
Inherited Developmental and Epileptic Encephalopathies


USL Centro Toscana, Neurology Unit, Nuovo Ospedale Santo Stefano, 59100 Prato, Italy
Neurol. Int. 2021, 13(4), 555-568; https://doi.org/10.3390/neurolint13040055
Submission received: 16 September 2021
/
Revised: 29 October 2021
/
Accepted: 2 November 2021
/
Published: 3 November 2021
(This article belongs to the Special Issue The Neurogenetics of Degenerative Disorders)
Abstract
:Epileptic encephalopathies often have a genetic etiology. The epileptic activity itself exerts a direct detrimental effect on neurodevelopment, which may add to the cognitive impairment induced by the underlying mutation (“developmental and epileptic encephalopathy”). The focus of this review is on inherited syndromes. The phenotypes of genetic disorders affecting ion channels, metabolic signalling, membrane trafficking and exocytosis, cell adhesion, cell growth and proliferation are discussed. Red flags suggesting family of genes or even specific genes are highlighted. The knowledge of the phenotypical spectrum can indeed prompt the clinician to suspect specific etiologies, expediting the diagnosis.
1. Introduction
An epileptic encephalopathy (EE) is a severe type of epilepsy in which the epileptic activity itself promotes cognitive and behavioural impairments above and beyond what might be expected from the underlying pathology alone [1].
Cognitive functions may be slowed, plateauing or regress, and patients often develop psychiatric and behavioural disturbances. A key component of the EE concept is that amelioration of the epileptiform activity may have the potential to improve the developmental consequences of the disorder [2]. We usually consider EE to exclusively occur in the paediatric population. Nevertheless, cognitive impairment may occur at any age and the concept of EE may be also applicable to syndromes arising in adulthood.
A genetic aetiology is often to be sought. A polygenic or complex genetic background can be involved, and modifier genes and epigenetics factors can also partially influence the phenotype [3].
However, in recent years, techniques of next-generation sequencing (NGS) applied to severe epilepsies have allowed to identify a growing number of single gene mutations that can promote seizures and affect neurodevelopment. Strikingly, some genetic syndromes may be characterized by early waning of epilepsy but persisting profound developmental consequences at long-term. Often it may not be possible to disentangle whether the epileptic or developmental component plays the major role.
To navigate through the nosological boundaries of these syndromes, different descriptors can be applied: “developmental encephalopathy” (DE), where there is just developmental impairment without frequent epileptic activity; “epileptic encephalopathy” (EE), where there is no pre-existing developmental delay, and the genetic mutation is not thought to cause slowing per se; and “developmental and epileptic encephalopathy” (DEE), where both factors play a role. To disclose a specific genetic aetiology in these syndromes is very important. Families are often relieved to find a cause after a distressing diagnostic conundrum; unnecessary testing can be avoided, and appropriate familial counselling can be established.
Most severe epilepsies result from de novo mutations, with a low recurrence risk due to germline mosaicism when parents are unaffected (estimated to be about 1%). The possibility of parental somatic mosaicism in apparent de novo variants have also to be considered, with reported figures up to 10% of families. The resulting recurrence risk cannot be calculated, but can approach 50% as observed in autosomal dominant disorders [4,5]. Familial clustering can also result from parental transmission with classical mendelian inheritance, especially for genes with variable penetrance and expressivity [6].
The spotlight of the current review will be on severe epilepsies in which the epileptic activity itself affects neurodevelopment (EE/DEE), especially focusing on inherited syndromes. For this purpose, the phenotype of inherited mutations affecting ion channels, metabolic signalling, membrane trafficking and exocytosis, cell adhesion, and cell growth and proliferation will be discussed.
2. Methods
The literature search was performed on the Medline and Embase databases. Articles published in English up to August 2021 were sought using Boolean logic with the following search terms: ‘inherited AND epil *’, ‘inherited AND encephalopathy’.
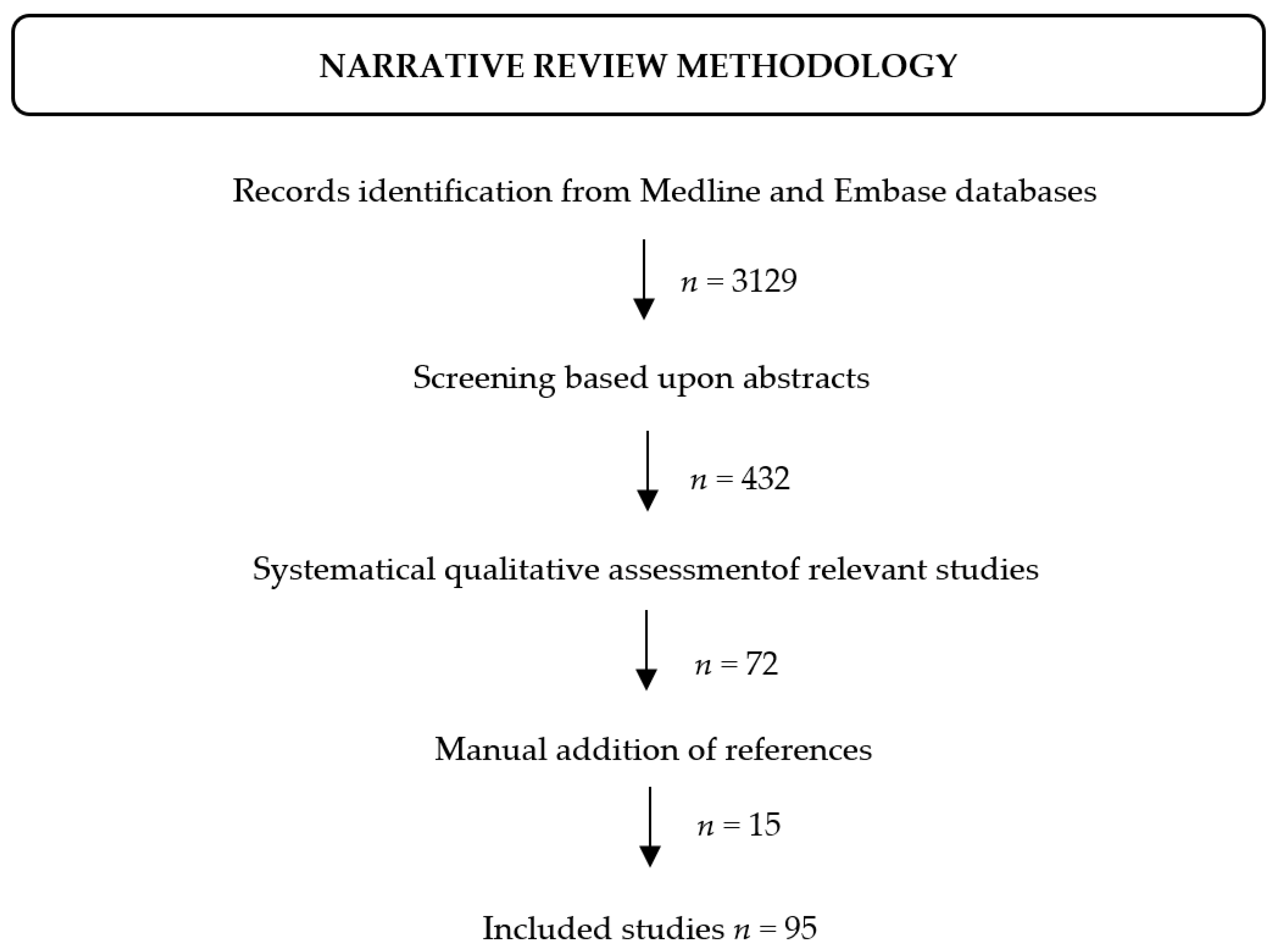
The retrieved articles have first been screened based upon the abstracts. The full text of those deemed to be relevant have then been systematically assessed. Additional references have been added after manual search on PubMed, Embase, the Human Gene Mutation Database (HGMD, http://www.hgmd.cf.ac.uk/ac/index.php), Google (https://www.google.com) and Google Scholar (https://scholar.google.com) search engines (accessed on 30 October 2021) (Figure 1).
3. Channelopathies
Ion channels are proteins located across cell membranes to form a pore through which ions move according to their electrochemical gradient.
Each channel exhibits a selective permeability to specific ions and is usually composed by a pore-forming subunit and accessory regulatory subunits (heteromeric structure). Channel gating can be regulated mostly by transmembrane electric potential (voltage-gated channels) or specific extracellular ligand molecules (ligand-gated channels).
Genetic channelopathies can promote systemic disorders, consistent with the ubiquitous expression of ion channels. The central nervous system is especially vulnerable, considering ion channels affect the generation, suppression, and spread of the action potential [7]. Most phenotypical traits are characterized by paroxysmal symptoms, especially seizures. Epilepsy may arise at any age [8] according to the different expression of ion channels during development [9].
Classical epilepsy syndromes (Table 1) and specific syndromes associated with single ion channel genes are well described.
The vast majority of genetic channelopathies have initially been reported in families with benign epilepsies. However, the large-scale application of NGS techniques has revealed the genotype–phenotype correlation may be extremely challenging. Ion channel genes initially associated only with benign phenotypes (e.g., KCNQ2, SCN2A) have also been demonstrated to be implicated in severe EE/DEE.
As a rule of thumb, inherited variants mostly provoke familial benign syndrome, and de novo mutations promote EE/DEE. There are remarkable exceptions: cases of EE/DEE can cluster within a family or co-occur with milder epilepsies as expression of variable expressivity.
3.1. Voltage-Gated Channels
Inherited EE/DEE have been reported to result from disruption of several voltage-gated channels.
Voltage-gated sodium channels (‘VGSCs’ or ‘Nav channels’) play a major role. A superfamily of genes clustering on chromosome 2 encodes for different isoforms of the pore-forming alpha subunit of this channel (Nav1.1–Nav1.9), whose opening results in a sodium inflow [10].
Mutations affecting three alpha subunit genes (SCN1A, SCN2A, and SCN8A) have been shown to cause epilepsies of variable severity [11].
SCN1A encodes for the alpha 1 subunit (Nav1.1), whose expression steadily augments from 4–5 months of age on [12]. The phenotypical spectrum of SCN1A mutations spans from mild to severe epilepsies, all characterized by fever-sensitive seizures. Dravet syndrome is the classical encephalopathic phenotype and mostly result from de novo heterozygous mutations (truncating, splice-site, frameshift intragenic deletions, gene duplications leading to haploinsufficiency, missense mutations affecting the pore-forming region of the protein) [13,14].
Inherited autosomal dominant mutations occur in 5–10% of cases and are usually missense. The syndrome can also be inherited for somatic or germline mosaic mutations in unaffected parents, with familial clustering in the offspring [13,15].
Dravet syndrome typically arises suddenly with unilateral and very prolonged febrile seizures at 5–8 months of age in previously healthy children [16]. This time window coincides with the period of mandatory vaccination in children; these patients were formerly considered to suffer an alleged ‘vaccine encephalopathy’ before the underlying SCN1A neurobiology was discovered [17,18]. During the first year of life, both febrile and afebrile seizures occur. From the second year of age, neurodevelopment plateaus and seizures flare as generalized tonic-clonic, alternating unilateral clonic, focal with impaired awareness, brief myoclonic, tonic (rare) or myoclonic non-convulsive status epilepticus described as ‘obtundation status’ [19,20,21]. Sodium-channel-blocker antiseizure medications (ASMs) (e.g., carbamazepine, phenytoin, lamotrigine) may precipitate seizures. The EEG background activity worsens over time, with generalized spike-wave and polyspike-wave activity, multifocal discharges, and sometimes photosensitivity [19,22]. Autistic traits and language impairment may establish themselves, and a wide-based ataxic gait sets in. After five years of age, epilepsy wanes with less frequent seizures [19,21,23]. In adulthood, myoclonic and absence seizures disappear; tonic/convulsive seizures occur mainly in sleep, and ataxia, dysarthria, intention tremor, and intellectual disability prevail [24,25]. Levodopa-responsive extrapyramidal signs and axonal motor neuropathy may also ensue [26].
The SCN2A gene encodes for the Nav1.2 channel, which is highly expressed from the birth [27]. Mutations of SCN2A have been increasingly recognized as a major cause of EE/DEE [16,28]. De novo missense and truncating variants are those usually associated with severe phenotypes. Inherited variants are mostly associated with milder epilepsies (e.g., benign familial neonatal-infantile seizure, autosomal dominant, missense mutations). Intrafamilial phenotypical heterogeneity is possible but rare. Cases of EE/DEE and mild epilepsy in the same family or febrile seizures are described suggesting variable expressivity [16]. In patients with SCN2A-related encephalopathy, seizures arise either from the first week of life or during childhood, accompanied by cognitive regression and autistic features [16,29]. The onset is abrupt, with focal seizures and spasms in clusters, possibly leading to specific syndromes such as Epilepsy of Infancy with Migrating Focal Seizures (EIMFS) and Ohtahara syndrome [16]. Recent findings suggest neonatal-onset seizures are provoked by gain-of-function missense mutations and respond to sodium-channel-blocker ASMs, whilst later onset epilepsies are associated to loss-of-function missense or truncating variants and do not respond these type of medications [29].
Heterozygous missense mutations of SCN8A have recently been associated with a wide spectrum of epilepsies. Inherited mutations almost exclusively yield benign infantile epilepsies; however SCN8A-related EE/DEE due to parental germline mosaicism has been described [30,31,32]. The SCN8A gene product (sodium channel subunit Nav1.6) is increasingly expressed from the neonatal period, progressively replacing the Nav1.2 channel in initial axonal segments. Accordingly, most patients with SCN8A mutations develop epilepsy in infancy (median age = 4 months) [32]. Patients with SCN8A-related encephalopathy suffer global developmental delay, sleep disorders, frequent startles and multiple intractable seizure types (focal, tonic with autonomic signs, clonic, myoclonic, absence, epileptic spasms) [32,33,34]. Dystonic/dyskinetic attacks may also occur [30,32].
Potassium channelopathies can also be responsible of EE/DEE, especially those affecting the voltage-gated ion channels (Kv) regulating the frequency and duration of action potential [35], such as those encoded by KCNQ2 and KCNQ3.
Mutations of these genes were initially identified in patients with autosomal dominant benign familial neonatal seizures [36,37]. Like many other channelopathies, the phenotypic spectrum now also includes severe epilepsies, including families with drug-resistant seizures and intellectual disability harboring inherited mutations of KCNQ2 [38] and KCNQ3 [39].
KCNQ2-related encephalopathy is characterized by drug-resistant tonic asymmetric, focal and clonic seizures in the first week of life, progressively waning in early infancy and remitting by the age of 1–3 years. The corresponding electroencephalographic patterns are neonatal burst-suppression evolving to multifocal epileptiform activity during follow-up. Sodium channel blockers ASMs can be effective. Early seizure freedom can ameliorate the neurodevelopmental outcome [40]. A dominant negative effect of KCNQ2 mutations has been demonstrated. The vast majority of mutations are missense and appear to cluster in four putative functional hotspots, affecting the conformation of the tetrameric subunits of the channel [41].
KCNQ3-related encephalopathy is similarly characterized by early onset drug-resistant seizures, centrotemporal EEG abnormalities, and intellectual disability at long-term [39].
Mutations affecting inwardly rectifying potassium (Kir) channels can provoke EE/DEE, especially missense mutations of KCNB1 disrupting the voltage sensor and the pore domain. The corresponding phenotype is characterized by predominant language difficulties, behavioral impairment, and seizures in 85% of cases. A single case with an inherited truncating variant has been identified in this syndrome (p.Arg583 *) [42].
Specific severe epileptic phenotypes can also result from the KCNT1 gene. Inherited missense mutations were initially reported in an unusual form of autosomal dominant sleep-related hypermotor epilepsy (SHE) complicated by intellectual disability and behavioural disturbances. Sporadic mutations mainly yield EIMFS: nearly continuous multifocal and migrating seizures in the first 6 months of life followed by persistent drug-resistant epilepsy with acquired microcephaly, intellectual disability, axial hypotonia, pyramidal, and extrapyramidal signs. However, both cases of SHE and EIMFS can co-occur within the same family. Irrespectively of the underlying syndrome, up to 30% of patients benefit from quinidine, an anti-arrhythmogenic potassium-channel blocker, for seizure control [43,44].
Eventually, calcium channelopathies may also be responsible of EE/DEE. These channels regulate the inflow of calcium ions according to their electrochemical gradient modulating gene transcription, neurotransmitter release, neurite outgrowth, and enzyme activity [45]. Heterozygous mutations affecting CACNA1A are those most often encountered in neurological phenotypes. This gene is involved in hemiplegic migraine, alternating hemiplegia, and episodic ataxia, both sporadic and familial. Different phenotypical traits may overlap in a CACNA1A family: each family member can exhibit seizures, episodes of hemiplegia, migraine, or ataxic disorders with variable penetrance and expressivity. Some degree of intellectual disability can also occur [7,46,47,48,49]. With regard to epilepsy, two main alternative phenotypes have been identified in these patients (i) early and recurrent status epilepticus followed by drug-resistant focal-onset epilepsy, early progressive cerebellar signs, hemiplegic accesses with consciousness impairment, severe intellectual disability (ii) generalized epilepsy with early-onset refractory absence seizures, stable cerebellar dysfunction, and milder intellectual disability [7].
Biallelic CACNA1A mutations inherited from unaffected parents have been disclosed in probands with early onset seizures, intellectual disability, autistic features, and progressive cerebral and optic nerve atrophy [50].
3.2. Ligand Gated Channels
Genetic dysfunction of GABA receptors can promote EE/DEE. GABRA1A is indeed a major causative gene for various epileptic encephalopathies, mostly sporadic but occasionally inherited through autosomal dominant mutations. Patients may develop Ohtahara syndrome, infantile spasms, Dravet syndrome-like phenotypes and myoclonic astatic epilepsy [51]. Red flags for suspecting GABRA1A mutations are infantile-onset epilepsy (from 1 day to 15 months of age), prominent tonic-clonic and myoclonic seizures, generalized spike-and-wave activity and photoparoxysmal response. No clear genotype-phenotype correlation has emerged, although the clinical presentation appears to be quite homogeneous within single families [51].
The ligand-gated NMDA receptor is a glutamate-dependent ion channel. Mutations of GRIN2A and GRIN2B, encoding for two subunits of the receptor, can yield several neurodevelopmental disorders and be inherited in an autosomal dominant manner. The phenotypical spectrum of GRIN2A mutations include isolated intellectual disability, idiopathic focal epilepsy, and epileptic encephalopathies. Language disorders are common and range from mild speech impairment with no seizures to the epilepsy aphasia spectrum (atypical rolandic epilepsy, continuous spikes and waves during slow-wave sleep, and Landau–Kleffner-Syndrome) [52]. Gain-of-function GRIN2B mutations cause West syndrome, as well as childhood onset focal epilepsy in association with intellectual disability. Cognitive skills depend upon the degree of channel function impairment [53].
4. Metabolic Disorders
Seizures and intellectual disability can also result from inborn errors of metabolism (IEM), often following an autosomal recessive pattern. Some of these disorders can be resolutely addressed with specific treatments (treatable IEM). A timely diagnosis may indeed limit or even prevent the devolvement of an overt encephalopathy as well as suggest investigating extra-cerebral comorbidities. Although the phenotype may be extremely complex, the clinician should be prompted to suspect metabolic disorders when encountering red flags such as dysmorphic features, organomegaly, positive family history and/or consanguinity, ophthalmological disorders, and metabolic acidosis with high anion gap. From the neurological standpoint, cognitive regression with worsening myoclonic seizures should ring a bell. Seizures may be triggered by external factors such as periods of stress, intercurrent illnesses, or metabolic complications (e.g., hypoglycemia) [54,55,56].
Seizures may also arise abruptly with unspecific characteristics, especially in the newborn. In this age range, we should especially investigate Pyridoxine dependency epilepsy and Pyridox(am)ine 5′phosphate oxidase (PNPO) deficiency.
Pyridoxine-dependent epilepsy begins with multifocal drug-resistant seizures since the first hours/days of life, namely, myoclonic jerks, tonic seizures, and focal motor seizures. It is provoked by autosomal recessive ALDH7A1 mutations. The biochemical defect consists in deficiency of α–aminoadipic semialdehyde dehydrogenase (antiquitin), which is involved in cerebral lysine metabolism. Patients should be promptly treated by pyridoxine to interrupt seizures and hamper cognitive deterioration (vitamin B6:100 mg intravenously, followed by oral supplementation 30 mg/kg/day in two divided doses for 3–7 days). Indeed, a trial of pyridoxine is recommended in any newborn with stormy onset seizures of unknown etiology given the dramatic change of prognosis it may bring. Diagnostic confirmation is done through demonstration of elevated levels of intermediary substrates in urine and/or plasma and cerebrospinal fluid [56,57].
Deficit of PNPO may give a similar phenotype. This enzyme is essential for the synthesis of pyridoxal phosphate (PLP), which is the active form of pyridoxin. These patients and those newborns not responding to pyridoxine should be expeditely treated by PLP supplementation. The definitive diagnosis can only be established by molecular genetic testing (autosomal recessive inheritance) [58].
In older children, a progressive EE/DEE can result from defects of cerebral glucose transporter (GLUT1). These are caused by mutations of SLC2A1 gene, mostly inherited as autosomal dominant or due to sporadic haploinsufficiency. Cases of autosomal recessive inheritance have been described.
The phenotype ranges from severe early onset drug-resistant seizures, developmental delay, and acquired microcephaly (De Vivo syndrome) to early onset absence epilepsy or Epilepsy with Myoclonic-Atonic Seizures [59]. Clinical clues to GLUT1 deficiency include an increase in seizures during fasting, progressive cognitive impairment, and paroxysmal exercise-induced dyskinesia. The diagnosis can result either from the finding of low CSF glucose levels or from molecular genetic testing. When suspecting GLUT1 deficiency, ketogenic diet should be introduced as this non-pharmacological treatment limits both seizures and neurological deterioration by providing alternative sources of energy to the brain (ketone bodies) [59].
Many other IEM can provoke seizures and intellectual disability at different ages with variable expressivity. For instance, neuronal ceroid-lipofuscinoses may affect the central nervous system of newborns, toddlers, children, or even adults, inducing progressive intellectual deterioration, vision impairment without organomegaly, and epilepsy that may follow a progressive myoclonic course [60,61]. The inheritance pattern can be autosomal dominant or recessive. A comprehension of the underlying molecular biology can have a striking impact on patient management. In 2019, a patient-customized rescue therapy was rapidly developed for a 6-year-old child with neuronal ceroid lipofuscinosis 7, after disclosing a pathogenic variant in the gene MFSD8 (inherited from the father) and a cryptic second hit affecting splice-site in intron 6 of the same gene (inherited from the mother). Researchers developed an antisense oligonucleotide (milasen) to block the splice-site mutation and obtained an objective improvement in seizure control and cognitive functioning [62].
Mitochondrial disorders similarly can arise at different ages and promote seizures with intellectual disability. The proteins of mitochondria are m encoded by nuclear DNA (nDNA) and in a small percentage by the mitochondrial DNA (mtDNA). Both mutations of nDNA (autosomal dominant or recessive) and mtDNA (maternally inherited) can affect the respiratory chain and the oxidative phosphorylation. Mitochondrial disorders tend to involve multiple organs with high energy demand (e.g., nervous system, skeletal and cardiac muscles, liver, kidneys, and endocrine system).
For instance, POLG-related disorders (nDNA) are characterized by progressive neurodegeneration, refractory epilepsy, movement disorder, neuropathy, and hepatic failure; seizures can be myoclonic, prolonged tonic-clonic, or focal motor (epilepsia partialis continua). Valproate is absolutely contraindicated [63,64].
Pyruvate dehydrogenase complex deficiency can result from mutations of different nDNA genes (PDHA, PDHB, LIAS, LIPT1, DLD, and PDH). In infancy, infantile spasms, clonic seizures, or refractory focal epilepsy can be observed, as well as developmental delay, ataxia, hypotonia, hypertonia, abnormal eye movements, dystonia, and axonal neuropathy; ketogenic diet may partially be effective in seizure control [65,66].
Leigh syndrome has a wide genotypical heterogeneity (>90 nDNA or mtDNA genes) and is typically characterized by focal drug-resistant seizures, developmental regression/delay, and basal ganglia/brainstem abnormalities on neuroimaging [67].
Myoclonic epilepsy with ragged red fibres (MERFF) (MTTL1 (80%): m.8344A > G; MTTK (10%): m.8356T > C, m.8363G > A, m.8361G > A) provoke an adult-onset progressive myoclonic epilepsy. Seizures can also often be generalized tonic, clonic or atonic. Cerebellar ataxia, cardiac arrhythmias, myopathy, diabetes, hearing loss, and dementia ensue [66,68].
Mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) (MTTL1 gen: m.3243A >G m.3271T > C; MTND5 gen: m.13513G > A) are mostly characterized by seizures during a stroke-like episode, often with focal status epilepticus and secondary encephalopathy [66,69].
To summarize, identifying a specific metabolic disorder in subjects with such complex phenotypes can be challenging. A tiered approach to the diagnosis of IEM can be usefully applied. A preliminary screening by history and physical exam and wide spectrum blood and urine biochemistry should be performed, after which targeted biochemical and molecular investigations can be undertaken [56].
5. Membrane Trafficking and Exocytosis
Genes involved in transmembrane trafficking may directly and indirectly affect neuronal excitability. Like channelopathies, the corresponding phenotypes span from benign to very severe epilepsies.
Mutations of STX1B are a relevant example. The STX1B gene encodes for syntaxin-1B, which composes the SNARE complex together with SNAP25 and synaptobrevin. This complex mediates the process of calcium-dependent synaptic vesicle release to enable neurotransmitter exocytosis and also interacts with the syntaxin binding protein 1 protein (encoded by STXBP1) [70].
Variants of STX1B provoke fever-associated epilepsies of variable severity including Epilepsy with Myoclonic-Atonic Seizures [71,72]. Intrafamilial clustering due to autosomal dominant mutations with variable expressivity also including EE/DEE has recently been reported [73].
Another gene involved in membrane trafficking is SYNGAP1, which regulates the postsynaptic density at excitatory glutamatergic neurons through the cytosolic protein SYNGAP1 (SYNaptic GTPase Activating Protein). Loss-of-function mutations causing haploinsufficiency lead to intellectual disability and severe epilepsy. This syndrome has indeed only been reported as sporadic, but a single inherited case has been due to parental mosaicism [74].
6. Cell Adhesion
The disruption of intercellular adhesion is especially detrimental for brain function.
Protocadherin 19 is a crucial molecule in this regard. It is a calcium dependent cell-cell adhesion molecule encoded by the PCDH19 gene, highly expressed during brain development, and involved in neuronal migration and in the establishment of synaptic connections. The gene is located on chromosome Xq22.3. Heterozygous mutations of PCDH19 typically cause a particular EE/DEE characterized by a singular mode of inheritance. Only heterozygous females are affected (X-linked dominant pattern). Hemizygous males are asymptomatic but can transmit the pathogenic variant to daughters. Mosaic males can exhibit complete or incomplete phenotype.
The condition results from loss-of-function variants causing cellular interference: in affected females and mosaic males, the presence of both wild-type and mutant cells interfere with one another due to the production of different surface proteins, whilst non-mosaic hemizygous males produce a homogenous population of cells [75,76,77]. Cases resulting from de novo mutations are also reported.
Typical features of PCDH19-related encephalopathy include generalized or focal seizures highly sensitive to fever, intellectual disability, autistic traits. Epilepsy may explode with brief recurrent seizure clusters (generally lasting 1–5 min and repeating multiple times a day) in late infancy lasting through childhood possibly leading to intensive care unit. The phenotype can resemble Dravet syndrome and according to some studies, PCDH19 variants are found in 25% of SCN1A-negative females exhibiting features of Dravet syndrome [75,78].
7. Cell Growth and Proliferation
The vast majority of the aforementioned EE/DEE exhibit a normal brain MRI or unspecific findings. Conversely, mutations affecting genes involved in cell growth and proliferation can disrupt brain morphology.
Mutations in the Aristaless-related homeobox gene (ARX) may promote an encephalopathic phenotype with either normal imaging (X-linked West syndrome, X-linked myoclonic epilepsy with spasticity and intellectual disability, Partington syndrome with mental retardation, ataxia and dystonia, and nonsyndromic forms of mental retardation) or associated to brain malformations (X-linked lissencephaly with abnormal genitalia). ARX-related conditions are inherited in an X-linked recessive manner or can be sporadic. Female carriers are generally asymptomatic or have mild phenotypes. Hence, it is always appropriate to test a seemingly unaffected mother of an affected proband, and skewed X-inactivation can occur [79,80].
The ARX protein helps to regulate the process by which cells differentiate and migrate. ARX-knockout mice are defective in neuroblast proliferation and in GABAergic interneuron migration [79,81].
Similarly, mutations of other genes involved in neurodevelopment can provoke malformative EE/DEE. Lissencephaly is the most common radiological pattern, but there are also peculiar MRI abnormalities that can suggest the involvement of specific genes (Table 2). Most mutations are de novo, yet familial cases are described as autosomal dominant (LIS1, DYNC1H1, tubulin-encoding TUBA1A, TUBB2A, TUBB2B, TUBB3, TUBB [TUBB5], and TUBG1 genes), X-linked dominant (DCX), or autosomal recessive (tubulin-encoding TUBA8).
Tubulinopathies may also have coloboma, heart defects, ichthyosiform dermatosis, mental retardation, and ear anomalies with hearing loss.
In most cases, global developmental delay is associated to epileptic spasms in the infancy, often followed by drug-resistant epilepsy in the long-term [82,83,84].
The gene CDKL5 is another important target of mutations provoking EE/DEE and exerts its functions within the AKT/GSK-3β signaling pathway, which is fundamental in processes of brain development, namely, neuronal precursor proliferation, survival, and maturation [87]. Mutations in CDKL5 are associated with a severe X-linked EE/DEE characterized by severe intellectual disability, generalized developmental delay, early-onset intractable seizures, infantile spasms, and Rett’s syndrome (RTT)-like features. The phenotype shares striking similarities with RTT: hyperventilation, hand stereotypies, and hypotonia affecting young females [88,89]. The majority of RTT cases are heterozygous for missense or nonsense mutations in the gene encoding for methyl-CpG-binding protein 2 (MeCP2) [90]. Several CDKL5 gene mutations, resulting in missense, nonsense, splice, and frameshift mutations or genomic deletions have been described in girls (heterozygous mutations) and a few boys [88]. In females, the phenotypic spectrum of the disease ranges from milder forms, which include the possibility of autonomous walking and drug-responsive seizures, to severe EE/DEE. In males, the phenotype is always severe [91].
Abnormalities in cell growth and proliferation can also lead to multisystem disorders. An outstanding example is Tuberous Sclerosis Complex (TSC), a neurocutaneous syndrome characterized by early onset EE/DEE and extracerebral involvement. Most cases are sporadic, about one third instead result from inherited autosomal dominant mutations. The causative genes are TSC1/TSC2, respectively encoding tuberin and hamartin, both of which physiologically inhibit the oncogenic mTOR pathway. Pathogenic mutations of these genes result in over-activation of the mTOR within all TSC-associated lesions yielding hamartomatous lesions and an increased risk of tumors.
Most children develop West syndrome. A specific ASM (vigabatrin) has been shown to bring a specific efficacy, but drug-resistance often accrues. The cognitive outcome is unfavorable, due to the presence of multiple malformations of cortical development (cortical tubers, subependymal nodules) and tumors (subependymal astrocytomas) that can render TSC patients surgical candidates. Extracerebral typical features are cutaneous (hypomelanotic macules, shagreen patches, angiofibromas), renal (renal cysts, hamartomas, angiomyolipomas), cardiac (rhabodmyoma), and pulmonary (lymphangioleiomyomatosis).
8. Conclusions
The clinical spectrum of the inherited EE/DEE range is very wide. Different mutations can apparently provoke common severe epilepsy syndromes. Next-generation sequencing techniques have revolutionized the landscape of molecular biology allowing a wide-spectrum screening of patients with EE/DEE. Despite this, the clinician’s assessment remains of paramount importance as suspecting a specific diagnosis can prompt appropriate testing and accelerate treatment. A punctual knowledge of the phenotypical spectrum can indeed constitute solid support to ameliorate the prognosis of patients and to accurately counsel family members.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The author declares no conflict of interest.
References
- Berg, A.T.; Berkovic, S.F.; Brodie, M.J.; Buchhalter, J.; Cross, J.H.; Boas, W.V.E.; Engel, J.; French, J.; Glauser, T.A.; Mathern, G.W.; et al. Revised terminology and concepts for organization of seizures and epilepsies: Report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia 2010, 51, 676–685. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshe, S.; et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McTague, A.; Howell, K.B.; Cross, J.H.; Kurian, M.; Scheffer, I. The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol. 2016, 15, 304–316. [Google Scholar] [CrossRef]
- Møller, R.S.; Liebmann, N.; Larsen, L.H.G.; Stiller, M.; Hentschel, J.; Kako, N.; Abdin, D.; Di Donato, N.; Pal, D.K.; Zacher, P.; et al. Parental mosaicism in epilepsies due to alleged de novo variants. Epilepsia 2019, 60, e63–e66. [Google Scholar] [CrossRef]
- Stosser, M.; Lindy, A.; Butler, E.; Retterer, K.; Piccirillo-Stosser, C.M.; Richard, G.; McKnight, D.A. High frequency of mosaic pathogenic variants in genes causing epilepsy-related neurodevelopmental disorders. Genet. Med. 2018, 20, 403–410. [Google Scholar] [CrossRef]
- Symonds, J.; McTague, A. Epilepsy and developmental disorders: Next generation sequencing in the clinic. Eur. J. Paediatr. Neurol. 2020, 24, 15–23. [Google Scholar] [CrossRef]
- Barros, J.; Damásio, J.; Tuna, A.; Alves, I.; Silveira, I.; Pereira-Monteiro, J.; Sequeiros, J.; Alonso, I.; Sousa, A.; Coutinho, P. Cerebellar ataxia, hemiplegic migraine, and related phenotypes due to a CACNA1A missense mutation: 12-year follow-up of a large Portuguese family. JAMA Neurol. 2013, 70, 235–240. [Google Scholar] [CrossRef] [Green Version]
- Bartolini, E.; Campostrini, R.; Kiferle, L.; Pradella, S.; Rosati, E.; Chinthapalli, K.; Palumbo, P. Epilepsy and brain channelopathies from infancy to adulthood. Neurol. Sci. 2019, 41, 749–761. [Google Scholar] [CrossRef]
- Ryan, D.P.; Ptáček, L.J. Episodic Neurological Channelopathies. Neuron 2010, 68, 282–292. [Google Scholar] [CrossRef] [Green Version]
- Catterall, W.A. Structure and function of voltage-gated sodium channels at atomic resolution: Voltage-gated sodium channels at atomic resolution. Exp. Physiol. 2014, 99, 35–51. [Google Scholar] [CrossRef] [Green Version]
- Escayg, A.; Goldin, A.L. Sodium channel SCN1A and epilepsy: Mutations and mechanisms: Sodium Channel SCN1A and Epilepsy. Epilepsia 2010, 51, 1650–1658. [Google Scholar] [CrossRef] [Green Version]
- Cheah, C.S.; Westenbroek, R.E.; Roden, W.H.; Kalume, F.; Oakley, J.C.; Jansen, L.A.; Catterall, W.A. Correlations in timing of sodium channel expression, epilepsy, and sudden death in Dravet syndrome. Channels 2013, 7, 468–472. [Google Scholar] [CrossRef] [Green Version]
- Claes, L.; Del Favero, J.; Ceulemans, B.; Lagae, L.; Van Broeckhoven, C.; De Jonghe, P. De Novo Mutations in the Sodium-Channel Gene SCN1A Cause Severe Myoclonic Epilepsy of Infancy. Am. J. Hum. Genet. 2001, 68, 1327–1332. [Google Scholar] [CrossRef] [Green Version]
- Marini, C.; Scheffer, I.E.; Nabbout, R.; Suls, A.; De Jonghe, P.; Zara, F.; Guerrini, R. The genetics of Dravet syndrome: Genetics of Dravet Syndrome. Epilepsia 2011, 52, 24–29. [Google Scholar] [CrossRef]
- Depienne, C.; Trouillard, O.; Gourfinkel-An, I.; Saint-Martin, C.; Bouteiller, D.; Graber, D.; Barthez-Carpentier, M.-A.; Gautier, A.; Villeneuve, N.; Dravet, C.; et al. Mechanisms for variable expressivity of inherited SCN1A mutations causing Dravet syndrome. J. Med. Genet. 2010, 47, 404–410. [Google Scholar] [CrossRef] [Green Version]
- Howell, K.B.; McMahon, J.M.; Carvill, G.L.; Tambunan, D.; Mackay, M.T.; Rodriguez-Casero, V.; Webster, R.; Clark, D.; Freeman, J.L.; Calvert, S.; et al. SCN2A encephalopathy: A major cause of epilepsy of infancy with migrating focal seizures. Neurology 2015, 85, 958–966. [Google Scholar] [CrossRef] [Green Version]
- McIntosh, A.; McMahon, J.; Dibbens, L.M.; Iona, X.; Mulley, J.C.; Scheffer, I.; Berkovic, S.F. Effects of vaccination on onset and outcome of Dravet syndrome: A retrospective study. Lancet Neurol. 2010, 9, 592–598. [Google Scholar] [CrossRef]
- Reyes, I.S.; Hsieh, D.T.; Laux, L.C.; Wilfong, A.A. Alleged Cases of Vaccine Encephalopathy Rediagnosed Years Later as Dravet Syndrome: TABLE 1. Pediatrics 2011, 128, e699–e702. [Google Scholar] [CrossRef]
- Dravet, C. The core Dravet syndrome phenotype: Core Dravet Syndrome. Epilepsia 2011, 52, 3–9. [Google Scholar] [CrossRef]
- Losito, E.; Kuchenbuch, M.; Chemaly, N.; Laschet, J.; Chiron, C.; Kaminska, A.; Nabbout, R. Age-related “Sleep/nocturnal” tonic and tonic clonic seizure clusters are underdiagnosed in patients with Dravet Syndrome. Epilepsy Behav. 2017, 74, 33–40. [Google Scholar] [CrossRef]
- Scheffer, I.E. Diagnosis and long-term course of Dravet syndrome. Eur. J. Paediatr. Neurol. 2012, 16, S5–S8. [Google Scholar] [CrossRef]
- Verbeek, N.E.; Wassenaar, M.; Van Campen, J.S.; Sonsma, A.C.M.; Gunning, B.; Knoers, N.V.A.M.; Lindhout, D.; Jansen, F.E.; Leijten, F.S.S.; Brilstra, E.H.; et al. Seizure precipitants in Dravet syndrome: What events and activities are specifically provocative compared with other epilepsies? Epilepsy Behav. 2015, 47, 39–44. [Google Scholar] [CrossRef]
- Guerrini, R. Dravet syndrome: The main issues. Eur. J. Paediatr. Neurol. 2012, 16, S1–S4. [Google Scholar] [CrossRef]
- Akiyama, M.; Kobayashi, K.; Yoshinaga, H.; Ohtsuka, Y. A long-term follow-up study of Dravet syndrome up to adulthood. Epilepsia 2009, 51, 1043–1052. [Google Scholar] [CrossRef] [PubMed]
- Genton, P.; Velizarova, R.; Dravet, C. Dravet syndrome: The long-term outcome. Epilepsia 2011, 52, 44–49. [Google Scholar] [CrossRef]
- Fasano, A.; Borlot, F.; Lang, A.E.; Andrade, D.M. Antecollis and levodopa-responsive parkinsonism are late features of Dravet syndrome. Neurology 2014, 82, 2250–2251. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Deprez, L.; Maljevic, S.; Pitsch, J.; Claes, L.; Hristova, D.; Jordanova, A.; Ala-Mello, S.; Bellan-Koch, A.; Blazevic, D.; et al. Molecular correlates of age-dependent seizures in an inherited neonatal-infantile epilepsy. Brain 2010, 133, 1403–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, K.; Kato, M.; Osaka, H.; Yamashita, S.; Nakagawa, E.; Haginoya, K.; Tohyama, J.; Okuda, M.; Wada, T.; Shimakawa, S.; et al. Clinical spectrum of SCN2A mutations expanding to Ohtahara syndrome. Neurology 2013, 81, 992–998. [Google Scholar] [CrossRef] [PubMed]
- Wolff, M.; Johannesen, K.M.; Hedrich, U.; Masnada, S.; Rubboli, G.; Gardella, E.; Lesca, G.; Ville, D.; Milh, M.; Villard, L.; et al. Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain 2017, 140, 1316–1336. [Google Scholar] [CrossRef]
- Gardella, E.; Marini, C.; Trivisano, M.; Fitzgerald, M.P.; Alber, M.; Howell, K.B.; Darra, F.; Siliquini, S.; Bölsterli, B.K.; Masnada, S.; et al. The phenotype of SCN8A developmental and epileptic encephalopathy. Neurology 2018, 91, e1112–e1124. [Google Scholar] [CrossRef] [Green Version]
- Johannesen, K.M.; Gardella, E.; Encinas, A.C.; Lehesjoki, A.; Linnankivi, T.; Petersen, M.B.; Lund, I.C.B.; Blichfeldt, S.; Miranda, M.J.; Pal, D.K.; et al. The spectrum of intermediate SCN8A-related epilepsy. Epilepsia 2019, 60, 830–844. [Google Scholar] [CrossRef] [Green Version]
- Larsen, J.; Carvill, G.L.; Gardella, E.; Kluger, G.; Schmiedel, G.; Barisic, N.; Depienne, C.; Brilstra, E.; Mang, Y.; Nielsen, J.E.K.; et al. The phenotypic spectrum of SCN8A encephalopathy. Neurology 2015, 84, 480–489. [Google Scholar] [CrossRef] [Green Version]
- Denis, J.; Villeneuve, N.; Cacciagli, P.; Mignon-Ravix, C.; Lacoste, C.; Lefranc, J.; Napuri, S.; Damaj, L.; Villega, F.; Pedespan, J.; et al. Clinical study of 19 patients with SCN8A-related epilepsy: Two modes of onset regarding EEG and seizures. Epilepsia 2019, 60, 845–856. [Google Scholar] [CrossRef]
- Trivisano, M.; Pavia, G.C.; Ferretti, A.; Fusco, L.; Vigevano, F.; Specchio, N. Generalized tonic seizures with autonomic signs are the hallmark of SCN8A developmental and epileptic encephalopathy. Epilepsy Behav. 2019, 96, 219–223. [Google Scholar] [CrossRef]
- Kuang, Q.; Purhonen, P.; Hebert, H. Structure of potassium channels. Cell. Mol. Life Sci. 2015, 72, 3677–3693. [Google Scholar] [CrossRef] [Green Version]
- Biervert, C.; Schroeder, B.C.; Kubisch, C.; Berkovic, S.F.; Propping, P.; Jentsch, T.J.; Steinlein, O.K. A Potassium Channel Mutation in Neonatal Human Epilepsy. Science 1998, 279, 403–406. [Google Scholar] [CrossRef] [Green Version]
- Charlier, C.; Singh, N.A.; Ryan, S.G.; Lewis, T.B.; Reus, B.E.; Leach, R.J.; Leppert, M. A pore mutation in a novel KQT-like potassium channel gene in an idiopathic epilepsy family. Nat. Genet. 1998, 18, 53–55. [Google Scholar] [CrossRef]
- Dedek, K.; Fusco, L.; Teloy, N.; Steinlein, O.K. Neonatal convulsions and epileptic encephalopathy in an Italian family with a missense mutation in the fifth transmembrane region of KCNQ2. Epilepsy Res. 2003, 54, 21–27. [Google Scholar] [CrossRef]
- Miceli, F.; Striano, P.; Soldovieri, M.V.; Fontana, A.; Nardello, R.; Robbiano, A.; Bellini, G.; Elia, M.; Zara, F.; Taglialatela, M.; et al. A novelKCNQ3mutation in familial epilepsy with focal seizures and intellectual disability. Epilepsia 2014, 56, e15–e20. [Google Scholar] [CrossRef]
- Weckhuysen, S.; Mandelstam, S.; Suls, A.; Audenaert, D.; Deconinck, T.; Claes, L.R.; Deprez, L.; Smets, K.; Hristova, D.; Yordanova, I.; et al. KCNQ2 encephalopathy: Emerging phenotype of a neonatal epileptic encephalopathy. Ann. Neurol. 2012, 71, 15–25. [Google Scholar] [CrossRef]
- Millichap, J.J.; Park, K.L.; Tsuchida, T.; Ben-Zeev, B.; Carmant, L.; Flamini, R.; Joshi, N.; Levisohn, P.M.; Marsh, E.; Nangia, S.; et al. KCNQ2 encephalopathy: Features, mutational hot spots, and ezogabine treatment of 11 patients. Neurol. Genet. 2016, 2, e96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bar, C.; Barcia, G.; Jennesson, M.; Le Guyader, G.; Schneider, A.; Mignot, C.; Lesca, G.; Breuillard, D.; Montomoli, M.; Keren, B.; et al. Expanding the genetic and phenotypic relevance of KCNB1 variants in developmental and epileptic encephalopathies: 27 new patients and overview of the literature. Hum. Mutat. 2020, 41, 69–80. [Google Scholar] [CrossRef]
- McTague, A.; Appleton, R.; Avula, S.; Cross, H.; King, M.D.; Jacques, T.S.; Bhate, S.; Cronin, A.; Curran, A.; Desurkar, A.; et al. Migrating partial seizures of infancy: Expansion of the electroclinical, radiological and pathological disease spectrum. Brain 2013, 136, 1578–1591. [Google Scholar] [CrossRef] [PubMed]
- Milligan, C.J.; Li, M.; Gazina, E.V.; Heron, S.E.; Nair, U.; Trager, C.; Reid, C.A.; Venkat, A.; Younkin, D.P.; Dlugos, D.J.; et al. KCNT1 gain of function in 2 epilepsy phenotypes is reversed by quinidine: KCNT1 and Human Epilepsy. Ann. Neurol. 2014, 75, 581–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simms, B.A.; Zamponi, G.W. Neuronal Voltage-Gated Calcium Channels: Structure, Function, and Dysfunction. Neuron 2014, 82, 24–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelini, C.; Van Gils, J.; Bigourdan, A.; Jouk, P.-S.; Lacombe, D.; Menegon, P.; Moutton, S.; Riant, F.; Sole, G.; Tournier-Lasserve, E.; et al. Major intra-familial phenotypic heterogeneity and incomplete penetrance due to a CACNA1A pathogenic variant. Eur. J. Med. Genet. 2019, 62, 103530. [Google Scholar] [CrossRef] [PubMed]
- Di Stefano, V.; Rispoli, M.G.; Pellegrino, N.; Graziosi, A.; Rotondo, E.; Napoli, C.; Pietrobon, D.; Brighina, F.; Parisi, P. Diagnostic and therapeutic aspects of hemiplegic migraine. J. Neurol Neurosurg. Psychiatry 2020, 91, 764–771. [Google Scholar] [CrossRef]
- Indelicato, E.; Boesch, S. From Genotype to Phenotype: Expanding the Clinical Spectrum of CACNA1A Variants in the Era of Next Generation Sequencing. Front. Neurol. 2021, 12, 639994. [Google Scholar] [CrossRef]
- Le Roux, M.; Barth, M.; Gueden, S.; de Cepoy, P.D.; Aeby, A.; Vilain, C.; Hirsch, E.; de Saint Martin, A.; des Portes, V.; Lesca, G.; et al. CACNA1A-associated epilepsy: Electroclinical findings and treatment response on seizures in 18 patients. Eur. J. Paediatr. Neurol. 2021, 33, 75–85. [Google Scholar] [CrossRef]
- Reinson, K.; Õiglane-Shlik, E.; Talvik, I.; Vaher, U.; Õunapuu, A.; Ennok, M.; Teek, R.; Pajusalu, S.; Murumets, Ü.; Tomberg, T.; et al. Biallelic CACNA1A mutations cause early onset epileptic encephalopathy with progressive cerebral, cerebellar, and optic nerve atrophy. Am. J. Med. Genet. A 2016, 170, 2173–2176. [Google Scholar] [CrossRef]
- Johannesen, K.; Marini, C.; Pfeffer, S.; Møller, R.S.; Dorn, T.; Niturad, C.E.; Gardella, E.; Weber, Y.; Søndergård, M.; Hjalgrim, H.; et al. Phenotypic spectrum of GABRA1: From generalized epilepsies to severe epileptic encephalopathies. Neurology 2016, 87, 1140–1151. [Google Scholar] [CrossRef]
- Carvill, G.L.; Regan, B.; Yendle, S.C.; O’Roak, B.; Lozovaya, N.; Bruneau, N.; Burnashev, N.; Khan, A.; Cook, J.; Geraghty, E.; et al. GRIN2A mutations cause epilepsy-aphasia spectrum disorders. Nat. Genet. 2013, 45, 1073–1076. [Google Scholar] [CrossRef] [Green Version]
- Lemke, J.R.; Hendrickx, R.; Geider, K.; Laube, B.; Schwake, M.; Harvey, R.J.; James, V.M.; Pepler, A.; Steiner, I.; Hörtnagel, K.; et al. GRIN2B mutations in west syndrome and intellectual disability with focal epilepsy: GRIN2B Mutations in Epilepsy. Ann. Neurol. 2014, 75, 147–154. [Google Scholar] [CrossRef] [Green Version]
- Almannai, M.; Al Mahmoud, R.A.; Mekki, M.; El-Hattab, A.W. Metabolic Seizures. Front. Neurol. 2021, 12, 985. [Google Scholar] [CrossRef]
- Van Konijnenburg, E.M.H.; Wortmann, S.B.; Koelewijn, M.J.; Tseng, L.A.; Houben, R.; Stöckler-Ipsiroglu, S.; Ferreira, C.R.; van Karnebeek, C.D. Metabolic Evaluation of Epilepsy: A Diagnostic Algorithm with Focus on Treatable Conditions. Front. Neurol. 2018, 9, 1016. [Google Scholar]
- Sharma, S.; Prasad, A.N. Inborn Errors of Metabolism and Epilepsy: Current Understanding, Diagnosis, and Treatment Approaches. Int. J. Mol. Sci. 2017, 18, E1384. [Google Scholar] [CrossRef] [Green Version]
- Van Karnebeek, C.D.M.; Jaggumantri, S. Current Treatment and Management of Pyridoxine-Dependent Epilepsy. Curr. Treat. Options Neurol. 2015, 17, 335. [Google Scholar] [CrossRef]
- Khayat, M.; Korman, S.H.; Frankel, P.; Weintraub, Z.; Hershckowitz, S.; Sheffer, V.F.; Ben Elisha, M.; Wevers, R.A.; Falik-Zaccai, T.C. PNPO deficiency: An under diagnosed inborn error of pyridoxine metabolism. Mol. Genet. Metab. 2008, 94, 431–434. [Google Scholar] [CrossRef]
- De Vivo, D.C.; Trifiletti, R.R.; Jacobson, R.I.; Ronen, G.M.; Behmand, R.A.; Harik, S.I. Defective glucose transport across the blood-brain barrier as a cause of persistent hypoglycorrhachia, seizures, and developmental delay. N. Engl. J. Med. 1991, 325, 703–709. [Google Scholar] [CrossRef]
- Ardicli, D.; Haliloglu, G.; Gocmen, R.; Gunbey, C.; Topcu, M. Unraveling neuronal ceroid lipofuscinosis type 2 (CLN2) disease: A tertiary center experience for determinants of diagnostic delay. Eur. J. Paediatr. Neurol. 2021, 33, 94–98. [Google Scholar] [CrossRef]
- Beltrán, L.; Valenzuela, G.R.; Loos, M.; Vargas, R.; Lizama, R.; Spinsanti, P.; Caraballo, R. Late-onset childhood neuronal ceroid lipofuscinosis: Early clinical and electroencephalographic markers. Epilepsy Res. 2018, 144, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Hu, C.; El Achkar, C.M.; Black, L.E.; Douville, J.; Larson, A.; Pendergast, M.K.; Goldkind, S.F.; Lee, E.A.; Kuniholm, A.; et al. Patient-Customized Oligonucleotide Therapy for a Rare Genetic Disease. N. Engl. J. Med. 2019, 381, 1644–1652. [Google Scholar] [CrossRef] [PubMed]
- Hikmat, O.; Naess, K.; Engvall, M.; Klingenberg, C.; Rasmussen, M.; Tallaksen, C.M.; Brodtkorb, E.; Ostergaard, E.; de Coo, I.F.M.; Pias-Peleteiro, L.; et al. Simplifying the clinical classification of polymerase gamma (POLG) disease based on age of onset; studies using a cohort of 155 cases. J. Inherit. Metab. Dis. 2020, 43, 726–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, S. Mitochondrial diseases and status epilepticus. Epilepsia 2018, 59, 70–77. [Google Scholar] [CrossRef]
- Ciara, E.; Rokicki, D.; Halat, P.; Karkucińska-Więckowska, A.; Piekutowska-Abramczuk, D.; Mayr, J.; Trubicka, J.; Szymańska-Dębińska, T.; Pronicki, M.; Pajdowska, M.; et al. Difficulties in recognition of pyruvate dehydrogenase complex deficiency on the basis of clinical and biochemical features. The role of next-generation sequencing. Mol. Genet. Metab. Rep. 2016, 7, 70–76. [Google Scholar] [CrossRef]
- Wesół-Kucharska, D.; Rokicki, D.; Jezela-Stanek, A. Epilepsy in Mitochondrial Diseases—Current State of Knowledge on Aetiology and Treatment. Children 2021, 8, 532. [Google Scholar] [CrossRef]
- Chang, X.; Wu, Y.; Zhou, J.; Meng, H.; Zhang, W.; Guo, J. A meta-analysis and systematic review of Leigh syndrome: Clinical manifestations, respiratory chain enzyme complex deficiency, and gene mutations. Medicine 2020, 99, e18634. [Google Scholar] [CrossRef]
- Lamperti, C.; Zeviani, M. Myoclonus epilepsy in mitochondrial disorders. Epileptic Disord. 2016, 18, 94–102. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Adesina, A.M.; Jones, J.; Scaglia, F. MELAS syndrome: Clinical manifestations, pathogenesis, and treatment options. Mol. Genet. Metab. 2015, 116, 4–12. [Google Scholar] [CrossRef]
- Smirnova, T.; Miniou, P.; Viegas-Péquignot, E.; Mallet, J. Assignment of the Human Syntaxin 1B Gene (STX) to Chromosome 16p11.2 by Fluorescence in Situ Hybridization. Genomics 1996, 36, 551–553. [Google Scholar] [CrossRef]
- Schubert, J.; EuroEPINOMICS RES Consortium; Siekierska, A.; Langlois, M.; May, P.; Huneau, C.; Becker, F.; Muhle, H.; Suls, A.; Lemke, J.R.; et al. Mutations in STX1B, encoding a presynaptic protein, cause fever-associated epilepsy syndromes. Nat. Genet. 2014, 46, 1327–1332. [Google Scholar] [CrossRef]
- Vlaskamp, D.R.; Rump, P.; Callenbach, P.M.; Vos, Y.J.; Sikkema-Raddatz, B.; van Ravenswaaij-Arts, C.M.; Brouwer, O.F. Haploinsufficiency of the STX1B gene is associated with myoclonic astatic epilepsy. Eur. J. Paediatr. Neurol. 2016, 20, 489–492. [Google Scholar] [CrossRef]
- Wolking, S.; May, P.; Mei, D.; Møller, R.S.; Balestrini, S.; Helbig, K.L.; Altuzarra, C.D.; Chatron, N.; Kaiwar, C.; Stöhr, K.; et al. Clinical spectrum of STX1B-related epileptic disorders. Neurology 2019, 92, e1238–e1249. [Google Scholar] [CrossRef] [Green Version]
- Myers, C.T.; Hollingsworth, G.; Muir, A.M.; Schneider, A.L.; Thuesmunn, Z.; Knupp, A.; King, C.; Lacroix, A.; Mehaffey, M.G.; Berkovic, S.F.; et al. Parental Mosaicism in “De Novo” Epileptic Encephalopathies. N. Engl. J. Med. 2018, 378, 1646–1648. [Google Scholar] [CrossRef]
- Depienne, C.; LeGuern, E. PCDH19-related infantile epileptic encephalopathy: An unusual X-linked inheritance disorder. Hum. Mutat. 2012, 33, 627–634. [Google Scholar] [CrossRef] [Green Version]
- Gecz, J.; Thomas, P.Q. Disentangling the paradox of the PCDH19 clustering epilepsy, a disorder of cellular mosaics. Curr. Opin. Genet. Dev. 2020, 65, 169–175. [Google Scholar] [CrossRef]
- Pederick, D.T.; Richards, K.L.; Piltz, S.G.; Kumar, R.; Mincheva-Tasheva, S.; Mandelstam, S.A.; Dale, R.C.; Scheffer, I.E.; Gecz, J.; Petrou, S.; et al. Abnormal Cell Sorting Underlies the Unique X-Linked Inheritance of PCDH19 Epilepsy. Neuron 2018, 97, 59–66.e5. [Google Scholar] [CrossRef] [Green Version]
- Depienne, C.; Bouteiller, D.; Keren, B.; Cheuret, E.; Poirier, K.; Trouillard, O.; Benyahia, B.; Quelin, C.; Carpentier, W.; Julia, S.; et al. Sporadic Infantile Epileptic Encephalopathy Caused by Mutations in PCDH19 Resembles Dravet Syndrome but Mainly Affects Females. PLoS Genet. 2009, 5, e1000381. [Google Scholar] [CrossRef]
- Friocourt, G.; Parnavelas, J. Mutations in ARX result in several defects involving GABAergic neurons. Front. Cell. Neurosci. 2010, 4, 4. [Google Scholar] [CrossRef] [Green Version]
- Poirier, K.; Abriol, J.; Souville, I.; Laroche-Raynaud, C.; Beldjord, C.; Gilbert, B.; Chelly, J.; Bienvenu, T. Maternal mosaicism for mutations in the ARX gene in a family with X linked mental retardation. Qual. Life Res. 2005, 118, 45–48. [Google Scholar] [CrossRef]
- Sherr, E.H. The ARX story (epilepsy, mental retardation, autism, and cerebral malformations): One gene leads to many phenotypes. Curr. Opin. Pediatr. 2003, 15, 567–571. [Google Scholar] [CrossRef]
- Bahi-Buisson, N.; Cavallin, M. Tubulinopathies Overview. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Mirzaa, G., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Jauhari, P.; Farmania, R.; Chakrabarty, B.; Kumar, A.; Gulati, S. Electrographic pattern recognition: A simple tool to predict clinical outcome in children with lissencephaly. Seizure 2020, 83, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Kolbjer, S.; Martin, D.A.; Pettersson, M.; Dahlin, M.; Anderlid, B.-M. Lissencephaly in an epilepsy cohort: Molecular, radiological and clinical aspects. Eur. J. Paediatr. Neurol. 2021, 30, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Barkovich, A.J.; Dobyns, W.B.; Guerrini, R. Malformations of Cortical Development and Epilepsy. Cold Spring Harb. Perspect. Med. 2015, 5, a022392. [Google Scholar] [CrossRef] [PubMed]
- Brock, S.; Cools, F.; Jansen, A.C. Neuropathology of genetically defined malformations of cortical development-A systematic literature review. Neuropathol. Appl. Neurobiol. 2021, 47, 585–602. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, C.; Trazzi, S.; Torricella, R.; Viggiano, R.; De Franceschi, M.; Amendola, E.; Gross, C.; Calza, L.; Bartesaghi, R.; Ciani, E. Loss of CDKL5 impairs survival and dendritic growth of newborn neurons by altering AKT/GSK-3β signaling. Neurobiol. Dis. 2014, 70, 53–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahi-Buisson, N.; Nectoux, J.; Rosas-Vargas, H.; Milh, M.; Boddaert, N.; Girard, B.; Cances, C.; Ville, D.; Afenjar, A.; Rio, M.; et al. Key clinical features to identify girls with CDKL5 mutations. Brain 2008, 131, 2647–2661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakimiec, M.; Paprocka, J.; Śmigiel, R. CDKL5 Deficiency Disorder—A Complex Epileptic Encephalopathy. Brain Sci. 2020, 10, 107. [Google Scholar] [CrossRef] [Green Version]
- Cheadle, J.P.; Gill, H.; Fleming, N.; Maynard, J.; Kerr, A.; Leonard, H.; Krawczak, M.; Cooper, D.N.; Lynch, S.; Thomas, N.; et al. Long-read sequence analysis of the MECP2 gene in Rett syndrome patients: Correlation of disease severity with mutation type and location. Hum. Mol. Genet. 2000, 9, 1119–1129. [Google Scholar] [CrossRef] [Green Version]
- Guerrini, R.; Parrini, E. Epilepsy in Rett syndrome, and CDKL5- and FOXG1-gene-related encephalopathies. Epilepsia 2012, 53, 2067–2078. [Google Scholar] [CrossRef]
- Caban, C.; Khan, N.; Hasbani, D.M.; Crino, P.B. Genetics of tuberous sclerosis complex: Implications for clinical practice. Appl. Clin. Genet. 2016, 10, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Franz, D.N.; Lawson, J.A.; Yapici, Z.; Brandt, C.; Kohrman, M.H.; Wong, M.; Milh, M.; Wiemer-Kruel, A.; Voi, M.; Coello, N.; et al. Everolimus dosing recommendations for tuberous sclerosis complex-associated refractory seizures. Epilepsia 2018, 59, 1188–1197. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Flow-chart depicting the literature review process.


{kind=link}
Table 1.
Classical epilepsy syndromes corresponding to EE/DEE (modified from https://www.epilepsydiagnosis.org/) (accessed on 30 October 2021).
Table 1.
Classical epilepsy syndromes corresponding to EE/DEE (modified from https://www.epilepsydiagnosis.org/) (accessed on 30 October 2021).
| Clinical Overview | Typical Age at Onset | |
|---|---|---|
| Early Myoclonic Encephalopathy | Myoclonic seizures are frequent, distinguishing this syndrome from Ohtahara syndrome | 1–3 months |
| Ohtahara syndrome | Tonic seizures predominate, myoclonic seizures are uncommon | 1–3 months |
| West syndrome | Epileptic spasms, hypsarrhythmia and global developmental delay | 3–12 months |
| Dravet syndrome | Normal child with prolonged, febrile and afebrile, focal and tonic-clonic seizures at onset | Around 6 months of age |
| Epilepsy Of Infancy With Migrating Focal Seizures | Focal seizures arise independently in both hemispheres and can migrate from one cortical region to another randomly but consecutively in the same seizure. | First 6 months |
| Epilepsy With Myoclonic-Atonic Seizures | Myoclonic-atonic seizures in an otherwise normal child who may have a history of febrile and/or afebrile seizures. | 6 months–6 years |
| Epileptic Encephalopathy With Continuous Spike-And-Wave During Sleep | Focal seizures, atypical absences, negative myoclonus/atonic seizures and neurocognitive regression | 2–12 years |
| Landau-Kleffner syndrome | Subacute onset of acquired aphasia. Seizures may not occur in all cases and when present are infrequent and self-limiting | 2–8 years |
Table 2.
Genes commonly associated to malformation of cortical development observed in families [85,86].
| Affected Gene | Main Imaging Phenotypes |
|---|---|
| ARX | X-linked lissencephaly with abnormal genitalia |
| DCX | Anteriorly predominant lissencephaly (males) Subcortical band heterotopia (females) |
| LIS1 | Posteriorly predominant lissencephaly |
| TUBA1A | Posteriorly predominant lissencephaly ± absent corpus callous, and cerebellar hypoplasia; polymicrogyria like pattern |
| TUBB2B | Posteriorly predominant lissencephaly ± cerebellar hypoplasia; polymicrogyria like |
| TUBB3 | Absent corpus callosum, polymicrogyria like pattern |
| TUBB [TUBB5] | Absent corpus callosum, polymicrogyria like pattern |
| TUBG1 | Posteriorly predominant lissencephaly |
| DYNC1H1 | Posteriorly predominant lissencephaly |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bartolini, E. Inherited Developmental and Epileptic Encephalopathies. Neurol. Int. 2021, 13, 555-568. https://doi.org/10.3390/neurolint13040055
AMA Style
Bartolini E. Inherited Developmental and Epileptic Encephalopathies. Neurology International. 2021; 13(4):555-568. https://doi.org/10.3390/neurolint13040055
Chicago/Turabian StyleBartolini, Emanuele. 2021. "Inherited Developmental and Epileptic Encephalopathies" Neurology International 13, no. 4: 555-568. https://doi.org/10.3390/neurolint13040055