74 | 3.38 | 3.3 |
| Citrus | ||||||||
| Recovery (%) ± SD | 92 ± 1.4 | 98 ± 8.8 | 101 ± 2.2 | 98 ± 2.3 | 96 ± 1.4 | 95 ± 1.7 | 94 ± 4.2 | 95 ± 3.19 |
| Precision (CV) | 1.58 | 7.79 | 2.39 | 2.59 | 3.35 | 1.9 | 3.39 | 1.79 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rizzo, S.; Russo, M.; Labra, M.; Campone, L.; Rastrelli, L. Determination of Chloramphenicol in Honey Using Salting-Out Assisted Liquid-Liquid Extraction Coupled with Liquid Chromatography-Tandem Mass Spectrometry and Validation According to 2002/657 European Commission Decision. Molecules 2020, 25, 3481. https://doi.org/10.3390/molecules25153481
Rizzo S, Russo M, Labra M, Campone L, Rastrelli L. Determination of Chloramphenicol in Honey Using Salting-Out Assisted Liquid-Liquid Extraction Coupled with Liquid Chromatography-Tandem Mass Spectrometry and Validation According to 2002/657 European Commission Decision. Molecules. 2020; 25(15):3481. https://doi.org/10.3390/molecules25153481
Chicago/Turabian StyleRizzo, Serena, Mariateresa Russo, Massimo Labra, Luca Campone, and Luca Rastrelli. 2020. "Determination of Chloramphenicol in Honey Using Salting-Out Assisted Liquid-Liquid Extraction Coupled with Liquid Chromatography-Tandem Mass Spectrometry and Validation According to 2002/657 European Commission Decision" Molecules 25, no. 15: 3481. https://doi.org/10.3390/molecules25153481