
Replacement of Chalcone-Ethers with Chalcone-Thioethers as Potent and Highly Selective Monoamine Oxidase-B Inhibitors and Their Protein-Ligand Interactions

 , , , ,
, , , , 


Abstract
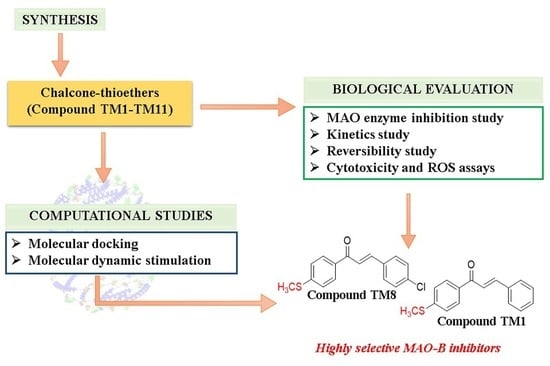
:
1. Introduction
2. Results
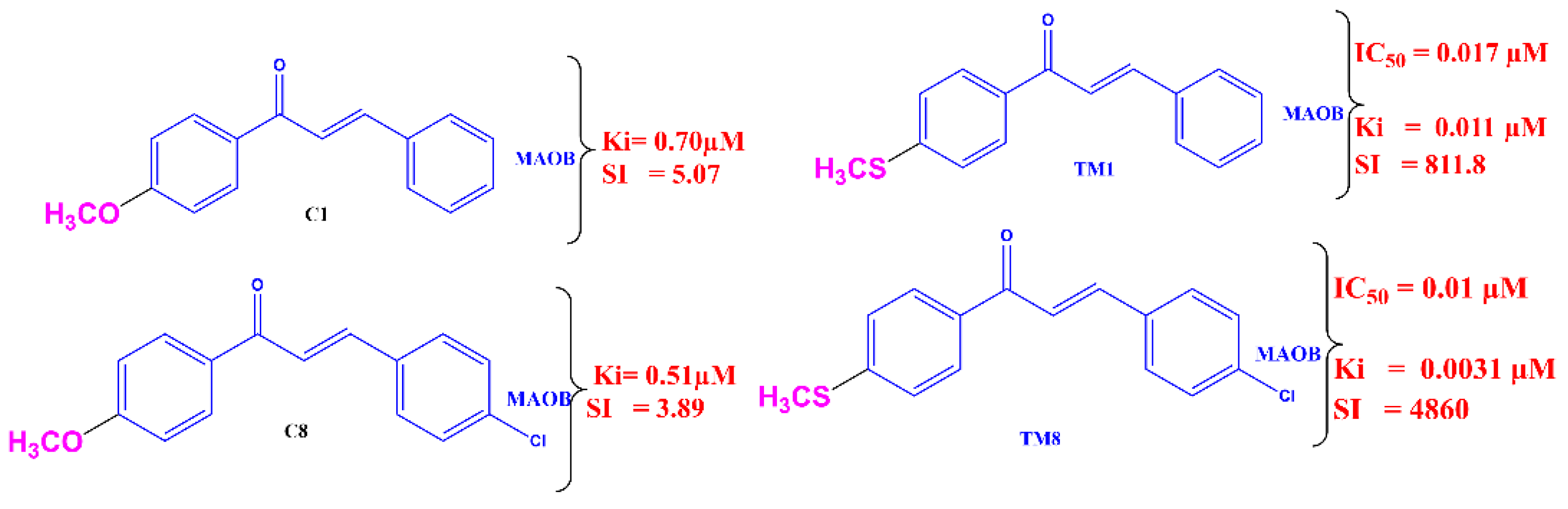
2.1. MAO Inhibition
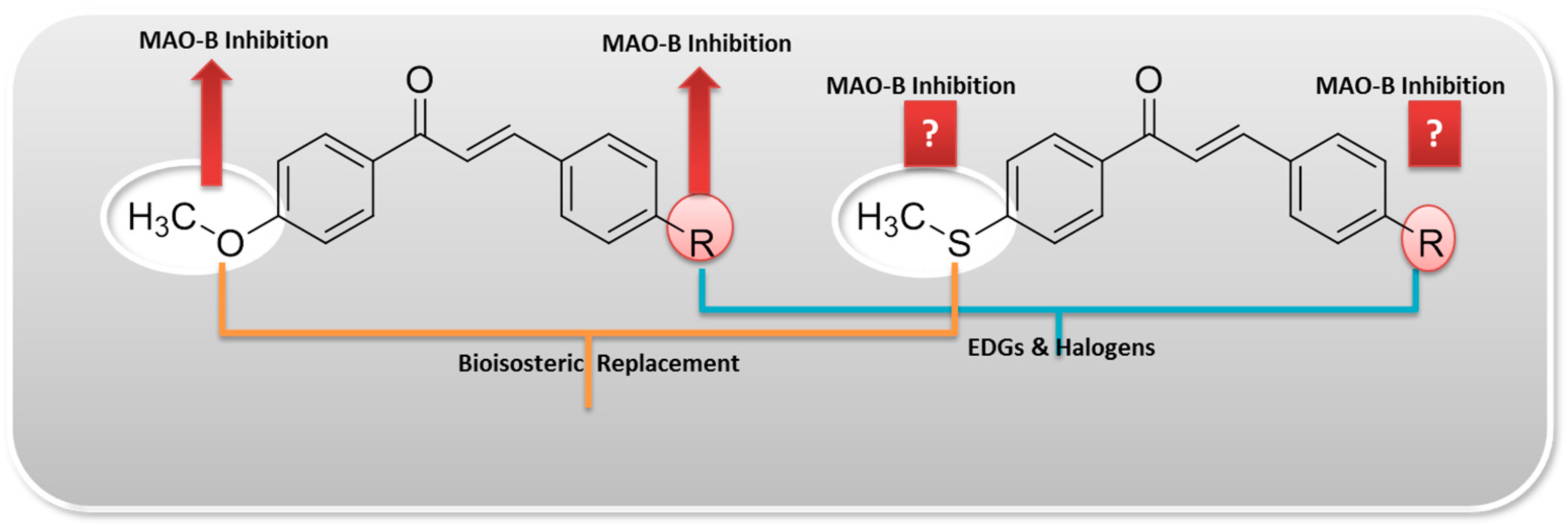
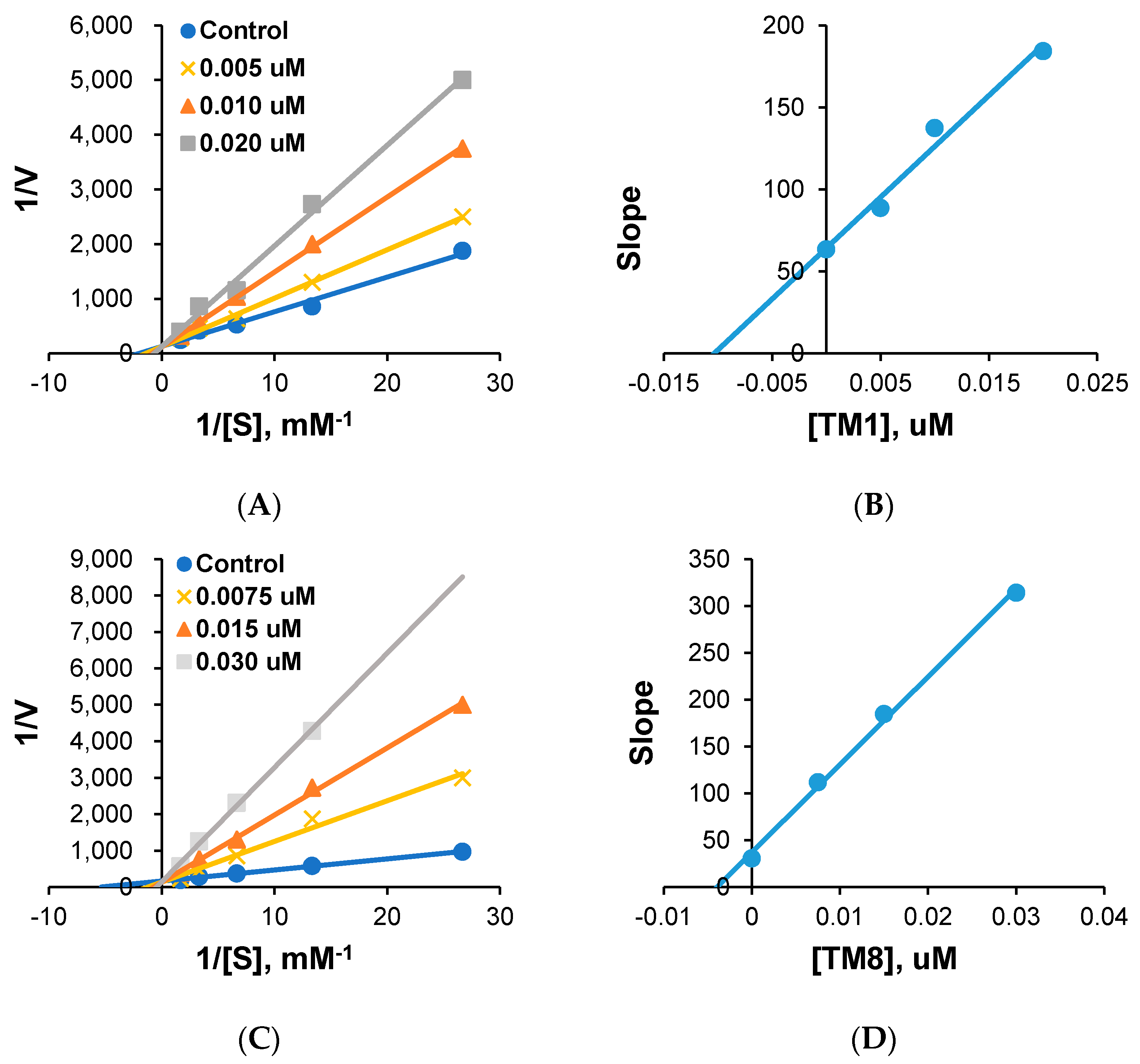
2.2. Kinetics
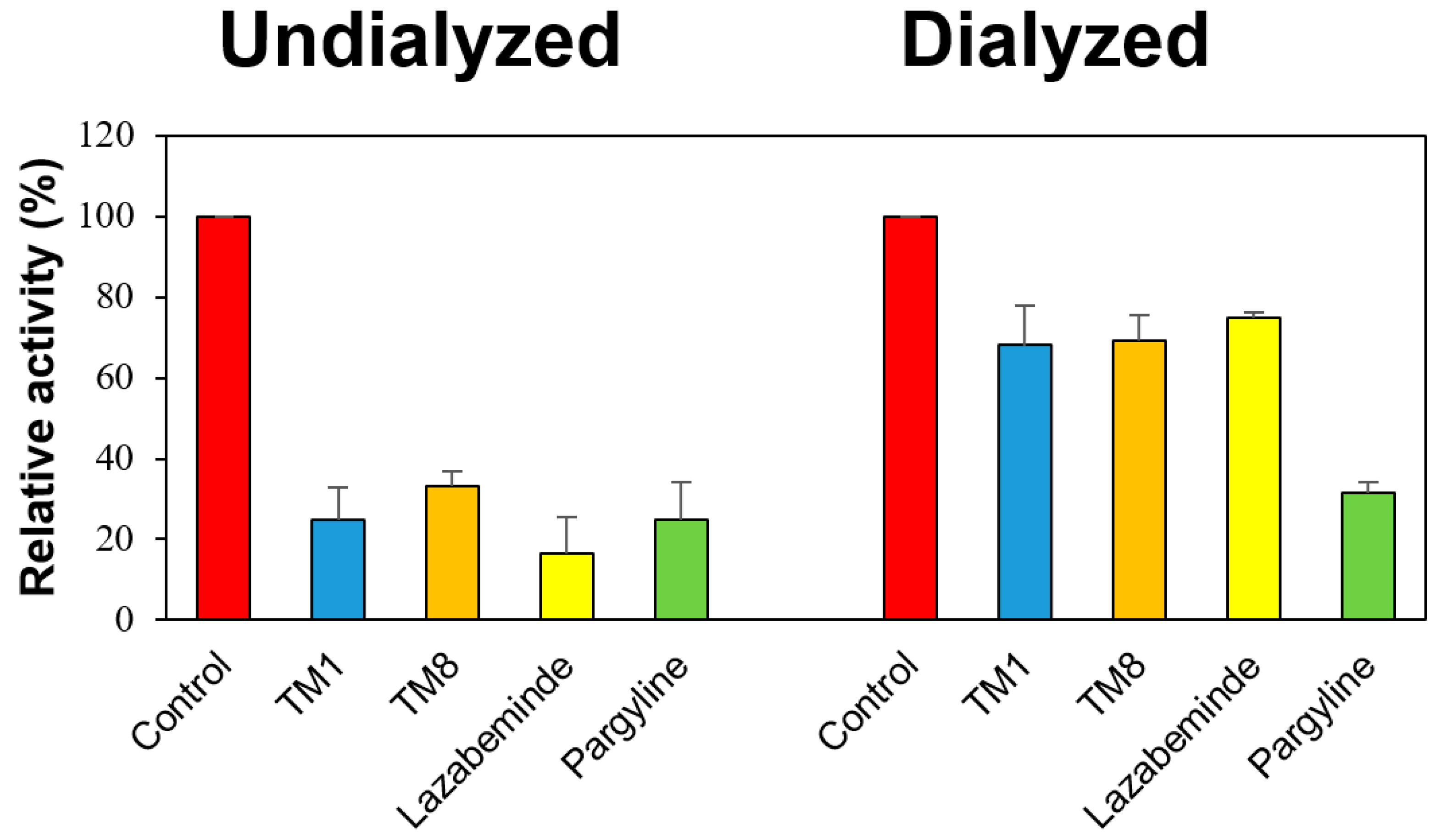
2.3. Reversibility
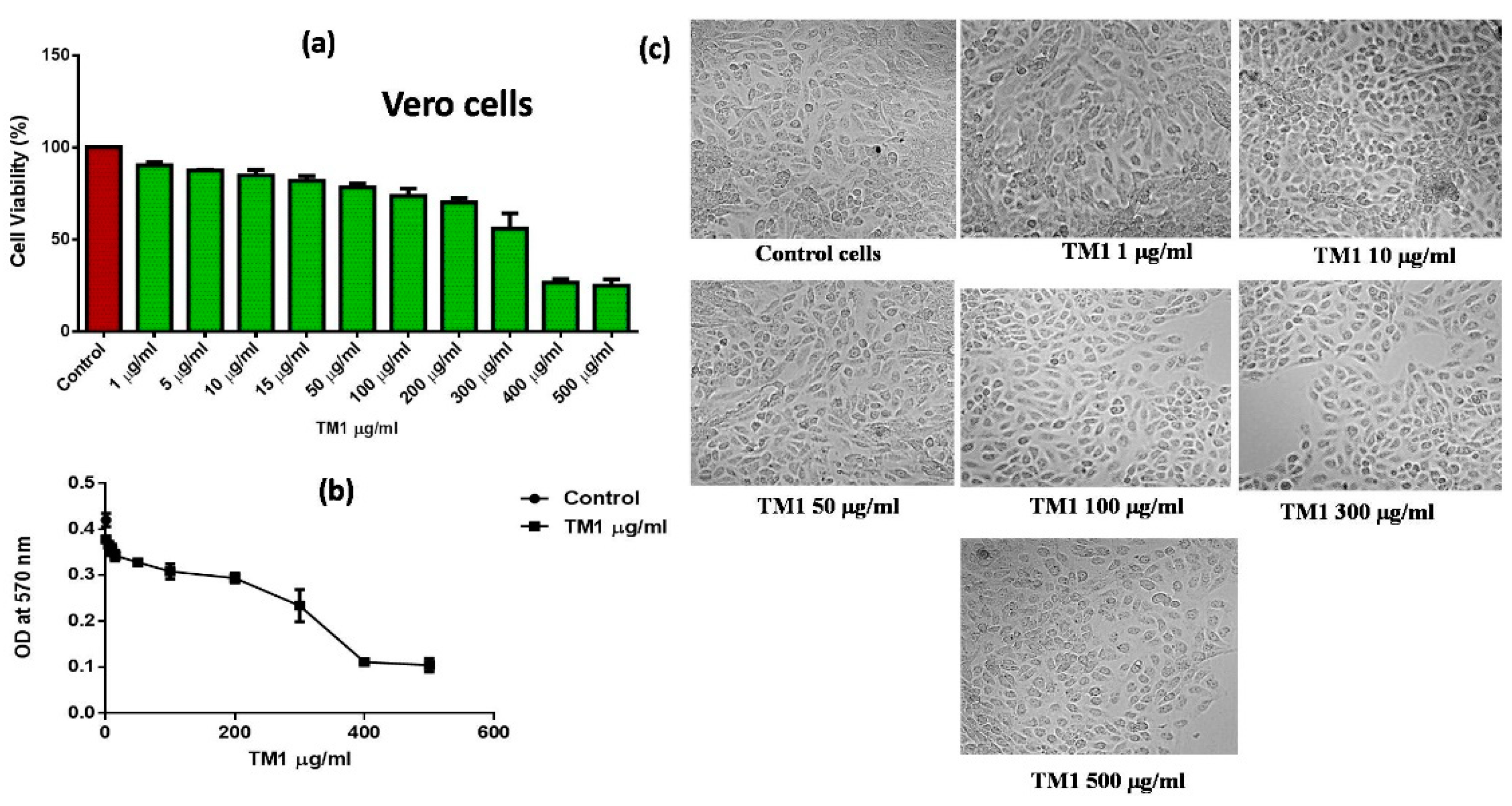
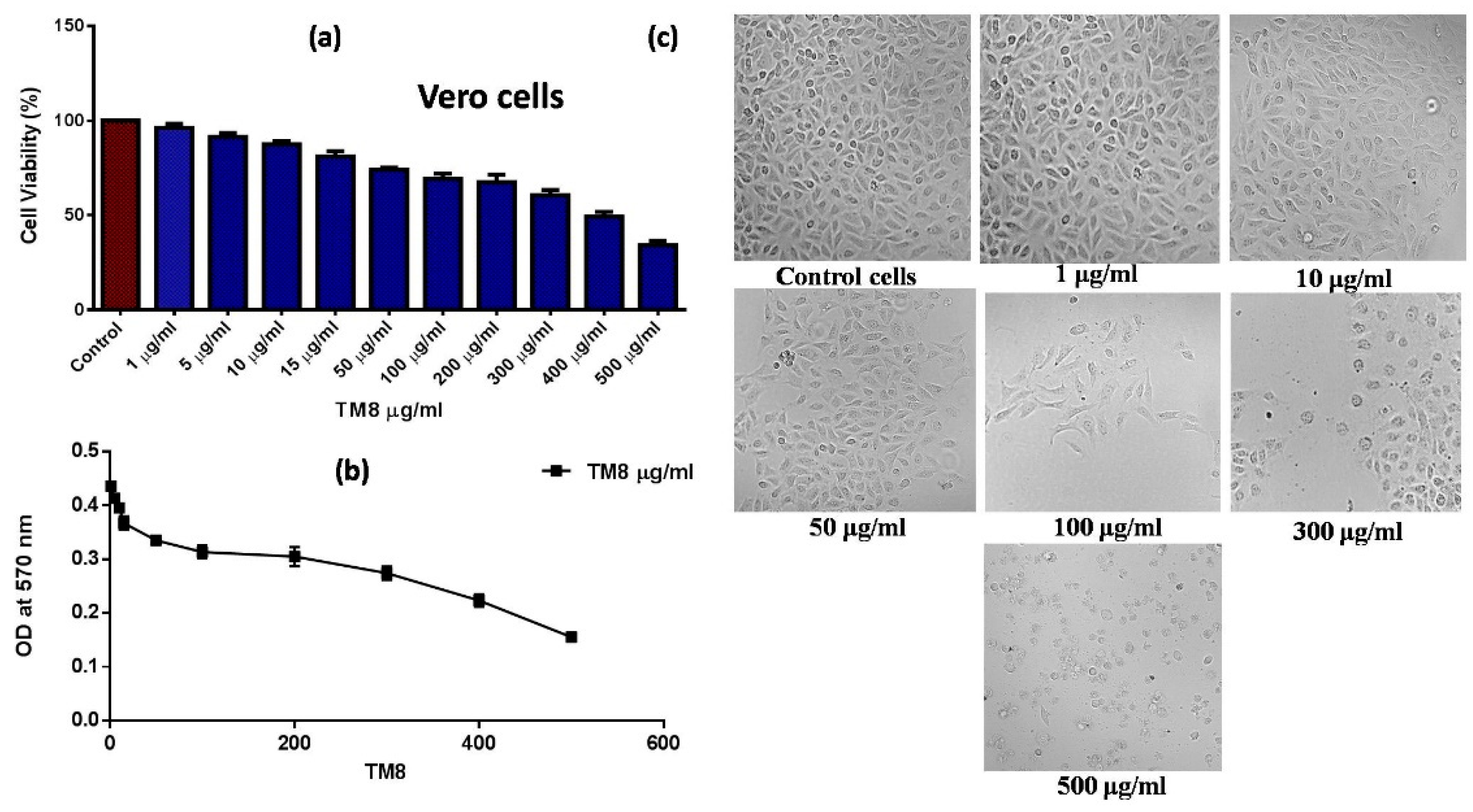
2.4. Cytotoxicity Evaluation
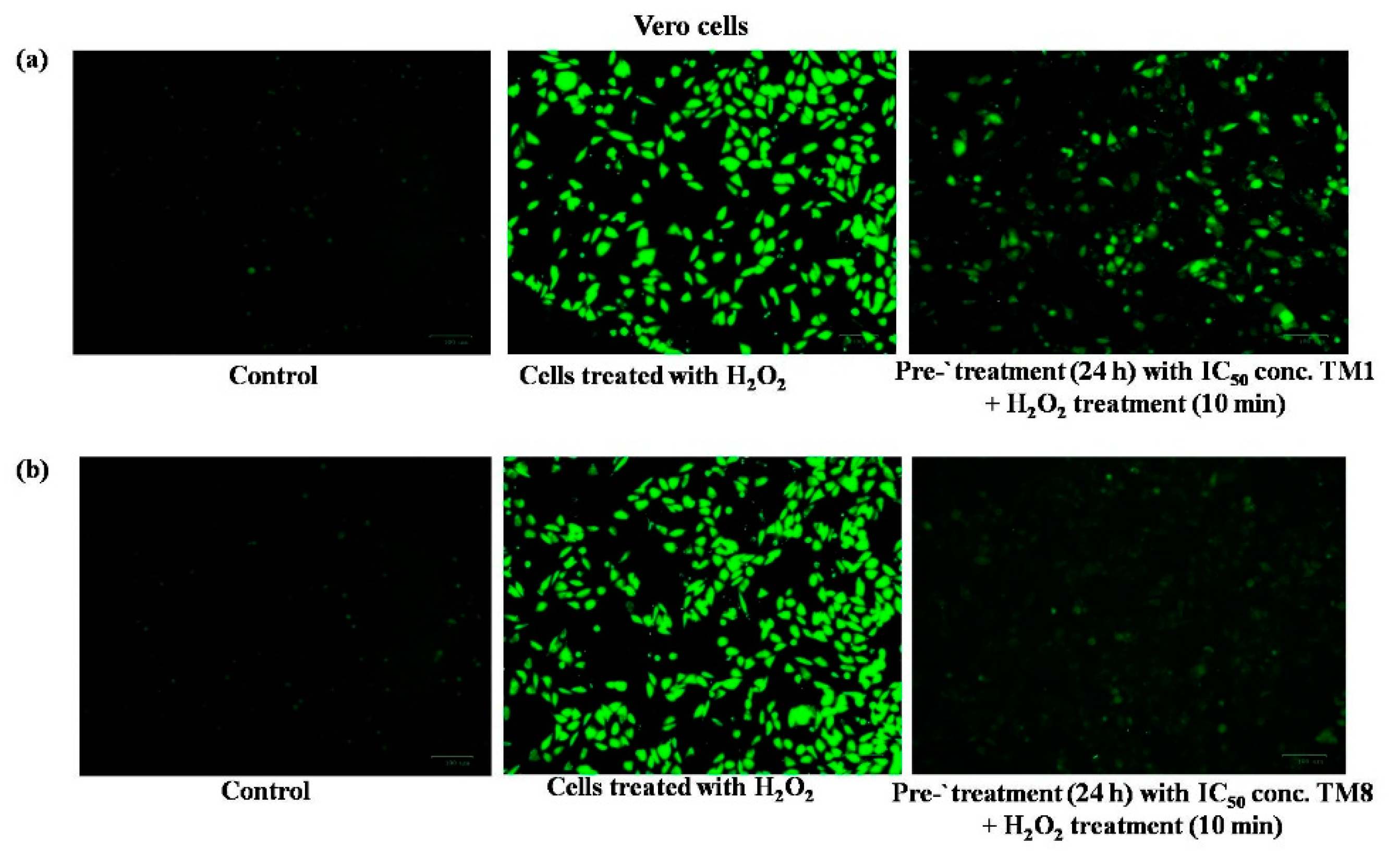
2.5. ROS Assay
2.6. Blood-Brain Barrier (BBB) Permeation Study by Parallel Artificial Membrane Permeability Assay (PAMPA)
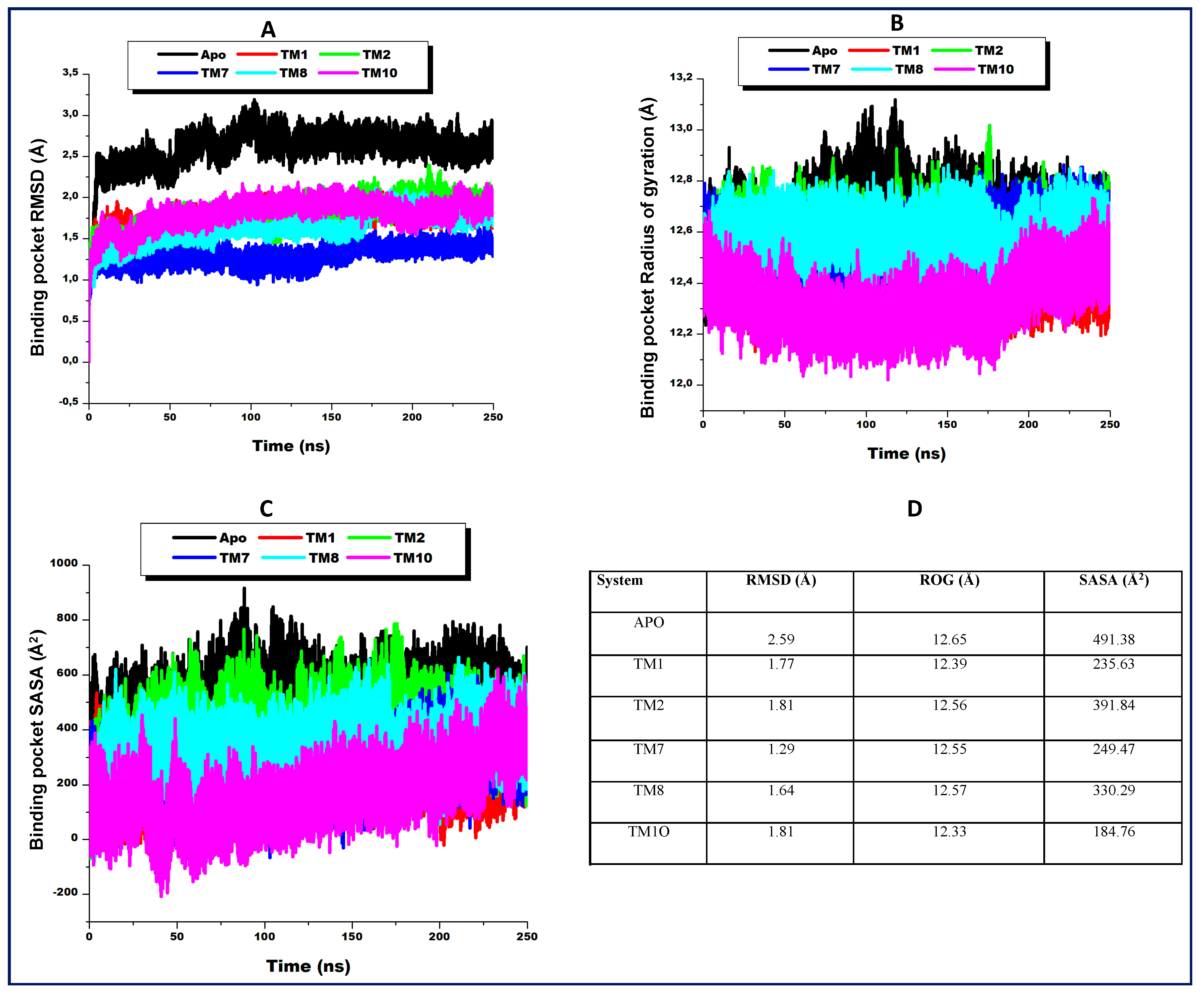
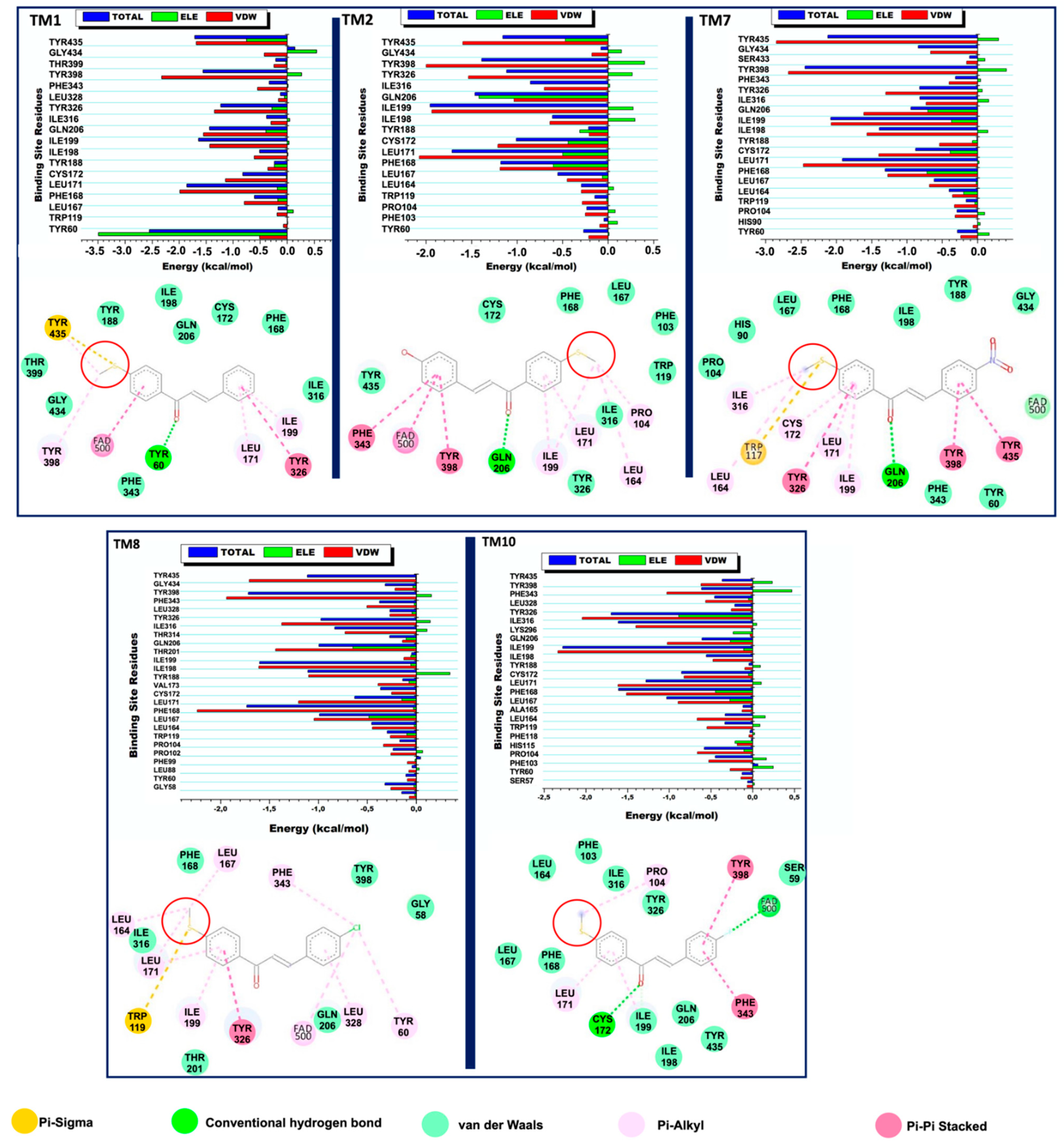
2.7. Inhibitor-Induced Binding Pocket Dynamics
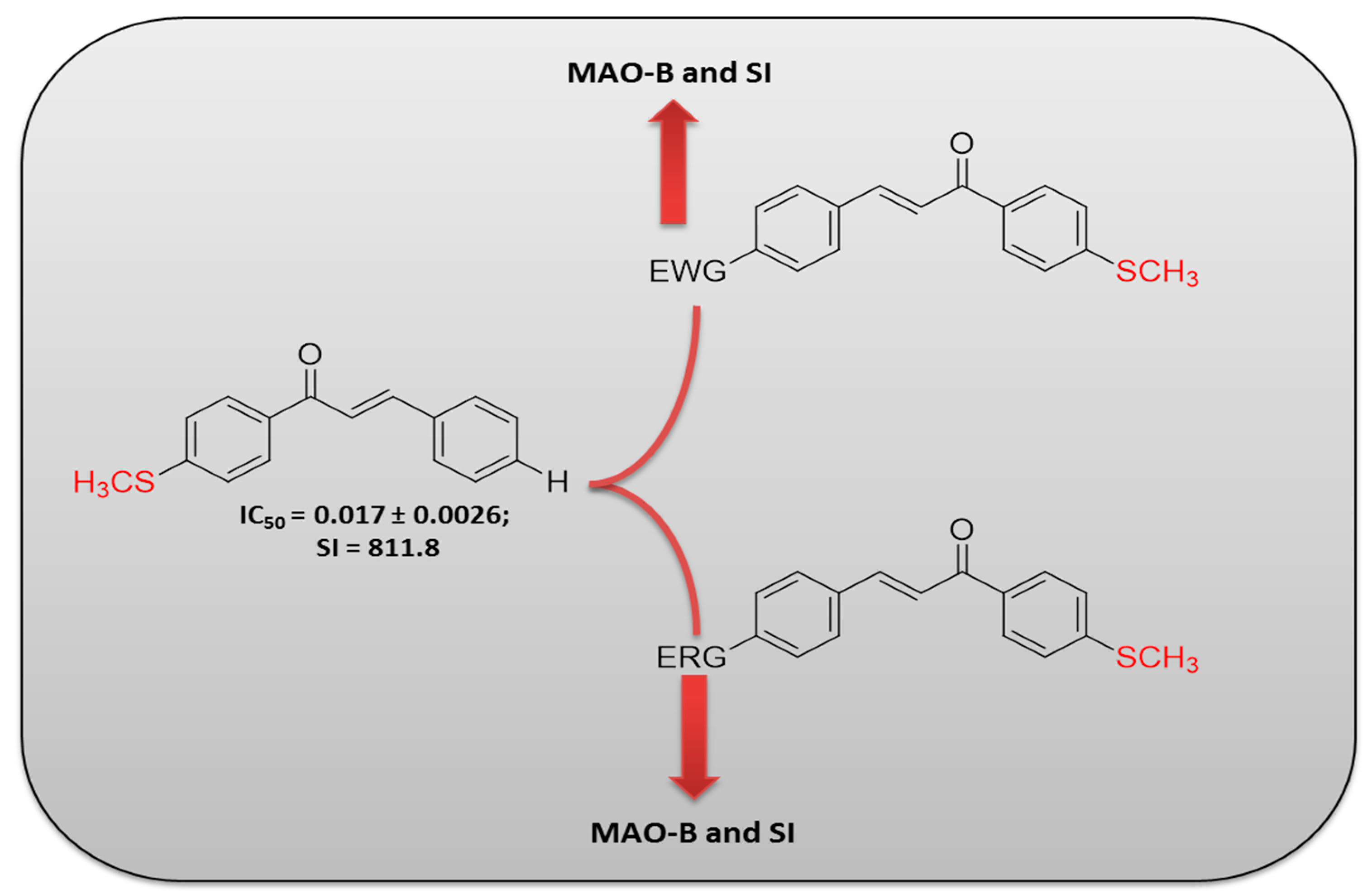
3. Discussion
4. Materials and Methods
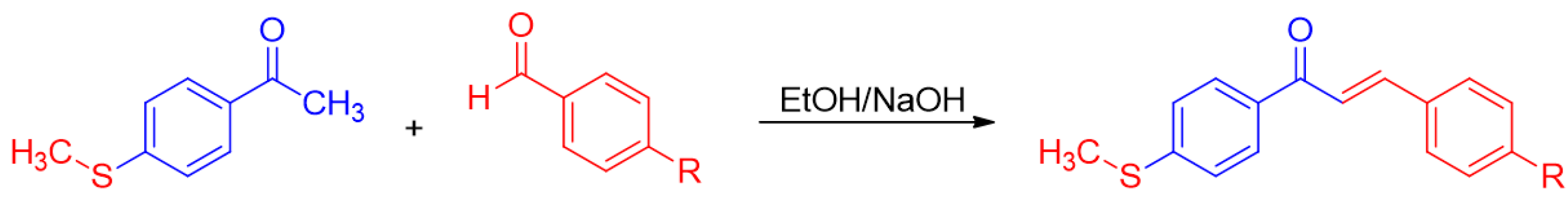
4.1. Synthesis
4.2. Biological Evaluations
4.2.1. MAO Enzyme Inhibition
4.2.2. Kinetics Study
4.2.3. Reversibility Studies
4.2.4. Cytotoxicity and ROS Assays
4.2.5. BBB Study by PAMPA Method
4.3. Computational Studies
4.3.1. System Preparation
4.3.2. Molecular Docking
4.3.3. Molecular Dynamics Simulation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Lau, L.M.L.; Breteler, M.M.B. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006, 5, 525–535. [Google Scholar] [CrossRef]
- Schapira, A.H.; Jenner, P. Etiology and pathogenesis of Parkinson’s disease. Mov. Disord 2011, 26, 1049–1055. [Google Scholar] [CrossRef]
- Rodriguez-Oroz, M.C.; Jahanshahi, M.; Krack, P.; Litvan, I.; Macias, R.; Bezard, E.; Obeso, J.A. Initial clinical manifestations of Parkinson’s disease: Features and pathophysiological mechanisms. Lancet Neurol. 2009, 8, 1128–1139. [Google Scholar] [CrossRef] [Green Version]
- Jankovic, J.; Aguilar, L.G. Current approaches to the treatment of Parkinson’s disease. Neuropsychiatr. Dis. Treat 2008, 4, 743–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tipton, K.F. 90 years of monoamine oxidase: Some progress and some confusion. J. Neural. Transm. 2018, 125, 1519–1551. [Google Scholar] [CrossRef]
- Carradori, S.; Secci, D.; Bolasco, A.; Chimenti, P.; D’Ascenzio, M. Patent-related survey on new monoamine oxidase inhibitors and their therapeutic potential. Expert Opin. Ther. Pat. 2012, 22, 909–939. [Google Scholar] [CrossRef] [PubMed]
- Youdim, M.B.; Edmondson, D.; Tipton, K.F. The therapeutic potential of monoamine oxidase inhibitors. Nat. Rev. Neurosci. 2006, 7, 295–309. [Google Scholar] [CrossRef]
- D’Amato, R.J.; Lipman, Z.P.; Snyder, S.H. Selectivity of the parkinsonian neurotoxin MPTP: Toxic metabolite MPP+ binds to neurtomelanin. Science 1986, 231, 987–989. [Google Scholar] [CrossRef] [PubMed]
- Foley, P.; Youdim, M.B.H.; Riederer, P. MAO-B inhibitors: Multiple roles in the therapy of neurodegenerative disorders? Parkinsonism Relat. Disord. 2000, 6, 25–47. [Google Scholar] [CrossRef]
- Ramsay, R.R. Inhibitor design for monoamine oxidases. Curr. Pharm. Des. 2013, 19, 2529–2539. [Google Scholar] [CrossRef]
- Mathew, B.; Parambi, D.G.T.; Mathew, G.E.; Uddin, M.S.; Inasu, S.T.; Kim, H. Emerging therapeutic potentials of dual-acting MAO and AChE inhibitors in Alzheimer’s and Parkinson’s diseases. Arch. Pharm. Chem. Life Sci. 2019, 352, e1900177. [Google Scholar] [CrossRef] [PubMed]
- Desideri, N.; Fioravanti, R.; Monaco, L.P.; Biava, M.; Yáñez, M.; Ortuso, F.; Alcaro, S. 1, 5-Diphenylpenta-2, 4-dien-1-ones as potent and selective monoamine oxidase-B inhibitors. Eur. J. Med. Chem. 2013, 59, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Hagenow, J.; Hagenow, S.; Grau, K.; Khanfar, M.; Hefke, L.; Proschak, E.; Stark, H. Reversible small molecule inhibitors of MAO A and MAO B with anilide motifs. Drug Des. Dev. Ther. 2020, 28, 317–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Nair, A.S.; Bhashkar, V.; Sudevan, S.T.; Koyiparambath, V.P.; Khames, A.; Abdelgawad, M.A.; Mathew, B. Navigating into the Chemical Space of Monoamine Oxidase Inhibitors by Artificial Intelligence and Cheminformatics Approach. ACS Omega. 2021, 6, 23399–23411. [Google Scholar] [CrossRef]
- Palakkathondi, A.; Oh, J.M.; Dev, S.; Rangrajan, T.M.; Kaipakasseri, S.; Kavuly, F.S.; Gmabacorta, N.; Nicolotti, O.; Kim, H.; Mathew, B. (Hetero-)(arylidene) arylhydrazides as Multitarget-Directed Monoamine Oxidase Inhibitors. ACS Comb. Sci. 2020, 22, 592–599. [Google Scholar] [CrossRef] [PubMed]
- Carradori, S.; Silvestri, R. New frontiers in selective human MAO-B inhibitors: Miniperspective. J. Med. Chem. 2015, 58, 6717–6732. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: Updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res. 2019, 47, W357–W3664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guglielmi, P.; Mathew, B.; Secci, D.; Carradori, S. Chalcones: Unearthing their therapeutic possibility as monoamine oxidase B inhibitors. Eur. J. Med. Chem. 2020, 205, 112650. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, C.; Zhang, W.; Sheng, C.; Zhang, W.; Xing, C.; Miao, Z. Chalcone: A privileged structure in medicinal chemistry. Chem. Rev. 2017, 17, 7762–7810. [Google Scholar] [CrossRef]
- Zhang, X.; Rakesh, K.P.; Bukhari, S.N.A.; Balakrishna, M.; Manukumar, H.M.; Ain, H.L. Multi-targetable chalcone analogs to treat deadly Alzheimer’s disease: Current view and upcoming advice. Bioorg. Chem. 2018, 80, 86–93. [Google Scholar] [CrossRef]
- Mathew, B.; Parambi, D.G.T.; Sivasankarapillai, V.S.; Uddin, M.D.S.; Suresh, J.; Mathew, G.E.; Joy, M.; Marathakam, A.; Gupta, S.V. Perspective design of chalcones for the management of CNS disorders: A mini-review. CNS Neurol. Disord. Drug Targets 2019, 18, 432–445. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, D.K.; Asati, V.; Bharti, S.K. An updated patent review of therapeutic applications of chalcone derivatives (2014-present). Expert Opin Ther. Pat. 2019, 29, 385–406. [Google Scholar] [CrossRef] [PubMed]
- Chimenti, F.; Fioravanti, R.; Bolasco, A.; Chimenti, P.; Secci, D.; Rossi, F.; Yáñez, M.; Oraalo, F.; Ortuso, F.; Alcaro, S. Chalcones: A valid scaffold for monoamine oxidases inhibitors. J. Med. Chem. 2009, 52, 2818–2824. [Google Scholar] [CrossRef] [PubMed]
- Mathew, B.; Uçar, G.; Mathew, G.E.; Mathew, S.; Purapurath, P.K.; Moolayil, F.; Mohan, S.; Gupta, S.V. Monoamine oxidase inhibitory activity: Methyl-versus chlorochalcone derivatives. Chem. Med. Chem. 2016, 11, 2649–2655. [Google Scholar] [CrossRef]
- Shalaby, R.; Petzer, J.P.; Petzer, A.; Ashraf, U.M.; Atari, E.; Alasmari, F.; Kumarasamy, S.; Sari, Y.; Khalil, A. SAR and molecular mechanism studies of monoamine oxidase inhibition by selected chalcone analogs. J. Enzym. Inhib. Med. Chem. 2019, 34, 863–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parambi, D.G.T.; Oh, J.M.; Baek, S.C.; Lee, J.P.; Tondo, A.R.; Nicolotti, O.; Kim, H. Design, synthesis and biological evaluation of oxygenated chalcones as potent and selective MAO-B inhibitors. Bioorg. Chem. 2019, 93, 103335. [Google Scholar] [CrossRef] [PubMed]
- Mathew, B.; Mathew, G.E.; Uçar, G.; Baysal, I.; Suresh, J.; Vilapurathu, J.K.; Prakasan, A.; Suresh, J.K.; Thomas, A. Development of fluorinated methoxylated chalcones as selective monoamine oxidase-B inhibitors: Synthesis, biochemistry and molecular docking studies. Bioorg. Chem. 2015, 62, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Morales-Camilo, N.; Salas, C.O.; Sanhueza, C.; Espinosa-Bustos, C.; Sepúlveda-Boza, S.; Reyes-Parada, M.; Gonzalez-Nio, F.; Caroli-Rezende, M.; Fierro, A. Synthesis, Biological Evaluation, and Molecular Simulation of Chalcones and Aurones as Selective MAO-B Inhibitors. Chem. Biol. Drug Des. 2015, 85, 685–695. [Google Scholar] [CrossRef]
- Hammuda, A.; Shalaby, R.; Rovida, S.; Edmondson, D.E.; Binda, C.; Khalil, A. Design and synthesis of novel chalcones as potent selective monoamine oxidase-B inhibitors. Eur. J. Med. Chem. 2016, 114, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Mathew, B.; Mathew, G.E.; Uçar, G.; Joy, M.; Nafna, E.K.; Lohidakshan, K.K.; Suresh, J. Monoamine oxidase inhibitory activity of methoxy-substituted chalcones. Int. J. Biol. Macromol. 2017, 104, 1321–1329. [Google Scholar] [CrossRef]
- Di, L.; Kerns, E.H.; Fan, K.; McConnell, O.J.; Carter, G.T. High throughput artificial membrane permeability assay for blood-brain barrier. Eur. J. Med. Chem. 2003, 38, 223–232. [Google Scholar] [CrossRef]
- Thiruvalluvar, A.; Subramanyam, M.; Butcher, R.J.; Karabasanagouda, T.; Adhikari, A.V. (E)-1-[4-(Methylsulfanyl) phenyl]-3-phenylprop-2-en-1-one. Acta Crystallogr. Sect. E Struct. Rep. 2008, 64, o1263. [Google Scholar] [CrossRef] [PubMed]
- Kavully, F.S.; Oh, J.M.; Dev, S.; Kaipakasseri, S.; Palakkathondi, A.; Vengamthodi, A.; Azeez, R.F.A.; Tondo, A.R.; Nicolotti, O.; Kim, H. Design of enamides as new selective monoamine oxidase-B inhibitors. J. Pharm. Pharmacol. 2020, 72, 916–926. [Google Scholar] [CrossRef]
- Baek, S.C.; Park, M.H.; Ryu, H.W.; Lee, J.P.; Kang, M.G.; Park, D.; Park, C.M.; Oh, S.R.; Kim, H. Rhamnocitrin isolated from Prunus padus var. seoulensis: A potent and selective reversible inhibitor of human monoamine oxidase A. Bioorg. Chem. 2018, 28, 317–325. [Google Scholar] [CrossRef]
- Mathew, B.; Baek, S.C.; Parambi, D.G.T.; Lee, J.P.; Joy, M.; Rilda, P.R.A.; Randev, R.V.; Nithyamol, P.; Vijayan, V.; Inasu, S.T.; et al. Selected aryl thiosemicarbazones as a new class of multi-targeted monoamine oxidase inhibitors. Med. Chem. Comm. 2018, 9, 1871–1881. [Google Scholar] [CrossRef] [PubMed]
- Park, C.; Cha, H.J.; Hong, S.H.; Kim, G.Y.; Kim, S.; Kim, H.S.; Kim, B.W.; Jeon, Y.J.; Choi, Y.H. Protective effect of phloroglucinol on oxidative stress-induced DNA damage and apoptosis through activation of the Nrf2/HO-1 signaling pathway in HaCaT human keratinocytes. Mar. Drugs 2019, 17, 225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez, W.I.; Soto, Y.; Ortíz, C.; Matta, J.; Meléndez, E. Ferrocenes as potential chemotherapeutic drugs: Synthesis, cytotoxic activity, reactive oxygen species production and micronucleus assay. Bioorg. Med. Chem. 2015, 23, 471–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knez, D.; Colettis, N.; Lacovino, L.G.; Sova, M.; Pišlar, A.; Konc, J.; Samo Lešnik, S.; Josefina Higgs, J.; Kamecki, F.; Mangialavori, I.; et al. Stereoselective activity of 1-propargyl-4-styrylpiperidine-like analogues that can discriminate between monoamine oxidase isoforms A and B. J. Med. Chem. 2020, 63, 1361–1387. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, H.G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Case, D.A.; Walker, R.C.; Huang, Y.; Lin, C.; Mermelstein, D.J.; Cheatham, T.E., III; Simmerling, C.; Li, P.; Roitberg, A.; Onufriev, A.; et al. AMBER 2018 Reference Manual. University of California, San Francisco. 2018. Available online: https://ambermd.org/doc12/Amber18.pdf (accessed on 10 October 2021).
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model 2006, 25, 247–260. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Residual Activity at 10 µM (%) | IC50 (µM) | SI b | ||
|---|---|---|---|---|---|
| MAO-A | MAO-B | MAO-A | MAO-B | ||
 TM1 | 62.6 ± 9.7 | 13.2 ± 0.7 | 13.80 ± 0.05 | 0.017 ± 0.003 | 811.8 |
 TM2 | 48.3 ± 2.4 | −5.7 ± 5.3 | 7.45 ± 0.25 | 0.021 ± 0.003 | 354.8 |
 TM3 | 44.6 ± 1.9 | 6.9 ± 0.0 | 8.82 ± 0.09 | 0.088 ± 0.011 | 100.2 |
 TM4 | 71.6 ± 3.8 | 14.7 ± 2.8 | 12.00 ± 0.99 | 0.058 ± 0.001 | 206.9 |
 TM5 | 47.9 ± 3.7 | 1.6 ± 1.1 | 5.91 ± 0.87 | 0.120 ± 0.011 | 49.3 |
 TM6 | 49.4 ± 8.7 | −9.5 ± 0.0 | 6.71 ± 0.75 | 0.130 ± 0.005 | 51.6 |
 TM7 | 66.1 ± 2.6 | 6.0 ± 1.9 | 30.00 ± 3.02 | 0.023 ± 0.001 | 1304.3 |
 TM8 | 77.4 ± 8.2 | 4.1 ± 0.5 | 48.60 ± 3.44 | 0.010 ± 0.005 | 4860.0 |
 TM9 | 74.7 ± 0.0 | 7.5 ± 2.1 | 33.60 ± 2.56 | 0.088 ± 0.006 | 381.8 |
 TM10 | 57.9 ± 6.0 | −0.4 ± 2.7 | 13.70 ± 1.22 | 0.026 ± 0.004 | 526.9 |
 TM11 | 79.5 ± 0.7 | 7.4 ± 1.2 | 25.20 ± 0.14 | 0.081 ± 0.032 | 311.1 |
| Toloxatone | 1.08 ± 0.03 | - | |||
| Lazabemide | - | 0.063 ± 0.015 | |||
| Clorgyline | 0.0070 ± 0.0007 | - | |||
| Pargyline | - | 0.028 ± 0.004 | |||
| Compounds | Bibliography [31] Pe (×10−6 cm·s−1) | Experimental Pe (×10−6 cm·s−1) | Prediction |
|---|---|---|---|
| TM1 | 15.54 ± 0.33 | CNS+ | |
| TM2 | 13.28 ± 0.80 | CNS+ | |
| TM7 | 15.53 ± 0.71 | CNS+ | |
| TM8 | 16.26 ± 0.26 | CNS+ | |
| TM10 | 15.17 ± 0.44 | CNS+ | |
| Progesterone | 9.3 | 9.32 ± 0.33 | CNS+ |
| Verapamil | 16.0 | 15.62 ± 0.42 | CNS+ |
| Piroxicam | 2.5 | 2.44 ± 0.26 | CNS± |
| Lomefloxacin | 1.1 | 1.22 ± 0.02 | CNS− |
| Dopamine | 0.2 | 0.22 ± 0.01 | CNS− |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mathew, B.; Oh, J.M.; Khames, A.; Abdelgawad, M.A.; Rangarajan, T.M.; Nath, L.R.; Agoni, C.; Soliman, M.E.S.; Mathew, G.E.; Kim, H. Replacement of Chalcone-Ethers with Chalcone-Thioethers as Potent and Highly Selective Monoamine Oxidase-B Inhibitors and Their Protein-Ligand Interactions. Pharmaceuticals 2021, 14, 1148. https://doi.org/10.3390/ph14111148
Mathew B, Oh JM, Khames A, Abdelgawad MA, Rangarajan TM, Nath LR, Agoni C, Soliman MES, Mathew GE, Kim H. Replacement of Chalcone-Ethers with Chalcone-Thioethers as Potent and Highly Selective Monoamine Oxidase-B Inhibitors and Their Protein-Ligand Interactions. Pharmaceuticals. 2021; 14(11):1148. https://doi.org/10.3390/ph14111148
Chicago/Turabian StyleMathew, Bijo, Jong Min Oh, Ahmed Khames, Mohamed A. Abdelgawad, T. M. Rangarajan, Lekshmi R. Nath, Clement Agoni, Mahmoud E. S. Soliman, Githa Elizabeth Mathew, and Hoon Kim. 2021. "Replacement of Chalcone-Ethers with Chalcone-Thioethers as Potent and Highly Selective Monoamine Oxidase-B Inhibitors and Their Protein-Ligand Interactions" Pharmaceuticals 14, no. 11: 1148. https://doi.org/10.3390/ph14111148