Identification of New Proteins and Potential Mitochondrial F1F0-ATPase Inhibitor Factor 1-Associated Mechanisms in Arabidopsis thaliana Using iTRAQ-Based Quantitative Proteomic Analysis


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Growth Conditions
2.2. Extraction, Quantification, and Digestion of Cellular Proteins
2.3. iTRAQ Labeling and Strong Cation Exchange Fractionation
2.4. Liquid Chromatography–Tandem Mass Spectrometry Analysis
2.5. Data and Bioinformatics Analysis
2.6. RNA Extraction and Real-Time Quantitative PCR
3. Results
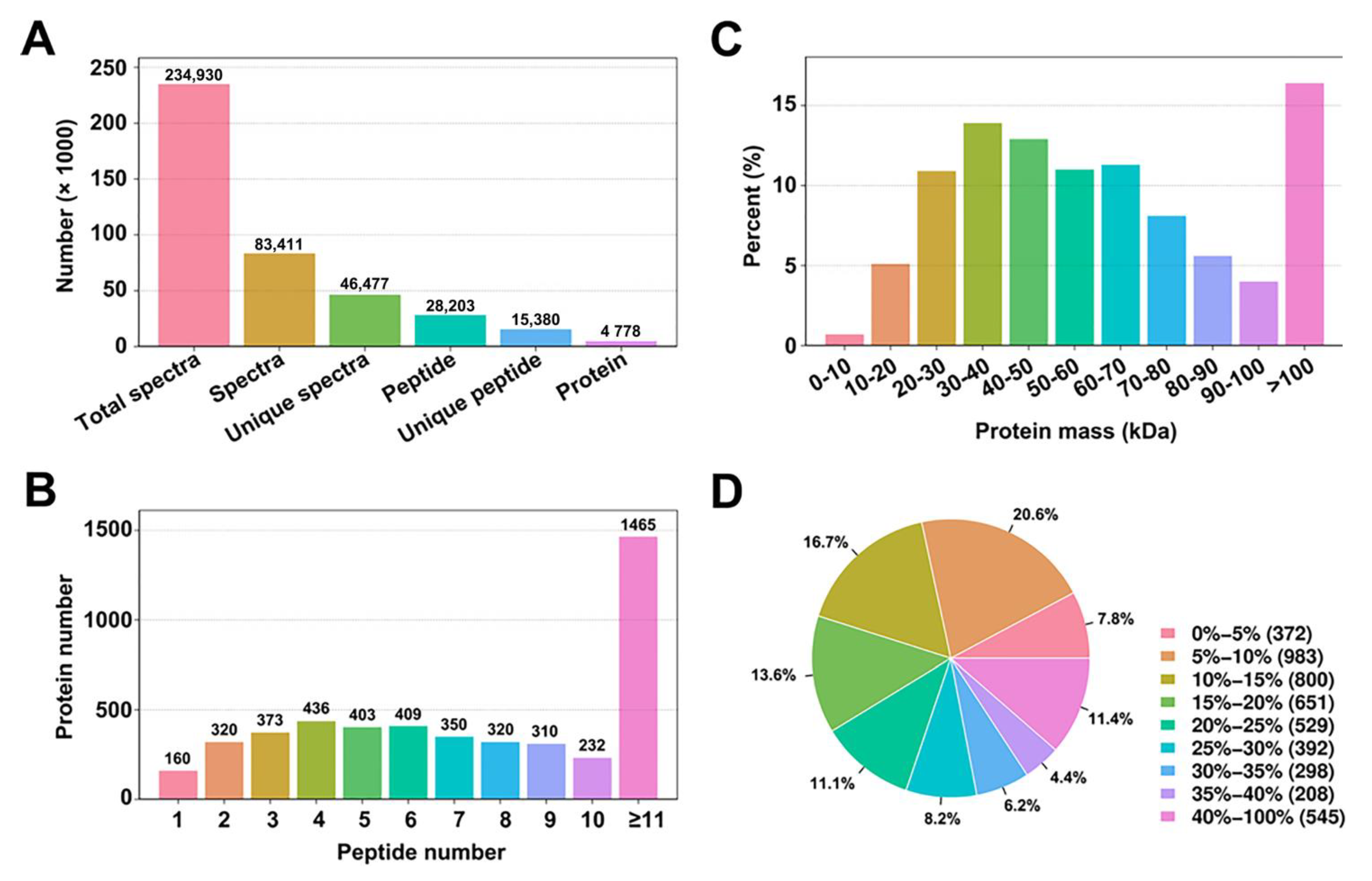
3.1. Analysis of A. thaliana Protein Profiles Using iTRAQ
3.2. Identification of Proteins Differentially Expressed between A. thaliana Wild-Type and the if1 Mutant Lines
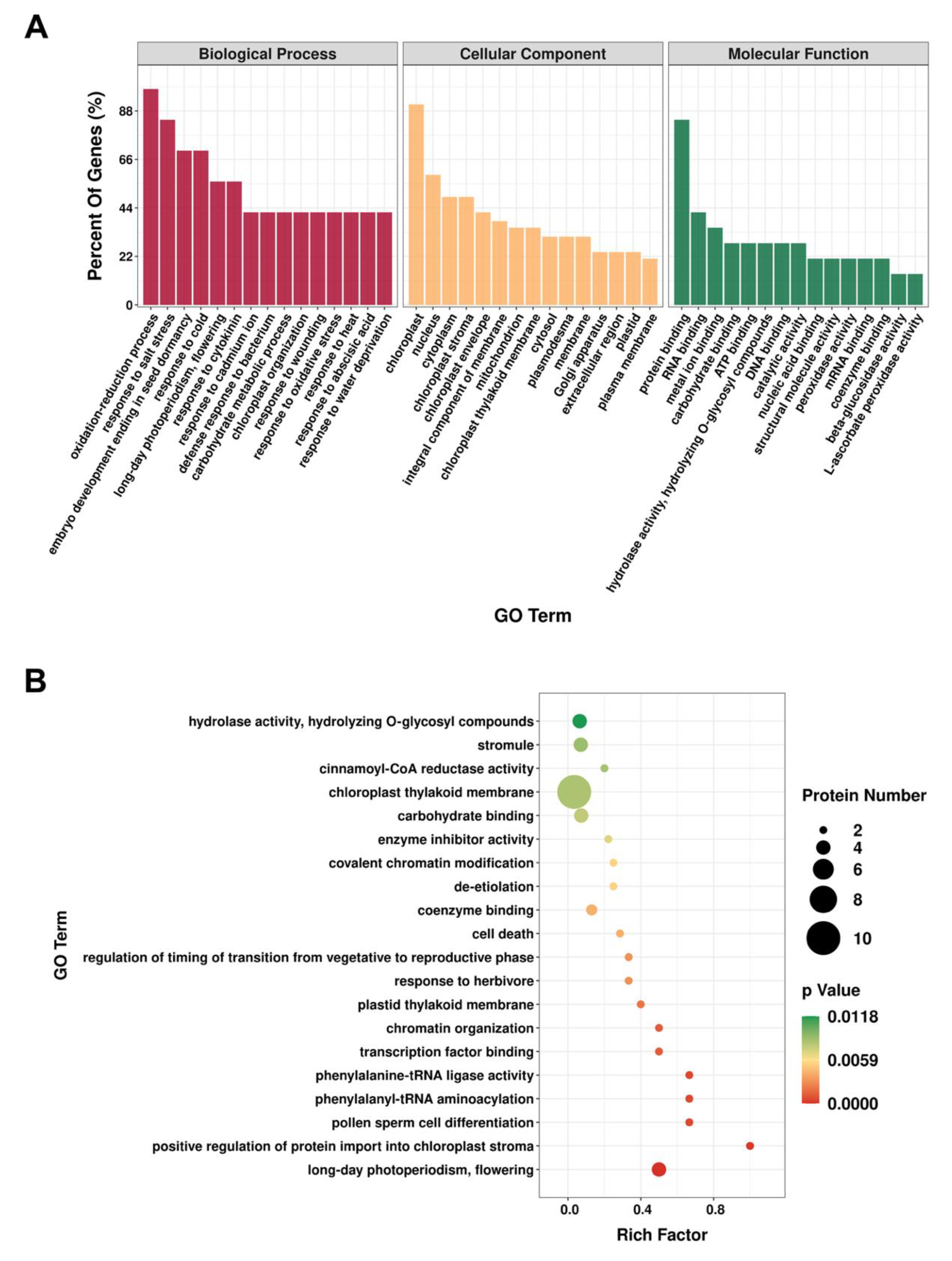
3.3. Functional Annotation of Identified Proteins with Differential Accumulation in A. thaliana Wild-Type and if1 Mutant Plants
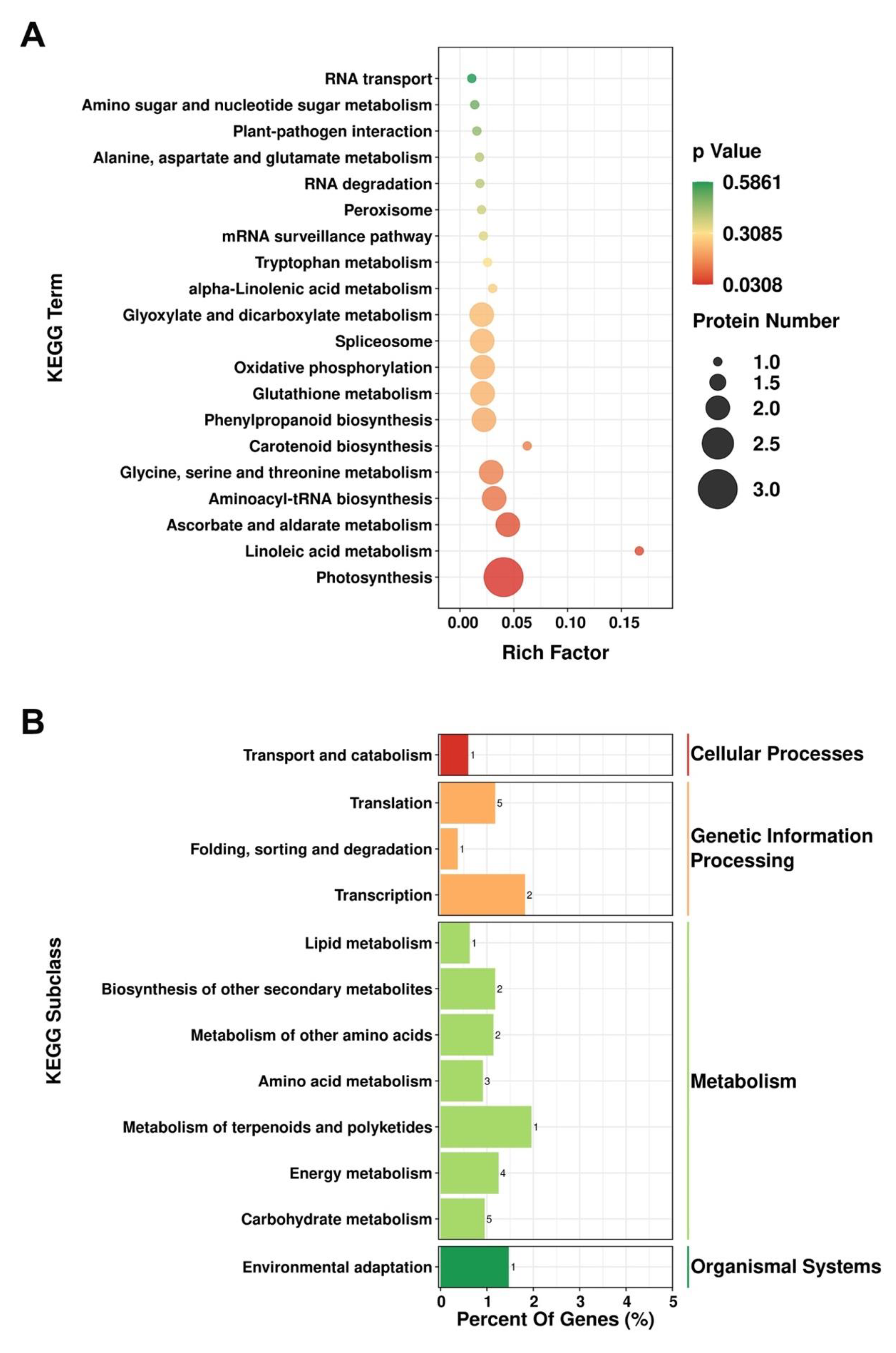
3.4. KEGG Pathway Analysis of the DEPs
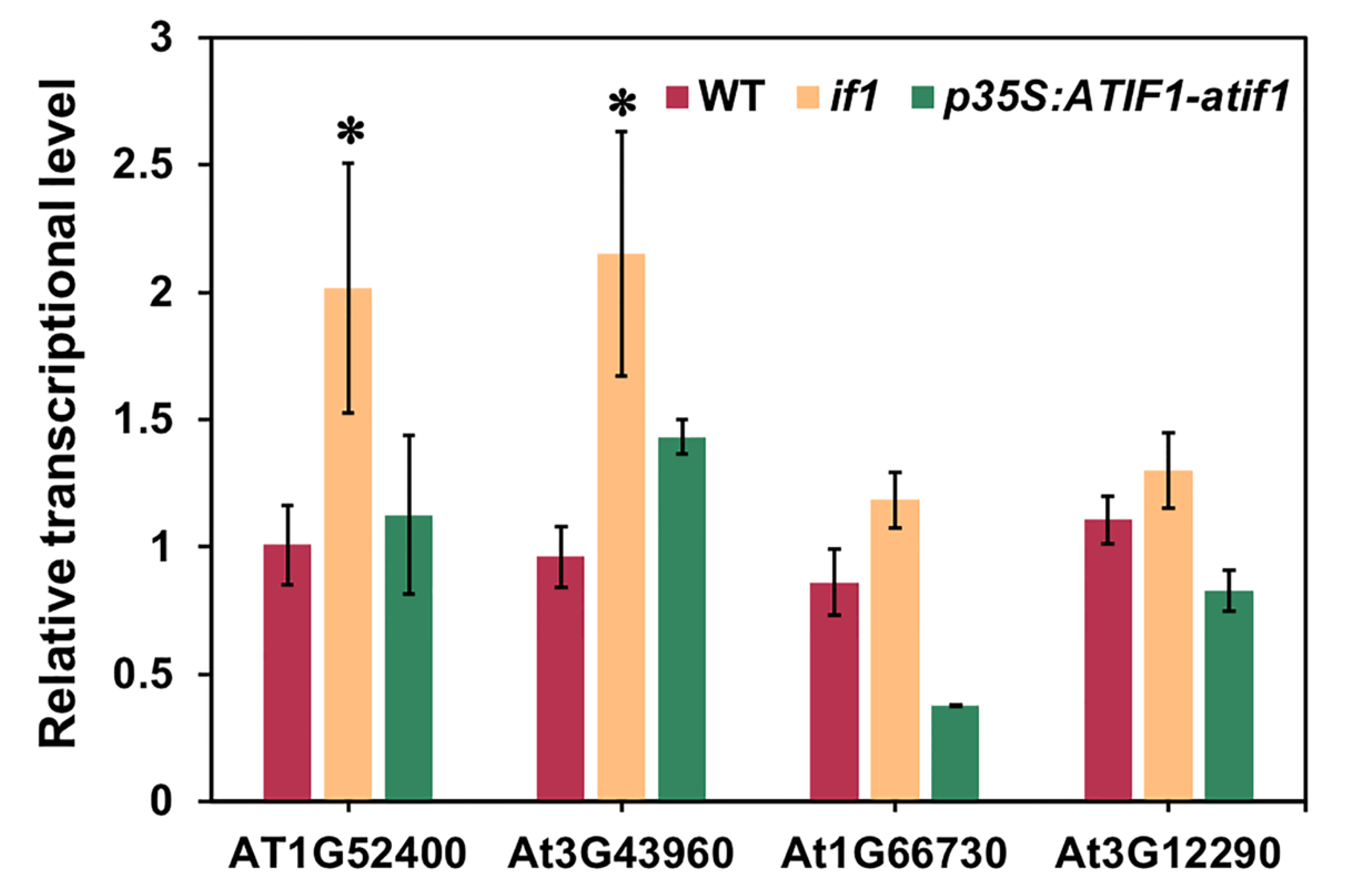
3.5. Analysis of the Transcriptional Expression of the DEPs
4. Discussion
4.1. Proteomic Changes between A. thaliana Wild-Type and if1 Mutant Seedlings
4.2. Identification of AtIF1-Regulated Proteins That Involved in Energy Metabolism
4.3. Identification of AtIF1-Regulated Proteins Involved in Reproductive Development
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Cabezon, E.; Butler, P.J.; Runswick, M.J.; Walker, J.E. Modulation of the oligomerization state of the bovine F1-ATPase inhibitor protein, IF1, by pH. J. Biol. Chem. 2020, 275, 25460–25464. [Google Scholar] [CrossRef] [Green Version]
- Cabezon, E.; Butler, P.J.; Runswick, M.J.; Carbajo, R.J.; Walker, J.E. Homologous and heterologous inhibitory effects of ATPase inhibitor proteins on F-ATPases. J. Biol. Chem. 2002, 277, 41334–41341. [Google Scholar] [CrossRef] [Green Version]
- Green, D.W.; Grover, G.J. The IF1 inhibitor protein of the mitochondrial F1F0-ATPase. Biochim. Biophys. Acta 2000, 1458, 343–355. [Google Scholar] [CrossRef] [Green Version]
- Campanella, M.; Parker, N.; Tan, C.H.; Hall, A.M.; Duchen, M.R. IF1: Setting the pace of the F1F0-ATP synthase. Trends Biochem. Sci. 2009, 34, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Faccenda, D.; Campanella, M. Molecular Regulation of the Mitochondrial F1F0-ATPsynthase: Physiological and Pathological Significance of the Inhibitory Factor 1 (IF1). Int. J. Cell Biol. 2012, 2012, 367934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabezón, E.; Runswick, M.J.; Leslie, A.G.; Walker, J.E. The structure of bovine IF (1), the regulatory subunit of mitochondrial F-ATPase. EMBO J. 2001, 20, 6990–6996. [Google Scholar] [CrossRef] [Green Version]
- Venard, R.; Brèthes, D.; Giraud, M.F.; Vaillier, J.; Velours, J.; Haraux, F. Investigation of the role and mechanism of IF1 and STF1 proteins, twin inhibitory peptides which interact with the yeast mitochondrial ATP synthase. Biochemistry 2003, 42, 7626–7636. [Google Scholar] [CrossRef]
- Norling, B.; Tourikas, C.; Hamasur, B.; Glaser, E. Evidence for an endogenous ATPase inhibitor protein in plant mitochondria. Purification and characterization. Eur. J. Biochem. 1990, 188, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Polgreen, K.E.; Featherstone, J.; Willis, A.C.; Harris, D.A. Primary structure and properties of the inhibitory protein of the mitochondrial ATPase (H+-ATP synthase) from potato. Biochim. Biophys. Acta 1995, 1229, 175–180. [Google Scholar] [CrossRef] [Green Version]
- Nakazono, M.; Imamura, T.; Tsutsumi, N.; Sasaki, T.; Hirai, A. Characterization of two cDNA clones encoding isozymes of the F1F0-ATPase inhibitor protein of rice mitochondria. Planta 2000, 210, 188–194. [Google Scholar] [CrossRef]
- Chen, C.; Meng, Y.; Shopan, J.; Whelan, J.; Hu, Z.; Yang, J.; Zhang, M. Identification and characterization of Arabidopsis thaliana mitochondrial F1F0-ATPase inhibitor factor 1. J. Plant Physiol. 2020, 254, 153264. [Google Scholar] [CrossRef]
- Chen, P.; Li, R.; Zhou, R. Comparative phosphoproteomic analysis reveals differentially phosphorylated proteins regulate anther and pollen development in kenaf cytoplasmic male sterility line. Amino Acids 2018, 50, 841–862. [Google Scholar] [CrossRef]
- McCormick, S. Male Gametophyte Development. Plant Cell 1993, 5, 1265–1275. [Google Scholar] [CrossRef]
- McCormick, S. Control of male gametophyte development. Plant Cell 2004, 16, S142–S153. [Google Scholar] [CrossRef]
- Lee, S.L.J.; Warmke, H.E. Organelle size and number in fertile and T-cytoplasmic male-sterile corn. Amer. J. Bot. 1979, 66, 141–148. [Google Scholar] [CrossRef]
- Hanson, M.R.; Bentolila, S. Interactions of mitochondrial and nuclear genes that affect male gametophyte development. Plant Cell 2004, 16, S154–S169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oeljeklaus, S.; Meyer, H.E.; Warscheid, B. Advancements in plant proteomics using quantitative mass spectrometry. J. Proteom. 2009, 72, 545–554. [Google Scholar] [CrossRef]
- Zieske, L. A perspective on the use of iTRAQ ™ reagent technology for protein complex and profiling studies. J. Exp. Bot. 2006, 57, 1501–1508. [Google Scholar] [CrossRef] [PubMed]
- Ross, P.L.; Huang, Y.N.; Marchese, J.N.; Williamson, B.; Parker, K.; Hattan, S.; Khainovski, N.; Pillai, S.; Dey, S.; Daniels, S.; et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell Proteom. 2005, 3, 1154–1169. [Google Scholar] [CrossRef] [Green Version]
- Tang, N.; Liu, W.; Zhang, W.; Tang, D. Integrative analysis of transcriptomic and proteomic changes related to male sterility in Tagetes erecta. Physiol. Mol. Biol. Plants 2020, 26, 2061–2074. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Liu, H.; Li, S.; Zhang, X.; Zhang, M.; Zhu, N.; Dufresne, C.P.; Chen, S.; Wang, Q. Regulation of BZR1 in fruit ripening revealed by iTRAQ proteomics analysis. Sci. Rep. 2016, 6, 33635. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.X.; Luo, Y.M.; Ye, Z.Q.; Cao, X.; Liang, J.N.; Wang, Q.; Wu, Y.; Wu, J.H.; Wang, H.Y.; Zhang, M.; et al. iTRAQ-based proteomics analysis of autophagy-mediated immune responses against the vascular fungal pathogen Verticillium dahliae in Arabidopsis. Autophagy 2018, 14, 598–618. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.Q.; Zhou, X.; Dong, L.; Guo, J.; Chen, Y.; Zhang, Y.; Wu, L.; Xu, M. iTRAQ-based analysis of the Arabidopsis proteome reveals insights into the potential mechanisms of anthocyanin accumulation regulation in response to phosphate deficiency. J. Proteom. 2018, 184, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Guo, Y.; Adil, M.F.; Sehar, S.; Cai, B.; Xiang, Z.; Tu, Y.; Zhao, D.; Shamsi, I.H. Comparative Proteomic Analysis by iTRAQ Reveals that Plastid Pigment Metabolism Contributes to Leaf Color Changes in Tobacco (Nicotiana tabacum) during Curing. Int. J. Mol. Sci. 2020, 21, 2394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.B.; Guo, K.; Liang, W.W.; Chen, Q.F.; Shi, J.; Shen, B. Quantitative proteomics analysis of proteins involved in leaf senescence of rice (Oryza sativa L.). Plant Growth Regul. 2018, 84, 341–349. [Google Scholar] [CrossRef]
- Zhang, L.; Dong, Y.; Wang, Q.; Du, C.; Xiong, W.; Li, X.; Zhu, S.; Li, Y. iTRAQ-Based Proteomics Analysis and Network Integration for Kernel Tissue Development in Maize. Int. J. Mol. Sci. 2017, 18, 1840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Y.; Wang, W.; Huang, X.; Liu, M.; Hebelstrup, K.H.; Yang, D.; Cai, J.; Wang, X.; Zhou, Q.; Cao, W.; et al. Nitrogen topdressing timing modifies the gluten quality and grain hardness related protein levels as revealed by iTRAQ. Food Chem. 2019, 277, 135–144. [Google Scholar] [CrossRef]
- Hu, G.; Koh, J.; Yoo, M.J.; Grupp, K.; Chen, S.; Wendel, J.F. Proteomic profiling of developing cotton fibers from wild and domesticated Gossypium barbadense. New Phytol. 2013, 200, 570–582. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Parker, J.; Koh, J.; Yoo, M.J.; Zhu, N.; Feole, M.; Yi, S.; Chen, S. Quantitative proteomics of tomato defense against Pseudomonas syringae infection. Proteomics 2013, 13, 1934–1946. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Xu, Y.; Feng, Z.; Yang, X.; Wang, X.; Gao, X. Multiple-platform data integration method with application to combined analysis of microarray and proteomic data. BMC Bioinform. 2012, 13, 320. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.; Cao, J.; Li, J.; Zhou, B.; Tang, J.; Miao, A. Phenotypic, histological and proteomic analyses reveal multiple differences associated with chloroplast development in yellow and variegated variants from Camellia sinensis. Sci. Rep. 2016, 6, 33369. [Google Scholar] [CrossRef] [Green Version]
- Fromme, P.; Grotjohann, I. Structure of Photosystems I and II. Results Probl. Cell Differ. 2008, 45, 33–72. [Google Scholar] [CrossRef]
- Tsunoyama, Y.; Morikawa, K.; Shiina, T.; Toyoshima, Y. Blue light specific and differential expression of a plastid sigma factor, Sig5 in Arabidopsis thaliana. FEBS Lett. 2002, 516, 225–228. [Google Scholar] [CrossRef] [Green Version]
- Mabbitt, P.D.; Wilbanks, S.M.; Eaton-Rye, J.J. Structure and function of the hydrophilic Photosystem II assembly proteins: Psb27, Psb28 and Ycf48. Plant Physiol. Biochem. 2014, 81, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.F.; Lu, J.; Yu, J.W.; Zhang, C.Q.; He, J.X.; Liu, Q.Q. The brassinosteroid-regulated transcription factors BZR1/BES1 function as a coordinator in multisignal-regulated plant growth. Biochim. Biophys. Acta Gene Regul. Mech. 2018, 1861, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Lv, M.; Wang, Y.; Wang, P.A.; Cui, Y.; Li, M.; Wang, R.; Gou, X.; Li, J. BES1 is activated by EMS1-TPD1-SERK1/2-mediated signaling to control tapetum development in Arabidopsis thaliana. Nat. Commun. 2019, 10, 4164. [Google Scholar] [CrossRef] [Green Version]
- Ye, Q.; Zhu, W.; Li, L.; Zhang, S.; Yin, Y.; Ma, H.; Wang, X. Brassinosteroids control male fertility by regulating the expression of key genes involved in Arabidopsis anther and pollen development. Proc. Natl. Acad. Sci. USA 2010, 107, 6100–6105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuda, N.; Tsuchiya, T.; Kishitani, S.; Tanaka, Y.; Toriyama, K. Partial male sterility in transgenic tobacco carrying antisense and sense PAL cDNA under the control of a tapetum-specific promoter. Plant Cell Physiol. 1996, 37, 215–222. [Google Scholar] [CrossRef]
- Liu, L.; Fan, X.D. Tapetum: Regulation and role in sporopollenin biosynthesis in Arabidopsis. Plant Mol. Biol. 2013, 83, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Cui, M.; Yang, L.; Kim, Y.J.; Zhang, D. Genetic and Biochemical Mechanisms of Pollen Wall Development. Trends Plant Sci. 2015, 20, 741–753. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession | Protein Description | Gene Name | Fold Change (if1/WT) |
|---|---|---|---|
| Up-regulated DEPs | |||
| A0A178W6T5_ARATH | HIP1.3 | At1g09240 | 1.21 |
| A0A0U5CLC7_9BRAS | Photosystem II D2 protein | psbD | 1.29 |
| A0A178V4M1_ARATH | RBP31 | At4g28660 | 1.29 |
| JAL28_ARATH | Nitrile-specifier protein 1 | NSP1 | 1.21 |
| BGL37_ARATH | Myrosinase 2 | TGG2 | 1.34 |
| A0A178W5I1_ARATH | L-ascorbate peroxidase | At1g07590 | 1.24 |
| D7KHW7_ARALL | L-ascorbate peroxidase | ARALYDRAFT_470824 | 1.24 |
| A0A178V395_ARATH | PAP_fibrillin domain-containing protein | At4g25850 | 1.22 |
| D7L023_ARALL | Alanine--glyoxylate aminotransferase | ARALYDRAFT_480305 | 1.23 |
| D7M155_ARALL | Plastid lipid-associated protein 1, chloroplast | ARALYDRAFT_911682 | 1.22 |
| LOX2_ARATH | Lipoxygenase 2, chloroplastic | LOX2 | 1.25 |
| JAL30_ARATH | PYK10-binding protein 1 | PBP1 | 1.2 |
| A0A178VSK5_ARATH | Uncharacterized protein | At2g41150 | 1.25 |
| A0A178VBA5_ARATH | Glutaredoxin-dependent peroxiredoxin | At3g05750 | 1.21 |
| RL61_ARATH | 60S ribosomal protein L6-1 | RPL6A | 1.26 |
| A0A178V6D3_ARATH | Phenylalanyl-tRNA synthetase | At3g52700 | 1.21 |
| A0A178WET2_ARATH | - | - | 1.22 |
| A0A178VVV5_ARATH | THF1 | At2g16340 | 1.21 |
| D7MSA7_ARALL | Uncharacterized protein | ARALYDRAFT_918349 | 1.47 |
| A0A178V4T2_ARATH | HMA domain-containing protein | At4g33520 | 2.02 |
| A0A178UEX5_ARATH | CDF1 | At5g22550 | 1.25 |
| D7M1M2_ARALL | Uncharacterized protein | GN=ARALYDRAFT_489157 | 1.25 |
| Q1H5F7_ARATH | Histone H2B | - | 1.24 |
| A0A178WJ12_ARATH | TLL1 | At1g40860 | 1.26 |
| A0A178UTL8_ARATH | XYL4 | At5g64200 | 1.21 |
| A0A178VST0_ARATH | JAL22 | At2g36220 | 1.26 |
| A0A178UDT2_ARATH | SAG15 | At5g49860 | 1.28 |
| BGL18_ARATH | Beta-D-glucopyranosyl abscisate beta-glucosidase | BGLU18 | 1.24 |
| FAX2_ARATH | Protein FATTY ACID EXPORT 2, chloroplastic | FAX2 | 1.21 |
| A0A178WJL7_ARATH | Phenylalanyl-tRNA synthetase beta subunit | At1g66730 | 1.2 |
| A0A178VZH7_ARATH | Histone H2B | At2g24830 | 1.27 |
| A0A178U9K8_ARATH | OPT4 | At5g64020 | 1.26 |
| A0A178UG56_ARATH | Uncharacterized protein | At5g46060 | 1.31 |
| A0A178W4H7_ARATH | Uncharacterized protein | At1g74710 | 1.22 |
| A0A178UH43_ARATH | - | - | 1.21 |
| A0A178WIZ0_ARATH | Lipase_GDSL domain-containing protein | At1g48430 | 1.21 |
| A0A178WEL1_ARATH | ATP-dependent Clp protease proteolytic subunit | At1g12090 | 1.25 |
| A0A178WD65_ARATH | ATARP4 | At1g19350 | 1.23 |
| D7KGE2_ARALL | Uncharacterized protein | ARALYDRAFT_472063 | 1.23 |
| A0A178V8P6_ARATH | Methionine aminopeptidase 2 | At3g54390 | 1.2 |
| D7KBN6_ARALL | Uncharacterized protein | ARALYDRAFT_472926 | 1.21 |
| A0A178W3R8_ARATH | Uncharacterized protein | At1g27300 | 1.21 |
| A0A178W7M1_ARATH | Uncharacterized protein | At1g29860 | 1.24 |
| A0A178UX37_ARATH | SGT1B | At4g12690 | 1.25 |
| D7LBM5_ARALL | tRNA pseudouridine synthase family protein | ARALYDRAFT_481943 | 1.83 |
| A0A178VSZ1_ARATH | JAL23 | At2g36240 | 1.27 |
| A0A178UZA3_ARATH | SSR16 | At4g39620 | 1.23 |
| A0A178UP34_ARATH | Epimerase domain-containing protein | At5g58490 | 1.25 |
| A0A178W827_ARATH | KTI1 | At1g67490 | 1.39 |
| A0A178WIQ3_ARATH | RZ-1b | At1g53730 | 1.23 |
| Q6NPL0_ARATH | At4g09040 | At4g09040 | 1.22 |
| A0A178VFH3_ARATH | Uncharacterized protein | At3g12290 | 1.24 |
| A0A178V9V7_ARATH | FTM4 | At3g12170 | 1.35 |
| A0A178WN71_ARATH | Plastocyanin | At1g70550 | 1.25 |
| D7MQE2_ARALL | Cinnamoyl-CoA reductase family | ARALYDRAFT_495968 | 1.29 |
| A0A178WC08_ARATH | SRZ21 | AXX17_At1g25030 | 1.33 |
| Y4554_ARATH | Uncharacterized protein At4g15545 | At4g15545 | 1.24 |
| D7KZQ1_ARALL | Leucine-rich repeat protein FLR1 | ARALYDRAFT_897426 | 1.26 |
| A0A178V6Y8_ARATH | BSD domain-containing protein | AXX17_At3g43960 | 1.22 |
| Down-regulated DEPs | |||
| A0A178WA55_ARATH | Uncharacterized protein | At1g50990 | 0.8 |
| A0A068CL28_ARALY | At4g21450p-like protein | - | 0.8 |
| A0A178V1L3_ARATH | PDE332 | At4g10860 | 0.82 |
| A0A178V0A8_ARATH | Nodulin-like domain-containing protein | At4g39930 | 0.68 |
| A0A178UJR2_ARATH | DegP10 | At5g33920 | 0.79 |
| A0A178VP21_ARATH | ANK_REP_REGION domain-containing protein | At3g03550 | 0.25 |
| D7MFW8_ARALL | Xyloglucan endotransglucosylase/hydrolase | ARALYDRAFT_354208 | 0.73 |
| A0A178WAG2_ARATH | Uncharacterized protein | AXX17_At1g08440 | 0.77 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.; Meng, Y.; Hu, Z.; Yang, J.; Zhang, M. Identification of New Proteins and Potential Mitochondrial F1F0-ATPase Inhibitor Factor 1-Associated Mechanisms in Arabidopsis thaliana Using iTRAQ-Based Quantitative Proteomic Analysis. Plants 2021, 10, 2385. https://doi.org/10.3390/plants10112385
Chen C, Meng Y, Hu Z, Yang J, Zhang M. Identification of New Proteins and Potential Mitochondrial F1F0-ATPase Inhibitor Factor 1-Associated Mechanisms in Arabidopsis thaliana Using iTRAQ-Based Quantitative Proteomic Analysis. Plants. 2021; 10(11):2385. https://doi.org/10.3390/plants10112385
Chicago/Turabian StyleChen, Cuiting, Yiqing Meng, Zhongyuan Hu, Jinghua Yang, and Mingfang Zhang. 2021. "Identification of New Proteins and Potential Mitochondrial F1F0-ATPase Inhibitor Factor 1-Associated Mechanisms in Arabidopsis thaliana Using iTRAQ-Based Quantitative Proteomic Analysis" Plants 10, no. 11: 2385. https://doi.org/10.3390/plants10112385