
Molecular Screening of Microorganisms Associated with Discolored Wood in Dead European Beech Trees Suffered from Extreme Drought Event Using Next Generation Sequencing

 , ,
, ,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Site and Sampling Methodology
2.2. DNA Extraction and Molecular Detection of Microbial Communities
2.3. Statistics
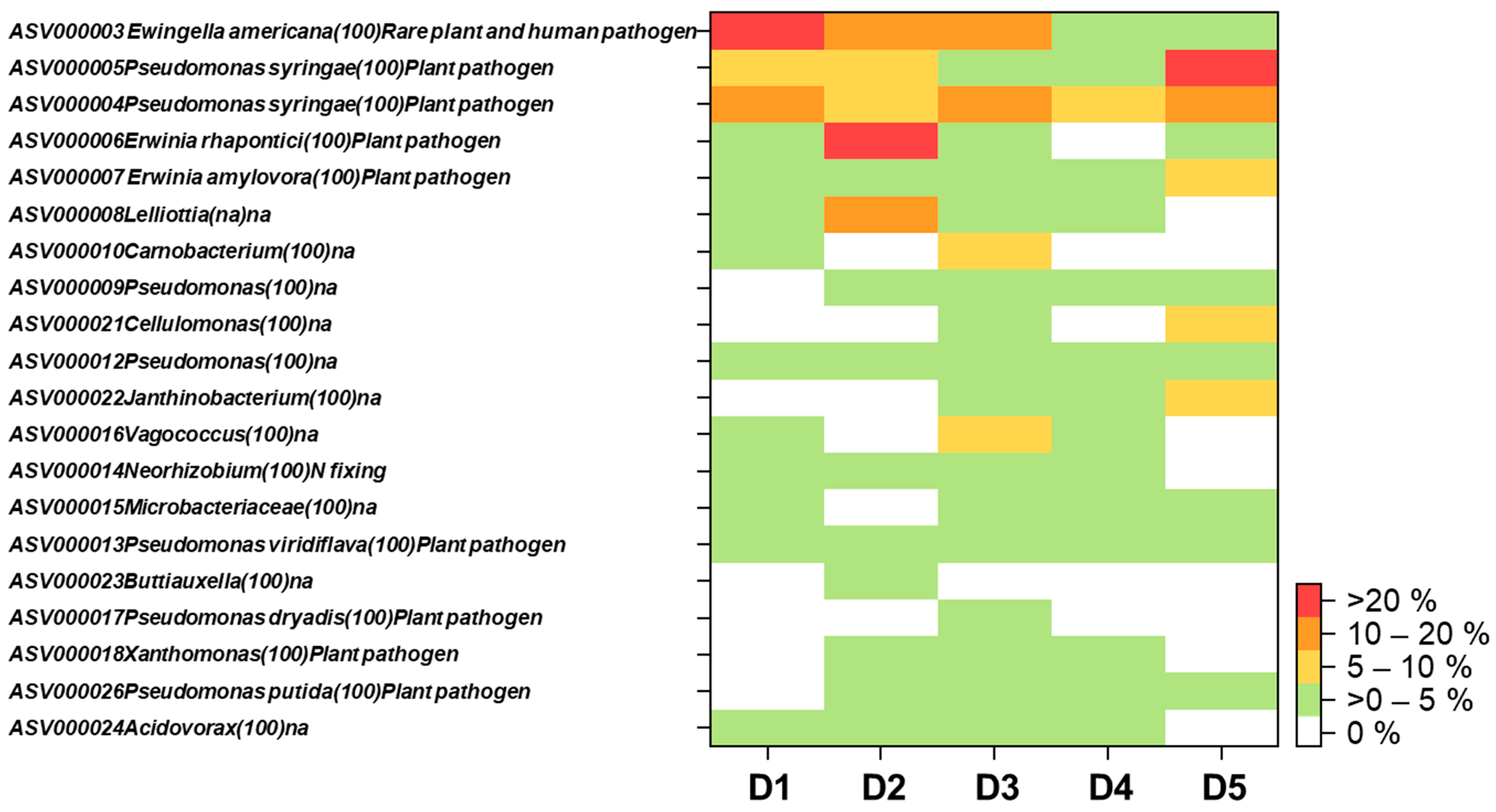
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cook, B.I.; Mankin, J.S.; Anchukaitis, K.J. Climate Change and Drought: From Past to Future. Curr. Clim. Chang. Rep. 2018, 4, 164–179. [Google Scholar] [CrossRef]
- Mukherjee, S.; Mishra, A.; Trenberth, K.E. Climate Change and Drought: A Perspective on Drought Indices. Curr. Clim. Chang. Rep. 2018, 4, 145–163. [Google Scholar] [CrossRef]
- Zhang, L.; Xiao, J.; Zhou, Y.; Zheng, Y.; Li, J.; Xiao, H. Drought Events and Their Effects on Vegetation Productivity in China. Ecosphere 2016, 7, e01591. [Google Scholar] [CrossRef]
- Grant, G.E.; Tague, C.L.; Allen, C.D. Watering the Forest for the Trees: An Emerging Priority for Managing Water in Forest Landscapes. Front. Ecol. Environ. 2013, 11, 314–321. [Google Scholar] [CrossRef] [Green Version]
- Schuldt, B.; Buras, A.; Arend, M.; Vitasse, Y.; Beierkuhnlein, C.; Damm, A.; Gharun, M.; Grams, T.E.E.; Hauck, M.; Hajek, P.; et al. A First Assessment of the Impact of the Extreme 2018 Summer Drought on Central European Forests. Basic Appl. Ecol. 2020, 45, 86–103. [Google Scholar] [CrossRef]
- McElrone, A.J.; Sherald, J.L.; Forseth, I.N. Interactive Effects of Water Stress and Xylem-limited Bacterial Infection on the Water Relations of a Host Vine. J. Exp. Bot. 2003, 54, 419–430. [Google Scholar] [CrossRef]
- Jayawardena, R.S.; Purahong, W.; Zhang, W.; Wubet, T.; Li, X.; Liu, M.; Zhao, W.; Hyde, K.D.; Liu, J.; Yan, J. Biodiversity of Fungi on Vitis vinifera L. Revealed by Traditional and High-Resolution Culture-Independent Approaches. Fungal Divers. 2018, 90, 1–84. [Google Scholar] [CrossRef] [Green Version]
- van Dijk, E.L.; Auger, H.; Jaszczyszyn, Y.; Thermes, C. Ten Years of Next-Generation Sequencing Technology. Trends Genet. 2014, 30, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Purahong, W.; Wubet, T.; Krüger, D.; Buscot, F. Molecular Evidence Strongly Supports Deadwood-Inhabiting Fungi Exhibiting Unexpected Tree Species Preferences in Temperate Forests. ISME J. 2017, 12, 289. [Google Scholar] [CrossRef] [PubMed]
- Dissanayake, A.J.; Purahong, W.; Wubet, T.; Hyde, K.D.; Zhang, W.; Xu, H.; Zhang, G.; Fu, C.; Liu, M.; Xing, Q.; et al. Direct Comparison of Culture-Dependent and Culture-Independent Molecular Approaches Reveal the Diversity of Fungal Endophytic Communities in Stems of Grapevine Vitis vinifera. Fungal Divers. 2018, 90, 85–107. [Google Scholar] [CrossRef]
- Hongsanan, S.; Jeewon, R.; Purahong, W.; Xie, N.; Liu, J.-K.; Jayawardena, R.S.; Ekanayaka, A.H.; Dissanayake, A.; Raspé, O.; Hyde, K.D.; et al. Can We Use Environmental DNA as Holotypes? Fungal Divers. 2018, 1–30. [Google Scholar] [CrossRef] [Green Version]
- Van Reckem, E.; De Vuyst, L.; Leroy, F.; Weckx, S. Amplicon-Based High-Throughput Sequencing Method Capable of Species-Level Identification of Coagulase-Negative Staphylococci in Diverse Communities. Microorganisms 2020, 8, 897. [Google Scholar] [CrossRef] [PubMed]
- Barba, M.; Hadidi, A. An Overview of Plant Pathology and Application of Next-Generation Sequencing Technologies. CAB Rev. 2015, 10, 1–21. [Google Scholar] [CrossRef]
- Purahong, W.; Orrù, L.; Donati, I.; Perpetuini, G.; Cellini, A.; Lamontanara, A.; Michelotti, V.; Tacconi, G.; Spinelli, F. Plant Microbiome and Its Link to Plant Health: Host Species, Organs and Pseudomonas syringae Pv. Actinidiae Infection Shaping Bacterial Phyllosphere Communities of Kiwifruit Plants. Front. Plant Sci. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Wahdan, S.; Hossen, S.; Tanunchai, B.; Sansupa, C.; Schädler, M.; Noll, M.; Wu, Y.-T.; Buscot, F.; Purahong, W. Life in the Wheat Litter: Effects of Future Climate on Microbiome and Function during the Early Phase of Decomposition. Microb. Ecol. 2021. [Google Scholar] [CrossRef]
- Francioli, D.; Lentendu, G.; Lewin, S.; Kolb, S. DNA Metabarcoding for the Characterization of Terrestrial Microbiota—Pitfalls and Solutions. Microorganisms 2021, 9, 361. [Google Scholar] [CrossRef]
- Riit, T.; Tedersoo, L.; Drenkhan, R.; Runno-Paurson, E.; Kokko, H.; Anslan, S. Oomycete-Specific ITS Primers for Identification and Metabarcoding. MycoKeys 2016, 14, 17–30. [Google Scholar] [CrossRef] [Green Version]
- Sillanpää, S.; Kramna, L.; Oikarinen, S.; Sipilä, M.; Rautiainen, M.; Aittoniemi, J.; Laranne, J.; Hyöty, H.; Cinek, O. Next-Generation Sequencing Combined with Specific PCR Assays to Determine the Bacterial 16S RRNA Gene Profiles of Middle Ear Fluid Collected from Children with Acute Otitis Media. mSphere 2 2017, 2, e00006-17. [Google Scholar] [CrossRef] [Green Version]
- ICP. Forst Conditions in Europe: The 2020 Assessment. ICP Forests Technical Report under the UNECE Convention on Long-Range Transboundary Air Pollution; Thünen Institute of Forest Ecosystems: Eberswalde, Germany, 2020; p. 95. [Google Scholar]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Weißbecker, C.; Schnabel, B.; Heintz-Buschart, A. Dadasnake, a Snakemake Implementation of DADA2 to Process Amplicon Sequencing Data for Microbial Ecology. GigaScience 2020, 9. [Google Scholar] [CrossRef]
- Vek, V.; Oven, P.; Ters, T.; Poljanšek, I.; Hinterstoisser, B. Extractives of Mechanically Wounded Wood and Knots in Beech. Holzforschung 2014, 68, 529–539. [Google Scholar] [CrossRef]
- Langer, G.J. Komplexe Schäden an Rotbuche (Fagus sylvatica) Und Auswirkungen Des Trockenen Und Heißen Sommers 2018 Auf Ältere Bestände (Waldschutzinfo Nr. 06/ 2019) 2019. Available online: https://www.nw-fva.de/fileadmin/nwfva/common/veroeffentlichen/waldschutzinfos/2019/NW-FVA_Waldschutzinfo_2019-06.pdf (accessed on 13 September 2021).
- Shortle, W.C.; Dudzik, K.R. Wood Decay in Living and Dead Trees: A Pictorial Overview; U.S. Department of Agriculture, Forest Service, Northern Research Station: Newtown Square, PA, USA, 2012; p. NRS-GTR-97. [Google Scholar]
- Purahong, W.; Hoppe, B.; Kahl, T.; Schloter, M.; Schulze, E.-D.; Bauhus, J.; Buscot, F.; Krüger, D. Changes within a Single Land-Use Category Alter Microbial Diversity and Community Structure: Molecular Evidence from Wood-Inhabiting Fungi in Forest Ecosystems. J. Environ. Manage. 2014, 139, 109–119. [Google Scholar] [CrossRef]
- Purahong, W.; Sadubsarn, D.; Tanunchai, B.; Wahdan, S.F.M.; Sansupa, C.; Noll, M.; Wu, Y.-T.; Buscot, F. First Insights into the Microbiome of a Mangrove Tree Reveal Significant Differences in Taxonomic and Functional Composition among Plant and Soil Compartments. Microorganisms 2019, 7, 585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global Patterns of 16S RRNA Diversity at a Depth of Millions of Sequences per Sample. Proc. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef] [Green Version]
- Ihrmark, K.; Bödeker, I.T.M.; Cruz-Martinez, K.; Friberg, H.; Kubartova, A.; Schenck, J.; Strid, Y.; Stenlid, J.; Brandström-Durling, M.; Clemmensen, K.E.; et al. New Primers to Amplify the Fungal ITS2 Region–Evaluation by 454-Sequencing of Artificial and Natural Communities. FEMS Microbiol. Ecol. 2012, 82, 666–677. [Google Scholar] [CrossRef] [PubMed]
- White, T.J.; Bruns, T.D.; Lee, S.; Taylor, J. Amplification and Direct Sequencing of Fungal Ribosomal RNA Genes for Phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Innis, M.A., Gelfand, D.H., Sninsky, J.J., White, T.J., Eds.; Academic Press: San Diego, CA, USA, 1990; Volume 38, pp. 315–322. [Google Scholar]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet.journal 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Kõljalg, U.; Nilsson, R.H.; Abarenkov, K.; Tedersoo, L.; Taylor, A.F.S.; Bahram, M.; Bates, S.T.; Bruns, T.D.; Bengtsson-Palme, J.; Callaghan, T.M.; et al. Towards a Unified Paradigm for Sequence-Based Identification of Fungi. Mol. Ecol. 2013, 22, 5271–5277. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Holmes, S.P. Exact Sequence Variants Should Replace Operational Taxonomic Units in Marker-Gene Data Analysis. ISME J. 2017, 11, 2639–2643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, R.H.; Larsson, K.-H.; Taylor, A.F.S.; Bengtsson-Palme, J.; Jeppesen, T.S.; Schigel, D.; Kennedy, P.; Picard, K.; Glöckner, F.O.; Tedersoo, L.; et al. The UNITE Database for Molecular Identification of Fungi: Handling Dark Taxa and Parallel Taxonomic Classifications. Nucleic Acids Res. 2019, 47, D259–D264. [Google Scholar] [CrossRef]
- Nguyen, N.H.; Song, Z.; Bates, S.T.; Branco, S.; Tedersoo, L.; Menke, J.; Schilling, J.S.; Kennedy, P.G. FUNGuild: An Open Annotation Tool for Parsing Fungal Community Datasets by Ecological Guild. Fungal Ecol. 2016, 20, 241–248. [Google Scholar] [CrossRef]
- Schena, L.; Duncan, J.M.; Cooke, D.E.L. Development and Application of a PCR-Based ‘Molecular Tool Box’ for the Identification of Phytophthora Species Damaging Forests and Natural Ecosystems. Plant Pathol. 2008, 57, 64–75. [Google Scholar] [CrossRef]
- Hammer, Ø.; Harper, D.A.T.; Ryan, P.D. PAST: Paleontological Statistics Software Package for Education and Data Analysis. Palaeontol. Electron. 2001, 4, 9. [Google Scholar]
- Hirooka, Y.; Rossman, A.Y.; Zhuang, W.-Y.; Salgado-Salazar, C.; Chaverri, P. Species Delimitation for Neonectria coccinea Group Including the Causal Agents of Beech Bark Disease in Asia, Europe, and North America. Mycosystema 2013, 32, 485–517. [Google Scholar]
- Domsch, K.H.; Gams, W.; Anderson, T.H. Compendium of Soil Fungi: Taxonomically Revised by W. Gams, 2nd ed.; IHW-Verlag: Eching, Germany, 2007; ISBN 978-3-930167-69-2. [Google Scholar]
- Lonsdale, D. Nectria coccinea infection of beech bark : Variations in disease in relation to predisposing factors. Ann. Sci. For. 1980, 37, 307–317. [Google Scholar] [CrossRef]
- Langer, G.J.; Bußkamp, J. Fungi Associated with Woody Tissues of European Beech and Their Impact on Tree Health. Front. Microbiol. 2021, 12, 2476. [Google Scholar] [CrossRef] [PubMed]
- Hilton, S.; Picot, E.; Schreiter, S.; Bass, D.; Norman, K.; Oliver, A.E.; Moore, J.D.; Mauchline, T.H.; Mills, P.R.; Teakle, G.R.; et al. Identification of Microbial Signatures Linked to Oilseed Rape Yield Decline at the Landscape Scale. Microbiome 2021, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Jung, T. Beech Decline in Central Europe Driven by the Interaction between Phytophthora Infections and Climatic Extremes. For. Pathol. 2009, 39, 73–94. [Google Scholar] [CrossRef]
- Matsiakh, I.; Kramarets, V.; Cleary, M. Occurrence and Diversity of Phytophthora Species in Declining Broadleaf Forests in Western Ukraine. For. Pathol. 2020, e12662. [Google Scholar] [CrossRef]
- Corcobado, T.; Cech, T.L.; Brandstetter, M.; Daxer, A.; Hüttler, C.; Kudláček, T.; Horta Jung, M.; Jung, T. Decline of European Beech in Austria: Involvement of Phytophthora spp. and Contributing Biotic and Abiotic Factors. Forests 2020, 11, 895. [Google Scholar] [CrossRef]
- Shang, Q.; Hyde, K.; Camporesi, E.; Maharachchikumbura, S.; Norphanphoun, C.; Brooks, S.; Liu, J.-K. Additions to the Genus Cytospora with Sexual Morph in Cytosporaceae. Mycosphere 2020, 11, 189–224. [Google Scholar] [CrossRef]
- Desprez-Loustau, M.-L.; Marçais, B.; Nageleisen, L.-M.; Piou, D.; Vannini, A. Interactive Effects of Drought and Pathogens in Forest Trees. Ann. For. Sci. 2006, 63, 597–612. [Google Scholar] [CrossRef] [Green Version]
- Hendry, S.J.; Boddy, L.; Lonsdale, D. Abiotic Variables Effect Differential Expression of Latent Infections in Beech (Fagus sylvatica). New Phytol. 2002, 155, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Agrios, G. Plant Pathology, 5th ed.; Academic Press: Cambridge, MA, USA, 2005; ISBN 978-0-12-044565-3. [Google Scholar]
- Lamichhane, J.R.; Varvaro, L.; Parisi, L.; Audergon, J.-M.; Morris, C.E. Chapter Four-Disease and Frost Damage of Woody Plants Caused by Pseudomonas syringae: Seeing the Forest for the Trees. In Advances in Agronomy; Sparks, D.L., Ed.; Academic Press: Cambridge, MA, USA, 2014; Volume 126, pp. 235–295. [Google Scholar]
- Palacio-Bielsa, A.; Roselló, M.; Llop, P.; López, M.M. Erwinia spp. from Pome Fruit Trees: Similarities and Differences among Pathogenic and Non-Pathogenic Species. Trees 2012, 26, 13–29. [Google Scholar] [CrossRef] [Green Version]
- Kamber, T. Characterization of Erwinia amylovora-Host Plant and-Biocontrol Agent Interactions. Ph.D. Thesis, University of Zurich, Zürich, Switzerland, 2013. [Google Scholar]
- Santander, R.D.; Català-Senent, J.F.; Figàs-Segura, À.; Biosca, E.G. From the Roots to the Stem: Unveiling Pear Root Colonization and Infection Pathways by Erwinia amylovora. FEMS Microbiol. Ecol. 2020, 96. [Google Scholar] [CrossRef] [PubMed]
- Gora, V.; König, J.; Lunderstädt, J. Population Dynamics of Beech Scale (Cryptococcus fagisuga) (Coccina, Pseudococcidae) Related to Physiological Defence Reactions of Attacked Beech Trees (Fagus sylvatica). Chemoecology 1996, 7, 112–120. [Google Scholar] [CrossRef]
- Langer, G.J.; Bußkamp, J.; Langer, E.J. Absterbeerscheinungen Bei Rotbuche Durch Wärme Und Trockenheit. AFZ Wald 2020, 75, 24–27. [Google Scholar]
- Brandt, J.; Albertsen, M. Investigation of Detection Limits and the Influence of DNA Extraction and Primer Choice on the Observed Microbial Communities in Drinking Water Samples Using 16S RRNA Gene Amplicon Sequencing. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Purahong, W.; Tanunchai, B.; Wahdan, S.F.M.; Buscot, F.; Schulze, E.-D. Molecular Screening of Microorganisms Associated with Discolored Wood in Dead European Beech Trees Suffered from Extreme Drought Event Using Next Generation Sequencing. Plants 2021, 10, 2092. https://doi.org/10.3390/plants10102092
Purahong W, Tanunchai B, Wahdan SFM, Buscot F, Schulze E-D. Molecular Screening of Microorganisms Associated with Discolored Wood in Dead European Beech Trees Suffered from Extreme Drought Event Using Next Generation Sequencing. Plants. 2021; 10(10):2092. https://doi.org/10.3390/plants10102092
Chicago/Turabian StylePurahong, Witoon, Benjawan Tanunchai, Sara Fareed Mohamed Wahdan, François Buscot, and Ernst-Detlef Schulze. 2021. "Molecular Screening of Microorganisms Associated with Discolored Wood in Dead European Beech Trees Suffered from Extreme Drought Event Using Next Generation Sequencing" Plants 10, no. 10: 2092. https://doi.org/10.3390/plants10102092