Morphology Dependence Degradation of Electro- and Magnetoactive Poly(3-hydroxybutyrate-co-hydroxyvalerate) for Tissue Engineering Applications

 ,
,  , ,
, ,  , and
, and 
Abstract
:1. Introduction
2. Experimental
2.1. Materials
2.2. Sample Processing
2.3. Degradation Assays
2.4. Morphological Analysis
2.5. Chemical Structure Analysis
2.6. Thermal Analysis
2.7. Elemental Surface Composition Analysis
2.8. Weight Loss Assessment
2.9. Cytotoxic Assay
3. Results and Discussion
3.1. Samples Macrostructure
3.2. Samples Microstructure
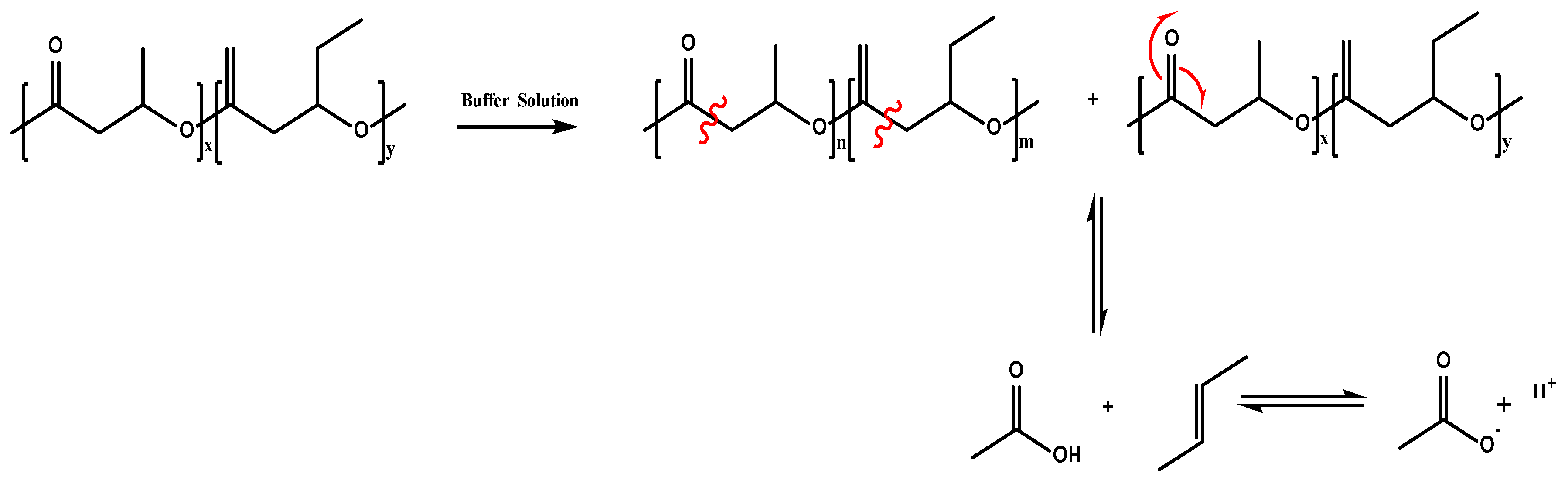
3.3. Chemical Analysis
3.4. Thermal Analysis
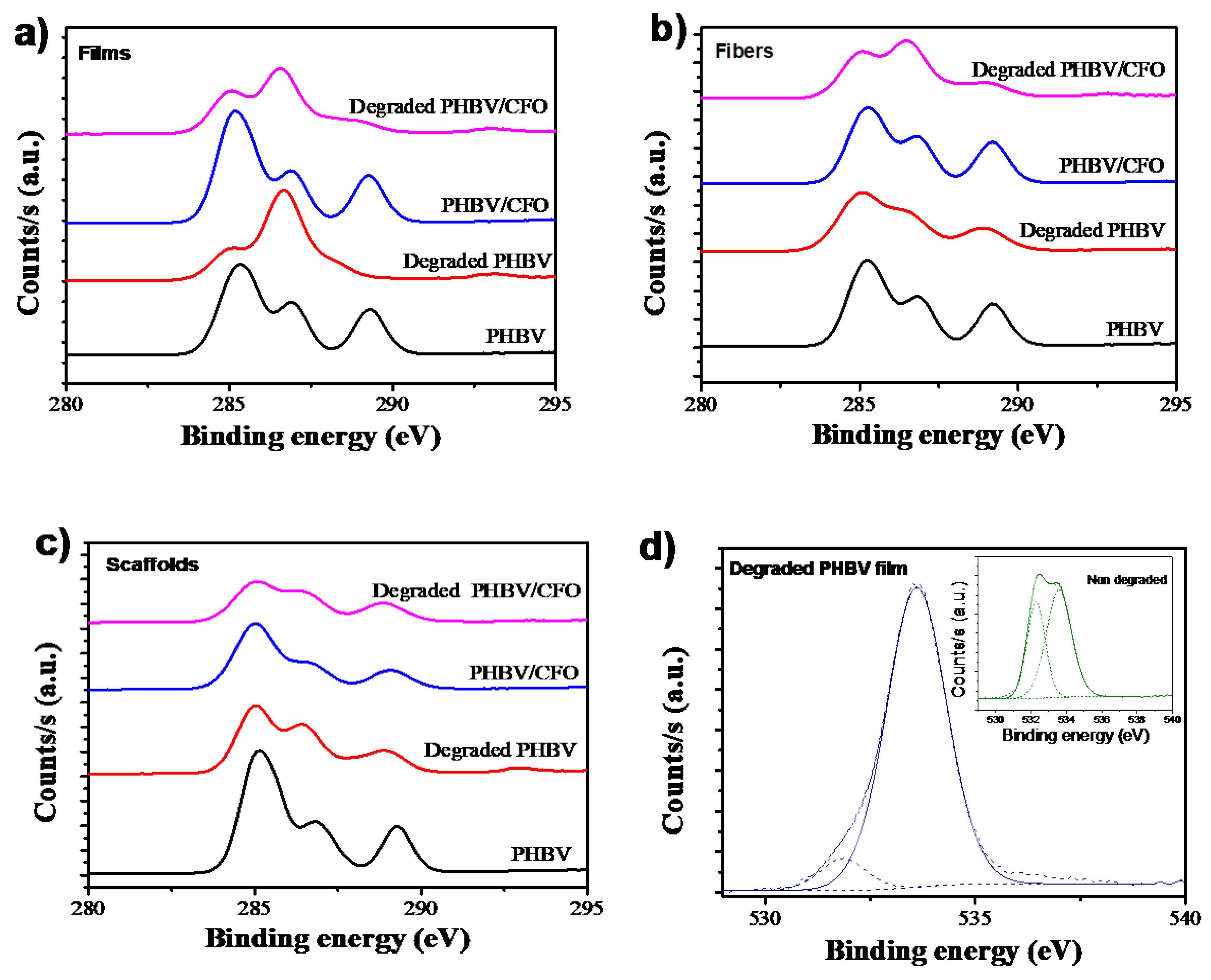
3.5. Elemental Surface Composition Analysis
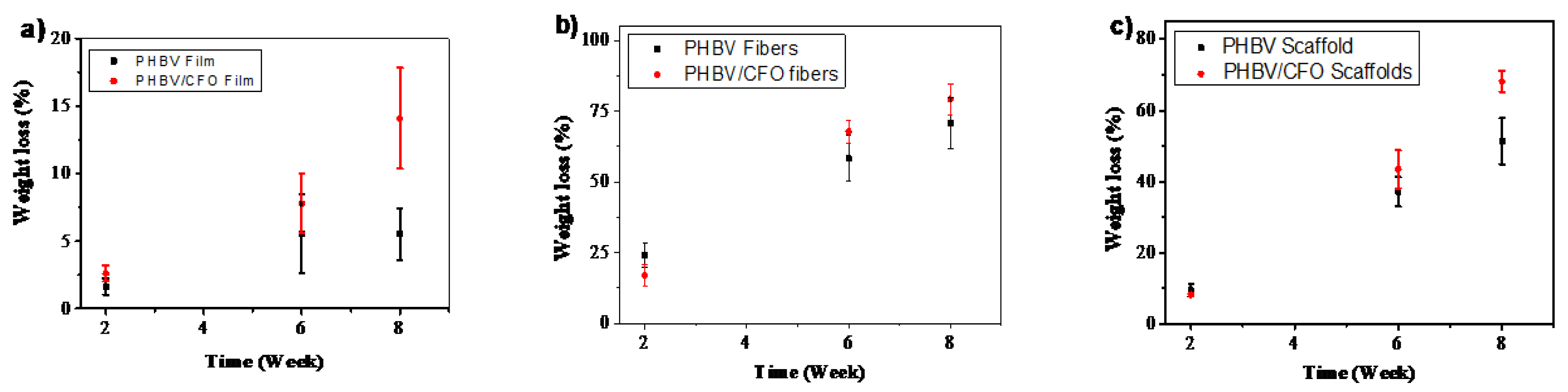
3.6. Weight Loss
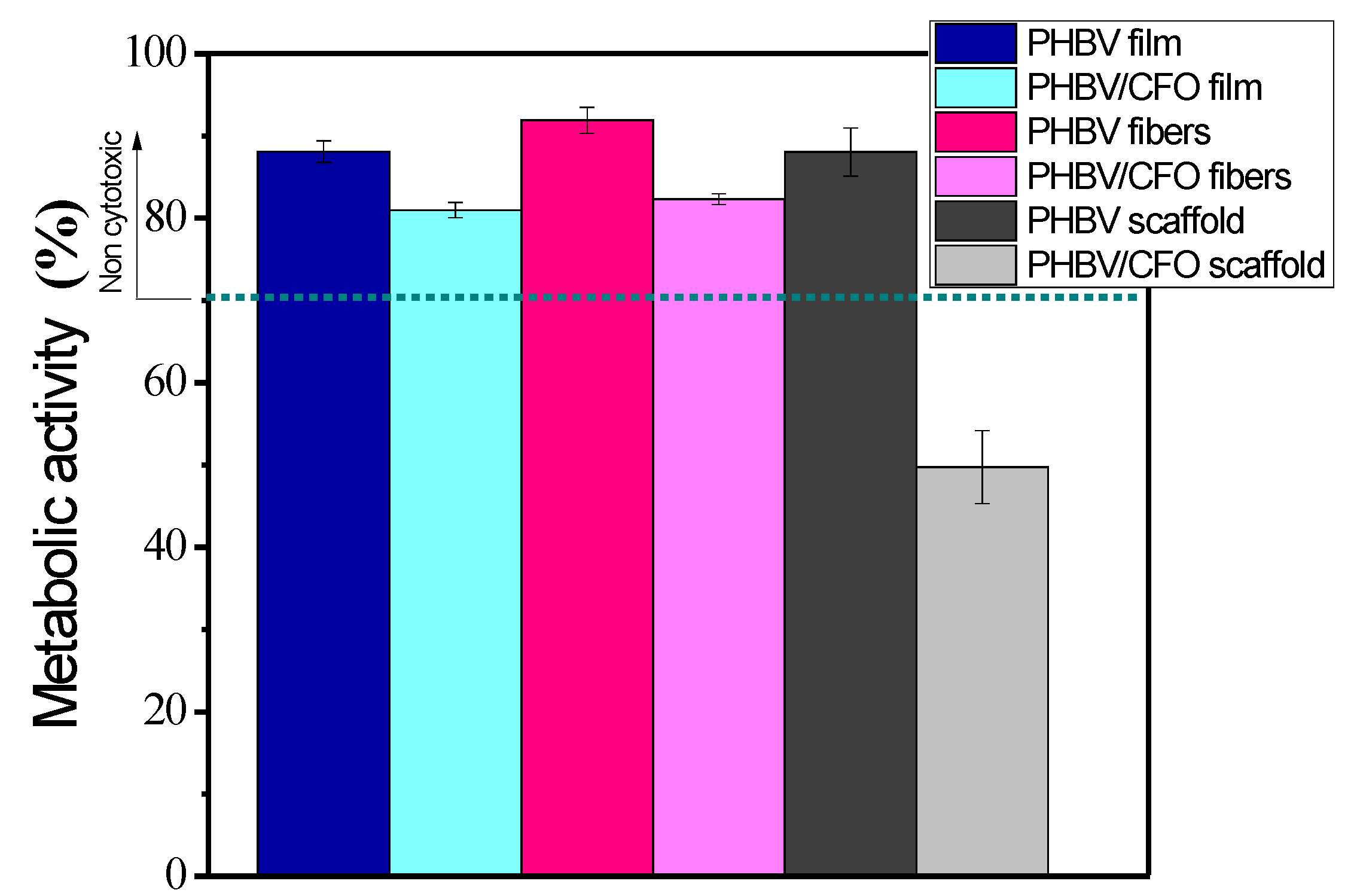
3.7. Cytotoxic Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kim, M.S.; Kim, J.H.; Min, B.H.; Chun, H.J.; Han, D.K.; Lee, H.B. Polymeric scaffolds for regenerative medicine. Polym. Rev. 2011, 51, 23–52. [Google Scholar] [CrossRef]
- Shin, H.; Jo, S.; Mikos, A.G. Biomimetic materials for tissue engineering. Biomaterials 2003, 24, 4353–4364. [Google Scholar] [CrossRef]
- Rezwan, K.; Chen, Q.Z.; Blaker, J.J.; Boccaccini, A.R. Biodegradable and bioactive porous polymer/inorganic composite scaffolds for bone tissue engineering. Biomaterials 2006, 27, 3413–3431. [Google Scholar] [CrossRef] [PubMed]
- Howard, D.; Buttery, L.D.; Shakesheff, K.M.; Roberts, S.J. Tissue engineering: Strategies, stem cells and scaffolds. J. Anat. 2008, 213, 66–72. [Google Scholar] [CrossRef]
- Barroca, N.; Marote, A.; Vieira, S.I.; Almeida, A.; Fernandes, M.H.V.; Vilarinho, P.M.; da Cruz e Silva, O.A.B. Electrically polarized PLLA nanofibers as neural tissue engineering scaffolds with improved neuritogenesis. Colloids Surf. B Biointerfaces 2018, 167, 93–103. [Google Scholar] [CrossRef]
- Correia, D.M.; Sencadas, V.; Ribeiro, C.; Martins, P.M.; Martins, P.; Gama, F.M.; Botelho, G.; Lanceros-Méndez, S. Processing and size range separation of pristine and magnetic poly(l-lactic acid) based microspheres for biomedical applications. J. Colloid Interface Sci. 2016, 476, 79–86. [Google Scholar] [CrossRef] [Green Version]
- Goonoo, N.; Bhaw-Luximon, A.; Passanha, P.; Esteves, S.R.; Jhurry, D. Third generation poly(hydroxyacid) composite scaffolds for tissue engineering. J. Biomed. Mater. Res. Part B Appl. Biomater. 2017, 105, 1667–1684. [Google Scholar] [CrossRef]
- Amaro, L.; Correia, D.M.; Marques-Almeida, T.; Martins, P.M.; Pérez, L.; Vilas, J.L.; Botelho, G.; Lanceros-Mendez, S.; Ribeiro, C. Tailored biodegradable and electroactive poly(Hydroxybutyrate-co-hydroxyvalerate) based morphologies for tissue engineering applications. Int. J. Mol. Sci. 2018, 19, 2149. [Google Scholar] [CrossRef] [Green Version]
- Gorodzha, S.N.; Muslimov, A.R.; Syromotina, D.S.; Timin, A.S.; Tcvetkov, N.Y.; Lepik, K.V.; Petrova, A.V.; Surmeneva, M.A.; Gorin, D.A.; Sukhorukov, G.B.; et al. A comparison study between electrospun polycaprolactone and piezoelectric poly(3-hydroxybutyrate-co-3-hydroxyvalerate) scaffolds for bone tissue engineering. Colloids Surf. B Biointerfaces 2017, 160, 48–59. [Google Scholar] [CrossRef]
- Ribeiro, C.; Correia, D.M.; Rodrigues, I.; Guardão, L.; Guimarães, S.; Soares, R.; Lanceros-Méndez, S. In vivo demonstration of the suitability of piezoelectric stimuli for bone reparation. Mater. Lett. 2017, 209, 118–121. [Google Scholar] [CrossRef]
- Hitscherich, P.; Wu, S.; Gordan, R.; Xie, L.H.; Arinzeh, T.; Lee, E.J. The effect of PVDF-TrFE scaffolds on stem cell derived cardiovascular cells. Biotechnol. Bioeng. 2016, 113, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, C.; Sencadas, V.; Correia, D.M.; Lanceros-Méndez, S. Piezoelectric polymers as biomaterials for tissue engineering applications. Colloids Surf. B Biointerfaces 2015, 136, 46–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribeiro, S.; Gomes, A.C.; Etxebarria, I.; Lanceros-Méndez, S.; Ribeiro, C. Electroactive biomaterial surface engineering effects on muscle cells differentiation. Mater. Sci. Eng. C 2018, 92, 868–874. [Google Scholar] [CrossRef]
- Martins, P.; Lopes, A.C.; Lanceros-Mendez, S. Electroactive phases of poly(vinylidene fluoride): Determination, processing and applications. Prog. Polym. Sci. 2014, 39, 683–706. [Google Scholar] [CrossRef]
- Fukada, E. Piezoelectricity in polymers and biological materials. Ultrasonics 1968, 6, 229–234. [Google Scholar] [CrossRef]
- Shamos, M.H.; Lavine, L.S. Piezoelectricity as a fundamental property of biological tissues. Nature 1967, 213, 267–269. [Google Scholar] [CrossRef]
- Martins, P.; Lanceros-Méndez, S. Polymer-based magnetoelectric materials: To be or not to be. Appl. Mater. Today 2019, 15, 558–561. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, G. Magnetoelectric Composites. Annu. Rev. Mater. Res. 2010, 40, 153–178. [Google Scholar] [CrossRef]
- Martins, P.; Lanceros-Méndez, S. Polymer-based magnetoelectric materials. Adv. Funct. Mater. 2013, 23, 3371–3385. [Google Scholar] [CrossRef]
- Cho, K.H.; Bichurin, M.I.; Petrov, V.M.; Bhalla, A.; Priya, S. Magnetoelectric laminate composite: Effect of piezoelectric layer on magnetoelectric properties. Ferroelectrics 2014, 473, 110–128. [Google Scholar] [CrossRef]
- Brito-Pereira, R.; Ribeiro, C.; Lanceros-Mendez, S.; Martins, P. Magnetoelectric response on Terfenol-D/P(VDF-TrFE) two-phase composites. Compos. Part B Eng. 2017, 120, 97–102. [Google Scholar] [CrossRef]
- Hermenegildo, B.; Ribeiro, C.; Pérez-Álvarez, L.; Vilas, J.L.; Learmonth, D.A.; Sousa, R.A.; Martins, P.; Lanceros-Méndez, S. Hydrogel-based magnetoelectric microenvironments for tissue stimulation. Colloids Surf. B Biointerfaces 2019, 181, 1041–1047. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, J.; Lasheras, A.; Martins, P.; Pereira, N.; Barandiarán, J.M.; Lanceros-Mendez, S. Metallic glass/PVDF magnetoelectric laminates for resonant sensors and actuators: A review. Sensors 2017, 17, 1251. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, C.; Correia, V.; Martins, P.; Gama, F.M.; Lanceros-Mendez, S. Proving the suitability of magnetoelectric stimuli for tissue engineering applications. Colloids Surf. B Biointerfaces 2016, 140, 430–436. [Google Scholar] [CrossRef] [Green Version]
- Brito-Pereira, R.; Correia, D.M.; Ribeiro, C.; Francesko, A.; Etxebarria, I.; Pérez-Álvarez, L.; Vilas, J.L.; Martins, P.; Lanceros-Mendez, S. Silk fibroin-magnetic hybrid composite electrospun fibers for tissue engineering applications. Compos. Part B Eng. 2018, 141, 70–75. [Google Scholar] [CrossRef]
- Freed, L.E.; Vunjak-Novakovic, G.; Biron, R.J.; Eagles, D.B.; Lesnoy, D.C.; Barlow, S.K.; Langer, R. Biodegradable polymer scaffolds for tissue engineering. Bio/Technol. 1994, 12, 689–693. [Google Scholar] [CrossRef]
- Ma, P.X. Biomimetic materials for tissue engineering. Adv. Drug Deliv. Rev. 2008, 60, 184–198. [Google Scholar] [CrossRef] [Green Version]
- Yamada, S.; Wang, Y.; Asakawa, N.; Yoshie, N.; Inoue, Y. Crystalline structural change of bacterial poly(3-hydroxybutyrate-co-3-hydroxyvalerate) with narrow compositional distribution. Macromolecules 2001, 34, 4659–4661. [Google Scholar] [CrossRef]
- Brunel, D.G.; Pachekoski, W.M.; Dalmolin, C.; Agnelli, J.A.M. Natural additives for poly (hydroxybutyrate -CO -hydroxyvalerate) -PHBV: Effect on mechanical properties and biodegradation. Mater. Res. 2014, 17, 1145–1156. [Google Scholar] [CrossRef] [Green Version]
- Xue, L.; Dai, S.; Li, Z. Biodegradable shape-memory block co-polymers for fast self-expandable stents. Biomaterials 2010, 31, 8132–8140. [Google Scholar] [CrossRef]
- Mutlu, G.; Calamak, S.; Ulubayram, K.; Guven, E. Curcumin-loaded electrospun PHBV nanofibers as potential wound-dressing material. J. Drug Deliv. Sci. Technol. 2018, 43, 185–193. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, X.; Wei, K.; Zhao, N.; Zhang, S.; Chen, J. Fabrication, characterization and long-term in vitro release of hydrophilic drug using PHBV/HA composite microspheres. Mater. Lett. 2007, 61, 1071–1076. [Google Scholar] [CrossRef]
- Tebaldi, M.L.; Maia, A.L.C.; Poletto, F.; de Andrade, F.V.; Soares, D.C.F. Poly(-3-hydroxybutyrate-co-3-hydroxyvalerate) (PHBV): Current advances in synthesis methodologies, antitumor applications and biocompatibility. J. Drug Deliv. Sci. Technol. 2019, 51, 115–126. [Google Scholar] [CrossRef]
- Hutmacher, D.W. Scaffolds in tissue engineering bone and cartilage. Biomaterials 2000, 21, 2529–2543. [Google Scholar] [CrossRef]
- Sultana, N.; Wang, M. PHBV/PLLA-based composite scaffolds fabricated using an emulsion freezing/freeze-drying technique for bone tissue engineering: Surface modification and in vitro biological evaluation. Biofabrication 2012, 4, 015003. [Google Scholar] [CrossRef]
- Smith, J.R.; Lamprou, D.A. Polymer coatings for biomedical applications: A review. Trans. IMF 2014, 92, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Jacob, J.; More, N.; Kalia, K.; Kapusetti, G. Piezoelectric smart biomaterials for bone and cartilage tissue engineering. Inflamm. Regen. 2018, 38, 2. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.Q.; Wu, Q. The application of polyhydroxyalkanoates as tissue engineering materials. Biomaterials 2005, 26, 6565–6578. [Google Scholar] [CrossRef]
- Huang, C.P.; Chen, X.M.; Chen, Z.Q. Biomimetic construction of poly(3-hydroxybutyrate-co-3-hydroxyvalerate)/apatite composite materials by an alternate incubation process. Mater. Lett. 2008, 62, 1499–1502. [Google Scholar] [CrossRef]
- Ragaert, P.; Buntinx, M.; Maes, C.; Vanheusden, C.; Peeters, R.; Wang, S.; D’Hooge, D.R.; Cardon, L. Polyhydroxyalkanoates for food packaging applications. In Reference Module in Food Science; Elsevier: Amsterdam, The Netherlands, 2019. [Google Scholar] [CrossRef]
- Ikada, Y.; Tsuji, H. Biodegradable polyesters for medical and ecological applications. Macromol. Rapid Commun. 2000, 21, 117–132. [Google Scholar] [CrossRef]
- Muthuraj, R.; Misra, M.; Mohanty, A.K. Biodegradable compatibilized polymer blends for packaging applications: A literature review. J. Appl. Polym. Sci. 2018, 135, 45726. [Google Scholar] [CrossRef] [Green Version]
- Correia, V.; Panadero, J.A.; Ribeiro, C.; Sencadas, V.; Rocha, J.G.; Gomez Ribelles, J.L.; Lanceros-Méndez, S. Design and validation of a biomechanical bioreactor for cartilage tissue culture. Biomech. Modeling Mechanobiol. 2016, 15, 471–478. [Google Scholar] [CrossRef]
- Wang, J.; Chu, L. Biological nitrate removal from water and wastewater by solid-phase denitrification process. Biotechnol. Adv. 2016, 34, 1103–1112. [Google Scholar] [CrossRef] [PubMed]
- O′Connor, D.; Hou, D.; Ok, Y.S.; Song, Y.; Sarmah, A.K.; Li, X.; Tack, F.M.G. Sustainable in situ remediation of recalcitrant organic pollutants in groundwater with controlled release materials: A review. J. Control. Release 2018, 283, 200–213. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, C.; Correia, D.M.; Ribeiro, S.; Fernandes, M.M.; Lanceros-Mendez, S. Piezo-and Magnetoelectric Polymers as Biomaterials for Novel Tissue Engineering Strategies. MRS Adv. 2018, 3, 1671–1676. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, M.; Correia, D.M.; Ribeiro, C.; Castro, N.; Correia, V.; Lanceros-Mendez, S. Bioinspired three-dimensional magneto-active scaffolds for bone tissue engineering. ACS Appl. Mater. Interfaces 2019. [Google Scholar] [CrossRef] [PubMed]
- Oyane, A.; Kim, H.M.; Furuya, T.; Kokubo, T.; Miyazaki, T.; Nakamura, T. Preparation and assessment of revised simulated body fluids. J. Biomed. Mater. Res. Part A 2003, 65, 188–195. [Google Scholar] [CrossRef]
- Fischer, D.; Li, Y.; Ahlemeyer, B.; Krieglstein, J.; Kissel, T. In vitro cytotoxicity testing of polycations: Influence of polymer structure on cell viability and hemolysis. Biomaterials 2003, 24, 1121–1131. [Google Scholar] [CrossRef]
- Leimann, F.V.; Biz, M.H.; Musyanovych, A.; Sayer, C.; Landfester, K.; Hermes de Araújo, P.H. Hydrolysis of poly(hydroxybutyrate-co-hydroxyvalerate) nanoparticles. J. Appl. Polym. Sci. 2013, 128, 3093–3098. [Google Scholar] [CrossRef]
- Jack, K.S.; Velayudhan, S.; Luckman, P.; Trau, M.; Grøndahl, L.; Cooper-White, J. The fabrication and characterization of biodegradable HA/PHBV nanoparticle-polymer composite scaffolds. Acta Biomater. 2009, 5, 2657–2667. [Google Scholar] [CrossRef]
- Yu, H.Y.; Qin, Z.Y.; Zhou, Z. Cellulose nanocrystals as green fillers to improve crystallization and hydrophilic property of poly(3-hydroxybutyrate-co-3-hydroxyvalerate). Prog. Nat. Sci. Mater. Int. 2011, 21, 478–484. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.C.; Masuoka, T. Degradation properties of PLA and PHBV films treated with CO2-plasma. React. Funct. Polym. 2009, 69, 287–292. [Google Scholar] [CrossRef]
- Wang, Y.J.; Lu, L.; Zheng, Y.D.; Chen, X.F. Improvement in hydrophilicity of PHBV films by plasma treatment. J. Biomed. Mater. Res. Part A 2006, 76A, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, B.M.P.; Pinheiro, L.M.P.; Nascente, P.A.P.; Ferreira, M.J.; Duek, E.A.R. Plasma surface treatments of poly(l-lactic acid) (PLLA) and poly(hydroxybutyrate-co-hydroxyvalerate) (PHBV). Mater. Sci. Eng. C 2009, 29, 806–813. [Google Scholar] [CrossRef]
- Abdalkarim, S.Y.H.; Yu, H.-Y.; Song, M.-L.; Zhou, Y.; Yao, J.; Ni, Q.-Q. In vitro degradation and possible hydrolytic mechanism of PHBV nanocomposites by incorporating cellulose nanocrystal-ZnO nanohybrids. Carbohydr. Polym. 2017, 176, 38–49. [Google Scholar] [CrossRef]
- Prabhakar, P.K.; Vijayaraghavan, S.; Philip, J.; Doble, M. Biocompatibility studies of functionalized CoFe2O4 magnetic nanoparticles. Curr. Nanosci. 2011, 7, 371–376. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Tm (°C) | ΔHm (J/g) | XC (%) | |
|---|---|---|---|---|
| Films | PHBV | 174 | 82 | 56 |
| PHBV/CFO | 177 | 70 | 48 | |
| PHBV (Degraded) | 173 | 40 | 27 | |
| PHBV/CFO (Degraded) | 173 | 66 | 45 | |
| Fibers | PHBV | 175 | 98 | 67 |
| PHBV/CFO | 177 | 67 | 46 | |
| PHBV (Degraded) | 171 | 49 | 33 | |
| PHBV/CFO (Degraded) | 172 | 66 | 45 | |
| Scaffolds | PHBV (Degraded) | 176 | 45 | 31 |
| PHBV/CFO (Degraded) | 173 | 37 | 25 |
| Samples | Elemental Composition (%) | |||
|---|---|---|---|---|
| C | O | O/C | ||
| Non-degraded | PHBV film | 69.4 | 30.6 | 0.44 |
| PHBV/CFO film | 71.9 | 28.1 | 0.39 | |
| PHBV fibers | 70.3 | 29.7 | 0.42 | |
| PHBV/CFO fibers | 69.0 | 31.0 | 0.45 | |
| PHBV scaffolds | 73.2 | 26.8 | 0.37 | |
| PHBV/CFO scaffolds | 73.3 | 26.7 | 0.36 | |
| Degraded | PHBV film | 66.8 | 33.3 | 0.49 |
| PHBV/CFO film | 63.7 | 36.0 | 0.56 | |
| PHBV fibers | 69.3 | 30.7 | 0.44 | |
| PHBV/CFO fibers | 66.5 | 33.4 | 0.50 | |
| PHBV scaffolds | 63.2 | 36.8 | 0.58 | |
| PHBV/CFO scaffolds | 68.3 | 31.7 | 0.46 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amaro, L.; Correia, D.M.; Martins, P.M.; Botelho, G.; Carabineiro, S.A.C.; Ribeiro, C.; Lanceros-Mendez, S. Morphology Dependence Degradation of Electro- and Magnetoactive Poly(3-hydroxybutyrate-co-hydroxyvalerate) for Tissue Engineering Applications. Polymers 2020, 12, 953. https://doi.org/10.3390/polym12040953
Amaro L, Correia DM, Martins PM, Botelho G, Carabineiro SAC, Ribeiro C, Lanceros-Mendez S. Morphology Dependence Degradation of Electro- and Magnetoactive Poly(3-hydroxybutyrate-co-hydroxyvalerate) for Tissue Engineering Applications. Polymers. 2020; 12(4):953. https://doi.org/10.3390/polym12040953
Chicago/Turabian StyleAmaro, Luis, Daniela M. Correia, Pedro M. Martins, Gabriela Botelho, Sónia A. C. Carabineiro, Clarisse Ribeiro, and Senentxu Lanceros-Mendez. 2020. "Morphology Dependence Degradation of Electro- and Magnetoactive Poly(3-hydroxybutyrate-co-hydroxyvalerate) for Tissue Engineering Applications" Polymers 12, no. 4: 953. https://doi.org/10.3390/polym12040953