
Design of Curcumin and Flavonoid Derivatives with Acetylcholinesterase and Beta-Secretase Inhibitory Activities Using in Silico Approaches

 ,
, 
 and
and
Abstract
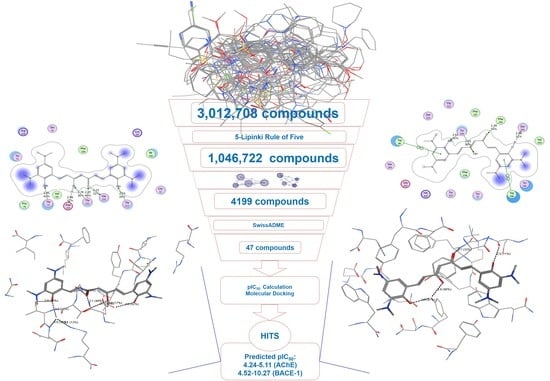
:
1. Introduction
2. Results
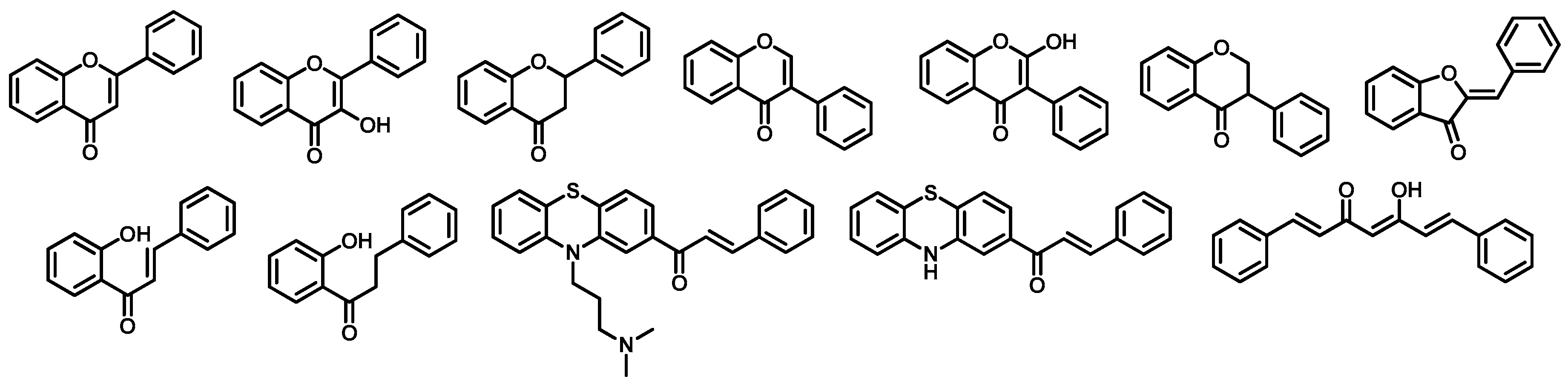
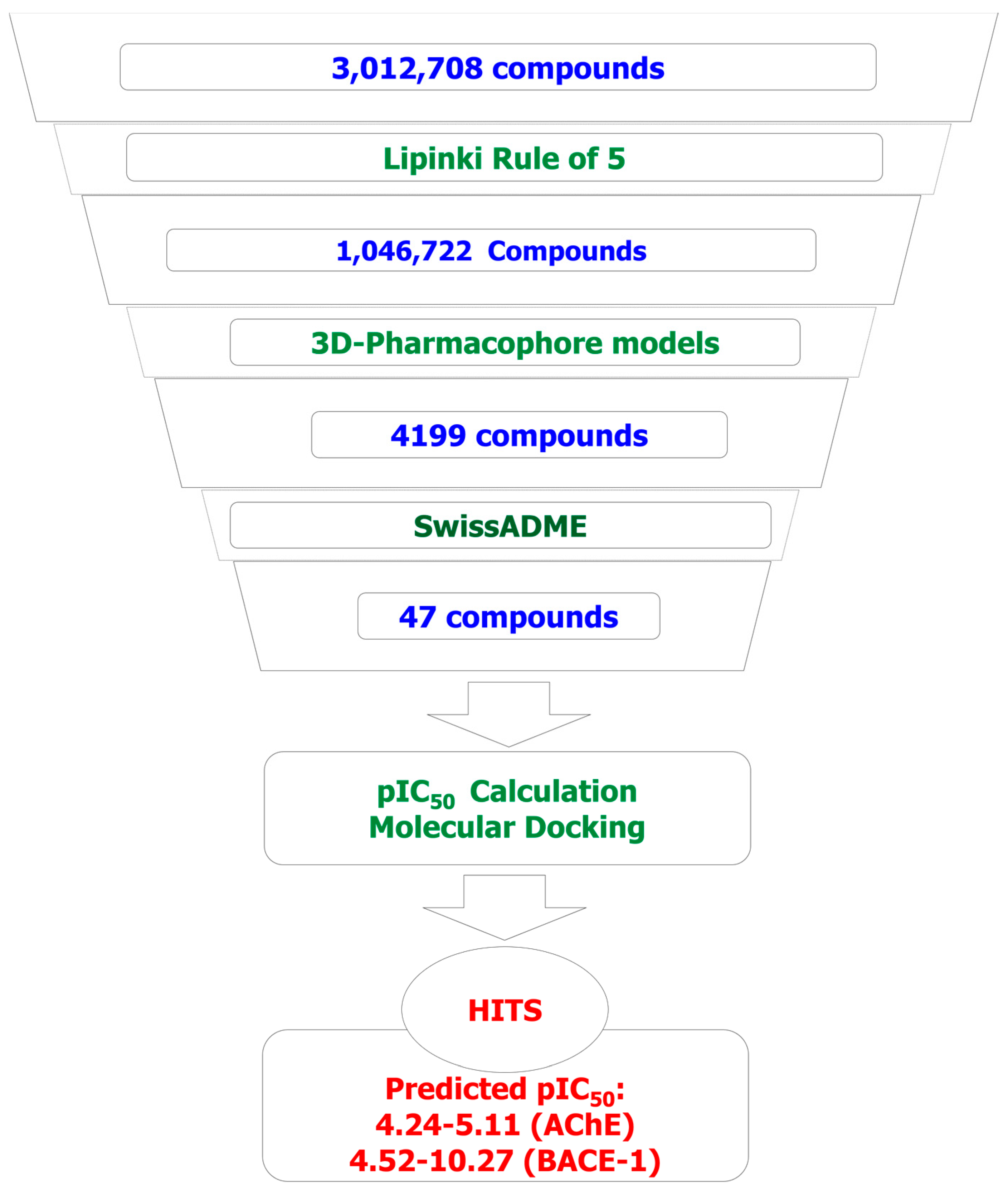
2.1. Combinatorial Library of Curcumin and Flavonoid Derivatives
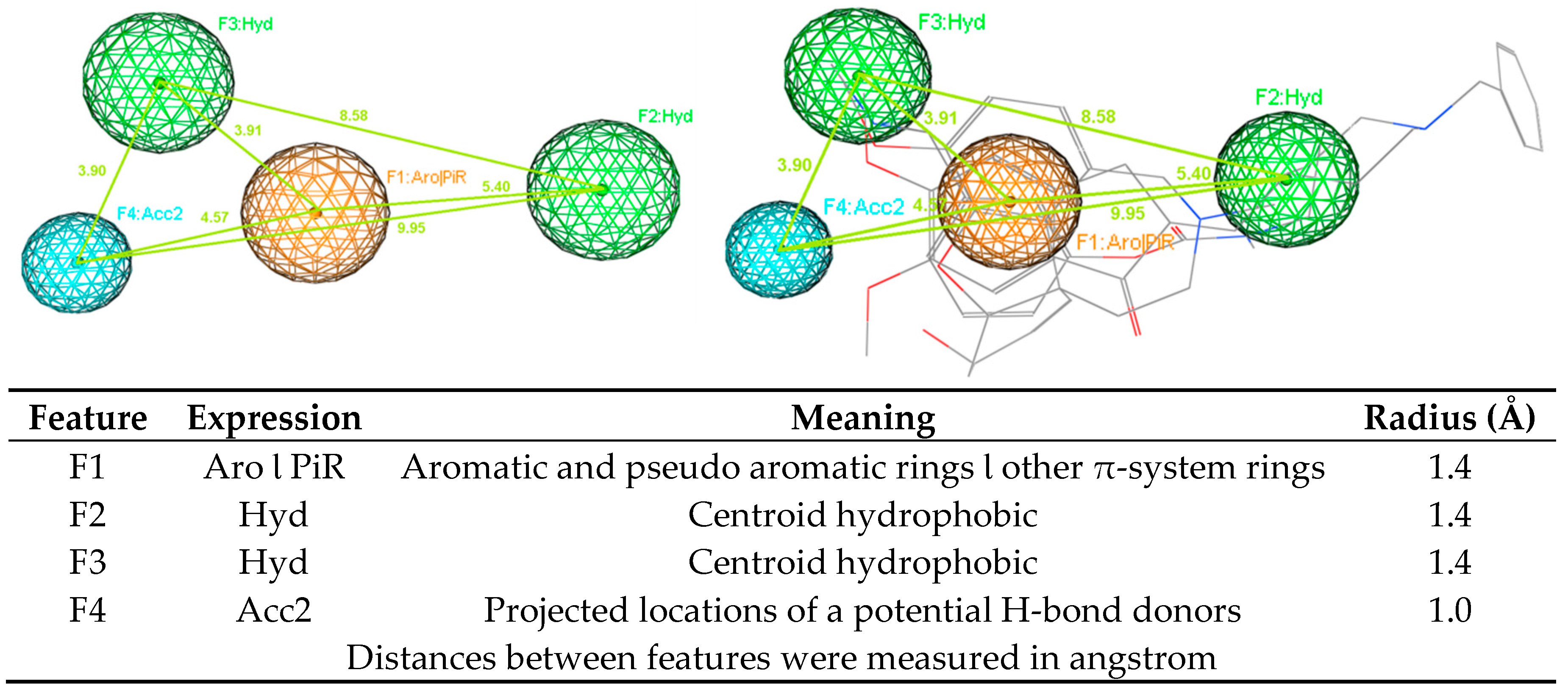
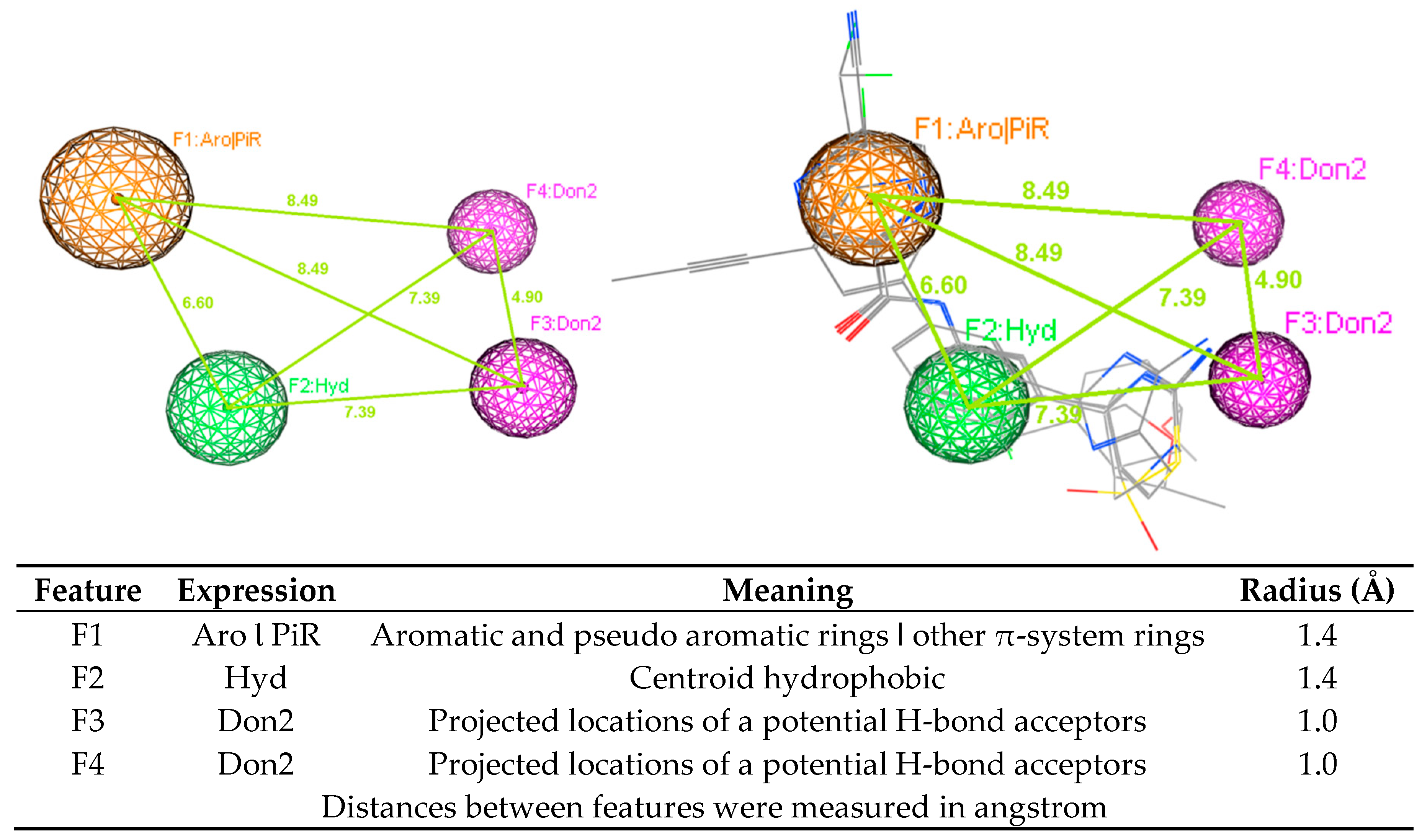
2.2. 3D-Pharmacophore Models
2.3. Virtual Screening
2.4. 2D-QSAR Models
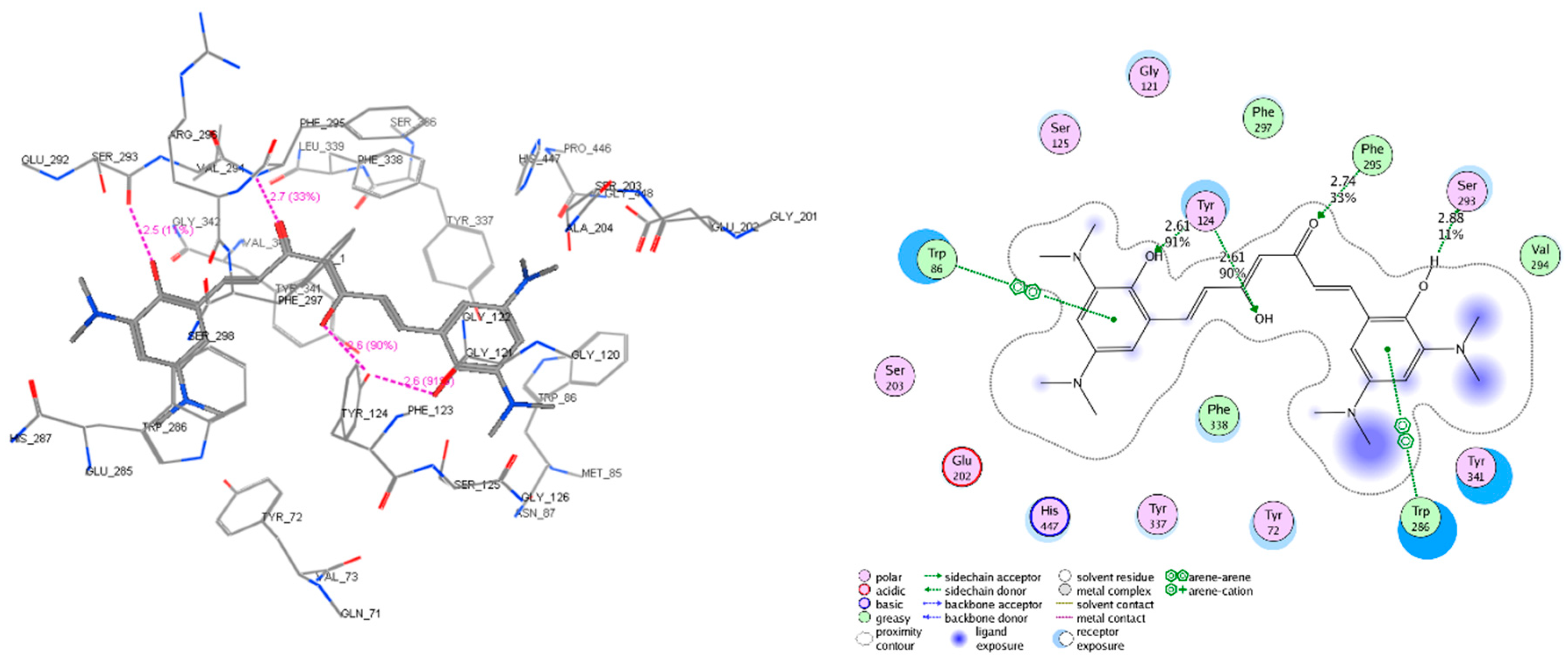
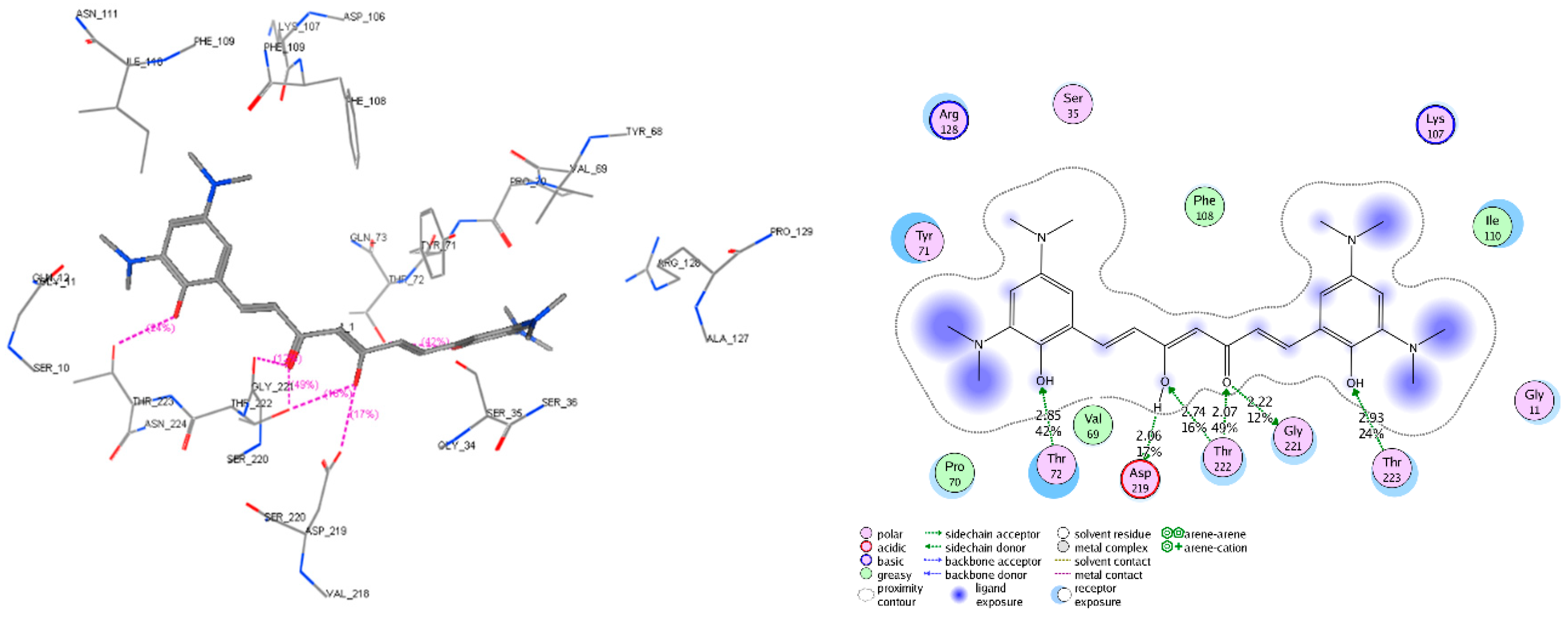
2.5. Molecular Docking
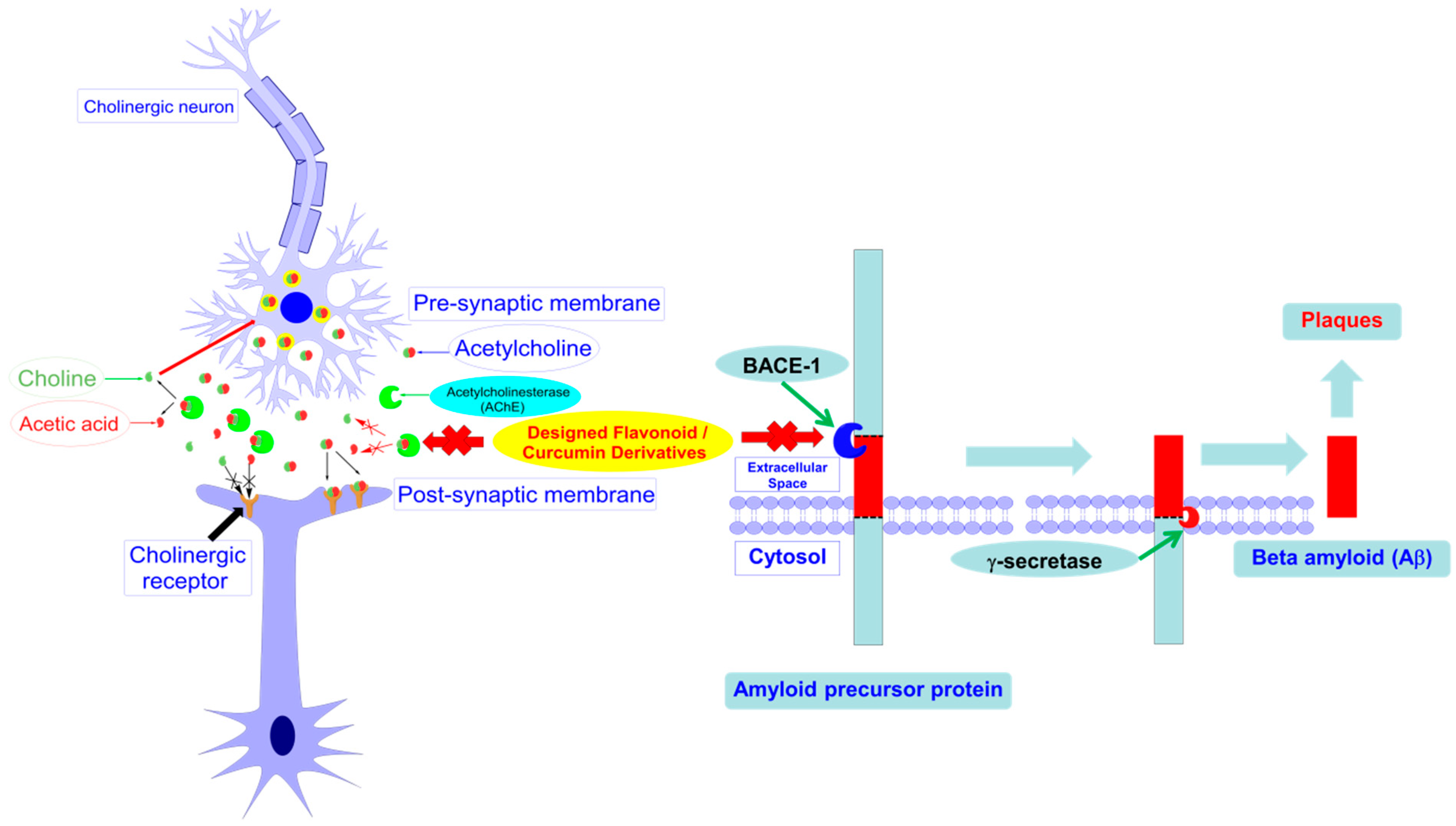
2.6. AChE and β-Secretase Inhibitory Activities
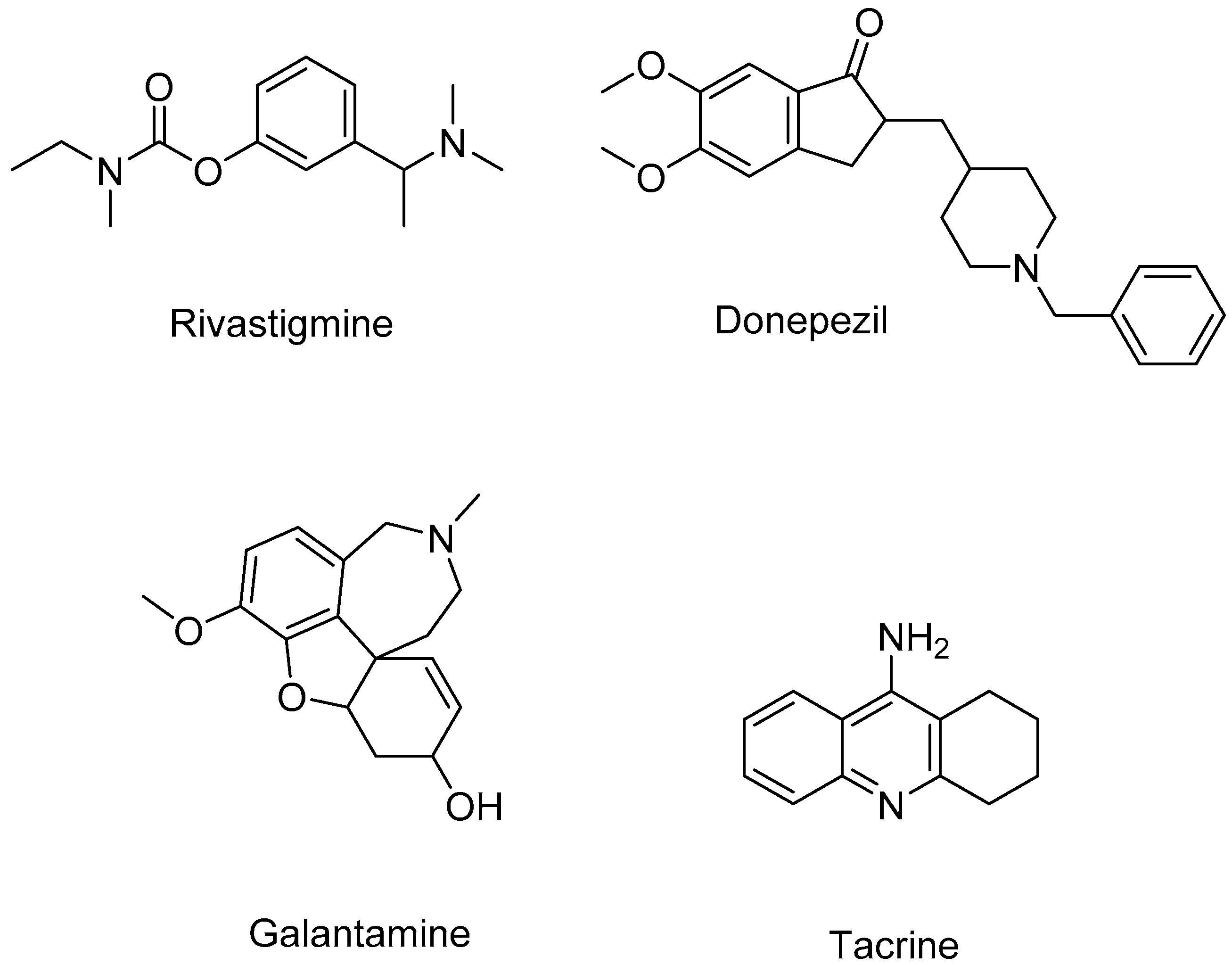
3. Discussion
4. Materials and Methods
4.1. Design a Combinatorial Library of Curcumin and Flavonoid Compounds
4.2. Building and Validating Pharmacophore Models
4.3. Building 2D-QSAR Models
4.3.1. Data Collection
4.3.2. Molecular Descriptors Calculation and Processing
4.3.3. Building and Validating of 2D-QSAR Models
4.4. Molecular Docking Procedure
4.4.1. Ligand Preparation
4.4.2. Docking and Results Evaluation
4.5. Chemistry
4.6. AChE Inhibition Assay
4.7. β-Secretase Inhibition Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Möller, H.J.; Graeber, M.B. The case described by Alois Alzheimer in 1911. Historical and conceptual perspectives based on the clinical record and neurohistological sections. Eur. Arch. Psychiatry Clin. Neurosci. 1998, 248, 111–122. [Google Scholar] [CrossRef]
- Kim, M.; Park, H.E.; Lee, S.-H.; Han, K.; Lee, J.H. Increased risk of Alzheimer’s disease in patients with psoriasis: A nationwide population−based cohort study. Sci. Rep. 2020, 10, 6454. [Google Scholar] [CrossRef] [Green Version]
- Lalut, J.; Payan, H.; Davis, A.; Lecoutey, C.; Legay, R.; Sopkova−de Oliveira Santos, J.; Claeysen, S.; Dallemagne, P.; Rochais, C. Rational design of novel benzisoxazole derivatives with acetylcholinesterase inhibitory and serotoninergic 5−HT4 receptors activities for the treatment of Alzheimer’s disease. Sci. Rep. 2020, 10, 3014. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; Getsios, D.; Revankar, N.; Xu, P.; Thompson, G.; Bobula, J.; Lacey, L.; Gaudig, M. Evaluating disease−modifying agents: A simulation framework for Alzheimer’s disease. Pharmacoeconomics 2014, 32, 1129–1139. [Google Scholar] [CrossRef]
- Jia, J.; Wei, C.; Chen, S.; Li, F.; Tang, Y.; Qin, W.; Zhao, L.; Jin, H.; Xu, H.; Wang, F.; et al. The cost of Alzheimer’s disease in China and re−estimation of costs worldwide. Alzheimers Dement. 2018, 14, 483–491. [Google Scholar] [CrossRef]
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s disease. Nat. Rev. Dis. Primers 2015, 1, 15056. [Google Scholar] [CrossRef]
- Vogel, J.W.; Iturria−Medina, Y.; Strandberg, O.T.; Smith, R.; Levitis, E.; Evans, A.C.; Hansson, O.; Weiner, M.; Aisen, P.; Petersen, R.; et al. Spread of pathological tau proteins through communicating neurons in human Alzheimer’s disease. Nat. Commun. 2020, 11, 2612. [Google Scholar] [CrossRef]
- Henstridge, C.M.; Hyman, B.T.; Spires−Jones, T.L. Beyond the neuron–cellular interactions early in Alzheimer disease pathogenesis. Nat. Rev. Neurosci. 2019, 20, 94–108. [Google Scholar] [CrossRef]
- Tampellini, D. Synaptic activity and Alzheimer’s disease: A critical update. Front. Neurosci. 2015, 9, 423. [Google Scholar] [CrossRef] [Green Version]
- Gong, C.X.; Liu, F.; Iqbal, K. Multifactorial Hypothesis and Multi−Targets for Alzheimer’s Disease. J. Alzheimers Dis. 2018, 64, S107–S117. [Google Scholar] [CrossRef]
- Rees, T.M.; Brimijoin, S. The role of acetylcholinesterase in the pathogenesis of Alzheimer’s disease. Drugs Today 2003, 39, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Vassar, R. BACE1: The beta−secretase enzyme in Alzheimer’s disease. J. Mol. Neurosci. 2004, 23, 105–114. [Google Scholar] [CrossRef]
- Tang, M.; Taghibiglou, C. The Mechanisms of Action of Curcumin in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 58, 1003–1016. [Google Scholar] [CrossRef]
- Ji, H.-F.; Zhang, H.-Y. Multipotent natural agents to combat Alzheimer’s disease. Functional spectrum and structural features. Acta. Pharmacol Sin. 2008, 29, 143–151. [Google Scholar] [CrossRef] [Green Version]
- Ayaz, M.; Sadiq, A.; Junaid, M.; Ullah, F.; Ovais, M.; Ullah, I.; Ahmed, J.; Shahid, M. Flavonoids as Prospective Neuroprotectants and Their Therapeutic Propensity in Aging Associated Neurological Disorders. Front. Aging Neurosci. 2019, 11, 155. [Google Scholar] [CrossRef] [Green Version]
- Xue, W.; Kim, J.-R.; Lee, S.-B.; Kim, Y.-J.; Jung, M.; Kwon, H.-W.; Ahn, Y.-J. Effects of curcuminoids identified in rhizomes of Curcuma longa on BACE−1 inhibitory and behavioral activity and lifespan of Alzheimer’s disease Drosophila models. BMC Complement. Altern. Med. 2014, 14, 88. [Google Scholar] [CrossRef] [Green Version]
- Williams, R.J.; Spencer, J.P. Flavonoids, cognition, and dementia: Actions, mechanisms, and potential therapeutic utility for Alzheimer disease. Free Radic Biol. Med. 2012, 52, 35–45. [Google Scholar] [CrossRef]
- Yang, C.; Wang, W.; Chen, L.; Liang, J.; Lin, S.; Lee, M.Y.; Ma, D.L.; Leung, C.H. Discovery of a VHL and HIF1alpha interaction inhibitor with in vivo angiogenic activity via structure−based virtual screening. Chem. Commun. 2016, 52, 12837–12840. [Google Scholar] [CrossRef]
- Kang, T.S.; Wang, W.; Zhong, H.J.; Liang, J.X.; Ko, C.N.; Lu, J.J.; Chen, X.P.; Ma, D.L.; Leung, C.H. A rhodium(III)−based inhibitor of autotaxin with antiproliferative activity. Biochim. Biophys. Acta. 2017, 1861, 256–263. [Google Scholar] [CrossRef]
- Zhong, H.J.; Wang, W.; Kang, T.S.; Yan, H.; Yang, Y.; Xu, L.; Wang, Y.; Ma, D.L. A Rhodium(III) Complex as an Inhibitor of Neural Precursor Cell Expressed, Developmentally Down−Regulated 8−Activating Enzyme with in Vivo Activity against Inflammatory Bowel Disease. J. Med. Chem. 2017, 60, 497–503. [Google Scholar] [CrossRef]
- Kumar, V.; Ojha, P.K.; Saha, A.; Roy, K. Exploring 2D−QSAR for prediction of beta−secretase 1 (BACE1) inhibitory activity against Alzheimer’s disease. SAR. QSAR. Environ. Res. 2020, 31, 87–133. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Roy, S.; Tripathi, S.; Sharma, A. Molecular docking based virtual screening of natural compounds as potential BACE1 inhibitors: 3D QSAR pharmacophore mapping and molecular dynamics analysis. J. Biomol. Struct. Dyn. 2016, 34, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.H.; Hsu, Y.C.; Chang, L.J.; Yang, J.M. An integrated approach with new strategies for QSAR models and lead optimization. BMC. Genom. 2017, 18, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abuhamdah, S.; Habash, M.; Taha, M.O. Elaborate ligand−based modeling coupled with QSAR analysis and in silico screening reveal new potent acetylcholinesterase inhibitors. J. Comput. Aided. Mol. Des. 2013, 27, 1075–1092. [Google Scholar] [CrossRef]
- MOE. 2008.10 edition. Chemical Computing Group Inc., 1010 Sherbrooke St. W, Suite 910, Montreal, Quebec, Canada H3A 2R7. Available online: https://www.chemcomp.com/ (accessed on 20 May 2019).
- Fei, J.; Zhou, L.; Liu, T.; Tang, X.-Y. Pharmacophore modeling, virtual screening, and molecular docking studies for discovery of novel Akt2 inhibitors. Int. J. Med. Sci. 2013, 10, 265–275. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug−likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A knowledge−based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Egan, W.J.; Merz, K.M., Jr.; Baldwin, J.J. Prediction of drug absorption using multivariate statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef]
- Muegge, I.; Heald, S.L.; Brittelli, D. Simple selection criteria for drug−like chemical matter. J. Med. Chem. 2001, 44, 1841–1846. [Google Scholar] [CrossRef] [PubMed]
- SciFinder. Available online: https://sso.cas.org/as/YpCJE/resume/as/authorization.ping (accessed on 30 June 2020).
- Wildman, S.A.; Crippen, G.M. Prediction of Physicochemical Parameters by Atomic Contributions. J. Chem. Inf. Comput. Sci. 1999, 39, 868–873. [Google Scholar] [CrossRef]
- Oprea, T.I. Property distribution of drug−related chemical databases. J. Comput. Aided Mol. Des. 2000, 14, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.; McDonald, J.J.; Kolodziej, S.A.; Kurumbail, R.G.; Williams, J.M.; Warren, C.J.; O’Neal, J.M.; Skepner, J.E.; Roberds, S.L. Discovery of potent inhibitors of soluble epoxide hydrolase by combinatorial library design and structure−based virtual screening. J. Med. Chem. 2011, 54, 1211–1222. [Google Scholar] [CrossRef]
- Roy, K.; Ambure, P.; Kar, S. How Precise Are Our Quantitative Structure–Activity Relationship Derived Predictions for New Query Chemicals? ACS Omega. 2018, 3, 11392–11406. [Google Scholar] [CrossRef] [Green Version]
- Niraj, R.R.; Saini, V.; Kumar, A. QSAR analyses of organophosphates for insecticidal activity and its in−silico validation using molecular docking study. Environ. Toxicol. Pharmacol. 2015, 40, 886–894. [Google Scholar] [CrossRef]
- Solomon, K.A.; Sundararajan, S.; Abirami, V. QSAR studies on N−aryl derivative activity towards Alzheimer’s disease. Molecules 2009, 14, 1448–1455. [Google Scholar] [CrossRef] [Green Version]
- Ambure, P.; Roy, K. Understanding the structural requirements of cyclic sulfone hydroxyethylamines as hBACE1 inhibitors against Aβ plaques in Alzheimer’s disease: A predictive QSAR approach. RSC Adv. 2016, 6, 28171–28186. [Google Scholar] [CrossRef]
- Jain, P.; Jadhav, H.R. Quantitative structure activity relationship analysis of aminoimidazoles as BACE−I inhibitors. Med. Chem. Res. 2013, 22, 1740–1746. [Google Scholar] [CrossRef]
- Hossain, T.; Islam, M.A.; Pal, R.; Saha, A. Exploring structural requirement and binding interactions of β−amyloid cleavage enzyme inhibitors using molecular modeling techniques. Med. Chem. Res. 2013, 22, 4766–4774. [Google Scholar] [CrossRef]
- Chakraborty, S.; Basu, S. Multi−functional activities of citrus flavonoid narirutin in Alzheimer’s disease therapeutics: An integrated screening approach and in vitro validation. Int. J. Biol. Macromol. 2017, 103, 733–743. [Google Scholar] [CrossRef] [PubMed]
- Son, M.; Park, C.; Rampogu, S.; Zeb, A.; Lee, K.W. Discovery of Novel Acetylcholinesterase Inhibitors as Potential Candidates for the Treatment of Alzheimer’s Disease. Int. J. Mol. Sci. 2019, 20, 1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhanjal, J.K.; Sharma, S.; Grover, A.; Das, A. Use of ligand−based pharmacophore modeling and docking approach to find novel acetylcholinesterase inhibitors for treating Alzheimer’s. Biomed. Pharmacother. 2015, 71, 146–152. [Google Scholar] [CrossRef] [PubMed]
- John, S.; Thangapandian, S.; Sakkiah, S.; Lee, K.W. Potent BACE−1 inhibitor design using pharmacophore modeling, in silico screening and molecular docking studies. BMC Bioinform. 2011, 12, S28. [Google Scholar] [CrossRef] [Green Version]
- Gay, M.; Evrard, C.; Descamps, F.; Carato, P.; Renault, N.; Coevoet, M.; Eddarkaoui, S.; Baud, C.; Larchanché, P.E.; Buée, L.; et al. A phenotypic approach to the discovery of compounds that promote non−amyloidogenic processing of the amyloid precursor protein: Toward a new profile of indirect β−secretase inhibitors. Eur. J. Med. Chem. 2018, 159, 104–125. [Google Scholar] [CrossRef]
- Beswick, P.; Charrier, N.; Clarke, B.; Demont, E.; Dingwall, C.; Dunsdon, R.; Faller, A.; Gleave, R.; Hawkins, J.; Hussain, I.; et al. BACE−1 inhibitors part 3: Identification of hydroxy ethylamines (HEAs) with nanomolar potency in cells. Bioorg. Med. Chem. Lett. 2008, 18, 1022–1026. [Google Scholar] [CrossRef]
- Charrier, N.; Clarke, B.; Cutler, L.; Demont, E.; Dingwall, C.; Dunsdon, R.; Hawkins, J.; Howes, C.; Hubbard, J.; Hussain, I.; et al. Second generation of BACE−1 inhibitors. Part 1: The need for improved pharmacokinetics. Bioorg. Med. Chem. Lett. 2009, 19, 3664–3668. [Google Scholar] [CrossRef]
- Charrier, N.; Clarke, B.; Cutler, L.; Demont, E.; Dingwall, C.; Dunsdon, R.; Hawkins, J.; Howes, C.; Hubbard, J.; Hussain, I.; et al. Second generation of BACE−1 inhibitors part 3: Towards non hydroxyethylamine transition state mimetics. Bioorg. Med. Chem. Lett. 2009, 19, 3674–3678. [Google Scholar] [CrossRef]
- Chen, S.H.; Lamar, J.; Guo, D.; Kohn, T.; Yang, H.C.; McGee, J.; Timm, D.; Erickson, J.; Yip, Y.; May, P.; et al. P3 cap modified Phe*−Ala series BACE inhibitors. Bioorg. Med. Chem. Lett. 2004, 14, 245–250. [Google Scholar] [CrossRef]
- Clarke, B.; Demont, E.; Dingwall, C.; Dunsdon, R.; Faller, A.; Hawkins, J.; Hussain, I.; MacPherson, D.; Maile, G.; Matico, R.; et al. BACE−1 inhibitors part 2: Identification of hydroxy ethylamines (HEAs) with reduced peptidic character. Bioorg. Med. Chem. Lett. 2008, 18, 1017–1021. [Google Scholar] [CrossRef]
- Ginman, T.; Viklund, J.; Malmstrom, J.; Blid, J.; Emond, R.; Forsblom, R.; Johansson, A.; Kers, A.; Lake, F.; Sehgelmeble, F.; et al. Core refinement toward permeable beta−secretase (BACE−1) inhibitors with low hERG activity. J. Med. Chem. 2013, 56, 4181–4205. [Google Scholar] [CrossRef] [PubMed]
- Hamada, Y.; Kiso, Y. Advances in the identification of beta−secretase inhibitors. Expert Opin. Drug Discov. 2013, 8, 709–731. [Google Scholar] [CrossRef] [PubMed]
- Ng, R.A.; Sun, M.; Bowers, S.; Hom, R.K.; Probst, G.D.; John, V.; Fang, L.Y.; Maillard, M.; Gailunas, A.; Brogley, L.; et al. Design and synthesis of hydroxyethylamine (HEA) BACE−1 inhibitors: Prime side chromane−containing inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 4674–4679. [Google Scholar] [CrossRef] [PubMed]
- Oehlrich, D.; Prokopcova, H.; Gijsen, H.J. The evolution of amidine−based brain penetrant BACE1 inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 2033–2045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, M.M.; Williamson, T.; Babu−Khan, S.; Bartberger, M.D.; Brown, J.; Chen, K.; Cheng, Y.; Citron, M.; Croghan, M.D.; Dineen, T.A.; et al. Design and preparation of a potent series of hydroxyethylamine containing beta−secretase inhibitors that demonstrate robust reduction of central beta−amyloid. J. Med. Chem. 2012, 55, 9009–9024. [Google Scholar] [CrossRef]
- Woltering, T.J.; Wostl, W.; Hilpert, H.; Rogers−Evans, M.; Pinard, E.; Mayweg, A.; Gobel, M.; Banner, D.W.; Benz, J.; Travagli, M.; et al. BACE1 inhibitors: A head group scan on a series of amides. Bioorg. Med. Chem. Lett. 2013, 23, 4239–4243. [Google Scholar] [CrossRef]
- Charrier, N.; Clarke, B.; Demont, E.; Dingwall, C.; Dunsdon, R.; Hawkins, J.; Hubbard, J.; Hussain, I.; Maile, G.; Matico, R.; et al. Second generation of BACE−1 inhibitors part 2: Optimisation of the non−prime side substituent. Bioorg. Med. Chem. Lett. 2009, 19, 3669–3673. [Google Scholar] [CrossRef]
- ChEMBL Database. Available online: https://www.ebi.ac.uk/chembl/ (accessed on 20 May 2019).
- Michael, A.D.; Andreas, G.K.J.; Khac−Minh, T.; Gerhard, F.E.; Wilfried, N.G. Predictive QSAR Models for Polyspecific Drug Targets: The Importance of Feature Selection. Curr. Comput. Aided Drug Des. 2008, 4, 91–110. [Google Scholar] [CrossRef]
- RapidMiner 5.3.013. Available online: https://rapidminer.com/ (accessed on 20 May 2019).
- Weka Software 3.8. Available online: https://waikato.github.io/weka−wiki/ (accessed on 20 May 2019).
- Khan, P.M.; Roy, K. Current approaches for choosing feature selection and learning algorithms in quantitative structure–activity relationships (QSAR). Expert Opin. Drug Discov. 2018, 13, 1075–1089. [Google Scholar] [CrossRef]
- Ngo, T.D.; Tran, T.D.; Le, M.T.; Thai, K.M. Computational predictive models for P−glycoprotein inhibition of in−house chalcone derivatives and drug−bank compounds. Mol. Divers. 2016, 20, 945–961. [Google Scholar] [CrossRef]
- Thai, K.-M.; Bui, Q.-H.; Tran, T.-D.; Huynh, T.-N.-P. QSAR modeling on benzo[c]phenanthridine analogues as topoisomerase I inhibitors and anti−cancer agents. Molecules 2012, 17, 5690–5712. [Google Scholar] [CrossRef] [PubMed]
- Chirico, N.; Gramatica, P. Real external predictivity of QSAR models: How to evaluate it? Comparison of different validation criteria and proposal of using the concordance correlation coefficient. J. Chem. Inf. Model. 2011, 51, 2320–2335. [Google Scholar] [CrossRef] [PubMed]
- Consonni, V.; Ballabio, D.; Todeschini, R. Comments on the definition of the Q2 parameter for QSAR validation. J. Chem. Inf. Model. 2009, 49, 1669–1678. [Google Scholar] [CrossRef] [PubMed]
- Todeschini, R.; Ballabio, D.; Grisoni, F. Beware of Unreliable Q(2)! A Comparative Study of Regression Metrics for Predictivity Assessment of QSAR Models. J. Chem. Inf. Model. 2016, 56, 1905–1913. [Google Scholar] [CrossRef]
- Sybyl X 2.0. Available online: http://www.tripos.com/index.php?family=modules,SimplePage,&page=SYBYL−X (accessed on 20 May 2019).
- Hevener, K.E.; Zhao, W.; Ball, D.M.; Babaoglu, K.; Qi, J.; White, S.W.; Lee, R.E. Validation of molecular docking programs for virtual screening against dihydropteroate synthase. J. Chem. Inf. Model. 2009, 49, 444–460. [Google Scholar] [CrossRef]
- Azam, F.; Madi, A.M.; Ali, H.I. Molecular Docking and Prediction of Pharmacokinetic Properties of Dual Mechanism Drugs that Block MAO−B and Adenosine A(2A) Receptors for the Treatment of Parkinson’s Disease. J. Young Pharm. 2012, 4, 184–192. [Google Scholar] [CrossRef] [Green Version]
- Protein Data Bank. Available online: https://www.rcsb.org/ (accessed on 20 May 2019).
- LeadIT 2.0.2. Available online: https://www.biosolveit.de/LeadIT/ (accessed on 20 May 2019).
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef]
- Pagadala, N.S.; Syed, K.; Tuszynski, J. Software for molecular docking: A review. Biophys. Rev. 2017, 9, 91–102. [Google Scholar] [CrossRef]
- Wang, Z. Comprehensive Organic Name Reactions and Reagents; John Wiley & Sons, Inc.: New York, NY, USA, 2010. [Google Scholar] [CrossRef]
- Bagal, D.B.; Qureshi, Z.S.; Dhake, K.P.; Khan, S.R.; Bhanage, B.M. An efficient and heterogeneous recyclable palladium catalyst for chemoselective conjugate reduction of α,β−unsaturated carbonyls in aqueous medium. Green Chem. 2011, 13, 1490–1494. [Google Scholar] [CrossRef]
- Tran, T.-D.; Nguyen, T.-C.; Nguyen, N.-S.; Nguyen, D.-M.; Nguyen, T.-T.; Le, M.-T.; Thai, K.-M. Synthesis of Novel Chalcones as Acetylcholinesterase Inhibitors. Appl. Sci. 2016, 6, 198. [Google Scholar] [CrossRef] [Green Version]
- Nuthakki, V.K.; Sharma, A.; Kumar, A.; Bharate, S.B. Identification of embelin, a 3−undecyl−1,4−benzoquinone from Embelia ribes as a Multitarg. anti−Alzheimer agent. Drug Dev. Res. 2019, 80, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Olasehinde, T.A.; Mabinya, L.V.; Olaniran, A.O.; Okoh, A.I. Chemical characterization, antioxidant properties, cholinesterase inhibitory and anti−amyloidogenic activities of sulfated polysaccharides from some seaweeds. Bioact. Carbohydr. Diet Fibre. 2019, 18, 100182. [Google Scholar] [CrossRef]
Sample Availability: Not available. |














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| -OH | -OCH3 | -OC2H5 | -OCOCH3 |
|---|---|---|---|
| -F | -Cl | -Br | -I |
| -NO2 | -NH2 | -N(CH3)2 | -NHCOCH3 |
| -CH2CHH=CH2 | -COOH | -COOCH3 | -COOC2H5 |
| -CN | -CONH2 | -SO2NH2 | -SH |
| -SCH3 | -SC2H5 | C6H5CH2O- |
| AChE | BACE-1 | ||||
|---|---|---|---|---|---|
| Training Set | Validation Sets | Training Set | Validation Sets | ||
| Active | Decoy | Active | Decoy | ||
| 04 | 655 | 26369 | 04 | 436 | 18217 |
| No. | Parameter | Pharmacophore Model | |
|---|---|---|---|
| A1 | B1 | ||
| 1 | Total molecules in database (D) | 27024 | 18653 |
| 2 | Total number of actives in database (A) | 655 | 436 |
| 3 | Total hits (Ht) | 914 | 438 |
| 4 | Active Hits (Ha) | 524 | 305 |
| 5 | %Yield of actives [(Ha/Ht) × 100] | 57.33 | 69.63 |
| 6 | %Ratio of actives [(Ha/A) × 100] | 80 | 69.95 |
| 7 | Enrichment factor (E), [(Ha × D)/(Ht × A)] | 23.65 | 29.79 |
| 8 | False negatives [A – Ha] | 131 | 131 |
| 9 | False positives [Ht − Ha] | 390 | 133 |
| 10 | Goodness-of-hit score (GH*) | 0.62 | 0.69 |
| AChE | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Model AT | Model AF: Full data set (n = 72) | |||||||||||||
| pIC50 = −0.92791 | pIC50 = −1.00890 | |||||||||||||
| +2.34847×BCUT_SLOGP_3 | +2.38027×BCUT_SLOGP_3 | |||||||||||||
| −0.14990×reactive | −0.11002×reactive | |||||||||||||
| −0.00355×PEOE_VSA+1 | −0.00391×PEOE_VSA+1 | |||||||||||||
| −0.00514×PEOE_VSA-3 | −0.00480×PEOE_VSA-3 | |||||||||||||
| −0.00219×SlogP_VSA2 | −0.00202×SlogP_VSA2 | |||||||||||||
| −0.00447×SMR_VSA2 | −0.00387×SMR_VSA2 | |||||||||||||
| Internal Validation | External Validation | |||||||||||||
| N | RMSE | R2 | RMSELOO | R2LOO | n | RMSE | R2 | R2(PRED) | CCC | |||||
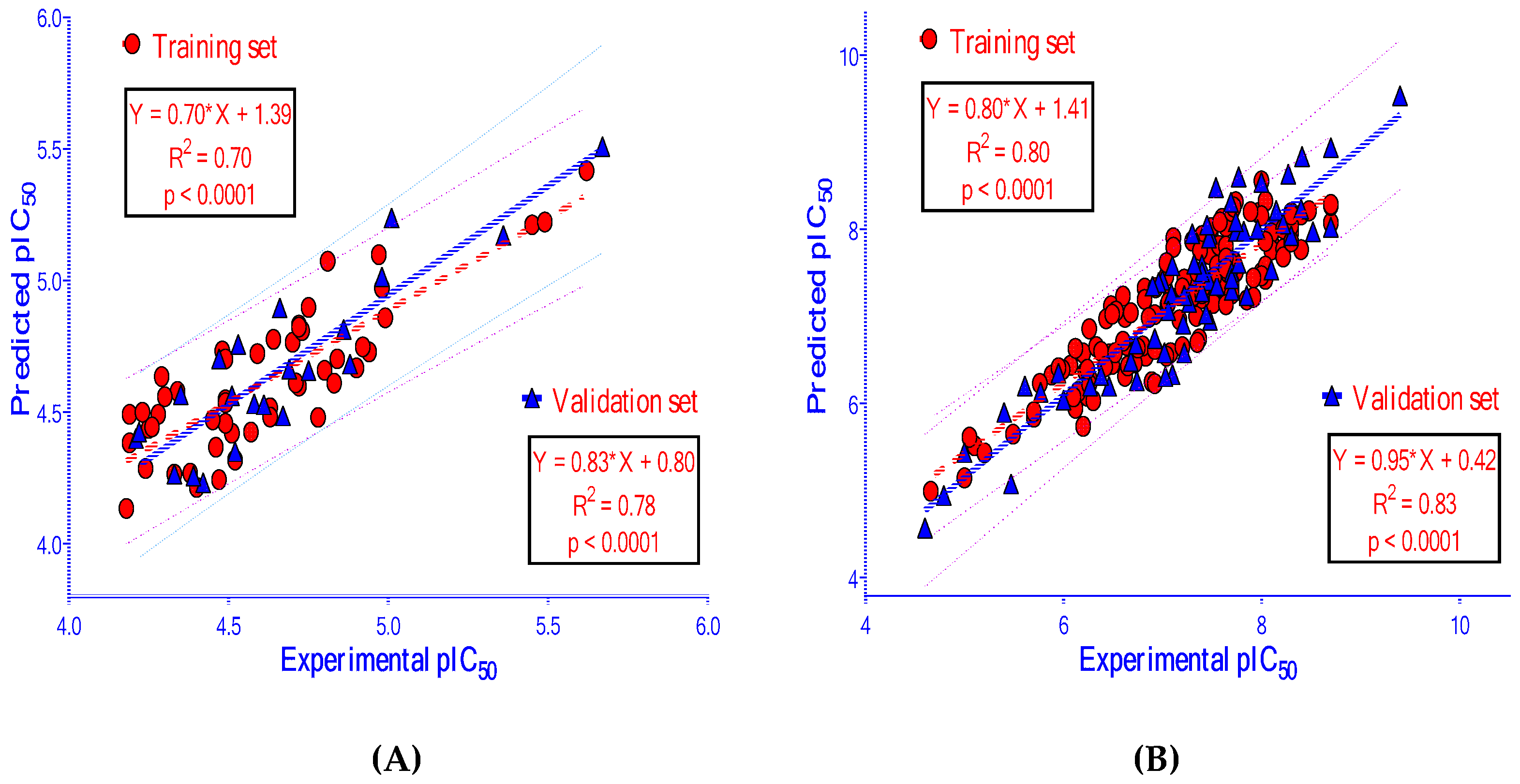
| 50 | 0.18 | 0.70 | 0.22 | 0.57 | 22 | 0.16 | 0.78 | 0.78 | 0.64 | 0.69 | 0.11 | 0.88 | 0.72 | |
| BACE-1 | ||||||||||||||
| Model BT | Model BF: Full Data Set (N = 215) | |||||||||||||
| pIC50 = 1.26826 | pIC50 = 1.01351 | |||||||||||||
| +0.87076×petitjean | +0.59775×petitjean | |||||||||||||
| +6.37086×BCUT_PEOE_1 | +4.85517×BCUT_PEOE_1 | |||||||||||||
| +3.30481×a_ICM | +3.13351×a_ICM | |||||||||||||
| −0.47753×chiral_u | −0.50839×chiral_u | |||||||||||||
| +0.08513×rings | +0.02540×rings | |||||||||||||
| +0.15746×a_nN | +0.16067×a_nN | |||||||||||||
| +0.00608×PEOE_VSA-0 | +0.00577×PEOE_VSA-0 | |||||||||||||
| +0.02183×PEOE_VSA-6 | +0.01771×PEOE_VSA-6 | |||||||||||||
| −0.25952×logS | −0.26227×logS | |||||||||||||
| +0.00893×SlogP_VSA3 | +0.00920×SlogP_VSA3 | |||||||||||||
| +0.00944×SlogP_VSA5 | +0.01101×SlogP_VSA5 | |||||||||||||
| Internal Validation | External Validation | |||||||||||||
| N | RMSE | R2 | RMSELOO | R2LOO | n | RMSE | R2 | R2(PRED) | CCC | |||||
| 150 | 0.37 | 0.80 | 0.40 | 0.77 | 65 | 0.41 | 0.83 | 0.81 | 0.79 | 0.76 | 0.05 | 0.91 | 0.76 | |
| Code | Category | Description |
|---|---|---|
| BCUT_SlogP_3 | Adjacency and distance matrix | A Burden’s parameter using atomic contribution to logP (using the Wildman and Crippen SlogP method [34]) instead of partial charge. |
| BCUT_PEOE_1 | Adjacency and distance matrix | A descriptor relating topological shape and partial charges. |
| petitjean | Adjacency and distance matrix | Value of (diameter-radius)/diameter. |
| reactive | Physical property | An indicator of the presence of reactive groups. A non-zero value indicates that the molecule contains a reactive group. The table of reactive groups is based on the Oprea set [35] and includes metals, phospho-, N/O/S-N/O/S single bonds, thiols, acyl halides, Michael Acceptors, azides, esters, etc. |
| logS | Physical property | The log of the aqueous solubility (mol/L). |
| PEOE_VSA-0, PEOE_VSA+1, PEOE_VSA-3, PEOE_VSA-6 | Partial charge | Sum of the proximate accessible van der Waals surface area (Å2), vi, calculation for each atom over all the atoms i, such that partial charge of atom i is in a specified range. |
| SlogP_VSA2, SlogP_VSA3, SlogP_VSA5 | Subdivided surface areas | Sum of the proximate accessible van der Waals surface area (Å2), vi, calculated for each atom over all the atoms, such that partition coefficient for atom i is in a specified range. |
| SMR_VSA2 | Subdivided surface areas | Sum of the proximate accessible van der Waals surface area (Å2), vi, calculation for each atom over all the atoms i, such that molar refractivity for atom i is in a specified range. |
| a_ICM | Atom counts and bond counts | The entropy of the element distribution in the molecule (including implicit hydrogens but not lone pair pseudo-atoms). |
| chiral_u | Atom counts and bond counts | The number of unconstrained chiral centers. |
| rings | Atom counts and bond counts | The number of rings. |
| a_Nn | Atom counts and bond counts | The number of nitrogen atoms. |
| AChE | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Ligand | Predicted pIC50 | In Vitro IC50 (µM) | In Vitro pIC50 | Docking Score (kJ.mol−1) | |||||||
| 1ACJ | 1DX6 | 1EVE | 1W6R | 4EY6 (chain A) | 4EY6 (chain B) | 4EY7 (chain A) | 4EY7 (chain B) | ||||
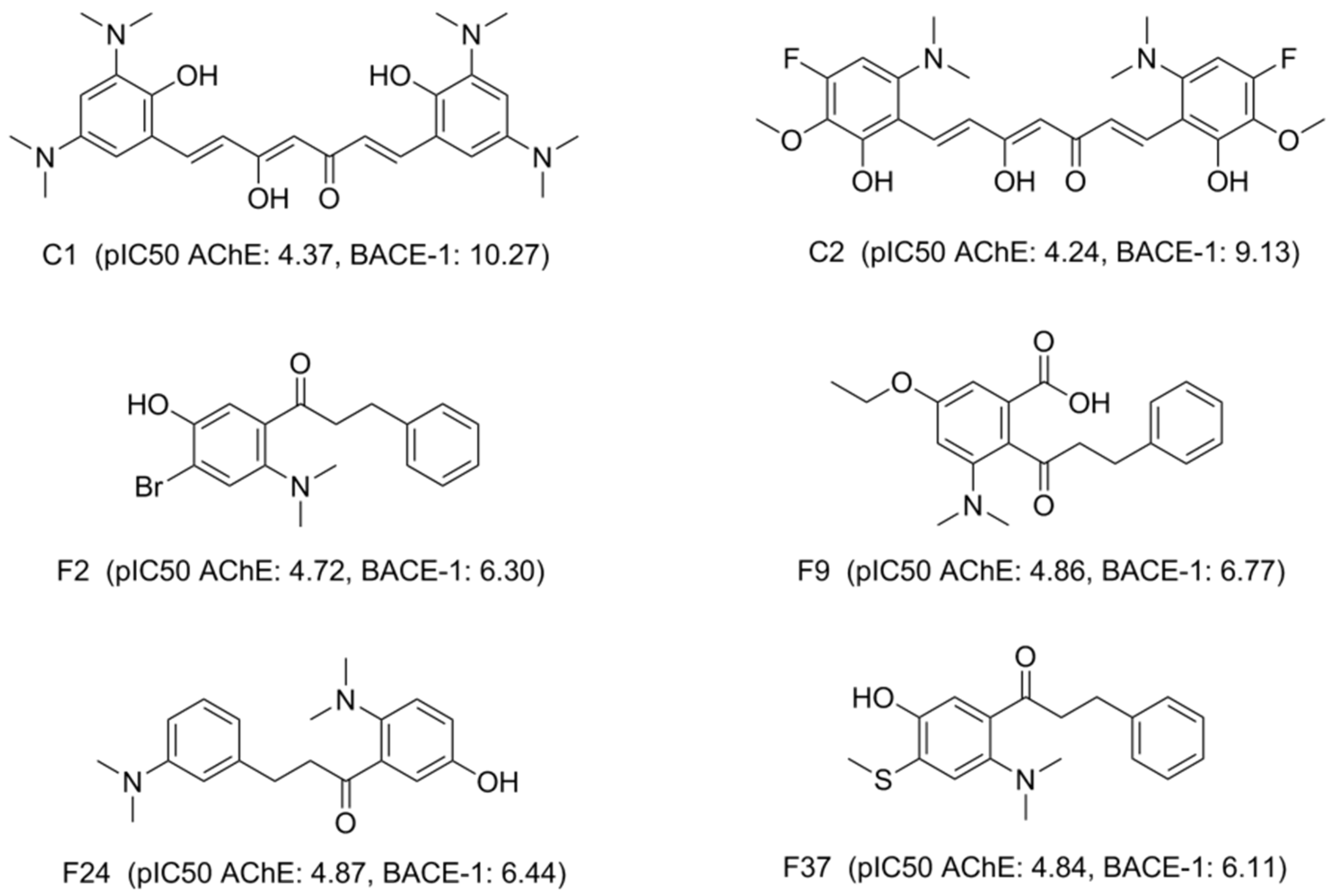
| C1 | 4.37 | - | - | Not docked | −24.13 | −25.62 | −25.47 | Not docked | Not docked | −34.38 | −36.23 |
| C2 | 4.24 | - | - | Not docked | −30.97 | −23.97 | −25.19 | −23.21 | −12.72 | −23.56 | −31.90 |
| F2 | 4.72 | - | - | −21.38 | −21.18 | −23.30 | -27.09 | −21.95 | −21.56 | −28.70 | −30.99 |
| F9 | 4.86 | 30.05 ± 1.24 | 4.52 ± 0.02 | −20.27 | −28.46 | −25.53 | −25.80 | −25.63 | −27.26 | −25.17 | −27.11 |
| F24 | 4.87 | 80.52 ± 3.07 | 4.09 ± 0.02 | −20.19 | −21.08 | −20.87 | −21.53 | −24.10 | −23.66 | −25.27 | −26.59 |
| F37 | 4.84 | - | - | −20.58 | −22.46 | −21.81 | −22.98 | −21.02 | −22.75 | −23.34 | −22.45 |
| BACE-1 | |||||||||||
| 3VEU | 4B78 | 5HU0 (chain A) | 5HU0 (chain B) | 5HU1 (chain A) | 5HU1 (chain B) | ||||||
| C1 | 10.27 | - | - | −24.28 | −10.23 | −17.28 | −22.09 | −17.39 | −14.74 | ||
| C2 | 9.13 | - | - | −24.04 | −24.64 | −27.00 | −16.51 | −25.78 | −17.79 | ||
| F2 | 6.30 | - | - | −19.51 | −13.11 | −14.79 | −14.92 | −16.47 | −17.20 | ||
| F9 | 6.77 | 1.85 ± 0.33 | 5.73 ± 0.08 | −21.34 | −15.98 | −18.53 | −17.83 | −20.32 | −19.49 | ||
| F24 | 6.44 | 3.52 ± 0.77 | 5.45 ± 0.10 | −22.39 | −14.36 | −18.58 | −17.07 | −15.09 | −16.19 | ||
| F37 | 6.11 | - | - | −21.87 | −13.66 | −17.39 | −15.80 | −16.61 | −15.09 | ||
| Source | Model | Training Set | Validation Set | |||
|---|---|---|---|---|---|---|
| N | R2 | Q2 | n | R2PRED | ||
| This study | PLS | 55 | 0.70 | 0.57 | 22 | 0.78 |
| Roy et al. 2018 [37] | MLR | 284 | 0.52–0.74 | 0.50–0.71 | 142 | 0.50–0.63 |
| Niraj et al. 2015 [38] | PLS | 24 | 0.78 | 0.70 | 11 | 0.66 |
| Abuhamdah et al. 2013 [24] | GFA−MLR | 68 | 0.94 | 0.92 | 17 | 0.84 |
| Solomon et al. [39] | GFA | 53 | 0.86 | 0.80 | 26 | 0.86 |
| Source | Model | Training Set | Validation Set | |||
|---|---|---|---|---|---|---|
| N | R2 | Q2 | n | R2PRED | ||
| This study | PLS | 150 | 0.80 | 0.77 | 65 | 0.81 |
| Kumar et al. 2019 [21] | PLS | 76 | 0.83 | 0.80 | 22 | 0.85 |
| Ambure et al. 2016 [40] | PLS | 52 | 0.83 | 0.76 | 22 | 0.81 |
| Ambure et al. 2016 [40] | MLR | 51 | 0.83 | 0.76 | 22 | 0.80 |
| Jain et al. 2013 [41] | MLR | 20 | 0.90 | 0.90 | 7 | 0.90 |
| Hossain et al. 2013 [42] | CoMFA | 71 | 1.00 | 0.77 | 35 | 0.77 |
| Hossain et al. 2013 [42] | CoMSIA | 71 | 1.00 | 0.73 | 35 | 0.71 |
| Hossain et al. 2013 [42] | PLS | 71 | 0.94 | 0.79 | 35 | 0.71 |
| Chakraborty et al. 2017 [43] | LHM | 20 | 0.94 | 0.91 | 10 | 0.86 |
| Roy et al. 2018 [37] | MLR | 51 | 0.76–0.83 | 0.71–0.76 | 23 | 0.75–0.91 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tran, T.-S.; Le, M.-T.; Tran, T.-D.; Tran, T.-H.; Thai, K.-M. Design of Curcumin and Flavonoid Derivatives with Acetylcholinesterase and Beta-Secretase Inhibitory Activities Using in Silico Approaches. Molecules 2020, 25, 3644. https://doi.org/10.3390/molecules25163644
Tran T-S, Le M-T, Tran T-D, Tran T-H, Thai K-M. Design of Curcumin and Flavonoid Derivatives with Acetylcholinesterase and Beta-Secretase Inhibitory Activities Using in Silico Approaches. Molecules. 2020; 25(16):3644. https://doi.org/10.3390/molecules25163644
Chicago/Turabian StyleTran, Thai-Son, Minh-Tri Le, Thanh-Dao Tran, The-Huan Tran, and Khac-Minh Thai. 2020. "Design of Curcumin and Flavonoid Derivatives with Acetylcholinesterase and Beta-Secretase Inhibitory Activities Using in Silico Approaches" Molecules 25, no. 16: 3644. https://doi.org/10.3390/molecules25163644