
Construction of Unusual Indole-Based Heterocycles from Tetrahydro-1H-pyridazino[3,4-b]indoles



Abstract
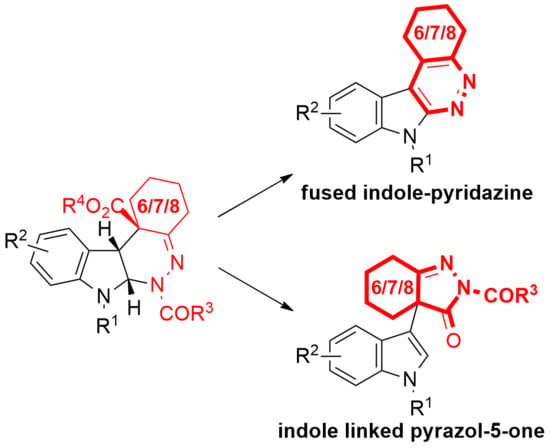
:
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. General
3.2. Two-Step Procedure for the Synthesis of 3
3.2.1. Procedure for the Oxidation of Indolines 1 to Indoles 2
3.2.2. Procedure for the Preparation of Fused Indole-Pyridazine 3 from 2
3.3. General Procedure for the Synthesis of Indole Linked Pyrazol-5-ones 4
3.4. One-Pot Procedure for the Synthesis of 4c
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhang, Y.-C.; Jiang, F.; Shi, F. Organocatalytic Asymmetric Synthesis of Indole-Based Chiral Heterocycles: Strategies, Reactions, and Outreach. Acc. Chem. Res. 2020, 53, 425–446. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Wang, Q.; Zhu, J. Metamorphosis of cycloalkenes for the divergent total synthesis of polycyclic indole alkaloids. Chem. Soc. Rev. 2018, 47, 7882–7898. [Google Scholar] [CrossRef] [PubMed]
- Zi, W.; Zuo, Z.; Ma, D. Intramolecular Dearomative Oxidative Coupling of Indoles: A Unified Strategy for the Total Synthesis of Indoline Alkaloids. Acc. Chem. Res. 2015, 48, 702–711. [Google Scholar] [CrossRef] [PubMed]
- Kochanowska-Karamyan, A.J.; Hamann, M.T. Marine Indole Alkaloids: Potential New Drug Leads for the Control of Depression and Anxiety. Chem. Rev. 2010, 110, 4489–4497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Festa, A.A.; Voskressensky, L.G.; Van der Eycken, E.V. Visible Light-Mediated Chemistry of Indoles and Related Heterocycles. Chem. Soc. Rev. 2019, 48, 4401–4423. [Google Scholar] [CrossRef]
- Stempel, E.; Gaich, T. Cyclohepta[b]indoles: A Privileged Structure Motif in Natural Products and Drug Design. Acc. Chem. Res. 2016, 49, 2390–2402. [Google Scholar] [CrossRef]
- Gribble, G.W.; Badenock, J.C. Heterocyclic Scaffolds II: Reactions and Applications of Indoles; Springer: Berlin/Heidelberg, Germany, 2010. [Google Scholar]
- Sonsona, I.G. Indole, a Privileged Structural Core Motif. Synlett 2015, 26, 2325–2326. [Google Scholar] [CrossRef] [Green Version]
- Melander, R.J.; Minvielle, M.J.; Melander, C. Controlling Bacterial Behavior with indole-containing Natural Products and Derivatives. Tetrahedron 2014, 70, 6363–6372. [Google Scholar] [CrossRef] [Green Version]
- Sharma, V.; Kumar, P.; Pathak, D. Biological Importance of the Indole Nucleus in Recent Years: A Comprehensive Review. J. Heterocyclic Chem. 2010, 47, 491–502. [Google Scholar] [CrossRef]
- Chadha, N.; Silakari, O. Indoles as Therapeutics of Interest in Medicinal Chemistry: Bird’s Eye View. Eur. J. Med. Chem. 2017, 134, 159–184. [Google Scholar] [CrossRef]
- Sravanthi, T.V.; Manju, S.L. Indoles—A Promising Scaffold for Drug Development. Eur. J. Pharm. Sci. 2016, 91, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, N.K.; Kaushik, N.; Attri, P.; Kumar, N.; Kim, C.H.; Verma, A.K.; Choi, E.H. Biomedical Importance of Indoles. Molecules 2013, 18, 6620–6662. [Google Scholar] [CrossRef]
- Wermuth, C.G. Are Pyridazines Privileged Structures? Med. Chem. Commun. 2011, 2, 935–941. [Google Scholar] [CrossRef]
- Neto, J.S.S.; Zeni, G. Ten Years of Progress in the Synthesis of Six-Membered N-heterocycles from Alkynes and Nitrogen Sources. Tetrahedron 2020, 76, 1–120. [Google Scholar] [CrossRef]
- Volonterio, A.; Moisan, L.; Rebek, J. Synthesis of Pyridazine-Based Scaffolds as α-Helix Mimetics. Org. Lett. 2007, 9, 3733–3736. [Google Scholar] [CrossRef]
- Jaballah, M.Y.; Serya, R.T.; Abouzid, K. Pyridazine Based Scaffolds as Privileged Structures in anti-Cancer Therapy. Drug Res. 2017, 67, 138–148. [Google Scholar] [CrossRef]
- Feraldi-Xypolia, A.; Pardo, D.G.; Cossy, J. Synthesis of α-(Trifluoromethyl)pyridazine Derivatives. Eur. J. Org. Chem. 2018, 3541–3553. [Google Scholar] [CrossRef]
- Butnariu, R.M.; Mangalagiu, I.I. New Pyridazine Derivatives: Synthesis, Chemistry and Biological Activity. Bioorg. Med. Chem. 2009, 17, 2823–2829. [Google Scholar] [CrossRef]
- Campagna, F.; Palluotto, F.; Mascia, M.P.; Maciocco, E.; Marra, C.; Carotti, A.; Carrieri, A. Synthesis and Biological Evaluation of Pyridazino[4,3-b]indoles and Indeno[1,2-c]pyridazines as New Ligands of Central and Peripheral Benzodiazepine Receptors. Il Farmaco 2003, 58, 129–140. [Google Scholar] [CrossRef]
- Cirrincione, G.; Almerico, A.M.; Barraja, P.; Diana, P.; Lauria, A.; Passannanti, A.; Musiu, C.; Pani, A.; Murtas, P.; Minnei, C.; et al. Derivatives of the New Ring System Indolo[1,2-c]benzo[1,2,3]triazine with Potent Antitumor and Antimicrobial Activity. J. Med. Chem. 1999, 42, 2561–2568. [Google Scholar] [CrossRef]
- Monge, A.; Font, M.; Parrado, P.; Fernandez-Alvarez, E. New Derivatives of 5H-Pyridazino (4,5-b) indole and 1,2,4-Triazino (4,5-a) indole and Related Compounds as Inhibitors of Blood Platelet Aggregation, Anti-hypertensive Agents and Thromboxane Synthetase inhibitors. Eur. J. Med. Chem. 1988, 23, 547–552. [Google Scholar] [CrossRef]
- El-Kashef, H.; Farghaly, A.A.H.; Haider, N.; Wobus, A. Unexpected Hydrazinolysis Behaviour of 1-Chloro-4-methyl-5Hpyridazino[4,5-b]indole and a Convenient Synthesis of New [1,2,4]-Triazolo[4′,3′:1,6]pyridazino[4,5-b]indoles. Molecules 2004, 9, 849–859. [Google Scholar] [CrossRef] [PubMed]
- González-Gómez, J.C.; Uriarte, U. A Convenient Preparation of 4-Carboxamide Derivatives of Pyridazino [4,5-b]indoles and Pyridazino[4,5-b]benzo[b]furans. Synlett 2002, 12, 2095–2097. [Google Scholar] [CrossRef]
- Panathur, N.; Gokhale, N.; Dalimba, U.; Koushik, P.V.; Yogeeswari, P.; Sriram, D. Synthesis of Novel 5-[(1,2,3-triazol-4-yl)methyl]-1-Methyl-3Hpyridazino[4,5-b]indol-4-one Derivatives by Click Reaction and Exploration of Their Anticancer Activity. Med. Chem. Res. 2016, 25, 135–148. [Google Scholar] [CrossRef]
- Bruel, A.; Bénéteau, R.; Chabanne, M.; Lozach, O.; Le Guevel, R.; Ravache, M.; Bénédetti, H.; Meijer, L.; Logé, C.; Robert, J.M. Synthesis of New Pyridazino[4,5-b]indol-4-ones and Pyridazin-3 (2H)-one Analogs as DYRK1A Inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 5037–5040. [Google Scholar] [CrossRef]
- Kobayashi, G.; Furukawa, S. Studies on Indole Derivatives. I. Synthesis of 3-Phenyl-9H-pyridazino[3,4-b]indole Derivatives. Chem. Pharm. Bull. 1964, 12, 1129–1135. [Google Scholar] [CrossRef] [Green Version]
- Shimoji, Y.; Tomita, K.; Karube, T.; Kamioka, T. Synthesis and Benzodiazepine Receptor Binding Studies of Pyridazino[3,4-b]indoles. Yakugaku Zasshi 1992, 112, 804–816. [Google Scholar] [CrossRef]
- Sharma, S.; Kaur, C.; Budhiraja, A.; Nepali, K.; Gupta, M.K.; Saxena, A.K.; Bedi, P.M.S. Chalcone Based Azacarboline Analogues as Novel Antitubulin Agents: Design, Synthesis, Biological Evaluation and Molecular Modeling Studies. Eur. J. Med. Chem. 2014, 85, 648–660. [Google Scholar] [CrossRef]
- Maity, P.; Adhikari, D.; Jana, A.K. An Overview on Synthetic Entries to Tetrahydro-β-carbolines. Tetrahedron 2019, 75, 965–1028. [Google Scholar] [CrossRef]
- Evans, B.E.; Rittle, K.E.; Bock, M.G.; DiPardo, R.M.; Freidinger, R.M.; Whitter, W.L.; Lundell, G.F.; Veber, D.F.; Anderson, P.S.; Chang, R.S.L.; et al. Methods for Drug Discovery: Development of Potent, Selective, Orally Effective Cholecystokinin Antagonists. J. Med. Chem. 1988, 31, 2235–2246. [Google Scholar] [CrossRef]
- Ciccolini, C.; Mari, G.; Gatti, F.G.; Gatti, G.; Giorgi, G.; Mantellini, F.; Favi, G. Synthesis of Polycylic Fused Indoline Scaffolds through a Substrate-Guided Reactivity Switch. J. Org. Chem. 2020. [Google Scholar] [CrossRef]
- Lee, S.; Lim, H.J.; Cha, K.; Sulikowski, G.A. Asymmetric Approaches to 1,2-disubstituted Mitosenes based on the Intramolecular Cyclization of Diazoesters. Tetrahedron 1997, 53, 16521–16532. [Google Scholar] [CrossRef]
- Soldatenkov, A.T.; Polyanskii, K.B.; Kolyadina, N.M.; Soldatova, S.A. Oxidation of Heterocyclic Compounds by Manganese Dioxide. Chem. Het. Comp. 2009, 45, 633–657. [Google Scholar] [CrossRef]
- Brucelle, F.; Renaud, P. Synthesis of Indolines, Indoles, and Benzopyrrolizidinones from Simple Aryl Azides. Org. Lett. 2012, 14, 3048–3051. [Google Scholar] [CrossRef] [PubMed]
- Campos, K.R.; Journet, M.; Lee, S.; Grabowski, E.J.J.; Tillyer, R.D. Asymmetric Synthesis of a Prostaglandin D2 Receptor Antagonist. J. Org. Chem. 2005, 70, 268–274. [Google Scholar] [CrossRef]
- Han, B.; Xiao, Y.-C.; Yao, Y.; Chen, Y.-C. Lewis Acid Catalyzed Intramolecular Direct Ene Reaction of Indoles. Angew. Chem. Int. Ed. 2010, 49, 10189–10191. [Google Scholar] [CrossRef]
- Pelcman, B.; Gribble, G.W. Total Synthesis of the Marine sponge Pigment Fascaplysin. Tetrahedron Lett. 1990, 31, 2381–2384. [Google Scholar] [CrossRef]
- Sridharan, V.; Menéndez, J.C. Cerium(IV) Ammonium Nitrate as a Catalyst in Organic Synthesis. Chem. Rev. 2010, 110, 3805–3849. [Google Scholar] [CrossRef]
- Dalvi, B.A.; Lokhande, P.D. Copper(II) Catalyzed Aromatization of Tetrahydrocarbazole: An unprecedented Protocol and its Utility towards the Synthesis of Carbazole Alkaloids. Tetrahedron Lett. 2018, 59, 2145–2149. [Google Scholar] [CrossRef]
- Thavanswaran, S.; Mccamley, K.; Scammells, P.J. N-Demethylation of Alkaloids. Nat. Prod. Commun. 2006, 1, 885–897. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.J.; Zhu, J.; Song, X.Y.; Wang, L.; Wang, S.R.; Tang, Y. Highly Enantioselective [3+2] Annulation of Indoles with Quinones to Access Structurally Diverse Benzofuroindolines. Angew. Chem. Int. Ed. 2018, 57, 3810–3814. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.Q.; Shen, Y.; Liu, J.X.; Tao, J.Y.; Shi, F. Enantioselective Direct α-Arylation of Pyrazol-5-ones with 2-Indolylmethanols via Organo-Metal Cooperative Catalysis. Org. Lett. 2017, 19, 1542–1545. [Google Scholar] [CrossRef] [PubMed]
- Attanasi, O.A.; De Crescentini, L.; Favi, G.; Filippone, P.; Mantellini, F.; Perrulli, F.R.; Santeusanio, S. Cultivating the Passion to Build Heterocycles from 1,2-Diaza-1,3-dienes: The Force of Imagination. Eur. J. Org. Chem. 2009, 3109–3127. [Google Scholar] [CrossRef]
- Lopes, S.M.M.; Cardoso, A.L.; Lemos, A.; PinhoeMelo, T.M.V.D. Recent Advances in the Chemistry of Conjugated Nitrosoalkenes and Azoalkenes. Chem. Rev. 2018, 118, 11324–11352. [Google Scholar] [CrossRef]
- Wei, L.; Shen, C.; Hu, Y.Z.; Taoa, H.Y.; Wang, C.J. Enantioselective Synthesis of Multi-nitrogencontaining Heterocycles Using Azoalkenes as Key Intermediates. Chem. Commun. 2019, 55, 6672–6684. [Google Scholar] [CrossRef]
- Sukhorukov, A.Y. Umpolung of Enamines: An Overview on Strategies and Synthons. Synlett 2020, 31, 439–449. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Oxidant | Solvent | Temp. (°C) | Time (h) | 2b(2b’)/4c | Yield (%) |
|---|---|---|---|---|---|---|
| 1 | DDQ (1 equiv) | dioxane | rt | 2 | 2b | trace |
| 2 | DDQ (1 equiv) | toluene | reflux | 24 | 2b | 21 |
| 3 | DDQ (1.5 equiv) | CH2Cl2 | rt | 15 | 2b | 27 |
| 4 | BQ (4 equiv) | toluene | reflux | 12 | – | – |
| 5 | NBS/TBPB (0.4 equiv) | CCl4 | reflux | 5 | – | – a |
| 6 | MnO2 (10 equiv) | benzene | 70 | 48 | 2b’ | 40 |
| 7 | MnO2 (25 equiv) | benzene | 70 | 24 | 2b’ | 55 |
| 8 | Na2Cr2O7 (1 equiv) | CHCl3 | reflux | 12 | – | – a |
| 9 | I2 (2 equiv) | CH3OH | reflux | 24 | – | – a |
| 10 | PCC (3.3 equiv) | CH2Cl2 | reflux | 2 | – | – |
| 11 | Pd/C (1 equiv) | AcOEt | reflux | 24 | – | – a |
| 12 | CAN (1 equiv) | CH2Cl2 | rt | 24 | 4c | 38 |
| 13 | CuCl2∙2H2O (0.1 equiv) | DMSO | 100 | 8 | 4c | 80 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciccolini, C.; De Crescentini, L.; Mantellini, F.; Mari, G.; Santeusanio, S.; Favi, G. Construction of Unusual Indole-Based Heterocycles from Tetrahydro-1H-pyridazino[3,4-b]indoles. Molecules 2020, 25, 4124. https://doi.org/10.3390/molecules25184124
Ciccolini C, De Crescentini L, Mantellini F, Mari G, Santeusanio S, Favi G. Construction of Unusual Indole-Based Heterocycles from Tetrahydro-1H-pyridazino[3,4-b]indoles. Molecules. 2020; 25(18):4124. https://doi.org/10.3390/molecules25184124
Chicago/Turabian StyleCiccolini, Cecilia, Lucia De Crescentini, Fabio Mantellini, Giacomo Mari, Stefania Santeusanio, and Gianfranco Favi. 2020. "Construction of Unusual Indole-Based Heterocycles from Tetrahydro-1H-pyridazino[3,4-b]indoles" Molecules 25, no. 18: 4124. https://doi.org/10.3390/molecules25184124