
Drug Amorphous Solid Dispersions Based on Poly(vinyl Alcohol): Evaluating the Effect of Poly(propylene Succinate) as Plasticizer

 ,
,  ,
,  and
and
Abstract
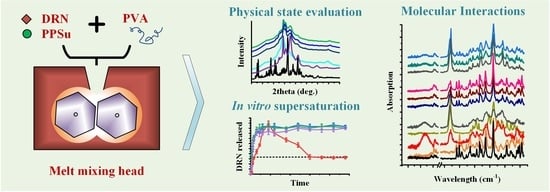
:
1. Introduction
2. Materials and Methods
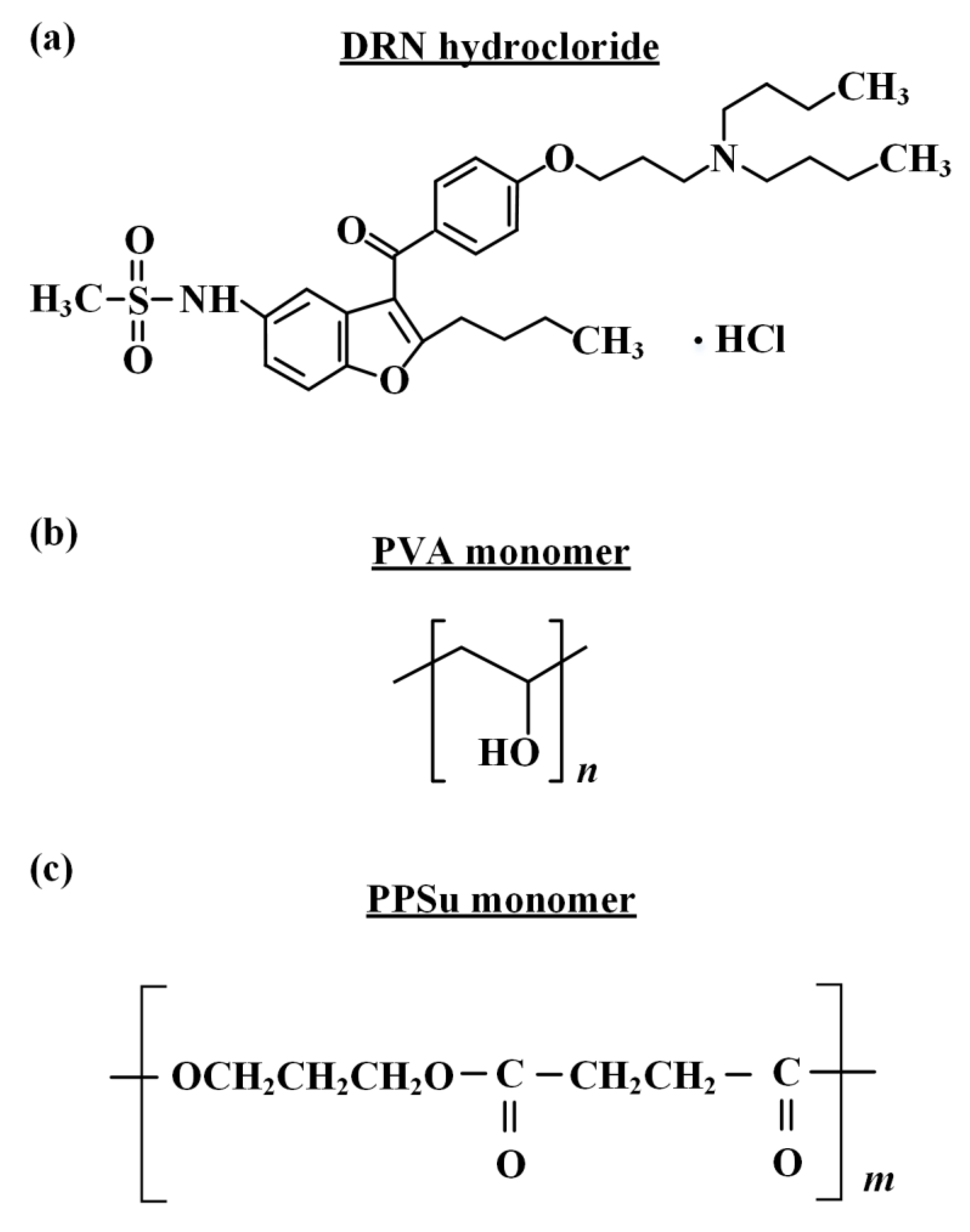
2.1. Materials
2.2. Preparation of PPSu
2.3. Preparation of ASDs
2.4. Hot Stage Polarized Microscopy (HSM)
2.5. Differential Scanning Calorimetry (DSC)
2.6. Thermogravimetric Analysis (TGA)
2.7. Powder X-ray Diffractometry (pXRD)
2.8. Attenuated Total Reflection Fourier Transform Infrared (ATR-FTIR) Spectroscopy
2.9. Supersaturation Dissolution Studies
2.10. Stability Studies
3. Results and Discussion
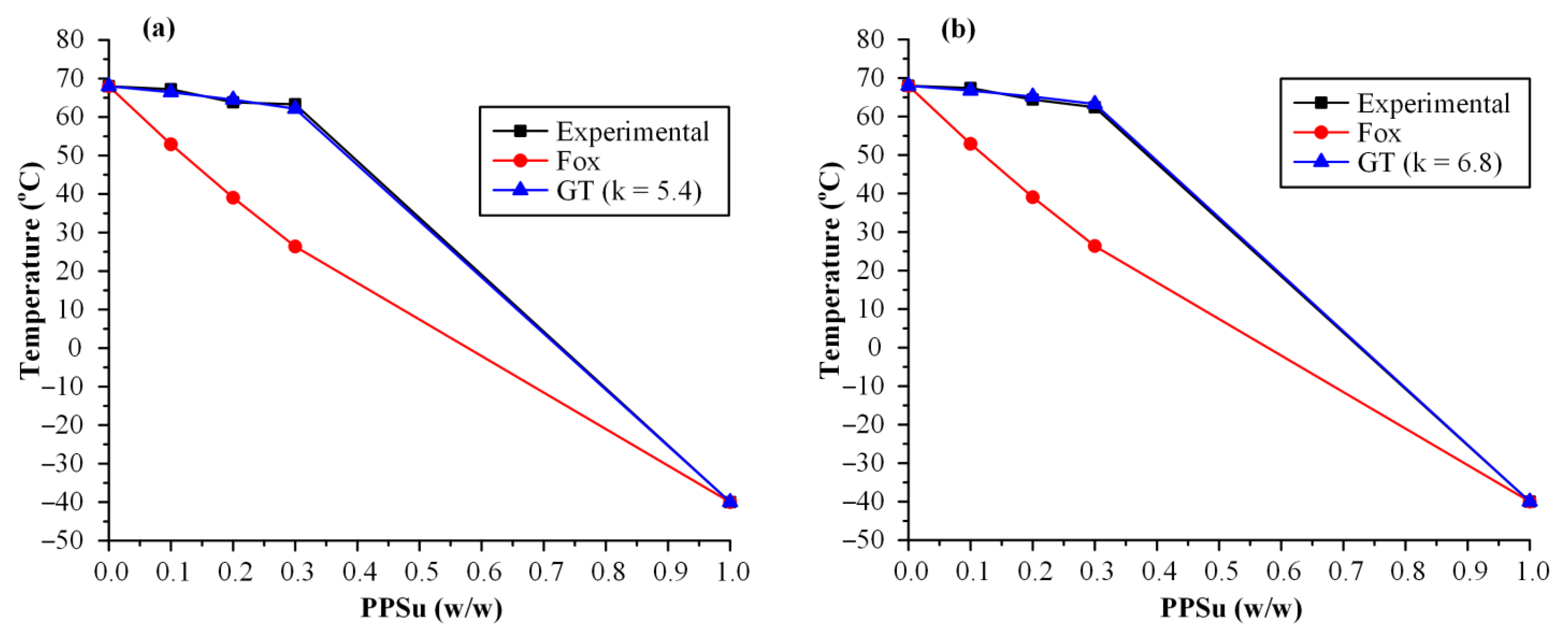
3.1. Evaluation of Melt-Mixing Time’s Effect on the Neat PVA-PPSu Matrices
3.2. DRN-Loaded PVA-PPSu ASDs
3.2.1. Thermal Stability via TGA
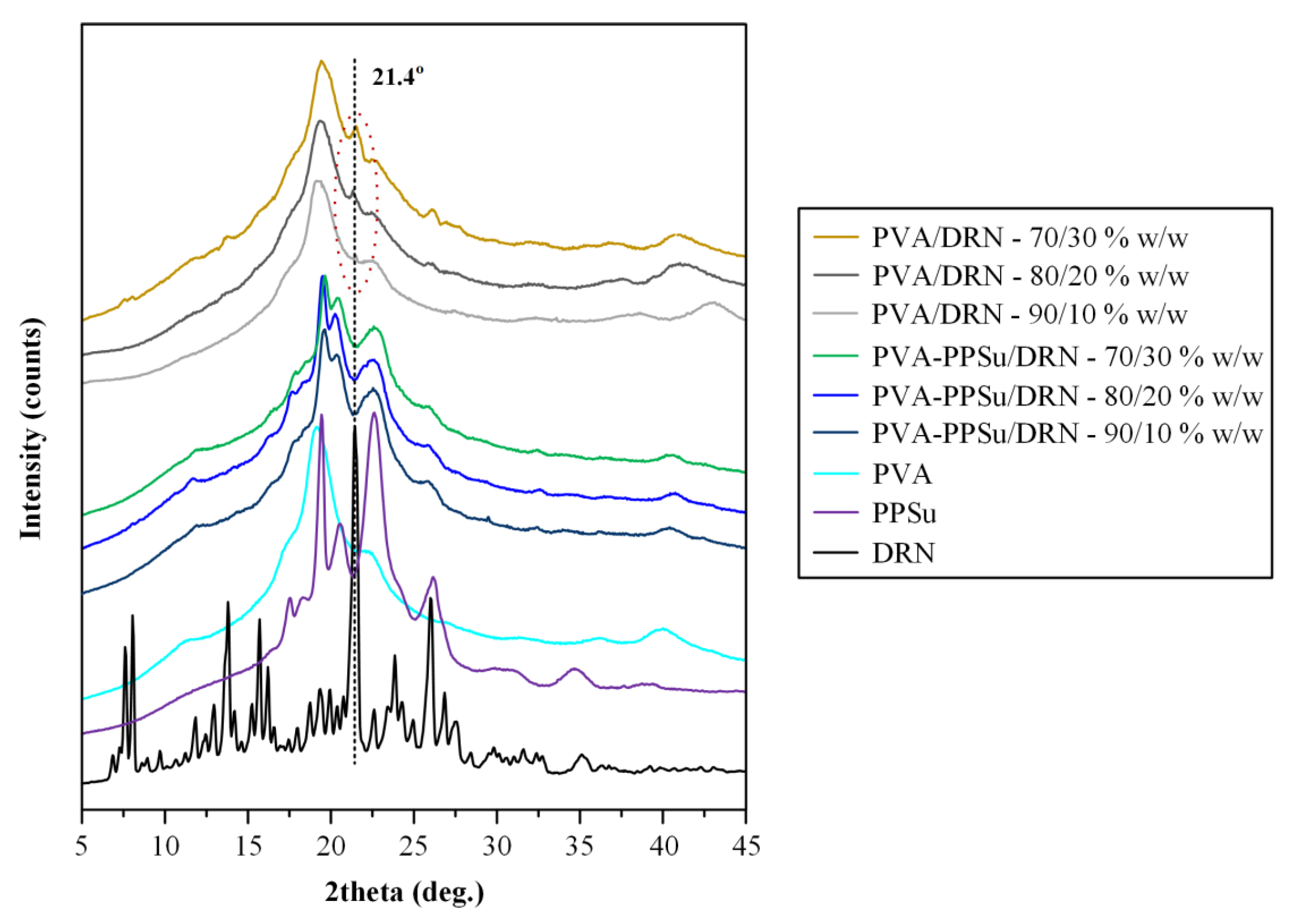
3.2.2. Physical State Evaluation via pXRD
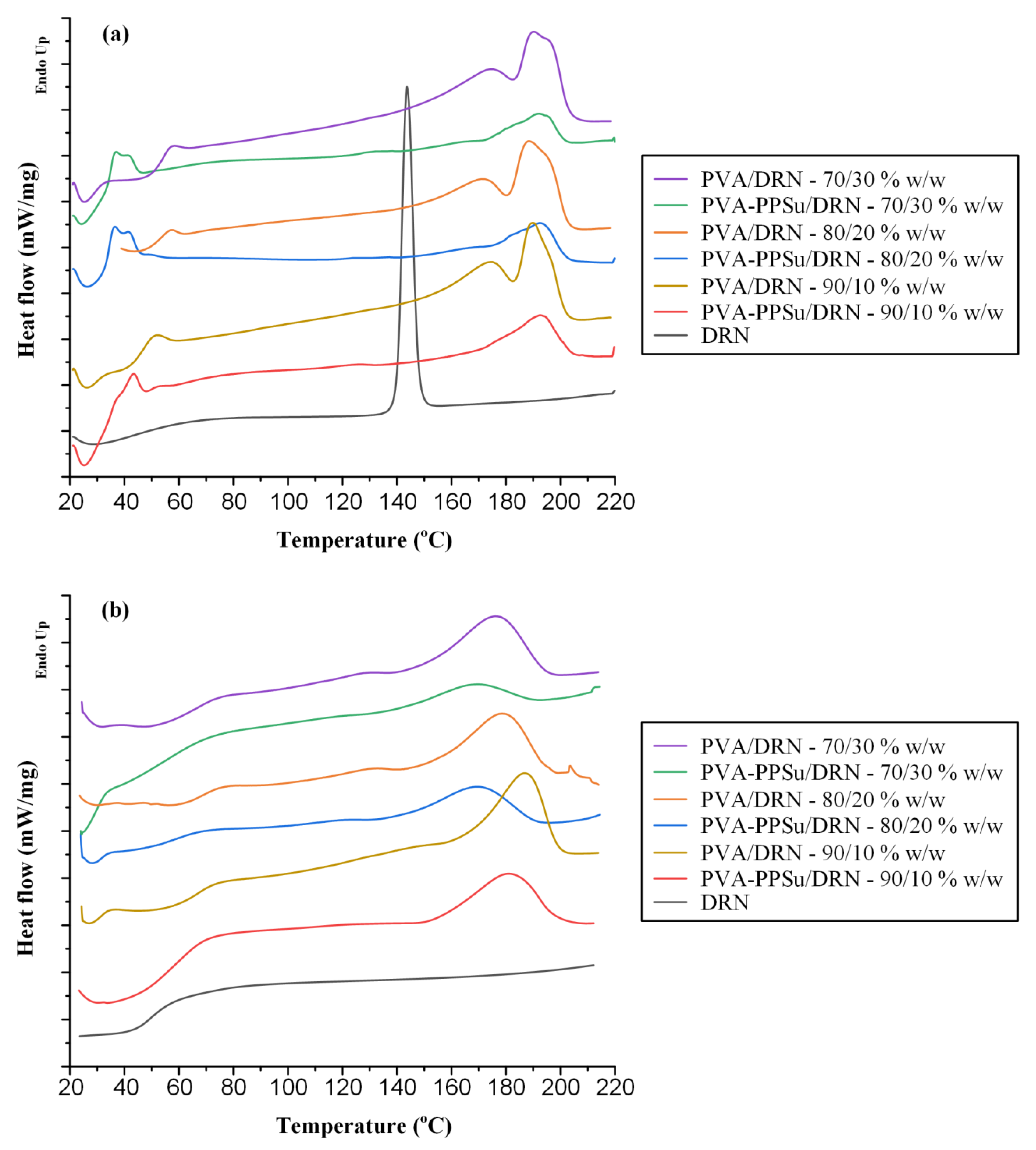
3.2.3. Evaluation of Thermal Properties
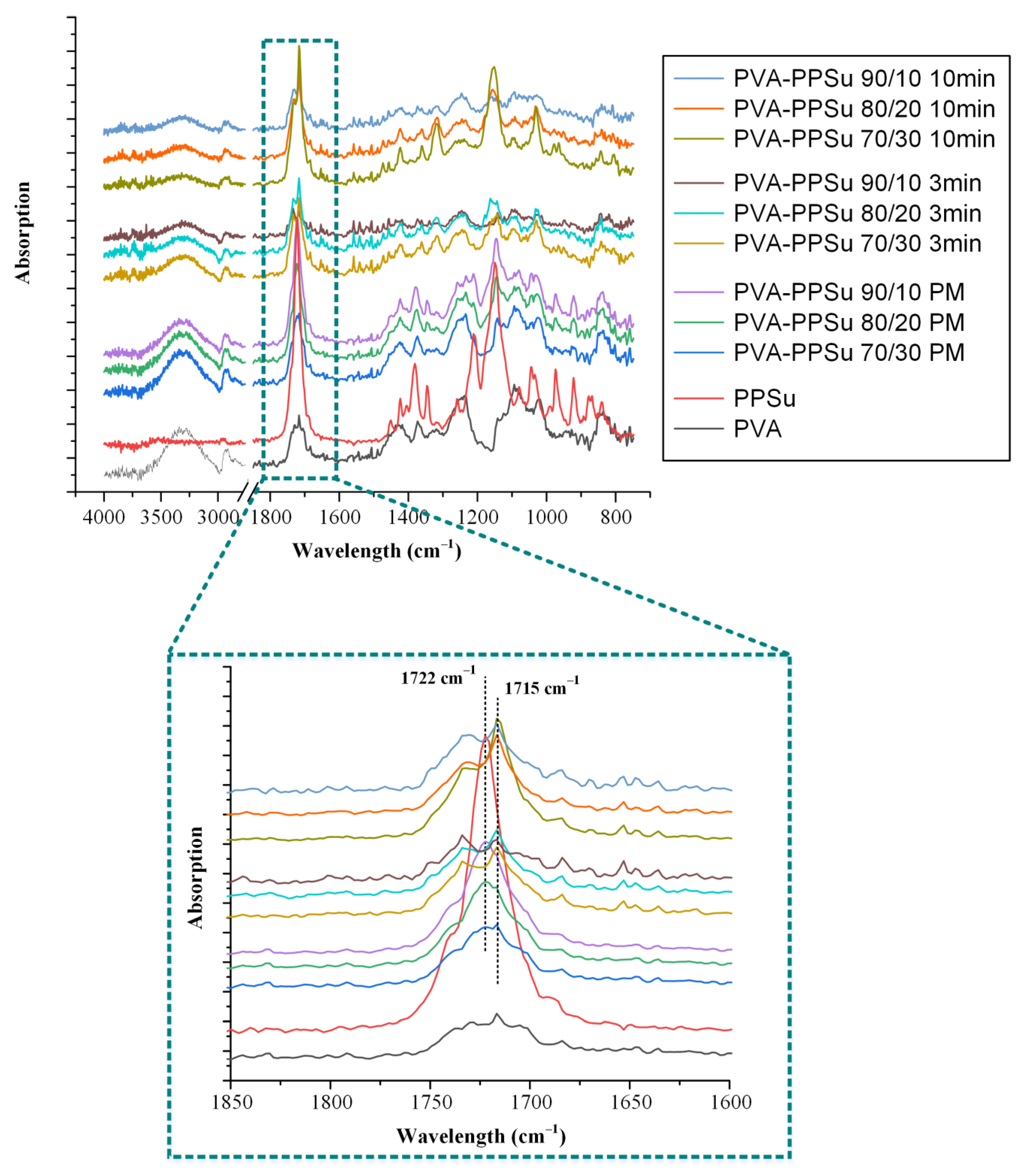
3.2.4. Evaluation of Molecular Interactions
3.2.5. Evaluation of Supersaturated Dissolution Studies
3.2.6. Storage Stability Results
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Teixeira, M.A.; Amorim, M.T.P.; Felgueiras, H.P. Poly(vinyl alcohol)-based nanofibrous electrospun scaffolds for tissue engineering applications. Polymers 2019, 12, 7. [Google Scholar] [CrossRef] [Green Version]
- Teodorescu, M.; Bercea, M.; Morariu, S. Biomaterials of pva and pvp in medical and pharmaceutical applications: Perspectives and challenges. Biotechnol. Adv. 2019, 37, 109–131. [Google Scholar] [CrossRef]
- Suzuki, A.; Sasaki, S. Swelling and mechanical properties of physically crosslinked poly(vinyl alcohol) hydrogels. Proc. Inst. Mech. Eng. Part H J. Eng. Med. 2015, 229, 828–844. [Google Scholar] [CrossRef] [PubMed]
- LaFountaine, J.S.; Jermain, S.V.; Prasad, L.K.; Brough, C.; Miller, D.A.; Lubda, D.; McGinity, J.W.; Williams, R.O. Enabling thermal processing of ritonavir–polyvinyl alcohol amorphous solid dispersions by kinetisol® dispersing. Eur. J. Pharm. Biopharm. 2016, 101, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Mori, Y.; Takahashi, M.; Ohno, Y.; Okura, R.; Ishida, M.; Higashi, T.; Motoyama, K.; Arima, H. Identification of molecular-interaction sites between lowly hydrolyzed polyvinyl alcohols and indomethacin by nmr spectroscopy. Int. J. Pharm. 2018, 549, 456–465. [Google Scholar] [CrossRef] [PubMed]
- Brough, C.; Miller, D.A.; Keen, J.M.; Kucera, S.A.; Lubda, D.; Williams, R.O. Use of polyvinyl alcohol as a solubility-enhancing polymer for poorly water soluble drug delivery (part 1). AAPS PharmSciTech 2016, 17, 167–179. [Google Scholar] [CrossRef] [Green Version]
- Aslam, M.; Kalyar, M.A.; Raza, Z.A. Polyvinyl alcohol: A review of research status and use of polyvinyl alcohol based nanocomposites. Polym. Eng. Sci. 2018, 58, 2119–2132. [Google Scholar] [CrossRef]
- DeMerlis, C.C.; Schoneker, D.R. Review of the oral toxicity of polyvinyl alcohol (pva). Food Chem. Toxicol. 2003, 41, 319–326. [Google Scholar] [CrossRef]
- Kelly, C.M.; DeMerlis, C.C.; Schoneker, D.R.; Borzelleca, J.F. Subchronic toxicity study in rats and genotoxicity tests with polyvinyl alcohol. Food Chem. Toxicol. 2003, 41, 719–727. [Google Scholar] [CrossRef]
- Wong, D.; Parasrampuria, J. Polyvinyl alcohol. In Analytical Profiles of Drug Substances and Excipients; Brittain, H.G., Ed.; Academic Press: Cambridge, MA, USA, 1996; Volume 24, pp. 397–441. [Google Scholar]
- Alexy, P.; Káchová, D.; Kršiak, M.; Bakoš, D.; Šimková, B. Poly(vinyl alcohol) stabilisation in thermoplastic processing. Polym. Degrad. Stab. 2002, 78, 413–421. [Google Scholar] [CrossRef]
- Tang, X.; Alavi, S. Recent advances in starch, polyvinyl alcohol based polymer blends, nanocomposites and their biodegradability. Carbohydr. Polym. 2011, 85, 7–16. [Google Scholar] [CrossRef]
- Macedo, J.; Samaro, A.; Vanhoorne, V.; Vervaet, C.; Pinto, J.F. Processability of poly(vinyl alcohol) based filaments with paracetamol prepared by hot-melt extrusion for additive manufacturing. J. Pharm. Sci. 2020, 109, 3636–3644. [Google Scholar] [CrossRef]
- Wlodarski, K.; Zhang, F.; Liu, T.; Sawicki, W.; Kipping, T. Synergistic effect of polyvinyl alcohol and copovidone in itraconazole amorphous solid dispersions. Pharm. Res. 2018, 35, 16. [Google Scholar] [CrossRef]
- Baghel, S.; Cathcart, H.; O’Reilly, N.J. Polymeric amorphous solid dispersions: A review of amorphization, crystallization, stabilization, solid-state characterization, and aqueous solubilization of biopharmaceutical classification system class ii drugs. J. Pharm. Sci. 2016, 105, 2527–2544. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Williams, R.O. Effects of the preparation process on the properties of amorphous solid dispersions. AAPS PharmSciTech 2018, 19, 1971–1984. [Google Scholar] [CrossRef]
- Kapourani, A.; Chatzitheodoridou, M.; Kontogiannopoulos, K.N.; Barmpalexis, P. Experimental, thermodynamic, and molecular modeling evaluation of amorphous simvastatin-poly(vinylpyrrolidone) solid dispersions. Mol. Pharm. 2020, 17, 2703–2720. [Google Scholar] [CrossRef]
- Kapourani, A.; Eleftheriadou, K.; Kontogiannopoulos, K.N.; Barmpalexis, P. Evaluation of rivaroxaban amorphous solid dispersions physical stability via molecular mobility studies and molecular simulations. Eur. J. Pharm. Sci. 2021, 157, 105642. [Google Scholar] [CrossRef]
- Meere, M.; Pontrelli, G.; McGinty, S. Modelling phase separation in amorphous solid dispersions. Acta Biomater. 2019, 94, 410–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palazi, E.; Karavas, E.; Barmpalexis, P.; Kostoglou, M.; Nanaki, S.; Christodoulou, E.; Bikiaris, D.N. Melt extrusion process for adjusting drug release of poorly water soluble drug felodipine using different polymer matrices. Eur. J. Pharm. Sci. 2018, 114, 332–345. [Google Scholar] [CrossRef] [PubMed]
- Medarević, D.; Djuriš, J.; Barmpalexis, P.; Kachrimanis, K.; Ibrić, S. Analytical and computational methods for the estimation of drug-polymer solubility and miscibility in solid dispersions development. Pharmaceutics 2019, 11, 372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellenberger, D.J.; Miller, D.A.; Kucera, S.U.; Williams, R.O. Improved vemurafenib dissolution and pharmacokinetics as an amorphous solid dispersion produced by kinetisol® processing. AAPS PharmSciTech 2018, 19, 1957–1970. [Google Scholar] [CrossRef] [PubMed]
- Lang, B.; McGinity, J.W.; Williams, R.O. Hot-melt extrusion—basic principles and pharmaceutical applications. Drug Dev. Ind. Pharm. 2014, 40, 1133–1155. [Google Scholar] [CrossRef] [PubMed]
- Sarabu, S.; Bandari, S.; Kallakunta, V.R.; Tiwari, R.; Patil, H.; Repka, M.A. An update on the contribution of hot-melt extrusion technology to novel drug delivery in the twenty-first century: Part II. Expert Opin. Drug Deliv. 2019, 16, 567–582. [Google Scholar] [CrossRef] [PubMed]
- Simões, M.F.; Pinto, R.M.A.; Simões, S. Hot-melt extrusion in the pharmaceutical industry: Toward filing a new drug application. Drug Discov. Today 2019, 24, 1749–1768. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.K.; Maniruzzaman, M.; Nokhodchi, A. Advanced pharmaceutical applications of hot-melt extrusion coupled with fused deposition modelling (fdm) 3d printing for personalised drug delivery. Pharmaceutics 2018, 10, 203. [Google Scholar] [CrossRef] [Green Version]
- Monschke, M.; Wagner, K.G. Impact of hpmcas on the dissolution performance of polyvinyl alcohol celecoxib amorphous solid dispersions. Pharmaceutics 2020, 12, 541. [Google Scholar] [CrossRef]
- Wu, W.; Tian, H.; Xiang, A. Influence of polyol plasticizers on the properties of polyvinyl alcohol films fabricated by melt processing. J. Polym. Environ. 2012, 20, 63–69. [Google Scholar] [CrossRef]
- Almeida, A.; Claeys, B.; Remon, J.P.; Vervaet, C. Hot-melt extrusion developments in the pharmaceutical industry. In Hot-Melt Extrusion: Pharmaceutical Applications; Douroumis, D., Ed.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2012; pp. 43–69. [Google Scholar]
- Katopodis, K.; Kapourani, A.; Vardaka, E.; Karagianni, A.; Chorianopoulou, C.; Kontogiannopoulos, K.N.; Bikiaris, D.N.; Kachrimanis, K.; Barmpalexis, P. Partially hydrolyzed polyvinyl alcohol for fusion-based pharmaceutical formulation processes: Evaluation of suitable plasticizers. Int. J. Pharm. 2020, 578, 119121. [Google Scholar] [CrossRef]
- Palamidi, A.; Kapourani, A.; Christodoulou, E.; Klonos, P.A.; Kontogiannopoulos, K.N.; Kyritsis, A.; Bikiaris, D.N.; Barmpalexis, P. Low molecular weight oligomers of poly(alkylene succinate) polyesters as plasticizers in poly(vinyl alcohol) based pharmaceutical applications. Polymers 2021, 13, 146. [Google Scholar] [CrossRef]
- Hohnloser, S.H.; Crijns, H.J.G.M.; van Eickels, M.; Gaudin, C.; Page, R.L.; Torp-Pedersen, C.; Connolly, S.J. Effect of dronedarone on cardiovascular events in atrial fibrillation. N. Engl. J. Med. 2009, 360, 668–678. [Google Scholar] [CrossRef] [Green Version]
- LE HEUZEY, J.-Y.; De Ferrari, G.M.; Radzik, D.; Santini, M.; Zhu, J.; Davy, J.-M. A short-term, randomized, double-blind, parallel-group study to evaluate the efficacy and safety of dronedarone versus amiodarone in patients with persistent atrial fibrillation: The dionysos study. J. Cardiovasc. Electrophysiol. 2010, 21, 597–605. [Google Scholar] [CrossRef]
- Sun, W.; Sarma, J.S.M.; Singh, B.N. Chronic and acute effects of dronedarone on the action potential of rabbit atrial muscle preparations: Comparison with amiodarone. J. Cardiovasc. Pharmacol. 2002, 39, 677–684. [Google Scholar] [CrossRef]
- Marcolino, A.I.P.; Macedo, L.B.; Nogueira-Librelotto, D.R.; Fernandes, J.R.; Bender, C.R.; Wust, K.M.; Frizzo, C.P.; Mitjans, M.; Vinardell, M.P.; Rolim, C.M.B. Preparation, characterization and in vitro cytotoxicity study of dronedarone hydrochloride inclusion complexes. Mater. Sci. Eng. C 2019, 100, 48–61. [Google Scholar] [CrossRef]
- Sun, D.D.; Ju, T.-c.R.; Lee, P.I. Enhanced kinetic solubility profiles of indomethacin amorphous solid dispersions in poly(2-hydroxyethyl methacrylate) hydrogels. Eur. J. Pharm. Biopharm. 2012, 81, 149–158. [Google Scholar] [CrossRef]
- Newman, A.; Zografi, G. Commentary: Considerations in the measurement of glass transition temperatures of pharmaceutical amorphous solids. AAPS PharmSciTech 2019, 21, 26. [Google Scholar] [CrossRef] [Green Version]
- Eguiazabal, J.I.; Iruin, J.J.; Cortazar, M.; Guzmán, G.M. Glass transition temperatures in blends of poly(n-vinyl-2-pyrrolidone) with a copolymer of bisphenol a and epichlorohydrin or with poly(vinyl butyral). Die Makromol. Chem. 1984, 185, 1761–1766. [Google Scholar] [CrossRef]
- Papageorgiou, G.Z.; Achilias, D.S.; Nanaki, S.; Beslikas, T.; Bikiaris, D. Pla nanocomposites: Effect of filler type on non-isothermal crystallization. Thermochim. Acta 2010, 511, 129–139. [Google Scholar] [CrossRef]
- Marom, E.; Rubnov, S. Amorphous Form of Dronedarone. WO2012001673A1, 05 January 2012. [Google Scholar]
- Baird, J.A.; Van Eerdenbrugh, B.; Taylor, L.S. A classification system to assess the crystallization tendency of organic molecules from undercooled melts. J. Pharm. Sci. 2010, 99, 3787–3806. [Google Scholar] [CrossRef]
- Kapourani, A.; Tzakri, T.; Valkanioti, V.; Kontogiannopoulos, K.N.; Barmpalexis, P. Drug crystal growth in ternary amorphous solid dispersions: Effect of surfactants and polymeric matrix-carriers. Int. J. Pharm. X 2021, 3, 100086. [Google Scholar] [PubMed]
- Kapourani, A.; Valkanioti, V.; Kontogiannopoulos, K.N.; Barmpalexis, P. Determination of the physical state of a drug in amorphous solid dispersions using artificial neural networks and atr-ftir spectroscopy. Int. J. Pharm. X 2020, 2, 100064. [Google Scholar] [PubMed]
- Sarode, A.L.; Sandhu, H.; Shah, N.; Malick, W.; Zia, H. Hot melt extrusion (hme) for amorphous solid dispersions: Predictive tools for processing and impact of drug–polymer interactions on supersaturation. Eur. J. Pharm. Sci. 2013, 48, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.T.D.; Tran, P.H.L. Molecular interactions in solid dispersions of poorly water-soluble drugs. Pharmaceutics 2020, 12, 745. [Google Scholar] [CrossRef]
- Van Nguyen, H.; Baek, N.; Lee, B.J. Enhanced gastric stability of esomeprazole by molecular interaction and modulation of microenvironmental ph with alkalizers in solid dispersion. Int. J. Pharm. 2017, 523, 189–202. [Google Scholar] [CrossRef]
- Dimitrios, B.; Vassilios, K.; Evangelos, K. Effectiveness of various drug carriers in controlled release formulations of raloxifene hcl prepared by melt mixing. Curr. Drug Deliv. 2009, 6, 425–436. [Google Scholar]
- Bevernage, J.; Brouwers, J.; Brewster, M.E.; Augustijns, P. Evaluation of gastrointestinal drug supersaturation and precipitation: Strategies and issues. Int. J. Pharm. 2013, 453, 25–35. [Google Scholar] [CrossRef]
- Sun, D.D.; Lee, P.I. Haste makes waste: The interplay between dissolution and precipitation of supersaturating formulations. AAPS J. 2015, 17, 1317–1326. [Google Scholar] [CrossRef] [Green Version]
- Augustijns, P.; Brewster, M.E. Supersaturating drug delivery systems: Fast is not necessarily good enough. J. Pharm. Sci. 2012, 101, 7–9. [Google Scholar] [CrossRef]
- Brouwers, J.; Brewster, M.E.; Augustijns, P. Supersaturating drug delivery systems: The answer to solubility-limited oral bioavailability? J. Pharm. Sci. 2009, 98, 2549–2572. [Google Scholar] [CrossRef] [PubMed]
- Han, S.D.; Jung, S.W.; Jang, S.W.; Jung, H.J.; Son, M.; Kim, B.M.; Kang, M.J. Preparation of solid dispersion of dronedarone hydrochloride with soluplus® by hot melt extrusion technique for enhanced drug release. Chem. Pharm. Bull. 2015, 63, 295–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alhalaweh, A.; Alzghoul, A.; Mahlin, D.; Bergström, C.A.S. Physical stability of drugs after storage above and below the glass transition temperature: Relationship to glass-forming ability. Int. J. Pharm. 2015, 495, 312–317. [Google Scholar] [CrossRef] [PubMed]
- Kapourani, A.; Vardaka, E.; Katopodis, K.; Kachrimanis, K.; Barmpalexis, P. Rivaroxaban polymeric amorphous solid dispersions: Moisture-induced thermodynamic phase behavior and intermolecular interactions. Eur. J. Pharm. Biopharm. 2019, 145, 98–112. [Google Scholar] [CrossRef] [PubMed]
- Rumondor, A.C.F.; Marsac, P.J.; Stanford, L.A.; Taylor, L.S. Phase behavior of poly(vinylpyrrolidone) containing amorphous solid dispersions in the presence of moisture. Mol. Pharm. 2009, 6, 1492–1505. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | DSC-Based Thermal Events | ||||||
|---|---|---|---|---|---|---|---|
| 1st Heating Scan | 2nd Heating Scan | ||||||
| PPSu | PVA | ||||||
| Tm,onset (°C) | ΔHf (J/g) | Tm,onset (°C) | ΔHf (J/g) | Tg (°C) | Tm,onset ** (°C) | ΔHf (J/g) | |
| PVA | - | - | 171.1 | 42.5 | 68.4 | 140.1 | 30.8 |
| PPSu | 45.3 | 51.9 | - | - | * | - | - |
| PVA/PPSu 90/10—3 min | - | - | 149.5 | 41.4 | 67.2 | 135.2 | 28.9 |
| PVA/PPSu 80/20—3 min | 36.9 | 9.8 | 123.8 | 39.7 | 63.8 | 132.1 | 38.5 |
| PVA/PPSu 70/30—3 min | 42.1 | 10.2 | 112.7 | 28.7 | 63.3 | 125.0 | 21.3 |
| PVA/PPSu 90/10—10 min | - | - | 119.3 | 23.4 | 67.4 | 128.8 | 14.7 |
| PVA/PPSu 80/20—10 min | - | - | 114.2 | 18.0 | 64.4 | 98.3 | 19.8 |
| PVA/PPSu 70/30—10 min | 36.0 | 6.9 | 113.7 | 14.6 | 62.4 | 89.8 | 17.9 |
| Samples | DSC-Based Thermal Events | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1st Heating Scan | 2nd Heating Scan | ||||||||
| PPSu | PVA | DRN | |||||||
| Tm,onset (°C) | ΔHf (J/g) | Tm,onset (°C) | ΔHf (J/g) | Tm,onset (°C) | ΔHf (J/g) | Tg (°C) | Tm,onset * (°C) | ΔHf (J/g) | |
| DRN | - | - | - | - | 141.6 | 92.0 | 49.5 | - | - |
| PVA/DRN 90/10 | - | - | # | # | 141.9 | # | 65.8 | 152.5 | 21.6 |
| PVA/DRN 80/20 | - | - | # | # | 142.0 | # | 65.5 | 140.9 | 19.3 |
| PVA/DRN 70/30 | - | - | # | # | 142.6 | # | 64.7 | 137.6 | 17.9 |
| PVA-PPSu/DRN 90/10 | 38.4 | 8.4 | 133.5 | 23.1 | - | - | 55.7 | 142.5 | 16.7 |
| PVA-PPSu/DRN 80/20 | 26.0 | 9.5 | 130.9 | 21.8 | - | - | 54.8 | 130.7 | 12.6 |
| PVA-PPSu/DRN 70/30 | 24.9 | 11.7 | 129.5 | 15.3 | - | - | 54.5 | 119.5 | 9.9 |
| Sample ID | AUC(0 -> t) (ppm min × 103) (Mean ± SD) | AUC(0 -> t) Ratio (Mean) |
|---|---|---|
| DRN form I crystals | 2.21 ± 0.05 | 1.00 |
| DRN amorphous | 4.94 ± 0.21 | 2.24 |
| PVA-PPSu/DRN 90/10 | 8.74 ± 0.28 | 3.96 |
| PVA-PPSu/DRN 80/20 | 8.60 ± 0.36 | 3.90 |
| PVA-PPSu/DRN 70/30 | 8.04 ± 0.40 | 3.64 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kapourani, A.; Palamidi, A.; Kontogiannopoulos, K.N.; Bikiaris, N.D.; Barmpalexis, P. Drug Amorphous Solid Dispersions Based on Poly(vinyl Alcohol): Evaluating the Effect of Poly(propylene Succinate) as Plasticizer. Polymers 2021, 13, 2922. https://doi.org/10.3390/polym13172922
Kapourani A, Palamidi A, Kontogiannopoulos KN, Bikiaris ND, Barmpalexis P. Drug Amorphous Solid Dispersions Based on Poly(vinyl Alcohol): Evaluating the Effect of Poly(propylene Succinate) as Plasticizer. Polymers. 2021; 13(17):2922. https://doi.org/10.3390/polym13172922
Chicago/Turabian StyleKapourani, Afroditi, Artemis Palamidi, Konstantinos N. Kontogiannopoulos, Nikolaos D. Bikiaris, and Panagiotis Barmpalexis. 2021. "Drug Amorphous Solid Dispersions Based on Poly(vinyl Alcohol): Evaluating the Effect of Poly(propylene Succinate) as Plasticizer" Polymers 13, no. 17: 2922. https://doi.org/10.3390/polym13172922