
Cytotoxic and Anti-Inflammatory Activities of Dihydroisocoumarin and Xanthone Derivatives from Garcinia picrorhiza

 , ,
, ,
Abstract
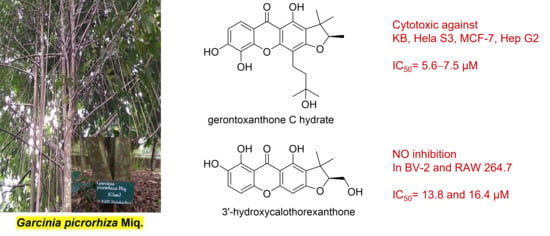
:
1. Introduction
2. Results and Discussion
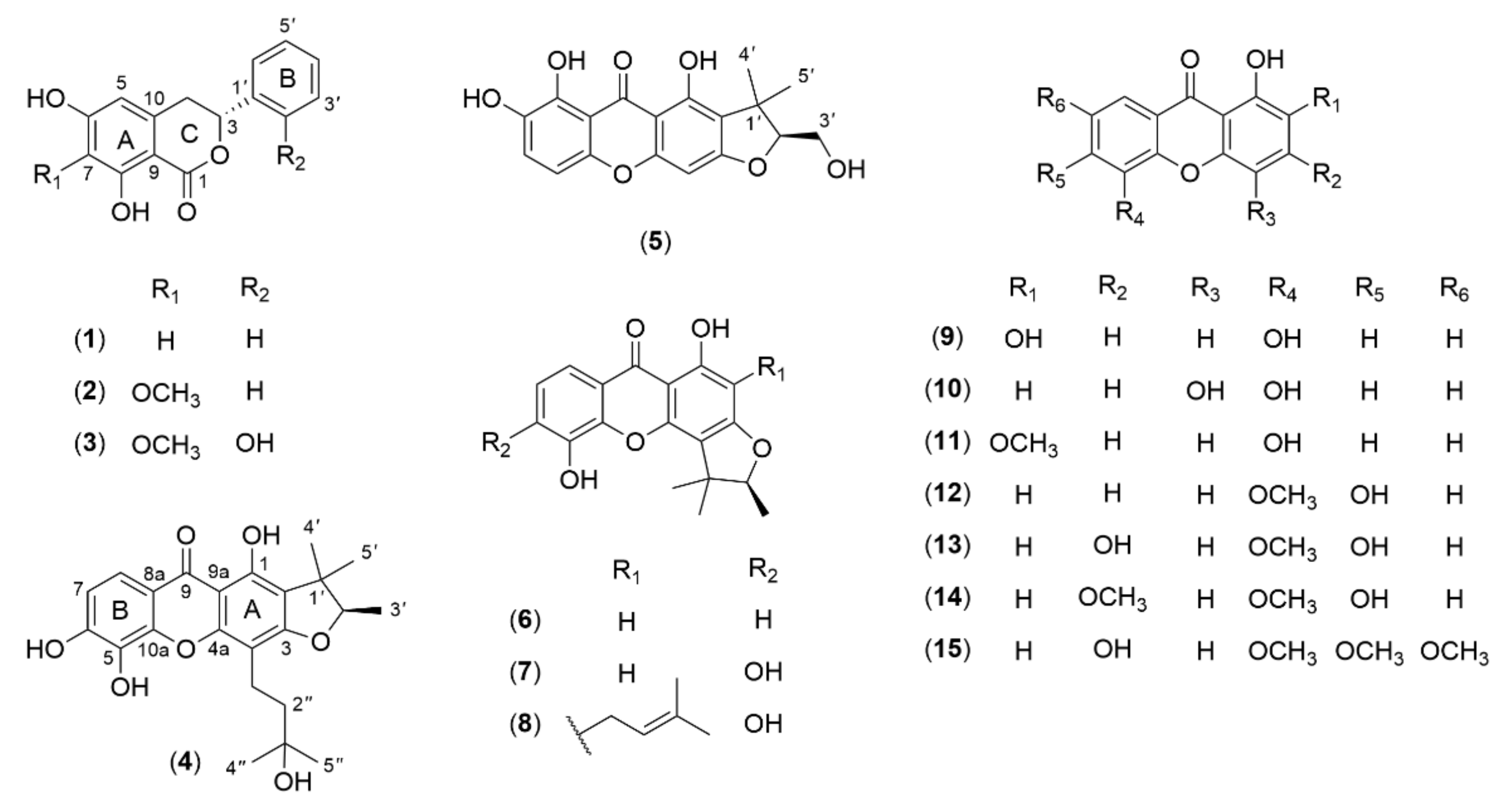
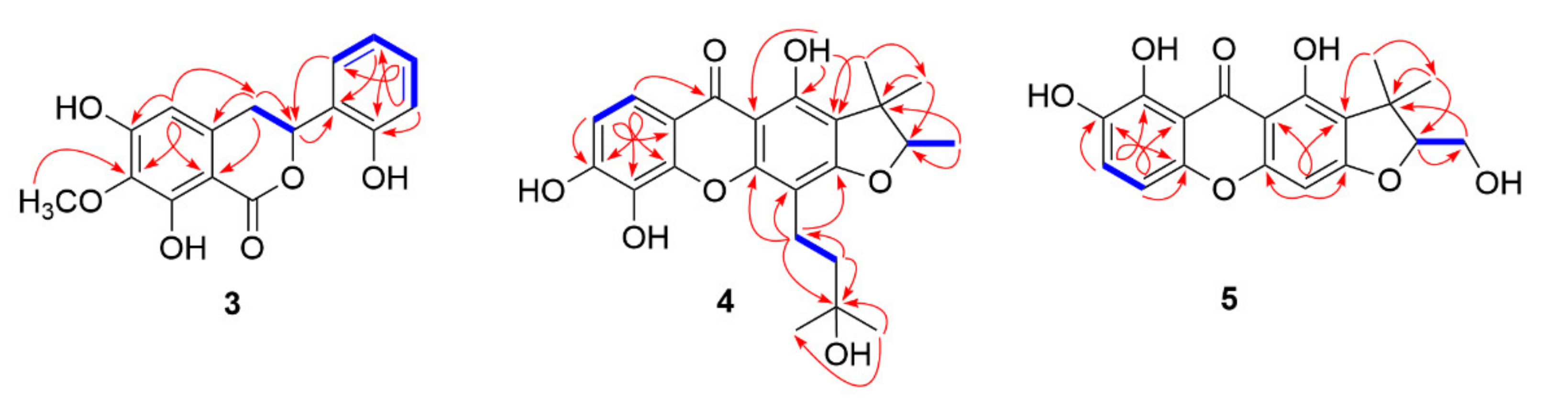
2.1. Structural Elucidation of the Isolated Compounds
2.2. Cytotoxic Activity against Human Cancer Cell Lines
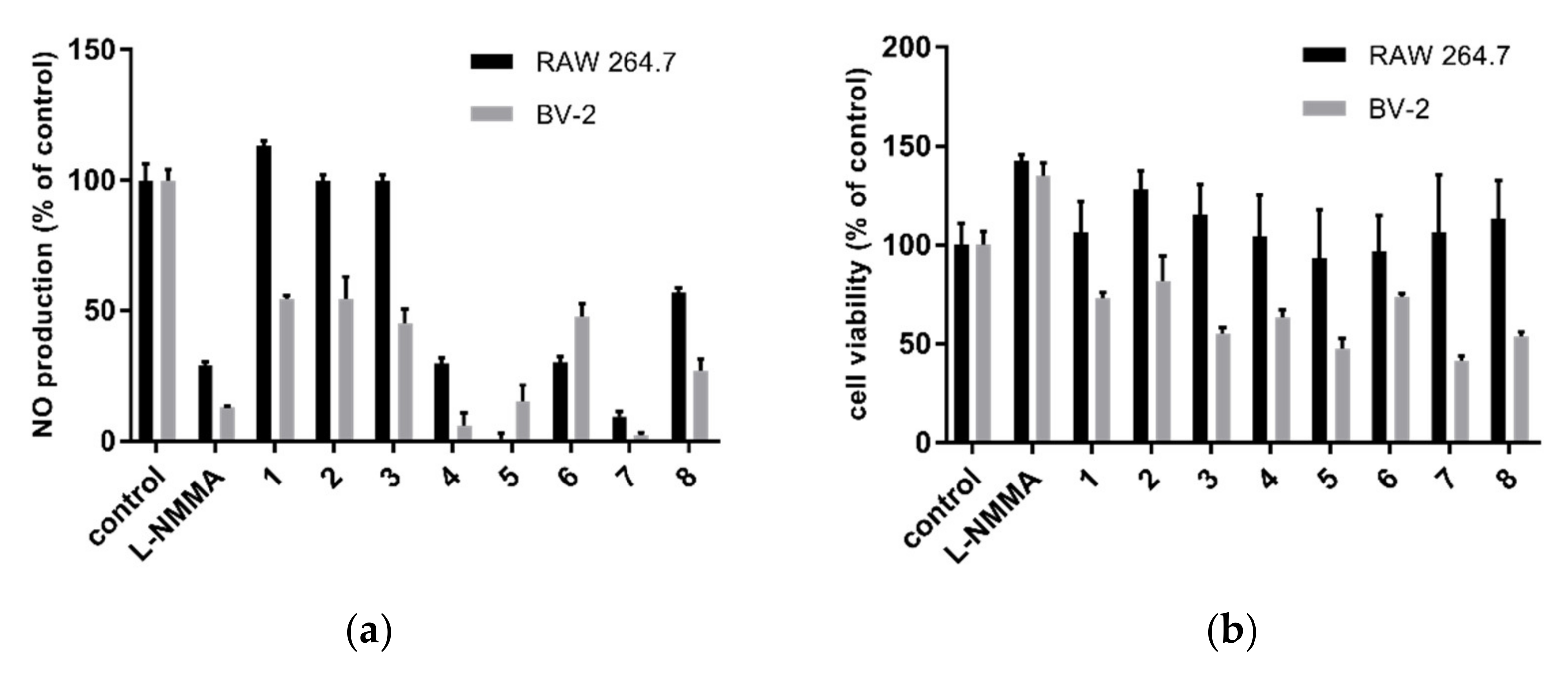
2.3. Nitric Oxide Inhibitory Activity
2.4. Cyclooxygenase (COX) Inhibitory Activity
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Cytotoxic Activity Assay
3.5. NO Production Inhibition Assay
3.6. COX Enzymes Inhibition Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Sweeney, P.W. Systematics and floral evolution in the plant genus Garcinia (Clusiaceae). Ph.D. Thesis, University of Missouri-St. Louis, St. Louis, MO, USA, 2008. [Google Scholar]
- Lim, T.K. Edible Medicinal and Non-Medicinal Plants: Volume 2, Fruits; Springer: New York, NY, USA, 2012; pp. 1–6. [Google Scholar]
- Hemshekhar, M.; Sunitha, K.; Santhosh, M.S.; Devaraja, S.; Kemparaju, K.; Vishwanath, B.S.; Niranjana, S.R.; Girish, K.S. An overview on genus Garcinia: Phytochemical and therapeutical aspects. Phytochem. Rev. 2011, 10, 325–351. [Google Scholar] [CrossRef]
- Xu, X.; Wu, Y.; Hu, M.; Li, X.; Bao, Q.; Bian, J.; You, Q.; Zhang, X. Novel natural product-like caged canthones bearing a carbamate moiety exhibit antitumor potency and anti-angiogenesis activity in vivo. Sci. Rep. 2016, 6, 1–14. [Google Scholar]
- Radji, M.; Sumiati, A.; Indani, N. Uji mutagenisitas dan anti kanker ekstrak aseton dan n-heksana dari kulit batang sesoot (Garcinia picrorrhiza Miq.). Pharm. Sci. Res. 2004, 1, 69–78. [Google Scholar] [CrossRef] [Green Version]
- Soemiati, A.; Kosela, S.; Hanafi, M.; Harrison, L. A novel cytotoxic polyisoprenylbenzophenone derivative compound from Garcinia picrorrhiza Miq. ITE Lett. Batteries New Technol. Med. 2006, 7, 1–5. [Google Scholar]
- Muharni, M.; Elfita, E.; Pertiwi, E. Antibacterial activity of xanthone from ethyl acetate extract of the steam bark of Garcinia picrorrhiza Miq. ALCHEMY J. Penel. Kim. 2017, 13, 252–263. [Google Scholar] [CrossRef] [Green Version]
- Sukandar, E.R.; Kaennakam, S.; Aree, T.; Nost, X.; Rassamee, K.; Bauer, R.; Siripong, P.; Ersam, T.; Tip-pyang, S. Picrorhizones A‒H, polyprenylated benzoylphloroglucinols from the stem bark of Garcinia picrorhiza. J. Nat. Prod. 2020, 10, 2102–2111. [Google Scholar] [CrossRef] [PubMed]
- Kaennakam, S.; Siripong, P.; Tip-pyang, S. Kaennacowanols A−C, three new xanthones and their cytotoxicity from the roots of Garcinia cowa. Fitoterapia 2015, 102, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Sukandar, E.R.; Siripong, P.; Khumkratok, S.; Tip-pyang, S. New depsidones and xanthone from the roots of Garcinia schomburgkiana. Fitoterapia 2016, 111, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Sukandar, E.R.; Kaennakam, S.; Rassamee, K.; Siripong, P.; Fatmawati, S.; Ersam, T.; Tip-pyang, S. Xanthones and biphenyls from the stems of Garcinia cylindrocarpa and their cytotoxicity. Fitoterapia 2018, 130, 112–117. [Google Scholar] [CrossRef]
- Sukandar, E.R.; Kaennakam, S.; Rassamee, K.; Ersam, T.; Siripong, P.; Tip-pyang, S. Tetrandraxanthones A-I, prenylated and geranylated xanthones from the stem bark of Garcinia tetrandra. J. Nat. Prod. 2019, 82, 1312–1318. [Google Scholar] [CrossRef] [PubMed]
- Jing, S.; Qu, Z.; Zhao, C.; Li, X.; Guo, L.; Liu, Z.; Zheng, Y.; Gao, W. Dihydroisocoumarins and dihydroisoflavones from the rhizomes of Dioscorea collettii with cytotoxic activity and structural revision of 2,2′-oxybis(1,4-di-tert-butylbenzene). Molecules 2021, 26, 5381. [Google Scholar] [CrossRef]
- Nedialkov, P.T.; Zheleva-Dimitrova, D.; Girreser, U.; Kitanov, G.M. A new isocoumarin from Hypericum annulatum. Nat. Prod. Res. 2007, 21, 1056–1060. [Google Scholar] [CrossRef]
- Tantapakul, C.; Maneerat, W.; Sripisut, T.; Ritthiwigrom, T.; Andersen, R.J.; Cheng, P.; Cheenpracha, S.; Raksat, A.; Laphookhieo, S. New benzophenones and xanthones from Cratoxylum sumatranum ssp. neriifolium and their antibacterial and antioxidant activities. J. Agric. Food Chem. 2016, 64, 8755–8762. [Google Scholar] [PubMed]
- Ito, C.; Miyamoto, Y.; Rao, K.S.; Furukawa, H. A novel Dibenzofuran and two new xanthones form Calophyllum panciflorum. Chem. Pharm. Bull. 1996, 44, 441–443. [Google Scholar] [CrossRef] [Green Version]
- Kainz, K.P.; Krenn, L.; Erdem, Z.; Kaehlig, H.; Zehl, M.; Bursch, W.; Berger, W.; Marian, B. 2-deprenyl-rheediaxanthone B isolated from Metaxya rostrata induces active cell death in colorectal tumor cells. PLoS ONE 2013, 8, e65745. [Google Scholar] [CrossRef]
- Boonsri, S.; Karalai, C.; Ponglimanont, C.; Kanjana-opas, A.; Chantrapromma, K. Antibacterial and cytotoxic xanthones from the roots of Cratoxylum formosum. Phytochemistry 2006, 67, 723–727. [Google Scholar] [CrossRef] [PubMed]
- Minami, H.; Kinoshita, M.; Fukuyama, Y.; Kodama, M.; Yoshidawa, T.; Sugiura, M.; Nakagawa, K.; Tago, H. Antioxidant xanthones from Garcinia subelliptica. Phytochemistry 1994, 36, 501–506. [Google Scholar] [CrossRef]
- Iinuma, M.; Tosa, H.; Tanaka, T.; Asai, F.; Shimano, R. Three xanthones from root bark of Garcinia subelliptica. Phytochemistry 1995, 38, 247–249. [Google Scholar] [CrossRef]
- Rath, G.; Potterat, O.; Mavi, S.; Hostettmann, K. Xanthones from Hypericum roeperanum. Phytochemistry 1996, 43, 513–520. [Google Scholar] [CrossRef]
- Zhang, Z.; ElSohly, H.N.; Jacob, M.R.; Pasco, D.S.; Walker, L.A.; Clark, A.M. Natural products inhibiting Candida albicans secreted aspartic proteases from Tovomita krukovii. Planta Med. 2002, 68, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Ghosal, S.; Chaudhuri, R.K.; Nath, A. Chemical constituents of Gentianaceae IV: New xanthones of Canscora decussata. J. Pharm. Sci. 1973, 62, 137–139. [Google Scholar] [CrossRef]
- Valentão, P.; Areias, F.; Amaral, J.; Andrade, P.; Seabra, R. Tetraoxygenated xanthones from Centaurium erythraea. Nat. Prod. Lett. 2000, 14, 319–323. [Google Scholar] [CrossRef]
- Imperato, F. A new xanthone from the fern Cystopterys fragilis. J. Nat. Prod. 1991, 54, 603–605. [Google Scholar] [CrossRef]
- Paraschos, S.; Magiatis, P.; Kalpoutzakis, E.; Harvala, C.; Skaltsounis, A.L. Three new dihydroisocoumarins from the Greek endemic species Scorzonera cretica. J. Nat. Prod. 2001, 64, 1585–1587. [Google Scholar] [CrossRef]
- Ito, C.; Mishina, Y.; Litaudon, M.; Cosson, J.P.; Furuawa, H. Xanthone and dihydroisocoumarin from Montrouziera sphaeroidea. Phytochemistry 2000, 53, 1043–1046. [Google Scholar] [CrossRef]
- Sukandar, E.R.; Ersam, T.; Fatmawati, S.; Siripong, S.; Aree, T.; Tip-pyang, S. Cylindroxanthones A–C, three new xanthones and their cytotoxicity from the stem bark of Garcinia cylindrocarpa. Fitoterapia 2016, 108, 62–65. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Lin, C.C.; Hattori, M.; Namba, T. Four prenylated xanthones from Cudrania cochinchinensis. Phytochemistry 1989, 28, 595–598. [Google Scholar] [CrossRef]
- Jo, Y.H.; Shin, B.; Liu, Q.; Lee, K.Y.; Oh, D.C.; Hwang, B.Y.; Lee, M.K. Antiproliferative prenylated xanthones and benzophenones from the roots of Cudrania tricuspidata in HSC-T6 cells. J. Nat. Prod. 2014, 77, 2361–2366. [Google Scholar] [CrossRef]
- Nguyen, L.T.T.; Nguyen, D.M.; Nguyen, L.H.D. A new xanthone from the bark of Calophyllum thorelii. Nat. Prod. Res. 2013, 27, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Rukachaisirikul, V.; Naklue, W.; Sukpondma, Y.; Phongpaichit, S. An antibacterial biphenyl derivative from Garcinia bancana Miq. Chem. Pharm. Bull. 2005, 53, 342–343. [Google Scholar] [CrossRef] [Green Version]
- Trisuwan, K.; Boonyaketgoson, S.; Rukachaisirikul, V.; Phongpaichit, S. Oxygenated xanthones and biflavanoids from the twigs of Garcinia xanthochymus. Tetrahedron Lett. 2014, 55, 3600–3602. [Google Scholar] [CrossRef]
- Zhang, Y.; Chao, L.; Ruan, J.; Zheng, C.; Yu, H.; Qu, L.; Han, L.; Wang, T. Bioactive constituents from the rhizomes of Dioscorea septemloba Thunb. Fitoterapia 2016, 115, 165–172. [Google Scholar] [CrossRef]
- Zhu, D.; Li, S.Q.; Yuan, Y.B.; Shao, Y.Y.; Yang, G.H.; Mao, X.; Wu, L.F.; Zhang, X.X.; Liang, W.Y.; Chen, W.J.; et al. Study on liposoluble compounds in rhizome of Dioscoreae hypoglaucae. Chin. Trad. Herb. Drugs 2016, 47, 379–382. [Google Scholar]
- Li, X.; Zhao, C.; Jing, S.; Sun, J.; Li, X.; Man, S.; Wang, Y.; Gao, W. Novel phenanthrene and isocoumarin from the rhizomes of Dioscorea nipponica Makino subsp. rosthornii (Prain et Burkill) C. T. Ting (Dioscoreaceae). Bioorg. Med. Chem. Lett. 2017, 27, 3595–3601. [Google Scholar] [CrossRef] [PubMed]
- Saeed, A. Isocoumarins, miraculous natural products blessed with diverse pharmacological activities. Eur. J. Med. Chem. 2016, 116, 290–317. [Google Scholar] [CrossRef]
- Siripong, P.; Kanokmedakul, K.; Piyaviriyagul, S.; Yahufai, J.; Chanpai, R.; Ruchirawat, S.; Oku, N. Antiproliferative naphthoquinone esters from Rhinacanthus nasutus Kurz. roots on various cancer cells. J. Trad. Med. 2006, 23, 166–172. [Google Scholar]
- Kaennakam, S.; Sukandar, E.R.; Juntagoot, T.; Siripong, P.; Tip-pyang, S. Four new xanthones and their cytotoxicity from the stems of Garcinia schomburgkiana. J. Nat. Med. 2021, in press. [Google Scholar]
- Tran, H.T.; Gao, X.; Kretschmer, N.; Pferschy-Wenzig, E.M.; Raab, P.; Pirker, T.; Temml, V.; Schuster, D.; Kunert, O.; Huynh, L.; et al. Anti-inflammatory and antiproliferative compounds from Sphaeranthus africanus. Phytomedicine 2019, 62, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lajter, I.; Pan, S.P.; Nikles, S.; Ortmann, S.; Vasas, A.; Csupor-Lottler, B.; Forgo, P.; Hohmann, J.; Bauer, R. Inhibition of COX-2 and NF-kappaB1 gene expression, NO production, 5-LOX, and COX-1 and COX-2 enzymes by extracts and constituents of Onopordum acanthium. Planta Med. 2015, 81, 1270–1276. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 3 | Position | 4 | 5 | |||
|---|---|---|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | ||
| 1 | 171.3 | 1 | 157.5 | 159.5 | |||
| O-2 | 2 | 116.9 | 117.9 | ||||
| 3 | 5.93, dd (11.6, 4.0) | 77.0 | 3 | 164.5 | 168.2 | ||
| 4a | 3.13, dd (16.4, 4.0) | 34.1 | 4 | 104.6 | 6.38, s | 90.4 | |
| 4b | 3.18, dd (16.4, 11.6) | 4a | 155.8 | 159.5 | |||
| 5 | 6.42, s | 107.3 | 10a | 146.9 | 149.8 | ||
| 6 | 157.7 | 5 | 133.4 | 6.90, d (8.8) | 107.0 | ||
| 7 | 134.8 | 6 | 151.9 | 7.33, d (8.8) | 124.6 | ||
| 8 | 157.7 | 7 | 6.96, d (8.4) | 113.9 | 141.4 | ||
| 9 | 102.7 | 8 | 7.61, d (8.4) | 117.3 | 148.0 | ||
| 10 | 137.4 | 8a | 114.7 | 108.3 | |||
| 1′ | 126.2 | 9 | 182.0 | 185.5 | |||
| 2′ | 155.0 | 9a | 104.4 | 103.4 | |||
| 3′ | 6.95, d (8.0) | 116.4 | 1′ | 44.6 | 43.6 | ||
| 4′ | 7.21, t (8.0) | 130.4 | 2′ | 4.53, q (6.4) | 91.5 | 4.51, t (5.6) | 96.0 |
| 5′ | 6.94, t (8.0) | 120.8 | 3′ | 1.41, d (6.4) | 14.8 | 3.91, d (5.6) | 61.4 |
| 6′ | 7.49, d (8.0) | 127.8 | 4′ | 1.24, s | 21.1 | 1.38, s | 20.6 |
| OH-8 | 11.44, brs | 5′ | 1.49, s | 25.7 | 1.57, s | 26.8 | |
| OCH3-7 | 3.84, s | 60.8 | 1″ | 2.91, m | 18.3 | ||
| 2″ | 1.81, m | 42.9 | |||||
| 3″ | 71.8 | ||||||
| 4″ | 1.32, s | 29.7 | |||||
| 5″ | 1.32, s | 29.7 | |||||
| OH-1 | 13.26, s | ||||||
| Compound | IC50 ± SEM (µM) a | ||||
|---|---|---|---|---|---|
| KB | HeLa S3 | MCF-7 | Hep G2 | HT-29 | |
| 4 | 7.5 ± 0.8 | 5.6 ± 0.1 | 5.7 ± 0.3 | 6.3 ± 0.6 | 20.3 ± 0.6 |
| 6b | 12.1 ± 0.1 | 20.7 ± 0.6 | 15.6 ± 0.3 | 22.8 ± 0.4 | inactive |
| 7b | 5.1 ± 0.4 | 6.0 ± 0.5 | 6.5 ± 0.2 | 10.0 ± 0.2 | inactive |
| 8b | 0.2 ± 0.1 | 0.3 ± 0.1 | 4.9 ± 0.4 | 3.8 ± 0.5 | 21.9 ± 1.2 |
| 13 | 11.4 ± 1.7 | 15.2 ± 1.3 | NT | NT | NT |
| Doxorubicin c | 0.02 ± 0.01 | 0.15 ± 0.02 | 1.29 ± 0.02 | 1.00 ± 0.17 | 0.59 ± 0.03 |
| Compound | RAW 264.7 | BV-2 | ||
|---|---|---|---|---|
| iNOS | Cytotoxicity | iNOS | Cytotoxicity | |
| 4 | 84.3 ± 3.5 | >200 | 20.0 ± 4.0 | 27.7 ± 6.4 |
| 5 | 16.4 ± 4.5 | >200 | 13.8 ± 1.6 | 74.7 ± 2.1 |
| 7 | 45.6 ± 6.5 | 85.6 ± 6.9 | 28.7 ± 2.3 | 31.7 ± 4.7 |
| Compound | % Inhibition at 20 µM | |
|---|---|---|
| COX-1 | COX-2 | |
| 2 | <10 | 10.4 ± 5.0 |
| 3 | 24.2 ± 12.3 | <10 |
| 4 | 32.4 ± 7.9 | <10 |
| 6 | 31.2 ± 21.7 | <10 |
| 7 | 15.4 ± 7.5 | <10 |
| 8 | 24.0 ± 15.0 | <10 |
| 9 | 22.6 ± 2.2 | <10 |
| 14 | 18.3 ± 12.1 | <10 |
| Indometacin f | 78.4 ± 4.1 | NT |
| Celecoxib g | NT | 83.5 ± 4.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sukandar, E.R.; Kaennakam, S.; Raab, P.; Nöst, X.; Rassamee, K.; Bauer, R.; Siripong, P.; Ersam, T.; Tip-pyang, S.; Chavasiri, W. Cytotoxic and Anti-Inflammatory Activities of Dihydroisocoumarin and Xanthone Derivatives from Garcinia picrorhiza. Molecules 2021, 26, 6626. https://doi.org/10.3390/molecules26216626
Sukandar ER, Kaennakam S, Raab P, Nöst X, Rassamee K, Bauer R, Siripong P, Ersam T, Tip-pyang S, Chavasiri W. Cytotoxic and Anti-Inflammatory Activities of Dihydroisocoumarin and Xanthone Derivatives from Garcinia picrorhiza. Molecules. 2021; 26(21):6626. https://doi.org/10.3390/molecules26216626
Chicago/Turabian StyleSukandar, Edwin R., Sutin Kaennakam, Pia Raab, Xuehong Nöst, Kitiya Rassamee, Rudolf Bauer, Pongpun Siripong, Taslim Ersam, Santi Tip-pyang, and Warinthorn Chavasiri. 2021. "Cytotoxic and Anti-Inflammatory Activities of Dihydroisocoumarin and Xanthone Derivatives from Garcinia picrorhiza" Molecules 26, no. 21: 6626. https://doi.org/10.3390/molecules26216626