
Ruthenium Complexes in the Fight against Pathogenic Microorganisms. An Extensive Review



Department of General and Inorganic Chemistry, Faculty of Pharmacy, “Carol Davila” University of Medicine and Pharmacy, 020956 Bucharest, Romania
*
Authors to whom correspondence should be addressed.
Pharmaceutics 2021, 13(6), 874; https://doi.org/10.3390/pharmaceutics13060874
Submission received: 1 May 2021
/
Revised: 7 June 2021
/
Accepted: 9 June 2021
/
Published: 13 June 2021
(This article belongs to the Special Issue Focus on Antibiotics – New Challenges and Steps Forward in Discovery and Development)
Abstract
:The widespread use of antibiotics has resulted in the emergence of drug-resistant populations of microorganisms. Clearly, one can see the need to develop new, more effective, antimicrobial agents that go beyond the explored ‘chemical space’. In this regard, their unique modes of action (e.g., reactive oxygen species (ROS) generation, redox activation, ligand exchange, depletion of substrates involved in vital cellular processes) render metal complexes as promising drug candidates. Several Ru (II/III) complexes have been included in, or are currently undergoing, clinical trials as anticancer agents. Based on the in-depth knowledge of their chemical properties and biological behavior, the interest in developing new ruthenium compounds as antibiotic, antifungal, antiparasitic, or antiviral drugs has risen. This review will discuss the advantages and disadvantages of Ru (II/III) frameworks as antimicrobial agents. Some aspects regarding the relationship between their chemical structure and mechanism of action, cellular localization, and/or metabolism of the ruthenium complexes in bacterial and eukaryotic cells are discussed as well. Regarding the antiviral activity, in light of current events related to the Covid-19 pandemic, the Ru (II/III) compounds used against SARS-CoV-2 (e.g., BOLD-100) are also reviewed herein.
1. Introduction
The alarming pace at which microorganisms are evading antibiotics constitutes a challenge for modern medicine [1]. The phenomenon of multidrug resistance has generated a sense of urgency around the development of new classes of antibiotics. Yet most of the drugs under clinical development for the treatment of bacterial infections are organic derivatives of currently used antibiotics, which suggests that these molecules are susceptible to in place mechanisms of bacterial resistance [2].
Although the pipeline for new antibiotics is running dry, the coordination chemistry field is still largely underexplored for antibacterial drug development, with limited clinical use for bismuth and silver-based antimicrobials. Bismuth compounds, for instance, are used for the treatment of H. pylori infections and diarrhea and in wound dressings [3], while silver compounds are used for wound healing applications and management of topical infections [4]. The focus of current research is directed towards the development of metal-based nanoparticles (NPs), with special interest being given to AgNPs following their introduction to the U.S. market in 2016 [5].
It is rather unfortunate that less attention is being given to metal complexes. It should be noted that metal-based compounds offer a vast structural diversity of three-dimensional (3D) scaffolds due to the variety of metal ions, ligands, and possible geometries [2,6,7]. While most organic fragments have linear (1D) or planar (2D) shapes, more complex 3D fragments are desirable for the molecular recognition by biomolecules and optimal interaction with intracellular targets [6]. Furthermore, increasing the 3D chemical topology of molecules has been correlated with a broader activity spectrum [7,8]. Therefore, metal complexes are ideal candidates for future drug discovery pursuits meant to access the underexplored 3D chemical space [6]. In addition, metal complexes possess unique mechanisms of action that are not readily available to organic compounds: ROS generation, redox activation, ligand exchange, and depletion of substrates involved in vital cellular processes [2,9,10]. When compared with solely organic molecules, metal-based compounds were found to display a significantly higher hit-rate against critical antibiotic-resistant pathogens (0.87% vs. 9.9%). Moreover, the percentages of toxic to healthy eukaryotic cells and/or hemolytic compounds in the two groups were found to be nearly identical. Therefore, a generally higher degree of toxicity cannot explain the remarkably high antimicrobial activity of the metal-based set of compounds compared with the organic molecules [2].
The potential of metal complexes has been acknowledged over the last two decades through several platinum-, ruthenium-, copper-, iron-, and gallium-based drugs, which have reached different stages in clinical trials for the treatment of cancer, neurodegenerative diseases, and malaria [11,12]. Several ruthenium (Ru) complexes have been evaluated in clinical trials for the treatment of cancer, namely NAMI-A [13,14], KP1019 [15,16] and its water-soluble sodium salt IT-139 (formerly KP1339) [17], and, more recently, TLD-1433 [18]. Previous knowledge of their chemical properties and biological behavior, gained from the research directed towards the development of novel anticancer compounds, has led to increased focus on tailoring ruthenium complexes as antimicrobial agents [1]. Moreover, a recent study screening 906 metal-containing compounds for antimicrobial activity identified ruthenium as the most frequent element found in active compounds that are nontoxic to eukaryotic cells, followed by silver, palladium, and iridium [2]. Therefore, ruthenium-based compounds hold promise for potential antimicrobial applications, which will be extensively reviewed in this paper.
In order to clarify the use of the terms ‘antibacterial’, ‘antibiotic’, and ‘antimicrobial’ in this manuscript, definitions are given below. The term antibacterial refers to substances, materials, or assemblies that kill or inhibit the growth of bacteria. WHO defines an antibiotic as a substance with a direct action on bacteria that is used for the treatment or prevention of infections or infectious diseases [19]. Although we recognize the distinction between these two terms, in order to avoid repetition, we have occasionally used the terms ‘antibiotic’ and ‘antibacterial’ interchangeably. Antimicrobials, on the other hand, will be used generically for compounds or materials that act against microorganisms (bacteria, fungi, viruses, protozoa, parasites, etc.). Consequently, antimicrobials will include antibacterials, antifungals, antivirals, antiprotozoals, and antiparasitics.
2. General Remarks on Bacterial Cell Structure. Gram-Positive vs. Gram-Negative Strains
The bacterial cell structure comes as a result of the extreme conditions they must survive in, which are inhospitable for eukaryotes. For instance, the rigid cell wall that covers the cell membrane is vital for protection from physical, chemical, and mechanical stressors. Based on the Gram staining procedure, bacteria are classified into two groups: Gram-positive and Gram-negative bacteria [1].
Gram-positive strains retain the Crystal Violet stain due to the presence of a thick layer of peptidoglycan in their cell walls, which is densely embedded with negatively charged glycopolymers called wall teichoic acids (Figure 1). The fairly porous cell wall structure generally allows for passage for exogenous molecules into the bacterial cells [20].
Gram-negative bacteria, however, have more complex cell wall structures (Figure 1). Due to the absence of inlaid teichoic acid molecules, their layer of peptidoglycan is thin, yet bound to an outer membrane coated with lipopolysaccharides (LPSs). LPSs are amphiphiles, consisting of a hydrophobic lipidic domain (lipid A) covalently bound to a polysaccharide, which comprises the O antigen and the inner and outer cores; these negatively charged (due to the presence of the phosphate and acid groups) macromolecules are stabilized by divalent cations such as calcium and magnesium. LPSs greatly decrease bacterial permeability to antibiotics and play a crucial role in the development of resistance mechanisms for many pathogenic Gram-negative bacteria [1,20].
Additionally, on the cell surface of some bacteria (e.g., Streptococcus pneumoniae) a slime layer or a capsule can offer additional protection against desiccation or phagocytosis by host cells. Flagella, fimbriae, and pili are external filamentous appendages that serve as organelles of locomotion or assist with bacterial attachment and adhesion to a surface or genetic exchange [1,21].
At physiological pH, the high content of zwitterionic phosphatidylcholine confers an overall neutral charge to the eukaryotic cell membranes. In contrast, bacterial outer cell walls and membranes are usually negatively charged due to the presence of negatively charged components (phospholipids, teichoic acids, and lipopolysaccharides) [1,23]. Hence, in order to increase selectivity, new antibacterial drugs (including ruthenium complexes) are generally designed so as to possess a cationic component.
3. Mechanisms of Action of Current Drugs
Antibiotics are classified into four major groups (Figure 2), based on their intracellular target and mechanism of action: (1) inhibition of bacterial cell wall synthesis (penicillin and its derivatives, cephalosporins, carbapenems, and glycopeptides—these drugs are more active against Gram-positive bacteria); (2) disruption of bacterial membranes (polymyxins—these are active against Gram-negative bacteria and considered a last-line therapy against Gram-negative ‘superbugs’); (3) inhibition of nucleic acid synthesis (quinolones, rifampicin, and sulphonamidesare—these are broad-spectrum synthetic antibiotics); and (4) inhibition of protein synthesis (tetracycline, aminoglycosides, chloramphenicol, and macrolides—these inhibit protein synthesis by targeting the RNA-rich surfaces of ribosomes) [1].
Several new classes of antibiotics have been discovered over the last two decades. Gepotidacin, for instance, belongs to a new chemical class of antibiotics called triazaacenaphthylene. It is a topoiosomerase inhibitor, which is currently being investigated in a phase III clinical study in patients with uncomplicated urinary tract infection and urogenital gonorrhoea [24]. Other current strategies include the use of phages (viruses that kill specific bacterial strains) [25], various types of engineered nanoparticles [25], and cationic materials, including cationic polypeptides, polymers, copolymers, and dendrimers [26]. Furthermore, several natural products, e.g., teixobactin, have been identified as lead compounds in the fight against antimicrobial resistance [27].
4. Mechanisms of Resistance to Antibiotics
Bacterial resistance to antibiotics can result from intrinsic or acquired antibiotic-resistant mechanisms. P. aeruginosa and other Gram-negative pathogens are intrinsically more resistant to antibiotics due to the reduced permeability of their outer membranes. These bacterial strains have porins of unusually low permeability. In addition, the outer membranes of mycobacteria have a high lipid content that allows for hydrophobic drugs such as fluoroquinolones to enter the cell but limits the access of hydrophilic drugs.
Acquired bacterial resistance is caused by alterations in microorganisms that result in drug inactivation or a decrease in therapeutic efficacy. Improper prescribing and overuse of antibiotics are factors that have contributed to the growing issue of microbial resistance. Consequently, infections have become increasingly difficult or even impossible to treat [28].
Bacterial resistance can emerge as a result of various biochemical mechanisms, including decreased drug uptake, modification of a specific bacterial target, enzymatic inactivation of the drug, and modifications to the bacterial efflux systems [1,28]. For instance, a common resistance mechanism is the alteration of the bacterial membrane permeability, resulting in limited uptake of an antibiotic. Modification of the drug’s target can involve mutations in DNA gyrase and topoisomerase IV or alterations in the structure and/or number of penicillin-binding proteins [5]. Drug inactivation occurs via mutations in genes coding for key enzymes, such as β-lactamases, acetyltransferases, adenylyltransferases, and aminoglycoside-3′-phosphotransferase. These mutations can occur either inside the bacterial chromosomal DNA or as a result of foreign genetic material acquisition. Acquisition of genetic material that confers resistance is possible through horizontal gene transfer, which is mediated either by plasmids or bacteriophages [28].
Another common mechanism of resistance used by many pathogens involves the association of multiple bacterial cells in matrices called biofilms. The bacterial cells within the biofilm have a slow metabolism rate and slow cell division. Therefore, antimicrobials targeting growing and dividing bacterial cells are rendered ineffective. Moreover, the thick biofilm extracellular matrix consists of bacterial polysaccharides, proteins, and DNA, which hinder access of the antimicrobial agent to the bacteria. It is also likely that the proximity of the bacterial cells facilitates horizontal gene transfer. Therefore, the antimicrobial resistance genes can be shared between the cells forming the biofilm [28,29,30].
Nosocomial infections or hospital-acquired infections are a growing threat worldwide and are often caused by multidrug-resistant bacteria. Interestingly, a small group of microorganisms, known as ESKAPE pathogens, are responsible for most antibiotic-resistant infections. These pathogens include: Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter spp., which possess innate resistance or can acquire resistance against multiple antibiotics [31].
5. Antibacterial and Antifungal Activities of Ruthenium Complexes
Based upon their chemical stability, Ru complexes can be classified as either stable, relatively inert compounds, and prodrugs. A metal complex is inert when the ligand framework remains unaltered in biological media. The ruthenium ion in these compounds acts merely as a central scaffold that carries the bioactive ligands to their target. Consequently, the properties of the coordinated ligands are essential to the antibacterial activity [32]. The presence of the ruthenium ion, however, provides the molecule with a positive charge, which aids in targeting the negatively charged cell wall structures of bacteria. The antibacterial activity of these complexes depends on their lipophilicity and charge, which in turn shape their ability to interact with specific targets (e.g., DNA, RNA, proteins, bacterial membranes).
Prodrugs are labile complexes that release the ligand/s when exposed to solvents and/or media and generate species that can bind to various biological targets or photoactivated drugs. The latter become active upon light irradiation and act as photosensitizers. Since this behavior is somewhat unconventional for the general understanding of the term ‘prodrug’ in the traditional medicinal chemistry sense, ‘prodrug-like molecules’ seems more appropriate to describe this type of metal complex. In the case of labile complexes, active species are released as a result of either partial or total ligand exchange in biological media. These active species are either ruthenium species resulting from ligand exchange with media components or the released ligands. In the latter case, the ruthenium compounds are called ‘carrier’ complexes; one such example is the Ru(II) chelate–chloroquine complex, [RuCl2(CQ)]2, where CQ = chloroquine (see 6. Antiparasitic activity of ruthenium complexes). In the following sections, ruthenium complexes will be classified based on their structure. Details and comments with regard to their mechanisms of action will be provided wherever such information is available.
5.1. Mononuclear Ruthenium (II) Complexes
Mononuclear polypyridylruthenium (II) complexes with antimicrobial activities were first reported in the 1950s and 1960s by Dwyer et al. [33,34]. With the general interest shifting towards discovering new analogues of existing classes of antibiotics, their impressive seminal work was unfortunately not further pursued. However, the advancement into clinical trials of NAMI-A, KP1019, and TLD1433 for the treatment of cancer and the urge to develop new classes of antibiotics have led, over the last two decades, to an increased focus on research and development of ruthenium-based antimicrobials [35].
Dwyer et al. made the first steps towards the development of kinetically inert Ru(II) complexes and the study of their in vitro and in vivo antimicrobial activities. The addition of methyl groups to the phenanthroline ligands enhanced lipophilicity and increased the activity of [Ru(Me4phen)3]2+ (Figure 3) against Gram-positive bacteria, as compared with [Ru(phen)3]2+ (Figure 3) [36]. More recent studies [37,38], however, have shown that these complexes are much less active against various antibiotic-resistant ESKAPE pathogens. Additionally, their activity in vivo has been proven to be unsatisfactory, as they caused severe neurotoxic effects when injected into mice [39].
Following up on this remarkable work, various heteroleptic mononuclear polypyridyl Ru (II) complexes were tested for antibacterial activity. Their activities (MIC values) against various bacterial strains, as well as toxicity towards healthy eukaryotic cells and modes of action, where available, are listed in Table 1.
5.1.1. Mononuclear Polypyridyl Ru (II) Complexes
R-825 (Figure 3) was shown to interfere with the iron acquisition systems in S. pneumoniae, which led to a dramatic decrease in intracellular iron, correlated with a bactericidal effect. In addition, R-825 was essentially non-toxic to human A549 non-small-cell lung cancer cells in vitro [41]. Iron is an essential nutrient for the development and survival of bacteria, as well as a key factor in host infection. In order to scavenge iron from their surroundings, bacteria make use of highly effective iron acquisition systems. In S. pneumoniae, the ABC transporters PiaABC, PiuABC, and PitABC play a major role in the acquisition of heme, ferrichrome, and ferric irons, respectively [72]. The deletion of the piuA gene in a mutant strain of S. pneumoniae resulted in a significant decrease in ruthenium uptake, leading to an increased resistance of the mutant to R-825 treatment. These results suggest that the mechanism of uptake for R-825 appears to involve active transport via the PiuABC iron uptake pathway [41]. Note that this mechanism of uptake is different than those used by the currently approved antibiotics. Generally, due to the chemical similarity between iron and ruthenium, the ability of novel antibiotics to interfere with iron acquisition systems in bacteria (including ABC transporters) is considered to be a viable strategy for the discovery of new antibacterial drugs.
A variety of mononuclear heteroleptic polypyridyl ruthenium (II) chelates bearing bpy, phen, dmp (4,4′-dimethyl-2,2′-bipyridine), or hdpa (2,2’-dipyridylamine) and other mono/bidentate ligands were active in various degrees against Gram-positive and Gram-negative bacteria and fungi [73,74,75,76,77,78,79,80,81]. Although their mechanisms of action have not been determined, all complexes were shown to interact with DNA duplexes and several exerted photoactivated cleavage of plasmid DNA in vitro [75,77,79,80,81] with singlet oxygen (1O2) probably playing a significant role in the cleavage mechanism.
Mononuclear Ru(II) Heteroleptic Complexes Bearing 2,2’-Bipyridine (bpy) Ligands
Numerous octahedral heteroleptic Ru(II) complexes containing 2,2’-bipyridine (bpy), with the general formula [Ru(bpy)2L]Yn (where L = a mono/bidentate ligand, note that when L is monodentate, the first coordination sphere of Ru(II) is saturated with chloride ions; Y = counterion) have been synthesized and tested against bacteria. Generally, these complexes showed moderate to high activity on Gram-positive bacteria, but were inactive against Gram-negative strains. X-03 (Figure 4), for instance, was active against several Gram-positive bacteria, S. pneumoniae, Listeria monocytogenes, and S. aureus, but showed no toxicity at the tested concentrations against Gram-negative microorganisms. X-03 appears to interfere with iron acquisition systems in S. pneumoniae cells, in a similar manner to R-825. Proteomic data revealed that X-03 caused the downregulation of several proteins involved in oxidative stress response and fatty acid biosynthesis, suggesting a mechanism of action based on increased susceptibility to oxidative stress and membrane damage. Additionally, X-03 displayed low toxicity even at a concentration 8 times higher than the MIC value to the A549 alveolar and HBE bronchial epithelial cell lines, indicating selective toxicity against bacteria [42].
Complexes with photolabile ligands, in which L is unidentately coordinated, L = 4-(4-chlorobenzoyl)pyridine (clbzpy), Y = PF6‾, n = 1 ([Ru(bpy)2Cl(clbzpy)]+, Figure 4), was moderately active against S. aureus and S. epidermidis. Additionally, the complex was shown to suffer blue light photolysis (453 nm) in aqueous solution and the resulting photoproduct, cis-[Ru(bpy)2(H2O)Cl]+, displayed high binding affinity towards DNA in vitro. The antibacterial activity, however, was not influenced by blue light irradiation, which indicates that the antibacterial activity is not due to DNA damage, but might be the result of bacterial membrane disruption [43]. Blue LED irradiation, however, has been shown to enhance the activity of [Ru(bpy)2(methionine)]2+, albeit not drastically, against S. aureus and S. epidermidis [44]. Methionine release and subsequent exchange with water molecules via photolysis at 453 and 505 nm in aqueous solution lead to cis-[Ru(bpy)2(H2O)2]2+, which can bind covalently to double-stranded DNA [44,82] and promote photocleavage [44].
[Ru(bpy)2L]Yn complexes, where L = BTPIP, ETPIP, CAPIP, Y = ClO4‾, n = 2, [Ru(dmb)2(ETPIP)]2+, and [Ru(phen)2(ETPIP)]2+ (see Figure 4 for the chemical structures and the IUPAC names of the ligands) displayed good activities against drug-susceptible S. aureus. [Ru(bpy)2(BTPIP)]2+ was the most active compound of the series (MIC = 0.016 mg/mL) and was shown to inhibit biofilm formation and, thus, prevent bacteria from developing drug resistance. [Ru(bpy)2(BTPIP)]2+ [46] and [Ru(phen)2(ETPIP)]2+ [45] increased the susceptibility of S. aureus to certain aminoglycosidic antibiotics (kanamycin and gentamicin). [Ru(phen)2(ETPIP)]2+ was found to suppress the gene regulatory activity of the catabolite control protein A (CcpA) in S. aureus, which can explain the synergistic effects observed for this complex and kanamycin [45]. Studies conducted on a murine skin infection model for Ru(bpy)2(BTPIP)]2+ showed that Ru(bpy)2(BTPIP)]2+ ointments were effective as topical products against skin infection [46]. These complexes, however, have proven to be cytotoxic to A549 cancer cell lines, with IC50 values lower than those required for the antibacterial activity [83,84,85,86], which might indicate poor selectivity towards bacteria. To the extent of our knowledge, no cytotoxic tests on normal cell lines have been performed.
The corresponding ruthenium(II) bipyridine complex in which L = curcumin and Y = PF6‾ (Figure 5) was tested against various ESKAPE pathogens. It displayed bactericidal activity against methicillin and vancomycin-resistant S. aureus strains (MIC = 1 µg/mL) and high selectivity towards bacteria as compared with eukaryotic Vero cells (SI > 80). Moreover, the complex strongly inhibited biofilm formation in S. aureus cells and displayed in vivo antibacterial activity against S. aureus comparable to that of vancomycin in a murine neutropenic thigh infection model. However, [Ru(bpy)2curcumin]+ was not toxic to the Gram-negative E. coli, K. pneumoniae, A. baumanii, and P. aeruginosa cells. In comparison, the corresponding Ru(II) complex, [Ru(phen)2curcumin]+, bearing 1,10-phenanthroline (Figure 5), was also active against the Gram-negative A. baumanii with a MIC value comparable to that of levofloxacin, in addition to its activity on the Gram-positive S. aureus bacteria and lack of toxicity against eukaryotic cells [47].
Mononuclear Ru(II) Heteroleptic Complexes Bearing 1,10-phenanthroline (phen)
Mononuclear Ru(II) complexes bearing phenanthroline ligands have also been investigated as potential antibacterial agents. Amongst these complexes, mono-bbn ([Ru(phen)2bbn]2+) (Figure 6), where bbn is bis[4(4’-methyl-2,2’-bipyridyl)]-1,n-alkane and n stands for the number of methylene groups in the alkane chain of bbn (n = 7 or 10), have been extensively investigated. Although mono-bb10 has a larger alkane chain and therefore is more lipophilic, it was less active than mono-bb7 against drug-susceptible S. aureus [38,87,88]. The bactericidal activity of mono-bb7 was linked to the extent of cellular accumulation, since its activity on Gram-negative strains is low and the uptake in Staphylococcus strains is much higher than in E. coli or P. aeruginosa [37,38]. Mono-bb7 caused membrane depolarization in S. aureus cells and increased membrane permeability, which might suggest the membrane damage as part of its mode of action [88]. Morphological changes indicative of membrane damage have also been reported for a similar complex, [Ru(phen)2(BPIP)]2+, where BPIP = 2-(4′-biphenyl)imidazo[4,5-f][1,10]phenanthroline (Figure 6), in Gram-positive (Micrococcus tetragenus and S. aureus) bacteria [76]. Mono-bb7 displayed selective activity against bacterial over healthy mammalian cells [38,89].
A complex in which the bb12 ligand is tetradentately bound to Ru (II), cis-α-[Ru(phen)bb12]2+ (Figure 7a, see for comparison the other isomers of the compound, depicted in Figure 7b,c), was found to be more active against the Gram-negative P. aeruginosa than the more lipophilic mono-bb7. The activity was found to be positively correlated with the uptake of the complex into the cells. Nonetheless, cis-α-[Ru(phen)bb12]2+ was still considerably more active against Gram-positive bacteria as compared with P. aeruginosa, the compound being more active against MRSA than ampicillin and gentamicin. Interestingly, cis-α-[Ru(phen)(bb12)]2+ was found to be two to four times more active than its geometric isomer, cis-β-[Ru(phen)(bb12)]2+, against the Gram-negative strains (E. coli and P. aeruginosa), while no difference in activity was found for the Gram-positive bacteria (S. aureus and MRSA). It is unclear why the cis-α isomer is more active, since no significant difference in cellular accumulation was observed for the two isomers. Moreover, both geometric isomers were shown to bind tightly and with similar potency to duplex DNA in vitro, but no correlation between the binding constants and activity was found [48]. It should be noted that DNA/RNA binding is a possible mechanism of action for these complexes, since several reports indicate that various inert Ru(II) polypyridyl complexes bearing phenanthroline ligands target DNA and RNA in bacterial and eukaryotic cells [76,90,91]. Notably, the similar complex cis-α-[Ru(Me4phen)(bb7)]2+ displayed similar activity towards Gram-positive and Gram-negative bacteria as cis-α-[Ru(phen)(bb12)]2+ and remarkably high DNA binding affinity (~107) [92].
Mononuclear Ru (II) Heteroleptic Complexes Bearing Pyridophenazine Ligands
[Ru(phen)2(dppz)]2+ (Figure 8), where dppz = dipyrido[3,2-a:2’,3’-c]phenazine and phen = 1,10-phenanthroline, displayed good bactericidal activity against M. smegmatis (MIC = 2 µg/mL). Its mechanism of action was suggested to be linked to ROS generation and DNA intercalation [93]. A similar complex, [Ru(2,9-Me2phen)2(dppz)]2+, was active against MRSA and B. subtilis, and displayed time–kill curves that were similar to those of currently used antibiotics, but displayed no activity against E. coli. The activity appeared to be correlated with the ability to intercalate into DNA double strands in vitro. In vivo antibacterial activity has been assessed using the nematode Caenorhabditis elegans infection model and [Ru(2,9-Me2phen)2(dppz)]2+ proved to be non-toxic to the nematodes [40].
[Ru(bb7)(dppz)]2+ (Figure 8) (bb7 = bis[4(4’-methyl-2,2’-bipyridyl)]-1,7-alkane) was 2–8 fold more active than its parent compound [Ru(phen)2(dppz)]2+ against both Gram-positive (S. aureus, MRSA) and Gram-negative bacteria (E. coli, P. aeruginosa). Although the two complexes have comparable lipophilicity, [Ru(bb7)(dppz)]2+ accumulated in P. aeruginosa to the same degree as in MRSA and was shown to permeabilize a model membrane system to a higher degree than [Ru(phen)2(dppz)]2+. Therefore, its higher cellular uptake might be responsible for the increase in activity. However, Ru(bb7)(dppz)]2+ was also ~3-fold more toxic to healthy eukaryotic cells than [Ru(phen)2(dppz)]2+, while still being more active against bacterial cells [49].
Complexes bearing tetrapyridophenazine (tpphz) are more lipophilic relative to their dppz analogues and generally more active. For instance, the luminescent, mononuclear ruthenium(II) complex bearing the tpphz ligand, [Ru(Me4phen)2(tpphz)]2+ (Figure 8), displayed a comparable activity to that of ampicillin and oxacillin in drug-sensitive strains and the activity was retained in resistant strains. The complex was taken up by both Gram-positive (E. faecalis, S. aureus) and Gram-negative (E. coli, A. baumannii, P. aeruginosa) bacteria in a glucose-independent manner and was shown to target chromosomal DNA in both Gram-positive and Gram-negative strains. Moreover, model toxicity screens showed that the compound is non-toxic to Galleria mellonella larvae at concentrations that are 3–25 times higher than the MIC values [50]. This complex represents the starting point for the kinetically inert dinuclear polypyridylruthenium(II) complex [Ru2(Me4phen)2(tpphz)]4+ (see below), which displayed higher antibacterial activity (Table 1), except against S. aureus. Unlike the dinuclear derivative, [Ru(Me4phen)2(tpphz)]2+ does not cause membrane damage.
5.1.2. Mononuclear Ru (II)–arene Complexes
Due to the promising anticancer activities of some representatives, the potential antibacterial properties of piano-stool Ru(II)-η6–arene complexes, with the general structure shown in Figure 9, have also been considered for antimicrobial applications [94,95,96,97,98,99,100,101,102,103]. While some of them displayed modest activity [76,79,80], complexes of the general formulae [Ru(η6-p-cymene)X2(PTA)] (RAPTA-C complexes), where X = Cl, Br, I, NCS (labile) and PTA = 1, 3, 5-triaza-7-phosphaadamantane, were active in different degrees against bacteria (E. coli, B. subtilis, P. aeruginosa) and fungi (Candida albicans, Cladosporium resinae, and Trichrophyton mentagrophytes). The PTA ligand was suggested to play a role in facilitating the uptake of the complex into bacterial cells, while the antimicrobial activity was suggested to be mediated by the interaction of the Ru(II) ion with intracellular proteins. Although the complexes were found to cause DNA damage in vitro, their affinity towards DNA was not correlated with their antibacterial activities. Interestingly, extracts from E. coli cells treated with a PTA derivative show specific protein–ruthenium interactions, suggesting that the intracellular proteins are most likely targets of these complexes [94].
Relying on potential interference with the iron-acquisition systems and in order to increase internalization of the complexes in bacteria, a Trojan Horse strategy was applied for three Ru (II)–arene complexes and one RAPTA-like complex bearing derivatives of deferoxamine B (DFO) (Figure 10) [104]. DFO is a commercially available siderophore, namely an iron chelator that is secreted by microorganisms to bind extracellular Fe (III) and aid in its transport across bacterial membranes inside the cells [105]. These compounds displayed only modest activity against three ESKAPE pathogens (S. aureus, K. pneumoniae, A. baumannii) and one fungal strain (C. albicans) when Fe (III) ions were present in the medium. Absence of iron in the media led to an increase in activity, particularly for K. pneumoniae. All Ru (II) complexes of this series, however, showed little to no activity against P. aeruginosa, E. coli, and C. neoformans, presumably because these bacterial and fungal strains are more susceptible to internalizing DFO. Antiproliferative studies on normal cells (HEK-293) showed that these complexes were essentially non-toxic towards normal eukaryotic cells in the presence of iron [104].
Various Ru(II)–arene complexes with thiosemicarbazone ligands were more active against Gram-positive bacteria than Gram-negative bacteria and/or fungi, but were still less active than the antibiotics used as controls (ampicillin, streptomycin, or ciprofloxacin) [95,98,100,106]. As was seen for other ruthenium complexes, they were shown to bind DNA and human serum albumin with significant affinity in vitro, suggesting that DNA and/or proteins are potential targets of these complexes in bacterial cells. Several complexes were shown to exert low cytotoxicity towards healthy cell lines [95].
Ru(II)-η6-p-cymene complexes bearing pyrazole derivatives containing N,S donor atoms exerted moderate antibacterial activity against Gram-positive strains, including S. aureus, S. epidermidis, and E. faecalis, while displaying very weak to no activity against Gram-negative bacteria (P. vulgaris, P. aeruginosa). Notably, the complexes were non-toxic against the healthy human fibroblast HFF-1 cells [107]. Other Ru(II)–arene complexes with various N,N- or N,O- bidendate ligands displayed moderate activity against various Gram-positive bacterial strains and, notably, were found to be more active against P. aeruginosa than various clinically used antibiotics used as controls [96,99].
While it is well known that Ru(II)–arene complexes have been widely investigated as potential anticancer agents, their clinical use as antibacterial drugs may be limited by their cytotoxic effects (and generally the poor selectivity for cancerous over healthy cells). Some of these complexes, however, exhibited dual antibacterial and anticancer activities [104]. This constitutes a desirable trait as current anticancer therapy weakens the immune system and often leaves patients susceptible to opportunistic infections. Conversely, patients suffering from a chronic infection are more prone to develop cancer due to certain defects in the immune response [108].
5.1.3. Other Mononuclear Ru Complexes
Various other Ru(II/III) complexes have been reported to possess antibacterial activity. However, microbiological studies for these complexes mainly involved disc diffusion assays or MIC testing, without any further research with regard to their modes of action [109,110,111,112,113,114,115,116,117,118,119,120]. These complexes were generally more active against Gram-positive strains, with little to no activity against Gram-negative or drug-resistant bacteria. However, a Ru(III) complex, [Ru(L)Cl2]Cl, where L is a N,N,N,N- tetradentate macrocyclic ligand derived from 2,6-diaminopyridine and 3-ethyl-2,4-pentanedione, was moderately active against the Gram-negative bacteria Xanthomonas campestris and P. aeruginosa and displayed higher activity than the corresponding Pd(II), Pt(II), and Ir(III) complexes [114]. Three ruthenium half-sandwich complexes containing phenyl hydrazone Schiff base ligands also displayed good activity against the Gram-negative P. aeruginosa, comparable to that of the positive control, gentamicin, and generally higher than the corresponding Ir(III) and Rh(III) complexes [111].
There are few examples of Ru(II) complexes that display antimycobacterial activity. However, ‘SCAR’ compounds, consisting of a series of Ru(II) complexes containing phosphine/picolinate/diimine ligands (Figure 11), had low MIC values against multidrug-resistant strains of M. tuberculosis [51,121,122]. Moreover, the SCAR complexes exerted synergistic interactions with first-line antibiotics, with the best overall synergistic activity observed with isoniazid [122]. Although these complexes displayed some selectivity towards bacterial over healthy eukaryotic cells, an increase in the toxic effects against bacteria was correlated with higher toxicity against eukaryotic cells. Cis-[RuCl2(dppb)(bpy)] (SCAR6), where dppb = 1,4-bis(diphenylphosphino)butane and bpy = 2,2’-bipyridine, the least active compound of the series, was found to be the least stable in aqueous solutions [121]. Upon dissolution in water, the chlorido ligands are released, and the resulting species was shown to bind covalently to DNA and induce DNA damage in a similar manner to cisplatin [51,121]. Moreover, the metabolic products of SCAR6 were responsible for the mutagenic effects of the compound observed in Salmonella typhimurium. In contrast, SCAR4 and SCAR5 did not display any mutagenic effect [51].
A biphosphinic ruthenium complex, cis-[Ru(dppb)(bqdi)Cl2]2+ (Figure 11, RuNN), where dppb = 1,4-bis(diphenylphosphino)butane and bqdi = o-benzoquinonediimine, displayed bacteriostatic and bactericidal activity against Gram-positive bacteria (S. aureus, including MRSA, and S. epidermidis). Time–kill kinetics studies indicated that RuNN displayed bactericidal activity in the first 1–5 h [52]. Note that this is a much shorter time than that reported for vancomycin or telavancin (24 h) [123]. Additionally, the combination treatment of RuNN and ampicillin (but not tetracycline) resulted in a dramatic increase in activity, highlighting the synergistic effect of the two drugs against Staphylococcus spp. For the drug-resistant S. epidermidis ATCC 35,984 strain, the MIC value for the RuNN + ampicillin treatment was 1/16 of that of ampicillin alone. Furthermore, RuNN inhibited the formation of S. aureus biofilms and reduced the total biomass of mature biofilms by ~50%. The complex displayed no hemolytic activity on erythrocytes [52].
Several ruthenium complexes with antibiotics have been reported. The activity of trimethoprim was, unfortunately, significantly decreased upon complexation with Ru(III) [124]. Complexes of the half-sandwich Ru(II)–arene complex [Ru(η6-p-cymene)] with a ciprofloxacin derivative, CipA, exhibited higher activity against E. coli and S. aureus than CipA. These complexes are labile in aqueous solutions and, therefore, their activity is probably the result of additive or synergistic effects of the [Ru(η6-p-cymene)] complex and CipA [125]. Ru(II) complexes with clotrimazole were active against mycobacteria, but were also found to be significantly toxic to mammalian cells [126]. Three Ru(III) complexes of ofloxacin, namely [Ru(OFL)2(Cl)2]Cl [Ru(OFL)(AA)(H2O)2]Cl2, where OFL = ofloxacin and AA is either glycine or alanine, were active against Gram-negative bacteria (E. coli and K. pneumoniae), but showed little to no activity on Gram-positive bacteria (S. epidermidis, S. aureus) [127]. This is unsurprising, given that fluoroquinolones are particularly effective against Gram-negative microorganisms [128].
Homo- and hetero-leptic ruthenium(II) complexes with ‘‘click’’ pyridyl-1,2,3-triazole ligands with various aliphatic and aromatic substituents (generally denoted as Ru-pytri and Ru-tripy, Figure 12) have been reported to possess good antibacterial activity. Generally, the most active complexes displayed high activity against Gram-positive strains, including MRSA (MIC = 1−8 µg/mL), but were less effective against Gram-negative bacteria (MIC = 8−128 µg/mL) [53,54]. The Ru-tripy series was generally more effective against Gram-negative bacteria than the Ru-pytri compounds [54]. Notably, the water-soluble chloride salts of the most active Ru-pytri complexes ([Ru(hexpytri)3]2+ and Ru(octpytri)3]2+, Figure 12) displayed higher activity than the gentamicin control against two strains of MRSA (MR 4393 and MR 4549). Moreover, the Ru-pytri complexes exhibited only modest cytotoxic effects at concentrations higher than the MIC values on Vero (African green monkey kidney epithelial) and human dermal keratinocyte cell lines [53]. For the Ru-tripy series, the activity appears to be closely linked to the length of the alkyl chain, with hexyl or heptyl substituents on the “click” ligands resulting in the highest activity of the corresponding homo- and hetero- leptic Ru(II) complexes. The MIC values for the most active complex of the Ru-tripy series, [Ru(hexyltripy)(heptyltripy)]Cl2, were 2 μg/mL and 8 μg/mL, respectively, against S. aureus and E. coli. Despite being generally more active than the Ru-pytri series, the Ru-tripy complexes demonstrated little to no selectivity for prokaryotic vs. eukaryotic cells (IC50 = 2–25 µM on eukaryotic cells lines—cancer and skin). With regard to their mechanism of action, transmission electron microscopy (TEM) experiments and propidium iodide assays identified cell wall/cytoplasmic membrane disruption as the main mechanism for the Ru-pytri complexes [53], while [Ru(hexyltripy)(heptyltripy)]Cl2 appears to cause abnormal cellular division [54].
Chitosan Schiff base derivatives conjugated to Ru(III) ions give polymers enhanced water solubility and antibacterial activity against Gram-positive (B. subtilis and S. aureus) and Gram-negative (E. coli, K. pneumoniae, and P. aeruginosa) bacteria [79].
5.2. Polynuclear Ruthenium (II) Complexes
5.2.1. Kinetically Inert Dinuclear Polypyridylruthenium (II) Complexes
The ruthenium polynuclear complexes, commonly known as Rubbn, are the most investigated ruthenium-based compounds with regard to their antimicrobial activities. Rubbn are kinetically inert dinuclear polypyridylruthenium (II) complexes with the general formula [(Ru(phen)2)2(μ-bbn)]4+ (Figure 13), where bbn = bis[4(4’-methyl-2-2’-bipyridyl)]-1, n-alkane. In the dinuclear Rubbn complexes, two mononuclear mono-bbn fragments (described above) are bridged by a flexible methylene linker, bbn, where n represents the number of methylene groups in the alkyl chain. Rubbn are moderately active against Gram-negative bacteria (E. coli, P. aeruginosa) and exhibit excellent activity against Gram-positive strains (including MRSA—MIC Rubb12/16 = 1 mg/L, while MIC gentamicin = 16 mg/L) [37,38]. The antibacterial activity appears to be closely linked to cellular uptake, which was, in turn, shown to be directly proportional to the length of the alkyl chain and therefore the lipophilicity of the compounds [38]. Of note, a follow-up study comparing the mononuclear [Ru(Me4phen)3]2+ (Figure 3) with the dinuclear Rubbn complexes reported significant differences in the cellular uptake and mode of action. While Rubbn are taken up by S. aureus cells via a passive transport mechanism, the cellular uptake of [Ru(Me4phen)3]2+ appears to be protein-mediated (active transport) [88]. In eukaryotic cells, however, Rubbn complexes are transported via either an active or a passive mechanism depending on the cell type and have been shown to localize to the mitochondria or the RNA-rich nucleolus [56,91,129].
The large positive charge (+4) and the hydrophobic alkyl chain are key structural features that contribute to the activity of the Rubbn complexes, allowing these compounds to pierce the bacterial cell walls and exert antibacterial activity. Based on the knowledge gained so far, two modes of action have been reported for dinuclear Rubbn complexes: membrane damage and/or interaction with nucleic acids, specifically ribosomal RNA.
Rubbn complexes were found to depolarize and permeabilize the membranes of S. aureus cells, while no membrane permeabilization was observed for [Ru(Me4phen)3]2+, although it did cause depolarization [88]. Additionally, Rubb12 was shown to embed via a pore-formation mechanism into negatively charged phospholipid multilamellar vesicles, an artificial model generally used to study drug–membrane interactions in vitro [130]. Interestingly, the corresponding Ir(III) complex, Irbb12 (with a formal charge of +6), was not taken up by cells and was inactive [60]. Molecular dynamics (MD) simulations showed that the bulky, positively charged Rubb12 spanned the bacterial membrane model at the negatively charged glycerol backbone and the bb12 linker threaded the hydrophobic core. It is yet to be determined whether the interaction with bacterial membranes results in a change of state (fluidity, charge) of the membrane and if it plays a part in the activity of Rubb12. It should be noted that the complex only interacted at the surface level with a neutrally charged eukaryotic membrane model, which could explain its lower toxicity towards healthy cells vs. bacteria (see below) [130]. This does not exclude the possibility of a protein-mediated transport of Rubb12 inside eukaryotic cells.
The bactericidal mechanism of these complexes [38] was originally presumed to be linked to their ability to bind DNA [131,132]. Indeed, the dinuclear polypyridyl complex [(phen)2Ru-(μ-tpphz)-Ru(phen)2]4+ [133] and Rubb7 [132] were found to localize to S. aureus chromosomal DNA. However, despite binding with reasonably high affinity to double-stranded DNA in vitro, Rubbn complexes prefer non-duplex structures such as bulges and hairpins[132,134,135]. Live cell microscopy experiments on E. coli cells showed that Rubb16 was found to localize at polysomes, with negligible binding to chromosomal DNA. Polysomes are formed when multiple ribosomes associate along the coding region of mRNA and therefore play an essential role in protein synthesis. The cationic charge of Rubb16 is thought to promote its interaction with the highly negatively charged polysomes. Furthermore, Rubb16 was found to induce condensation of the polysomes, an effect which is thought to hinder protein production and therefore inhibit bacterial growth [90]. Rubbn also displayed high affinity towards the serum transport proteins albumin and transferrin in vitro, which suggests that these complexes could potentially target intracellular proteins [88].
As was shown for Rubb16, targeting ribosomal RNA (rRNA) in bacteria can be advantageous for the development of selective antibacterial agents, since there are significant differences between prokaryotic and eukaryotic rRNA [136]. Moreover, in vitro experiments and MD simulations have shown that Rubb12 only interacts at a surface level with a neutral membrane bilayer mimic of a eukaryotic membrane [130]. Indeed, these inert Ru(II) complexes generally display selectivity for bacteria over normal eukaryotic cells. Although toxic to cancer cells, Rubb12/16 were much less active (up to 100-fold) against healthy cell lines [89,90,129]. In spite of the fact that Rubb16 is slightly more active against bacteria than Rubb12, the higher in vitro toxicity of Rubb16 to both healthy eukaryotic cells and red blood cells makes Rubb12 a more promising drug candidate [37].
Rubb12 injected intramuscularly was not toxic to mice at concentrations up to 64 mg/kg. Moreover, pharmacokinetic experiments have shown that 30 min post-administration, serum concentrations of Rubb12 are higher than the MIC values for Gram-positive bacteria and were maintained for more than 3 h [55]. Encapsulation of Rubb12 in cucurbit[10]uril (Rubb12⊂Q[10]) resulted in a two-fold decrease in toxicity (free Rubb12—1 mg/kg, Rubb12⊂Q[10]—2 mg/kg) when administered intravenously to mice. Interestingly, while free Rubb12 accumulated predominantly in the liver, Rubb12⊂Q[10] was found to be distributed in comparable amounts in both the liver and kidneys. A substantial reduction (∼2-fold) in the ruthenium concentrations (quantified using Inductively Coupled Plasma Mass Spectrum, ICP-MS) found in the liver was reflected by an increase (∼4-fold) in the kidneys. The significant increase in kidney accumulation is the result of the renal excretion of Rubb12⊂Q[10]. The encapsulation in cucurbit[10]uril resulted in higher cellular accumulation, lower toxicity, and faster clearance of Rubb12 [137].
As opposed to Rubbn, which bear flexible linkers, systems bridged by a rigid, extended aromatic ligand possess a property that is rather unusual for this class of complexes, that is a generally higher activity against pathogenic Gram-negative as compared with Gram-positive bacteria. The more rigid structure of these complexes is thought to play an essential role in their activity against Gram-negative strains, as well as the presence of potentially ionizable nitrogen sites and the more complex 3D structure when compared with typical drug architectures [57,138]. Thus, a range of luminescent dinuclear Ru(II) complexes bearing tetrapyridophenazine (tpphz) (Figure 13) were found to be more active against Gram-negative (both a wild-type and a multidrug-resistant strain of E. coli) than Gram-positive (a vancomycin resistant strain of E. faecalis) bacteria. [(Ru2(5-Mephen)2)2(tpphz)]4+ was the least active compound of the series, most likely due to its low water solubility. For the other three complexes, a direct, positive relationship was observed between lipophilicity and activity. The lead compound of the series, [Ru2(Me4phen)2(tpphz)]4+, was also non-toxic to healthy eukaryotic cells (Table 1). Of note, all complexes showed appreciable activity against the ESKAPE pathogens and [Ru2(Me4phen)2(tpphz)]4+ even displayed higher activity than ampicillin against the wild-type strain of E. coli and against E. faecalis. Selectivity towards the Gram-negative strains has also been observed for the mononuclear parent compound, [Ru(phen)2(tpphz)]2+, even though it was found to be significantly less active than its dinuclear derivatives against all bacterial strains [57].
[Ru2(Me4phen)2(tpphz)]4+ was shown to be actively taken up into Gram-negative bacterial cells and to disrupt the bacterial membrane structure before internalization [57], results which were further substantiated by transcriptomic analysis. Thus, the complex caused a significant downregulation of genes involved in membrane transport and the tricarboxylic acid cycle and upregulation of the spy gene [58]. The spy gene, encoding a periplasmic chaperone, is involved in zinc homeostasis and in maintaining the homeostasis of protein folding under cellular stress [139]. Thus, overexpression of the spy gene in the [Ru2(Me4phen)2(tpphz)]4+-stressed cells indicates protein damage in the outer membrane. Moreover, multi-drug resistant E. coli cells developed resistance to [Ru2(Me4phen)2(tpphz)]4+ much slower, and only in low levels, in comparison with various clinically available antibiotics. Encouragingly, [Ru2(Me4phen)2(tpphz)]4+ was active at low micromolar concentrations against other Gram-negative ESKAPE pathogens, including P. aeruginosa and A. baumannii [58].
A similar mode of action involving membrane and DNA damage was reported in the less susceptible, Gram-positive S. aureus cells. However, [Ru2(Me4phen)2(tpphz)]4+ was found to accumulate to a lower extent in Gram-positive when compared with Gram-negative bacteria, which may account for the lower efficacy of these complexes against the former. This was shown to be related to a resistance mechanism developed by Gram-positive bacteria against cationic species, which involves upregulation of the mprF gene. Overexpression of this gene leads to the accumulation of positively charged phospholipids on the outer leaflet of the cytoplasmic membrane, which repel cationic molecules, such as metal complexes. Consequently, it was found that [Ru2(Me4phen)2(tpphz)]4+ was more active against a mprF-deficient S. aureus strain and in mutant S. aureus strains missing, or with altered, wall teichoic acids [59].
This class of compounds, particularly [Ru2(Me4phen)2(tpphz)]4+, shows remarkable promise for the treatment of infections caused by Gram-negative pathogens. In addition, the lead compound displays good kinetic solubility, which suggests good bioavailability and possible oral administration [58]. Clearly, animal experiments are needed to further assess the efficacy of this class of compounds as novel antibacterial agents in vivo.
5.2.2. Chlorido Dinuclear Polypyridylruthenium (II) Complexes
A range of symmetrical dinuclear polypyridylruthenium(II) complexes with the general formula [(Ru(terpy)Cl)2(μ-bbn)]2+ (where terpy = 2,2’:6’,2’’-terpyridine) have been reported [55,60]. These labile complexes are commonly denoted as Cl-Rubbn-Cl (Figure 14). These complexes have a positive charge of +2; however, upon dissolution in water followed by the substitution of the chloride ions with solvent molecules, their charge increases to +4 [60]. The Cl-Rubb7/12/16-Cl complexes exert bactericidal activity against Gram-positive strains (S. aureus and MRSA), E. coli, and P. aeruginosa, with Cl-Rubb12-Cl being the lead compound of the series. Cl-Rubb7/12-Cl are more active than their dinuclear inert analogues; however, the Cl-Rubb16-Cl complex was significantly less active than Rubb16 [60]. It is uncertain why this variation occurs, but a possible reason is speculated to be that the enhanced cellular uptake of the Cl-Rubb7/12-Cl complexes can compensate for a reduction in activity. Since Rubb16 readily accumulates into cells, the addition of chlorido groups only results in a lower activity.
Asymmetrical chloride-containinig dinuclear polypyridylruthenium(II) complexes, Rubb7/12/16-Cl (Figure 14), have also been reported. The Rubbn-Cl complexes contain two ruthenium centers bridged by a flexible methylene linker. One ruthenium center bears a labile chlorido ligand, while the second is kinetically inert. The MIC values calculated for these complexes are comparable with those reported for the previously described Cl-Rubb7/12/16-Cl series. Furthermore, with the exception of Rubb16-Cl, Rubbn-Cl complexes exert superior antibacterial activities as compared with their inert dinuclear analogues, with Rubb12-Cl being the most active against both Gram-positive and Gram-negative strains. Rubb12-Cl was found to be 30- to 80-fold more toxic to the bacteria than to eukaryotic cell lines—two healthy cell lines (baby hamster BHK and embryonic HEK-293 kidney) and one cancerous cell line (liver carcinoma HepG2). Interestingly, large differences were found in the cytotoxic activity of Rubb7-Cl as compared with Rubb12/16-Cl. It was significantly more active towards the Gram-negative E. coli than against the Gram-positive S. aureus and MRSA, significantly more toxic to HepG2 (IC50 = 3.7 µM), and far less toxic to BHK (IC50 = 238 µM) cells than Rubb12/16-Cl. Cellular localization studies in HepG2 cells suggest that all complexes of this series were shown to accumulate preferentially in the rRNA-rich nucleolus. In addition, the large differences in the toxicity profile of the Rubbn-Cl complexes might be related to the fact that Rubb7-Cl binds to chromosomal DNA to a greater extent than Rubb12/16-Cl [56].
5.2.3. Tri-/Tetra-Nuclear Polypyridylruthenium(II) Complexes
Generally, the more lipohilic tri- and tetra- nuclear complexes, Rubb7/10/12/16-tri and Rubb7/10/12/16-tetra (Figure 15), displayed higher activities against Gram-positive and Gram-negative strains, as well as a range of bacterial clinical isolates, than the dinuclear Rubbn complexes [37,61]. The linear tetranuclear [Rubbn-tetra]8+ complexes were more active, with MIC values < 1 µM against Gram-positive bacteria, than their non-linear trinuclear [Rubbn-tri]6+ analogues. Time–kill curve experiments showed that Rubb12-tri and Rubb12-tetra exert bactericidal activity and kill bacteria within 3–8 h [55].
As opposed to the inert dinuclear Rubbn complexes, there is no apparent relationship between the antibacterial activities of the Rubbn-tri and Rubbn-tetra complexes and lipophilicity or cellular uptake. The more active tetranuclear [Rubbn-tetra]8+ complexes are less lipophilic than their trinuclear [Rubbn-tri]6+ analogues, despite the additional non-polar bbn ligand. This is unsurprising, considering the difference in the overall charge of the tri- and tetra- nuclear complexes. Moreover, even though Rubbn-tri and Rubbn-tetra complexes were more active against Gram-positive bacteria, they were shown to accumulate to a greater extent in Gram-negative bacteria [61]. Within eukaryotic Hep-G2 cells, Rubb12-tri and Rubb12-tetra have been shown to accumulate preferentially in the RNA-rich nucleolus, as was previously described for the dinuclear Rubbn complexes [91].
The mechanism of action of the Rubbn-tri and Rubbn-tetra complexes is yet to be determined, but it is thought to be related to their abilities to bind to nucleic acids and/or proteins [1,61]. The Rubbn-tri and Rubbn-tetra complexes have been shown to interact with single-stranded oligonucletides and proteins in vitro, with significantly higher affinities than their dinuclear analogues [87,88,91]. The mechanism underlying their interactions with the DNA backbone may differ for the linear tetranuclear and the three dimensional non-linear trinuclear species [87].
Two inert polypyridylruthenium(II) tetranuclear complexes, containing the bridging ligand bis[4(4’-methyl-2,2’-bipyridyl)]-1,7-heptane, with linear (Rubb7-tetra or Rubb7-TL) and non-linear (Rubb7-TNL) (Figure 15) structures, displayed good antibacterial activity against both Gram-positive (S. aureus, MRSA) and Gram-negative (E. coli, P. aeruginosa) bacteria. The non-linear (branched) species displayed slightly higher activity than the corresponding linear analogue and accumulated in the nucleolus and cytoplasm but not in the mitochondria [62].
Rubbn-tri and Rubbn-tetra complexes were found to be more toxic than Rubbn to carcinoma and healthy mammalian cell lines in vitro, with IC50 values lower than or comparable to that of cisplatin [55,89,91]. However, the tri- and tetra- nuclear complexes were still ~50-fold more toxic to Gram-positive bacteria and 25 times more toxic to the susceptible Gram-negative strains than to eukaryotic cells [55]. Rubb7-TNL was slightly less toxic to healthy eukaryotic BHK cells than its linear analogue (Table 1), yet still more toxic than cisplatin [62]. In comparison, the dinuclear ΔΔ-Rubb12 complex was ~100-fold more toxic to Gram-positive bacteria. The cytotoxic effects of the tri- and tetra- nuclear ruthenium complexes towards eukaryotic cells suggest that merely increasing the lipophilicity and charge is likely to result in decreased selectivity. Therefore, the general goal now is the development of new ruthenium complexes that are highly selective towards bacteria.
5.2.4. Other Polynuclear Complexes
The dinuclear [Ru2L3]4+ ruthenium(II) triply stranded helicate, bearing bidentate ‘‘click’’ pyridyl-1,2,3-triazole ligands, displayed modest antimicrobial activity (MIC > 256 µg/mL) [140] as compared with similar mononuclear Ru(II) complexes bearing ‘‘click’’ ligands [53,54]. However, in contrast to the similarly structured [Fe2L3]4+ helicates, the more kinetically inert [Ru2L3]4+ system proved stable over a period of at least 3 days in DMSO solutions [140].
The binuclear ruthenium (III) complexes [RuX3L]2 (X = Cl, X = Br), [RuX3L1.5]2 (X = Br), and [RuX3L2]2 (X = Br), where L stands for 2-substituted benzimidazole derivatives, were moderately active against Gram-negative bacteria (E. coli and S. typhi) as tested by the agar diffusion method. The activity on the Gram-positive bacteria S. aureus and Bacillus aureus was, however, low when compared with the standard antibiotics ampicillin and fluconazole [141].
5.3. Hetero-bi/tri-Metallic Complexes
The organometallic complex containing ruthenocene (Compound 1, Figure 16a) was moderately active against MRSA, admittedly less so than the ferrocene derivative (Compound 2, Figure 16a). The organometallic complex containing ferrocene (2) (Figure 16a) was found to generate ROS, in contrast to 1, as indicated by oxidative stress assays. Consequently, the difference in activity was suggested to result from their differing abilities to generate ROS [142].
Incorporation of ferrocene as well as ruthenium in a half-sandwich heterobimetallic complex bearing a ferrocenyl–salicylaldimine moiety (Figure 16b) showed promising activity against Mycobacterium tuberculosis. Due to the observed glycerol-dependent antimycobacterial activity, a possible mechanism of action involves disruption of glycerol metabolism and accumulation of toxic intermediate metabolites. The complex was found to possess relatively low cytotoxicity in vitro against normal microbial flora, which also suggests selectivity [143].
A Ru(II)–Pt(II) bimetallic complex, [RuCl(tpy)(dpp)PtCl2](PF6), where dpp = 2,3- bis(2-pyridyl)pyrazine and tpy = 2,2’:6’,2’’-terpyridine, was reported to inhibit the growth of E. coli cells (albeit at a relatively high concentration of 400 µM). In contrast, its monometallic Ru(II) precursor, [RuCl(tpy)(dpp)](PF6), was inactive against E. coli. The improved activity of the Ru(II)/Pt(II) heteronuclear complex was attributed to the cis-PtCl2 moiety, although the heterobimetallic complex was still less effective than cisplatin alone [144]. Although [RuCl(tpy)(dpp)PtCl2](PF6) was reported in a follow-up study to induce DNA photocleavage, the effect of light irradiation on its antibacterial activity was not assessed [145].
5.4. Ruthenium-Based Carbon-Monoxide-Releasing Molecules (CORMs)
With a unique mode of action involving ligand exchange, carbon-monoxide-releasing molecules (CORMs) represent an emerging class of biologically active organometallic derivatives. Although their mechanism of action is fairly complex and not yet fully understood (Figure 17), CORMs release carbon monoxide (CO) to bind to intracellular targets, which is partially responsible for their activity. The chemistry and antimicrobial activity of ruthenium-based CORMs have already been reviewed in several excellent works [32,146,147,148,149]. The reader can find in the following pages a summary of what has already been reviewed, as well as references to more recent research.
CORM-2, a highly lipophilic dinuclear ruthenium(II) complex with the formula [Ru(CO)3Cl2]2 (Figure 18), and the water-soluble mononuclear CORM-3, [Ru(CO)3Cl(glycinate)] (Figure 18), have been intensely investigated over the last two decades. Various reports have shown that CORM-2 and CORM-3 exhibit broad-spectrum antibacterial activity against several strains and clinical isolates of Gram-positive (S. aureus, Lactobacillus lactis) and Gram-negative (E. coli, H. pylori, Campylobacter jejuni, P. aeruginosa, Salmonella enterica serovar Typhimurium) bacteria [63,64,66,67,150,151,152,153]. Notably, CORM-3 displays bactericidal activity against antibiotic-resistant P. aeruginosa [66], H. pylori [64], and E. coli [67] strains.
Studies in various buffers indicated that significant ligand exchange is likely to occur in biological media. Both the Cl‾ and glycinate ligands of CORM-3 are labile and undergo partial or full displacement by either water molecules or other counter-ions (e.g., phosphate) existing in the buffer or growth medium constituents [154].
5.4.1. Mechanisms of Action
The Role of CO
CO is an inorganic compound that can bind hemoglobin with highly toxic effects. CO is produced endogenously as a result of heme breakdown by heme oxygenase. It is generally known to interact with metalloproteins due to its affinity towards transition metals (for instance, the ferrous ions in hemoglobin). Despite its notorious toxic effects, CO acts as a signaling molecule with important therapeutical properties, which include anti-inflammatory, anti-apoptotic and anti-proliferative effects [146]. The possibility of limiting its intrinsic severe toxicity and enhancing the biological activity has been explored with pro-drugs acting as CO-releasing molecules (CORMs), including transition metal (Mn, Ru, Fe, Mo) carbonyl complexes.
Ru-carbonyl CORMs were initially thought to act merely as vectors designed to deliver the toxic CO gas inside bacterial cells and, hence, the respiratory chain was presumed to be the main target of these molecules. The antibacterial activity of Ru-based CORMs was attributed to their ability to release CO in certain microenvironments of the cell, effecting an increase in the ratio of CO relative to O2, which eventually impedes the oxygen metabolism [35,142,150]. Indeed, there is substantial evidence in the literature that CORM-2 and CORM-3 impair aerobic respiration in E. coli [152,155,156], P. aeruginosa [66,157], H. pylori [64], C. jejuni [150], and S. enterica [152]. However, administration of BSA-Ru(II)(CO)2, an adduct formed between BSA and the hydrolytic decomposition products of CORM-3 in vitro, was demonstrated to release CO in a controlled manner in tumor-bearing mice, but did not produce any significant effect on bacterial growth in E. coli cells [158]. Additionally, in physiological conditions CORM-3 was found to release low amounts of CO inside bacterial cells (for 100 µM CORM-3, the concentration of CO detected in cells was < 0.1 µM) [159]. Thus, the toxicity of CO alone appears to be insufficient to explain the antibacterial activity of these compounds.
ROS Generation
ROS-induced oxidative stress has also been assessed as a possible mechanism of action responsible for the antimicrobial activity of CORMs. This assumption was based on the positive correlation observed in E. coli between the bactericidal activity and the ROS levels generated upon treatment with CORMs [35,160]. In vitro studies performed in aqueous solutions indicated that CORM-2 and CORM-3 are able to generate OH• [160,161] and O2•‾ [151,161] radicals. However, the amount of superoxide ions was measured to be only ~1% of the total CORM-3 concentration, which does not account for the bactericidal activity of the compound [151]. In airway smooth muscle cells, CORM-2 stimulated ROS production through inhibition of cytochromes on both NAD(P)H oxidase and the respiratory chain [162,163]. Furthermore, E. coli mutant strains in which genes encoding catalases and superoxide dismutases (SODs) have been deleted are more susceptible to CORM-2 treatment due to an increase in intracellular ROS content; this effect is alleviated upon supplementation of the culture medium with antioxidants (reduced glutathione or cysteine) [160]. For CORM-3, however, addition of catalase or SOD did not have any significant impact on its respiratory effects in E. coli, implying that peroxide or superoxide are not involved in the activity of CORM-3 in these cells [152,155]. In C. jejuni, however, CORM-3 was shown to inhibit respiration and generate hydrogen peroxide, although no effect on cell growth was observed even at concentrations as high as 500 µM [150]. Addition of various sulfur-containing antioxidants, namely cysteine, N-acetyl cysteine (NAC), or glutathione (GSH), abolished the respiratory and growth inhibitory effects of ruthenium–carbonyl CORMs in E. coli and P. aeruginosa [66,151,157,160,164]. However, this effect is presumed to be independent of the antioxidant activity of CORMs, based on two reports showing that NAC strongly inhibits the uptake of CORMs in E. coli cells [155] and a NAC–CORM-2 complex displays no activity against bacterial cells [165]. It is more likely that the Ru(II) species derived from CORMs in biological environments form adducts with exogenous compounds bearing thiol groups, which cannot be readily internalized into bacteria and are therefore less potent antibacterial agents. Non-thiol antioxidants do not alleviate the inhibitory effects of CORMs on respiration [155]. Moreover, CORM-3 was shown to impair the tricarboxylic acid (TCA) cycle, also known as the Krebs cycle, in E. coli cells treated under anaerobic conditions, suggesting that its activity extends beyond ROS generation [67]. Hence, it is unlikely that ROS-induced oxidative stress represents the main mechanism behind the CORMs’ bactericidal activity, although ROS generation probably plays some part in inhibiting the growth and respiration of CORM-2 on E. coli cells.
Membrane Damage
The bactericidal activity of CORM-3 has also been linked to membrane damage in E. coli cells, as penetration of propidium iodide [156] and N-phenyl-1-napthylamine [166], fluorescent dyes that cannot pierce healthy membranes, is allowed after CORM-3 treatments. Clearly, loss of membrane integrity can occur in the aftermath of cell death; therefore, it is not necessarily part of the antibacterial mechanism.
The Role of the Ru(II) ion Interactions with Proteins and DNA
In ruthenium-based CORMs, the Ru ion was assumed to have more of a structural role. This paradigm was based on the assumption that ruthenium–carbonyl CORMs were stable enough to reach the intracellular environment, where reducing agents (e.g., sulfites) would trigger CO release [35]. However, more recent research suggests that CORM-2 and CORM-3 undergo ligand exchange and interact with serum proteins in vivo to form protein–Ru(CO)2 adducts. CO release occurs following decomposition of these adducts [158,167,168,169,170]. Additionally, no CO release was detected in vitro upon addition of CORM-2 and CORM-3 in phosphate buffers and cell culture media in the absence of sulfur-containing reducing agents [159].
Therefore, CO release cannot be solely responsible for the cytotoxic effects of CORMs, which is further inferred by the fact that CORM-3 is toxic even for heme-deficient cells [166]. Moreover, Ru-carbonyl CORMs are considerably more active than other non-ruthenium-based CORMs [63,102,157] and inhibit aerobic respiration and bacterial growth more potently than CO gas alone [66,156]. Taking into account all of the above-mentioned arguments, it stands to reason that the ruthenium ion plays an essential role in the antimicrobial activity of these metal complexes.
The Ru(II) ion in CORM-3 was found to bind tightly to thiols. Addition of various compounds containing thiol groups in growth media protected both bacterial and mammalian cells against CORM-3. The binding affinities of CORM-3 for the compounds tested vary in the order cysteine ≈ GSH >> histidine > methionine. Moreover, a direct positive correlation was found between the protective effects of these compounds and the dissociation constants of the complexes formed between CORM-3 and the respective thiol compounds. Other amino acids (alanine and aspartate) did not exert significant protective effects. Southam et al. suggest a mechanism in which CORM-3 undergoes ligand displacement reactions in buffers or media to generate complex species in which the Ru(II) centers are readily available to bind to intracellular components such as glutathione. Another mode of action for CORMs is therefore presumed to involve Ru(II) binding to intracellular targets, impairment of glutathione-dependent systems, and disruption of redox homeostasis [159].
Indeed, CORMs have been shown to interact with various intracellular or membrane-bound proteins. CORM-3 has been shown to interact in vitro with the serum proteins myoglobin, hemoglobin, transferrin, and albumin, forming protein–Ru(II)(CO)2 adducts [167,168]. As described above, CORM-3 possesses two labile ancillary ligands (Cl‾ and glycinate), which can be readily released in aqueous media, allowing further interaction with serum proteins to occur [168]. With BSA, CORM-3 forms in vitro a [BSA-(Ru(II)(CO)2)16] complex, in which the Ru(II)(CO)2 adducts bind to histidine residues exposed on the surface of the protein. As stated above, the CO-releasing protein–Ru(II)(CO)2 complex did not have any significant effect on bacterial growth in E. coli cells [158]. The reason is unknown. In addition, CORM-2 has also been shown to inhibit urease activity in H. pylori [64] and lactate dehydrogenase in primary rat cardiocytes [171]. H. pylori urease is essential to the survival of the bacterium in the acidic gastric milieu [172]; therefore, its inhibition can represent a viable strategy against H. pylori infections. The histidine-rich active site involved in coordination of Ni(II) ions is presumed to be the target of CORM-2. It is uncertain whether urease inhibition occurs via direct binding of the Ru(II) ion to the active site accompanied by Ni(II) displacement, or CO binding to the Ni(II) ion in the active site.
Soft and borderline transition metals have been shown to bind to Fe–S clusters, which are important cofactors of various enzymes including several pertaining to the Krebs (or TCA) cycle [173]. CO is also reported to bind to iron–sulfur clusters in a redox-dependent manner [174]. Therefore, Fe–S enzymes have been studied as potential targets for CORMs. Indeed, treatment of E. coli cells with CORM-2 resulted in an increase in intracellular iron, suggesting degradation of the Fe–S clusters. This assumption was further supported by the significant inhibition of two Fe–S proteins, aconitase B and glutamate synthase, following exposure of E. coli extracts to CORM-2. Although the presence of intracellular Fe–S clusters was shown to correlate with the antimicrobial activity of CORM-2, it was not clearly determined whether the Ru(II) ion of CORM-2 binds directly to Fe–S clusters, or if the degradation of the clusters occurs indirectly as a result of other processes [160]. However, a cell extract from E. coli overexpressing aconitase B displayed a 50% decrease in the activity of the enzyme after incubation with CORM-3, relative to untreated cells, suggesting that the protein–CORM-3 complex occurs at a post-translational level. Additionally, recent metabolomics studies in E. coli cells revealed that CORM-3 inhibits the activity of several Fe–S proteins, namely the glutamate synthase GOGAT and enzymes of the TCA cycle (aconitase B, isocitrate dehydrogenase, and fumarase). In response to the severe imbalance in the energy and redox homeostasis caused by the Ru-carbonyl complex, activation of the glycolysis pathway was detected in the CORM-3-stressed cells. Notably, other non-CO-releasing Ru(II) species, used as controls, were non-toxic to E. coli cells and had no effect on the Fe–S enzymes at the concentration used in this study (120 µM—a growth inhibitory but nonlethal concentration of CORM-3) [67].
Although numerous cytotoxic ruthenium complexes developed as anticancer agents have been shown to interact with DNA, no in vitro or in vivo studies clearly demonstrate whether Ru-based CORMs bind directly to DNA or not. However, CORM-2 has been shown to induce DNA damage and increase the expression of a double-strand break repair gene, recA, in E. coli [65,160]. DNA damage can be the result of CORM-2-induced generation of intracellular ROS, although this has not been clearly established [65].
Effects on Gene Expression
Transcriptome studies on E. coli revealed that CORM treatments under either aerobic or anaerobic conditions trigger complex transcriptional responses of gene expression [151,156,166,175,176] that exceed those induced by CO alone [177]. CORM-2 and CORM-3 downregulate genes involved in aerobic respiration, energy metabolism, and biosynthesis pathways and upregulate those involved in the SOS response and DNA damage and repair mechanisms. A recent gene profiling study analyzed the effects induced by CORM-2 exposure on a multidrug-resistant extended-spectrum beta-lactamase (ESBL)-producing uropathogenic E. coli clinical isolate [65]. Numerous genes encoding the NADH dehydrogenase complex were repressed by CORM-2 [65], as was previously shown for CORM-3 in the E. coli K12 strain [156]. Transcriptomics analysis of E. coli cells treated with CORM-3 indicated altered expression of the cytochrome genes cyoABCDE and cydAB [151,156]. However, CORM-2 had no effect on the expression of cytochrome genes, which could be attributed to the differences in the growth media [65].
Exposure to CORM-2 and CORM-3 increased the expression of genes coding for proteins with roles in stress response and adaptation, e.g., ibBA, ibpA, and spy [65,151,156,166,176]. The spy gene appears to be one of the main non-heme targets for CORMs. Several genes coding for multidrug efflux pump proteins were also upregulated by CORM-2 [65] and CORM-3 [166]. Upregulation of multidrug efflux pump systems has been shown to lead to the development of resistant phenotypes over time [178]. However, the growth inhibitory activity of CORM-2 was not diminished by repeated exposure (20 times), neither in the multidrug-resistant ESBL-producing E. coli strain, nor in two other antibiotic-susceptible E. coli strains [65].
Significant upregulation has also been found for genes involved in metal homeostasis, such as iron or zinc [151,156,166], and genes involved in the uptake and/or metabolism of sulfur compounds (sulphate-thiosulphate, methionine, cysteine, glutathione) and the sulfur starvation response [67,151,166,176]. In agreement with the already-discussed inhibitory effects of CORMs on Fe–S enzymes [67,160], genes involved in Fe–S cluster biosynthesis and repair are also upregulated by CORM-2 and CORM-3 [151,166,176]. Transcriptomic data, therefore, correlate well with the in vitro observation that sulfur species represent intracellular targets of Ru(II)-based CORM [151].
5.4.2. Ruthenium-Based CORM Polymers
Encapsulation of drugs into polymers is a modern therapeutic strategy that makes use of building blocks with 3D structures that enable controlled ligand exchange [179]. Conjugation to lipophilic polymers reduces the access of water molecules to CORMs, causing the solvent-assisted ligand exchange reactions to occur at a slower, sustainable pace. A Ru-based CORM was conjugated to the side chain of polymeric fibers bearing different thiol moieties, yielding the three water-soluble CO-releasing macromolecules CORM-polymers 1–3 (Figure 19). The resulting polymers have been shown to exhibit bactericidal activity against P. aeruginosa and to prevent biofilm formation more efficiently than CORM-2, most likely due to their high CO-loading capacity, controlled release of CO, and prolonged half-lives. Notably, the antimicrobial activity was not directly proportional to the half-lives of the complexes, since CORM-polymer 2 was the most active compound of the series, while CORM-polymer 1 had the longest half-life [180].
5.4.3. Cellular Uptake
The mechanisms of uptake of CORM-2 and CORM-3 complexes are unknown. It is also unclear which are the Ru-CORM-derived species that pierce the bacterial membranes and it is likely that different species use different mechanisms of uptake. However, ruthenium species (quantified using ICP-MS) have been found to accumulate to high levels in E. coli cells treated with either CORM-2 or CORM-3 [155,156]. CORM-3 was found to be rapidly taken up by E. coli cells at an initial rate of 85 µM/min over the first 2.5 minutes after treatment, with intracellular Ru levels reaching a plateau after ∼40 min [151]. CORM-3 accumulated to higher levels in S. enterica serovar Typhimurium than in E. coli, and at a faster initial uptake rate (>120 µM/min). This may explain, at least partially, why Salmonella strains are more susceptible to CORM-3 than E. coli [153]. Notably, simultaneous addition of NAC and CORM-2 or CORM-3 reduced the ruthenium accumulation inside bacterial cells, which is probably why exogenous thiols, such as NAC, are able to interfere with the antibacterial activity of both CORM-2 and CORM-3 [155]. These findings also suggest that the bactericidal effects of these compounds are dependent on the ruthenium uptake by the bacterial cells [151]. It has been suggested that Ru–based CORMs could be transported actively, or diffuse, inside the cells via an unidentified route against the concentration gradient and, due to the reactions that occur inside the cells, uptake can continue passively [153].
5.4.4. Toxicity and Pharmacokinetics
The more lipophilic, DMSO-soluble, CORM-2 is generally more toxic to mammalian cells than the water-soluble CORM-3. It has been suggested that the use of DMSO is at least partially accountable for the increased cytotoxicity [102,171,181]. Toxic concentrations reported for eukaryotic cells are considerably higher than the MIC values [66,102,153,164,169,170,171,181]. For instance, the bactericidal effects against P. aeruginosa occurred at concentrations of CORM-3 that are 50-fold lower than the toxic concentrations for macrophages [66]. However, survival of the mammalian cells was drastically increased by the presence of exogenous thiols in the growth media. For instance, treatment with 25 µM CORM-3 in phosphate-buffered saline (PBS) decreased survival relative to untreated human colon carcinoma RKO cells in PBS by 92%, compared with only 23% in RPMI-1640 growth medium, whilst in DMEM the survival rate was enhanced relative to untreated controls [159].
In vivo studies revealed that a two-week CORM-3 treatment (with increasing doses from 7.5 to 22.5 mg/kg) caused no mortality or any apparent side effects to healthy mice [66]. In contrast, consecutive administrations of 15–37 mg CORM-3/kg in rats caused severe liver and kidney damage after 21 days of treatment. Biodistribution studies in CORM-3-treated mice concluded that the ruthenium species derived from CORM-3 mostly accumulated in the blood for the first hour after the intravenous administration and then were slowly distributed to the kidneys, liver, lungs, and heart. Notably, only trace amounts of ruthenium were found in the brain, suggesting that the complex and its derived species did not cross the blood–brain barrier. Both ruthenium and elevated levels of protein were found in the urine of the CORM-3-treated mice, indicating kidney damage. Moreover, the RuII center was oxidized to RuIII in vivo by enzymes such as cytochrome P450 [170].
5.4.5. In Vivo Studies Regarding the Antibacterial Activity of CORMs
Several studies have reported the in vivo antibacterial activity of Ru–carbonyl CORMs [66,157,182]. In a murine model of polymicrobial sepsis, treatment with 10 μM CORM-2/kg 12 and 2 h before the inoculation of bacteria resulted in a significant decrease in bacterial counts relative to the vehicle-treated mice. CORM-2 improved the survival rates of heme oxygenase (HO)-1 null mice, mutants that are more susceptible to polymicrobial infection, even when administered intraperitoneally after the onset of sepsis [182]. Administration of CORM-2 (12.8 mg/kg) was also shown to significantly increase the survival rates of BALB/c mice infected with P. aeruginosa [157].
Injections of CORM-3 (7.5–22.5 mg/kg) in the murine model of P. aeruginosa infection reduced bacterial counts in the spleen and increased the survival rates of the infected mice (from 20% in the vehicle-treated group to 100% in the CORM-3-treated mice). Moreover, treatment with CORM-3 reduced bacterial counts in the spleen of immunosuppressed mice to a similar degree to in immunocompetent mice, suggesting a direct, rather than host-mediated, antibacterial effect of CORM-3 [66].
The modes of action in vivo of these compounds are still unknown and require further assessment. The promising results of these studies, however, pave the way for a more extensive preclinical evaluation of the antibacterial efficacy of Ru-based CORMs.
5.5. Ruthenium Complexes in Antimicrobial Photodynamic Therapy
Photodynamic therapy (PDT) is a therapeutic strategy that makes use of a combination of photosensitive molecules, light, and molecular oxygen. PDT has been investigated against a range of medical conditions, including atherosclerosis, psoriasis, and malignant cancers [183,184]. Antimicrobial photodynamic therapy (aPDT) has been used against a variety of microbial pathogens (bacteria, fungi, and viruses). It relies on the ability of a compound, a photosensitizer, to generate singlet oxygen (1O2) and other ROS upon light irradiation, causing bacteria inactivation [185].
Ru(II) complexes, particularly Ru(II)–polypyridyl complexes, have been intensively investigated for PDT applications against malignant cancers due to their optical properties, such as the long luminescence lifetimes of the triplet metal-to-ligand charge transfer (MLCT) excited state [184,186]. The remarkable potential of Ru complexes as PDT agents has been confirmed by TLD-1433 [18], which is currently undergoing phase II clinical studies as a photosensitizer for PDT against bladder cancer.
Taking into account the remarkable success of Ru(II)-based PDT agents in the treatment of cancer, several Ru(II) complexes have been considered as potential photosensitizers for aPDT. For instance, [Ru(dmob)3]2+ (Figure 20), where dmob = 4,4’-dimethoxy-2,2’-bipyridine, was more active than the corresponding complexes bearing bpy and phen ligands against S. aureus, P. aeruginosa, and C. albicans strains. The enhanced activity was attributed to the increased lipophilicity of the complex due to the presence of methoxy groups in its structure, which can translate to enhanced uptake by the bacterial cells [68].
[Ru(bpy)2(dppn)]2+ (Figure 20), where bpy = 2,2’-bipyridine and dppn = 4,5,9,16-tetraazadibenzo[a,c]-napthacene, was shown to cause potent photoinactivation of E. coli cells, while dark incubation with the compound had no effect on the viability of the microorganism. Treatment with [Ru(bpy)2(dppn)]2+ led to a 70% CFU decrease at 0.1 µM and complete inactivation at 0.5 µM following light activation [187].
The ruthenium(II) complex cis-[Ru(bpy)2(INH)2]2+ (Figure 20), where INH = isoniazid, has been shown to undergo stepwise photoactivation in aqueous media after irradiation with 465 nm blue light. The resulting products of this process are two equivalents of the antituberculosis drug isoniazid and cis-[Ru(bpy)2(H2O)2]2+. cis-[Ru(bpy)2(INH)2]2+ was inactive in the dark; however, upon photoactivation, it was 5.5-fold more efficient against Mycobacterium smegmatis in comparison with isoniazid. Notably, cis-[Ru(bpy)2(INH)2]2+ displayed high selectivity towards mycobacteria over healthy MRC-5 human lung cells in vitro [69].
A heterobimetallic complex [Ru(Ph2phen)2(dpp)PtCl2]2+ (Figure 20), where Ph2phen = 4,7-diphenyl-1,10-phenanthroline and dpp = 2,3-bis(2-pyridyl)pyrazine, has been reported to induce photocytotoxic effects in E. coli cells in the presence of oxygen and visible light. The dose required for complete cell growth inhibition under visible light irradiation was 5 µM, as opposed to 20 µM in the dark [70]. In comparison, cisplatin induced complete cell growth inhibition at 5 µM in the dark, but a similar complex, [RuCl(tpy)(dpp)PtCl2]+ (see above), had the same effect at 200 µM [145]. Inside the cells, photoactivated [Ru(Ph2phen)2(dpp)PtCl2]2+ was shown to bind to chromosomal DNA [70]. Further experiments are needed to assess the nature of the DNA binding, as well as what species are responsible for the activity.
Incorporation in or conjugation with biocompatible polymers has been used as an efficient strategy to increase the ability of ruthenium complexes to penetrate bacterial cell walls and therefore their antimicrobial activity. A Ru(II)–polypyridyl complex, [Ru(bpy)2-dppz-7-hydroxymethyl][PF6]2 (RuOH), where bpy = 2,2’-bipyridine and dppz = dipyrido[3,2-a:2;2’,3’-c]phenazine, was found to be inactive against Gram-positive and Gram-negative bacteria. This lack of activity was thought to stem from its low uptake by bacterial cells. In order to solve this issue, RuOH was conjugated to the end-chain of a hydrophobic polylactide (PLA) polymer to form ruthenium–polylactide (RuPLA) nanoconjugates (Figure 20). Although RuPLA nanoconjugates displayed superior photophysical properties, including luminescence and enhanced 1O2 generation, they were only moderately active against Gram-positive (S. aureus, S. epidermidis) bacteria, with MIC values of 25 µM. The RuPLA nanoconjugates remained non-toxic to the Gram-negative (E. coli and P. aeruginosa) bacterial strains and displayed phototoxicity against human cervical carcinoma cells (IC50 = 4.4 µM) [185].
In a recent study, the antibacterial activity of the purely inorganic polymer [Ru(CO)2Cl2]n (Figure 20), with repeating dicarbonyldichlororuthenium (II) monomers, was studied against E. coli and S. aureus. Significant inhibitory effects were observed on both strains at concentrations as low as 6.6 ng/mL after irradiation with 365 nm UV light. Interestingly, the polymer displayed stronger photobactericidal activity against the Gram-negative E. coli (MIC ~33 ng/mL) than the Gram-positive S. aureus (MIC ~166 ng/mL) bacteria. In addition, [Ru(CO)2Cl2]n remained non-toxic to human dermal fibroblasts and red blood cells at concentrations much higher than the MIC values. Although the complex was considerably toxic to both bacterial strains in the dark, the antibacterial activity of [Ru(CO)2Cl2]n was significantly increased upon photoirradiation, which can be attributed to the enhanced generation of ROS under UV light. SEM analysis revealed that its mode of action might involve disruption of bacterial membranes. Moreover, [Ru(CO)2Cl2]n was able to cause morphological changes to biofilm structures and to disassemble the biofilm matrix [71]. It should be noted that although the structure of [Ru(CO)2Cl2]n is similar to that of CORMs, there is no information available in the literature on the ability of the polymer to release CO or undergo ligand exchange in aqueous media.
The antibacterial photosensitizing activity towards a panel of bacterial strains has been assessed for seventeen homo- or heteroleptic polypyridyl Ru(II) complexes with the following formulae: [Ru(Phen)3](PF6)2, [Ru(Phen)2(Phen-X)](PF6)2, [Ru(Phen)(Phen-X)2](PF6)2, [Ru(Phen-X)3](PF6)2, [Ru(Phen-X)2Cl2], or [Ru(Phen)2Cl2] (Figure 21), varying due to the number and the nature of the substituents. With regard to the most active complexes, 2, 5, and 6 stood out, 5 was highly efficient against MRSA N315 even without light irradiation, and 2 demonstrated activity against four S. aureus strains, one E. coli strain, and three P. aeruginosa strains. However, 2 and 5 were more toxic towards eukaryotic cells upon light irradiation, with 6 being non-toxic. The counterion (PF6‾ vs. Cl‾) did not appear to have a significant effect on the antibacterial activity. In contrast, a dicationic charge was vital to the activity, taking into account that the two neutral Ru(II) complexes, 16 and 17, were inactive. Surprisingly, the best photosensitizers for 1O2 production (8, 9, 10, 15) did not correspond to the most efficient aPDT agents (2, 5, 6). The ability of the complexes to interact efficiently with bacteria seems to be crucial for aPDT activity, considering the short half-life of ROS generated upon light irradiation. Thus, solely increasing 1O2 production is not sufficient to yield more efficient aPDT agents. Parameters impacting the interactions with bacteria, such as lipophilicity and ability to form aggregates, should also be considered in the development of optimized future compounds for aPDT [184].
6. Antiparasitic Activity of Ruthenium Complexes
Parasitic infections, including malaria (Plasmodium sp.), Chagas’ disease (Trypanosoma cruzi), African trypanosomiasis (Trypanosoma brucei), and leishmaniasis (Leishmania sp.), mainly affect the tropical and subtropical regions of Africa and Asia and only a narrow spectrum of effective drugs is available for treatment. In this context, several ruthenium complexes have been reported as efficient antiparasitic agents active against malaria, leishmaniasis, trypanosomiasis, and Chagas’ disease [188]. Generally, the enhanced antiplasmodial activity of these complexes when compared to the free ligands is thought to be related to their increased lipophilicity, which translates to increased uptake into the parasite’s cells and/or increased ability to evade the parasite’s drug efflux pumps.
6.1. Antiplasmodial Activity
Malaria is a highly infectious parasitic disease, with over 40% of the world’s population living in an endemic region. Malaria parasites belong to the genus Plasmodium, the most virulent strain being P. falciparum [189,190]. Conventional treatment strategies use either quinoline-based drugs, such as chloroquine (CQ)) and its derivatives, or fixed-dose combination therapies containing a derivative of the Chinese natural product artemisinin. The increasing widespread resistance to these compounds requires urgent attention to the development of new therapeutic strategies [190,191].
An organometallic complex, [RuCl2(CQ)]2 (Figure 22a), where CQ = chloroquine, displayed 2–5-fold increased activity against P. falciparum compared with chloroquine diphosphate in vitro [192]. Moreover, the complex was significantly more active when compared with its organic derivative in mice infected with Plasmodium berghei, with no apparent signs of acute toxicity up to 30 days after treatment [193]. [RuCl2(CQ)]2 was shown to bind to hematin and inhibit aggregation of β-hematin (synthetic hemozoin—a target of the malaria parasite) in vitro, albeit to a slightly lower extent than chloroquine diphosphate. However, the heme aggregation inhibitory activity of the complex is significantly higher than that displayed by chloroquine, suggesting that the main target of the complex is the heme aggregation process. [RuCl2(CQ)]2 was shown to be significantly more lipophilic than chloroquine diphosphate, suggesting that the addition of Ru(II) induced drastic changes in the pharmacokinetic profile of the organometallic compound. One chlorido ligand from each of the two Ru(II) centers is displaced by water molecules upon addition to aqueous solutions. The resulting species, [RuCl(OH2)3(CQ)]2[Cl]2, is considered to be the active species in vitro and in vivo. The enhanced activity of the complex against CQ-resistant strains of P. falciparum was suggested to relate to its lipophilicity. This can be explained by the fact that the parasite efflux pump, usually involved in the resistance mechanism to chloroquine, has a lower ability to bind to highly lipophilic drugs [192].
CQ has also been used as a chelating ligand in a series of organoruthenium complexes with the general formula [RuCQ(η6-C10H14)(N–H)]2+, where η6-C10H14 is α-phellandrene and N–H is either 2’-bipyridine (BCQ), 5,5’-dimethyl-2,2’-bipyridine (MCQ), 1,10-phenanthroline (FCQ), or 4,7-diphenyl-1,10-phenanthroline (FFCQ). As was previously shown for [RuCl2(CQ)]2, the Ru–CQ bonds are stable, and CQ is not released upon aquation. The organoruthenium complexes displayed intraerythrocytic activity against CQ-sensitive and -resistant strains of P. falciparum. Unlike CQ, the complexes exerted moderate activity against the liver stage and potent activity against the sexual stage of the parasite, suggesting that they operate via a different mechanism than that of CQ. It has been shown that [RuCQ(η6-C10H14)(N–H)]2+ induces oxidative stress in the parasite, which might be linked to their mode of action. In addition, the organoruthenium complexes displayed low mammalian cytotoxicity and inhibited parasitemia in mice infected with P. berghei [194].
A range of Ru(II)–arene complexes were developed in the knowledge that increasing the lipophilic properties of a drug is likely to increase passive diffusion through membranes and hence the antiplasmodial activity. For instance, a series of half-sandwich Ru(II) complexes with aryl-functionalized organosilane thiosemicarbazone ligands were more active against P. falciparum at low micromolar concentrations (2.29–6.66 µM) and less cytotoxic to the Chinese Hamster Ovarian (CHO) cell line in comparison with the corresponding free ligands. It should be stated that the activity of the complexes was still much lower than that of both CQ and artesunate, which were used as controls. However, the complexes also displayed much lower resistance index values relative to the control drugs, which suggests that the parasites are less likely to develop cross-resistance to the metal complexes [195].
An enhancement of the antiplasmodial activity has also been observed for cyclometallated Ru(II) complexes of 2-phenylbenzimidazoles (Figure 22b), when compared with the free ligands. These complexes were found to be active against CQ-sensitive and multidrug-resistant P. falciparum strains, with IC50 values in the low to submicromolar range (0.12–3.02 µM). The nature of the substituent on the η6-p-cymene moiety does not seem to influence the activity to a great extent. Although CQ was still more active than the cyclometallated complexes against both strains, the latter displayed lower resistance index values relative to CQ. In addition, the metal complexes displayed relatively low cytotoxicity against the mammalian CHO cells. Notably, the Ru(II) complexes were found to be more active than the Ir(III) analogues on the resistant strain [196], which was also reported for other Ru(II)–arene complexes [197]. PTA-derived ruthenium(II) quinoline complexes (Figure 22c) were, however, generally less effective against CQ-sensitive and resistant strains of P. falciparum than their Ir(III) correspondents, but were also much less toxic to the CHO cells. In addition, these RAPTA-like complexes inhibited β-hematin formation, suggesting that their mechanism of action is similar to that of CQ [198].
Di- and tri- nuclear Ru(II)-η6-p-cymene complexes (Figure 23a), in which the ruthenium centers are bridged by pyridyl aromatic ether ligands, were evaluated against CQ-sensitive and -resistant P. falciparum strains. While the dinuclear derivative displayed only moderate activity, the trinuclear complex proved to be highly active in both strains, displaying activities in the nanomolar range (IC50 = 240 nM and 670 nM for the CQ-sensitive and -resistant P. falciparum strains, respectively). The trinuclear complex was also able to inhibit more efficiently β-hematin formation in vitro, in comparison with the dinuclear derivative, which suggests that hemozoin might be a target of the complexes in vivo. Notably, the trinuclear Ru(II) complex was only slightly more toxic than the corresponding tripyridyl ether ligand, indicating that it was the incorporation of a triazine moiety that had a more significant impact on activity [197]. This was confirmed by the fact that trinuclear Ru(II)-η6-p-cymene complexes, in which the ruthenium centers are bridged by pyridyl aromatic ester ligands lacking the triazine moiety (Figure 23b), are much less efficient antiparasitic agents [199].
Using ‘old’ drugs to assist in the search for new agents that are more efficient for either common or rare diseases is the scope of a relatively new therapeutic strategy called drug repositioning/repurposing. For instance, a series of ferrocenyl and ruthenocenyl derivatives incorporating tamoxifen-based compounds were tested against CQ-resistant P. falciparum blood forms. Tamoxifen (Figure 24) is an anticancer agent used in current treatment plans to prevent and treat breast cancer. The results within this series indicated that the ruthenocenyl-containing complexes (Figure 24) were more active than their ferrocenyl analogues, but still only displayed moderate activity (IC50 = 4.7–16.5 mM) against P. falciparum. The ruthenocenyl complexes were considered nontoxic to HepG2 cells [200].
6.2. Antitrypanosomal Activity
Chagas’ disease (American trypanosomiasis) affects millions of people worldwide, mainly in Central and South America, where the disease is endemic. It is a life-threatening disease caused by the parasite Trypanosoma cruzi. No vaccines are currently available, and treatment options are limited to only two drugs, benznidazole and nirfurtimox [201]. Sleeping sickness (African trypanosomiasis) predominantly affects people living in sub-Saharan Africa and is transmitted by the bite of the tsetse fly. The disease is caused by the insect-borne T. brucei parasite [202].
Two Ru(II)–NO donor compounds, namely trans-[Ru(NO)(NH3)4(isn)](BF4)3 (Figure 25) where isn = isonicotinamide and trans-[Ru(NO)(NH3)4(imN)](BF4)3 (Figure 25) where imN = imidazole, displayed significant activity against T. cruzi both in vitro and in vivo. NO release upon reduction of the ruthenium nitrosyls in culture cells and animal models is thought to play an essential role in the antiproliferative and trypanocidal activities. Notably, trans-[Ru(NO)(NH3)4(imN)](BF4)3 allowed for survival of up to 80% of infected mice at a much lower dose (100 nmol/kg/day) than that required for benznidazole (385 μmol/kg/day) [203,204]. Ru(II) complexes with the formulae cis-[Ru(NO)(bpy)2(imN)](PF6)3 and cis-[Ru(NO)(bpy)2SO3]PF6 displayed inhibitory effects on the T. cruzi glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (IC50 = 89 and 153 μM, respectively), which is a potential molecular target. These compounds exhibited in vitro and in vivo trypanocidal activities at doses up to 1000-fold lower than the clinical dose for benznidazole [205]. Furthermore, in a series of nitro/nitrosyl-Ru(II) complexes, cis, trans-[RuCl(NO)(dppb)(5,5’-mebipy)](PF6)2, where 5,5′-mebipy = 5,5′-dimethyl-2,2′-bipyridine and dppb = 1,4-bis(diphenylphosphino)butane, was the most active compound. The complex displayed an IC50 of 2.1 µM against trypomastigotes (the form of the parasite during the acute stage of the disease) and an IC50 of 1.3 µM against amastigotes (the form of the parasite during the chronic stage of the disease), while it was less toxic to macrophages. Moreover, the complex exerted synergistic activity with benznidazole in vitro against trypomastigotes and in vivo in infected mice [206].
A series of symmetric trinuclear ruthenium complexes bearing azanaphthalene ligands with the general formula [Ru3O(CH3COO)6(L)3]PF6 (Figure 26), where L = (1) quinazoline (qui), (2) 5-nitroisoquinoline (5-nitroiq), (3) 5-bromoisoquinoline (5-briq), (4) isoquinoline (iq), (5) 5-aminoisoquinoline (5- amiq), and (6) 5,6,7,8-tetrahydroisoquinoline (thiq), were developed. All complexes presented in vitro trypanocidal activity, complex 6 being the lead compound of the series, with IC50 values of 1.39 µM against trypomastigotes and 1.06 µM against amastigotes. Complex 6 was up to 10 times more effective than benznidazole, while being essentially non-toxic to healthy mammalian cells (SI trypomastigote: 160, SI amastigote: 209) [207].
A range of Ru (II)–cyclopentadienyl thiosemicarbazone complexes displayed sub- or micromolar IC50 values against T. cruzi and T. brucei. Notably, [RuCp(PPh3)L] (Figure 27), where HL is the N-methyl derivative of 5-nitrofuryl containing thiosemicarbazone and Cp is cyclopentadienyl, exhibited high (IC50 T. cruzi = 0.41 µM; IC50 T. brucei = 3.5 µM) and selective activity (SI T. cruzi > 49 and SI T. brucei SI > 6). These complexes had the ability to interact with DNA in vitro, but no correlation with the biological activity was observed [208]. A Ru(II)–cyclopentadienyl clotrimazole complex, [RuCp(PPh3)2(CTZ)](CF3SO3) (Figure 27), where Cp = cyclopentadienyl and CTZ = clotrimazole, was more cytotoxic on T. cruzi than nifurtimox. With regard to its mechanism of action, the complex was shown to impair the sterol biosynthetic pathway in T. cruzi [209]. In another series of Ru(II)–cyclopentadienyl clotrimazole complexes, [RuII(p-cymene)(bpy)(CTZ)][BF4]2 (Figure 27) was found to be the most active compound, increasing the activity of CTZ by a factor of 58 against T. cruzi (IC50 = 0.1 µM), with no appreciable toxicity to human osteoblasts [210].
6.3. Antileishmaniasis Activity
Leishmaniasis is a disease caused by protozoan parasites of the genus Leishmania and is characterized by high morbidity. It is estimated that more than 1 billion people live in endemic areas, with more than 1 million new cases of leishmaniasis occurring annually. Current treatment for leishmaniasis relies on the use of pentavalent antimonials and other drugs, such as pentamidine isethionate, amphotericin B, and miltefosine. However, antileishmanial treatment cannot provide a sterile cure, and the parasite can cause a relapse when the human body is immunosuppressed [211,212].
An improved antiplasmodial activity in comparison with that of the free ligand has been reported for Ru(II)–lapachol complexes. [RuCl2(Lap)(dppb)] was active against L. amazonensis promastigotes and infected macrophages, with submicromolar IC50 values comparable with that of the reference drug, amphotericin B. In addition, the complex was not toxic to macrophages at concentrations much higher than the IC50 values [212].
[RuII(p-cymene)(bpy)(CTZ)][BF4]2 (Figure 27) was found to be active against promastigotes of L. major at nanomolar concentrations (IC50 = 15 nM) and displayed no appreciable toxicity against human osteoblasts (SI > 500). Moreover, in L. major-infected mice macrophages, the complex caused a significant inhibition of the proliferation of intracellular amastigotes (IC70 = 29 nM) [210].
7. Antiviral Activity of Ruthenium Complexes
7.1. Anti-HIV Activity
The mixed-valent tetranuclear ruthenium–oxo oxalato cluster Na7[Ru4(µ3-O)4(C2O4)6] exerted promising anti-HIV-1 activity with over 98% inhibition of viral replication toward the R5-tropic HIV-1 strain at a 5 µM concentration and similar inhibitory activity toward X4-tropic viral replication. Moreover, the ruthenium–oxo oxalato cluster displayed selective anti-viral activity, with over 90% survival of the host cells registered at concentrations up to 50 µM. Notably, Na7[Ru4(µ3-O)4(C2O4)6] was 10-fold more effective against HIV-1 reverse transcriptase (IC50 = 1.9 nM) than the commonly used HIV-1 RT inhibitor 3’-azido-3’-deoxythmidine-5’-triphosphate (IC50 = 68 nM) [213].
Another ruthenium cluster, [H4Ru4(η6-p-benzene)4]2+, displayed selective activity against Polio virus, without inhibiting the growth of healthy human cells. It has been suggested that the complex might only be cytotoxic in Polio-infected cells, as the virus alters cell membrane permeability, facilitating passage for the cluster [94].
[Ru(bpy)2eilatin]2+ (Figure 28), where eilatin = dibenzotetraazaperylene, inhibited HIV-1 replication in CD4+ HeLa cells and in human peripheral blood monocytes (IC50 values ~1 µM). Eilatin is a fused, heptacyclic aromatic alkaloid that was isolated from the sea squirts belonging to Eudistoma sp., reported to possess cytotoxic and antiproliferative activities. [Ru(bpy)2eilatin]2+ is a kinetically inert complex and in vitro studies suggest that its mechanism of action relies upon inhibition of key protein–RNA interactions. The planar structure of the bidentate ligand, eilatin, was found to be essential for the activity of the complex [214].
7.2. Anti-SARS-Cov-2 Activity
In spite of the extensive vaccination campaigns that are currently ongoing, the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is spreading at an alarming pace across the world. The large number of mutations rendered several new variants less susceptible to treatment options, and possibly to vaccines. Thus, there is still an urgent need for the development of new drugs with a broader spectrum of activity [215,216].
BOLD-100 (sodium trans-[tetrachlorobis(1H-indazole)ruthenate(III)], KP1339, Figure 29), developed as an anticancer agent, was shown to selectively inhibit stress-induced upregulation of 78-kDa glucose-regulated protein (GRP78) [217,218,219], which is a master chaperone protein serving critical functions in the endoplasmic reticulum of normal cells [220,221]. The interaction of SARS-CoV-2 spike protein with the GRP78 protein located on the cell membrane can mediate viral entry. Therefore, disruption of this interaction may be used to develop novel therapeutic strategies against coronavirus [222]. Indeed, BOLD-100 was reported to reduce viral loads in various COVID-19 variants, including the more virulent B.1.1.7, originally identified in the United Kingdom. Unlike vaccines, which are more effective against certain viral variants, BOLD-100, with a broad antiviral mechanism of action, appears to remain active on all mutant strains [223]. In vivo studies are currently in progress.
BOLD-100 has a tolerable safety profile (minimal neurological or hematological effects), as was shown in a recently completed phase I clinical study involving 41 patients with advanced cancer. Moreover, it is currently undergoing clinical trials in combination with FOLFOX chemotherapy (which includes folinic acid, 5-fluorouracil, and oxaliplatin) for gastrointestinal solid tumors [224]. Therefore, BOLD-100 has already been successfully developed as a clinical-stage product, which suggests its potential for rapid further clinical development against COVID-19.
Additionally, [Ru(bpy)3]2+ is used in the Elecsys® Anti-SARS-CoV-2, a chemiluminescence immunoassay intended for qualitative detection of antibodies to SARS-CoV-2 in human serum and plasma, which has been approved worldwide. In this assay, the SARS-CoV-2-specific recombinant antigen is labeled with the ruthenium complex [225,226]. Other metal complexes identified as potential anti-SARS-Cov-2 agents include auranofin [227,228] and Re(I) tricarbonyl complexes [229].
8. Conclusions
Ruthenium-based antimicrobial agents have a fairly complex mode of action involving multiple mechanisms acting in synergy. The knowledge gained so far in this area suggests that the activity of ruthenium compounds against microbial cells is based upon their ability to induce oxidative stress, interact with the genetic material, proteins, or other intracellular targets, and/or damage the cell membranes. The complex interplay between these modes of action is likely responsible for the activity of some ruthenium-based compounds against drug-resistant strains.
Generally, ruthenium complexes exert excellent activity against Gram-positive bacteria (e.g., S. aureus and MRSA) and, with some exceptions (see, for instance, the dinuclear polypyridylruthenium(II) complexes and ruthenium-based CORMs), display lower activity towards Gram-negative strains (e.g., E. coli and P. aeruginosa). With regard to their activity against Gram-negative bacteria, one can notice a trend towards higher efficacy against E. coli when compared with P. aeruginosa and K. pneumoniae. For most classes of compounds, activity towards both Gram-negative and Gram-positive strains has been correlated to the uptake of the complex into the cells.
Additionally, this work highlights recent advances in ruthenium-based compounds that are active against neglected tropical diseases caused by parasites, such as malaria, Chagas’ disease, and leishmaniasis. Notably, several complexes possess excellent activity, at submicromolar concentrations, results that raise awareness about the potential use of ruthenium compounds as effective antiparasitic agents. Moreover, the antiviral activity of ruthenium complexes, particularly the anti-HIV and anti-SARS-Cov-2 activities, has been reviewed herein. It is worth noting that BOLD-100 (formerly denoted KP1339) displays a broad antiviral mechanism of action and appears to remain active on all mutant strains of the SARS-Cov-2 virus.
In general terms, ruthenium complexes have been shown to display low levels of toxicity towards healthy eukaryotic cells in vitro and in vivo. This finding underlines the potential of these compounds for future clinical development, since selective toxicity against microbial over host cells in vitro and in vivo is imperative for a potential drug to advance in clinical trials. More in vivo studies are clearly needed in order to provide proof beyond a reasonable doubt that ruthenium complexes are strong candidates in the field of antimicrobial drug discovery.
In conclusion, this work aimed to highlight the potential of ruthenium-based compounds as novel antimicrobial agents due to the diverse range of complex 3D structures and modes of action they provide. Given that the pipeline of new antibiotics is running dry, the ruthenium species with high activity and selectivity presented herein may represent the starting point for a much-needed new class of antimicrobial agents. Therefore, we hope that this review will succeed in raising awareness about the potential of ruthenium complexes for antimicrobial applications and spur further research into their development.
Author Contributions
A.-C.M. performed a literature search, conceptualized the manuscript, and contributed to the writing of, corrections to, and the final shape of the manuscript. V.U. performed a literature search and contributed to the writing of and corrections to the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by The Executive Unit for the Financing of Higher Education, Research, Development, and Innovation (UEFISCDI), Project No. 383PED/2020 and Project No. PD 219/2020.
Acknowledgments
We thank Joseph Cowell for proofreading the manuscript and improving the use of English throughout the paper.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Li, F.; Collins, J.G.; Keene, F.R. Ruthenium complexes as antimicrobial agents. Chem. Soc. Rev. 2015, 44, 2529–2542. [Google Scholar] [CrossRef] [Green Version]
- Frei, A.; Zuegg, J.; Elliott, A.G.; Baker, M.; Braese, S.; Brown, C.; Chen, F.G.; Dowson, C.; Dujardin, G.; Jung, N.; et al. Metal complexes as a promising source for new antibiotics. Chem. Sci. 2020, 11, 2627–2639. [Google Scholar] [CrossRef] [Green Version]
- Keogan, D.M.; Griffith, D.M. Current and potential applications of bismuth-based drugs. Molecules 2014, 19, 15258–15297. [Google Scholar] [CrossRef] [Green Version]
- Silver, S. Bacterial silver resistance: Molecular biology and uses and misuses of silver compounds. FEMS Microbiol. Rev. 2003, 27, 341–353. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-López, E.; Gomes, D.; Esteruelas, G.; Bonilla, L.; Lopez-Machado, A.L.; Galindo, R.; Cano, A.; Espina, M.; Ettcheto, M.; Camins, A.; et al. Metal-based nanoparticles as antimicrobial agents: An overview. Nanomaterials 2020, 10, 292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, C.N.; Prosser, K.E.; Stokes, R.W.; Cordes, A.; Metzler-Nolte, N.; Cohen, S.M. Expanding medicinal chemistry into 3D space: Metallofragments as 3D scaffolds for fragment-based drug discovery. Chem. Sci. 2020, 11, 1216–1225. [Google Scholar] [CrossRef] [Green Version]
- Hung, A.W.; Ramek, A.; Wang, Y.; Kaya, T.; Wilson, J.A.; Clemons, P.A.; Young, D.W. Route to three-dimensional fragments using diversity-oriented synthesis. Proc. Natl. Acad. Sci. USA 2011, 108, 6799–6804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galloway, W.R.J.D.; Isidro-Llobet, A.; Spring, D.R. Diversity-oriented synthesis as a tool for the discovery of novel biologically active small molecules. Nat. Commun. 2010, 1, 80. [Google Scholar] [CrossRef] [Green Version]
- Frei, A. Metal complexes, an untapped source of antibiotic potential? Antibiotics 2020, 9, 90. [Google Scholar] [CrossRef] [Green Version]
- Anthony, E.J.; Bolitho, E.M.; Bridgewater, H.E.; Carter, O.W.L.; Donnelly, J.M.; Imberti, C.; Lant, E.C.; Lermyte, F.; Needham, R.J.; Palau, M.; et al. Metallodrugs are unique: Opportunities and challenges of discovery and development. Chem. Sci. 2020, 11, 12888–12917. [Google Scholar] [CrossRef]
- Claudel, M.; Schwarte, J.V.; Fromm, K.M. New Antimicrobial Strategies Based on Metal Complexes. Chemistry 2020, 2, 849–899. [Google Scholar]
- Munteanu, A.-C.; Notaro, A.; Jakubaszek, M.; Cowell, J.; Tharaud, M.; Goud, B.; Uivarosi, V.; Gasser, G. Synthesis, Characterization, Cytotoxic Activity, and Metabolic Studies of Ruthenium(II) Polypyridyl Complexes Containing Flavonoid Ligands. Inorg. Chem. 2020, 59, 4424–4434. [Google Scholar] [CrossRef]
- Rademaker-Lakhai, J.M.; van den Bongard, D.; Pluim, D.; Beijnen, J.H.; Schellens, J.H.M. A Phase I and Pharmacological Study with Imidazolium-trans-DMSO-imidazole-tetrachlororuthenate, a Novel Ruthenium Anticancer Agent. Clin. Cancer Res. 2004, 10, 3717–3727. [Google Scholar] [CrossRef] [Green Version]
- Leijen, S.; Burgers, S.A.; Baas, P.; Pluim, D.; Tibben, M.; Van Werkhoven, E.; Alessio, E.; Sava, G.; Beijnen, J.H.; Schellens, J.H.M. Phase I/II study with ruthenium compound NAMI-A and gemcitabine in patients with non-small cell lung cancer after first line therapy. Invest. New Drugs 2015, 33, 201–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartinger, C.G.; Jakupec, M.A.; Zorbas-Seifried, S.; Groessl, M.; Egger, A.; Berger, W.; Zorbas, H.; Dyson, P.J.; Keppler, B.K. KP1019, A New Redox-Active Anticancer Agent – Preclinical Development and Results of a Clinical Phase I Study in Tumor Patients. Chem. Biodivers. 2008, 5, 2140–2155. [Google Scholar] [CrossRef] [PubMed]
- Lentz, F.; Drescher, A.; Lindauer, A.; Henke, M.; Hilger, R.A.; Hartinger, C.G.; Scheulen, M.E.; Dittrich, C.; Keppler, B.K.; Jaehde, U. Pharmacokinetics of a novel anticancer ruthenium complex (KP1019, FFC14A) in a phase I dose-escalation study. Anticancer. Drugs 2009, 20, 97–103. [Google Scholar] [CrossRef]
- Trondl, R.; Heffeter, P.; Kowol, C.R.; Jakupec, M.A.; Berger, W.; Keppler, B.K. NKP-1339, the first ruthenium-based anticancer drug on the edge to clinical application. Chem. Sci. 2014, 5, 2925–2932. [Google Scholar] [CrossRef] [Green Version]
- Monro, S.; Colón, K.L.; Yin, H.; Roque, J.; Konda, P.; Gujar, S.; Thummel, R.P.; Lilge, L.; Cameron, C.G.; McFarland, S.A. Transition Metal Complexes and Photodynamic Therapy from a Tumor-Centered Approach: Challenges, Opportunities, and Highlights from the Development of TLD1433. Chem. Rev. 2019, 119, 797–828. [Google Scholar] [CrossRef]
- Antibiotic Resistance. Available online: https://www.who.int/news-room/fact-sheets/detail/antibiotic-resistance (accessed on 20 April 2021).
- Silhavy, T.J.; Kahne, D.; Walker, S. The bacterial cell envelope. Cold Spring Harb. Perspect. Biol. 2010, 2, a000414. [Google Scholar] [CrossRef] [PubMed]
- Pizarro-Cerdá, J.; Cossart, P. Bacterial Adhesion and Entry into Host Cells. Cell 2006, 124, 715–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huan, Y.; Kong, Q.; Mou, H.; Yi, H. Antimicrobial Peptides: Classification, Design, Application and Research Progress in Multiple Fields. Front. Microbiol. 2020, 11, 582779. [Google Scholar] [CrossRef] [PubMed]
- Uivarosi, V.; Munteanu, A.-C.; Nițulescu, G.M. Chapter 2—An Overview of Synthetic and Semisynthetic Flavonoid Derivatives and Analogues: Perspectives in Drug Discovery. In Studies in Natural Products Chemistry; Atta-ur-Rahman, Ed.; Elsevier: Amsterdam, The Netherlands, 2019; Volume 60, pp. 29–84. ISBN 1572-5995. [Google Scholar]
- Provenzani, A.; Hospodar, A.R.; Meyer, A.L.; Leonardi Vinci, D.; Hwang, E.Y.; Butrus, C.M.; Polidori, P. Multidrug-resistant gram-negative organisms: A review of recently approved antibiotics and novel pipeline agents. Int. J. Clin. Pharm. 2020, 42, 1016–1025. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Kim, E.S.; Yoo, Y.-J.; Bae, H.-W.; Chung, I.-Y.; Cho, Y.-H. Phage-Derived Antibacterials: Harnessing the Simplicity, Plasticity, and Diversity of Phages. Viruses 2019, 11, 268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfei, S.; Schito, A.M. Positively charged polymers as promising devices against multidrug resistant gram-negative bacteria: A Review. Polymers 2020, 12, 1195. [Google Scholar] [CrossRef]
- Guo, C.; Mandalapu, D.; Ji, X.; Gao, J.; Zhang, Q. Chemistry and Biology of Teixobactin. Chem. – A Eur. J. 2018, 24, 5406–5422. [Google Scholar] [CrossRef]
- Reygaert, W.C. An overview of the antimicrobial resistance mechanisms of bacteria. AIMS Microbiol. 2018, 4, 482–501. [Google Scholar] [CrossRef]
- Raafat, D.; Otto, M.; Reppschläger, K.; Iqbal, J.; Holtfreter, S. Fighting Staphylococcus aureus Biofilms with Monoclonal Antibodies. Trends Microbiol. 2019, 27, 303–322. [Google Scholar] [CrossRef]
- Stewart, P.S. Mechanisms of antibiotic resistance in bacterial biofilms. Int. J. Med. Microbiol. 2002, 292, 107–113. [Google Scholar] [CrossRef]
- Gholizadeh, P.; Köse, Ş.; Dao, S.; Ganbarov, K.; Tanomand, A.; Dal, T.; Aghazadeh, M.; Ghotaslou, R.; Ahangarzadeh Rezaee, M.; Yousefi, B.; et al. How CRISPR-Cas System Could Be Used to Combat Antimicrobial Resistance. Infect. Drug Resist. 2020, 13, 1111–1121. [Google Scholar] [CrossRef] [Green Version]
- Southam, H.M.; Butler, J.A.; Chapman, J.A.; Poole, R.K. The Microbiology of Ruthenium Complexes. In Advances in Microbial Physiology; Poole, R.K., Ed.; Elsevier: London, UK, 2017; Volume 71, pp. 1–96. ISBN 0065-2911. [Google Scholar]
- Dwyer, F.P.; Gyarfas, E.C.; Rogers, W.P.; Koch, J.H. Biological Activity of Complex Ions. Nature 1952, 170, 190–191. [Google Scholar] [CrossRef]
- Dwyer, F.P.; Mayhew, E.; Roe, E.M.F.; Shulman, A. Inhibition of Landschütz Ascites Tumour Growth by Metal Chelates Derived from 3,4,7,8-Tetramethyl-1,10-phenanthroline. Br. J. Cancer 1965, 19, 195–199. [Google Scholar] [CrossRef]
- Abd-El-Aziz, A.S.; Agatemor, C.; Etkin, N. Antimicrobial resistance challenged with metal-based antimicrobial macromolecules. Biomaterials 2017, 118, 27–50. [Google Scholar] [CrossRef]
- Shulman, A.; Dwyer, F.P. Metal Chelates in Biological Systems. In Chelating Agents and Metal Chelates; Dwyer, F.P., Mellor, D.P., Eds.; Academic Press: Cambridge, MA, USA, 1964; ISBN 978-0-12-395499-2. [Google Scholar]
- Li, F.; Mulyana, Y.; Feterl, M.; Warner, J.M.; Collins, J.G.; Keene, F.R. The antimicrobial activity of inert oligonuclear polypyridylruthenium(II) complexes against pathogenic bacteria, including MRSA. Dalt. Trans. 2011, 40, 5032–5038. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Feterl, M.; Mulyana, Y.; Warner, J.M.; Collins, J.G.; Keene, F.R. In vitro susceptibility and cellular uptake for a new class of antimicrobial agents: Dinuclear ruthenium(II) complexes. J. Antimicrob. Chemother. 2012, 67, 2686–2695. [Google Scholar] [CrossRef] [Green Version]
- Dwyer, F.P.; Gyarfas, E.C.; Wright, R.D.; Shulman, A. Effect of Inorganic Complex Ions on Transmission at a Neuromuscular Junction. Nature 1957, 179, 425–426. [Google Scholar] [CrossRef]
- Bolhuis, A.; Hand, L.; Marshall, J.E.; Richards, A.D.; Rodger, A.; Aldrich-Wright, J. Antimicrobial activity of ruthenium-based intercalators. Eur. J. Pharm. Sci. 2011, 42, 313–317. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.-Y.; Sun, B.; Zhang, L.; Li, N.; Han, J.; Zhang, J.; Sun, X.; He, Q.-Y. Chemical Interference with Iron Transport Systems to Suppress Bacterial Growth of Streptococcus pneumoniae. PLoS One 2014, 9, e105953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.-Y.; Zhang, L.; Liu, J.; Li, N.; Yu, G.; Cao, K.; Han, J.; Zeng, G.; Pan, Y.; Sun, X.; et al. Proteomic analysis on the antibacterial activity of a Ru(II) complex against Streptococcus pneumoniae. J. Proteomics 2015, 115, 107–116. [Google Scholar] [CrossRef]
- de Sousa, A.P.; Ellena, J.; Gondim, A.C.S.; Lopes, L.G.F.; Sousa, E.H.S.; de Vasconcelos, M.A.; Teixeira, E.H.; Ford, P.C.; Holanda, A.K.M. Antimicrobial activity of cis-[Ru(bpy)2(L)(L′)]n+ complexes, where L = 4-(4-chlorobenzoyl)pyridine or 4-(benzoyl)pyridine and L′ = Cl− or CO. Polyhedron 2018, 144, 88–94. [Google Scholar] [CrossRef]
- De Sousa, A.P.; Gondim, A.C.S.; Sousa, E.H.S.; de Vasconcelos, M.A.; Teixeira, E.H.; Bezerra, B.P.; Ayala, A.P.; Martins, P.H.R.; Lopes, L.G.d.F.; Holanda, A.K.M. An unusual bidentate methionine ruthenium(II) complex: Photo-uncaging and antimicrobial activity. J. Biol. Inorg. Chem. 2020, 25, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Jiang, G.; Wang, J.; Duan, X.; Liao, Z.; Lin, X.; Shen, J.; Xiong, Y.; Jiang, G. Two ruthenium polypyridyl complexes functionalized with thiophen: Synthesis and antibacterial activity against Staphylococcus aureus. New J. Chem. 2020, 44, 17215–17221. [Google Scholar] [CrossRef]
- Bu, S.; Jiang, G.; Jiang, G.; Liu, J.; Lin, X.; Shen, J.; Xiong, Y.; Duan, X.; Wang, J.; Liao, X. Antibacterial activity of ruthenium polypyridyl complexes against Staphylococcus aureus and biofilms. J. Biol. Inorg. Chem. 2020, 25, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, P.; Shukla, M.; Kaul, G.; Chopra, S.; Patra, A.K. Rationally designed curcumin based ruthenium(ii) antimicrobials effective against drug-resistant Staphylococcus aureus. Dalt. Trans. 2019, 48, 11822–11828. [Google Scholar] [CrossRef] [PubMed]
- Gorle, A.K.; Feterl, M.; Warner, J.M.; Primrose, S.; Constantinoiu, C.C.; Keene, F.R.; Collins, J.G. Mononuclear Polypyridylruthenium(II) Complexes with High Membrane Permeability in Gram-Negative Bacteria—in particular Pseudomonas aeruginosa. Chem. – A Eur. J. 2015, 21, 10472–10481. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Sun, B.; Kell, R.E.M.; Southam, H.M.; Butler, J.A.; Li, X.; Poole, R.K.; Keene, F.R.; Collins, J.G. The Antimicrobial Activity of Mononuclear Ruthenium(II) Complexes Containing the dppz Ligand. Chempluschem 2018, 83, 643–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smitten, K.L.; Thick, E.J.; Southam, H.M.; Bernardino de la Serna, J.; Foster, S.J.; Thomas, J.A. Mononuclear ruthenium(ii) theranostic complexes that function as broad-spectrum antimicrobials in therapeutically resistant pathogens through interaction with DNA. Chem. Sci. 2020, 11, 8828–8838. [Google Scholar] [CrossRef]
- De Grandis, R.A.; Resende, F.A.; da Silva, M.M.; Pavan, F.R.; Batista, A.A.; Varanda, E.A. In vitro evaluation of the cyto-genotoxic potential of Ruthenium(II) SCAR complexes: a promising class of antituberculosis agents. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2016, 798–799, 11–18. [Google Scholar] [CrossRef] [Green Version]
- Andrade, A.L.; de Vasconcelos, M.A.; Arruda, F.V.d.F.; do Nascimento Neto, L.G.; Carvalho, J.M.d.S.; Gondim, A.C.S.; Lopes, L.G.d.F.; Sousa, E.H.S.; Teixeira, E.H. Antimicrobial activity and antibiotic synergy of a biphosphinic ruthenium complex against clinically relevant bacteria. Biofouling 2020, 36, 442–454. [Google Scholar] [CrossRef]
- Kumar, S.V.; Scottwell, S.O.; Waugh, E.; McAdam, C.J.; Hanton, L.R.; Brooks, H.J.L.; Crowley, J.D. Antimicrobial Properties of Tris(homoleptic) Ruthenium(II) 2-Pyridyl-1,2,3-triazole “click” Complexes against Pathogenic Bacteria, Including Methicillin-Resistant Staphylococcus aureus (MRSA). Inorg. Chem. 2016, 55, 9767–9777. [Google Scholar] [CrossRef]
- van Hilst, Q.V.C.; Vasdev, R.A.S.; Preston, D.; Findlay, J.A.; Scottwell, S.; Giles, G.I.; Brooks, H.J.L.; Crowley, J.D. Synthesis, Characterisation and Antimicrobial Studies of some 2,6-bis(1,2,3-Triazol-4-yl)Pyridine Ruthenium(II) “Click” Complexes. Asian J. Org. Chem. 2019, 8, 496–505. [Google Scholar] [CrossRef]
- Gorle, A.K.; Li, X.; Primrose, S.; Li, F.; Feterl, M.; Kinobe, R.T.; Heimann, K.; Warner, J.M.; Keene, F.R.; Collins, J.G. Oligonuclear polypyridylruthenium(II) complexes: Selectivity between bacteria and eukaryotic cells. J. Antimicrob. Chemother. 2016, 71, 1547–1555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Heimann, K.; Li, F.; Warner, J.M.; Richard Keene, F.; Grant Collins, J. Dinuclear ruthenium(II) complexes containing one inert metal centre and one coordinatively-labile metal centre: Syntheses and biological activities. Dalt. Trans. 2016, 45, 4017–4029. [Google Scholar] [CrossRef] [Green Version]
- Smitten, K.L.; Southam, H.M.; de la Serna, J.B.; Gill, M.R.; Jarman, P.J.; Smythe, C.G.W.; Poole, R.K.; Thomas, J.A. Using Nanoscopy To Probe the Biological Activity of Antimicrobial Leads That Display Potent Activity against Pathogenic, Multidrug Resistant, Gram-Negative Bacteria. ACS Nano 2019, 13, 5133–5146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varney, A.M.; Smitten, K.L.; Thomas, J.A.; McLean, S. Transcriptomic Analysis of the Activity and Mechanism of Action of a Ruthenium(II)-Based Antimicrobial That Induces Minimal Evolution of Pathogen Resistance. ACS Pharmacol. Transl. Sci. 2021, 4, 168–178. [Google Scholar] [CrossRef]
- Smitten, K.L.; Fairbanks, S.D.; Robertson, C.C.; Bernardino de la Serna, J.; Foster, S.J.; Thomas, J.A. Ruthenium based antimicrobial theranostics – using nanoscopy to identify therapeutic targets and resistance mechanisms in Staphylococcus aureus. Chem. Sci. 2020, 11, 70–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandrala, M.; Li, F.; Feterl, M.; Mulyana, Y.; Warner, J.M.; Wallace, L.; Keene, F.R.; Collins, J.G. Chlorido-containing ruthenium(II) and iridium(III) complexes as antimicrobial agents. Dalt. Trans. 2013, 42, 4686–4694. [Google Scholar] [CrossRef]
- Gorle, A.K.; Feterl, M.; Warner, J.M.; Wallace, L.; Keene, F.R.; Collins, J.G. Tri- and tetra-nuclear polypyridyl ruthenium(II) complexes as antimicrobial agents. Dalt. Trans. 2014, 43, 16713–16725. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.; Sundaraneedi, M.K.; Southam, H.M.; Poole, R.K.; Musgrave, I.F.; Keene, F.R.; Collins, J.G. Synthesis and biological properties of tetranuclear ruthenium complexes containing the bis[4(4′-methyl-2,2′-bipyridyl)]-1,7-heptane ligand. Dalt. Trans. 2019, 48, 14505–14515. [Google Scholar] [CrossRef]
- Nobre, L.S.; Seixas, J.D.; Romão, C.C.; Saraiva, L.M. Antimicrobial Action of Carbon Monoxide-Releasing Compounds. Antimicrob. Agents Chemother. 2007, 51, 4303–4307. [Google Scholar] [CrossRef] [Green Version]
- Tavares, A.F.; Parente, M.R.; Justino, M.C.; Oleastro, M.; Nobre, L.S.; Saraiva, L.M. The bactericidal activity of carbon monoxide-releasing molecules against Helicobacter pylori. PLoS One 2013, 8, e83157. [Google Scholar] [CrossRef] [Green Version]
- Sahlberg Bang, C.; Demirel, I.; Kruse, R.; Persson, K. Global gene expression profiling and antibiotic susceptibility after repeated exposure to the carbon monoxide-releasing molecule-2 (CORM-2) in multidrug-resistant ESBL-producing uropathogenic Escherichia coli. PLoS One 2017, 12, e0178541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desmard, M.; Davidge, K.S.; Bouvet, O.; Morin, D.; Roux, D.; Foresti, R.; Ricard, J.D.; Denamur, E.; Poole, R.K.; Montravers, P.; et al. A carbon monoxide-releasing molecule (CORM-3) exerts bactericidal activity against Pseudomonas aeruginosa and improves survival in an animal model of bacteraemia. FASEB J. 2009, 23, 1023–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, S.M.; Marques, J.; Romão, C.C.; Saraiva, L.M. Metabolomics of Escherichia coli Treated with the Antimicrobial Carbon Monoxide-Releasing Molecule CORM-3 Reveals Tricarboxylic Acid Cycle as Major Target. Antimicrob. Agents Chemother. 2019, 63, e00643–e00719. [Google Scholar] [CrossRef]
- Donnelly, R.F.; Fletcher, N.C.; McCague, P.J.; Donnelly, J.; McCarron, P.A.; Tunney, M.M. Design, Synthesis and Photodynamic Antimicrobial Activity of Ruthenium Trischelate Diimine Complexes. Lett. Drug Des. Discov. 2007, 4, 175–179. [Google Scholar] [CrossRef]
- Smith, N.A.; Zhang, P.; Greenough, S.E.; Horbury, M.D.; Clarkson, G.J.; McFeely, D.; Habtemariam, A.; Salassa, L.; Stavros, V.G.; Dowson, C.G.; et al. Combatting AMR: Photoactivatable ruthenium(II)-isoniazid complex exhibits rapid selective antimycobacterial activity. Chem. Sci. 2017, 8, 395–404. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, S.L.; Stepanyan, L.; Vahidi, N.; Jain, A.; Winkel, B.S.J.; Brewer, K.J. Visible light induced antibacterial properties of a Ru(II)–Pt(II) bimetallic complex. Inorganica Chim. Acta 2017, 454, 229–233. [Google Scholar] [CrossRef]
- Ghosh, S.; Amariei, G.; Mosquera, M.E.G.; Rosal, R. Polymeric ruthenium precursor as a photoactivated antimicrobial agent. J. Hazard. Mater. 2021, 402, 123788. [Google Scholar] [CrossRef]
- Yang, X.-Y.; He, K.; Du, G.; Wu, X.; Yu, G.; Pan, Y.; Zhang, G.; Sun, X.; He, Q.-Y. Integrated Translatomics with Proteomics to Identify Novel Iron–Transporting Proteins in Streptococcus pneumoniae. Front. Microbiol. 2016, 7, 78. [Google Scholar] [CrossRef] [PubMed]
- Srishailam, A.; Gabra, N.M.; Kumar, Y.P.; Reddy, K.L.; Devi, C.S.; Anil Kumar, D.; Singh, S.S.; Satyanarayana, S. Synthesis, characterization; DNA binding and antitumor activity of ruthenium(II) polypyridyl complexes. J. Photochem. Photobiol. B Biol. 2014, 141, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.R.; Reddy, P.V.; Kumar, Y.P.; Srishailam, A.; Nambigari, N.; Satyanarayana, S. Synthesis, Characterization, DNA Binding, Light Switch “On and Off”, Docking Studies and Cytotoxicity, of Ruthenium(II) and Cobalt(III) Polypyridyl Complexes. J. Fluoresc. 2014, 24, 803–817. [Google Scholar] [CrossRef]
- Mallepally, R.R.; Putta, V.R.; Chintakuntla, N.; Vuradi, R.K.; Kotha, L.R.; Sirasani, S. DNA Binding Behavior, Sensor Studies, Antimicrobial, Photocleavage and In vitro Cytotoxicity of Synthesized Ru(II) Complexes with Assorted Intercalating Polypyridyl Ligands. J. Fluoresc. 2016, 26, 1101–1113. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Zhang, W.; Lv, M.; Yang, E.; Zhao, Q.; Wang, W. Antibacterial activity of ruthenium(II) polypyridyl complex manipulated by membrane permeability and cell morphology. Bioorg. Med. Chem. Lett. 2015, 25, 2068–2073. [Google Scholar] [CrossRef] [PubMed]
- Putta, V.R.; Chintakuntla, N.; Mallepally, R.R.; Avudoddi, S.; K, N.; Nancherla, D.; V V N, Y.; R S, P.; Surya, S.S.; Sirasani, S. Synthesis and Evaluation of In Vitro DNA/Protein Binding Affinity, Antimicrobial, Antioxidant and Antitumor Activity of Mononuclear Ru(II) Mixed Polypyridyl Complexes. J. Fluoresc. 2016, 26, 225–240. [Google Scholar] [CrossRef]
- Ravi Kumar, V.; Nagababu, P.; Srinivas, G.; Rajender Reddy, M.; Vinoda Rani, M.; Ravi, M.; Satyanarayana, S. Investigation of DNA/BSA binding of three Ru(II) complexes by various spectroscopic methods, molecular docking and their antimicrobial activity. J. Coord. Chem. 2017, 70, 3790–3809. [Google Scholar] [CrossRef]
- Vadivel, T.; Dhamodaran, M. Synthesis, characterization and antibacterial studies of ruthenium(III) complexes derived from chitosan schiff base. Int. J. Biol. Macromol. 2016, 90, 44–52. [Google Scholar] [CrossRef]
- Ashwini Kumar, K.; Laxma Reddy, K.; Satyanarayana, S. Study of the interaction between ruthenium(II) complexes and CT-DNA: synthesis, characterisation, photocleavage and antimicrobial activity studies. Supramol. Chem. 2010, 22, 629–643. [Google Scholar] [CrossRef]
- Ashwini Kumar, K.; Reddy, K.L.; Vidhisha, S.; Satyanarayana, S. Synthesis, characterization and DNA binding and photocleavage studies of [Ru(bpy)2BDPPZ]2+, [Ru(dmb)2BDPPZ]2+ and [Ru(phen)2BDPPZ]2+ complexes and their antimicrobial activity. Appl. Organomet. Chem. 2009, 23, 409–420. [Google Scholar] [CrossRef]
- Garner, R.N.; Gallucci, J.C.; Dunbar, K.R.; Turro, C. [Ru(bpy)2(5-cyanouracil)2]2+ as a Potential Light-Activated Dual-Action Therapeutic Agent. Inorg. Chem. 2011, 50, 9213–9215. [Google Scholar] [CrossRef] [Green Version]
- Jiang, G.-B.; Zhang, W.-Y.; He, M.; Gu, Y.-Y.; Bai, L.; Wang, Y.-J.; Yi, Q.-Y.; Du, F. New ruthenium polypyridyl complexes functionalized with fluorine atom or furan: Synthesis, DNA-binding, cytotoxicity and antitumor mechanism studies. Spectrochim. Acta. A. Mol. Biomol. Spectrosc. 2020, 227, 117534. [Google Scholar] [CrossRef]
- Jiang, G.-B.; Zhang, W.-Y.; He, M.; Gu, Y.-Y.; Bai, L.; Wang, Y.-J.; Yi, Q.-Y.; Du, F. Design and synthesis of new ruthenium polypyridyl complexes with potent antitumor activity in vitro. Spectrochim. Acta. A. Mol. Biomol. Spectrosc. 2019, 220, 117132. [Google Scholar] [CrossRef]
- Jiang, G.-B.; Zhang, W.-Y.; He, M.; Gu, Y.-Y.; Bai, L.; Wang, Y.-J.; Yi, Q.-Y.; Du, F. Development of four ruthenium polypyridyl complexes as antitumor agents: Design, biological evaluation and mechanism investigation. J. Inorg. Biochem. 2020, 208, 111104. [Google Scholar] [CrossRef]
- Jiang, G.-B.; Zhang, W.-Y.; He, M.; Gu, Y.-Y.; Bai, L.; Wang, Y.-J.; Yi, Q.-Y.; Du, F. Anticancer activity of two ruthenium(II) polypyridyl complexes toward Hepatocellular carcinoma HepG-2 cells. Polyhedron 2019, 169, 209–218. [Google Scholar] [CrossRef]
- Mulyana, Y.; Weber, D.K.; Buck, D.P.; Motti, C.A.; Collins, J.G.; Keene, F.R. Oligonuclear polypyridylruthenium(II) complexes incorporating flexible polar and non-polar bridges: synthesis, DNA-binding and cytotoxicity. Dalt. Trans. 2011, 40, 1510–1523. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Feterl, M.; Warner, J.M.; Keene, F.R.; Collins, J.G. Dinuclear polypyridylruthenium(II) complexes: Flow cytometry studies of their accumulation in bacteria and the effect on the bacterial membrane. J. Antimicrob. Chemother. 2013, 68, 2825–2833. [Google Scholar] [CrossRef] [Green Version]
- Pisani, M.J.; Fromm, P.D.; Mulyana, Y.; Clarke, R.J.; Körner, H.; Heimann, K.; Collins, J.G.; Keene, F.R. Mechanism of cytotoxicity and cellular uptake of lipophilic inert dinuclear polypyridylruthenium(II) complexes. ChemMedChem 2011, 6, 848–858. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Harry, E.J.; Bottomley, A.L.; Edstein, M.D.; Birrell, G.W.; Woodward, C.E.; Keene, F.R.; Collins, J.G. Dinuclear ruthenium(II) antimicrobial agents that selectively target polysomes in vivo. Chem. Sci. 2014, 5, 685–693. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Gorle, A.K.; Ainsworth, T.D.; Heimann, K.; Woodward, C.E.; Collins, J.G.; Keene, F.R. RNA and DNA binding of inert oligonuclear ruthenium(II) complexes in live eukaryotic cells. Dalt. Trans. 2015, 44, 3594–3603. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.; Southam, H.M.; Butler, J.A.; Poole, R.K.; Burgun, A.; Tarzia, A.; Keene, F.R.; Collins, J.G. Synthesis, isomerisation and biological properties of mononuclear ruthenium complexes containing the bis[4(4′-methyl-2,2′-bipyridyl)]-1,7-heptane ligand. Dalt. Trans. 2018, 47, 2422–2434. [Google Scholar] [CrossRef] [Green Version]
- Liao, G.; Ye, Z.; Liu, Y.; Fu, B.; Fu, C. Octahedral ruthenium (II) polypyridyl complexes as antimicrobial agents against mycobacterium. PeerJ 2017, 5, e3252. [Google Scholar] [CrossRef] [Green Version]
- Allardyce, C.S.; Dyson, P.J.; Ellis, D.J.; Salter, P.A.; Scopelliti, R. Synthesis and characterisation of some water soluble ruthenium(II)–arene complexes and an investigation of their antibiotic and antiviral properties. J. Organomet. Chem. 2003, 668, 35–42. [Google Scholar] [CrossRef]
- Beckford, F.A.; Stott, A.; Gonzalez-Sarrías, A.; Seeram, N.P. Novel microwave synthesis of half-sandwich [(η6-C6H6)Ru] complexes and an evaluation of the biological activity and biochemical reactivity. Appl. Organomet. Chem. 2013, 27, 425–434. [Google Scholar] [CrossRef]
- Gichumbi, J.M.; Friedrich, H.B.; Omondi, B.; Singh, M.; Naicker, K.; Chenia, H.Y. Synthesis, characterization, and cytotoxic and antimicrobial activities of ruthenium(II) arene complexes with N,N-bidentate ligands. J. Coord. Chem. 2016, 69, 3531–3544. [Google Scholar] [CrossRef]
- Singh, K.S.; Devi, P.; Sawant, S.G.; Kaminsky, W. Arene ruthenium(II) complexes with 2-acetamidothiazole derived ligands: Synthesis, structural studies, antifouling and antibacterial properties. Polyhedron 2015, 100, 321–325. [Google Scholar] [CrossRef]
- Obradović, D.; Nikolić, S.; Milenković, I.; Milenković, M.; Jovanović, P.; Savić, V.; Roller, A.; Đorđić Crnogorac, M.; Stanojković, T.; Grgurić-Šipka, S. Synthesis, characterization, antimicrobial and cytotoxic activity of novel half-sandwich Ru(II) arene complexes with benzoylthiourea derivatives. J. Inorg. Biochem. 2020, 210, 111164. [Google Scholar] [CrossRef] [PubMed]
- Turan, N.; Buldurun, K.; Alan, Y.; Savci, A.; Çolak, N.; Mantarcı, A. Synthesis, characterization, antioxidant, antimicrobial and DNA binding properties of ruthenium(II), cobalt(II) and nickel(II) complexes of Schiff base containing o-vanillin. Res. Chem. Intermed. 2019, 45, 3525–3540. [Google Scholar] [CrossRef]
- Shadap, L.; Banothu, V.; Adepally, U.; Adhikari, S.; Kollipara, M.R. Variable structural bonding modes and antibacterial studies of thiosemicarbazone ligands of ruthenium, rhodium, and iridium metal complexes. J. Coord. Chem. 2020, 73, 175–187. [Google Scholar] [CrossRef]
- Lapasam, A.; Banothu, V.; Addepally, U.; Kollipara, M.R. Synthesis, structural and antimicrobial studies of half-sandwich ruthenium, rhodium and iridium complexes containing nitrogen donor Schiff-base ligands. J. Mol. Struct. 2019, 1191, 314–322. [Google Scholar] [CrossRef]
- Nobre, L.S.; Jeremias, H.; Romão, C.C.; Saraiva, L.M. Examining the antimicrobial activity and toxicity to animal cells of different types of CO-releasing molecules. Dalt. Trans. 2016, 45, 1455–1466. [Google Scholar] [CrossRef]
- Dkhar, L.; Banothu, V.; Kaminsky, W.; Kollipara, M.R. Synthesis of half sandwich platinum group metal complexes containing pyridyl benzothiazole hydrazones: Study of bonding modes and antimicrobial activity. J. Organomet. Chem. 2020, 914, 121225. [Google Scholar] [CrossRef]
- Laurent, Q.; Batchelor, L.K.; Dyson, P.J. Applying a Trojan Horse Strategy to Ruthenium Complexes in the Pursuit of Novel Antibacterial Agents. Organometallics 2018, 37, 915–923. [Google Scholar] [CrossRef]
- Chhabra, R.; Saha, A.; Chamani, A.; Schneider, N.; Shah, R.; Nanjundan, M. Iron Pathways and Iron Chelation Approaches in Viral, Microbial, and Fungal Infections. Pharmaceuticals 2020, 13, 275. [Google Scholar] [CrossRef] [PubMed]
- Yildirim, H.; Guler, E.; Yavuz, M.; Ozturk, N.; Kose Yaman, P.; Subasi, E.; Sahin, E.; Timur, S. Ruthenium (II) complexes of thiosemicarbazone: Synthesis, biosensor applications and evaluation as antimicrobial agents. Mater. Sci. Eng. C 2014, 44, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Namiecińska, E.; Sadowska, B.; Więckowska-Szakiel, M.; Dołęga, A.; Pasternak, B.; Grazul, M.; Budzisz, E. Anticancer and antimicrobial properties of novel η6-p-cymene ruthenium(ii) complexes containing a N,S-type ligand, their structural and theoretical characterization. RSC Adv. 2019, 9, 38629–38645. [Google Scholar] [CrossRef] [Green Version]
- Felício, M.R.; Silva, O.N.; Gonçalves, S.; Santos, N.C.; Franco, O.L. Peptides with Dual Antimicrobial and Anticancer Activities. Front. Chem. 2017, 5, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appelt, P.; Fagundes, F.D.; Facchin, G.; Gabriela Kramer, M.; Back, D.F.; Cunha, M.A.A.; Sandrino, B.; Wohnrath, K.; de Araujo, M.P. Ruthenium (II) complexes containing 2-mercaptothiazolinates as ligands and evaluation of their antimicrobial activity. Inorganica Chim. Acta 2015, 436, 152–158. [Google Scholar] [CrossRef]
- Beloglazkina, E.K.; Manzheliy, E.A.; Moiseeva, A.A.; Maloshitskaya, O.A.; Zyk, N.V.; Skvortsov, D.A.; Osterman, I.A.; Sergiev, P.V.; Dontsova, O.A.; Ivanenkov, Y.A.; et al. Synthesis, characterisation, cytotoxicity and antibacterial activity of ruthenium(II) and rhodium(III) complexes with sulfur-containing terpyridines. Polyhedron 2016, 107, 27–37. [Google Scholar] [CrossRef]
- Lapasam, A.; Dkhar, L.; Joshi, N.; Poluri, K.M.; Kollipara, M.R. Antimicrobial selectivity of ruthenium, rhodium, and iridium half sandwich complexes containing phenyl hydrazone Schiff base ligands towards B. thuringiensis and P. aeruginosa bacteria. Inorganica Chim. Acta 2019, 484, 255–263. [Google Scholar] [CrossRef]
- Elnagar, M.M.; Samir, S.; Shaker, Y.M.; Abdel-Shafi, A.A.; Sharmoukh, W.; Abdel-Aziz, M.S.; Abou-El-Sherbini, K.S. Synthesis, characterization, and evaluation of biological activities of new 4′-substituted ruthenium (II) terpyridine complexes: Prospective anti-inflammatory properties. Appl. Organomet. Chem. 2021, 35, 1–16. [Google Scholar] [CrossRef]
- Nyawade, E.A.; Friedrich, H.B.; Omondi, B.; Chenia, H.Y.; Singh, M.; Gorle, S. Synthesis and characterization of new α,α′-diaminoalkane-bridged dicarbonyl(η5-cyclopentadienyl)ruthenium(II) complex salts: Antibacterial activity tests of η5-cyclopentadienyl dicarbonyl ruthenium(II) amine complexes. J. Organomet. Chem. 2015, 799–800, 138–146. [Google Scholar] [CrossRef]
- Rani, S.; Kumar, S.; Chandra, S. Synthesis, structural, spectral, thermal and antimicrobial studies of palladium(II), platinum(II), ruthenium(III) and iridium(III) complexes derived from N,N,N,N-tetradentate macrocyclic ligand. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2011, 78, 1507–1514. [Google Scholar] [CrossRef]
- Balasubramian, K.P.; Raju, V.V.; Chinnusamy, V. Synthesis, characteristic, catalytic and antimicrobial activities of imidazolo substituted benzylidene imines with ruthenium(II) complexes. J. Indian Chem. Soc. 2009, 86, 570–576. [Google Scholar]
- Thilagavathi, N.; Manimaran, A.; Priya, N.P.; Sathya, N.; Jayabalakrishnan, C. Synthesis, characterization, electrochemical, catalytic and antimicrobial activity studies of hydrazone Schiff base ruthenium(II) complexes. Appl. Organomet. Chem. 2010, 24, 301–307. [Google Scholar] [CrossRef]
- Thilagavathi, N.; Manimaran, A.; Padma Priya, N.; Sathya, N.; Jayabalakrishnan, C. Synthesis, spectroscopic, redox, catalytic and antimicrobial properties of some ruthenium(II) Schiff base complexes. Transit. Met. Chem. 2009, 34, 725–732. [Google Scholar] [CrossRef]
- Thilagavathi, N.; Jayabalakrishnan, C. Synthesis, characterization, catalytic and antimicrobial studies of ruthenium(III) complexes. Cent. Eur. J. Chem. 2010, 8, 842–851. [Google Scholar] [CrossRef]
- Thangadurai, T.D.; Ihm, S.-K. Ruthenium(II) Complexes Derived from Substituted Cyclobutane and Substituted Thiazole Schiff Base Ligands: Synthetic, Spectral, Catalytic and Antimicrobial Studies. Synth. React. Inorganic, Met. Nano-Metal Chem. 2005, 35, 499–507. [Google Scholar] [CrossRef]
- Matshwele, J.T.P.; Nareetsile, F.; Mapolelo, D.; Matshameko, P.; Leteane, M.; Nkwe, D.O.; Odisitse, S. Synthesis of Mixed Ligand Ruthenium (II/III) Complexes and Their Antibacterial Evaluation on Drug-Resistant Bacterial Organisms. J. Chem. 2020, 2020, 2150419. [Google Scholar] [CrossRef]
- Pavan, F.R.; Poelhsitz, G.V.; Barbosa, M.I.F.; Leite, S.R.A.; Batista, A.A.; Ellena, J.; Sato, L.S.; Franzblau, S.G.; Moreno, V.; Gambino, D.; et al. Ruthenium(II) phosphine/diimine/picolinate complexes: Inorganic compounds as agents against tuberculosis. Eur. J. Med. Chem. 2011, 46, 5099–5107. [Google Scholar] [CrossRef] [PubMed]
- Pavan, F.R.; Poelhsitz, G.V.; da Cunha, L.V.P.; Barbosa, M.I.F.; Leite, S.R.A.; Batista, A.A.; Cho, S.H.; Franzblau, S.G.; de Camargo, M.S.; Resende, F.A.; et al. In Vitro and In Vivo Activities of Ruthenium(II) Phosphine/Diimine/Picolinate Complexes (SCAR) against Mycobacterium tuberculosis. PLoS One 2013, 8, e64242. [Google Scholar]
- Rolston, K.V.; Wang, W.; Nesher, L.; Smith, J.R.; Rybak, M.J.; Prince, R.A. Time-kill determination of the bactericidal activity of telavancin and vancomycin against clinical methicillin-resistant Staphylococcus aureus isolates from cancer patients. Diagn. Microbiol. Infect. Dis. 2017, 87, 338–342. [Google Scholar] [CrossRef]
- Demirezen, N.; Tarınç, D.; Polat, D.; Çeşme, M.; Gölcü, A.; Tümer, M. Synthesis of trimethoprim metal complexes: Spectral, electrochemical, thermal, DNA-binding and surface morphology studies. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2012, 94, 243–255. [Google Scholar] [CrossRef]
- Ude, Z.; Romero-Canelón, I.; Twamley, B.; Fitzgerald Hughes, D.; Sadler, P.J.; Marmion, C.J. A novel dual-functioning ruthenium(II)–arene complex of an anti-microbial ciprofloxacin derivative — Anti-proliferative and anti-microbial activity. J. Inorg. Biochem. 2016, 160, 210–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colina-Vegas, L.; Dutra, J.L.; Villarreal, W.; Neto, J.H.d.A.; Cominetti, M.R.; Pavan, F.; Navarro, M.; Batista, A.A. Ru(II)/clotrimazole/diphenylphosphine/bipyridine complexes: Interaction with DNA, BSA and biological potential against tumor cell lines and Mycobacterium tuberculosis. J. Inorg. Biochem. 2016, 162, 135–145. [Google Scholar] [CrossRef] [Green Version]
- Naglah, A.M.; Al-Omar, M.A.; Almehizia, A.A.; Obaidullah, A.J.; Bhat, M.A.; Al-Shakliah, N.S.; Belgacem, K.; Majrashi, B.M.; Refat, M.S.; Adam, A.M.A. Synthesis, Spectroscopic, and Antimicrobial Study of Binary and Ternary Ruthenium(III) Complexes of Ofloxacin Drug and Amino Acids as Secondary Ligands. Crystals 2020, 10, 225. [Google Scholar] [CrossRef] [Green Version]
- Măciucă, A.-M.; Munteanu, A.-C.; Uivarosi, V. Quinolone Complexes with Lanthanide Ions: An Insight into their Analytical Applications and Biological Activity. Molecules 2020, 25, 1347. [Google Scholar] [CrossRef] [Green Version]
- Pisani, M.J.; Weber, D.K.; Heimann, K.; Collins, J.G.; Keene, F.R. Selective mitochondrial accumulation of cytotoxic dinuclear polypyridyl ruthenium(II) complexes. Metallomics 2010, 2, 393–396. [Google Scholar] [CrossRef] [PubMed]
- Weber, D.K.; Sani, M.-A.; Downton, M.T.; Separovic, F.; Keene, F.R.; Collins, J.G. Membrane Insertion of a Dinuclear Polypyridylruthenium(II) Complex Revealed by Solid-State NMR and Molecular Dynamics Simulation: Implications for Selective Antibacterial Activity. J. Am. Chem. Soc. 2016, 138, 15267–15277. [Google Scholar] [CrossRef] [PubMed]
- Morgan, J.L.; Spillane, C.B.; Smith, J.A.; Buck, D.P.; Collins, J.G.; Keene, F.R. Dinuclear ruthenium(II) complexes with flexible bridges as non-duplex DNA binding agents. Dalt. Trans. 2007, 4333–4342. [Google Scholar] [CrossRef]
- Li, F.; Weber, D.K.; Morgan, J.L.; Collins, J.G.; Keene, F.R. An approach to therapeutic agents through selective targeting of destabilised nucleic acid duplex sequences. Dalt. Trans. 2012, 41, 6528–6535. [Google Scholar] [CrossRef]
- Gill, M.R.; Garcia-Lara, J.; Foster, S.J.; Smythe, C.; Battaglia, G.; Thomas, J.A. A ruthenium(II) polypyridyl complex for direct imaging of DNA structure in living cells. Nat. Chem. 2009, 1, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Millimouno, F.M.; Dong, J.; Yang, L.; Li, J.; Li, X. Targeting Apoptosis Pathways in Cancer and Perspectives with Natural Compounds from Mother Nature. Cancer Prev. Res. 2014, 7, 1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buck, D.P.; Paul, J.A.; Pisani, M.J.; Collins, J.G.; Keene, F.R. Binding of a Flexibly-linked Dinuclear Ruthenium(II) Complex to Adenine-bulged DNA Duplexes. Aust. J. Chem. 2010, 63, 1365–1375. [Google Scholar] [CrossRef]
- Recht, M.I.; Douthwaite, S.; Puglisi, J.D. Basis for prokaryotic specificity of action of aminoglycoside antibiotics. EMBO J. 1999, 18, 3133–3138. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Gorle, A.K.; Ranson, M.; Vine, K.L.; Kinobe, R.; Feterl, M.; Warner, J.M.; Keene, F.R.; Collins, J.G.; Day, A.I. Probing the pharmacokinetics of cucurbit[7, 8 and 10]uril: And a dinuclear ruthenium antimicrobial complex encapsulated in cucurbit[10]uril. Org. Biomol. Chem. 2017, 15, 4172–4179. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.G.; May-Dracka, T.L.; Gagnon, M.M.; Tommasi, R. Trends and exceptions of physical properties on antibacterial activity for Gram-positive and Gram-negative pathogens. J. Med. Chem. 2014, 57, 10144–10161. [Google Scholar] [CrossRef]
- Kitamura, Y.; Ihara, T.; Tsujimura, Y.; Osawa, Y.; Sasahara, D.; Yamamoto, M.; Okada, K.; Tazaki, M.; Jyo, A. Template-directed formation of luminescent lanthanide complexes: Versatile tools for colorimetric identification of single nucleotide polymorphism. J. Inorg. Biochem. 2008, 102, 1921–1931. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.V.; Lo, W.K.C.; Brooks, H.J.L.; Crowley, J.D. Synthesis, structure, stability and antimicrobial activity of a ruthenium(II) helicate derived from a bis-bidentate “click” pyridyl-1,2,3-triazole ligand. Inorganica Chim. Acta 2015, 425, 1–6. [Google Scholar] [CrossRef]
- Roopashree, B.; Gayathri, V.; Gopi, A.; Devaraju, K.S. Syntheses, characterizations, and antimicrobial activities of binuclear ruthenium(III) complexes containing 2-substituted benzimidazole derivatives. J. Coord. Chem. 2012, 65, 4023–4040. [Google Scholar] [CrossRef]
- Wenzel, M.; Patra, M.; Senges, C.H.R.; Ott, I.; Stepanek, J.J.; Pinto, A.; Prochnow, P.; Vuong, C.; Langklotz, S.; Metzler-Nolte, N.; et al. Analysis of the mechanism of action of potent antibacterial hetero-tri-organometallic compounds: a structurally new class of antibiotics. ACS Chem. Biol. 2013, 8, 1442–1450. [Google Scholar] [CrossRef]
- Stringer, T.; Seldon, R.; Liu, N.; Warner, D.F.; Tam, C.; Cheng, L.W.; Land, K.M.; Smith, P.J.; Chibale, K.; Smith, G.S. Antimicrobial activity of organometallic isonicotinyl and pyrazinyl ferrocenyl-derived complexes. Dalt. Trans. 2017, 46, 9875–9885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, A.; Winkel, B.S.J.; Brewer, K.J. In vivo inhibition of E. coli growth by a Ru(II)/Pt(II) supramolecule [(tpy)RuCl(dpp)PtCl2](PF6). J. Inorg. Biochem. 2007, 101, 1525–1528. [Google Scholar] [CrossRef]
- Jain, A.; Wang, J.; Mashack, E.R.; Winkel, B.S.J.; Brewer, K.J. Multifunctional DNA Interactions of Ru−Pt Mixed Metal Supramolecular Complexes with Substituted Terpyridine Ligands. Inorg. Chem. 2009, 48, 9077–9084. [Google Scholar] [CrossRef]
- Wareham, L.K.; Poole, R.K.; Tinajero-Trejo, M. CO-releasing Metal Carbonyl Compounds as Antimicrobial Agents in the Post-antibiotic Era. J. Biol. Chem. 2015, 290, 18999–19007. [Google Scholar] [CrossRef] [Green Version]
- Ling, K.; Men, F.; Wang, W.-C.; Zhou, Y.-Q.; Zhang, H.-W.; Ye, D.-W. Carbon Monoxide and Its Controlled Release: Therapeutic Application, Detection, and Development of Carbon Monoxide Releasing Molecules (CORMs). J. Med. Chem. 2018, 61, 2611–2635. [Google Scholar] [CrossRef]
- Adach, W.; Błaszczyk, M.; Olas, B. Carbon monoxide and its donors - Chemical and biological properties. Chem. Biol. Interact. 2020, 318, 108973. [Google Scholar] [CrossRef]
- Yan, H.; Du, J.; Zhu, S.; Nie, G.; Zhang, H.; Gu, Z.; Zhao, Y. Emerging Delivery Strategies of Carbon Monoxide for Therapeutic Applications: From CO Gas to CO Releasing Nanomaterials. Small 2019, 15, 1904382. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.; Mann, B.E.; Motterlini, R.; Poole, R.K. The carbon monoxide-releasing molecule, CORM-3 (Ru(CO)3Cl(glycinate)), targets respiration and oxidases in Campylobacter jejuni, generating hydrogen peroxide. IUBMB Life 2011, 63, 363–371. [Google Scholar] [CrossRef] [PubMed]
- McLean, S.; Begg, R.; Jesse, H.E.; Mann, B.E.; Sanguinetti, G.; Poole, R.K. Analysis of the bacterial response to Ru(CO)3Cl(Glycinate) (CORM-3) and the inactivated compound identifies the role played by the ruthenium compound and reveals sulfur-containing species as a major target of CORM-3 action. Antioxid. Redox Signal. 2013, 19, 1999–2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, J.L.; Jesse, H.E.; Hughes, B.; Lund, V.; Naylor, K.; Davidge, K.S.; Cook, G.M.; Mann, B.E.; Poole, R.K. Ru(CO)3Cl(Glycinate) (CORM-3): A carbon monoxide-releasing molecule with broad-spectrum antimicrobial and photosensitive activities against respiration and cation transport in Escherichia coli. Antioxidants Redox Signal. 2013, 19, 497–509. [Google Scholar] [CrossRef] [Green Version]
- Rana, N.; McLean, S.; Mann, B.E.; Poole, R.K. Interaction of the carbon monoxide-releasing molecule Ru(CO)3Cl(glycinate) (CORM-3) with Salmonella enterica serovar Typhimurium: In situ measurements of carbon monoxide binding by integrating cavity dual-beam spectrophotometry. Microbiology 2014, 160, 2771–2779. [Google Scholar] [CrossRef]
- Johnson, T.R.; Mann, B.E.; Teasdale, I.P.; Adams, H.; Foresti, R.; Green, C.J.; Motterlini, R. Metal carbonyls as pharmaceuticals? [Ru(CO)3Cl(glycinate)], a CO-releasing molecule with an extensive aqueous solution chemistry. Dalt. Trans. 2007, 1500–1508. [Google Scholar] [CrossRef]
- Jesse, H.E.; Nye, T.L.; McLean, S.; Green, J.; Mann, B.E.; Poole, R.K. Cytochrome bd-I in Escherichia coli is less sensitive than cytochromes bd-II or bo′ to inhibition by the carbon monoxide-releasing molecule, CORM-3: N-acetylcysteine reduces CO-RM uptake and inhibition of respiration. Biochim. Biophys. Acta - Proteins Proteomics 2013, 1834, 1693–1703. [Google Scholar] [CrossRef] [Green Version]
- Davidge, K.S.; Sanguinetti, G.; Yee, C.H.; Cox, A.G.; McLeod, C.W.; Monk, C.E.; Mann, B.E.; Motterlini, R.; Poole, R.K. Carbon monoxide-releasing antibacterial molecules target respiration and global transcriptional regulators. J. Biol. Chem. 2009, 284, 4516–4524. [Google Scholar] [CrossRef] [Green Version]
- Desmard, M.; Foresti, R.; Morin, D.; Dagouassat, M.; Berdeaux, A.; Denamur, E.; Crook, S.H.; Mann, B.E.; Scapens, D.; Montravers, P.; et al. Differential antibacterial activity against Pseudomonas aeruginosa by carbon monoxide-releasing molecules. Antioxid. Redox Signal. 2012, 16, 153–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaves-Ferreira, M.; Albuquerque, I.S.; Matak-Vinkovic, D.; Coelho, A.C.; Carvalho, S.M.; Saraiva, L.M.; Romão, C.C.; Bernardes, G.J.L. Spontaneous CO Release from RuII(CO)2–Protein Complexes in Aqueous Solution, Cells, and Mice. Angew. Chemie Int. Ed. 2015, 54, 1172–1175. [Google Scholar] [CrossRef] [Green Version]
- Southam, H.M.; Smith, T.W.; Lyon, R.L.; Liao, C.; Trevitt, C.R.; Middlemiss, L.A.; Cox, F.L.; Chapman, J.A.; El-Khamisy, S.F.; Hippler, M.; et al. A thiol-reactive Ru(II) ion, not CO release, underlies the potent antimicrobial and cytotoxic properties of CO-releasing molecule-3. Redox Biol. 2018, 18, 114–123. [Google Scholar] [CrossRef]
- Tavares, A.F.N.; Teixeira, M.; Romão, C.C.; Seixas, J.D.; Nobre, L.S.; Saraiva, L.M. Reactive oxygen species mediate bactericidal killing elicited by carbon monoxide-releasing molecules. J. Biol. Chem. 2011, 286, 26708–26717. [Google Scholar] [CrossRef] [Green Version]
- Marazioti, A.; Bucci, M.; Coletta, C.; Vellecco, V.; Baskaran, P.; Szabó, C.; Cirino, G.; Marques, A.R.; Guerreiro, B.; Gonçalves, A.M.L.; et al. Inhibition of nitric oxide-stimulated vasorelaxation by carbon monoxide-releasing molecules. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2570–2576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taillé, C.; El-Benna, J.; Lanone, S.; Boczkowski, J.; Motterlini, R. Mitochondrial respiratory chain and NAD(P)H oxidase are targets for the antiproliferative effect of carbon monoxide in human airway smooth muscle. J. Biol. Chem. 2005, 280, 25350–25360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuckerbraun, B.S.; Chin, B.Y.; Bilban, M.; D’Avila, J.d.C.; Rao, J.; Billiar, T.R.; Otterbein, L.E. Carbon monoxide signals via inhibition of cytochrome c oxidase and generation of mitochondrial reactive oxygen species. FASEB J. 2007, 21, 1099–1106. [Google Scholar] [CrossRef] [Green Version]
- Murray, T.S.; Okegbe, C.; Gao, Y.; Kazmierczak, B.I.; Motterlini, R.; Dietrich, L.E.P.; Bruscia, E.M. The Carbon Monoxide Releasing Molecule CORM-2 Attenuates Pseudomonas aeruginosa Biofilm Formation. PLoS One 2012, 7, e35499. [Google Scholar] [CrossRef]
- Seixas, J.D.; Chaves-Ferreira, M.; Montes-Grajales, D.; Gonçalves, A.M.; Marques, A.R.; Saraiva, L.M.; Olivero-Verbel, J.; Romão, C.C.; Bernardes, G.J.L. An N-Acetyl Cysteine Ruthenium Tricarbonyl Conjugate Enables Simultaneous Release of CO and Ablation of Reactive Oxygen Species. Chem. Eur. J. 2015, 21, 14708–14712. [Google Scholar] [CrossRef] [Green Version]
- Wilson, J.L.; Wareham, L.K.; McLean, S.; Begg, R.; Greaves, S.; Mann, B.E.; Sanguinetti, G.; Poole, R.K. CO-Releasing Molecules Have Nonheme Targets in Bacteria: Transcriptomic, Mathematical Modeling and Biochemical Analyses of CORM-3 [Ru(CO)3Cl(glycinate)] Actions on a Heme-Deficient Mutant of Escherichia coli. Antioxid. Redox Signal. 2015, 23, 148–162. [Google Scholar]
- Santos-Silva, T.; Mukhopadhyay, A.; Seixas, J.D.; Bernardes, G.J.L.; Romão, C.C.; Romão, M.J. CORM-3 Reactivity toward Proteins: The Crystal Structure of a Ru(II) Dicarbonyl−Lysozyme Complex. J. Am. Chem. Soc. 2011, 133, 1192–1195. [Google Scholar] [CrossRef] [PubMed]
- Santos-Silva, T.; Mukhopadhyay, A.; Seixas, J.D.; Bernardes, G.J.L.; Romão, C.C.; Romão, M.J. Towards improved therapeutic CORMs: Understanding the reactivity of CORM-3 with proteins. Curr. Med. Chem. 2011, 18, 3361–3366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seixas, J.D.; Santos, M.F.A.; Mukhopadhyay, A.; Coelho, A.C.; Reis, P.M.; Veiros, L.F.; Marques, A.R.; Penacho, N.; Gonçalves, A.M.L.; Romão, M.J.; et al. A contribution to the rational design of Ru(CO)3Cl2L complexes for in vivo delivery of CO. Dalt. Trans. 2015, 44, 5058–5075. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Liu, H.; Zhao, Q.; Chen, Y.; Liu, B.; Zhang, B.; Zheng, Q. Syntheses and evaluation of drug-like properties of CO-releasing molecules containing ruthenium and group 6 metal. Eur. J. Med. Chem. 2014, 74, 199–215. [Google Scholar] [CrossRef]
- Winburn, I.C.; Gunatunga, K.; McKernan, R.D.; Walker, R.J.; Sammut, I.A.; Harrison, J.C. Cell damage following carbon monoxide releasing molecule exposure: Implications for therapeutic applications. Basic Clin. Pharmacol. Toxicol. 2012, 111, 31–41. [Google Scholar] [CrossRef]
- Stingl, K.; De Reuse, H. Staying alive overdosed: How does Helicobacter pylori control urease activity? Int. J. Med. Microbiol. 2005, 295, 307–315. [Google Scholar] [CrossRef]
- Lemire, J.A.; Harrison, J.J.; Turner, R.J. Antimicrobial activity of metals: Mechanisms, molecular targets and applications. Nat. Rev. Microbiol. 2013, 11, 371–384. [Google Scholar] [CrossRef]
- Arnett, C.H.; Chalkley, M.J.; Agapie, T. A Thermodynamic Model for Redox-Dependent Binding of Carbon Monoxide at Site-Differentiated, High Spin Iron Clusters. J. Am. Chem. Soc. 2018, 140, 5569–5578. [Google Scholar] [CrossRef] [Green Version]
- Wilson, J.L.; McLean, S.; Begg, R.; Sanguinetti, G.; Poole, R.K. Analysis of transcript changes in a heme-deficient mutant of Escherichia coli in response to CORM-3 [Ru(CO)3Cl(glycinate)]. Genomics Data 2015, 5, 231–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nobre, L.S.; Al-Shahrour, F.; Dopazo, J.; Saraiva, L.M. Exploring the antimicrobial action of a carbon monoxide-releasing compound through whole-genome transcription profiling of Escherichia coli. Microbiology 2009, 155, 813–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wareham, L.K.; Begg, R.; Jesse, H.E.; Van Beilen, J.W.A.; Ali, S.; Svistunenko, D.; McLean, S.; Hellingwerf, K.J.; Sanguinetti, G.; Poole, R.K. Carbon Monoxide Gas Is Not Inert, but Global, in Its Consequences for Bacterial Gene Expression, Iron Acquisition, and Antibiotic Resistance. Antioxid. Redox Signal. 2016, 24, 1013–1028. [Google Scholar] [CrossRef] [Green Version]
- Li, X.-Z.; Nikaido, H. Efflux-mediated drug resistance in bacteria: An update. Drugs 2009, 69, 1555–1623. [Google Scholar] [CrossRef]
- Villemin, E.; Ong, Y.C.; Thomas, C.M.; Gasser, G. Polymer encapsulation of ruthenium complexes for biological and medicinal applications. Nat. Rev. Chem. 2019, 3, 261–282. [Google Scholar] [CrossRef]
- Nguyen, D.; Nguyen, T.-K.; Rice, S.A.; Boyer, C. CO-Releasing Polymers Exert Antimicrobial Activity. Biomacromolecules 2015, 16, 2776–2786. [Google Scholar] [CrossRef]
- Bang, C.S.; Kruse, R.; Demirel, I.; Önnberg, A.; Söderquist, B.; Persson, K. Multiresistant uropathogenic extended-spectrum β-lactamase (ESBL)-producing Escherichia coli are susceptible to the carbon monoxide releasing molecule-2 (CORM-2). Microb. Pathog. 2014, 66, 29–35. [Google Scholar] [CrossRef]
- Chung, S.W.; Liu, X.; Macias, A.A.; Baron, R.M.; Perrella, M.A. Heme oxygenase-1-derived carbon monoxide enhances the host defense response to microbial sepsis in mice. J. Clin. Invest. 2008, 118, 239–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Josefsen, L.B.; Boyle, R.W. Photodynamic Therapy and the Development of Metal-Based Photosensitisers. Met. Based. Drugs 2008, 2008, 276109. [Google Scholar] [CrossRef] [Green Version]
- Le Gall, T.; Lemercier, G.; Chevreux, S.; Tücking, K.-S.; Ravel, J.; Thétiot, F.; Jonas, U.; Schönherr, H.; Montier, T. Ruthenium(II) Polypyridyl Complexes as Photosensitizers for Antibacterial Photodynamic Therapy: A Structure-Activity Study on Clinical Bacterial Strains. ChemMedChem 2018, 13, 2229–2239. [Google Scholar] [CrossRef]
- Soliman, N.; Sol, V.; Ouk, T.-S.; Thomas, C.M.; Gasser, G. Encapsulation of a Ru(II) Polypyridyl Complex into Polylactide Nanoparticles for Antimicrobial Photodynamic Therapy. Pharmaceutics 2020, 12, 961. [Google Scholar] [CrossRef]
- Rensmo, H.; Lunell, S.; Siegbahn, H. Absorption and electrochemical properties of ruthenium(II) dyes, studied by semiempirical quantum chemical calculations. J. Photochem. Photobiol. A Chem. 1998, 114, 117–124. [Google Scholar] [CrossRef]
- Lei, W.; Zhou, Q.; Jiang, G.; Zhang, B.; Wang, X. Photodynamic inactivation of Escherichia coli by Ru(II) complexes. Photochem. Photobiol. Sci. 2011, 10, 887–890. [Google Scholar] [CrossRef] [PubMed]
- Allardyce, C.; Dyson, P. Ruthenium in Medicine: Current Clinical Uses and Future Prospects. Platin. Met. Rev. 2001, 45, 62–69. [Google Scholar]
- Wassmer, S.C.; Grau, G.E.R. Severe malaria: What’s new on the pathogenesis front? Int. J. Parasitol. 2017, 47, 145–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baartzes, N.; Stringer, T.; Smith, G.S. Targeting Sensitive-Strain and Resistant-Strain Malaria Parasites Through a Metal-Based Approach. In Advances in Bioorganometallic Chemistry; Hirao, T., Moriuchi, T., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 193–213. ISBN 978-0-12-814197-7. [Google Scholar]
- Miotto, O.; Almagro-Garcia, J.; Manske, M.; Macinnis, B.; Campino, S.; Rockett, K.A.; Amaratunga, C.; Lim, P.; Suon, S.; Sreng, S.; et al. Multiple populations of artemisinin-resistant Plasmodium falciparum in Cambodia. Nat. Genet. 2013, 45, 648–655. [Google Scholar] [CrossRef]
- Martínez, A.; Rajapakse, C.S.K.; Naoulou, B.; Kopkalli, Y.; Davenport, L.; Sánchez-Delgado, R.A. The mechanism of antimalarial action of the ruthenium(II)-chloroquine complex [RuCl(2)(CQ)] (2). J. Biol. Inorg. Chem. 2008, 13, 703–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Delgado, R.A.; Navarro, M.; Pérez, H.; Urbina, J.A. Toward a Novel Metal-Based Chemotherapy against Tropical Diseases. 2. Synthesis and Antimalarial Activity in Vitro and in Vivo of New Ruthenium− and Rhodium−Chloroquine Complexes. J. Med. Chem. 1996, 39, 1095–1099. [Google Scholar] [CrossRef]
- Macedo, T.S.; Colina-Vegas, L.; DA Paixão, M.; Navarro, M.; Barreto, B.C.; Oliveira, P.C.M.; Macambira, S.G.; Machado, M.; Prudêncio, M.; D’Alessandro, S.; et al. Chloroquine-containing organoruthenium complexes are fast-acting multistage antimalarial agents. Parasitology 2016, 143, 1543–1556. [Google Scholar] [CrossRef] [Green Version]
- Adams, M.; de Kock, C.; Smith, P.J.; Land, K.M.; Liu, N.; Hopper, M.; Hsiao, A.; Burgoyne, A.R.; Stringer, T.; Meyer, M.; et al. Improved antiparasitic activity by incorporation of organosilane entities into half-sandwich ruthenium(II) and rhodium(III) thiosemicarbazone complexes. Dalt. Trans. 2015, 44, 2456–2468. [Google Scholar] [CrossRef] [Green Version]
- Rylands, L.-I.; Welsh, A.; Maepa, K.; Stringer, T.; Taylor, D.; Chibale, K.; Smith, G.S. Structure-activity relationship studies of antiplasmodial cyclometallated ruthenium(II), rhodium(III) and iridium(III) complexes of 2-phenylbenzimidazoles. Eur. J. Med. Chem. 2019, 161, 11–21. [Google Scholar] [CrossRef]
- Chellan, P.; Land, K.M.; Shokar, A.; Au, A.; An, S.H.; Taylor, D.; Smith, P.J.; Chibale, K.; Smith, G.S. Di- and Trinuclear Ruthenium-, Rhodium-, and Iridium-Functionalized Pyridyl Aromatic Ethers: A New Class of Antiparasitic Agents. Organometallics 2013, 32, 4793–4804. [Google Scholar] [CrossRef]
- Stringer, T.; Quintero, M.A.S.; Wiesner, L.; Smith, G.S.; Nordlander, E. Evaluation of PTA-derived ruthenium(II) and iridium(III) quinoline complexes against chloroquine-sensitive and resistant strains of the Plasmodium falciparum malaria parasite. J. Inorg. Biochem. 2019, 191, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Chellan, P.; Land, K.M.; Shokar, A.; Au, A.; An, S.H.; Taylor, D.; Smith, P.J.; Riedel, T.; Dyson, P.J.; Chibale, K.; et al. Synthesis and evaluation of new polynuclear organometallic Ru(ii), Rh(iii) and Ir(iii) pyridyl ester complexes as in vitro antiparasitic and antitumor agents. Dalt. Trans. 2014, 43, 513–526. [Google Scholar] [CrossRef] [Green Version]
- De Souza, N.B.; Aguiar, A.C.C.; de Oliveira, A.C.; Top, S.; Pigeon, P.; Jaouen, G.; Goulart, M.O.F.; Krettli, A.U. Antiplasmodial activity of iron(II) and ruthenium(II) organometallic complexes against Plasmodium falciparum blood parasites. Mem. Inst. Oswaldo Cruz 2015, 110, 981–988. [Google Scholar] [CrossRef] [Green Version]
- Lidani, K.C.F.; Andrade, F.A.; Bavia, L.; Damasceno, F.S.; Beltrame, M.H.; Messias-Reason, I.J.; Sandri, T.L. Chagas Disease: From Discovery to a Worldwide Health Problem. Front. public Heal. 2019, 7, 166. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, P.G.E. Update on human African trypanosomiasis (sleeping sickness). J. Neurol. 2019, 266, 2334–2337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, J.J.N.; Osakabe, A.L.; Pavanelli, W.R.; Silva, J.S.; Franco, D.W. In vitro and in vivo antiproliferative and trypanocidal activities of ruthenium NO donors. Br. J. Pharmacol. 2007, 152, 112–121. [Google Scholar] [CrossRef] [Green Version]
- Silva, J.J.N.; Pavanelli, W.R.; Pereira, J.C.M.; Silva, J.S.; Franco, D.W. Experimental chemotherapy against Trypanosoma cruzi infection using ruthenium nitric oxide donors. Antimicrob. Agents Chemother. 2009, 53, 4414–4421. [Google Scholar] [CrossRef] [Green Version]
- Silva, J.J.N.; Guedes, P.M.M.; Zottis, A.; Balliano, T.L.; Nascimento Silva, F.O.; França Lopes, L.G.; Ellena, J.; Oliva, G.; Andricopulo, A.D.; Franco, D.W.; et al. Novel ruthenium complexes as potential drugs for Chagas’s disease: Enzyme inhibition and in vitro/in vivo trypanocidal activity. Br. J. Pharmacol. 2010, 160, 260–269. [Google Scholar] [CrossRef] [Green Version]
- Bastos, T.M.; Barbosa, M.I.F.; da Silva, M.M.; Júnior, J.W.d.c.; Meira, C.S.; Guimaraes, E.T.; Ellena, J.; Moreira, D.R.M.; Batista, A.A.; Soares, M.B.P. Nitro/nitrosyl-ruthenium complexes are potent and selective anti-Trypanosoma cruzi agents causing autophagy and necrotic parasite death. Antimicrob. Agents Chemother. 2014, 58, 6044–6055. [Google Scholar] [CrossRef] [Green Version]
- Possato, B.; Carneiro, Z.A.; de Albuquerque, S.; Nikolaou, S. New uses for old complexes: The very first report on the trypanocidal activity of symmetric trinuclear ruthenium complexes. J. Inorg. Biochem. 2017, 176, 156–158. [Google Scholar] [CrossRef] [PubMed]
- Fernández, M.; Arce, E.R.; Sarniguet, C.; Morais, T.S.; Tomaz, A.I.; Azar, C.O.; Figueroa, R.; Diego Maya, J.; Medeiros, A.; Comini, M.; et al. Novel ruthenium(II) cyclopentadienyl thiosemicarbazone compounds with antiproliferative activity on pathogenic trypanosomatid parasites. J. Inorg. Biochem. 2015, 153, 306–314. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez Arce, E.; Sarniguet, C.; Moraes, T.S.; Vieites, M.; Tomaz, A.I.; Medeiros, A.; Comini, M.A.; Varela, J.; Cerecetto, H.; González, M.; et al. A new ruthenium cyclopentadienyl azole compound with activity on tumor cell lines and trypanosomatid parasites. J. Coord. Chem. 2015, 68, 2923–2937. [Google Scholar] [CrossRef]
- Martínez, A.; Carreon, T.; Iniguez, E.; Anzellotti, A.; Sánchez, A.; Tyan, M.; Sattler, A.; Herrera, L.; Maldonado, R.A.; Sánchez-Delgado, R.A. Searching for new chemotherapies for tropical diseases: Ruthenium-clotrimazole complexes display high in vitro activity against Leishmania major and Trypanosoma cruzi and low toxicity toward normal mammalian cells. J. Med. Chem. 2012, 55, 3867–3877. [Google Scholar] [CrossRef] [Green Version]
- Sundar, S.; Chakravarty, J. Leishmaniasis: an update of current pharmacotherapy. Expert Opin. Pharmacother. 2013, 14, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, M.I.F.; Corrêa, R.S.; de Oliveira, K.M.; Rodrigues, C.; Ellena, J.; Nascimento, O.R.; Rocha, V.P.C.; Nonato, F.R.; Macedo, T.S.; Barbosa-Filho, J.M.; et al. Antiparasitic activities of novel ruthenium/lapachol complexes. J. Inorg. Biochem. 2014, 136, 33–39. [Google Scholar] [CrossRef]
- Wong, E.L.-M.; Sun, R.W.-Y.; Chung, N.P.-Y.; Lin, C.-L.S.; Zhu, N.; Che, C.-M. A Mixed-Valent Ruthenium−Oxo Oxalato Cluster Na7[Ru4(μ3-O)4(C2O4)6] with Potent Anti-HIV Activities. J. Am. Chem. Soc. 2006, 128, 4938–4939. [Google Scholar] [CrossRef]
- Luedtke, N.W.; Hwang, J.S.; Glazer, E.C.; Gut, D.; Kol, M.; Tor, Y. Eilatin Ru(II) complexes display anti-HIV activity and enantiomeric diversity in the binding of RNA. Chembiochem 2002, 3, 766–771. [Google Scholar] [CrossRef]
- van Oosterhout, C.; Hall, N.; Ly, H.; Tyler, K.M. COVID-19 evolution during the pandemic – Implications of new SARS-CoV-2 variants on disease control and public health policies. Virulence 2021, 12, 507–508. [Google Scholar] [CrossRef]
- Mahase, E. Covid-19: What new variants are emerging and how are they being investigated? BMJ 2021, 372, n158. [Google Scholar] [CrossRef] [PubMed]
- Neuditschko, B.; Legin, A.A.; Baier, D.; Schintlmeister, A.; Reipert, S.; Wagner, M.; Keppler, B.K.; Berger, W.; Meier-Menches, S.M.; Gerner, C. Interaction with Ribosomal Proteins Accompanies Stress Induction of the Anticancer Metallodrug BOLD-100/KP1339 in the Endoplasmic Reticulum. Angew. Chem. Int. Ed. Engl. 2021, 60, 5063–5068. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Xiao, Y.; Li, D.; Jiang, Q.; Zhu, L.; Lin, D.; Jiang, H.; Chen, W.; Wang, L.; Liu, C.; et al. PGK1 and GRP78 overexpression correlates with clinical significance and poor prognosis in Chinese endometrial cancer patients. Oncotarget 2017, 9, 680–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakewell, S.J.; Rangel, D.F.; Ha, D.P.; Sethuraman, J.; Crouse, R.; Hadley, E.; Costich, T.L.; Zhou, X.; Nichols, P.; Lee, A.S. Suppression of stress induction of the 78-kilodalton glucose regulated protein (GRP78) in cancer by IT-139, an anti-tumor ruthenium small molecule inhibitor. Oncotarget 2018, 9, 29698–29714. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, I.M.; Abdelmalek, D.H.; Elfiky, A.A. GRP78: A cell’s response to stress. Life Sci. 2019, 226, 156–163. [Google Scholar] [CrossRef]
- Ha, D.P.; Van Krieken, R.; Carlos, A.J.; Lee, A.S. The stress-inducible molecular chaperone GRP78 as potential therapeutic target for coronavirus infection. J. Infect. 2020, 81, 452–482. [Google Scholar] [CrossRef]
- Ibrahim, I.M.; Abdelmalek, D.H.; Elshahat, M.E.; Elfiky, A.A. COVID-19 spike-host cell receptor GRP78 binding site prediction. J. Infect. 2020, 80, 554–562. [Google Scholar] [CrossRef]
- Bold Therapeutics Potential to Fight COVID-19. Available online: https://www.bold-therapeutics.com/COVID-19 (accessed on 18 April 2021).
- O’Kane, G.M.; Spratlin, J.L.; Kavan, P.; Goodwin, R.A.; McWhirter, E.; Thompson, D.; Jones, M.; McAllister, E.R.; Machado, A.; Lemmerick, Y.; et al. BOLD-100-001 (TRIO039): A phase Ib dose-escalation study of BOLD-100 in combination with FOLFOX chemotherapy in patients with advanced gastrointestinal solid tumors. J. Clin. Oncol. 2021, 39, TPS145. [Google Scholar] [CrossRef]
- Lippi, G.; Salvagno, G.L.; Pegoraro, M.; Militello, V.; Caloi, C.; Peretti, A.; De Nitto, S.; Bovo, C.; Lo Cascio, G. Preliminary evaluation of Roche Cobas Elecsys Anti-SARS-CoV-2 chemiluminescence immunoassay. Clin. Chem. Lab. Med. 2020, 58, e251–e253. [Google Scholar] [CrossRef]
- Riester, E.; Findeisen, P.; Hegel, J.K.; Kabesch, M.; Ambrosch, A.; Rank, C.M.; Langen, F.; Laengin, T.; Niederhauser, C. Performance evaluation of the Roche Elecsys Anti-SARS-CoV-2 S immunoassay. medRxiv 2021. [Google Scholar] [CrossRef]
- Rothan, H.A.; Stone, S.; Natekar, J.; Kumari, P.; Arora, K.; Kumar, M. The FDA-approved gold drug auranofin inhibits novel coronavirus (SARS-COV-2) replication and attenuates inflammation in human cells. Virology 2020, 547, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Marzo, T.; Messori, L. A Role for Metal-Based Drugs in Fighting COVID-19 Infection? The Case of Auranofin. ACS Med. Chem. Lett. 2020, 11, 1067–1068. [Google Scholar] [CrossRef] [PubMed]
- Karges, J.; Kalaj, M.; Gembicky, M.; Cohen, S.M. ReI Tricarbonyl Complexes as Coordinate Covalent Inhibitors for the SARS-CoV-2 Main Cysteine Protease. Angew. Chemie Int. Ed. 2021, 60, 10716. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Comparison between Gram-negative and Gram-positive bacteria cell walls. Adapted from [22] with permission. Copyright © 2020 Huan, Kong, Mou and Yi.
Figure 1.
Comparison between Gram-negative and Gram-positive bacteria cell walls. Adapted from [22] with permission. Copyright © 2020 Huan, Kong, Mou and Yi.

Figure 2.
Mechanisms of action of currently used antibiotics (Image by Kendrick Johnson, licensed under the Creative Commons Attribution-Share Alike 3.0 Unported license).
Figure 2.
Mechanisms of action of currently used antibiotics (Image by Kendrick Johnson, licensed under the Creative Commons Attribution-Share Alike 3.0 Unported license).

Figure 3.
Examples of inert structural mononuclear polypyridylruthenium (II) complexes.

Figure 4.
Chemical structures of heteroleptic Ru(II) complexes bearing 2,2’-bipyridine (bpy) ligands. BTPIP = (2-(4-(benzo[b]thiophen-2-yl)phenyl)-1H-imidazo [4,5-f][1,10]phenanthroline); ETPIP = 2-(4-(thiophen-2-ylethynyl)phenyl)-1H-imidazo[4,5-f][1,10]phenanthroline); CAPIP = (E)-2-(2-(furan-2-yl)vinyl)-1H-imidazo[4,5-f][1,10]phenanthroline; dmp = 4,4’-dimethyl-2,2’-bipyridine; bpy = 2,2’-bipyridine; phen = 1,10-phenanthroline.
Figure 4.
Chemical structures of heteroleptic Ru(II) complexes bearing 2,2’-bipyridine (bpy) ligands. BTPIP = (2-(4-(benzo[b]thiophen-2-yl)phenyl)-1H-imidazo [4,5-f][1,10]phenanthroline); ETPIP = 2-(4-(thiophen-2-ylethynyl)phenyl)-1H-imidazo[4,5-f][1,10]phenanthroline); CAPIP = (E)-2-(2-(furan-2-yl)vinyl)-1H-imidazo[4,5-f][1,10]phenanthroline; dmp = 4,4’-dimethyl-2,2’-bipyridine; bpy = 2,2’-bipyridine; phen = 1,10-phenanthroline.

Figure 5.
[Ru(N-N)2curcumin]+, where N-N is either 2,2’-bypiridine (bpy) or 1,10-phenanthroline (phen).
Figure 5.
[Ru(N-N)2curcumin]+, where N-N is either 2,2’-bypiridine (bpy) or 1,10-phenanthroline (phen).

Figure 6.
Chemical structures of heteroleptic Ru(II) complexes bearing 1,10-phenanthroline (phen) ligands.
Figure 6.
Chemical structures of heteroleptic Ru(II) complexes bearing 1,10-phenanthroline (phen) ligands.

Figure 7.
The ligand bbn and the possible isomeric forms of the mononuclear complex [Ru(phen)(bbn)]2+ with bbn as a tetradentate ligand: (a) cis-α isomer, (b) cis-β isomer, and (c) a form in which the central polymethylene chain spans the trans. Reproduced from [48] with permission. Copyright © 2015 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
Figure 7.
The ligand bbn and the possible isomeric forms of the mononuclear complex [Ru(phen)(bbn)]2+ with bbn as a tetradentate ligand: (a) cis-α isomer, (b) cis-β isomer, and (c) a form in which the central polymethylene chain spans the trans. Reproduced from [48] with permission. Copyright © 2015 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.

Figure 8.
Chemical structures of heteroleptic Ru (II) complexes bearing pyridophenazine ligands.

Figure 9.
Representative ‘piano stool’ RuII-η6–arene complex, where X, Y, and/or Z is a labile ligand.
Figure 9.
Representative ‘piano stool’ RuII-η6–arene complex, where X, Y, and/or Z is a labile ligand.

Figure 10.
General structure of deferoxamine B (DFO)-derived Trojan Horse antibacterial drugs and some DFO-derived Ruthenium(II)–Arene Complexes [104].
Figure 10.
General structure of deferoxamine B (DFO)-derived Trojan Horse antibacterial drugs and some DFO-derived Ruthenium(II)–Arene Complexes [104].

Figure 11.
Chemical structures of selected SCAR complexes and RuNN.

Figure 12.
Chemical structures of ruthenium(II) complexes with ‘‘click’’ pyridyl-1,2,3-triazole ligands with various aliphatic and aromatic substituents (generally denoted as Ru-pytri [53] and Ru-tripy [54]). Adapted with permission from [53], Copyright © 2016, American Chemical Society and [54], © 2019 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.
Figure 12.
Chemical structures of ruthenium(II) complexes with ‘‘click’’ pyridyl-1,2,3-triazole ligands with various aliphatic and aromatic substituents (generally denoted as Ru-pytri [53] and Ru-tripy [54]). Adapted with permission from [53], Copyright © 2016, American Chemical Society and [54], © 2019 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.

Figure 13.
Chemical structures of the inert dinuclear Rubbn ([Ru2(phen)2(tpphz)]4+, [Ru2(5-Mephen)2(tpphz)]4+, [Ru2(2,9-Me2phen)2(tpphz)]4+, and [Ru2(Me4phen)2(tpphz)]4+) and mononuclear ([Ru(phen)2(tpphz)]2+) complexes.
Figure 13.
Chemical structures of the inert dinuclear Rubbn ([Ru2(phen)2(tpphz)]4+, [Ru2(5-Mephen)2(tpphz)]4+, [Ru2(2,9-Me2phen)2(tpphz)]4+, and [Ru2(Me4phen)2(tpphz)]4+) and mononuclear ([Ru(phen)2(tpphz)]2+) complexes.

Figure 14.
Chemical structures of labile dinuclear Cl-Rubbn-Cl and Rubbn-Cl complexes, where n = 7, 12, 16.
Figure 14.
Chemical structures of labile dinuclear Cl-Rubbn-Cl and Rubbn-Cl complexes, where n = 7, 12, 16.

Figure 15.
Chemical structures of inert tri- and tetra- nuclear ruthenium complexes, where n = 7, 10, 12 or 16.
Figure 15.
Chemical structures of inert tri- and tetra- nuclear ruthenium complexes, where n = 7, 10, 12 or 16.

Figure 16.
Chemical structures of hetero- (a) trimetallic complexes bearing ruthenocene or ferrocene moieties and (b) bimetallic complex bearing a ferrocenyl–salicylaldimine moiety.
Figure 16.
Chemical structures of hetero- (a) trimetallic complexes bearing ruthenocene or ferrocene moieties and (b) bimetallic complex bearing a ferrocenyl–salicylaldimine moiety.

Figure 17.
Modes of action and intracellular targets of CORMs. The bacterial membrane includes the inner membrane (IM), the outer membrane (OM), and periplasm (P), which are represented at the top. 1. CORMs enter bacteria by unknown pathways and mechanisms; CO enters the cells by diffusion. 2. After they enter the cell, CORMs release CO, forming inactivated CORM (iCORM). 3. CO, CORM, and iCORM are detected by transcription factors (TFs), causing transcriptional changes. 4. TFs are activated by ROS that may be generated directly by CORMs or can be generated as a result of the interaction of CORMs with the respiratory chain. 5. A simplified aerobic respiratory chain of bacteria is represented, consisting of a flavin-containing NADH dehydrogenase, a ubiquinone (Q) pool, and a terminal heme-containing quinol oxidase. 6. CO binds to the heme-containing quinol oxidase active site, competing with oxygen and impeding respiration. 7. Impairment of ATP generation by ATP synthase. 8. CO or CORM may directly or indirectly interact with IM transporters. 9. Diverse cellular responses to CO and CORM. Question marks represent unknown targets, effects, or mechanisms: transport into (or out of) cells; intracellular mechanisms of CO release from CORMs; interaction with TFs and modification of gene expression by CORMs; effects of CORMs on membrane transporters. Figure reproduced from [146].
Figure 17.
Modes of action and intracellular targets of CORMs. The bacterial membrane includes the inner membrane (IM), the outer membrane (OM), and periplasm (P), which are represented at the top. 1. CORMs enter bacteria by unknown pathways and mechanisms; CO enters the cells by diffusion. 2. After they enter the cell, CORMs release CO, forming inactivated CORM (iCORM). 3. CO, CORM, and iCORM are detected by transcription factors (TFs), causing transcriptional changes. 4. TFs are activated by ROS that may be generated directly by CORMs or can be generated as a result of the interaction of CORMs with the respiratory chain. 5. A simplified aerobic respiratory chain of bacteria is represented, consisting of a flavin-containing NADH dehydrogenase, a ubiquinone (Q) pool, and a terminal heme-containing quinol oxidase. 6. CO binds to the heme-containing quinol oxidase active site, competing with oxygen and impeding respiration. 7. Impairment of ATP generation by ATP synthase. 8. CO or CORM may directly or indirectly interact with IM transporters. 9. Diverse cellular responses to CO and CORM. Question marks represent unknown targets, effects, or mechanisms: transport into (or out of) cells; intracellular mechanisms of CO release from CORMs; interaction with TFs and modification of gene expression by CORMs; effects of CORMs on membrane transporters. Figure reproduced from [146].

Figure 18.
Chemical structures of CORM-2 and CORM-3.

Figure 19.
Chemical structures of ruthenium-based CORM-polymers 1–3.

Figure 20.
Chemical structures of ruthenium complexes developed for Antimicrobial Photodynamic Therapy.
Figure 20.
Chemical structures of ruthenium complexes developed for Antimicrobial Photodynamic Therapy.

Figure 21.
Chemical structures of the homo- or heteroleptic polypyridyl Ru(II) complexes (1)–(17) with the general formulae [Ru(Phen)3](PF6)2, [Ru(Phen)2(Phen-X)](PF6)2, [Ru(Phen)(Phen-X)2](PF6)2, [Ru(Phen-X)3](PF6)2, [Ru(Phen-X)2Cl2], or [Ru(Phen)2Cl2]. The core structures of the complexes (1)–(17) correspond to either (a) or (b), as denoted in the top right corner of the figure. The fluorene unit was bonded to the 1,10-phenanthroline moiety ligand either directly (Fluorenyl, bottom right corner) or via a triple bond (T-Fluorenyl).
Figure 21.
Chemical structures of the homo- or heteroleptic polypyridyl Ru(II) complexes (1)–(17) with the general formulae [Ru(Phen)3](PF6)2, [Ru(Phen)2(Phen-X)](PF6)2, [Ru(Phen)(Phen-X)2](PF6)2, [Ru(Phen-X)3](PF6)2, [Ru(Phen-X)2Cl2], or [Ru(Phen)2Cl2]. The core structures of the complexes (1)–(17) correspond to either (a) or (b), as denoted in the top right corner of the figure. The fluorene unit was bonded to the 1,10-phenanthroline moiety ligand either directly (Fluorenyl, bottom right corner) or via a triple bond (T-Fluorenyl).

Figure 22.
Chemical structures of ruthenium complexes with antiplasmodial activity. (a) [RuCl2(CQ)]2, (b) cyclometallated Ru(II) complexes of 2-phenylbenzimidazoles, and (c) PTA-derived ruthenium(II) quinoline complexes.
Figure 22.
Chemical structures of ruthenium complexes with antiplasmodial activity. (a) [RuCl2(CQ)]2, (b) cyclometallated Ru(II) complexes of 2-phenylbenzimidazoles, and (c) PTA-derived ruthenium(II) quinoline complexes.

Figure 23.
Di- and tri- nuclear Ru(II)-η6-p-cymene complexes in which the ruthenium centers are bridged by (a) pyridyl aromatic ether ligands and (b) pyridyl aromatic ester ligands.
Figure 23.
Di- and tri- nuclear Ru(II)-η6-p-cymene complexes in which the ruthenium centers are bridged by (a) pyridyl aromatic ether ligands and (b) pyridyl aromatic ester ligands.

Figure 24.
Chemical structures of tamoxifen and the ruthenocenyl complexes incorporating tamoxifen-based ligands.
Figure 24.
Chemical structures of tamoxifen and the ruthenocenyl complexes incorporating tamoxifen-based ligands.

Figure 25.
Chemical structures of the ruthenium NO-donor complexes trans-[Ru(NO)(NH3)4(isn)]3+ and trans-[Ru(NO)(NH3)4(imN)]3+.
Figure 25.
Chemical structures of the ruthenium NO-donor complexes trans-[Ru(NO)(NH3)4(isn)]3+ and trans-[Ru(NO)(NH3)4(imN)]3+.

Figure 26.
Chemical structures of symmetric trinuclear ruthenium complexes bearing azanaphthalene ligands with the general formula [Ru3O(CH3COO)6(L)3]PF6.
Figure 26.
Chemical structures of symmetric trinuclear ruthenium complexes bearing azanaphthalene ligands with the general formula [Ru3O(CH3COO)6(L)3]PF6.

Figure 27.
Chemical structures of Ru(II)–arene complexes with antitrypanosomal activity.

Figure 28.
Chemical structure of [Ru(bpy)2eilatin]2+.

Figure 29.
Chemical structure of BOLD-100 (sodium trans-[tetrachlorobis(1H-indazole)ruthenate(III)], KP1339).
Figure 29.
Chemical structure of BOLD-100 (sodium trans-[tetrachlorobis(1H-indazole)ruthenate(III)], KP1339).


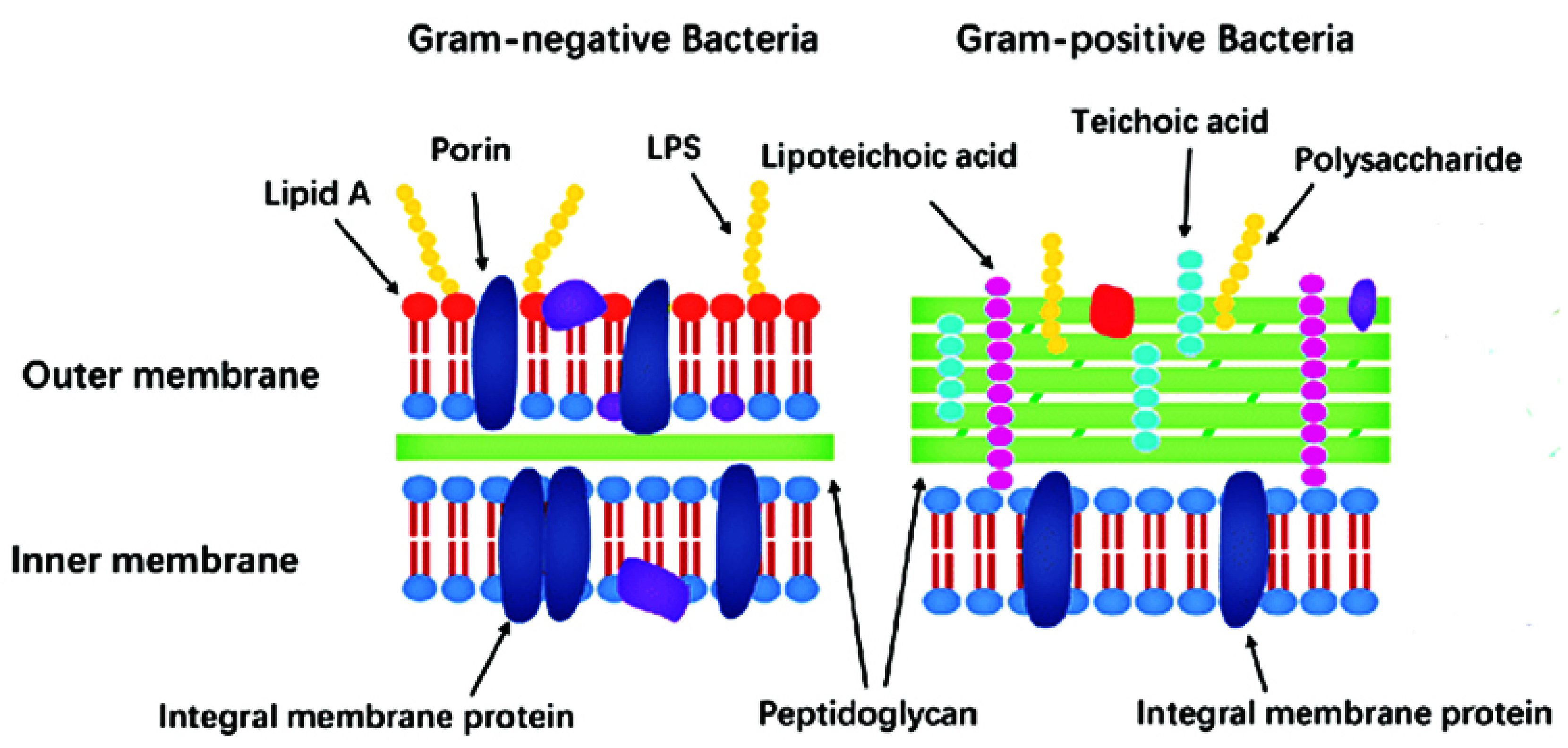{kind=link}
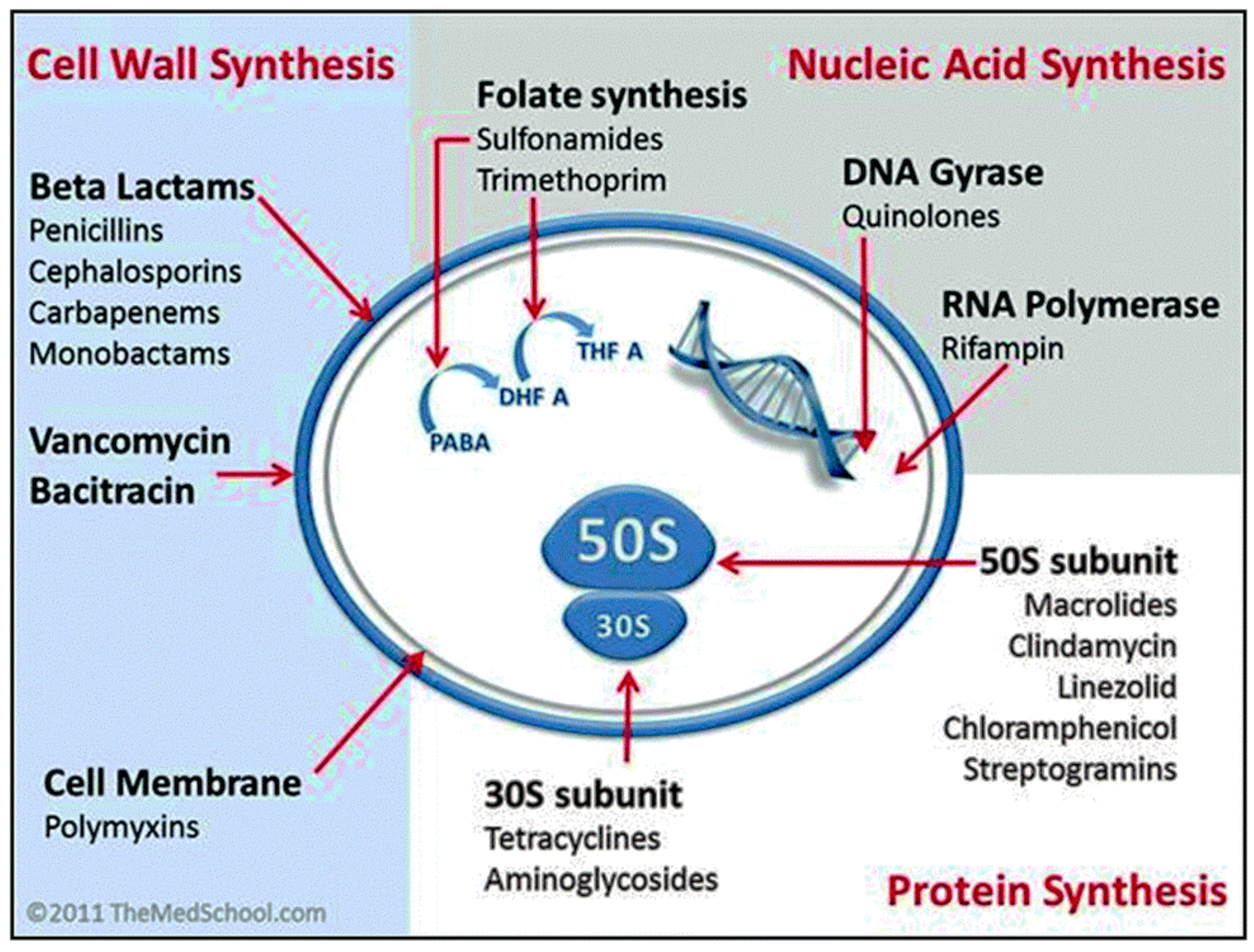{kind=link}
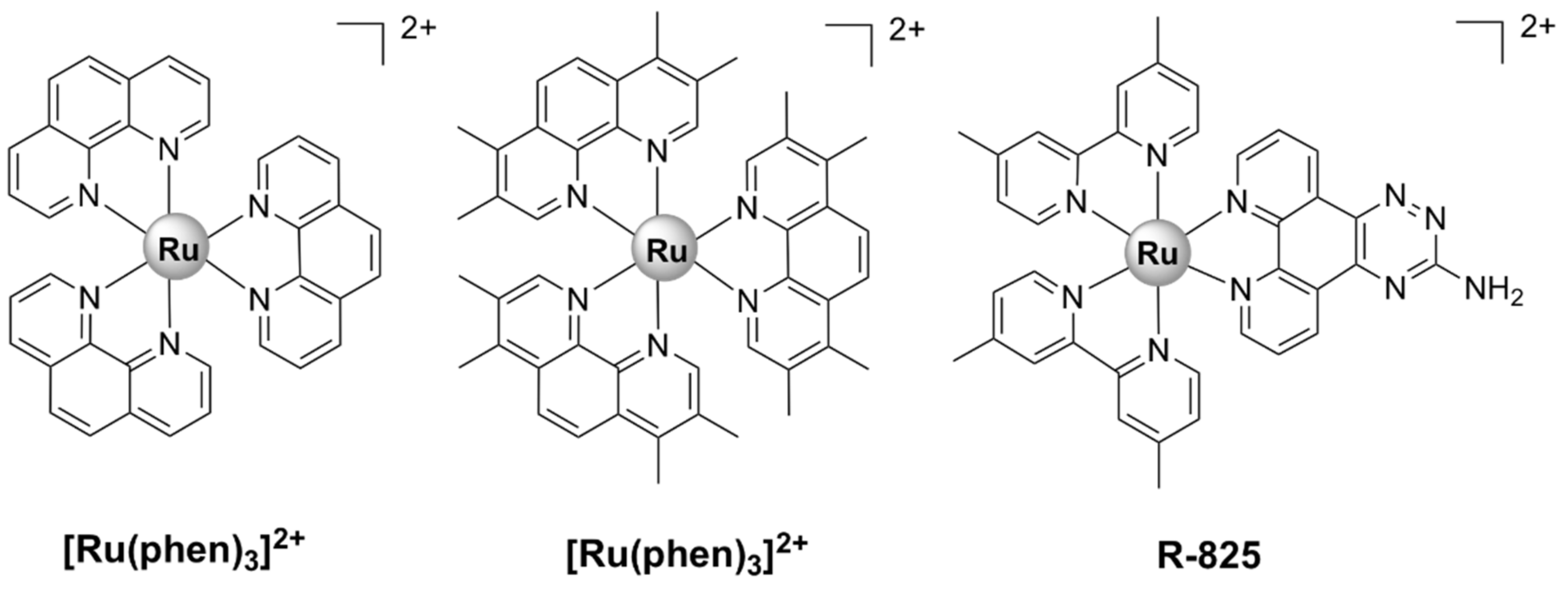{kind=link}
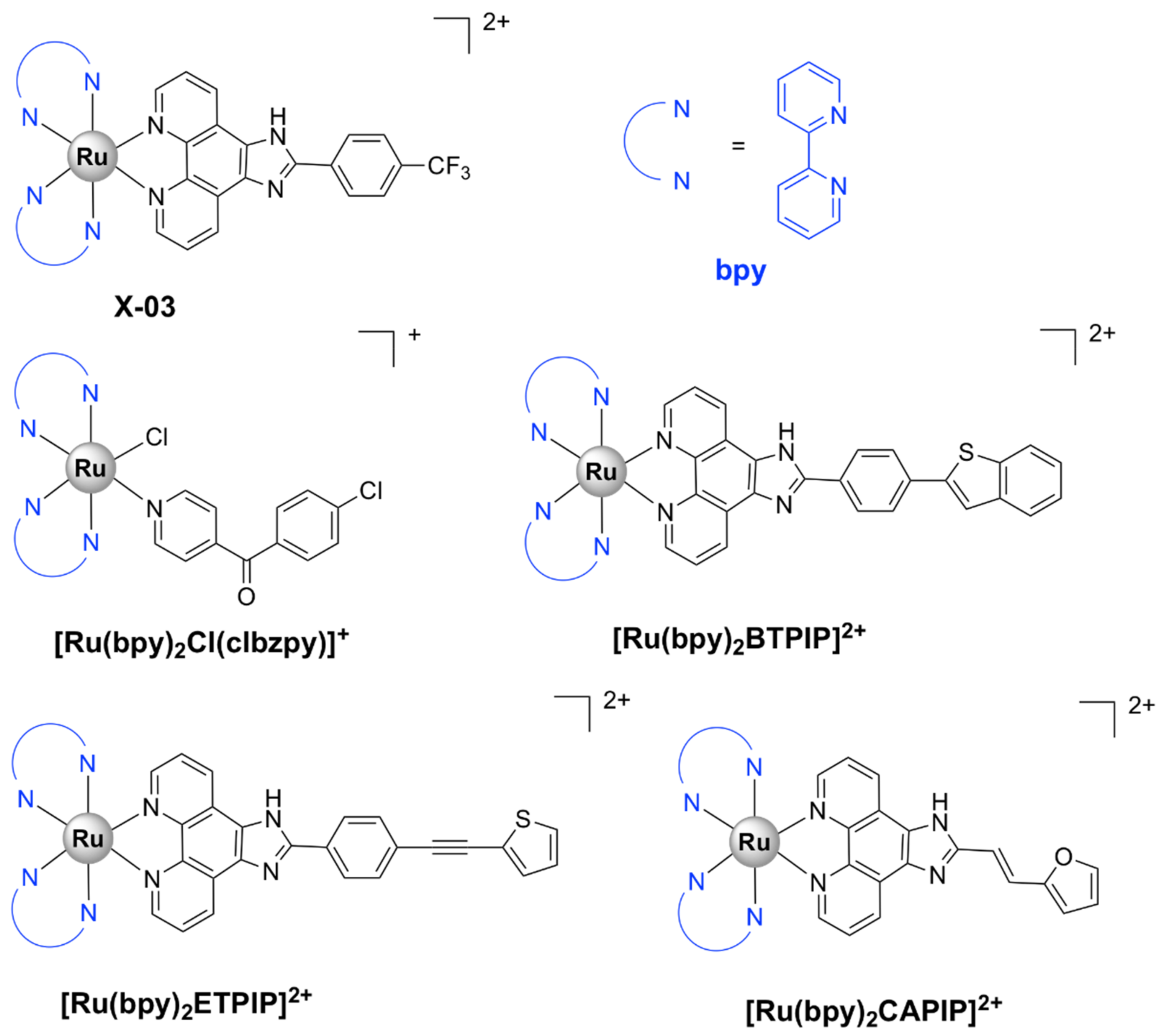{kind=link}
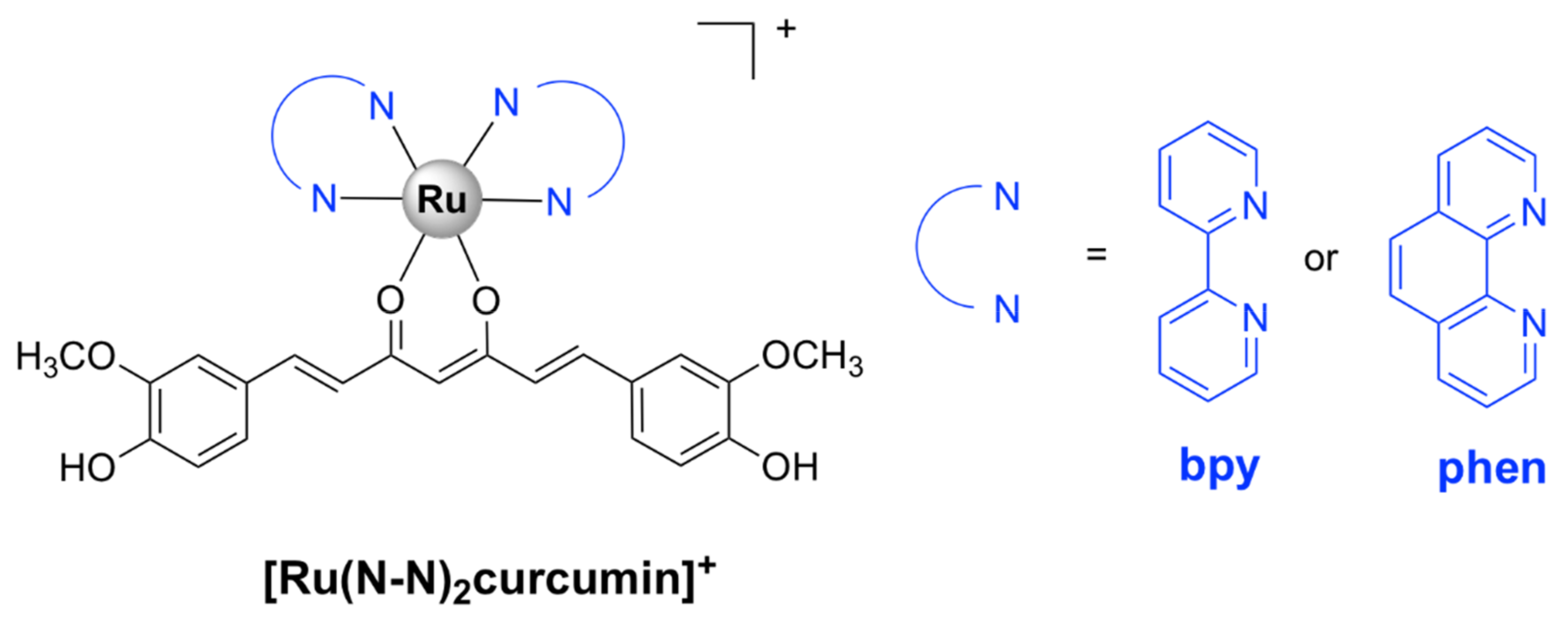{kind=link}
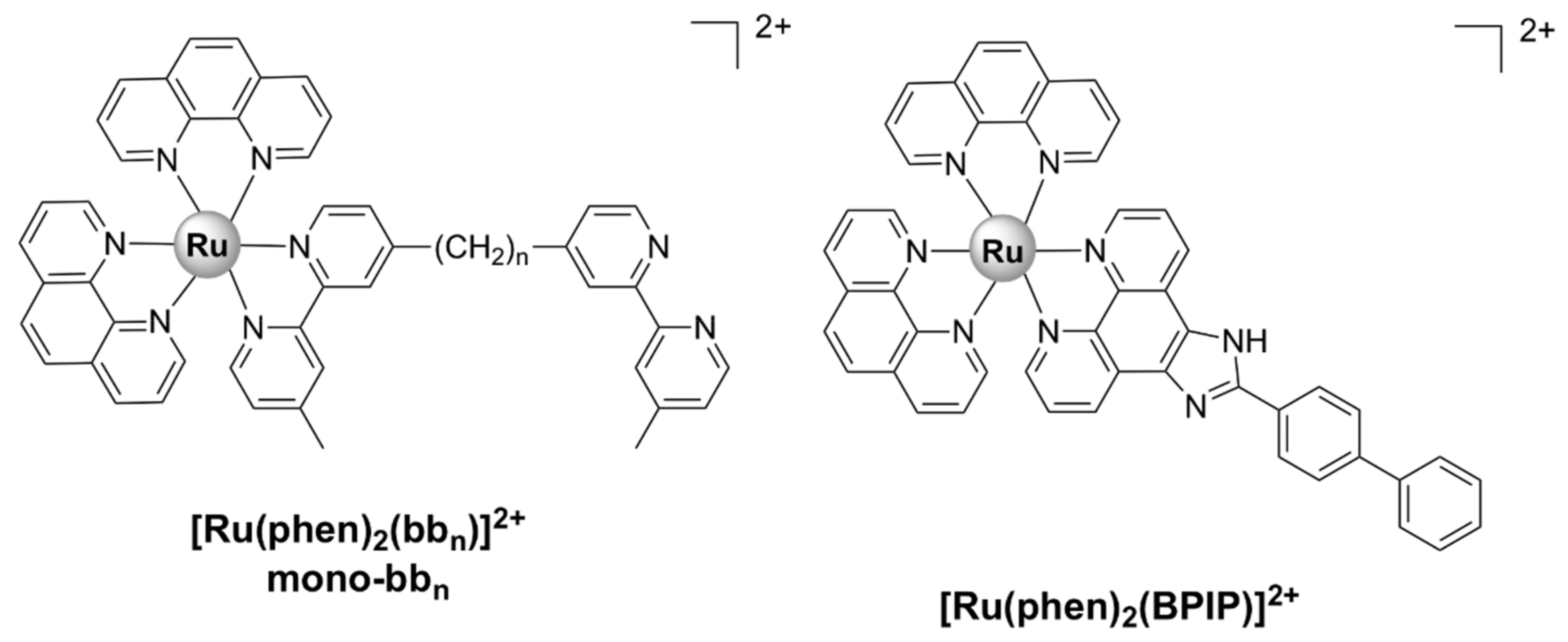{kind=link}
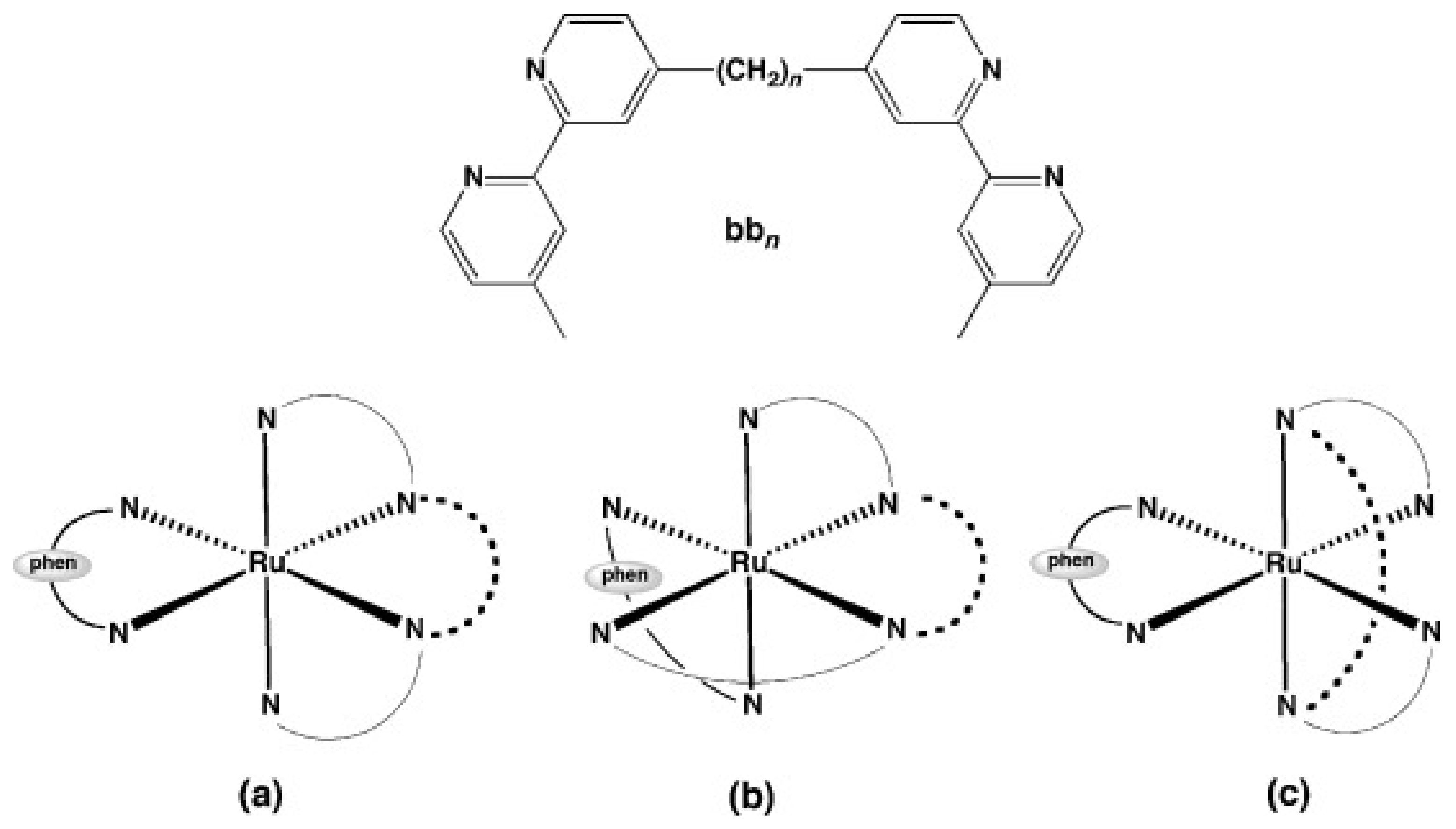{kind=link}
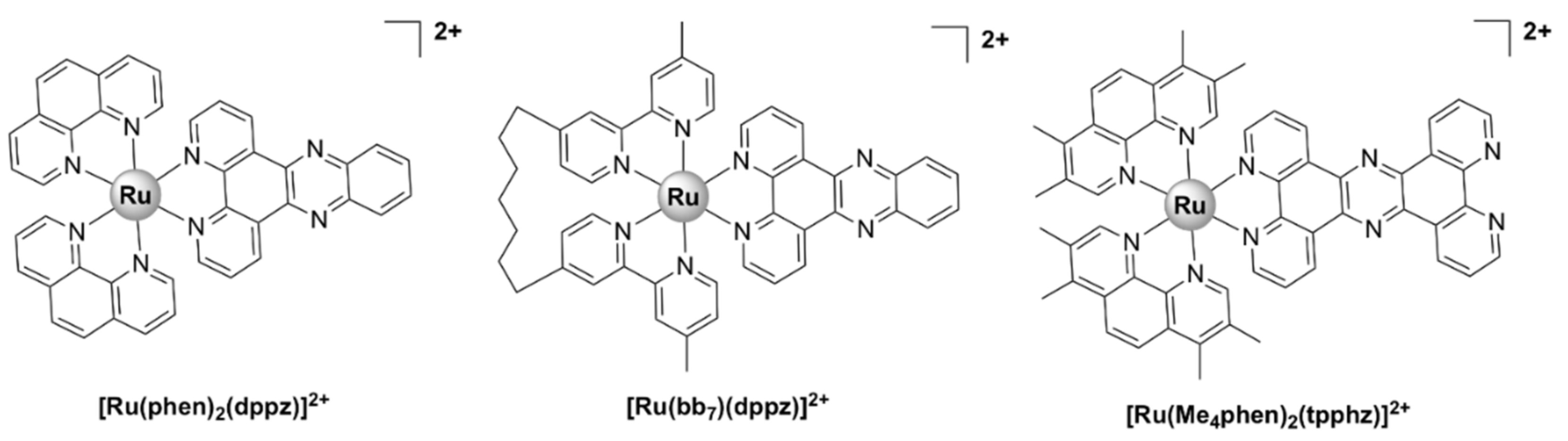{kind=link}
{kind=link}
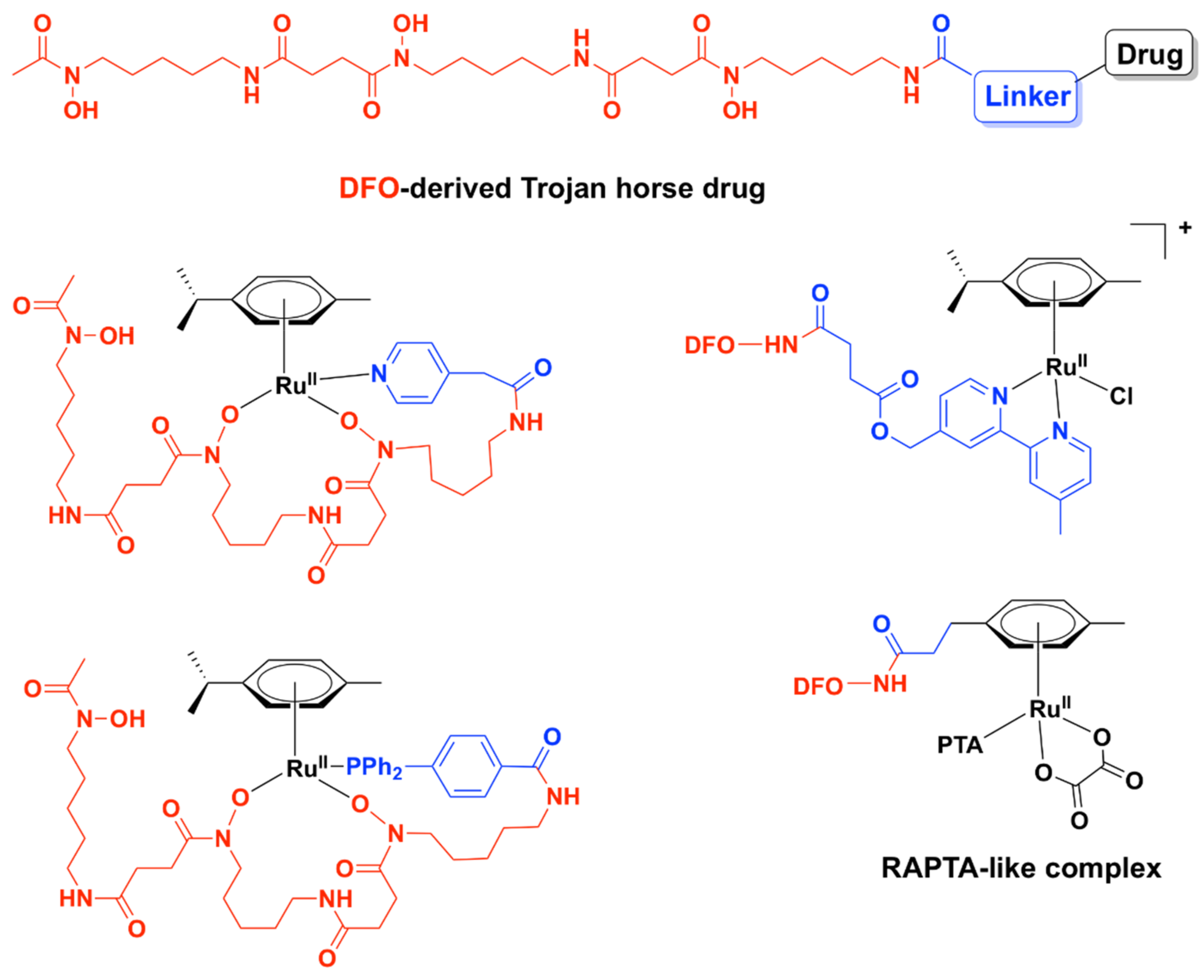{kind=link}
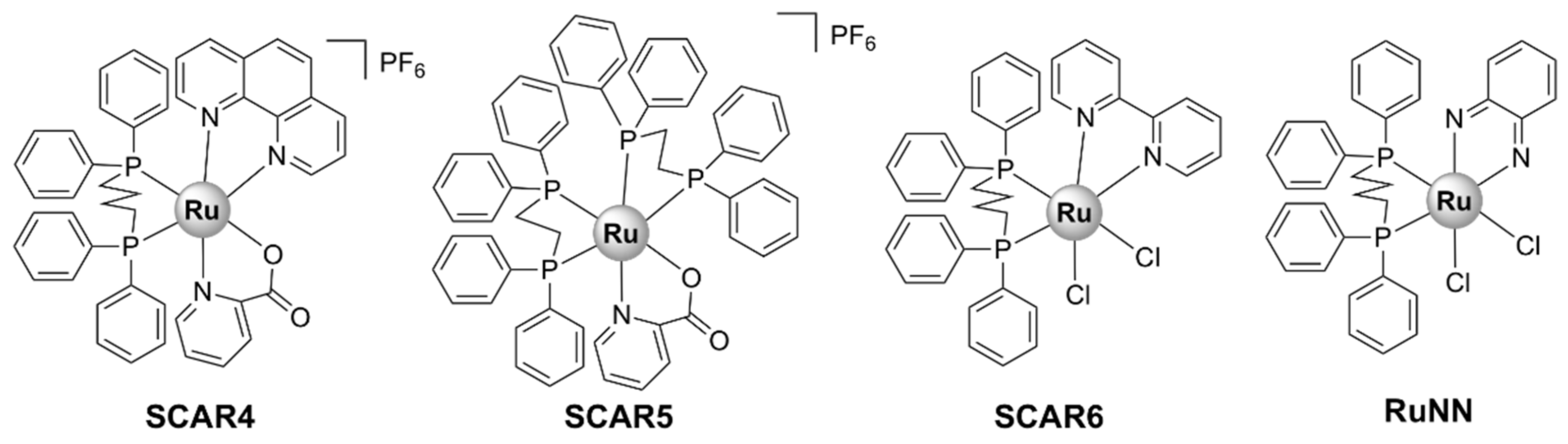{kind=link}
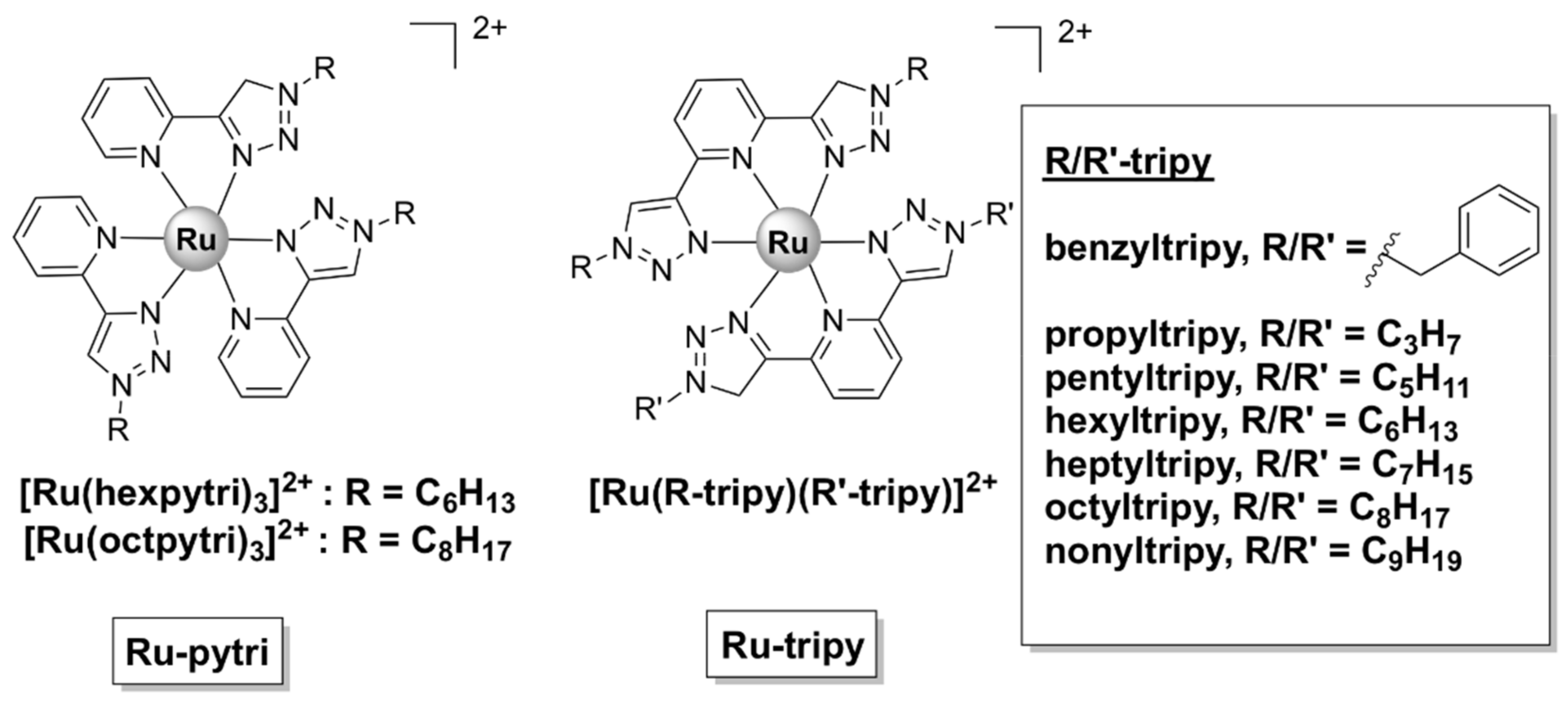{kind=link}
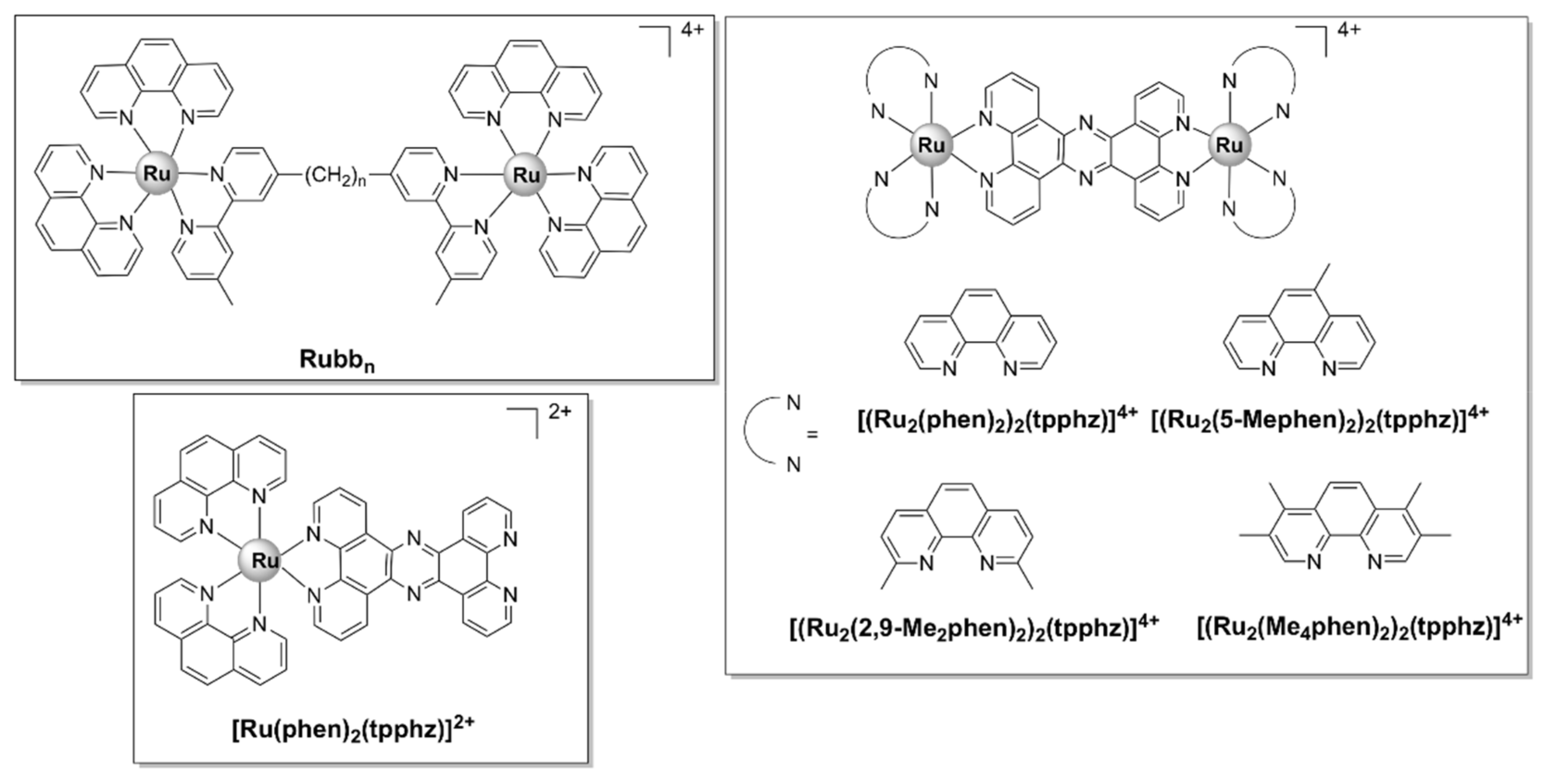{kind=link}
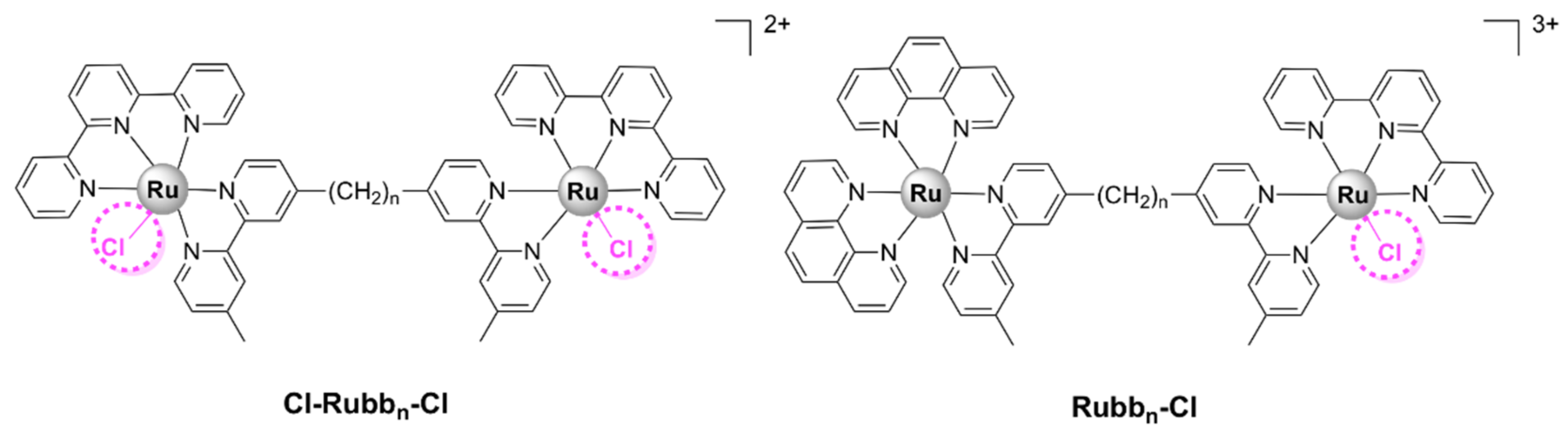{kind=link}
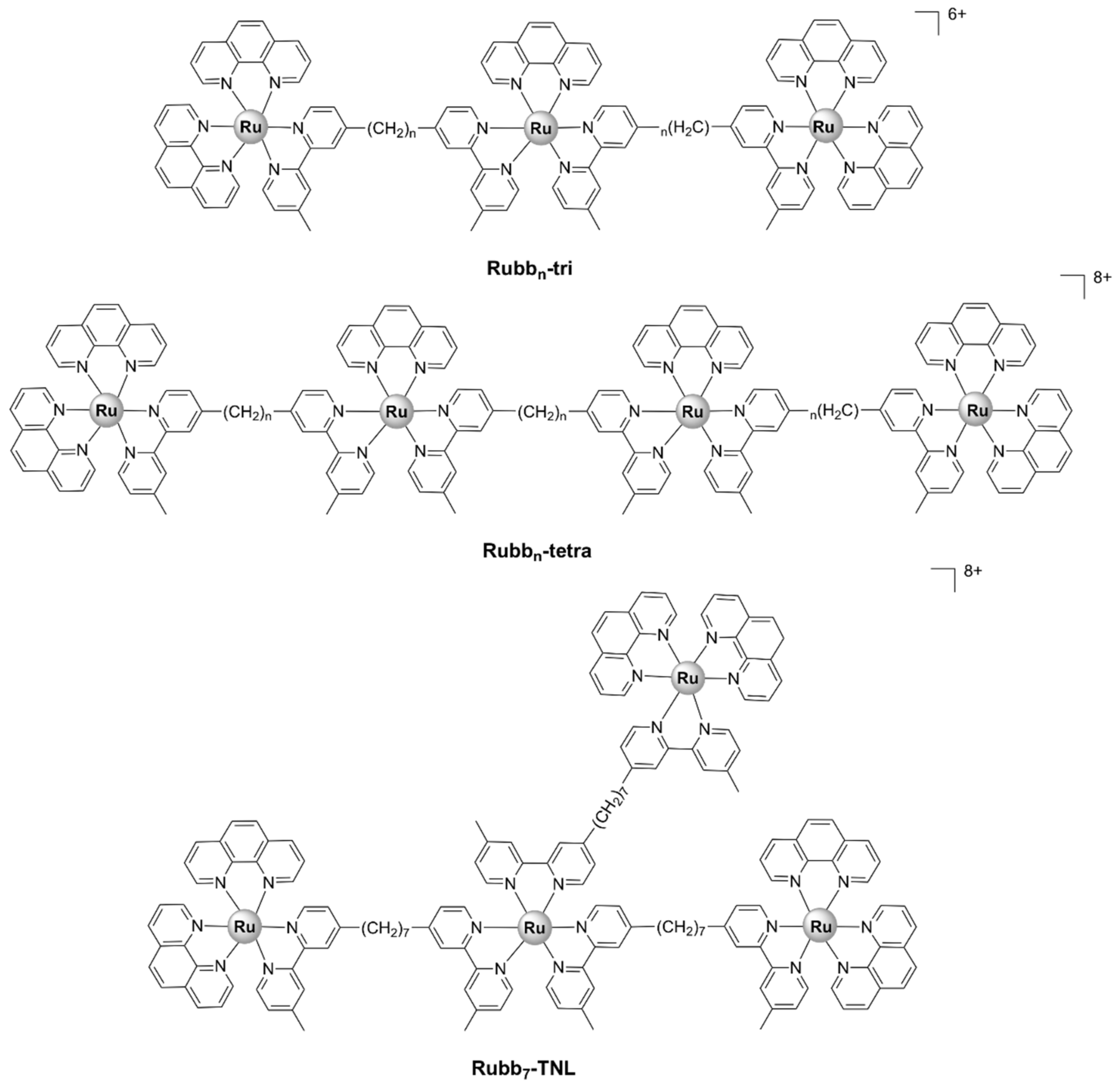{kind=link}
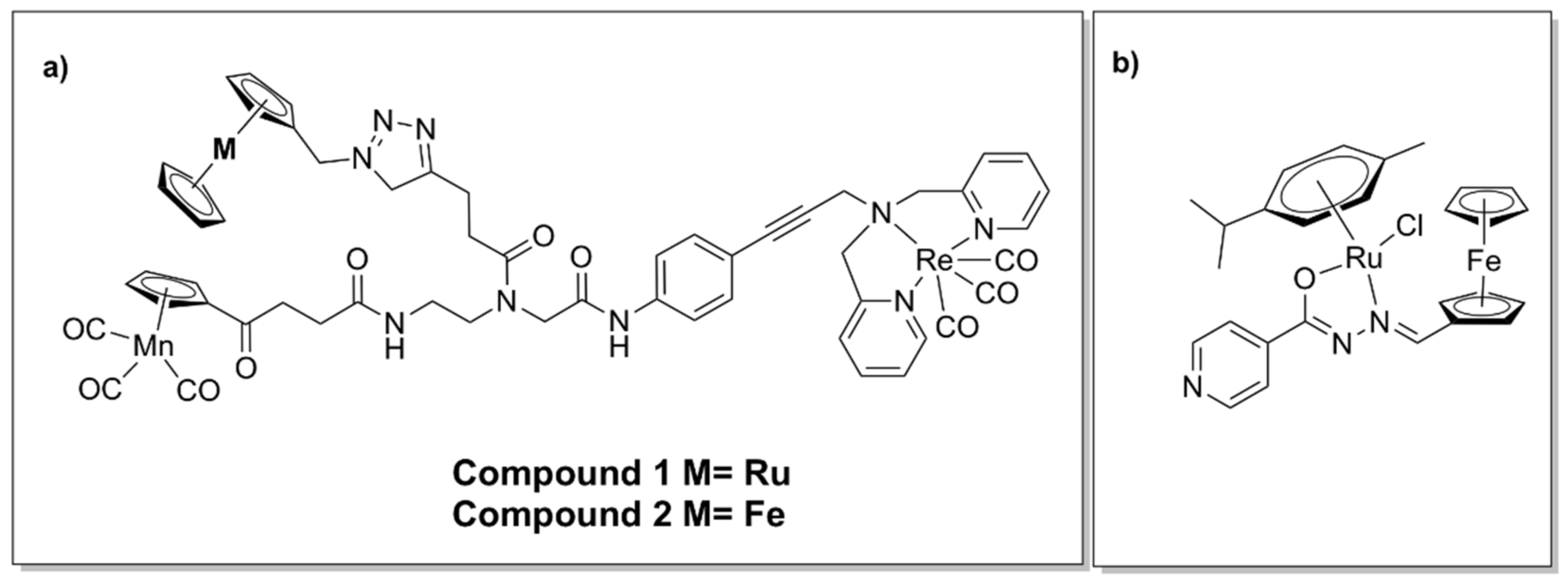{kind=link}
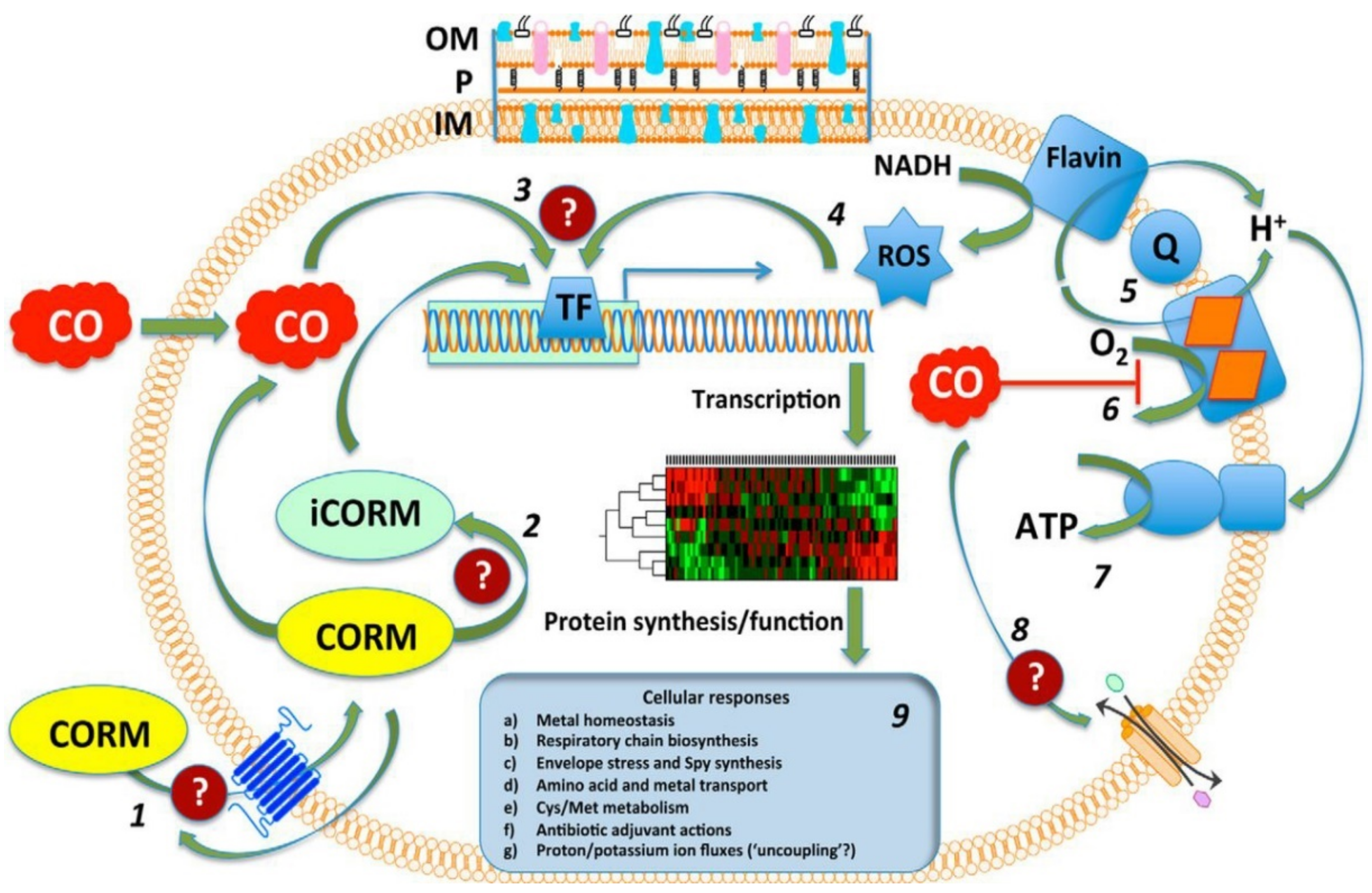{kind=link}
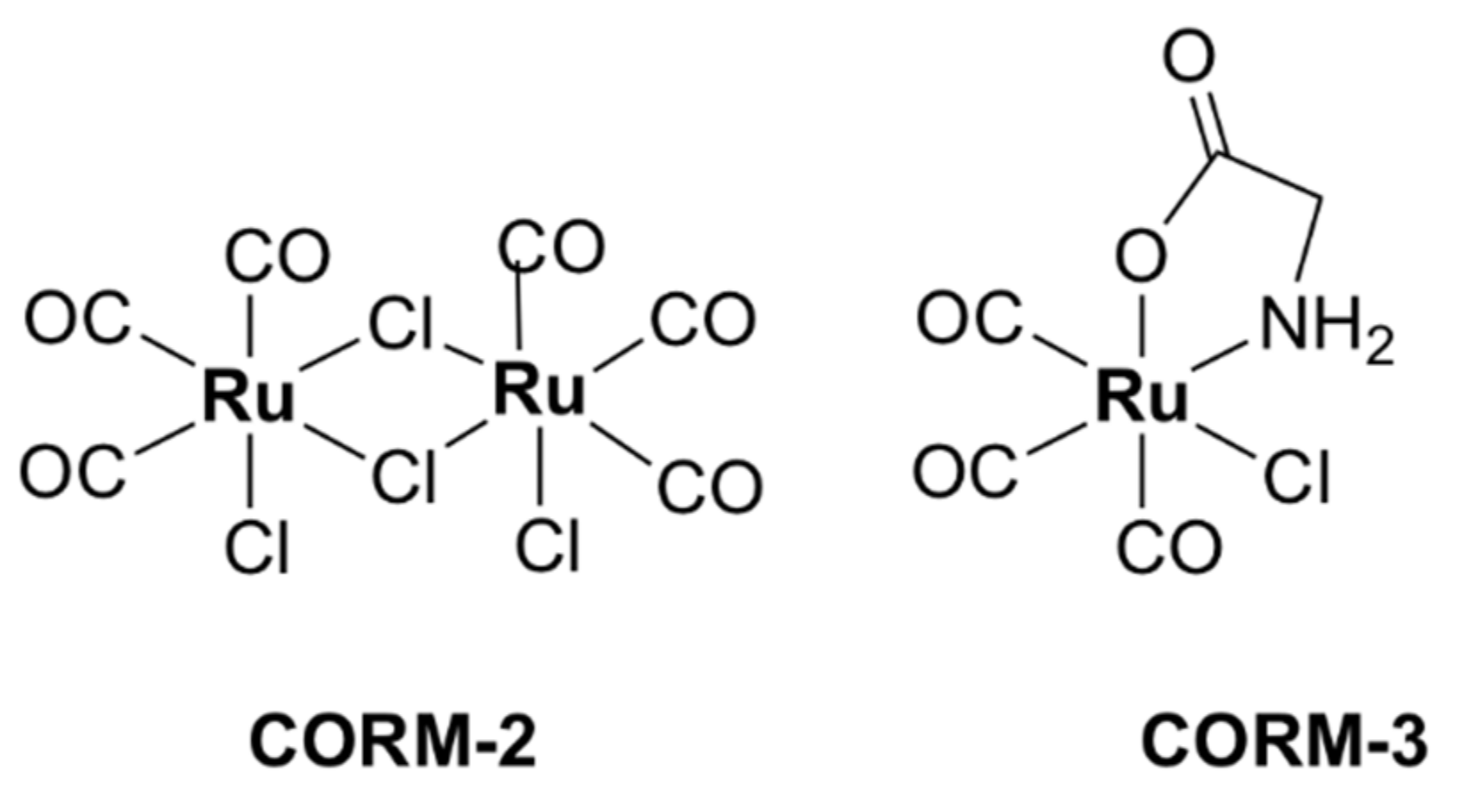{kind=link}
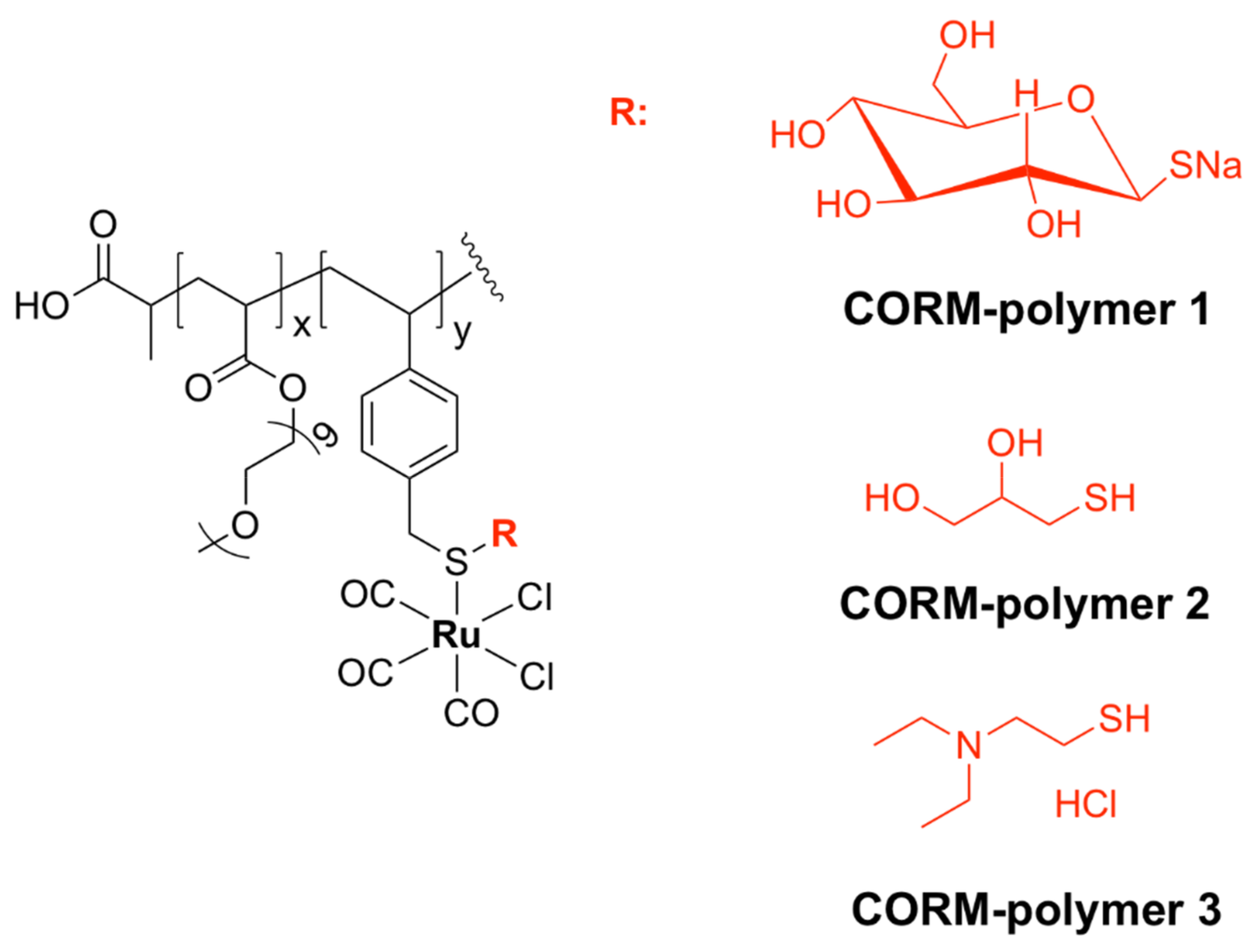{kind=link}
{kind=link}
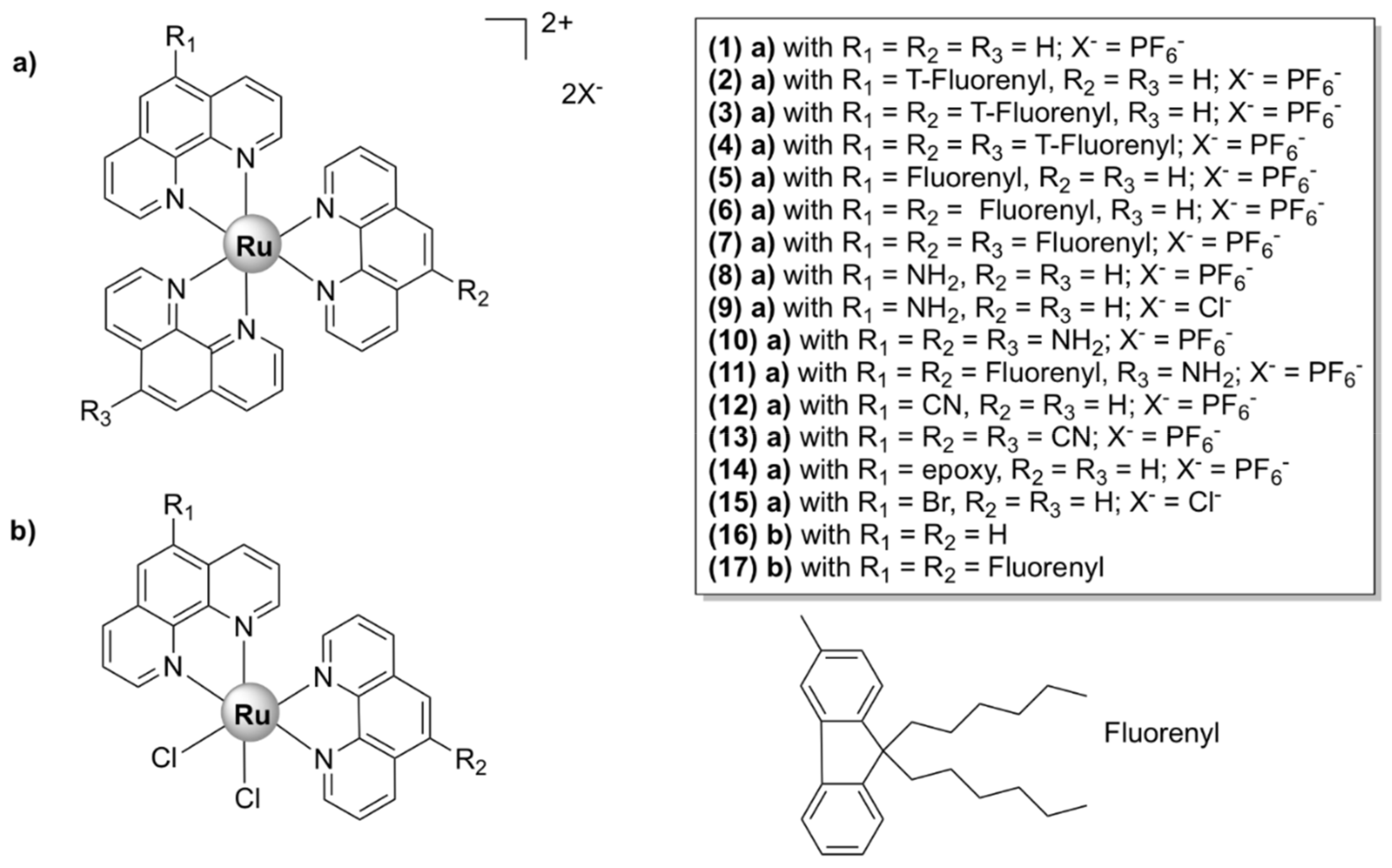{kind=link}
{kind=link}
{kind=link}
{kind=link}
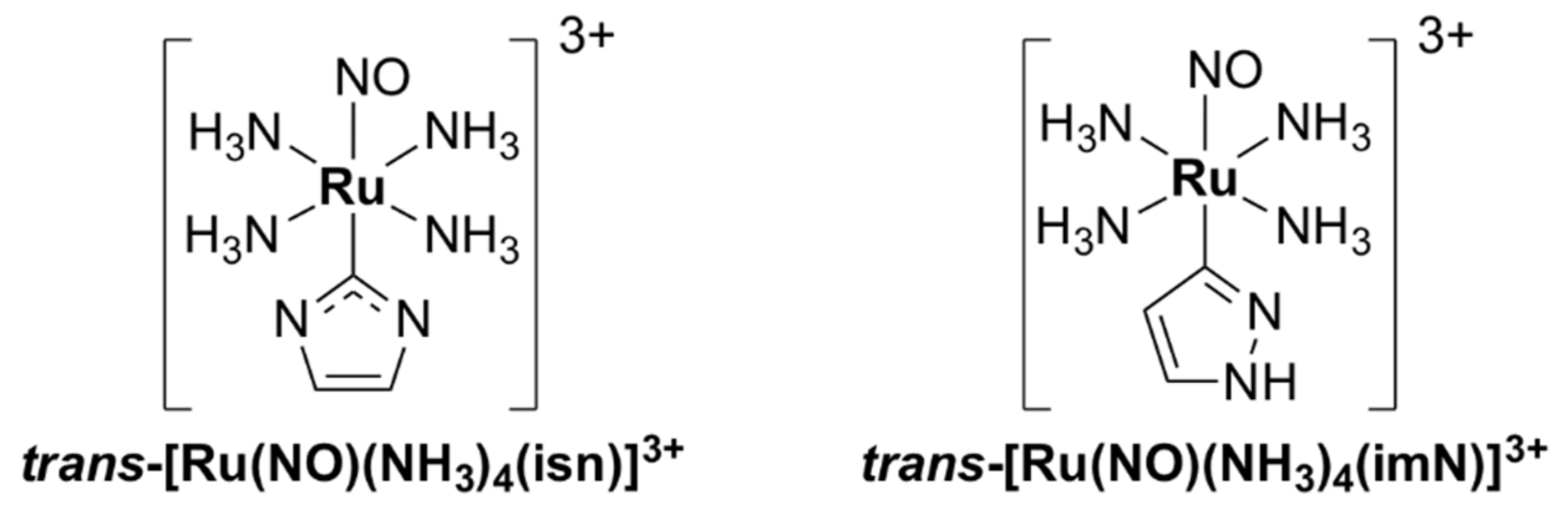{kind=link}
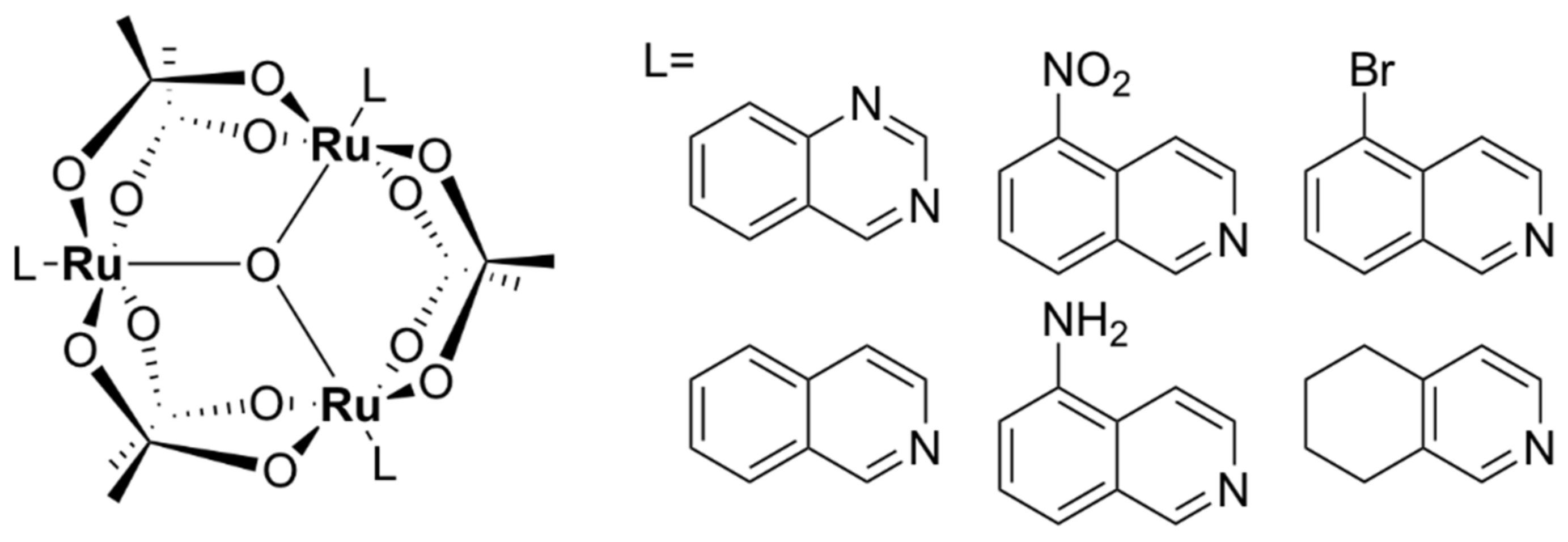{kind=link}
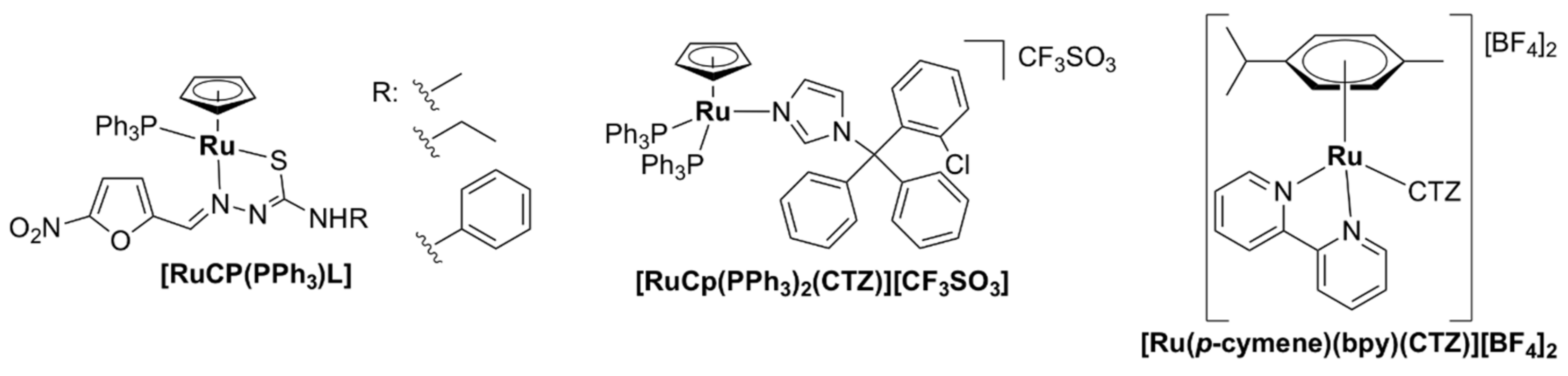{kind=link}
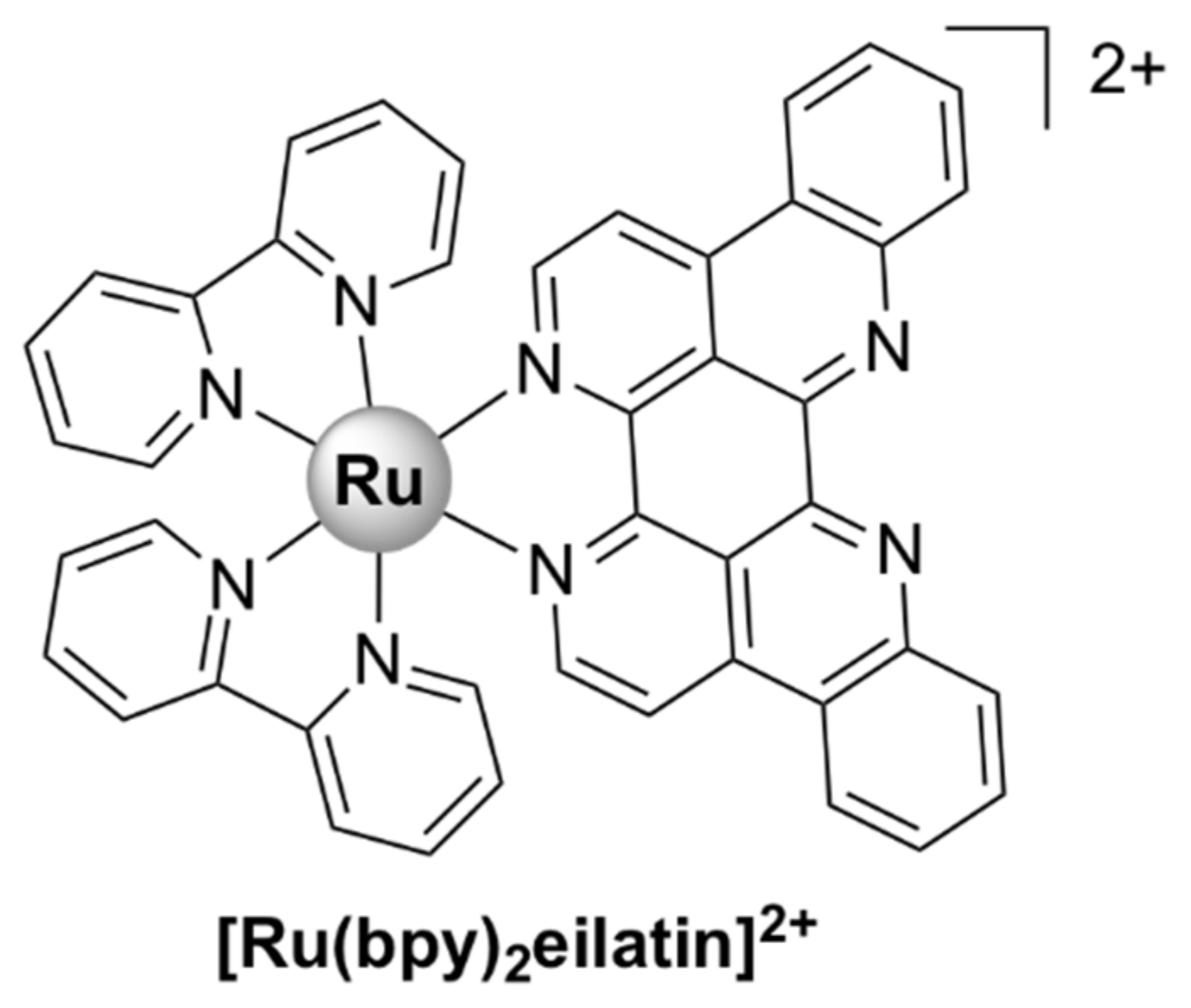{kind=link}
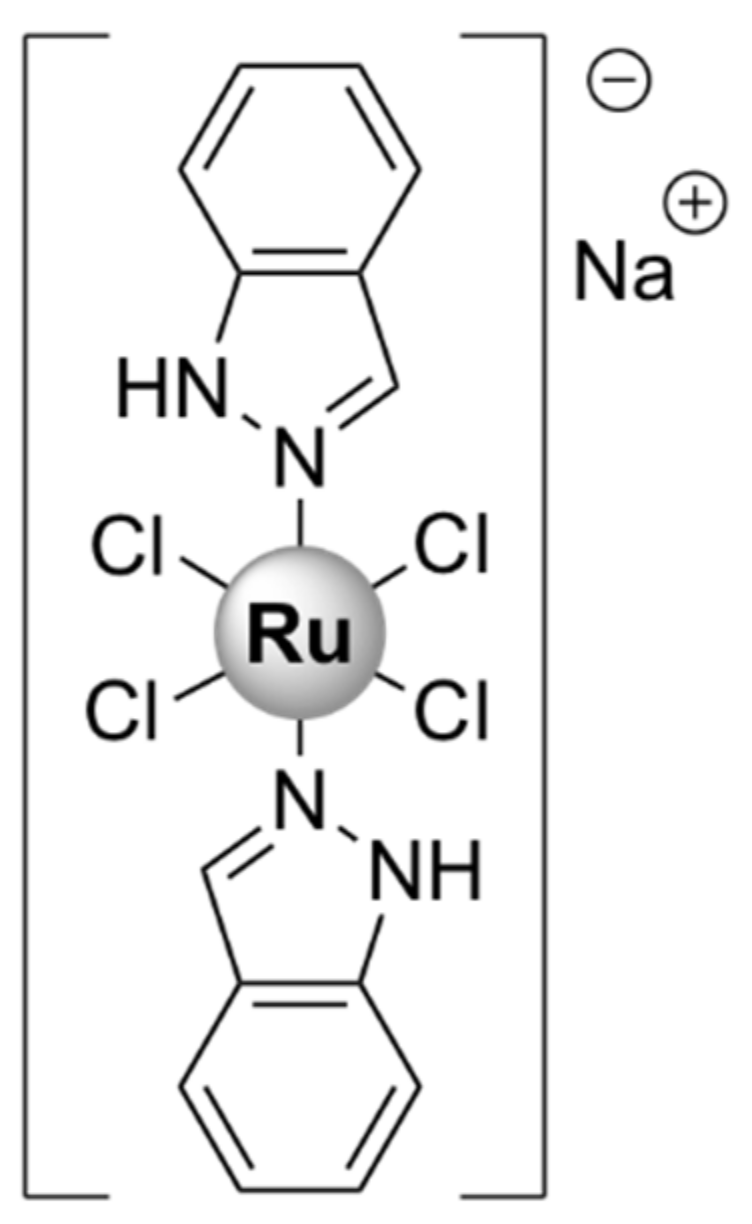{kind=link}
Table 1.
Activities of selected ruthenium complexes against bacteria, toxicity to healthy mammalian cells, and mode of action.
Table 1.
Activities of selected ruthenium complexes against bacteria, toxicity to healthy mammalian cells, and mode of action.
| Complex [Reference] | Activity Strain: MIC Values (µg/mL) | Toxicity to Healthy Mammalian Cells (IC50, µg/mL, 24 h, unless Stated Otherwise) | Modes of Action | |
|---|---|---|---|---|
| Gram-Positive Strains | Gram-Negative Strains | |||
| Polypyridylruthenium (II) complexes | ||||
| [Ru(2,9-Me2phen)2(dppz)]2+ [40] | S. aureus MRSA252: 2, MRSA41: 4, MSSA160: 8, B. subtilis 168: 4 | Not active on E. coli MC4100 | - | bactericidal; DNA intercalation |
| R-825 [41] | S. pneumoniae D39 WT: 27.5 piuA mutant: 55 | - | Not toxic to human alveolar epithelial A549 cells up to 480 µM | interference with iron acquisition systems in S. pneumoniae cells |
| X-03 [42] | S. pneumoniae D39: 25, Streptococcus suis 05ZYH33: 100, S. pyogenes MGAS5005: 25, Listeria monocytogenes 19,117: 25, S. aureus 29,213: 50 | E. coli K12: > 200, Vibrio alginolyticus V12G01: > 200, Vibrio parahaemolyticus RIMD 2,210,633: > 200, A. baumanii 19,606: > 200 | Not toxic to human alveolar A549 and bronchial HBE epithelial cells up to 100 µg/mL | interference with iron acquisition systems in S. pneumoniae cells; oxidative stress, membrane damage |
| [Ru(bpy)2Cl(clbzpy)]+ [43] | S. aureus ATCC 25,923: 500, S. epidermidis ATCC 12,228: 250 | P. aeruginosa ATCC 10,145: not active | - | membrane damage |
| [Ru(bpy)2(methionine)]2+ [44] | upon blue LED irradiation S. aureus ATCC 25,923: 62.5, S. epidermidis ATCC 12,228: 125 | P. aeruginosa ATCC 10,145: not active E. coli ATCC 11,303: 500 | - | DNA photodamage |
| [Ru(dmb)2(ETPIP)]2+ [45] | S. aureus Newman: 50 | - | - | - |
| [Ru(phen)2(ETPIP)]2+ [45] | S. aureus Newman: 25 | - | - | inhibits biofilm formation; interacts with intracellular thiols |
| [Ru(bpy)2(BTPIP)]2+ [46] | S. aureus Newman: 16 | - | - | inhibits biofilm formation |
| [Ru(bpy)2curcumin]+ [47] | S. aureus ATCC 29,213: 1 | A. baumanii BAA-1605: > 64, E. coli ATCC 25,922: > 64, K. pneumoniae BAA-1705: > 64, P. aeruginosa ATCC 27,853: > 64 | Vero (African green monkey kidney epithelial) cells: > 80 | bactericidal; inhibits biofilm formation |
| [Ru(phen)2curcumin]+ [47] | S. aureus ATCC 29,213: 1 | A. baumanii BAA-1605: 8–16, E. coli ATCC 25,922: > 64, K. pneumoniae BAA-1705: > 64, P. aeruginosa ATCC 27,853: > 64 | Vero (African green monkey kidney epithelial) cells: > 80 | - |
| Mono-bb7 [38] | S. aureus MSSA ATCC 25,923: 4 MRSA (JCU culture collection): 16 | E. coli ATCC 25,922: 16 P. aeruginosa ATCC 27,853: > 128 | - | bactericidal; membrane damage |
| Mono-bb10 [37,38] | S. aureus MSSA ATCC 25,923: 4 MRSA (JCU culture collection): 16 | E. coli ATCC 25,922: 16 P. aeruginosa ATCC 27,853: 32 | - | bactericidal |
| Mono-bb16 [37] | S. aureus MSSA ATCC 25,923: 16 MRSA (JCU culture collection): 16 | E. coli ATCC 25,922: 64 P. aeruginosa ATCC 27,853: 64 | - | - |
| cis-α-[Ru(phen)bb12]2+ [48] | S. aureus MSSA ATCC 25,923: 0.5 MRSA (JCU culture collection): 4 | E. coli ATCC 25,922: 8 P. aeruginosa ATCC 27,853: 8 | - | DNA binding |
| cis-β-[Ru(phen)(bb12)]2+ [48] | S. aureus MSSA ATCC 25,923: 0.5 MRSA (JCU culture collection): 4 | E. coli ATCC 25,922: 16 P. aeruginosa ATCC 27,853: 32 | - | DNA binding |
| [Ru(bb7)(dppz)]2+ [49] | S. aureus SH 1000: 2 MRSA USA 300 LAC JE2: 2 | E. coli avian pathogenic: 8 uropathogenic: 8 E. coli MG1655: 8 P. aeruginosa PAO1: 16 | human embryonic kidney HEK-293 cells: 27 (48 h), human fetal hepatocyte L02 cells: 64 (48 h) | bactericidal, DNA binding |
| [Ru(Me4phen)2(dppz)]2+ [50] | S. aureus SH1000: 9.7, E. faecalis V583: 38.8 | E. coli MG1655: 4.9, EC958: 4.9, P. aeruginosa PA2017: 9.7 A. baumannii AB184: 9.7 | - | bactericidal, chromosomal DNA binding |
| SCAR4 [51] | M. tuberculosis H37Rv ATCC 27,294 (neither G+, nor G-): 0.63 | - | Mouse monocyte macrophage J774A.1 cell line: 19.5 | covalent binding to DNA |
| SCAR5 [51] | M. tuberculosis H37Rv ATCC 27,294 (neither G+, nor G-): 0.26 | - | J774A.1: 3.9 | covalent binding to DNA |
| SCAR6 [51] | M. tuberculosis H37Rv ATCC 27,294 (neither G+, nor G-): 3.90 | - | J774A.1: 78.2 | covalent binding to DNA |
| RuNN [52] | S. aureus ATCC 25,923: 15.6, S. aureus ATCC 700,698 (MRSA): 62.5 S. epidermidis ATCC 12,228: 31.2, S. epidermidis ATCC 358,983: 62.5 | - | no cytotoxic effect against human erythrocytes | bactericidal; inhibits biofilm formation |
| [Ru(hexpytri)3](PF6)2 [53] | S. aureus MSSA ATCC 25,923: 8, S. aureus MSSA NZRM 9653: 1, S. aureus MRSA MR 9519: 4, S. pyogenes: 4 | E. coli ATCC 25,922: non-active | Vero cells: IC50 > 128 (48h) | cell wall/cytoplasmic membrane damage |
| [Ru(hexyltripy) (heptyltripy)]Cl2 [54] | S. aureus ATCC 25,923: 2 | E. coli ATCC 25,922: 8 | HDFa (skin cells): 16.4 | abnormal cellular division |
| ΔΔ-Rubb7 [37,38] | S. aureus MSSA ATCC 25,923: 16 MRSA (JCU culture collection): 16 | E. coli ATCC 25,922: 16 P. aeruginosa ATCC 27853: 128 | Red blood cells: > 1024 | bactericidal; membrane damage, interaction with ribosomal RNA |
| ΔΔ-Rubb12 [55,56] | S. aureus MSSA ATCC 25,923: 1 MRSA (JCU culture collection): 1 | E. coli ATCC 25,922: 2 P. aeruginosa ATCC 27,853: 16 | Baby hamster kidney (BHK): 113.9, HEK-293: 82.2 | bactericidal; membrane damage, interaction with ribosomal RNA |
| ΔΔ-Rubb16 [56] | S. aureus MSSA ATCC 25,923: 1 MRSA (JCU culture collection): 1 | E. coli ATCC 25,922: 4 P. aeruginosa ATCC 27,853: 8 | Red blood cells: 22, BHK: 49.8, HEK-293: 35.1 | bactericidal; membrane damage, interaction with ribosomal RNA |
| [Ru2(Me4phen)2(tpphz)]4+ [57,58,59] | S. aureus MSSA SH1000: 86, Enterococcus faecalis V583: 1 | E. coli WT G1655: 2.5, EC958 ST131 (multi-drug-resistant, clinical isolate): 3.5, P. aeruginosa (clinical isolate): 4, K. pneumoniae (clinical isolate): 3.5, A. baumannii (clinical isolate): 3.5 | HEK-293: 270 | membrane and DNA damage |
| Cl-Rubb7-Cl [55,60] | S. aureus MSSA ATCC 25,923: 8 MRSA (JCU culture collection): 8 | E. coli ATCC 25,922: 8 P. aeruginosa ATCC 27,853: 32 | - | bactericidal |
| Cl-Rubb12-Cl [55,60] | S. aureus MSSA ATCC 25,923: 1 MRSA (JCU culture collection): 1 | E. coli ATCC 25,922: 2 P. aeruginosa ATCC 27,853: 8 | - | bactericidal |
| Cl-Rubb16-Cl [55,60] | S. aureus MSSA ATCC 25,923: 8 MRSA (JCU culture collection): 8 | E. coli ATCC 25,922: 8 P. aeruginosa ATCC 27,853: > 128 | - | bactericidal |
| Rubb7-Cl [56] | S. aureus MSSA ATCC 25,923: 8 MRSA (JCU culture collection): 16 | E. coli ATCC 25,922: 1 P. aeruginosa ATCC 27,853: 16 | BHK: 337.5, HEK-293: 98 | interaction with chromosomal DNA and ribosomal RNA |
| Rubb12-Cl [56] | S. aureus MSSA ATCC 25,923: 1 MRSA (JCU culture collection): 1 | E. coli ATCC 25,922: 1 P. aeruginosa ATCC 27,853: 16 | BHK: 70.6, HEK-293: 87.3 | interaction with chromosomal DNA and ribosomal RNA |
| Rubb16-Cl [56] | S. aureus MSSA ATCC 25,923: 1 MRSA (JCU culture collection): 2 | E. coli ATCC 25,922: 4 P. aeruginosa ATCC 27,853: 64 | BHK: 34.9, HEK-293: 63.5 | interaction with chromosomal DNA and ribosomal RNA |
| Rubb7-tri [37,61] | S. aureus MSSA ATCC 25,923: 4 MRSA (JCU culture collection): 4 | E. coli ATCC 25,922: 16 P. aeruginosa ATCC 27,853: 2 | - | interaction with DNA |
| Rubb7-tetra (Rubb7-TL) [62] | S. aureus MSSA ATCC 25,923: 8 MRSA (JCU culture collection): 16 | E. coli avian pathogenic: 16 uropathogenic: 16 E. coli MG1655: 16 P. aeruginosa PAO1: 32 | BHK: 176 (24 h) BHK: 36.4 (72 h) | interaction with proteins |
| Rubb7-TNL [62] | S. aureus MSSA ATCC 25,923: 4 MRSA (JCU culture collection): 8 | E. coli avian pathogenic: 16 uropathogenic: 16 E. coli MG1655: 8 P. aeruginosa PAO1: 16 | BHK: 276 (24 h) BHK: 81.6 (72 h) | interaction with proteins |
| Rubb12-tri [37,55,61] | S. aureus: 1 MRSA (JCU culture collection): 1 | E. coli: 4 P. aeruginosa: 32 | BHK: 50.9 (72 h), HEK-293: 21.8 (72 h) | bactericidal, interaction with DNA |
| Rubb12-tetra [37,55,61] | S. aureus: 2 MRSA (JCU culture collection): 2 | E. coli: 2 P. aeruginosa: 16 | BHK: 43.7 (72 h), HEK-293: 21.3 (72 h) | bactericidal, interaction with DNA |
| Rubb16-tri [37,55,61] | S. aureus: 2 MRSA (JCU culture collection): 2 | E. coli: 8 P. aeruginosa: 32 | BHK: 25.1 (72 h), HEK-293: 20.2 (72 h) | bactericidal, interaction with DNA |
| Rubb16-tetra [37,55,61] | S. aureus: 2 MRSA (JCU culture collection): 2 | E. coli: 8 P. aeruginosa: 32 | BHK: 19.8 (72 h), HEK-293: 15.8 (72 h) | bactericidal, interaction with DNA |
| Ruthenium-based CORMs | ||||
| CORM-2 [63,64,65] | Growth inhibitory effects on S. aureus (MIC value not reported) | E. coli avian pathogenic: 250, uropathogenic: 250, E. coli MG1655: 250, P. aeruginosa PAO1: 3.8 H. pylori strains (including antibiotic resistant): 100–200 | Murine RAW264.7 monocyte macrophages: > 50 (DMEM culture medium) | Bactericidal, inhibition of aerobic respiration, inhibition of biofilm formation and disruption of mature biofilms, ROS generation, interaction with chromosomal DNA and intracellular proteins, interference with iron homeostasis |
| CORM-3 [64,66,67] | Growth inhibitory effects on S. aureus, Lactobacillus lactis (MIC value not reported) | E. coli MG1655: 4 (minimal GDMM medium) and > 512 (in rich MH-II medium) H. pylori 26,695: 420 (antibiotic resistant strains) | L929 murine fibroblast cells: 63 (RPMI culture medium), RAW264.7: > 30 (DMEM culture medium) | |
| Ruthenium complexes in Antimicrobial Photodynamic Therapy | ||||
| [Ru(dmob)3]2+ [68] | S. aureus NCTC 10788: 12.5 | P. aeruginosa NCTC 8626: 50 | - | Light activation |
| cis-[Ru(bpy)2(INH)2]2+ [69] | Mycobacterium smegmatis: 4 | human lung fibroblast MRC-5 cell line: > 200 | 465 nm blue light activation | |
| [Ru(Ph2phen)2(dpp) PtCl2]2+ [70] | - | E. coli JM109: 8 | - | visible light activation, binding to chromosomal DNA |
| [Ru(CO)2Cl2]n [71] | S. aureus CETC 240, coincident with ATCC 6538 P: 0.033 | E. coli CET 516, coincident with ATCC 8739: 0.0066 | human dermal fibroblasts hDF: > 3.33 | 365 nm UV light activation, ROS generation, biofilm inhibition |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Munteanu, A.-C.; Uivarosi, V. Ruthenium Complexes in the Fight against Pathogenic Microorganisms. An Extensive Review. Pharmaceutics 2021, 13, 874. https://doi.org/10.3390/pharmaceutics13060874
AMA Style
Munteanu A-C, Uivarosi V. Ruthenium Complexes in the Fight against Pathogenic Microorganisms. An Extensive Review. Pharmaceutics. 2021; 13(6):874. https://doi.org/10.3390/pharmaceutics13060874
Chicago/Turabian StyleMunteanu, Alexandra-Cristina, and Valentina Uivarosi. 2021. "Ruthenium Complexes in the Fight against Pathogenic Microorganisms. An Extensive Review" Pharmaceutics 13, no. 6: 874. https://doi.org/10.3390/pharmaceutics13060874
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.