
Current Advances of Nitric Oxide in Cancer and Anticancer Therapeutics
by
 , , ,
, , ,
Joel Mintz
1,†, 
Anastasia Vedenko
2,†,
Omar Rosete
3 ,
,
Khushi Shah
4,
Gabriella Goldstein
5 ,
,
Joshua M. Hare
2,6,7,
Ranjith Ramasamy
3,6,* and
Himanshu Arora
2,3,6,* 1
Dr. Kiran C. Patel College of Allopathic Medicine, Nova Southeastern University, Davie, FL 33328, USA
2
John P Hussman Institute for Human Genomics, Miller School of Medicine, University of Miami, Miami, FL 33136, USA
3
Department of Urology, Miller School of Medicine, University of Miami, Miami, FL 33136, USA
4
College of Arts and Sciences, University of Miami, Miami, FL 33146, USA
5
College of Health Professions and Sciences, University of Central Florida, Orlando, FL 32816, USA
6
The Interdisciplinary Stem Cell Institute, Miller School of Medicine, University of Miami, Miami, FL 33136, USA
7
Department of Medicine, Cardiology Division, Miller School of Medicine, University of Miami, Miami, FL 33136, USA
*
Authors to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Vaccines 2021, 9(2), 94; https://doi.org/10.3390/vaccines9020094
Submission received: 5 December 2020
/
Revised: 18 January 2021
/
Accepted: 20 January 2021
/
Published: 27 January 2021
(This article belongs to the Special Issue Cancer Immunotherapy: Advances and Future Prospects)
Abstract
:Nitric oxide (NO) is a short-lived, ubiquitous signaling molecule that affects numerous critical functions in the body. There are markedly conflicting findings in the literature regarding the bimodal effects of NO in carcinogenesis and tumor progression, which has important consequences for treatment. Several preclinical and clinical studies have suggested that both pro- and antitumorigenic effects of NO depend on multiple aspects, including, but not limited to, tissue of generation, the level of production, the oxidative/reductive (redox) environment in which this radical is generated, the presence or absence of NO transduction elements, and the tumor microenvironment. Generally, there are four major categories of NO-based anticancer therapies: NO donors, phosphodiesterase inhibitors (PDE-i), soluble guanylyl cyclase (sGC) activators, and immunomodulators. Of these, NO donors are well studied, well characterized, and also the most promising. In this study, we review the current knowledge in this area, with an emphasis placed on the role of NO as an anticancer therapy and dysregulated molecular interactions during the evolution of cancer, highlighting the strategies that may aid in the targeting of cancer.
1. Introduction
Nitric oxide (NO) is a molecule with a very short half-life, produced by the action of nitric oxide synthases. Since NO was first discovered as being identical to endothelium-relaxing factor, the number of biochemical and physiological processes that undergo some form of NO signaling has continued to grow. When NO was observed to influence the development, growth, and metastasis of tumor cells, many studies emerged that were in direct conflict with one another. For many years, debate raged within the community about whether NO was tumoricidal or carcinogenic. However, as the body of scientific literature grew, the role of nitric oxide within carcinogenesis has been more clearly defined. Unfortunately for those seeking to make therapeutics, nitric oxide appears to have the capability to be both tumor-promoting and tumoricidal. NO’s bimodal effects on different cancer types is a phenomenon best termed as the Yin and Yang of NO [1,2,3]. Determining which effect predominates is complex and often depends upon the tissue NO exerts its effects, the concentration of NO administered, and tumor microenvironment. Nevertheless, these discoveries have led to a wide number of proposed uses for NO as an anticancer agent, either alone or in combination with other treatment modalities [4]. Here, we seek to outline the complexities of NO signaling within carcinogenesis and tumor progression at the biochemical and physiological levels. Furthermore, we also discuss the impact of NO in cancer therapy and outline its role as an emerging anticancer agent.
2. Physiology of NO
2.1. Chemical Properties of NO
Nitric oxide is a diatomic free radical molecule with high reactivity across a wide range of biomolecules. High output of nitric oxide leads to effects such as nitration, nitrosation, and oxidation, which can then affect cellular functioning. Nitric oxide can interact with oxygen or oxide ions to form reactive nitrogen species such as dinitrogen trioxide and peroxynitrite [5]. It can also react with nitrogen dioxide to form dinitrogen trioxide and can react with superoxide to form peroxynitrite [6]. Both molecules can cause DNA damage through nitrosative and oxidative stress. Both molecules can cause DNA damage through nitrosative and oxidative stress. Dinitrogen trioxide can lead to formation of N-nitrosamines through nitrosation of amines and then alkylate DNA. This alkylation of primary amines can lead to the formation of diazonium ions and further deamination [7]. Peroxynitrites can oxidize and nitrate DNA and also potentially cause single-strand DNA breaks due to an attack on the sugar-phosphate backbone [7]. The biological effects of nitric oxide are dependent upon myriad factors such as the formation of the molecule, its metabolism, types of nitric oxide synthases present, and concentration of nitric oxide present.
2.2. Synthesis of NO
The primary means by which cells synthesize NO (Figure 1) is through the conversion of l-arginine to l-citrulline via the enzymatic action of nitric oxide synthases (NOS) [8]. There are three isoforms of the NOS family—inducible NOS (iNOS/NOS2), endothelial NOS (eNOS/NOS3), and neuronal NOS (nNOS/NOS1). The eNOS and nNOS isoforms are constitutively expressed in a variety of cell types and can be activated as a result of calmodulin-binding due to a rise in intracellular calcium. The constitutive NOS isoforms become activated or inhibited by phosphorylation from protein kinases. Unlike the constitutive NOS isoforms, iNOS displays a higher affinity for calmodulin, and therefore its activation is not calcium-dependent. Among the three isoforms, iNOS, along with being calcium-independent, also produces high concentrations of NO in a shorter time frame [8].
Another more controversial mechanism of NO formation is through the nitrite-nitrate-nitric oxide pathway. Nitrates and nitrites are physiologically recycled in the blood and tissues to produce NO and other bioactive nitrogen oxides [9,10,11,12]. In the body, nitrate, primarily from diet and oxidation of NOS-derived NO, is actively taken up by the salivary glands and reduced to nitrite anion by commensal bacteria in the mouth [13,14]. Nitrite is further metabolized in blood and tissues into a variety of bioactive nitrogen oxides. This reduction is catalyzed through numerous pathways involving myoglobin/hemoglobin, ascorbate, xanthine oxireductase, and polyphenols [12,15,16,17,18,19,20,21,22]. The production of NO from these pathways is enhanced by hypoxic and acidotic conditions [23,24,25]. However, not all studies support the hypothesis that nitrite-nitrate derived nitric oxide contributes meaningfully as a biologically relevant signaling mode for downstream NO effects.
2.3. NO-Mediated Post-Translational Modifications
The dominant mode of NO signaling specificity occurs through post-translational modification of specific proteins, which regulates function in a manner analogous to post-translational phosphorylation [26]. S-nitrosylation is achieved through the covalent attachment of NO to the thiol side of a cysteine residue [27].
Proteins from almost all functional classes are substrates for S-nitrosylation. However, protein S-nitrosylation shows spatiotemporal specificity for certain cysteine residues within a protein [28]. While only a few cysteine residues are targeted at physiological amounts of NO, the modifications are generally enough to change the protein function, activity or interaction specificity [29]. For example, S-nitrosylation of cardiac ion channels modifies the channel’s dynamics and activity profile [30]. S-nitrosylation is a dynamic bidirectional process, and the relative balance of nitrosylated and de-nitrosylated proteins can serve as a biofeedback mechanism for physiological functions [30,31]. In the heart and vasculature, protein de-nitrysolation via S-nitrosoglutathione reductase (GSNO-R) regulates vascular tone and β adrenergic activity [31], the processes of S-nitrosylation and de-nitrosylation are important regulatory mechanisms for carcinogenesis and metastasis and may be responsible for ischemic episodes and more [32,33,34,35]. Importantly, hemoglobin is a key target of S-nitrosylation, whereby S-nitrosylated thiols participate in the allosteric shift that regulates the process of oxygen loading and unloading [36]. S-nitrosylation is critical for NO to exert its pro-cancer effect in many malignancies, as the downstream consequences of S-nitrosylation can cause aberrant signaling, which over time may lead to unchecked growth, angiogenesis, and metastasis [37].
Another type of post-translational modification mediated by NO is S-glutathionylation. S-glutathionylation is a reversible process that involves the addition of a proximal donor of glutathione to thiolate anions of cysteines in the target proteins. This modification alters the mass, charge, structure, and/or function of the protein and may also prevent degradation via sulfhydryl overoxidation or proteolysis [38]. Presence of s-glutathionylated serum proteins can be used as a biomarker in individuals exposed to oxidative or nitrosative stress-causing agents [38]. This process is a candidate mechanism for controlling the generation of reactive oxygen and nitrogen species associated with stress signaling and functional responses [26,38].
A third type of post-translational modification is mediated by NO via protein tyrosine nitration, although this is not widely accepted. This modification is caused by NO-derived oxidants like peroxynitrite and involves the formation of an intermediate tyrosine radical [39]. Nitration of protein tyrosine residues happens when the hydrogen at the third position in the phenolic ring is substituted by a nitro group, forming 3-nitrotyrosine. The formation of this product shows an oxidative modification that favors pro-oxidant processes [40]. The two major mechanisms leading to tyrosine nitration in vivo are the formation of peroxynitrite and the production of nitrogen dioxide by heme proteins [41]. Overall, tyrosine nitration is proposed to cause profound structural and functional changes in proteins and might serve as a marker for nitrosative stress [42,43].
2.4. NO cGMP Signaling Pathway
The nitric oxide-cyclic GMP-protein kinase G (PKG) cascade is recognized as an endogenous apoptotic pathway in many cancer types [44]. Two distinct types of guanylate cyclases (GCs) catalyze the conversion of guanosine triphosphate (GTP) to cyclic guanosine monophosphate (cGMP). Particulate GCs are transmembrane receptors for natriuretic peptides, whereas cytosolic-soluble GC serves as a target receptor for NO [44,45]. NO, at nanomolecular levels, binds to prosthetic heme on the β subunit of soluble GC, forming a NO-GC complex [46]. NO-GC binding then increases conversion of GTP to cGMP. The increase in cGMP initiates downstream effectors including two serine-threonine kinases, PKG-I and PKG-II, which share common targets with protein kinase A (PKA) but activate PKA-independent pathways as well [47]. The NO-cGMP signaling pathway is the primary means by which NO serves as a vasodilator, as increasing cGMP concentration inhibits calcium influx into the cytoplasm, preventing smooth muscle contraction (Figure 2) [48].
2.5. NO and Redox Balance
The effect of NO is often balanced against reactive oxygen species (ROS), whereby localized changes in the concentration often inversely affect the other, termed nitroso–redox balance, which has been suggested to play a major role in heart failure [35,49]. Thus, elevations in superoxide, a ROS, lead to decreases in NO and vice versa. Low basal levels of NO are important for maintaining the redox state of the tissues, whereby reactions of NO and superoxide produce substrates that bind to S-nitrosylation sites on proteins [35,36]. Furthermore, at physiological concentrations NO also performs a host of other antioxidant functions, which can help prevent aberrant signaling [50]. However, pathophysiological processes upregulate NOSs, resulting in excess NO production, which can lead to abnormal signaling [51]. When NO production becomes elevated, it can cause nitrosative stress. Nitrosative stress, acting similarly in a manner to oxidative stress, can then affect homeostasis and alter protein function [35,50]. A full review of the mechanisms that underlie this process and how it contributes to human disease was previously published by Hare and Stamler [35].
3. NO and the Immune Response
3.1. NO and the Antimicrobial Response
NO was first discovered to play a role in immunological function as a byproduct of macrophage activation [52]. Traditional experiments focused on the role of iNOS, which was found to be upregulated in the alveolar macrophages of patients with tuberculosis and other infections [53]. Multiple studies reported that NO can contribute to host antimicrobial resistance, such as iNOS-deficient mice often, but not always, display exacerbated infections, and NO is upregulated in response to markers of infection such as lipopolysaccharide [53,54,55]. One proposed mechanism for NO’s antimicrobial effect is the creation of powerful free radicals. These free radicals, such as peroxynitrite, can directly damage pathogens through phagosomal redox chemistry, although they can also affect S-nitrosylated pathogen proteins, modifying critical components required for proper cellular functioning [56,57]. Additionally, NO is found to bind to a wide array of intracellular molecules, including transcription factors and inorganic molecules, which may aid in infection clearance [52]. However, inhibition of iNOS may actually aid in recovery from certain infections, and NO was implicated in influenza-mediated pulmonary injury [53,58]. Evidence also suggests that macrophages manage their own internal concentration of NO and that too much NO can have potentially harmful intracellular effects [57,59,60]. These self-protective mechanisms may be enhanced by iNOS compartmentalization to phagosomes, preventing iNOS from acting on other compartments [61]. These results provide evidence that NO mediates immune responses, although NO’s exact effect can be variable and is tightly regulated intracellularly.
3.2. Pro-Inflammatory Response
NO’s function in inflammation is complex, and NO has been implicated in both immunosuppressive and immunopathological effects [52]. iNOS is often upregulated in response to inflammation, as one study of sepsis suggested that NO levels were elevated in response to higher concentrations of pro-inflammatory cytokines, such as TNF-α, which in turn could directly upregulate iNOS through nuclear factor kappa B (NF-κB) signaling [62,63]. Additionally, anti-inflammatory cytokines, such as IL-10, were associated with lower NO levels [62]. NO is consistently elevated during chronic inflammatory states, which can lead to cellular apoptosis through some combination of cytochrome c release, p53 activation, or Bcl-2 associated X /Bcl-2 homologous antagonist killer protein (BAX/BAK) recruitment [64]. However, if NO levels become inordinate, NO can also lead to necrosis [64]. The difference between NO leading to apoptosis or necrosis is hypothesized to be due to a peroxynitrite concentration-dependent mechanism, with overwhelming levels of peroxynitrite inevitably leading to necrosis [64]. The discovery of NO’s central role in inflammation led to the hypothesis that NO and iNOS may contribute to the pathogenesis of many different diseases, including those in the heart, pulmonary system, vascular dynamics, and the blood [35]. Indeed, elevated concentrations of NO and inflammatory cytokines are found in a plethora of conditions, which provides evidence that NO may be essential for some pathogenic effects, although this is controversial [64,65,66,67].
3.3. Anti-Inflammatory Effects
Such controversy has endured because NO is involved in immune autoregulation across many different cell types and intracellular processes. At the molecular level, NO has been shown to downregulate its own production through inhibition of NF-κB [68]. Furthermore, NO has also been shown to attenuate cytokine activating molecules such as IL-1β-converting enzyme and interferon-γ (IFN-γ)-inducing factor, suggesting that NO can simultaneously aid in the immune response and tune the immune response [69]. Interestingly, T lymphocytes do not express iNOS directly, but inhibition of iNOS directly affects their functioning [70]. NO downregulates T-helper cell 1 (Th1) response through selective inhibition of IFN-γ, IL-2, P-selection, and the cellular adhesion molecules intracellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule (VCAM) [71]. NO also increases the production of IL-4, prostaglandin-E2, and IL-12, which selectively support T-helper cell 2 (Th2) production [71]. When combined with prior work which found that NO can selectively encourage immune cell apoptosis and downregulate antigen presentation, these findings strongly implicate NO as a key mediator of autoimmune responses [52,71,72]. However, these findings remain controversial as many labs have found null or opposite results [72]. Still, a number of animal models suggest that iNOS inhibition is directly correlated with decreased suppression of autoimmune diseases and some exhibited increased severity, providing system-level evidence of NO’s immunoregulatory function [71].
4. NO and Carcinogenesis
4.1. Carcinogenesis Overview
Similar to its other immune functions, NO has been shown to simultaneously be tumoricidal and tumor-promoting (Figure 2) [73]. NO is highly reactive with nucleic acids and can induce mutations upon chronic exposure [74]. One hypothesis suggests that NO-induced mutations from iNOS upregulation may drive carcinogenesis from chronic inflammatory processes [74]. However, these mutations may then induce p53 mediated growth arrest, DNA repair enzyme activation, and iNOS downregulation [7]. Therefore, p53 mutations may be a critical inflection point that steers NO away from growth arrest and apoptosis towards carcinogenesis [74]. Importantly, cell types inside a tumor may not be homogenous, and they contain a vast network of connective tissue, blood vessels, and immune cells, termed the tumor microenvironment (TME), which may also be differentially regulated by NO [74,75]. Furthermore, NO may modify tumor cell metabolism inside the microenvironment by promoting the Warburg effect and chemotherapeutic resistance [76]. Upon tumor formation, NO promotes angiogenesis, downregulates immune surveillance and encourages metastasis with gross pathological specimens demonstrating markedly elevated levels of NO or iNOS in a wide variety of malignant tumors. Increased iNOS expression has been linked to poor patient outcomes for multiple types of malignancies and thus has become an important target for future therapeutic delivery [73].
4.2. NO Biochemistry within the TME
NO is an important immune modulator of the TME. Innate immune cells such as macrophages and NK cells upregulate production of NO during tumor cell invasion [77,78]. Within the TME, NO is produced mainly by iNOS, expressed in macrophages and tumor cells, and to a much lesser extent by eNOS and nNOS. NO is the main chemical macrophages utilize to mount an immune attack against tumor antigens, and functions to activate apoptosis [79]. NO can bind to the heme copper center of cytochrome c oxidase, thereby oxidizing the enzyme and inhibiting the mitochondrial respiratory chain, leading to permeability and escape of cytochrome c into the cytoplasm [80,81]. Additional mechanisms of apoptosis activation include phosphorylation of p53, a tumor suppressor antigen, p53 retention in the nucleus, and s-nitrosylation of NF-kB, an anti-apoptotic factor, inhibiting its ability to bind to the DNA [80,81]. However, NO might not only exert cytotoxic effects but is actually more dichotomous in nature with both tumor-promoting and tumor-suppressing effects. At high concentrations, NO is able to induce death, while at low concentrations it may actually protect cells from death. At 50–100 nM, NO is able to phosphorylate extracellular signal regulated kinase (ERK) activate the AKT pathway, and stabilize HIF1a, while at the 300 nM–1 μM range, NO induces DNA damage, p53 activation, and causes nitrosative stress [56].
4.3. NO and Angiogenesis in the TME
Angiogenesis occurs in hypoxic core regions of the tumor due to lack of oxygen and nutrients, which may be intricately regulated by the presence of NO. Hypoxia-inducible factor 1 and 2 activate the expression of pro-angiogenic growth factors such as VEGF, FGF-2, IL-8, PDGF, IGF2, and TGFβ. HIF1α and HIF1β are usually degraded in normoxic conditions, but are stabilized in hypoxic conditions (<5% oxygen) as well as by low levels of NO and reactive nitrogen species (RNS) [82,83,84]. Low levels of NO may also regulate endothelial cell fate by activating GC and increasing cGMP levels (Figure 3), which acts directly on the endothelium to cause its reorganization into vessel-like formations [85]. Additionally, low levels of NO are able to activate matrix metalloproteinases (MMP-1, -9, -13), which degrade the components of the extracellular matrix and paves the way for tumor cell dissemination outside of the tumor and endothelial cells into the tumor core for blood vessel formation [86,87]. Thus, low NO is beneficial for ensuring upregulation of pro-angiogenic factors.
4.4. NO and Immune Cells within the TME
Macrophages can switch between different phenotypes and perform completely opposite functions in the TME based on their inflammatory potential. For instance, M1 macrophages are induced by pro-inflammatory cytokines (tumor necrosis factor-alpha TNFα, interferon-gamma INF-γ, Interleukin-1b IL-1β), activate Th1 immune response, and induce their cytotoxic effects by upregulation of iNOS that produces toxic concentrations of NO [77,78]. In fact, NOS2 expression is a hallmark of M1 macrophages. Alternatively, activated M2 macrophages are less immunologically inflamed and often act as housekeeping cells, which sweep apoptosis debris after pathogen clearance. These are activated by signals from anti-inflammatory cytokines such as tumor growth factor-beta (TGFβ) and anti-inflammatory interleukins (IL-10, IL-13), which downregulate iNOS production and reduce the activity of M1 macrophages and T helper cells [88,89]. Given their anti-tumor potential and ease of isolation, exogenous infusion of M1 macrophages has been investigated as a potential autologous therapy approach [90,91]. One study utilized ex vivo LPS-stimulated macrophages to autologously re-infuse into patients; however, results showed no significant clinical improvement with minor side-effects [92,93]. Another study using INF-γ-stimulated macrophages showed reduced metastasis and tumor growth but no significant tumor regression in mouse studies [93,94] and subsequent human clinical trials [95]. These conflicting results may need to account for the presence of anti-inflammatory mediators (TGFβ, IL-10, PGE2) and the hypoxic microenvironment that inhibits iNOS expression, which neutralize M1 infusions or skew them back to the immunologically suppressive M2 type [96]. Thus, finding ways to increase production of NO by tumor cells and macrophages, thereby activating immunity toward immunologically active M1 macrophages, might be necessary to effectively eradicate the tumor (Figure 4).
Aside from their NO-producing ability, macrophages are among the most prominent immune cells in the TME, as they are able to infiltrate deep into the tumor and can account for almost 50% of the tumor mass [91,97]. Tumor-associated macrophages (TAMs) reside in the most hypoxic and necrotic regions of the tumor, where they are known to assist in tumor progression [98]. TAMs express pro-angiogenic growth factors and matrix metalloproteinases MMPs (VEGF, PDGF, FGF-2, MMP-7, MMP-12), as well as create an environment that makes M1 macrophages and CD4+ and CD8+ T cells unresponsive to tumor antigens. Another type of macrophage, Tie2-expressing monocytes (TEMs), resides close to blood vessels, where they function in a similar way to TAMs by promoting angiogenesis via upregulation of VEGF and MMPs [99]. Depletion of TEMs results in marked inhibition of angiogenesis, so these are required for metastasis [100]. Thus, tumor-infiltrating macrophages are more similar to the anti-inflammatory M2 type, which suggests that the majority of macrophages in established tumors are anti-inflammatory and thrive in immunologically suppressed conditions.
Chronic inflammation has been linked to multiple forms of malignant transformation, and NO produced by different immune cells during acute infections plays a significant role in this process. Since it is extremely small and lipophilic, NO is able to quickly diffuse through cell membranes, oxidize DNA, deaminate bases, and deactivate proteins in the DNA repair machinery through s-nitrosylation. Additionally, long-term this genomic instability might activate oncogenes and inhibit tumor suppressor genes [101,102]. In a p53 knockout mouse model, increased NO production due to iNOS upregulation through a negative-feedback loop accelerated spontaneous tumor formation [103]. In addition, high amounts of NO in a breast cancer line activated expression of c-Myc, a potent oncogene [104]. Furthermore, deactivation of retinoblastoma (Rb) tumor suppressor gene by NO-driven hyperphosphorylation was shown in a mouse model of colitis [105]. Thus, prolonged episodes of acute infection can lead to overall genomic instability and activation of undesirable oncogenes as well as inhibition of tumor suppressor genes.
In the immunogenically inactive tumors, usually occurring during escape of tumor cells and metastasis phase, NO production is lowered to ensure decreased immunogenicity. This occurs by several processes. Anti-inflammatory cytokines in the TME actively lower the expression of iNOS and degradation of iNOS mRNA by hypoxic conditions within the tumor core [106]. Moreover, NO produced by TAMs is often captured by circulating erythrocytes, where it oxidizes hemoglobin iron centers and thus reduces circulating NO even further [107]. Regardless of which mechanism is responsible, immunologically suppressed tumors show reduced expression of NO. These low amounts of NO usually modulate apoptosis protein cascades through s-nitrosylation of caspase-3 and Bcl-2 [108,109]. Thus maintaining low/transient levels of NO and ensuring that apoptotic pathways are effectively inhibited is one potential mechanism by which tumors are able to increase their proliferation capacity.
4.5. NO as a Biomarker in Carcinogenesis
Estimation of iNOS expression in solid tumors has attracted interest for biomarker use; however, there are multiple levels of iNOS regulation that must first be considered. Different iNOS isoforms are under transcriptional regulation due to multiple transcription binding sites within the promoter of the gene, and these sites are susceptible to regulation by various cytokine mediators [110,111]. iNOS promoters are also different among species; in the mouse, induction with lipopolysaccharide (LPS) and pro-inflammatory cytokines (TNFα, INFγ, IL-1b) is enough to upregulate iNOS expression, while a more complex scheme is needed to achieve the same effect in humans [112]. The stability of iNOS mRNA is also under tight regulation by various cytokines that upregulate RNA binding proteins which compete for binding to the 3′-UTR region, thus stabilizing or destabilizing mRNA transcript [111]. Additionally, iNOS transcripts might be targeted through cytokine signals (SOCS-1) by small non-coding RNA molecules (miRNA) such as miR-155 [113] and miR-146 [94,114], resulting in translational inhibition. Finally, ready availability of L-arginine as a substrate for NO production is crucial, as it is mostly supplemented through the diet, and the bioavailability of necessary co-factors for proper enzymatic activity such as tetrahydrobiopterin (BH4) may also influence iNOS activity [111]. Thus, the use of iNOS expression as a prognostic biomarker, though promising, is currently controversial.
What complicates the issue further is that iNOS expression might not be as well correlated with cancer progression as previously thought and finding methods that show an accurate estimation of NO production is currently a difficult task. Expression of iNOS can be measured using Western blot analysis, qPCR, or immunohistochemistry, though iNOS mRNA is subject to degradation in paraffin-embedded blocks. Moreover, iNOS expression is not always indicative of appreciable NO production, depending on the tumor niche and cytokine content, as already mentioned. Thus, measuring the activity of the enzyme might be more beneficial; however, this is limited to the availability of fresh tissue. Different techniques have been developed, such as estimation of nitrates and nitrites [115], measuring conversion of radiolabeled (H3) L-arginine to (H3)-L-citrulline [116,117], or immunohistochemical detection of nitrotyrosylated proteins [118]. Needless to say, these techniques are tedious and expensive to implement, and might not be a direct measure of NO production. Several studies have shown a correlation between iNOS expression and tumor stage progression in malignant melanoma [119,120], poor survival in colorectal cancer [118,121], poor prognosis in estrogen receptor (ER)-negative breast cancer patients [122,123], and lymph node metastasis in pancreatic cancer [124]. However, in large hypoxic tumors, NO production was shown to be reduced when compared to smaller immunologically active tumors [94]; thus, iNOS expression by overall tumor mass might indicate a gradient of concentrations, which further hinders its prognostic potential [115]. Interestingly, in the lungs, excess exhaled NO from iNOS has been proposed as a biomarker of lung cancer, which was significantly higher in those with lung cancer [125,126,127]. However, exhaled NO is notably upregulated in other inflammatory airway conditions, particularly in asthma and heart failure, and its specificity and sensitivity for lung cancer remain under investigation [126,128,129]. Therefore, iNOS biomarker potential, though attractive, necessitates the development of better tools and a clearer understanding of its expression patterns within solid tumors.
5. NO in Different Cancer Types
5.1. Lung Cancer
Lung cancer has the second-highest incidence amongst all cancer types, with a poor 5-year survival rate of 4–17% [130,131]. Due to its high incidence and often aggressive course, it is no surprise that NO has been shown to function in the pathogenesis of lung cancer. In fact, population-based studies of NO and NO metabolites suggest that increased accumulation of NO metabolites were associated with an increased risk of lung cancer, even after controlling for relevant confounding factors such as smoking [132]. Furthermore, cigarette smoking, a notable risk factor for lung cancer, often contains NO and other ROS [133]. Such compounds may encourage the development of NO-mediated cellular changes, which can eventually accumulate in malignant lung tissue (Figure 5) [133]. Excess exposure of lung tissue to NO results in the accumulation of nitrosylated proteins, which were significantly higher in those with lung cancer [134]. Often, this effect is mediated via iNOS, which is often shown to be upregulated in lung cancer cell lines, similar to other cancers [125,126].
Excess NO in lung tissues may contribute to a number of effects that synergistically act to encourage cancer formation. For example, one study noted that gaseous nitric oxide and inducible NOS produced an increase of 8-nitroguanine, which may increase DNA damage and encourage mutagenesis [135]. Although protein nitrosylation is often viewed as a marker of oxidative stress, one group proposed that protein nitrosylation may also impair antioxidant proteins and those involved in cellular metabolism, which may further contribute to the development of non-small-cell lung cancer (NSCLC) [136]. Furthermore, elevated iNOS in NSCLC cells are linked to p53 mutations, removing growth checkpoint inhibition and creating cells with unlimited replicative potential, another notable effect of NO [137,138]. Within lung cancer, the antiapoptotic effect is hypothesized function through BCL-2 upregulation and modifications to FAS death-ligand signaling, which encourages aggressive growth [139]. Once established, NO also modulates integrin expression amongst NSCLC cells via protein kinase B (AKT) activity, and increased AKT activity is correlated with a poor prognosis and chemotherapeutic resistance [140]. Increased exposure to nitric oxide over time is itself correlated with enhanced cellular migration and chemotherapy resistance [141,142]. Nevertheless, a number of NO-donating compounds have been reported to have efficacy at inhibiting some of these effects [143,144,145]. As such, NO-donating drugs may raise the concentration of NO to cytotoxic levels, bypassing the physiological mechanisms underlying tumorigenesis and actually serve to inhibit cancer growth.
5.2. Breast Cancer
Amongst all non-skin cancer sites in females, breast cancer has the highest incidence and second-highest mortality, although the 5-year survival rate is over 90% [129]. Similar to NSCLC and other types of cancer, elevated levels of iNOS were noted from tissue samples of patients with breast cancer, which was significantly greater than normal breast tissue and benign breast diseases such as fibroadenomas [123]. NO was also shown to function through the same p53 and AKT pathways as those of lung cancer cells, suggesting common pathways for NO signaling to induce cancer growth and encourage progression [146]. Such pathways may work synergistically with other NO pathways in breast cancer, such as the epidermal growth factor receptor (EGFR)-mediated activation of ERK [147]. Furthermore, a number of studies suggest that the activity of NO pathways in breast cancer are concentration-dependent, which is backed by other NO studies. NO concentration of less than 100 nM activates cGMP-dependent pathways, while concentrations of 200–600 nM activate cGMP independent pathways [146]. Above these concentrations, NO has been suggested to phosphorylate p53 and halt the function of DNA repair enzymes [146]. cGMP dependent pathways were noted to be dysregulated within breast cancer, with lower concentration of cGMP being correlated to malignant disease, suggesting low physiological concentration of NO-mediated cGMP signaling may actually provide protection against breast cancer [148]. cGMP independent pathways have been discussed previously and are not unique to breast cancer. Nevertheless, NO-mediated HIF-1a stabilization, nitrosylation of metabolic enzymes, and induction of VEGF are major pathways of breast cancer growth and metastasis [146]. Additionally, NO-mediated activation of matrix metalloproteinases (MMP) and inhibition of tissue inhibitor of matrix metalloproteinases-1 (TIMP-1) may reorganize the tumor microenvironment to increase metastatic potential, while further stabilizing downstream cGMP-independent NO pathways [146]. Numerous studies have found that many of these NO-mediated effectors, such as VEGF and CXCR4, are correlated to lymph node metastasis and overall prognosis [149,150]. The growth arrest and p53 phosphorylation exhibited by supraphysiological concentrations of NO may explain the effect that NO-conjugated drugs have at inhibiting breast cancer growth and inducing apoptosis [151]. The underlying mechanisms behind how nitric oxide becomes upregulated in breast cancer remain unknown; however, similar to other cancer types, systemic effects from environmental, hereditary, and physiological processes all may play a role. For example, iNOS was noted to be upregulated in response to glucocorticoids, a stress hormone, suggesting multisystem processes may contribute to the pathogenesis of breast cancer [152]. Furthermore, cyclooxygenase-2 (COX-2) and NO are strongly linked in breast cancer, with one multivariate analysis suggesting that those with COX-2 and iNOS-positive tumors were associated with extremely poor prognosis [153]. Nevertheless, the exact role of NO in breast cancer remains controversial, especially in the context of estrogen- and progesterone-mediated signaling.
Estrogen and progesterone have also been shown to modulate nitric oxide expression, through the actions of eNOS and iNOS [154,155]. Estrogen is thought to downregulate the expression of iNOS and upregulate the expression of eNOS, which in turn activates phosphoinositide 3-kinase (PI3K) and AKT through direct activation and increased transcription, which have been shown to be critical for cancer proliferation [156]. Furthermore, upregulation of eNOS appears to be a unique feature of estrogen-dependent tumors, which are lacking in ER- breast cancers [157]. Interestingly, progesterone’s function within nitric oxide synthesis appears to be more controversial, with several studies suggesting that progesterone can affect eNOS production and others suggesting null or contradictory effects [156,158,159]. For example, one study suggested that progesterone-induced iNOS expression in vitro, which then induced cell apoptosis [160]. Additionally, progesterone has been proposed to affect and downregulate estrogen-mediated nitric oxide production [156]. These results suggest that breast cancer tumor progesterone status is a favorable prognostic factor, an effect that is backed by clinical studies [161]. Although it is currently unknown exactly how the surface marker human epidermal growth factor receptor 2 (HER2/neu) affects nitric oxide production, one group suggests that HER2/neu downregulates nitric oxide production, ablating the apoptotic effect of chemotherapeutics in vitro, potentially through a COX-2 mechanism [162,163]. Nevertheless, triple-negative breast cancer, the most aggressive form, appears to also be strongly correlated to iNOS [164]. Thus, nitric oxide expression appears to be central to the pathogenesis of breast cancer, regardless of receptor phenotype expression.
5.3. Prostate Cancer
Prostate cancer is highly prevalent in men and has an age-adjusted incidence of 453.8 per 100,000 and is highest amongst those 65–74 [165]. Similar to other cancers, iNOS expression was also significantly increased in prostate adenocarcinoma when compared to healthy prostate tissue [166]. Furthermore, previous work demonstrated that iNOS expression was greatest amongst those with metastasis and high Gleason scores, and one meta-analysis found that tumor iNOS expression may serve prognostic value [167,168]. These findings are backed by clinical studies, which suggest greater nitrosative stress in those with prostate cancer as compared to benign prostatic hyperplasia (BPH) and controls [169]. However, eNOS also appears to be linked to prostate cancer, as some but not all studies have shown that genetic polymorphisms of iNOS and eNOS carry an increased risk of high Gleason score prostate cancer [170,171,172]. eNOS may in turn upregulate pleiotrophin (PTN), expression through ERK activity, increasing tumor and endothelial migration, laying the groundwork for metastatic disease [173]. Additionally, the eNOS complex can interact with the estrogen receptor to transcriptionally alter other important gene products, such as Glutathione transferase P1, which are correlated to disease pathogenesis [174].
Prostate cancer is often initially androgen-sensitive, which is useful in androgen deprivation therapy, and is often a first-line therapy for advanced and metastatic disease [175]. This advanced form of prostate cancer is termed castration-resistant (CRPC) and is associated with poor outcomes [176]. Previous studies have implicated NO as a potential mediator of androgen resistance by androgen receptor transcriptional suppression and direct androgen receptor inhibition, which were mediated by iNOS and eNOS, respectively [177,178]. iNOS induction may encourage tumor growth, as two studies from the same lab found that NO promotes survival and accelerates tumor growth after oxidative stress [179,180]. However, testosterone, an androgen, also increases NO concentration and survival of prostate cancer cells, suggesting that NO’s mechanism in androgen sensitivity and resistance remains elusive [1,2,181,182]. These androgen-specific mechanisms may interact with previously defined effects of NO on hypoxia-induced HIF-1a and tumor angiogenesis, compounding the effect of nitric oxide on prostate cancer [183].
5.4. Gastrointestinal Cancers
When summed, digestive system cancers have the highest incidence amongst all organ systems with colorectal cancer having the third-highest incidence amongst all non-skin types, regardless of gender [129]. Interestingly, one analysis of gastrointestinal cancers suggested that most had adherent iNOS expression, although whether iNOS expression was upregulated or downregulated was dependent upon cancer type [184]. Similar to other cancers, gastrointestinal cancers may also use previously discussed NO signaling pathways such as PI3-K, p53, AKT, PTEN, NF-kB, MMPs, and HIF-1a in their carcinogenic pathogenesis [184]. Malignant transformation of colorectal and other cancers are often dependent upon the epithelial–mesenchymal transition (EMT) in which cancer cells express genes that are normally associated with connective tissue [185]. Although a number of cell signals can stimulate this pathway, one particularly important player is the APC/Wnt/β-catenin pathway, which is often dysregulated in colorectal cancer [186]. In normal healthy colonic tissue, APC downregulates B-catenin, which prevents polyp formation [186]. NO has also been shown to upregulate the Wnt/β-catenin pathways, potentially through negative feedback NF-κB response elements on a Dickkopf-1 gene promoter that reduces gene silencing [187]. iNOS has been previously suggested to influence the function of NF-κB, and these findings correlate to the increased iNOS expression, which is often noted in colorectal carcinomas. However, increased iNOS expression is not a ubiquitous finding across all studies [188]. In addition, APC and the wingless-related integration site (WNT)/B-catenin pathways also serve to regulate COX-2 in a similar manner to NO, suggesting that NO may work in synchrony with COX-2 to promote its pro-cancer effects [184,189]. COX-2 has been previously linked to many of the same pro-cancer effects as NO [190]. Nevertheless, the underlying mechanisms linking COX-2 to NO remain elusive, although both undergo NF-kB regulation [191]. Taken together, these findings suggest that regulation of NO and APC may occur simultaneously, and treatment strategies targeting these pathways may prevent WNT/B-catenin upregulation, preventing the development of cancer.
Similar to cigarette smoking, the processing of digested metabolites and chronic infectious agents may also lead to cancer through nitric oxide-mediated mechanisms. For example, one of the major risk factors for gastric cancer is dietary consumption of nitro-compounds, with a relative risk of 1.31 (95% confidence interval (CI), 1.13–1.52) for nitrites, and 1.34 (95% CI, 1.02–1.76) for NDMA, although nitrate consumption was associated with a lower risk of gastric cancer 0.80 (95% CI, 0.69–0.93) [192]. Dietary consumption of salivary nitrites exposes nitrites to stomach acid and ascorbic acid, which then produces nitric oxide, which can diffuse rapidly to surrounding tissue [193]. A similar increase in NO production was noted with chronic Helicobacte pylori infection, suggesting that enhanced nitric oxide exposure from environmental and infectious agents may be responsible for the development of cancer, especially when there is high or chronic levels of exposure to these agents [194]. Viruses were also not immune to NO, as Hepatitis B was shown to be inhibited by NO via INF-γ, and chronic infection with Hepatitis B may induce increased NO, which can predispose hepatocytes to mutagenesis, potentially through a c-jun, an n-terminal protein kinase (JNK) [195,196]. Furthermore, Hepatitis C was also shown to induce DNA damage through upregulation of iNOS and nitrosylation of DNA glycosylase [196,197]. The effects of chronic viral-induced upregulation of NO may be compounded by mutations to proteins regulating nitrosylation, like GSNOR, which can cause the buildup of formaldehyde, a well-known carcinogen [196], increasing the risk of liver cancer. Evidence also suggests that iNOS and NO may be dysregulated upon alcohol exposure, as iNOS and NO are upregulated in response to ingested toxins [196,198]. When combined with studies on NO, such results suggest that increased nitric oxide dysregulation in response to cigarettes and alcohol may be one mechanism for the increased cancer risk amongst patients who engage in these behaviors.
5.5. Other Cancers
NO dysregulation is linked to a wide range of malignancies, including those of the brain [199], genitourinary system [200,201], skin [202], thyroid [203], and head and neck [204,205] cancers. Due to the complexity of nitric oxide and its wide number of potential interactions, it is suggested that the multifactorial effects of NO are cancer-specific. Although NO may often work through a common pathway such as p53 to induce mutagenesis, the exact mechanisms often differ between cancer types. Furthermore, in cases of sporadic cancers with minimal risk factors and no underlying conditions, it is unknown exactly what causes the underlying upregulation of NO or if the elevated NO is in response to another effect. However, chronic unchecked NO signaling is clearly beneficial to tumors and can encourage progression and metastasis. Therefore, therapies that can control NO growth signals have great promise, although delivering such therapies to the tumor without affecting nearby or distant healthy cells remains a significant problem.
6. NO in Anticancer Therapy
6.1. NO Donors
NO donors function by increasing NO or NO isoforms (NO- or NO+) without the need for endogenous production [206]. NO donors work through a number of different mechanisms; however, the end result is often still the same, i.e., an increase in NO concentration within tissue beds [207,208]. Such modulation of NO concentration has lead to a number of proposed clinical uses for NO donors outside of cancer (Table 1) [209,210,211,212,213,214,215,216,217,218,219,220,221,222]. The blood vessels are particularly sensitive to the effect of NO, as the systemic veno- and vasodilator effects are useful for treating angina, acute coronary syndromes, and other cardiovascular diseases [223]. In cancer therapy, NO donors are particularly helpful as chemo- and radiotherapeutic sensitizing agents [224]. Advanced malignancies are often characterized by incomplete vascularization, which induces localized hypoxia. The resulting hypoxia stimulates the hypoxia-inducible factor1α (HIF-1α) pathway, priming cancer cells for survival against a variety of cellular death mechanisms induced by radio- or chemotherapy including autophagy, apoptosis, and DNA damage [224,225]. NO donors attempt to reverse this effect by increasing tumor perfusion, enhancing the effect of antitumor therapy [224].
Multiple clinical studies (Table 2) have demonstrated the efficacy of nitroglycerin (GTN), an organic nitrate, within the treatment of cancer. For example, in one phase II clinical trial, transdermal GTN improved outcomes for patients with advanced non-squamous cell lung cancer [226]. Not only has GTN shown efficacy in treating NSCLC, but GTN has demonstrated clinical effectiveness in liver, colorectal, and prostate cancer as well [227,228,229,230,231,232,233]. Furthermore, other preparations utilizing isosorbide mononitrate have also been attempted, although this approach did not show efficicacy in two clinical studies [234,235]. Although arginine, a nitric oxide precursor, is not traditionally viewed as a nitric oxide donor, a number of clinical trials have been completed examining the effectiveness of nutritional arginine for cancer (NCT04564521, NCT00559156, NCT02655081, NCT02844387). However, none currently have shown results.
HIF-1α-mediated resistance has been demonstrated to be vital for tumor resistance to a number of chemotherapeutics, although a number of specific mechanisms have been described that are often tumor-type-dependent [225]. NO donors may be similarly useful in the treatment of other cancers by targeting the underlying HIF-1α-mediated chemotherapy resistance, although no active clinical trials that test the effectiveness of adding a NO donor alone to chemotherapy are underway [225]. Importantly, the exact effectiveness of NO donors may vary by the type of cancer being treated and the NO donor being administered, making the ideal therapeutic to use NO donors in cancer therapy elusive. Nevertheless, an incredible number of preclinical in vitro and in vivo studies have demonstrated considerable efficacy in using a wide range of NO donors to treat cancer (Table 3). Amongst all NO donors that have not undergone clinical studies, diazeniumdiolates have shown the most promise. For example, DETA/NO demonstrated reversal of chemotherapeutic resistance for 5-fluorouracil (5-FU), doxorubicin, cisplatin, and fludarabine, and sensitized prostate cancer cells to TRAIL-mediated apoptosis and, when combined with a farnesyltransferase inhibitor, led to selective apoptosis in breast cancer cells [224,236,237,238]. Furthermore, the combination of PROLI/NO with carboplatin led to improved prognosis amongst rats with 6-c gliomas, which was thought to be due to improved chemotherapeutic delivery through the blood–brain barrier [239]. Preclinical studies also suggest that NO donors do not always have to be adjuvant to chemotherapy to demonstrate anticancer properties. One diazeniumdiolate, DETANONOate, was shown to inhibit the mesenchymal-to-epithelial transition and reverse the metastatic properties of the tumor [240]. A synthetic NO metal donor, ruthenium nitrosyl complex trans-[Ru(NO)(NH3)4(py)](PF6)3](pyNO), demonstrated considerable mitochondrial inhibition and an increase of ROS within the tumor, encouraging caspase-mediated cell death in liver cancer cells [239]. Arora et al. found that treating CRPC with GSNO, an S-nitrosothiol, reduced the expression of M2 macrophage expression within the TME, an important component of tumor progression [182]. Furthermore, other markers of cancer progression and resistance were suppressed, namely VEGF, the androgen receptor, and Androgen Receptor Splice Variant 7, while cytotoxicity increased through a greater number of M1 cytotoxic macrophages [182]. However, it should be noted that the potential mechanisms of NO donors as an anticancer agent are numerous (Table 3). Such studies suggest that NO donors have a number of novel mechanisms that may be beneficial in cancer therapy. However, NO often can affect multiple tissue beds simultaneously, including those outside of the tumor, and so consistent delivery of cytotoxic NO presents a major obstacle for researchers [207].
6.2. Phosphodiesterase-Inhibitors
Phosphodiesterases (PDEs) function as metallohydralses that catalyze the breakdown of cyclic adenosine monophosphate (cAMP) or cGMP into their inactive forms, 5′-AMP or GMP [264]. PDEs are thought to be involved with cancer progression and tumor growth because of the positive association they have with increasing tumor grade and stage as well as the decrease of cAMP and cGMP noted in many tumors [265]. Elevated PDE-5 levels have been documented in various types of human carcinomas including prostate, pancreatic, lung, colon adenocarcinoma, breast, and bladder squamous carcinoma [266]. PDE-5 inhibitors (PDE-5i) blunt the function of this critical recycling enzyme and block the breakdown of cGMP in 5′-GMP, which enhances the NO/cGMP signaling pathway [44]. Thus, PDE-5is function similarly to NO donors by enhancing the effect of NO on tissue; however, they rely on endogenous sources to maintain their effect rather than the exogenous NO provided by an NO donor [267].
PDE-5is are commonly used clinically to treat erectile dysfunction (ED); however, a wide range of PDE-is have shown anticancer activity, with many tested in clinical trials (Table 4) [265]. The primary differences between PDEs and the corresponding inhibitors that determine their functional significance are their different tissue bed distributions in addition to different regulatory feedback mechanisms and affinities for cGMP, cAMP, or both [265]. For example, thymoquinone, a natural herb with PDE-1i activity, has shown efficacy at inhibiting the growth of acute lymphoblastic lymphoma, cervical, and malignant central nervous system tumor cells, and an active clinical trial investigating the efficacy of thymoquinone to treat premalignant leukoplakia has recently been completed (CT03208790) [268,269,270,271,272]. Numerous other phosphodiesterase inhibitors (PDE-is) have also shown similar clinical effectiveness in preclinical studies [265]. Nevertheless, PDE-is may be limited by dose toxicity and systemic side effects unrelated to the primary tumor site. One large epidemiological study of 15,000 American men suggested that use of PDE-5is was associated with an increased incidence of melanoma; however, a retrospective meta-analysis found that although 4 out of 7 studies showed an increased risk of melanoma development with PDE-5i use, they failed to account for major confounders, and there was no linkage between the PDE-5i use and melanoma [273,274]. Interestingly, one report also suggests that PDE-5is can prevent the progression and development of prostate cancer, although other studies have shown null and even contradictory results [44,275,276,277]. Furthermore, the effectiveness of PDE-is as chemotherapeutics may be linked to their ability to enhance chemotherapy. Similarly to PROLI/NO, PDE-5is also demonstrated that they were able to increase the transport of doxorubicin across the blood–brain barrier in a rat brain tumor model, increasing the effect of chemotherapy [278]. In another study, the addition of sildenafil, a PDE-5i, with or without roflumilast, a PDE-4i, and theophylline, a methylxanthine, to lung cancer cell lines showed increased apoptosis and growth inhibition when given alongside a platinum chemotherapeutic [279]. Importantly, the same regime was not effective when combined with docetaxel, a taxane [279]. Taxanes function by disrupting microtubule disassembly, whereas platinum agents generate DNA double-stranded breaks through ROS generation. PDE-is may amplify NO’s downstream signaling cascade and increase the production of free radicals, which may then augment the free radicals produced from a platinum agent, thus increasing the efficacy of cancer. The potential anticancer mechanisms of PDE-is are numerous, and their anticancer properties were recently reviewed by Peng et al. [265]. Nevertheless, the addition of PDE-5i as adjuvant chemotherapy sensitizing agents may work through multiple mechanisms and is not strictly limited to one class of chemotherapeutic agents. Emerging evidence suggests that the sildenafil also improved the efficacy of docetaxel at treating CRPC, which the authors hypothesized was due to improved action of docetaxel on effector pathways, namely cGMP-mediated apoptosis and ERK/JNK downregulation [280]. Although numerous preclinical studies outline the potential roles for PDE-is in anticancer therapy, clinical trials (Table 4) have only begun to show evidence of efficacy [265,281]. Large, multicenter studies are needed before widespread clinical adoption of PDE-is in anticancer regimens.
6.3. Soluble Guanylate Cyclase Activators
Soluble guanylyl cyclase (sGC) is the receptor for NO, which binds to the ferrous (Fe2+) heme at histidine 105 of the β1 subunit [294]. Upon binding of NO, a remarkable increase in sGC activity is observed and cGMP production increases at least 200-fold [294]. In cancer, activation of sGC is notably impaired in a number of cancer cell lines including prostate, breast, and glioma, and restoration of sGC may decrease disease progression [294,295,296,297]. sGC activators, usually riociguat or vericiguat, stimulate the sGC irrespective of endogenous NO [298]. Although current clinical uses of sGC activators are limited to pulmonary hypertension and heart failure, other promising results exist for a host of other diseases including chronic kidney disease, systemic sclerosis, COPD, and even cancer [298,299,300,301,302,303,304,305].
Efficacy of sGC activators in cancer is not well studied, although two preclinical studies suggest that sGC can mitigate platinum chemotherapeutic resistance in oropharyngeal squamous cell carcinoma [304,305]. Nevertheless, some sGC activators were noted to be metabolites of pro-carcinogenic organic compounds, which the authors hypothesized could be partly responsible for carcinogenesis [306]. Furthermore, other studies suggested that peptides against sGC may actually be useful in treating androgen-independent prostate cancer [307]. Although of potential benefit for some cancers, the contradictory results of sGC activators provide further evidence of the dual-nature of NO in cancer pathogenesis. More preclinical studies are needed to demonstrate the efficacy or non-efficacy of sGC activator compounds in other cancers to make conclusions on their future potential as therapeutics.
6.4. Immunity Activators: PD-L1, PD-1, CSF1, and CSF1R
Immunotherapy is a developing area of cancer therapy with extremely promising results [308]. One such immunotherapy targets the programmed cell death-1 signaling cascade, at either the ligand (PD-L1) or the receptor (PD-1). PD-1 is commonly upregulated in response to immune activation and can be seen on the surface of CD4+, CD8+, and natural killer T cells, in addition to dendritic cells and B cells [309]. Meanwhile, the PD-L1 ligand is expressed on healthy peripheral tissues and serves to downregulate immunological response by decreasing proliferation, cytokine signaling, and overall survival of T-cells upon binding to PD-1 [309]. Lack of PD-1 or its ligand contributes to autoimmunity, as T-cells cells are free from peripheral immune surveillance feedback to continuously attack host tissues [309]. However, in cancer, the overexpression of PD-L1 allows the cancer to escape peripheral immune surveillance entirely, contributing to cell growth, immortality, and cancer progression [310]. Blockage of PD-L1 on cancer cells or PD-1 on immune cells via monoclonal antibodies has shown considerable benefit in clinical trials [311,312]. To date, three PD-L1 antibodies, atezolizumab, avelumab, and durvalumab, as well as two PD-1 antibodies, nivolumab, and pembrolizumab, have been approved for various malignancies [311]. The PD-1/PD-1L therapy appears to increase NO release, likely through increased immunological activity. In one study, exhaled NO significantly increased after administration of Nivolumab, suggesting increased lung inflammation, and increases in exhaled NO were especially notable in patients with COPD [190,313]. However, Nivolumab was not associated with an increased risk of COPD exacerbations or spirometry decline [313]. Interestingly, increased expression of PD-1L on cancer cells is thought to be mediated by a HIF-1α mechanism [314,315]. Treatment with a NO donor in combination with a PD-1/PD-1L therapy can potentially reverse this effect of HIF-1α on PD-L1 accumulation within the tumor [314]. Furthermore, the transcription factor YY1 is shown to further mediate the PD-1L expression, which NO can inhibit, further enhancing the effect of immunological therapy [314,316,317]. Inhibition of YY1 by NO may also activate apoptotic pathways, further increasing the effect of immunological therapy and chemotherapy. Preclinical models that target this pathway are promising, as NO modulation combined with PD-L1 therapy shows efficacy against breast cancer and melanoma in vivo [318,319]. However, current ongoing clinical trials (NCT03236935, NCT04095689) are utilizing L-NMMA, a non-selective NOS inhibitor, combined with PD-L1 therapy so the clinical outcomes of PD-1/PD-L1 augmentation with NO are unknown.
Similar to PD1 therapy, other immunotherapies also have critical interactions with NO. One such immunotherapy is anti-Colony Stimulating Factor-1 Receptor (CSF1R). CSF1R responds to its ligands, colony-stimulating factor-1 (CSF1), and interleukin-34 (IL-34) and functions to control macrophage proliferation and survival [320]. CSF1 is one of many macrophage growth factors; however, previous studies found that CSF1R was released during late-stage inflammatory reactions, which the authors suggested was related to the development of M2 macrophages [321]. CSF1R accumulation has been associated with poor patient prognosis [322]. Similarly, TAM M2 anti-inflammatory macrophages have also been associated with cancer progression and poor patient prognosis [323]. Therefore, targeting this pathway with immunotherapy has been suggested to be of great clinical benefit, especially for patients with advanced malignancies [322]. CSF1 application to breast cancer cells showed significant induction of iNOS activity and a corresponding increase in NO [324]. Furthermore, treatment with CSF1 antibodies reversed this effect, suggesting that iNOS was central to the function of the enzyme [324]. Interestingly, this observation clashes with previous studies suggesting that M2 macrophage activation decreases NO-mediated reactive species production [79,325]. The role of NO within this pathway remains poorly understood, and no active clinical trials are ongoing with CSF1R/CSF1 monoclonal antibodies and NO modulators.
7. Future Directions
Current therapeutic NO applications are limited by pharmacodynamics, pharmacokinetics, or systemic absorption and toxicity [207,326]. To help overcome these challenges, a number of NO-releasing and -containing compounds have been developed that enhance the pharmacodynamic properties of NO as an anticancer agent. One avenue that has shown efficacy is the conjugation of NO to a number of different molecules, such as non-steriodal anti-inflammatory drugs (NSAIDS) and chemotherapeutics. For example, NO conjugated with NSAIDS and doxorubicin either enhanced cytotoxicity or increased intracellular accumulation of chemotherapeutics within the tumor [207,327,328]. NO hybridization may also be effective in increasing the cytotoxicity of drugs not traditionally regarded as chemotherapeutics, such as lopinavir, an antiretroviral agent [329]. Nevertheless, NO or NO-modifying hybrid drugs may still lack the necessary specificity to engage in tumor-specific targeting and may still be limited by systemic toxicity.
Multiple interesting and innovative mechanisms have been proposed to aid in tumor-specific targeting. One approach is conjugating NO donors and PDE-is to designer antibodies for immunotherapy, which have shown efficacy [330,331,332]. For example, one study designed a NO-releasing antibody against CD24+, a widely expressed hepatocellular cancer marker, and was found to have high cellular uptake and apoptotic activity [332]. Another study validated NO-donating metal complexes conjugated to polyclonal antibodies in vitro and found an 80% increase in cytotoxicity with the NO donor used as an antibody-drug complex (ADC) [331]. Other ADC-utilizing PDE-4is have also shown promise, although the efficacy of novel NO ADCs largely remains unknown, especially when compared to existing treatment regimens [330]. Other approaches utilize specific environmental triggers to encourage NO release from a conjugated compound via an environmental trigger. For example, NO can be conjugated to doxorubicin such that it becomes released when exposed to wavelengths of visible light [333]. Further modification of the NO-drug-releasing compounds to nanoparticles and liposomes has been shown to increase the tumor selectivity, half-life, or wavelength responsiveness of NO-donating drugs combined with macromolecules [207,334]. Other organic and inorganic polymers or porous materials have also been proposed as NO-releasing agents, although their efficacy and biocompatibility are still under study [207,326]. Although preclinical studies of NO-donating macromolecules are promising, few studies have tested the efficacy of these compounds in vivo, suggesting that clinical applications of such compounds are far off. Thus, the future of NO therapy may lie upon clinicians finding the right mechanism to deliver NO alongside chemotherapy.
8. Conclusions
The discovery of multiple NO-mediated pathways within cancer has unlocked a number of novel NO-based therapies. Many of these novel therapies center around delivery of NO directly to the tumor and TME. Such localized increases in NO may reverse chemotherapeutic and radiotherapeutic resistance mediated by HIF-1α, although preclinical trials have suggested the efficacy with a number of other mechanisms. However, a primary limitation is the controlled delivery of NO directly to the tumor that tightly regulates localized NO concentration while minimizing side effects. Multiple promising studies have supported the efficacy of NO-releasing biomolecules to aid in NO delivery. Advances in biomaterials, combined with multiple clinical trials demonstrating the efficacy of NO-related therapies alongside radio-, immuno-, and chemotherapy, suggest that the future of NO as an anticancer agent has only begun.
Author Contributions
Conceptualization, J.M., A.V., J.M.H., R.R., and H.A.; methodology, J.M., A.V.; formal analysis/literature review, J.M., A.V., K.S., O.R.; investigation, J.M., A.V., K.S., G.G., O.R.; re-sources, J.M., A.V., K.S., O.R.; data curation, J.M., A.V., K.S., O.R.; writing—original draft preparation, J.M., A.V., K.S., O.R.; writing—review and editing, J.M.; visualization, G.G.; supervision, J.M.H., R.R., and H.A.; project administration, J.M.H., R.R., and H.A.; funding acquisi-tion, J.M.H., R.R., and H.A. All authors have read and agreed to the published version of the manuscript.
Funding
We would like to thank the American Urological Association Research Scholar Award for H.A. and the American Cancer Society for R.R. J.M.H. is supported by NIH grants 1R01 HL137355, 1R01 HL107110, 1R01 HL134558, 5R01 CA136387, and 5UM1 HL113460, and the Soffer Family Foundation.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Disclosures
J.M.H. discloses a relationship with Vestion Inc. that includes equity, board membership, and consulting. J.M.H. is the Chief Scientific Officer, a compensated consultant and advisory board member for Longeveron, and holds equity in Longeveron. J.M.H. is also the co-inventor of intellectual property licensed to Longeveron.
Abbreviations
cGMP: cyclic guanosine monophosphate; GTP: guanosine triphosphate; NADP+/NADPH: Nicotinamide adenine dinucleotide phosphate; O2: Oxygen; H2O: Water; PPi: Pyrophosphate; GMP: guanosine monophosphate; PDE-5 phosphodiesterase 5; NO: Nitric oxide; sGC: soluble guanyl cyclase; STAT3: Signal transducer and activator of transcription 3; NO: Nitric Oxide; CNS: Central Nervous System; HEENT: Head, eyes, ears, neck and throat; AKT: Protein kinase B; FAS-L: Tumor necrosis factor ligand superfamily member 6; BCL-2: B-cell lymphoma 2; HIF-1α: Hypoxia inducible factor 1α; iNOS: inducible nitric oxide synthase; eNOS: endothelial nitric oxide synthase; nNOS: neuronal nitric oxide synthase; PTN: pleiotrophin; ERK: extracellular signal regulated kinase; VEGF: vascular endothelial growth factor; CXCR4: C-X-C chemokine receptor type 4; CRPC: castration-resistant prostate cancer; MMP: Matrix metalloproteinase; TIMP-1: metallopeptidase inhibitor 1; PI3-k: phosphatidylinositol 3-kinase; WNT: Wingless-related integration site; COX-2: Cyclooxygenase-2; NF-κB: Nuclear factor kappa B.
References
- Soni, Y.; Softness, K.; Arora, H.; Ramasamy, R. The Yin Yang Role of Nitric Oxide in Prostate Cancer. Am. J. Mens. Health 2020, 14, 1557988320903191. [Google Scholar] [CrossRef] [PubMed]
- Burke, A.J.; Sullivan, F.J.; Giles, F.J.; Glynn, S.A. The yin and yang of nitric oxide in cancer progression. Carcinogenesis 2013, 34, 503–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bentz, B.G.; Simmons, R.L.; Haines, G.K., 3rd; Radosevich, J.A. The yin and yang of nitric oxide: Reflections on the physiology and pathophysiology of NO. Head Neck 2000, 22, 71–83. [Google Scholar] [CrossRef]
- Kamm, A.; Przychodzen, P.; Kuban-Jankowska, A.; Jacewicz, D.; Dabrowska, A.M.; Nussberger, S.; Wozniak, M.; Gorska-Ponikowska, M. Nitric oxide and its derivatives in the cancer battlefield. Nitric Oxide 2019, 93, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Subapriya, R.; Kumaraguruparan, R.; Ramachandran, C.R.; Nagini, S. Oxidant-antioxidant status in patients with oral squamous cell carcinomas at different intraoral sites. Clin. Biochem. 2002, 35, 489–493. [Google Scholar] [CrossRef]
- Habib, A.A.S. Biochemistry of Nitric Oxide. Indian J. Clin. Biochem. 2011, 26, 3–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiming, X.U.; Liu, L.Z.; Loizidou, M.; Ahmed, M.; Charles, I.G. The role of nitric oxide in cancer. Cell Res. 2002, 12, 311–320. [Google Scholar]
- Moncada, S.; Palmer, R.M.J.; Higgs, E.A. Biosynthesis of nitric oxide from l-arginine. Biochem. Pharmacol. 1989, 38, 1709–1715. [Google Scholar] [CrossRef]
- Benjamin, N.; O’Driscoll, F.; Dougall, H.; Duncan, C.; Smith, L.; Golden, M.; McKenzie, H. Stomach NO synthesis. Nature 1994, 368, 502. [Google Scholar] [CrossRef]
- Lundberg, J.O.; Weitzberg, E.; Lundberg, J.M.; Alving, K. Intragastric nitric oxide production in humans: Measurements in expelled air. Gut 1994, 35, 1543–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zweier, J.L.; Wang, P.; Samouilov, A.; Kuppusamy, P. Enzyme-independent formation of nitric oxide in biological tissues. Nat. Med. 1995, 1, 804–809. [Google Scholar] [CrossRef] [PubMed]
- Cosby, K.; Partovi, K.S.; Crawford, J.H.; Patel, R.P.; Reiter, C.D.; Martyr, S.; Yang, B.K.; Waclawiw, M.A.; Zalos, G.; Xu, X.; et al. Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nat. Med. 2003, 9, 1498–1505. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.M.; Wu, Z.F.; Pang, B.X.; Jin, L.Y.; Qin, L.Z.; Wang, S.L. From Nitrate to Nitric Oxide: The Role of Salivary Glands and Oral Bacteria. J. Dent. Res. 2016, 95, 1452–1456. [Google Scholar] [CrossRef] [PubMed]
- Duncan, C.; Dougall, H.; Johnston, P.; Green, S.; Brogan, R.; Leifert, C.; Smith, L.; Golden, M.; Benjamin, N. Chemical generation of nitric oxide in the mouth from the enterosalivary circulation of dietary nitrate. Nat. Med. 1995, 1, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Gardner, P.R.; Gardner, A.M.; Martin, L.A.; Salzman, A.L. Nitric oxide dioxygenase: An enzymic function for flavohemoglobin. Proc. Natl. Acad. Sci. USA 1998, 95, 10378–10383. [Google Scholar] [CrossRef] [Green Version]
- Rassaf, T.; Flögel, U.; Drexhage, C.; Hendgen-Cotta, U.; Kelm, M.; Schrader, J. Nitrite reductase function of deoxymyoglobin: Oxygen sensor and regulator of cardiac energetics and function. Circ. Res. 2007, 100, 1749–1754. [Google Scholar] [CrossRef]
- Shiva, S.; Huang, Z.; Grubina, R.; Sun, J.; Ringwood, L.A.; MacArthur, P.H.; Xu, X.; Murphy, E.; Darley-Usmar, V.M.; Gladwin, M.T. Deoxymyoglobin is a nitrite reductase that generates nitric oxide and regulates mitochondrial respiration. Circ. Res. 2007, 100, 654–661. [Google Scholar] [CrossRef]
- Helms, C.; Kim-Shapiro, D.B. Hemoglobin-mediated nitric oxide signaling. Free Radic. Biol. Med. 2013, 61, 464–472. [Google Scholar] [CrossRef] [Green Version]
- Sibmooh, N.; Piknova, B.; Schechter, A.N. Ascorbic Acid Catalyzes Nitric Oxide Production from Nitrite Ions. Blood 2006, 108, 1574. [Google Scholar] [CrossRef]
- Carlsson, S.; Wiklund, N.P.; Engstrand, L.; Weitzberg, E.; Lundberg, J.O. Effects of pH, nitrite, and ascorbic acid on nonenzymatic nitric oxide generation and bacterial growth in urine. Nitric Oxide 2001, 5, 580–586. [Google Scholar] [CrossRef]
- Godber, B.L.; Doel, J.J.; Sapkota, G.P.; Blake, D.R.; Stevens, C.R.; Eisenthal, R.; Harrison, R. Reduction of nitrite to nitric oxide catalyzed by xanthine oxidoreductase. J. Biol. Chem. 2000, 275, 7757–7763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocha, B.S.; Gago, B.; Barbosa, R.M.; Laranjinha, J. Dietary polyphenols generate nitric oxide from nitrite in the stomach and induce smooth muscle relaxation. Toxicology 2009, 265, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Millar, T.M.; Stevens, C.R.; Benjamin, N.; Eisenthal, R.; Harrison, R.; Blake, D.R. Xanthine oxidoreductase catalyses the reduction of nitrates and nitrite to nitric oxide under hypoxic conditions. FEBS Lett. 1998, 427, 225–228. [Google Scholar] [CrossRef] [Green Version]
- Gladwin, M.T.; Raat, N.J.H.; Shiva, S.; Dezfulian, C.; Hogg, N.; Kim-Shapiro, D.B.; Patel, R.P. Nitrite as a vascular endocrine nitric oxide reservoir that contributes to hypoxic signaling, cytoprotection, and vasodilation. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H2026–H2035. [Google Scholar] [CrossRef] [PubMed]
- Nagababu, E.; Ramasamy, S.; Abernethy, D.R.; Rifkind, J.M. Active nitric oxide produced in the red cell under hypoxic conditions by deoxyhemoglobin-mediated nitrite reduction. J. Biol. Chem. 2003, 278, 46349–46356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klatt, P.; Lamas, S. Regulation of protein function by S-glutathiolation in response to oxidative and nitrosative stress. Eur. J. Biochem. 2000, 267, 4928–4944. [Google Scholar] [CrossRef]
- Stamler, J.S.; Lamas, S.; Fang, F.C. Nitrosylation. Cell 2001, 106, 675–683. [Google Scholar] [CrossRef] [Green Version]
- Tannenbaum, S.R.; White, F.M. Regulation and specificity of S-nitrosylation and denitrosylation. ACS Chem. Biol. 2006, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Divya Seth, J.S.S. The SNO-proteome: Causation and Classifications. Curr. Opin. Chem. Biol. 2011, 15, 129. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, D.R.; Treuer, A.; Sun, Q.A.; Stamler, J.S.; Hare, J.M. S-Nitrosylation of cardiac ion channels. J. Cardiovasc. Pharmacol. 2009, 54. [Google Scholar] [CrossRef] [Green Version]
- Beigi, F.; Gonzalez, D.R.; Minhas, K.M.; Sun, Q.-A.; Foster, M.W.; Khan, S.A.; Treuer, A.V.; Dulce, R.A.; Harrison, R.W.; Saraiva, R.M.; et al. Dynamic denitrosylation via S-nitrosoglutathione reductase regulates cardiovascular function. Proc. Natl. Acad. Sci. USA 2012, 109, 4314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, C.; Li, Y.; Huang, X.; Wei, M.; Huang, Y.; Tang, Z.; Huang, H.; Zhou, W.; Wang, Y.; Hu, J. Extensive protein S-nitrosylation associated with human pancreatic ductal adenocarcinoma pathogenesis. Cell Death Dis. 2019, 10, 914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanhueza, C.; Bennett, J.C.; Valenzuela-Valderrama, M.; Contreras, P.; Lobos-González, L.; Campos, A.; Wehinger, S.; Lladser, Á.; Kiessling, R.; Leyton, L.; et al. Caveolin-1-Mediated Tumor Suppression Is Linked to Reduced HIF1α S-Nitrosylation and Transcriptional Activity in Hypoxia. Cancers 2020, 12, 2349. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Huang, M.; Xu, P.; Wang, M.; Ye, S.; Wang, Q.; Zeng, S.; Chen, X.; Gao, W.; Chen, J.; et al. Microcystins-LR induced apoptosis via S-nitrosylation of GAPDH in colorectal cancer cells. Ecotoxicol. Environ. Saf. 2020, 190, 110096. [Google Scholar] [CrossRef] [PubMed]
- Hare, J.M.; Stamler, J.S. NO/redox disequilibrium in the failing heart and cardiovascular system. J. Clin. Investig. 2005, 115, 509–517. [Google Scholar] [CrossRef] [Green Version]
- Foster, M.W.; McMahon, T.J.; Stamler, J.S. S-nitrosylation in health and disease. Trends Mol. Med. 2003, 9, 160–168. [Google Scholar] [CrossRef]
- Wang, Z. Protein S-nitrosylation and cancer. Cancer Lett. 2012, 320, 123–129. [Google Scholar] [CrossRef]
- Grek, C.L.; Zhang, J.; Manevich, Y.; Townsend, D.M.; Tew, K.D. Causes and consequences of cysteine S-glutathionylation. J. Biol. Chem. 2013, 288, 26497–26504. [Google Scholar] [CrossRef] [Green Version]
- Radi, R. Nitric oxide, oxidants, and protein tyrosine nitration. Proc. Natl. Acad. Sci. USA 2004, 101, 4003–4008. [Google Scholar] [CrossRef] [Green Version]
- Bartesaghi, S.; Radi, R. Fundamentals on the biochemistry of peroxynitrite and protein tyrosine nitration. Redox Biol. 2018, 14, 618–625. [Google Scholar] [CrossRef]
- Aulak, K.S.; Koeck, T.; Crabb, J.W.; Stuehr, D.J. Dynamics of protein nitration in cells and mitochondria. Am. J. Physiol. Heart Circ. Physiol. 2004, 286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radi, R. Protein tyrosine nitration: Biochemical mechanisms and structural basis of functional effects. Acc. Chem. Res. 2013, 46, 550–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ischiropoulos, H. Protein tyrosine nitration—An update. Arch. Biochem. Biophys. 2009, 484, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Peak, T.C.; Richman, A.; Gur, S.; Yafi, F.A.; Hellstrom, W.J.G. The Role of PDE5 Inhibitors and the NO/cGMP Pathway in Cancer. Sex. Med. Rev. 2016, 4, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Derbyshire, E.R.; Marletta, M.A. Structure and regulation of soluble guanylate cyclase. Annu. Rev. Biochem. 2012, 81, 533–559. [Google Scholar] [CrossRef] [Green Version]
- Stone, J.R.; Marletta, M.A. Spectral and kinetic studies on the activation of soluble guanylate cyclase by nitric oxide. Biochemistry 1996, 35, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Francis, S.H.; Busch, J.L.; Corbin, J.D.; Sibley, D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol. Rev. 2010, 62, 525–563. [Google Scholar] [CrossRef]
- Carvajal, J.A.; Germain, A.M.; Huidobro-Toro, J.P.; Weiner, C.P. Molecular mechanism of cGMP-mediated smooth muscle relaxation. J. Cell. Physiol. 2000, 184, 409–420. [Google Scholar] [CrossRef]
- Hare, J.M. Nitroso-redox balance in the cardiovascular system. N. Engl. J. Med. 2004, 351. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, D.R.; Treuer, A.V.; Castellanos, J.; Dulce, R.A.; Hare, J.M. Impaired S-nitrosylation of the ryanodine receptor caused by xanthine oxidase activity contributes to calcium leak in heart failure. J. Biol. Chem. 2010, 285. [Google Scholar] [CrossRef] [Green Version]
- Lind, M.; Hayes, A.; Caprnda, M.; Petrovic, D.; Rodrigo, L.; Kruzliak, P.; Zulli, A. Inducible nitric oxide synthase: Good or bad? Biomed. Pharmacother. 2017, 93, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Bogdan, C. Nitric oxide and the immune response. Nat. Immunol. 2001, 2, 907–916. [Google Scholar] [CrossRef] [PubMed]
- MacMicking, J.; Xie, Q.W.; Nathan, C. Nitric oxide and macrophage function. Annu. Rev. Immunol. 1997, 15, 323–350. [Google Scholar] [CrossRef] [PubMed]
- Miwa, M.; Stuehr, D.J.; Marletta, M.A.; Wishnok, J.S.; Tannenbaum, S.R. Nitrosation of amines by stimulated macrophages. Carcinogenesis 1987, 8, 955–958. [Google Scholar] [CrossRef] [PubMed]
- Stuehr, D.J.; Marletta, M.A. Induction of nitrite/nitrate synthesis in murine macrophages by BCG infection, lymphokines, or interferon-gamma. J. Immunol. 1987, 139, 518–525. [Google Scholar] [PubMed]
- Wink, D.A.; Hines, H.B.; Cheng, R.Y.S.; Switzer, C.H.; Flores-Santana, W.; Vitek, M.P.; Ridnour, L.A.; Colton, C.A. Nitric oxide and redox mechanisms in the immune response. J. Leukoc. Biol. 2011, 89, 873–891. [Google Scholar] [CrossRef] [Green Version]
- Hernansanz-Agustín, P.; Izquierdo-Álvarez, A.; García-Ortiz, A.; Ibiza, S.; Serrador, J.M.; Martínez-Ruiz, A. Nitrosothiols in the immune system: Signaling and protection. Antioxid. Redox Signal. 2013, 18, 288–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karupiah, G.; Chen, J.H.; Mahalingam, S.; Nathan, C.F.; MacMicking, J.D. Rapid interferon gamma-dependent clearance of influenza A virus and protection from consolidating pneumonitis in nitric oxide synthase 2-deficient mice. J. Exp. Med. 1998, 188, 1541–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Hausladen, A.; Zeng, M.; Que, L.; Heitman, J.; Stamler, J.S. A metabolic enzyme for S-nitrosothiol conserved from bacteria to humans. Nature 2001, 410, 490–494. [Google Scholar] [CrossRef] [PubMed]
- Stoyanovsky, D.A.; Tyurina, Y.Y.; Tyurin, V.A.; Anand, D.; Mandavia, D.N.; Gius, D.; Ivanova, J.; Pitt, B.; Billiar, T.R.; Kagan, V.E. Thioredoxin and lipoic acid catalyze the denitrosation of low molecular weight and protein S-nitrosothiols. J. Am. Chem. Soc. 2005, 127, 15815–15823. [Google Scholar] [CrossRef]
- Davis, A.S.; Vergne, I.; Master, S.S.; Kyei, G.B.; Chua, J.; Deretic, V. Mechanism of inducible nitric oxide synthase exclusion from mycobacterial phagosomes. PLoS Pathog. 2007, 3, e186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groeneveld, P.H.; Kwappenberg, K.M.; Langermans, J.A.; Nibbering, P.H.; Curtis, L. Relation between pro- and anti-inflammatory cytokines and the production of nitric oxide (NO) in severe sepsis. Cytokine 1997, 9, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Madrigal, J. The Increase in TNF-α Levels Is Implicated in NF-κB Activation and Inducible Nitric Oxide Synthase Expression in Brain Cortex after Immobilization Stress. Neuropsychopharmacology 2002, 26, 155–163. [Google Scholar] [CrossRef]
- Murphy, M.P. Nitric oxide and cell death. Biochim. Biophys. Acta (BBA)-Bioenerg. 1999, 1411, 401–414. [Google Scholar] [CrossRef] [Green Version]
- Spiller, F.; Formiga, R.O.; da Silva Coimbra, J.F.; Alves-Filho, J.C.; Cunha, T.M.; Cunha, F.Q. Targeting nitric oxide as a key modulator of sepsis, arthritis and pain. Nitric Oxide 2019, 89, 32–40. [Google Scholar] [CrossRef]
- Kröncke, K.D.; Fehsel, K.; Kolb-Bachofen, V. Inducible nitric oxide synthase in human diseases. Clin. Exp. Immunol. 1998, 113, 147–156. [Google Scholar] [CrossRef]
- McInnes, I.B.; Leung, B.P.; Field, M.; Wei, X.Q.; Huang, F.P.; Sturrock, R.D.; Kinninmonth, A.; Weidner, J.; Mumford, R.; Liew, F.Y. Production of nitric oxide in the synovial membrane of rheumatoid and osteoarthritis patients. J. Exp. Med. 1996, 184, 1519–1524. [Google Scholar] [CrossRef]
- Matthews, J.R.; Botting, C.H.; Panico, M.; Morris, H.R.; Hay, R.T. Inhibition of NF-kappaB DNA binding by nitric oxide. Nucleic Acids Res. 1996, 24, 2236–2242. [Google Scholar] [CrossRef]
- Zamora, R.; Vodovotz, Y.; Billiar, T.R. Inducible Nitric Oxide Synthase and Inflammatory Diseases. Mol. Med. 2000, 6, 347–373. [Google Scholar] [CrossRef] [Green Version]
- Nagy, G.; Clark, J.M.; Buzás, E.I.; Gorman, C.L.; Cope, A.P. Nitric oxide, chronic inflammation and autoimmunity. Immunol. Lett. 2007, 111, 1–5. [Google Scholar] [CrossRef]
- Kolb, H.; Kolb-Bachofen, V. Nitric oxide in autoimmune disease: Cytotoxic or regulatory mediator? Immunol. Today 1998, 19, 556–561. [Google Scholar] [CrossRef]
- Coleman, J.W. Nitric oxide in immunity and inflammation. Int. Immunopharmacol. 2001, 1, 1397–1406. [Google Scholar] [CrossRef]
- Choudhari, S.K.; Chaudhary, M.; Bagde, S.; Gadbail, A.R.; Joshi, V. Nitric oxide and cancer: A review. World J. Surg. Oncol. 2013, 11, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lala, P.K.; Chakraborty, C. Role of nitric oxide in carcinogenesis and tumour progression. Lancet Oncol. 2001, 2, 149–156. [Google Scholar] [CrossRef]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- PeÑarando, J.; Aranda, E.; RodrÍguez-Ariza, A. Immunomodulatory roles of nitric oxide in cancer: Tumor microenvironment says “NO” to antitumor immune response. Transl. Res. 2019, 210, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.M. The many faces of macrophage activation. J. Leukoc. Biol. 2003, 73, 209–212. [Google Scholar] [CrossRef]
- Mosser, D.M.; Zhang, X. Activation of murine macrophages. Curr. Protoc. Immunol. 2008, 83, 14.2.1–14.2.8. [Google Scholar] [CrossRef]
- Weigert, A.; Brüne, B. Nitric oxide, apoptosis and macrophage polarization during tumor progression. Nitric Oxide 2008, 19, 95–102. [Google Scholar] [CrossRef]
- Brüne, B. Nitric oxide: NO apoptosis or turning it ON? Cell Death Differ. 2003, 10, 864–869. [Google Scholar] [CrossRef]
- Jeannin, J.-F.; Leon, L.; Cortier, M.; Sassi, N.; Paul, C.; Bettaieb, A. Nitric oxide-induced resistance or sensitization to death in tumor cells. Nitric Oxide 2008, 19, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Nizet, V.; Johnson, R.S. Interdependence of hypoxic and innate immune responses. Nat. Rev. Immunol. 2009, 9, 609–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walmsley, S.R.; Chilvers, E.R.; Whyte, M.K.B. Hypoxia. Hypoxia, hypoxia inducible factor and myeloid cell function. Arthritis Res. Ther. 2009, 11, 219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahat, M.A.; Bitterman, H.; Lahat, N. Molecular mechanisms regulating macrophage response to hypoxia. Front. Immunol. 2011, 2, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridnour, L.A.; Thomas, D.D.; Donzelli, S.; Espey, M.G.; Roberts, D.D.; Wink, D.A.; Isenberg, J.S. The biphasic nature of nitric oxide responses in tumor biology. Antioxid. Redox Signal. 2006, 8, 1329–1337. [Google Scholar] [CrossRef] [PubMed]
- Ziche, M.; Morbidelli, L. Molecular regulation of tumour angiogenesis by nitric oxide. Eur. Cytokine Netw. 2009, 20, 164–170. [Google Scholar] [CrossRef]
- Ridnour, L.A.; Windhausen, A.N.; Isenberg, J.S.; Yeung, N.; Thomas, D.D.; Vitek, M.P.; Roberts, D.D.; Wink, D.A. Nitric oxide regulates matrix metalloproteinase-9 activity by guanylyl-cyclase-dependent and -independent pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 16898–16903. [Google Scholar] [CrossRef] [Green Version]
- Martinez, F.O.; Helming, L.; Gordon, S. Alternative activation of macrophages: An immunologic functional perspective. Annu. Rev. Immunol. 2009, 27, 451–483. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S.; Martinez, F.O. Alternative activation of macrophages: Mechanism and functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef] [Green Version]
- Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. The Yin-Yang of tumor-associated macrophages in neoplastic progression and immune surveillance. Immunol. Rev. 2008, 222, 155–161. [Google Scholar] [CrossRef]
- Murdoch, C.; Giannoudis, A.; Lewis, C.E. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood 2004, 104, 2224–2234. [Google Scholar] [CrossRef] [PubMed]
- Hennemann, B.; Beckmann, G.; Eichelmann, A.; Rehm, A.; Andreesen, R. Phase I trial of adoptive immunotherapy of cancer patients using monocyte-derived macrophages activated with interferon gamma and lipopolysaccharide. Cancer Immunol. Immunother. 1998, 45, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Andreesen, R.; Hennemann, B.; Krause, S.W. Adoptive immunotherapy of cancer using monocyte-derived macrophages: Rationale, current status, and perspectives. J. Leukoc. Biol. 1998, 64, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Perske, C.; Lahat, N.; Levin, S.S.; Bitterman, H.; Hemmerlein, B.; Rahat, M.A. Loss of inducible nitric oxide synthase expression in the mouse renal cell carcinoma cell line RENCA is mediated by microRNA miR-146a. Am. J. Pathol. 2010, 177, 2046–2054. [Google Scholar] [CrossRef] [PubMed]
- Monnet, I.; Breau, J.-L.; Moro, D.; Lena, H.; Eymard, J.-C.; Ménard, O.; Vuillez, J.-P.; Chokri, M.; Romet-Lemonne, J.-L.; Lopez, M. Intrapleural infusion of activated macrophages and gamma-interferon in malignant pleural mesothelioma: A phase II study. Chest 2002, 121, 1921–1927. [Google Scholar] [CrossRef] [PubMed]
- Kees, T.; Egeblad, M. Innate immune cells in breast cancer--from villains to heroes? J. Mammary Gland Biol. Neoplasia 2011, 16, 189–203. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Coffelt, S.B.; Hughes, R.; Lewis, C.E. Tumor-associated macrophages: Effectors of angiogenesis and tumor progression. Biochim. Biophys. Acta 2009, 1796, 11–18. [Google Scholar] [CrossRef]
- Venneri, M.A.; De Palma, M.; Ponzoni, M.; Pucci, F.; Scielzo, C.; Zonari, E.; Mazzieri, R.; Doglioni, C.; Naldini, L. Identification of proangiogenic TIE2-expressing monocytes (TEMs) in human peripheral blood and cancer. Blood 2007, 109, 5276–5285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Palma, M.; Venneri, M.A.; Galli, R.; Sergi, L.; Politi, L.S.; Sampaolesi, M.; Naldini, L. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell 2005, 8, 211–226. [Google Scholar] [CrossRef] [Green Version]
- Lechner, M.; Lirk, P.; Rieder, J. Inducible nitric oxide synthase (iNOS) in tumor biology: The two sides of the same coin. Semin. Cancer Biol. 2005, 15, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Kundu, J.K.; Surh, Y.-J. Inflammation: Gearing the journey to cancer. Mutat. Res. 2008, 659, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.P.; He, P.; Subleski, J.; Hofseth, L.J.; Trivers, G.E.; Mechanic, L.; Hofseth, A.B.; Bernard, M.; Schwank, J.; Nguyen, G.; et al. Nitric oxide is a key component in inflammation-accelerated tumorigenesis. Cancer Res. 2008, 68, 7130–7136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glynn, S.A.; Boersma, B.J.; Dorsey, T.H.; Yi, M.; Yfantis, H.G.; Ridnour, L.A.; Martin, D.N.; Switzer, C.H.; Hudson, R.S.; Wink, D.A.; et al. Increased NOS2 predicts poor survival in estrogen receptor-negative breast cancer patients. J. Clin. Investig. 2010, 120, 3843–3854. [Google Scholar] [CrossRef] [PubMed]
- Ying, L.; Hofseth, A.B.; Browning, D.D.; Nagarkatti, M.; Nagarkatti, P.S.; Hofseth, L.J. Nitric oxide inactivates the retinoblastoma pathway in chronic inflammation. Cancer Res. 2007, 67, 9286–9293. [Google Scholar] [CrossRef] [Green Version]
- Melillo, G.; Taylor, L.S.; Brooks, A.; Cox, G.W.; Varesio, L. Regulation of inducible nitric oxide synthase expression in IFN-gamma-treated murine macrophages cultured under hypoxic conditions. J. Immunol. 1996, 157, 2638–2644. [Google Scholar]
- Heller, A. Apoptosis-inducing high (.)NO concentrations are not sustained either in nascent or in developed cancers. ChemMedChem 2008, 3, 1493–1499. [Google Scholar] [CrossRef]
- Leon, L.; Jeannin, J.-F.; Bettaieb, A. Post-translational modifications induced by nitric oxide (NO): Implication in cancer cells apoptosis. Nitric Oxide 2008, 19, 77–83. [Google Scholar] [CrossRef]
- Iyer, A.K.V.; Azad, N.; Wang, L.; Rojanasakul, Y. Role of S-nitrosylation in apoptosis resistance and carcinogenesis. Nitric Oxide 2008, 19, 146–151. [Google Scholar] [CrossRef] [Green Version]
- Taylor, B.S.; de Vera, M.E.; Ganster, R.W.; Wang, Q.; Shapiro, R.A.; Morris, S.M., Jr.; Billiar, T.R.; Geller, D.A. Multiple NF-kappaB enhancer elements regulate cytokine induction of the human inducible nitric oxide synthase gene. J. Biol. Chem. 1998, 273, 15148–15156. [Google Scholar] [CrossRef] [Green Version]
- Pautz, A.; Art, J.; Hahn, S.; Nowag, S.; Voss, C.; Kleinert, H. Regulation of the expression of inducible nitric oxide synthase. Nitric Oxide 2010, 23, 75–93. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Nathan, C. The high-output nitric oxide pathway: Role and regulation. J. Leukoc. Biol. 1994, 56, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhao, Q.; Matta, R.; Meng, X.; Liu, X.; Liu, C.-G.; Nelin, L.D.; Liu, Y. Inducible nitric-oxide synthase expression is regulated by mitogen-activated protein kinase phosphatase-1. J. Biol. Chem. 2009, 284, 27123–27134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, R.; Phillips, R.A.; Zhang, Y.; Khan, D.; Crasta, O.; Ahmed, S.A. Suppression of LPS-induced Interferon-gamma and nitric oxide in splenic lymphocytes by select estrogen-regulated microRNAs: A novel mechanism of immune modulation. Blood 2008, 112, 4591–4597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cianchi, F.; Cortesini, C.; Fantappiè, O.; Messerini, L.; Sardi, I.; Lasagna, N.; Perna, F.; Fabbroni, V.; Di Felice, A.; Perigli, G.; et al. Cyclooxygenase-2 activation mediates the proangiogenic effect of nitric oxide in colorectal cancer. Clin. Cancer Res. 2004, 10, 2694–2704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, E.; Noh, S.H.; Lee, Y.D.; Lee, H.Y.; Han, J.-W.; Lee, H.W.; Hong, S. Differential expression of nitric oxide synthase in human stomach cancer. Cancer Lett. 1999, 146, 173–180. [Google Scholar] [CrossRef]
- Franchi, A.; Massi, D.; Santucci, M.; Masini, E.; Degl’Innocenti, D.R.; Magnelli, L.; Fanti, E.; Naldini, A.; Ardinghi, C.; Carraro, F.; et al. Inducible nitric oxide synthase activity correlates with lymphangiogenesis and vascular endothelial growth factor-C expression in head and neck squamous cell carcinoma. J. Pathol. 2006, 208, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Gochman, E.; Mahajna, J.; Shenzer, P.; Dahan, A.; Blatt, A.; Elyakim, R.; Reznick, A.Z. The expression of iNOS and nitrotyrosine in colitis and colon cancer in humans. Acta Histochem. 2012, 114, 827–835. [Google Scholar] [CrossRef]
- Massi, D.; Franchi, A.; Sardi, I.; Magnelli, L.; Paglierani, M.; Borgognoni, L.; Reali, U.M.; Santucci, M. Inducible nitric oxide synthase expression in benign and malignant cutaneous melanocytic lesions. J. Pathol. 2001, 194, 194–200. [Google Scholar] [CrossRef]
- Ekmekcioglu, S.; Ellerhorst, J.A.; Prieto, V.G.; Johnson, M.M.; Broemeling, L.D.; Grimm, E.A. Tumor iNOS predicts poor survival for stage III melanoma patients. Int. J. Cancer 2006, 119, 861–866. [Google Scholar] [CrossRef]
- Zafirellis, K.; Zachaki, A.; Agrogiannis, G.; Gravani, K. Inducible nitric oxide synthase expression and its prognostic significance in colorectal cancer. APMIS 2010, 118, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Vakkala, M.; Kahlos, K.; Lakari, E.; Pääkkö, P.; Kinnula, V.; Soini, Y. Inducible nitric oxide synthase expression, apoptosis, and angiogenesis in in situ and invasive breast carcinomas. Clin. Cancer Res. 2000, 6, 2408–2416. [Google Scholar] [PubMed]
- Bulut, A.S.; Erden, E.; Sak, S.D.; Doruk, H.; Kursun, N.; Dincol, D. Significance of inducible nitric oxide synthase expression in benign and malignant breast epithelium: An immunohistochemical study of 151 cases. Virchows Arch. 2005, 447, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Kasper, H.-U.; Wolf, H.; Drebber, U.; Wolf, H.-K.; Kern, M.-A. Expression of inducible nitric oxide synthase and cyclooxygenase-2 in pancreatic adenocarcinoma: Correlation with microvessel density. World J. Gastroenterol. 2004, 10, 1918–1922. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Wang, C.H.; Chen, T.C.; Lin, H.C.; Yu, C.T.; Kuo, H.P. Increased level of exhaled nitric oxide and up-regulation of inducible nitric oxide synthase in patients with primary lung cancer. Br. J. Cancer 1998, 78, 534–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.-F.; Zhao, D.-H.; Qi, Y.; Wang, J.-G.; Zhao, M.; Xiao, K.; Xie, L.-X. The clinical value of exhaled nitric oxide in patients with lung cancer. Clin. Respir. J. 2018, 12, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.P.; Lewis, C.; Thomas, P.S. Exhaled breath analysis: Novel approach for early detection of lung cancer. Lung Cancer 2009, 63, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Dweik, R.A.; Boggs, P.B.; Erzurum, S.C.; Irvin, C.G.; Leigh, M.W.; Lundberg, J.O.; Olin, A.-C.; Plummer, A.L.; Taylor, D.R. American Thoracic Society Committee on Interpretation of Exhaled Nitric Oxide Levels (FENO) for Clinical Applications An official ATS clinical practice guideline: Interpretation of exhaled nitric oxide levels (FENO) for clinical applications. Am. J. Respir. Crit. Care Med. 2011, 184, 602–615. [Google Scholar] [CrossRef] [Green Version]
- Hare, J.M.; Nguyen, G.C.; Massaro, A.F.; Drazen, J.M.; Stevenson, L.W.; Colucci, W.S.; Fang, J.C.; Johnson, W.; Givertz, M.M.; Lucas, C. Exhaled nitric oxide: A marker of pulmonary hemodynamics in heart failure. J. Am. Coll. Cardiol. 2002, 40. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, F.R.; Scagliotti, G.V.; Mulshine, J.L.; Kwon, R.; Curran, W.J., Jr.; Wu, Y.-L.; Paz-Ares, L. Lung cancer: Current therapies and new targeted treatments. Lancet 2017, 389, 299–311. [Google Scholar] [CrossRef]
- American Cancer Society. Cancer Facts & Figures 2020; American Cancer Society: Atlanta, GA, USA, 2020. [Google Scholar]
- Gào, X.; Xuan, Y.; Benner, A.; Anusruti, A.; Brenner, H.; Schöttker, B. Nitric Oxide Metabolites and Lung Cancer Incidence: A Matched Case-Control Study Nested in the ESTHER Cohort. Oxid. Med. Cell. Longev. 2019, 2019, 6470950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masri, F. Role of nitric oxide and its metabolites as potential markers in lung cancer. Ann. Thorac. Med. 2010, 5, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Pignatelli, B.; Li, C.Q.; Boffetta, P.; Chen, Q.; Ahrens, W.; Nyberg, F.; Mukeria, A.; Bruske-Hohlfeld, I.; Fortes, C.; Constantinescu, V.; et al. Nitrated and oxidized plasma proteins in smokers and lung cancer patients. Cancer Res. 2001, 61, 778–784. [Google Scholar] [PubMed]
- Hsieh, Y.S.; Wang, H.C.; Tseng, T.H.; Chang, W.C.; Wang, C.J. Gaseous nitric oxide-induced 8-nitroguanine formation in human lung fibroblast cells and cell-free DNA. Toxicol. Appl. Pharmacol. 2001, 172, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Masri, F.A.; Comhair, S.A.A.; Koeck, T.; Xu, W.; Janocha, A.; Ghosh, S.; Dweik, R.A.; Golish, J.; Kinter, M.; Stuehr, D.J.; et al. Abnormalities in nitric oxide and its derivatives in lung cancer. Am. J. Respir. Crit. Care Med. 2005, 172, 597–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yongsanguanchai, N.; Pongrakhananon, V.; Mutirangura, A.; Rojanasakul, Y.; Chanvorachote, P. Nitric oxide induces cancer stem cell-like phenotypes in human lung cancer cells. Am. J. Physiol. Cell Physiol. 2015, 308, C89–C100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimoto, H.; Sasaki, J.; Matsumoto, M.; Suga, M.; Ando, Y.; Iggo, R.; Tada, M.; Saya, H.; Ando, M. Significant correlation of nitric oxide synthase activity and p53 gene mutation in stage I lung adenocarcinoma. Jpn. J. Cancer Res. 1998, 89, 696–702. [Google Scholar] [CrossRef] [PubMed]
- Luanpitpong, S.; Chanvorachote, P. Nitric Oxide and Aggressive Behavior of Lung Cancer Cells. Anticancer Res. 2015, 35, 4585–4592. [Google Scholar] [PubMed]
- Saisongkorh, V.; Maiuthed, A.; Chanvorachote, P. Nitric oxide increases the migratory activity of non-small cell lung cancer cells via AKT-mediated integrin αv and β1 upregulation. Cell. Oncol. 2016, 39, 449–462. [Google Scholar] [CrossRef]
- Wongvaranon, P.; Pongrakhananon, V.; Chunhacha, P.; Chanvorachote, P. Acquired resistance to chemotherapy in lung cancer cells mediated by prolonged nitric oxide exposure. Anticancer Res. 2013, 33, 5433–5444. [Google Scholar]
- Sanuphan, A.; Chunhacha, P.; Pongrakhananon, V.; Chanvorachote, P. Long-term nitric oxide exposure enhances lung cancer cell migration. Biomed. Res. Int. 2013, 2013, 186972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maciag, A.E.; Chakrapani, H.; Saavedra, J.E.; Morris, N.L.; Holland, R.J.; Kosak, K.M.; Shami, P.J.; Anderson, L.M.; Keefer, L.K. The Nitric Oxide Prodrug JS-K Is Effective against Non–Small-Cell Lung Cancer Cells In Vitro and In Vivo: Involvement of Reactive Oxygen Species. J. Pharmacol. Exp. Ther. 2011, 336, 313–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.M.; Upadhyaya, P.; Kassie, F. Nitric oxide-donating aspirin (NO-Aspirin) suppresses lung tumorigenesis in vitro and in vivo and these effects are associated with modulation of the EGFR signaling pathway. Carcinogenesis 2018, 39, 911–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, S.; Yang, W.; Wang, Y.; Ou, W.; Ma, Q.; Yu, C.; Ren, J.; Zhong, G.; Shi, H.; Yuan, Z.; et al. Cationic liposome-mediated nitric oxide synthase gene therapy enhances the antitumor effects of cisplatin in lung cancer. Int. J. Mol. Med. 2013, 31, 33–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basudhar, D.; Somasundaram, V.; de Oliveira, G.A.; Kesarwala, A.; Heinecke, J.L.; Cheng, R.Y.; Glynn, S.A.; Ambs, S.; Wink, D.A.; Ridnour, L.A. Nitric Oxide Synthase-2-Derived Nitric Oxide Drives Multiple Pathways of Breast Cancer Progression. Antioxid. Redox Signal. 2017, 26, 1044–1058. [Google Scholar] [CrossRef]
- Garrido, P.; Shalaby, A.; Walsh, E.M.; Keane, N.; Webber, M.; Keane, M.M.; Sullivan, F.J.; Kerin, M.J.; Callagy, G.; Ryan, A.E.; et al. Impact of inducible nitric oxide synthase (iNOS) expression on triple negative breast cancer outcome and activation of EGFR and ERK signaling pathways. Oncotarget 2017, 8, 80568–80588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Windham, P.F.; Tinsley, H.N. cGMP signaling as a target for the prevention and treatment of breast cancer. Semin. Cancer Biol. 2015, 31, 106–110. [Google Scholar] [CrossRef]
- Yasuoka, H.; Tsujimoto, M.; Yoshidome, K.; Nakahara, M.; Kodama, R.; Sanke, T.; Nakamura, Y. Cytoplasmic CXCR4 expression in breast cancer: Induction by nitric oxide and correlation with lymph node metastasis and poor prognosis. BMC Cancer 2008, 8, 340. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, Y.; Yasuoka, H.; Tsujimoto, M.; Yoshidome, K.; Nakahara, M.; Nakao, K.; Nakamura, M.; Kakudo, K. Nitric oxide in breast cancer: Induction of vascular endothelial growth factor-C and correlation with metastasis and poor prognosis. Clin. Cancer Res. 2006, 12, 1201–1207. [Google Scholar] [CrossRef] [Green Version]
- Nath, N.; Chattopadhyay, M.; Rodes, D.B.; Nazarenko, A.; Kodela, R.; Kashfi, K. Nitric Oxide-Releasing Aspirin Suppresses NF-κB Signaling in Estrogen Receptor Negative Breast Cancer Cells in Vitro and in Vivo. Molecules 2015, 20, 12481–12499. [Google Scholar] [CrossRef] [Green Version]
- Flaherty, R.L.; Intabli, H.; Falcinelli, M.; Bucca, G.; Hesketh, A.; Patel, B.A.; Allen, M.C.; Smith, C.P.; Flint, M.S. Stress hormone-mediated acceleration of breast cancer metastasis is halted by inhibition of nitric oxide synthase. Cancer Lett. 2019, 459, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Davila-Gonzalez, D.; Chang, J.C.; Billiar, T.R. NO and COX2: Dual targeting for aggressive cancers. Proc. Natl. Acad. Sci. USA 2017, 114, 13591–13593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duckles, S.P.; Miller, V.M. Hormonal modulation of endothelial NO production. Pflug. Arch. 2010, 459, 841–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yallampalli, C.; Dong, Y.L. Estradiol-17beta inhibits nitric oxide synthase (NOS)-II and stimulates NOS-III gene expression in the rat uterus. Biol. Reprod. 2000, 63, 34–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pance, A. Nitric oxide and hormones in breast cancer: Allies or enemies? Future Oncol. 2006, 2, 275–288. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.H.J.; Alalami, O.; van den Berg, H.W. Reduced expression of endothelial and inducible nitric oxide synthase in a human breast cancer cell line which has acquired estrogen independence. Cancer Lett. 1999, 144, 65–74. [Google Scholar] [CrossRef]
- Vakkala, M.; Paakko, P.; Soini, Y. eNOS expression is associated with the estrogen and progesterone receptor status in invasive breast carcinoma. Int. J. Oncol. 2000, 17, 667–671. [Google Scholar] [CrossRef]
- Simoncini, T.; Fu, X.-D.; Caruso, A.; Garibaldi, S.; Baldacci, C.; Giretti, M.S.; Mannella, P.; Flamini, M.I.; Sanchez, A.M.; Genazzani, A.R. Drospirenone increases endothelial nitric oxide synthesis via a combined action on progesterone and mineralocorticoid receptors. Hum. Reprod. 2007, 22, 2325–2334. [Google Scholar] [CrossRef] [Green Version]
- Bentrari, F.; Arnould, L.; Jackson, A.P.; Jeannin, J.-F.; Pance, A. Progesterone enhances cytokine-stimulated nitric oxide synthase II expression and cell death in human breast cancer cells. Lab. Investig. 2005, 85, 624–632. [Google Scholar] [CrossRef]
- Bardou, V.-J.; Arpino, G.; Elledge, R.M.; Osborne, C.K.; Clark, G.M. Progesterone receptor status significantly improves outcome prediction over estrogen receptor status alone for adjuvant endocrine therapy in two large breast cancer databases. J. Clin. Oncol. 2003, 21, 1973–1979. [Google Scholar] [CrossRef] [Green Version]
- Simeone, A.-M.; Broemeling, L.D.; Rosenblum, J.; Tari, A.M. HER2/neu reduces the apoptotic effects of N-(4-hydroxyphenyl)retinamide (4-HPR) in breast cancer cells by decreasing nitric oxide production. Oncogene 2003, 22, 6739–6747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simeone, A.-M.; Li, Y.-J.; Broemeling, L.D.; Johnson, M.M.; Tuna, M.; Tari, A.M. Cyclooxygenase-2 is essential for HER2/neu to suppress N- (4-hydroxyphenyl)retinamide apoptotic effects in breast cancer cells. Cancer Res. 2004, 64, 1224–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granados-Principal, S.; Liu, Y.; Guevara, M.L.; Blanco, E.; Choi, D.S.; Qian, W.; Patel, T.; Rodriguez, A.A.; Cusimano, J.; Weiss, H.L.; et al. Inhibition of iNOS as a novel effective targeted therapy against triple-negative breast cancer. Breast Cancer Res. 2015, 17, 1169. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Siegel, D.A.; King, J.B. Stage-specific incidence rates and trends of prostate cancer by age, race, and ethnicity, United States, 2004-2014. Ann. Epidemiol. 2018, 28, 328–330. [Google Scholar] [CrossRef] [PubMed]
- Uotila, P.; Valve, E.; Martikainen, P.; Nevalainen, M.; Nurmi, M.; Härkönen, P. Increased expression of cyclooxygenase-2 and nitric oxide synthase-2 in human prostate cancer. Urol. Res. 2001, 29, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Aaltoma, S.H.; Lipponen, P.K.; Kosma, V.M. Inducible nitric oxide synthase (iNOS) expression and its prognostic value in prostate cancer. Anticancer Res. 2001, 21, 3101–3106. [Google Scholar]
- Liao, W.; Ye, T.; Liu, H. Prognostic Value of Inducible Nitric Oxide Synthase (iNOS) in Human Cancer: A Systematic Review and Meta-Analysis. BioMed Res. Int. 2019, 2019, 6304851. [Google Scholar] [CrossRef] [Green Version]
- Arsova-Sarafinovska, Z.; Eken, A.; Matevska, N.; Erdem, O.; Sayal, A.; Savaser, A.; Banev, S.; Petrovski, D.; Dzikova, S.; Georgiev, V.; et al. Increased oxidative/nitrosative stress and decreased antioxidant enzyme activities in prostate cancer. Clin. Biochem. 2009, 42, 1228–1235. [Google Scholar] [CrossRef]
- Lee, K.-M.; Kang, D.; Park, S.K.; Berndt, S.I.; Reding, D.; Chatterjee, N.; Chanock, S.; Huang, W.-Y.; Hayes, R.B. Nitric oxide synthase gene polymorphisms and prostate cancer risk. Carcinogenesis 2009, 30, 621–625. [Google Scholar] [CrossRef] [Green Version]
- Dianat, S.S.; Margreiter, M.; Eckersberger, E.; Finkelstein, J.; Kuehas, F.; Herwig, R.; Ayati, M.; Lepor, H.; Djavan, B. Gene polymorphisms and prostate cancer: The evidence. BJU Int. 2009, 104, 1560–1572. [Google Scholar] [CrossRef]
- Jacobs, E.J.; Hsing, A.W.; Bain, E.B.; Stevens, V.L.; Wang, Y.; Chen, J.; Chanock, S.J.; Zheng, S.L.; Xu, J.; Thun, M.J.; et al. Polymorphisms in angiogenesis-related genes and prostate cancer. Cancer Epidemiol. Biomark. Prev. 2008, 17, 972–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polytarchou, C.; Hatziapostolou, M.; Poimenidi, E.; Mikelis, C.; Papadopoulou, A.; Parthymou, A.; Papadimitriou, E. Nitric oxide stimulates migration of human endothelial and prostate cancer cells through up-regulation of pleiotrophin expression and its receptor protein tyrosine phosphatase beta/zeta. Int. J. Cancer 2009, 124, 1785–1793. [Google Scholar] [CrossRef] [PubMed]
- Re, A.; Aiello, A.; Nanni, S.; Grasselli, A.; Benvenuti, V.; Pantisano, V.; Strigari, L.; Colussi, C.; Ciccone, S.; Mazzetti, A.P.; et al. Silencing of GSTP1, a prostate cancer prognostic gene, by the estrogen receptor-β and endothelial nitric oxide synthase complex. Mol. Endocrinol. 2011, 25, 2003–2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharifi, N. Androgen Deprivation Therapy for Prostate Cancer. JAMA 2005, 294, 238. [Google Scholar] [CrossRef] [PubMed]
- Kirby, M.; Hirst, C.; Crawford, E.D. Characterising the castration-resistant prostate cancer population: A systematic review. Int. J. Clin. Pract. 2011, 65, 1180–1192. [Google Scholar] [CrossRef]
- Cronauer, M.V.; Ince, Y.; Engers, R.; Rinnab, L.; Weidemann, W.; Suschek, C.V.; Burchardt, M.; Kleinert, H.; Wiedenmann, J.; Sies, H.; et al. Nitric oxide-mediated inhibition of androgen receptor activity: Possible implications for prostate cancer progression. Oncogene 2007, 26, 1875–1884. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Jia, L.; Zhang, Y.; Wu, D.; Xu, Z.; Ng, C.-F.; To, K.K.W.; Huang, Y.; Chan, F.L. Increased expression of activated endothelial nitric oxide synthase contributes to antiandrogen resistance in prostate cancer cells by suppressing androgen receptor transactivation. Cancer Lett. 2013, 328, 83–94. [Google Scholar] [CrossRef]
- Bhowmick, R.; Girotti, A.W. Pro-survival and pro-growth effects of stress-induced nitric oxide in a prostate cancer photodynamic therapy model. Cancer Lett. 2014, 343, 115–122. [Google Scholar] [CrossRef] [Green Version]
- Fahey, J.M.; Girotti, A.W. Accelerated migration and invasion of prostate cancer cells after a photodynamic therapy-like challenge: Role of nitric oxide. Nitric Oxide 2015, 49, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Thomas, L.N.; Morehouse, T.J.; Too, C.K.L. Testosterone and prolactin increase carboxypeptidase-D and nitric oxide levels to promote survival of prostate cancer cells. Prostate 2012, 72, 450–460. [Google Scholar] [CrossRef]
- Arora, H.; Panara, K.; Kuchakulla, M.; Kulandavelu, S.; Burnstein, K.L.; Schally, A.V.; Hare, J.M.; Ramasamy, R. Alterations of tumor microenvironment by nitric oxide impedes castration-resistant prostate cancer growth. Proc. Natl. Acad. Sci. USA 2018, 115, 11298–11303. [Google Scholar] [CrossRef] [Green Version]
- Stewart, G.D.; Ross, J.A.; McLaren, D.B.; Parker, C.C.; Habib, F.K.; Riddick, A.C.P. The relevance of a hypoxic tumour microenvironment in prostate cancer. BJU Int. 2010, 105, 8–13. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, G.A.; Cheng, R.Y.S.; Ridnour, L.A.; Basudhar, D.; Somasundaram, V.; McVicar, D.W.; Monteiro, H.P.; Wink, D.A. Inducible Nitric Oxide Synthase in the Carcinogenesis of Gastrointestinal Cancers. Antioxid. Redox Signal. 2017, 26, 1059–1077. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, H.P.; Rodrigues, E.G.; Reis, A.K.C.A.; Longo, L.S., Jr.; Ogata, F.T.; Moretti, A.I.S.; da Costa, P.E.; Teodoro, A.C.S.; Toledo, M.S.; Stern, A. Nitric oxide and interactions with reactive oxygen species in the development of melanoma, breast, and colon cancer: A redox signaling perspective. Nitric Oxide 2019, 89, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Bienz, M.; Clevers, H. Linking Colorectal Cancer to Wnt Signaling. Cell 2000, 103, 311–320. [Google Scholar] [CrossRef] [Green Version]
- Du, Q.; Zhang, X.; Liu, Q.; Zhang, X.; Bartels, C.E.; Geller, D.A. Nitric oxide production upregulates Wnt/β-catenin signaling by inhibiting Dickkopf-1. Cancer Res. 2013, 73, 6526–6537. [Google Scholar] [CrossRef] [Green Version]
- Ambs, S.; Merriam, W.G.; Bennett, W.P.; Felley-Bosco, E.; Ogunfusika, M.O.; Oser, S.M.; Klein, S.; Shields, P.G.; Billiar, T.R.; Harris, C.C. Frequent nitric oxide synthase-2 expression in human colon adenomas: Implication for tumor angiogenesis and colon cancer progression. Cancer Res. 1998, 58, 334–341. [Google Scholar]
- Araki, Y.; Okamura, S.; Hussain, S.P.; Nagashima, M.; He, P.; Shiseki, M.; Miura, K.; Harris, C.C. Regulation of cyclooxygenase-2 expression by the Wnt and ras pathways. Cancer Res. 2003, 63, 728–734. [Google Scholar]
- Goradel, N.H.; Najafi, M.; Salehi, E.; Farhood, B.; Mortezaee, K. Cyclooxygenase-2 in cancer: A review. J. Cell. Physiol. 2019, 234, 5683–5699. [Google Scholar] [CrossRef]
- Tak, P.P.; Firestein, G.S. NF-kappaB: A key role in inflammatory diseases. J. Clin. Investig. 2001, 107, 7–11. [Google Scholar] [CrossRef]
- Song, P.; Wu, L.; Guan, W. Dietary Nitrates, Nitrites, and Nitrosamines Intake and the Risk of Gastric Cancer: A Meta-Analysis. Nutrients 2015, 7, 9872–9895. [Google Scholar] [CrossRef]
- Iijima, K.; Grant, J.; McElroy, K.; Fyfe, V.; Preston, T.; McColl, K.E.L. Novel mechanism of nitrosative stress from dietary nitrate with relevance to gastro-oesophageal junction cancers. Carcinogenesis 2003, 24, 1951–1960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rieder, G.; Hofmann, J.A.; Hatz, R.A.; Stolte, M.; Enders, G.A. Up-regulation of inducible nitric oxide synthase in Helicobacter pylori-associated gastritis may represent an increased risk factor to develop gastric carcinoma of the intestinal type. Int. J. Med. Microbiol. 2003, 293, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, L.G.; McClary, H.; Loudis, J.M.; Chisari, F.V. Nitric oxide inhibits hepatitis B virus replication in the livers of transgenic mice. J. Exp. Med. 2000, 191, 1247–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwakiri, Y.; Kim, M.Y. Nitric oxide in liver diseases. Trends Pharmacol. Sci. 2015, 36, 524–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, M.; LaRusso, N.F.; Shapiro, R.A.; Billiar, T.R.; Gores, G.J. Nitric oxide-mediated inhibition of DNA repair potentiates oxidative DNA damage in cholangiocytes. Gastroenterology 2001, 120, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Toh, Y.; Oki, E.; Ohgaki, K.; Sakamoto, Y.; Ito, S.; Egashira, A.; Saeki, H.; Kakeji, Y.; Morita, M.; Sakaguchi, Y.; et al. Alcohol drinking, cigarette smoking, and the development of squamous cell carcinoma of the esophagus: Molecular mechanisms of carcinogenesis. Int. J. Clin. Oncol. 2010, 15, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Altieri, R.; Fontanella, M.; Agnoletti, A.; Panciani, P.P.; Spena, G.; Crobeddu, E.; Pilloni, G.; Tardivo, V.; Lanotte, M.; Zenga, F.; et al. Role of Nitric Oxide in Glioblastoma Therapy: Another Step to Resolve the Terrible Puzzle? Transl. Med. UniSa 2015, 12, 54–59. [Google Scholar] [PubMed]
- Thomsen, L.L.; Lawton, F.G.; Knowles, R.G.; Beesley, J.E.; Riveros-Moreno, V.; Moncada, S. Nitric oxide synthase activity in human gynecological cancer. Cancer Res. 1994, 54, 1352–1354. [Google Scholar] [PubMed]
- Seabra, A.B.; Durán, N. Nitric oxide donors for prostate and bladder cancers: Current state and challenges. Eur. J. Pharmacol. 2018, 826, 158–168. [Google Scholar] [CrossRef]
- Yarlagadda, K.; Hassani, J.; Foote, I.P.; Markowitz, J. The role of nitric oxide in melanoma. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Choe, W.; Kim, S.; Hwang, T.S.; Lee, S.S. Expression of inducible nitric oxide synthase in thyroid neoplasms: Immunohistochemical and molecular analysis. Pathol. Int. 2003, 53, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Gallo, O.; Masini, E.; Morbidelli, L.; Franchi, A.; Fini-Storchi, I.; Vergari, W.A.; Ziche, M. Role of nitric oxide in angiogenesis and tumor progression in head and neck cancer. J. Natl. Cancer Inst. 1998, 90, 587–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Utispan, K.; Koontongkaew, S. High Nitric Oxide Adaptation in Isogenic Primary and Metastatic Head and Neck Cancer Cells. Anticancer Res. 2020, 40, 2657–2665. [Google Scholar] [CrossRef] [PubMed]
- Toda, N.; Ayajiki, K.; Okamura, T. Nitric oxide and penile erectile function. Pharmacol. Ther. 2005, 106, 233–266. [Google Scholar] [CrossRef] [PubMed]
- Alimoradi, H.; Greish, K.; Gamble, A.B.; Giles, G.I. Controlled Delivery of Nitric Oxide for Cancer Therapy. Pharm. Nanotechnol. 2019, 7, 279–303. [Google Scholar] [CrossRef] [PubMed]
- Megson, I.L.; Webb, D.J. Nitric oxide donor drugs: Current status and future trends. Expert Opin. Investig. Drugs 2002, 11, 587–601. [Google Scholar] [PubMed]
- Grosse, G.M.; Schwedhelm, E.; Worthmann, H.; Choe, C.-U. Arginine Derivatives in Cerebrovascular Diseases: Mechanisms and Clinical Implications. Int. J. Mol. Sci. 2020, 21, 1798. [Google Scholar] [CrossRef] [Green Version]
- Howell, K.; Costello, C.M.; Sands, M.; Dooley, I.; McLoughlin, P. L-Arginine promotes angiogenesis in the chronically hypoxic lung: A novel mechanism ameliorating pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, L1042–L1050. [Google Scholar] [CrossRef] [Green Version]
- Csont, T.; Ferdinandy, P. Cardioprotective effects of glyceryl trinitrate: Beyond vascular nitrate tolerance. Pharmacol. Ther. 2005, 105, 57–68. [Google Scholar] [CrossRef]
- Ferreira, J.C.B.; Mochly-Rosen, D. Nitroglycerin Use in Myocardial Infarction Patients. Circ. J. 2012, 76, 15–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- den Uil, C.A.; Brugts, J.J. Impact of intravenous nitroglycerin in the management of acute decompensated heart failure. Curr. Heart Fail. Rep. 2015, 12, 87–93. [Google Scholar] [CrossRef] [PubMed]
- van Wezel, H.B.; Bovill, J.G.; Visser, C.A.; Koolen, J.J.; Janse, M.J.; Meijne, N.G.; Barendse, G.A.M.; Floor, L.M.; Djamin, R. Myocardial metabolism, catecholamine balance, and left ventricular function during coronary artery surgery: Effects of nitroprusside and nifedipine. J. Cardiothorac. Anesth. 1987, 1, 408–417. [Google Scholar] [CrossRef]
- Hess, W.; Schulte-Sasse, U.; Tarnow, J. Nifedipine versus nitroprusside for controlling hypertensive episodes during coronary artery bypass surgery. Eur. Heart J. 1984, 5, 140–145. [Google Scholar] [CrossRef]
- Miller, R.R.; Awan, N.A.; Joye, J.A.; Maxwell, K.S.; DeMaria, A.N.; Amsterdam, E.A.; Mason, D.T. Combined dopamine and nitroprusside therapy in congestive heart failure. Greater augmentation of cardiac performance by addition of inotropic stimulation to afterload reduction. Circulation 1977, 55, 881–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franciosa, J.; Limas, C.; Guiha, N.; Rodriguera, E.; Cohn, J. Improved left ventricular function during nitroprusside infusion in acute myocardial infarction. Lancet 1972, 299, 650–654. [Google Scholar] [CrossRef]
- Shapiro, B.; Fig, L.M. Management of pheochromocytoma. Endocrinol. Metab. Clin. North. Am. 1989, 18, 443–481. [Google Scholar] [CrossRef]
- Souto, S.; Palma, P.; Fregonesi, A.; Palma, T.; Reis, L.O. Vascular modifications of the clitoris induced by topic nitric oxide donor gel--preliminary study. J. Sex. Med. 2011, 8, 484–488. [Google Scholar] [CrossRef] [PubMed]
- Salas, E.; Langford, E.J.; Marrinan, M.T.; Martin, J.F.; Moncada, S.; de Belder, A.J. S-nitrosoglutathione inhibits platelet activation and deposition in coronary artery saphenous vein grafts in vitro and in vivo. Heart 1998, 80, 146–150. [Google Scholar] [CrossRef] [Green Version]
- Kaposzta, Z.; Clifton, A.; Molloy, J.; Martin, J.F.; Markus, H.S. S-nitrosoglutathione reduces asymptomatic embolization after carotid angioplasty. Circulation 2002, 106, 3057–3062. [Google Scholar] [CrossRef] [Green Version]
- Snyder, A.H.; McPherson, M.E.; Hunt, J.F.; Johnson, M.; Stamler, J.S.; Gaston, B. Acute Effects of Aerosolized S -Nitrosoglutathione in Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2002, 165, 922–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellisarii, F.I.; Radico, F.; Muscente, F.; Horowitz, J.; De Caterina, R. Nitrates and other nitric oxide donors in cardiology: Current positioning and perspectives. Cardiovasc. Drugs Ther. 2012, 26, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Fu, J.; Zhang, Y. Nitric Oxide Donor-Based Cancer Therapy: Advances and Prospects. J. Med. Chem. 2017, 60, 7617–7635. [Google Scholar] [CrossRef] [PubMed]
- Jing, X.; Yang, F.; Shao, C.; Wei, K.; Xie, M.; Shen, H.; Shu, Y. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol. Cancer 2019, 18, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasuda, H.; Yamaya, M.; Nakayama, K.; Sasaki, T.; Ebihara, S.; Kanda, A.; Asada, M.; Inoue, D.; Suzuki, T.; Okazaki, T.; et al. Randomized phase II trial comparing nitroglycerin plus vinorelbine and cisplatin with vinorelbine and cisplatin alone in previously untreated stage IIIB/IV non-small-cell lung cancer. J. Clin. Oncol. 2006, 24, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-S.; Chuang, M.-T.; Tsai, Y.-S.; Tsai, H.-M.; Lin, X.-Z. Nitroglycerine use in transcatheter arterial (chemo)embolization in patients with hepatocellular carcinoma and dual-energy CT assessment of Lipiodol retention. Eur. Radiol. 2012, 22, 2193–2200. [Google Scholar] [CrossRef] [PubMed]
- Siemens, D.R.; Heaton, J.P.W.; Adams, M.A.; Kawakami, J.; Graham, C.H. Phase II study of nitric oxide donor for men with increasing prostate-specific antigen level after surgery or radiotherapy for prostate cancer. Urology 2009, 74, 878–883. [Google Scholar] [CrossRef] [PubMed]
- Illum, H.; Wang, D.H.; Dowell, J.E.; Hittson, W.J.; Torrisi, J.R.; Meyer, J.; Huerta, S. Phase I dose escalation trial of nitroglycerin in addition to 5-fluorouracil and radiation therapy for neoadjuvant treatment of operable rectal cancer. Surgery 2015, 158, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Arrieta, O.; Blake, M.; de la Mata-Moya, M.D.; Corona, F.; Turcott, J.; Orta, D.; Alexander-Alatorre, J.; Gallardo-Rincón, D. Phase II study. Concurrent chemotherapy and radiotherapy with nitroglycerin in locally advanced non-small cell lung cancer. Radiother. Oncol. 2014, 111, 311–315. [Google Scholar] [CrossRef]
- Huerta, S.; Chilka, S.; Bonavida, B. Huerta Nitric Oxide donors: Novel cancer therapeutics (Review). Int. J. Oncol. 2008, 909–927. [Google Scholar] [CrossRef] [Green Version]
- Dingemans, A.-M.C.; Groen, H.J.M.; Herder, G.J.M.; Stigt, J.A.; Smit, E.F.; Bahce, I.; Burgers, J.A.; van den Borne, B.E.E.M.; Biesma, B.; Vincent, A.; et al. A randomized phase II study comparing paclitaxel-carboplatin-bevacizumab with or without nitroglycerin patches in patients with stage IV nonsquamous nonsmall-cell lung cancer: NVALT12 (NCT01171170)†. Ann. Oncol. 2015, 26, 2286–2293. [Google Scholar] [CrossRef] [PubMed]
- Reinmuth, N.; Meyer, A.; Hartwigsen, D.; Schaeper, C.; Huebner, G.; Skock-Lober, R.; Bier, A.; Gerecke, U.; Held, C.-P.; Reck, M. Randomized, double-blind phase II study to compare nitroglycerin plus oral vinorelbine plus cisplatin with oral vinorelbine plus cisplatin alone in patients with stage IIIB/IV non-small cell lung cancer (NSCLC). Lung Cancer 2014, 83, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Han, J.Y.; Nam, B.H.; Kim, H.Y.; Yoon, S.J.; Kim, H.T.; Lee, J.S. A randomized phase II study of irinotecan plus cisplatin versus irinotecan plus capecitabine with or without isosorbide-5-mononitrate in advanced non-small-cell lung cancer. Ann. Oncol. 2012, 23, 2925–2930. [Google Scholar] [CrossRef] [PubMed]
- Downie, I.P.; Umar, T.; Boote, D.J.; Mellor, T.K.; Hoffman, G.R.; Brennan, P.A. Does administration of isosorbide mononitrate affect cellular proliferation in oral squamous cell carcinoma? A prospective randomized clinical study. J. Oral Maxillofac. Surg. 2004, 62, 1064–1068. [Google Scholar] [CrossRef]
- Pervin, S.; Singh, R.; Gau, C.L.; Edamatsu, H.; Tamanoi, F.; Chaudhuri, G. Potentiation of nitric oxide-induced apoptosis of MDA-MB-468 cells by farnesyltransferase inhibitor: Implications in breast cancer. Cancer Res. 2001, 61, 4701–4706. [Google Scholar]
- Huerta-Yepez, S.; Vega, M.; Jazirehi, A.; Garban, H.; Hongo, F.; Cheng, G.; Bonavida, B. Nitric oxide sensitizes prostate carcinoma cell lines to TRAIL-mediated apoptosis via inactivation of NF-kappa B and inhibition of Bcl-xl expression. Oncogene 2004, 23, 4993–5003. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Wang, L.; Mollica, M.; Re, A.T.; Wu, S.; Zuo, L. Nitric oxide in cancer metastasis. Cancer Lett. 2014, 353, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Weyerbrock, A.; Walbridge, S.; Pluta, R.M.; Saavedra, J.E.; Keefer, L.K.; Oldfield, E.H. Selective opening of the blood-tumor barrier by a nitric oxide donor and long-term survival in rats with C6 gliomas. J. Neurosurg. 2003, 99, 728–737. [Google Scholar] [CrossRef] [PubMed]
- Baritaki, S.; Huerta-Yepez, S.; Sahakyan, A.; Karagiannides, I.; Bakirtzi, K.; Jazirehi, A.; Bonavida, B. Mechanisms of nitric oxide-mediated inhibition of EMT in cancer: Inhibition of the metastasis-inducer Snail and induction of the metastasis-suppressor RKIP. Cell Cycle 2010, 9, 4931–4940. [Google Scholar] [CrossRef] [Green Version]
- Pipili-Synetos, E.; Papageorgiou, A.; Sakkoula, E.; Sotiropoulou, G.; Fotsis, T.; Karakiulakis, G.; Maragoudakis, M.E. Inhibition of angiogenesis, tumour growth and metastasis by the NO-releasing vasodilators, isosorbide mononitrate and dinitrate. Br. J. Pharmacol. 1995, 116, 1829–1834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kseibati, M.O.; Shehatou, G.S.G.; Sharawy, M.H.; Eladl, A.E.; Salem, H.A. Nicorandil ameliorates bleomycin-induced pulmonary fibrosis in rats through modulating eNOS, iNOS, TXNIP and HIF-1α levels. Life Sci. 2020, 246, 117423. [Google Scholar] [CrossRef]
- Porshneva, K.; Papiernik, D.; Psurski, M.; Łupicka-Słowik, A.; Matkowski, R.; Ekiert, M.; Nowak, M.; Jarosz, J.; Banach, J.; Milczarek, M.; et al. Temporal inhibition of mouse mammary gland cancer metastasis by CORM-A1 and DETA/NO combination therapy. Theranostics 2019, 9, 3918. [Google Scholar] [CrossRef] [PubMed]
- Kaliyaperumal, K.; Sharma, A.K.; McDonald, D.G.; Dhindsa, J.S.; Yount, C.; Singh, A.K.; Won, J.S.; Singh, I. S-Nitrosoglutathione-mediated STAT3 regulation in efficacy of radiotherapy and cisplatin therapy in head and neck squamous cell carcinoma. Redox Biol. 2015, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chénais, B.; Yapo, A.; Lepoivre, M.; Tenu, J.P. N omega-hydroxy-L-arginine, a reactional intermediate in nitric oxide biosynthesis, induces cytostasis in human and murine tumor cells. Biochem. Biophys. Res. Commun. 1993, 196. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Diao, Y.; Liu, Y.; Gao, N.; Gao, D.; Wan, Y.; Zhong, J.; Jin, G. Synergistic apoptosis-inducing effect of aspirin and isosorbide mononitrate on human colon cancer cells. Mol. Med. Rep. 2015, 12. [Google Scholar] [CrossRef] [Green Version]
- Yao, X.; Wu, Y.; Zhu, M.; Qian, H.; Chen, Y. Nitric oxide/cyclic guanosine monophosphate inducers sodium nitroprusside and L-arginine inhibit the proliferation of gastric cancer cells via the activation of type II cyclic guanosine monophosphate-dependent protein kinase. Oncol. Lett. 2015, 10. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Lan, C.; Fang, Y.; Zhang, Y.; Wang, J.; Guo, J.; Wan, S.; Yang, S.; Wang, R.; Fang, D. Sodium nitroprusside (SNP) sensitizes human gastric cancer cells to TRAIL-induced apoptosis. Int. Immunopharmacol. 2013, 17. [Google Scholar] [CrossRef]
- Kielbik, M.; Klink, M.; Brzezinska, M.; Szulc, I.; Sulowska, Z. Nitric oxide donors: Spermine/NO and diethylenetriamine/NO induce ovarian cancer cell death and affect STAT3 and AKT signaling proteins. Nitric Oxide 2013, 35. [Google Scholar] [CrossRef]
- Yamada, M.; Momose, K.; Richelson, E.; Yamada, M. Sodium nitroprusside-induced apoptotic cellular death via production of hydrogen peroxide in murine neuroblastoma N1E-115 cells. J. Pharmacol. Toxicol. Methods 1996, 35. [Google Scholar] [CrossRef]
- Rim, D.E.; Yoo, H.J.; Lee, J.H.; Kwon, O.J.; Jeong, S.W. Role of GS28 in sodium nitroprusside-induced cell death in cervical carcinoma cells. J. Biochem. Mol. Toxicol. 2019, 33. [Google Scholar] [CrossRef]
- Quan, Y.Y.; Liu, Y.H.; Lin, C.M.; Wang, X.P.; Chen, T.S. Peroxynitrite dominates sodium nitroprusside-induced apoptosis in human hepatocellular carcinoma cells. Oncotarget 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verovski, V.N.; den Berge DL, V.; Soete, G.A.; Bols, B.L.; Storme, G.A. Intrinsic radiosensitivity of human pancreatic tumour cells and the radiosensitising potency of the nitric oxide donor sodium nitroprusside. Br. J. Cancer 1996, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayevska, O.; Chen, O.; Karatsai, O.; Bobak, Y.; Barska, M.; Lyniv, L.; Pavlyk, I.; Rzhepetskyy, Y.; Igumentseva, N.; Redowicz, M.J.; et al. Nitric oxide donor augments antineoplastic effects of arginine deprivation in human melanoma cells. Exp. Cell Res. 2017, 355. [Google Scholar] [CrossRef] [PubMed]
- Cardaci, S.; Filomeni, G.; Rotilio, G.; Ciriolo, M.R. Reactive oxygen species mediate p53 activation and apoptosis induced by sodium nitroprusside in SH-SY5Y cells. Mol. Pharmacol. 2008, 74. [Google Scholar] [CrossRef] [PubMed]
- Terwel, D.; Nieland, L.J.; Schutte, B.; Reutelingsperger, C.P.; Ramaekers, F.C.; Steinbusch, H.W. S-nitroso-N-acetylpenicillamine and nitroprusside induce apoptosis in a neuronal cell line by the production of different reactive molecules. Eur. J. Pharmacol. 2000, 400. [Google Scholar] [CrossRef] [Green Version]
- Pervin, S.; Singh, R.; Chaudhuri, G. Nitric oxide-induced cytostasis and cell cycle arrest of a human breast cancer cell line (MDA-MB-231): Potential role of cyclin D1. Proc. Natl. Acad. Sci. USA 2001, 98, 3583–3588. [Google Scholar] [CrossRef] [Green Version]
- Martinez, L.; Thames, E.; Kim, J.; Chaudhuri, G.; Singh, R.; Pervin, S. Increased sensitivity of African American triple negative breast cancer cells to nitric oxide-induced mitochondria-mediated apoptosis. BMC Cancer 2016, 16. [Google Scholar] [CrossRef] [Green Version]
- Markowitz, J.; Wang, J.; Vangundy, Z.; You, J.; Yildiz, V.; Yu, L.; Foote, I.P.; Branson, O.E.; Stiff, A.R.; Brooks, T.R.; et al. Nitric oxide mediated inhibition of antigen presentation from DCs to CD4+ T cells in cancer and measurement of STAT1 nitration. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Lu, S.; Dong, Z. Regulation of TGF-beta1 gene transcription in human prostate cancer cells by nitric oxide. Prostate 2007, 67, 1825–1833. [Google Scholar] [CrossRef]
- Jeon, H.K.; Choi, S.U.; Jung, N.P. Association of the ERK1/2 and p38 kinase pathways with nitric oxide-induced apoptosis and cell cycle arrest in colon cancer cells. Cell Biol. Toxicol. 2005, 21. [Google Scholar] [CrossRef]
- Furuhashi, S.; Sugita, H.; Takamori, H.; Horino, K.; Nakahara, O.; Okabe, H.; Miyake, K.; Tanaka, H.; Beppu, T.; Baba, H. NO donor and MEK inhibitor synergistically inhibit proliferation and invasion of cancer cells. Int. J. Oncol. 2012, 40. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Sun, C.; Xiao, G.; Shan, H.; Tang, L.; Yi, Y.; Yu, W.; Gu, Y. S-nitrosylation of the Peroxiredoxin-2 promotes S-nitrosoglutathione-mediated lung cancer cells apoptosis via AMPK-SIRT1 pathway. Cell Death Dis. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neves-Zaph, S.R. Phosphodiesterase Diversity and Signal Processing Within cAMP Signaling Networks. Adv. Neurobiol. 2017, 17, 3–14. [Google Scholar] [PubMed]
- Peng, T.; Gong, J.; Jin, Y.; Zhou, Y.; Tong, R.; Wei, X.; Bai, L.; Shi, J. Inhibitors of phosphodiesterase as cancer therapeutics. Eur. J. Med. Chem. 2018, 150, 742–756. [Google Scholar] [CrossRef] [Green Version]
- Das, A.; Durrant, D.; Salloum, F.N.; Xi, L.; Kukreja, R.C. PDE5 inhibitors as therapeutics for heart disease, diabetes and cancer. Pharmacol. Ther. 2015, 147, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Chuang, A.T.; Strauss, J.D.; Murphy, R.A.; Steers, W.D. Sildenafil, a type-5 CGMP phosphodiesterase inhibitor, specifically amplifies endogenous cGMP-dependent relaxation in rabbit corpus cavernosum smooth muscle in vitro. J. Urol. 1998, 160, 257–261. [Google Scholar] [CrossRef]
- Gali-Muhtasib, H.; Roessner, A.; Schneider-Stock, R. Thymoquinone: A promising anti-cancer drug from natural sources. Int. J. Biochem. Cell Biol. 2006, 38, 1249–1253. [Google Scholar] [CrossRef]
- Abusnina, A.; Alhosin, M.; Keravis, T.; Muller, C.D.; Fuhrmann, G.; Bronner, C.; Lugnier, C. Down-regulation of cyclic nucleotide phosphodiesterase PDE1A is the key event of p73 and UHRF1 deregulation in thymoquinone-induced acute lymphoblastic leukemia cell apoptosis. Cell. Signal. 2011, 23, 152–160. [Google Scholar] [CrossRef]
- Ichwan, S.J.A.; Al-Ani, I.M.; Bilal, H.G.; Suriyah, W.H.; Taher, M.; Ikeda, M.A. Apoptotic activities of thymoquinone, an active ingredient of black seed (Nigella sativa), in cervical cancer cell lines. Chin. J. Physiol. 2014, 57, 249–255. [Google Scholar] [CrossRef]
- Arafa, E.-S.A.; Zhu, Q.; Shah, Z.I.; Wani, G.; Barakat, B.M.; Racoma, I.; El-Mahdy, M.A.; Wani, A.A. Thymoquinone up-regulates PTEN expression and induces apoptosis in doxorubicin-resistant human breast cancer cells. Mutat. Res. 2011, 706, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Farkhondeh, T.; Samarghandian, S.; Hozeifi, S.; Azimi-Nezhad, M. Therapeutic effects of thymoquinone for the treatment of central nervous system tumors: A review. Biomed. Pharmacother. 2017, 96, 1440–1444. [Google Scholar] [CrossRef]
- Li, W.-Q.; Qureshi, A.A.; Robinson, K.C.; Han, J. Sildenafil use and increased risk of incident melanoma in US men: A prospective cohort study. JAMA Intern. Med. 2014, 174, 964–970. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Z.; Le, S.; Alexanian, C.; Boddu, S.; Merleev, A.; Marusina, A.; Maverakis, E. No Causal Link between Phosphodiesterase Type 5 Inhibition and Melanoma. World J. Mens. Health 2019, 37, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Chavez, A.H.; Coffield, K.S.; Rajab, M.H.; Jo, C. Incidence rate of prostate cancer in men treated for erectile dysfunction with phosphodiesterase type 5 inhibitors: Retrospective analysis. Asian J. Androl. 2013, 15, 246–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michl, U.; Molfenter, F.; Graefen, M.; Tennstedt, P.; Ahyai, S.; Beyer, B.; Budäus, L.; Haese, A.; Heinzer, H.; Oh, S.J.; et al. Use of phosphodiesterase type 5 inhibitors may adversely impact biochemical recurrence after radical prostatectomy. J. Urol. 2015, 193, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Gallina, A.; Bianchi, M.; Gandaglia, G.; Cucchiara, V.; Suardi, N.; Montorsi, F.; Briganti, A. A Detailed Analysis of the Association Between Postoperative Phosphodiesterase Type 5 Inhibitor Use and the Risk of Biochemical Recurrence After Radical Prostatectomy. Eur. Urol. 2015, 68, 750–753. [Google Scholar] [CrossRef] [PubMed]
- Black, K.L.; Yin, D.; Ong, J.M.; Hu, J.; Konda, B.M.; Wang, X.; Ko, M.K.; Bayan, J.-A.; Sacapano, M.R.; Espinoza, A.; et al. PDE5 inhibitors enhance tumor permeability and efficacy of chemotherapy in a rat brain tumor model. Brain Res. 2008, 1230, 290–302. [Google Scholar] [CrossRef] [Green Version]
- Domvri, K.; Zarogoulidis, K.; Zogas, N.; Zarogoulidis, P.; Petanidis, S.; Porpodis, K.; Kioseoglou, E.; Hohenforst-Schmidt, W. Potential synergistic effect of phosphodiesterase inhibitors with chemotherapy in lung cancer. J. Cancer 2017, 8, 3648–3656. [Google Scholar] [CrossRef] [Green Version]
- Muniyan, S.; Rachagani, S.; Parte, S.; Halder, S.; Seshacharyulu, P.; Kshirsagar, P.; Siddiqui, J.A.; Vengoji, R.; Rauth, S.; Islam, R.; et al. Sildenafil Potentiates the Therapeutic Efficacy of Docetaxel in Advanced Prostate Cancer by Stimulating NO-cGMP Signaling. Clin. Cancer Res. 2020, 26. [Google Scholar] [CrossRef]
- Weed, D.T.; Vella, J.L.; Reis, I.M.; De la Fuente, A.C.; Gomez, C.; Sargi, Z.; Nazarian, R.; Califano, J.; Borrello, I.; Serafini, P. Tadalafil reduces myeloid-derived suppressor cells and regulatory T cells and promotes tumor immunity in patients with head and neck squamous cell carcinoma. Clin. Cancer Res. 2015, 21, 39–48. [Google Scholar] [CrossRef] [Green Version]
- Weed, D.T.; Zilio, S.; Reis, I.M.; Sargi, Z.; Abouyared, M.; Gomez-Fernandez, C.R.; Civantos, F.J.; Rodriguez, C.P.; Serafini, P. The Reversal of Immune Exclusion Mediated by Tadalafil and an Anti-tumor Vaccine Also Induces PDL1 Upregulation in Recurrent Head and Neck Squamous Cell Carcinoma: Interim Analysis of a Phase I Clinical Trial. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Califano, J.A.; Khan, Z.; Noonan, K.A.; Rudraraju, L.; Zhang, Z.; Wang, H.; Goodman, S.; Gourin, C.G.; Ha, P.K.; Fakhry, C.; et al. Tadalafil augments tumor specific immunity in patients with head and neck squamous cell carcinoma. Clin. Cancer Res. 2015, 21, 30–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masters, G.A.; Li, S.; Dowlati, A.; Madajewicz, S.; Langer, C.; Schiller, J.; Johnson, D. A Phase II Trial of Carboplatin and Gemcitabine with Exisulind (IND #65,056) in Patients with Advanced Non-small Cell Lung Cancer: An Eastern Cooperative Oncology Group Study (E1501). J. Thorac. Oncol. 2006, 1, 673–678. [Google Scholar] [PubMed]
- Pusztai, L.; Zhen, J.H.; Arun, B.; Rivera, E.; Whitehead, C.; Thompson, W.J.; Nealy, K.M.; Gibbs, A.; Symmans, W.F.; Esteva, F.J.; et al. Phase I and II study of exisulind in combination with capecitabine in patients with metastatic breast cancer. J. Clin. Oncol. 2003, 21. [Google Scholar] [CrossRef] [PubMed]
- Witta, S.E.; Gustafson, D.L.; Pierson, A.S.; Menter, A.; Holden, S.N.; Basche, M.; Persky, M.; O’Bryant, C.L.; Zeng, C.; Baron, A.; et al. A phase I and pharmacokinetic study of exisulind and docetaxel in patients with advanced solid tumors. Clin. Cancer Res. 2004, 10, 7229–7237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, C.W.; Stadler, W.M.; Vogelzang, N.J. A phase I/II dose-escalation study of exisulind and docetaxel in patients with hormone-refractory prostate cancer. BJU Int. 2005, 95. [Google Scholar] [CrossRef] [PubMed]
- Sinibaldi, V.J.; Elza-Brown, K.; Schmidt, J.; Eisenberger, M.A.; Rosenbaum, E.; Denmeade, S.R.; Pili, R.; Walczak, J.; Baker, S.D.; Zahurak, M.; et al. Phase II evaluation of docetaxel plus exisulind in patients with androgen independent prostate carcinoma. Am. J. Clin. Oncol. 2006, 29. [Google Scholar] [CrossRef]
- Sun, W.; Stevenson, J.P.; Gallo, J.M.; Redlinger, M.; Haller, D.; Algazy, K.; Giantonio, B.; Alila, H.; O’Dwyer, P.J. Phase I and pharmacokinetic trial of the proapoptotic sulindac analog CP-461 in patients with advanced cancer. Clin. Cancer Res. 2002, 8, 3100–3104. [Google Scholar]
- Kelly, K.; Mejia, A.; Suhasini, A.N.; Lin, A.P.; Kuhn, J.; Karnad, A.B.; Weitman, S.; Aguiar, R.C. Safety and Pharmacodynamics of the PDE4 Inhibitor Roflumilast in Advanced B-cell Malignancies. Clin. Cancer Res. 2017, 23. [Google Scholar] [CrossRef] [Green Version]
- Zacharski, L.R.; Moritz, T.E.; Baczek, L.A.; Rickles, F.R.; Edwards, R.L.; Forman, W.B.; Forcier, R.J.; Cornell, C.J.; Haakenson, C.M.; Ballard, H.S. Effect of mopidamol on survival in carcinoma of the lung and colon: Final report of Veterans Administration Cooperative Study No. 188. J. Natl. Cancer Inst. 1988, 80. [Google Scholar] [CrossRef]
- Isacoff, W.H.; Bendetti, J.K.; Barstis, J.J.; Jazieh, A.R.; Macdonald, J.S.; Philip, P.A. Phase II trial of infusional fluorouracil, leucovorin, mitomycin, and dipyridamole in locally advanced unresectable pancreatic adenocarcinoma: SWOG S9700. J. Clin. Oncol. 2007, 25. [Google Scholar] [CrossRef] [PubMed]
- Wiernik, P.H.; Paietta, E.; Goloubeva, O.; Lee, S.J.; Makower, D.; Bennett, J.M.; Wade, J.L.; Ghosh, C.; Kaminer, L.S.; Pizzolo, J.; et al. Phase II study of theophylline in chronic lymphocytic leukemia: A study of the Eastern Cooperative Oncology Group (E4998). Leukemia 2004, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bian, K.; Ghassemi, F.; Sotolongo, A.; Siu, A.; Shauger, L.; Kots, A.; Murad, F. NOS-2 signaling and cancer therapy. IUBMB Life 2012, 64, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.-J.; Vetter, M.; Che, D.; Liu, S.; Tsai, M.-L.; Chang, C.-H. The bradykinin/soluble guanylate cyclase signaling pathway is impaired in androgen-independent prostate cancer cells. Cancer Lett. 2002, 177, 181–187. [Google Scholar] [CrossRef]
- Wen, H.-C.; Chuu, C.-P.; Chen, C.-Y.; Shiah, S.-G.; Kung, H.-J.; King, K.-L.; Su, L.-C.; Chang, S.-C.; Chang, C.-H. Elevation of soluble guanylate cyclase suppresses proliferation and survival of human breast cancer cells. PLoS ONE 2015, 10, e0125518. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Li, J.T.; Zheng, F.; Martin, E.; Kots, A.Y.; Krumenacker, J.S.; Choi, B.-K.; McCutcheon, I.E.; Weisbrodt, N.; Bögler, O.; et al. Restoring Soluble Guanylyl Cyclase Expression and Function Blocks the Aggressive Course of Glioma. Mol. Pharmacol. 2011, 80, 1076–1084. [Google Scholar] [CrossRef] [Green Version]
- Sandner, P. From molecules to patients: Exploring the therapeutic role of soluble guanylate cyclase stimulators. Biol. Chem. 2018, 399, 679–690. [Google Scholar] [CrossRef]
- Paul, T.; Salazar-Degracia, A.; Peinado, V.I.; Tura-Ceide, O.; Blanco, I.; Barreiro, E.; Barberà, J.A. Soluble guanylate cyclase stimulation reduces oxidative stress in experimental Chronic Obstructive Pulmonary Disease. PLoS ONE 2018, 13, e0190628. [Google Scholar] [CrossRef] [Green Version]
- Ghofrani, H.-A.; Galiè, N.; Grimminger, F.; Grünig, E.; Humbert, M.; Jing, Z.-C.; Keogh, A.M.; Langleben, D.; Kilama, M.O.; Fritsch, A.; et al. Riociguat for the treatment of pulmonary arterial hypertension. N. Engl. J. Med. 2013, 369, 330–340. [Google Scholar] [CrossRef] [Green Version]
- Gheorghiade, M.; Greene, S.J.; Butler, J.; Filippatos, G.; Lam, C.S.P.; Maggioni, A.P.; Ponikowski, P.; Shah, S.J.; Solomon, S.D.; Kraigher-Krainer, E.; et al. Effect of Vericiguat, a Soluble Guanylate Cyclase Stimulator, on Natriuretic Peptide Levels in Patients With Worsening Chronic Heart Failure and Reduced Ejection Fraction: The SOCRATES-REDUCED Randomized Trial. JAMA 2015, 314, 2251–2262. [Google Scholar] [CrossRef]
- Stasch, J.-P.; Schlossmann, J.; Hocher, B. Renal effects of soluble guanylate cyclase stimulators and activators: A review of the preclinical evidence. Curr. Opin. Pharmacol. 2015, 21, 95–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandner, P.; Zimmer, D.P.; Milne, G.T.; Follmann, M.; Hobbs, A.; Stasch, J.-P. Soluble Guanylate Cyclase Stimulators and Activators. Handb. Exp. Pharmacol. 2019. online ahead of print. [Google Scholar] [CrossRef] [Green Version]
- Tuttle, T.R.; Takiar, V.; Kumar, B.; Kumar, P.; Ben-Jonathan, N. Soluble guanylate cyclase stimulators increase sensitivity to cisplatin in head and neck squamous cell carcinoma cells. Cancer Lett. 2017, 389, 33–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.-R.; Lin, C.; Lu, C.-C.; Kuo, S.-C.; Tsao, J.-W.; Juan, Y.-N.; Chiu, H.-Y.; Lee, F.-Y.; Yang, J.-S.; Tsai, F.-J. YC-1 induces G 0 /G 1 phase arrest and mitochondria-dependent apoptosis in cisplatin-resistant human oral cancer CAR cells. Biomedicine 2017, 7, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeRubertis, F.R.; Craven, P.A. Stimulation of soluble guanylate cyclase activity and cellular accumulation of cyclic guanosine 3′,5′-monophosphate by the carcinogen 4-nitroquinoline 1-oxide: Brief communication. J. Natl. Cancer Inst. 1977, 59, 1741–1745. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Hsieh, C.-L.; Bhansali, M.; Kannan, A.; Shemshedini, L. A peptide against soluble guanylyl cyclase α1: A new approach to treating prostate cancer. PLoS ONE 2013, 8, e64189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Francisco, L.M.; Sage, P.T.; Sharpe, A.H. The PD-1 pathway in tolerance and autoimmunity. Immunol. Rev. 2010, 236, 219–242. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar] [PubMed]
- Wang, Y.; Zhou, S.; Yang, F.; Qi, X.; Wang, X.; Guan, X.; Shen, C.; Duma, N.; Vera Aguilera, J.; Chintakuntlawar, A.; et al. Treatment-Related Adverse Events of PD-1 and PD-L1 Inhibitors in Clinical Trials: A Systematic Review and Meta-analysis. JAMA Oncol. 2019, 5, 1008–1019. [Google Scholar] [CrossRef]
- Akinleye, A.; Rasool, Z. Immune checkpoint inhibitors of PD-L1 as cancer therapeutics. J. Hematol. Oncol. 2019, 12, 677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, Y.; Inui, N.; Karayama, M.; Imokawa, S.; Yamada, T.; Yokomura, K.; Asada, K.; Kusagaya, H.; Kaida, Y.; Matsuda, H.; et al. Effect of PD-1 inhibitor on exhaled nitric oxide and pulmonary function in non-small cell lung cancer patients with and without COPD. Int. J. Chron. Obstruct. Pulmon. Dis. 2019, 14, 1867–1877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hays, E.; Bonavida, B. Nitric Oxide-Mediated Enhancement and Reversal of Resistance of Anticancer Therapies. Antioxidants 2019, 8, 407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, C.; Barsoum, I.; Kim, J.; Black, M.; Siemens, R.D. Mechanisms of Hypoxia-Induced Immune Escape in Cancer and Their Regulation by Nitric Oxide. Redox Biol. 2015, 5, 417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hays, E.; Bonavida, B. YY1 regulates cancer cell immune resistance by modulating PD-L1 expression. Drug Resist. Updat. 2019, 43, 10–28. [Google Scholar] [CrossRef] [PubMed]
- Hongo, F.; Garban, H.; Huerta-Yepez, S.; Vega, M.; Jazirehi, A.R.; Mizutani, Y.; Miki, T.; Bonavida, B. Inhibition of the transcription factor Yin Yang 1 activity by S-nitrosation. Biochem. Biophys. Res. Commun. 2005, 336, 692–701. [Google Scholar] [CrossRef] [PubMed]
- Knox, S.J.; Ning, S.; Peehl, D.; Oronsky, B.; Scicinski, J. Abstract C181: RRx-001 combined with anti-PD-L1 antibody increases the complete response rate in a preclinical myeloma model. In Proceedings of the Therapeutic Agents: Other; American Association for Cancer Research: Philadelphia, PA, USA, 2015; p. C181. [Google Scholar]
- Davila-Gonzalez, D.; Rosato, R.R.; Dave, B.; Choi, D.S.; Qian, W.; Kozielski, A.J.; Chen, W.; Ensor, J.E.; Chang, J.C. Abstract A202: Evaluating the combination of anti-PD-1 and nitric oxide synthase inhibition therapy in 12 triple-negative breast cancer patient-derived xenografts using a human-derived immune system model. In Proceedings of the Immune Checkpoints; American Association for Cancer Research: Philadelphia, PA, USA, 2018; p. A202. [Google Scholar]
- Hume, D.A.; Caruso, M.; Ferrari-Cestari, M.; Summers, K.M.; Pridans, C.; Irvine, K.M. Phenotypic impacts of CSF1R deficiencies in humans and model organisms. J. Leukoc. Biol. 2020, 107, 205–219. [Google Scholar] [CrossRef] [Green Version]
- Lenzo, J.C.; Turner, A.L.; Cook, A.D.; Vlahos, R.; Anderson, G.P.; Reynolds, E.C.; Hamilton, J.A. Control of macrophage lineage populations by CSF-1 receptor and GM-CSF in homeostasis and inflammation. Immunol. Cell Biol. 2012, 90, 429–440. [Google Scholar] [CrossRef]
- Cannarile, M.A.; Weisser, M.; Jacob, W.; Jegg, A.-M.; Ries, C.H.; Rüttinger, D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J. Immunother. Cancer 2017, 5, 53. [Google Scholar] [CrossRef]
- Lin, Y.; Xu, J.; Lan, H. Tumor-associated macrophages in tumor metastasis: Biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019, 12, 76. [Google Scholar] [CrossRef]
- Lin, C.-W.; Shen, S.-C.; Ko, C.-H.; Lin, H.-Y.; Chen, Y.-C. Reciprocal activation of macrophages and breast carcinoma cells by nitric oxide and colony-stimulating factor-1. Carcinogenesis 2010, 31, 2039–2048. [Google Scholar] [CrossRef] [PubMed]
- Rahat, M.A.; Hemmerlein, B. Macrophage-tumor cell interactions regulate the function of nitric oxide. Front. Physiol. 2013, 4, 144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, R.V.; Fernandes, A.C.; Antunes, F.; Lin, Z.; Rocha, J.; Pires, J.; Pinto, M.L. New generation of nitric oxide-releasing porous materials: Assessment of their potential to regulate biological functions. Nitric Oxide 2019, 90, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Kashfi, K.; Rigas, B. Molecular targets of nitric-oxide-donating aspirin in cancer. Biochem. Soc. Trans. 2005, 33, 701–704. [Google Scholar] [CrossRef] [PubMed]
- Chegaev, K.; Riganti, C.; Lazzarato, L.; Rolando, B.; Guglielmo, S.; Campia, I.; Fruttero, R.; Bosia, A.; Gasco, A. Nitric oxide donor doxorubicins accumulate into Doxorubicin-resistant human colon cancer cells inducing cytotoxicity. ACS Med. Chem. Lett. 2011, 2, 494–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paskas, S.; Mazzon, E.; Basile, M.S.; Cavalli, E.; Al-Abed, Y.; He, M.; Rakocevic, S.; Nicoletti, F.; Mijatovic, S.; Maksimovic-Ivanic, D. Lopinavir-NO, a nitric oxide-releasing HIV protease inhibitor, suppresses the growth of melanoma cells in vitro and in vivo. Investig. New Drugs 2019, 37, 1014–1028. [Google Scholar] [CrossRef]
- Yu, S.; Pearson, A.D.; Lim, R.K.; Rodgers, D.T.; Li, S.; Parker, H.B.; Weglarz, M.; Hampton, E.N.; Bollong, M.J.; Shen, J.; et al. Targeted Delivery of an Anti-inflammatory PDE4 Inhibitor to Immune Cells via an Antibody-drug Conjugate. Mol. Ther. 2016, 24, 2078–2089. [Google Scholar] [CrossRef] [Green Version]
- Ramos, L.C.B.; Rodrigues, F.P.; Biazzotto, J.C.; de Paula Machado, S.; Slep, L.D.; Hamblin, M.R.; da Silva, R.S. Targeting the mitochondrial VDAC in hepatocellular carcinoma using a polyclonal antibody-conjugated to a nitrosyl ruthenium complex. J. Biol. Inorg. Chem. 2018, 23, 903. [Google Scholar] [CrossRef] [Green Version]
- Sun, F.; Wang, Y.; Luo, X.; Ma, Z.; Xu, Y.; Zhang, X.; Lv, T.; Zhang, Y.; Wang, M.; Huang, Z.; et al. Anti-CD24 Antibody–Nitric Oxide Conjugate Selectively and Potently Suppresses Hepatic Carcinoma. Cancer Res. 2019, 79, 3395–3405. [Google Scholar] [CrossRef] [Green Version]
- Chegaev, K.; Fraix, A.; Gazzano, E.; Abd-Ellatef, G.E.F.; Blangetti, M.; Rolando, B.; Conoci, S.; Riganti, C.; Fruttero, R.; Gasco, A.; et al. Light-Regulated NO Release as a Novel Strategy to Overcome Doxorubicin Multidrug Resistance. ACS Med. Chem. Lett. 2017, 8, 361–365. [Google Scholar] [CrossRef]
- Yoshikawa, T.; Mori, Y.; Feng, H.; Phan, K.Q.; Kishimura, A.; Kang, J.-H.; Mori, T.; Katayama, Y. Rapid and continuous accumulation of nitric oxide-releasing liposomes in tumors to augment the enhanced permeability and retention (EPR) effect. Int. J. Pharm. 2019, 565, 481–487. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Major synthesis and effector pathways of NO. Synthesis of NO primarily occurs through the action of NOS, although the conversion of dietary nitrogen containing compounds has also been proposed. Once produced, the function of NO can directly upregulate cGMP second messenger signaling pathways and directly modify the function of proteins through nitration. However, in the presence of other ROS such as superoxide, NO may form more reactive intermediates which can further alter the functionality of proteins. The balance of NO with ROS is critical to maintaining proper cellular function and the NO/ROS imbalance is implicated in the pathogenesis of many different diseases.
Figure 1.
Major synthesis and effector pathways of NO. Synthesis of NO primarily occurs through the action of NOS, although the conversion of dietary nitrogen containing compounds has also been proposed. Once produced, the function of NO can directly upregulate cGMP second messenger signaling pathways and directly modify the function of proteins through nitration. However, in the presence of other ROS such as superoxide, NO may form more reactive intermediates which can further alter the functionality of proteins. The balance of NO with ROS is critical to maintaining proper cellular function and the NO/ROS imbalance is implicated in the pathogenesis of many different diseases.

Figure 2.
NO and PDE5 inhibitors control cGMP levels, thereby lowering vascular pressure. NO binds Scheme 5. which is responsible for hydrolysis of cGMP. NO, nitric oxide; PDE5, phosphodiesterase-5; cGMP, cyclic guanosine monophosphate.
Figure 2.
NO and PDE5 inhibitors control cGMP levels, thereby lowering vascular pressure. NO binds Scheme 5. which is responsible for hydrolysis of cGMP. NO, nitric oxide; PDE5, phosphodiesterase-5; cGMP, cyclic guanosine monophosphate.

Figure 3.
Concentration-dependent effects of NO in cancer. Low NO improves molecular processes that maintain normal physiology but may influence cancer progression of already established cancers, such as proliferation, angiogenesis, metastasis, and switch to immunologically suppressive immune cell types, such as M2 macrophages. High NO influx leads to DNA damage, p53 activation, and nitrosative stress, which may promote carcinogenesis initially, but in already-established cancers, high NO promotes processes that activate immunity and improve chemotherapeutic efficacy. NO, nitric oxide.
Figure 3.
Concentration-dependent effects of NO in cancer. Low NO improves molecular processes that maintain normal physiology but may influence cancer progression of already established cancers, such as proliferation, angiogenesis, metastasis, and switch to immunologically suppressive immune cell types, such as M2 macrophages. High NO influx leads to DNA damage, p53 activation, and nitrosative stress, which may promote carcinogenesis initially, but in already-established cancers, high NO promotes processes that activate immunity and improve chemotherapeutic efficacy. NO, nitric oxide.

Figure 4.
NO promotes inflammatory tumor microenvironment by increasing polarization of M1 macrophages, which in turn produce NO through upregulation of iNOS, and other immune cells that can effecTable 1. macrophages and other pro-inflammatory cell types, to immunologically suppressed tumors that favor M2 macrophage switch which in turn downregulate iNOS production and promote immunosuppression, angiogenesis and are resistant to immunotherapy. TNFα, tumor necrosis factor-alpha; INFγ, interferon-gamma; IL-1β, Interleukin-1b.
Figure 4.
NO promotes inflammatory tumor microenvironment by increasing polarization of M1 macrophages, which in turn produce NO through upregulation of iNOS, and other immune cells that can effecTable 1. macrophages and other pro-inflammatory cell types, to immunologically suppressed tumors that favor M2 macrophage switch which in turn downregulate iNOS production and promote immunosuppression, angiogenesis and are resistant to immunotherapy. TNFα, tumor necrosis factor-alpha; INFγ, interferon-gamma; IL-1β, Interleukin-1b.

Figure 5.
Diversity of NO functioning in cancer. Multiple NO-mediated cancer pathways contribute to cancer growth and metastasis. Common pathways of NO-mediated mutagenesis and cancer growth, include p53 mutation, AKT upregulation, and VEGF induction, among others. Aberrant NO signaling also occurs in response to common carcinogens, including viruses, alcohol, and tobacco, suggesting that NO may lie in a common carcinogenic pathway shared by these compounds. In hormone-sensitive tumors, NO paradoxically functions to transmit the hormonal growth signals and can make the tumor hormone-insensitive.
Figure 5.
Diversity of NO functioning in cancer. Multiple NO-mediated cancer pathways contribute to cancer growth and metastasis. Common pathways of NO-mediated mutagenesis and cancer growth, include p53 mutation, AKT upregulation, and VEGF induction, among others. Aberrant NO signaling also occurs in response to common carcinogens, including viruses, alcohol, and tobacco, suggesting that NO may lie in a common carcinogenic pathway shared by these compounds. In hormone-sensitive tumors, NO paradoxically functions to transmit the hormonal growth signals and can make the tumor hormone-insensitive.


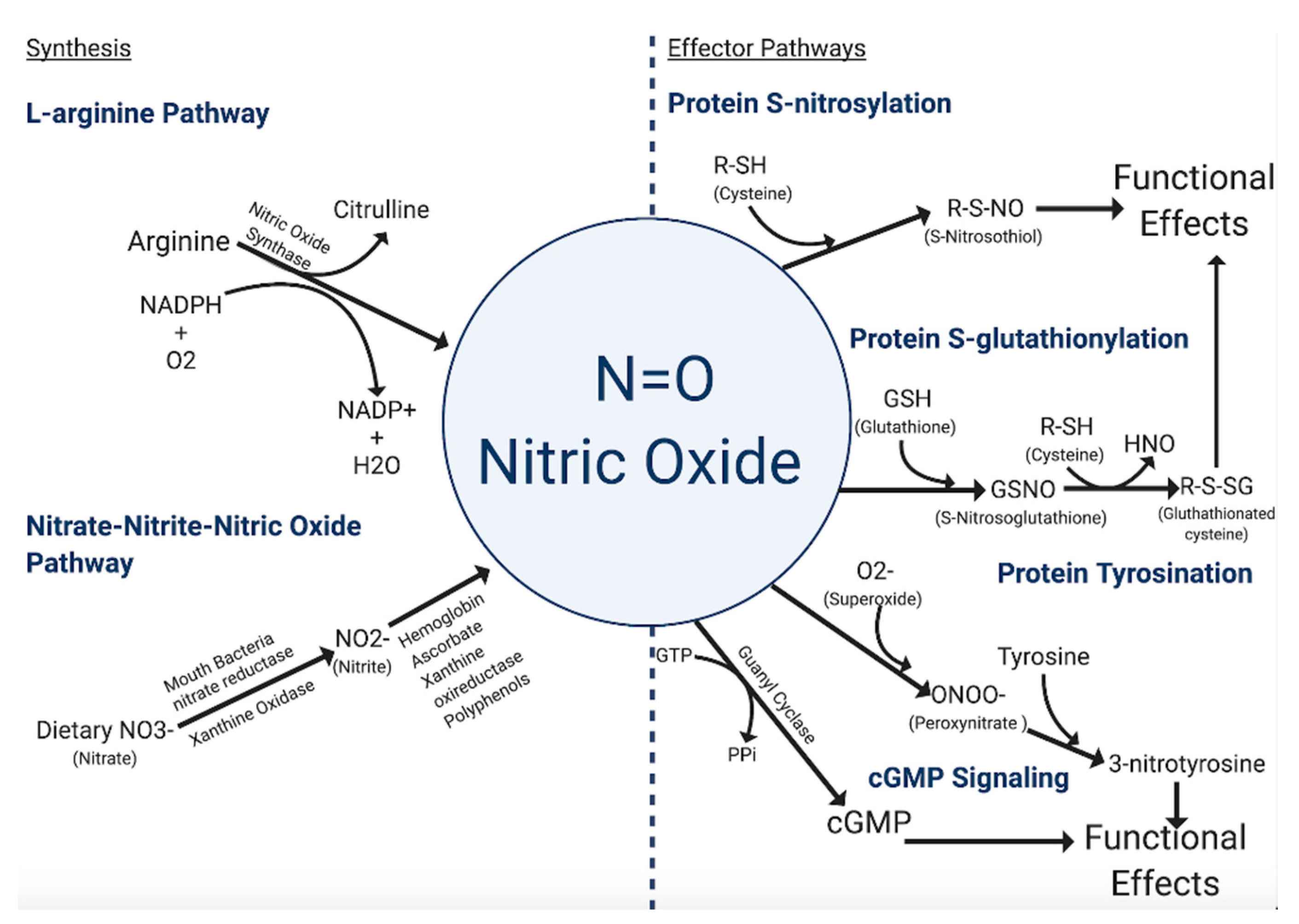{kind=link}
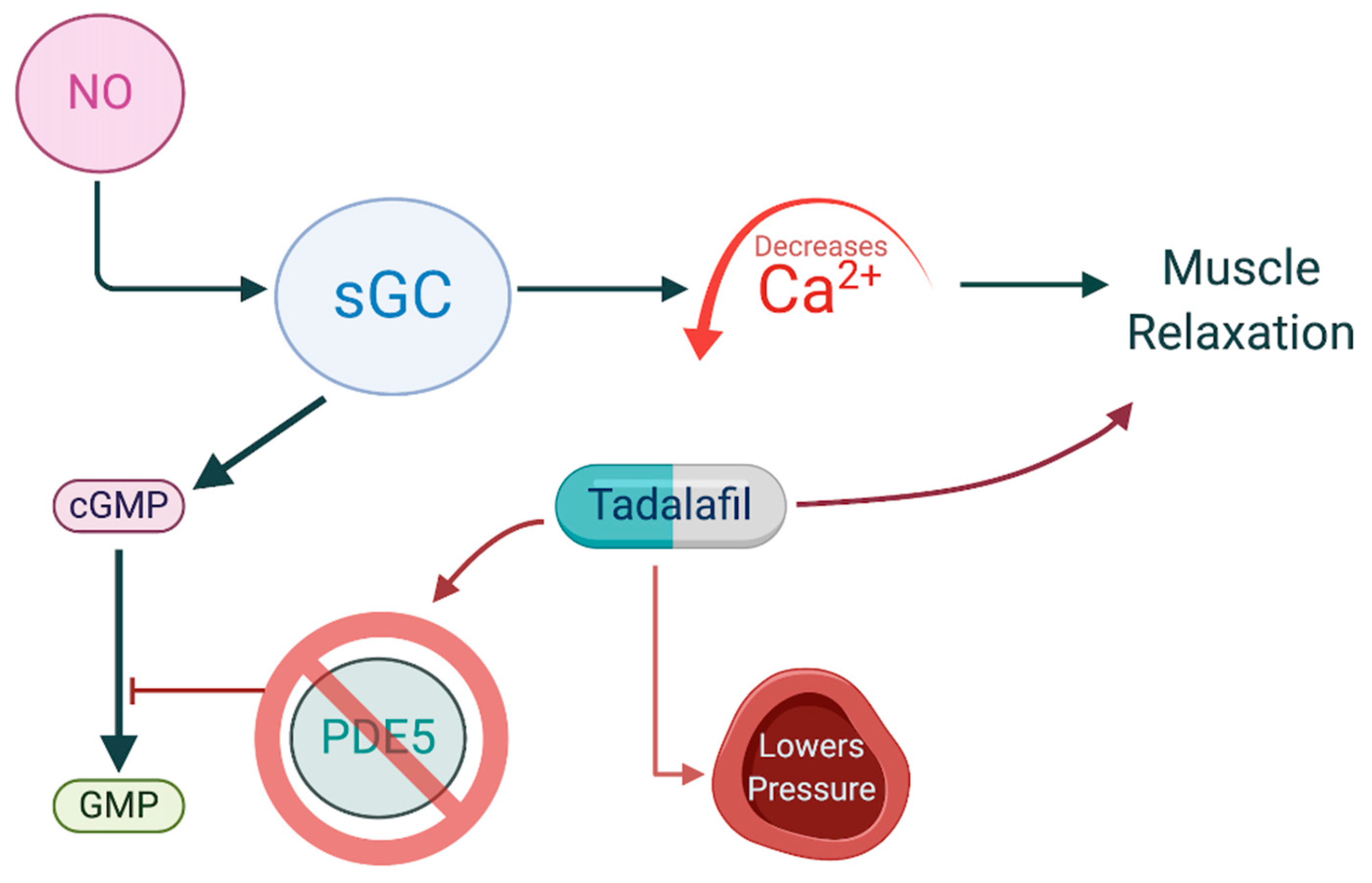{kind=link}
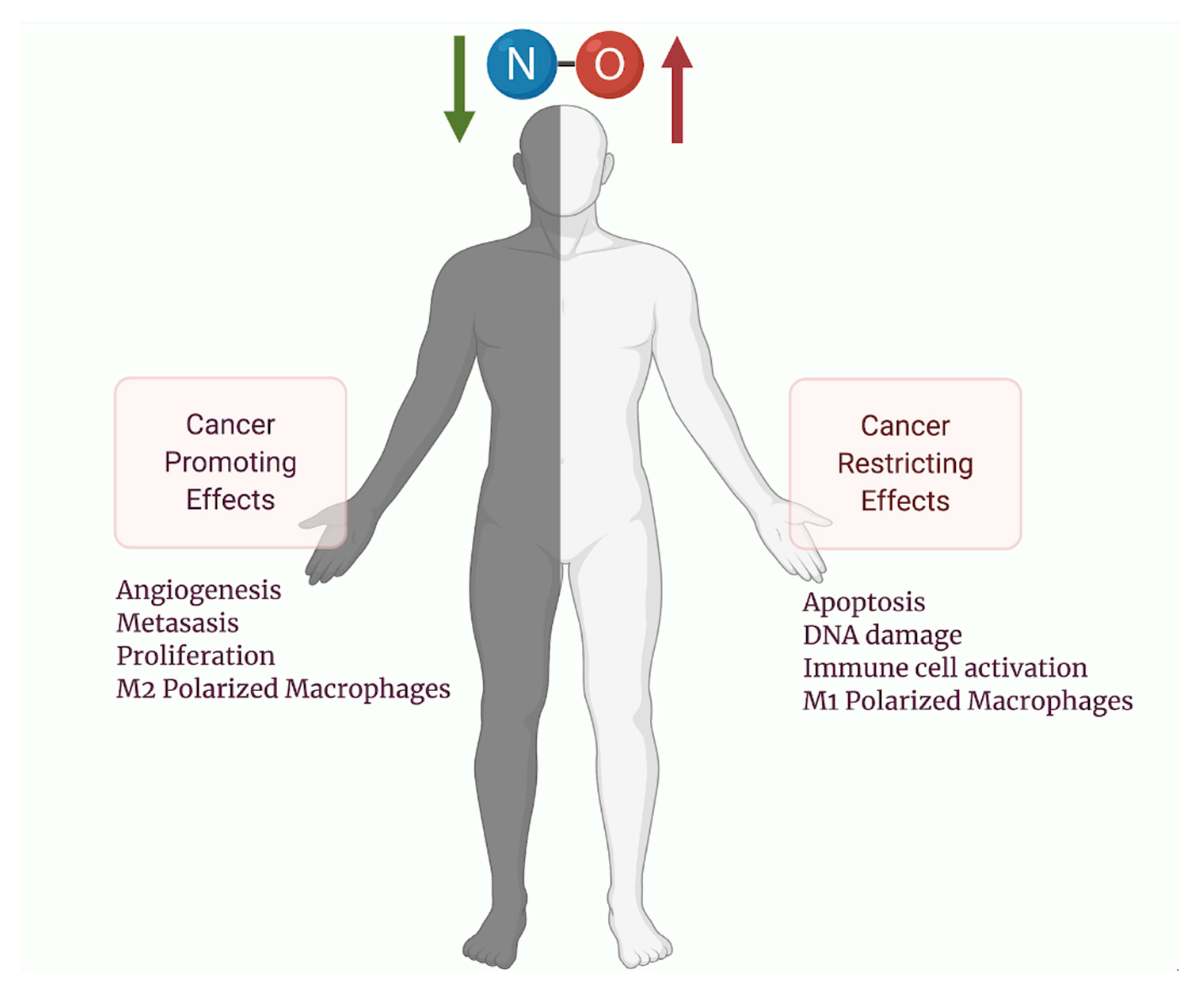{kind=link}
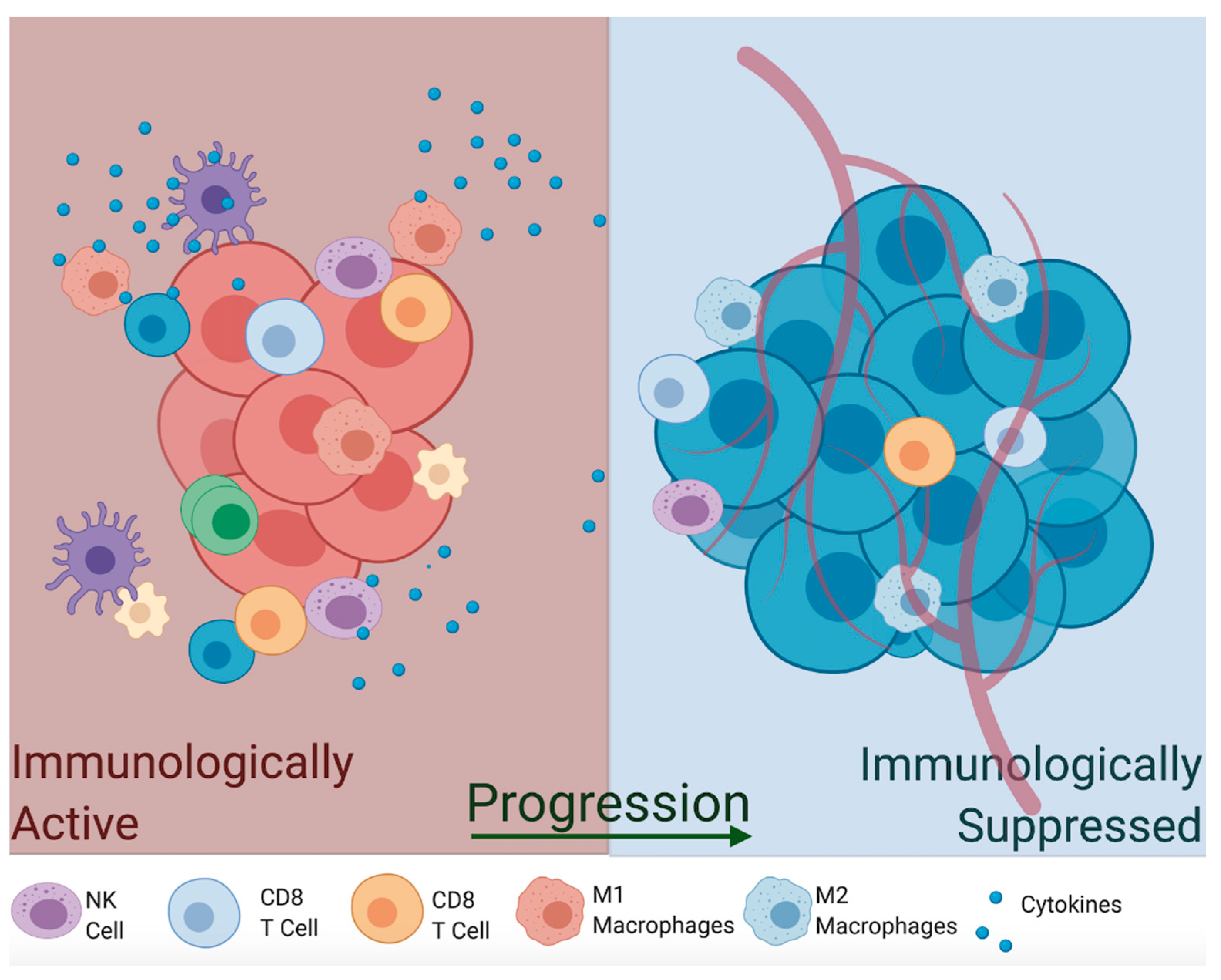{kind=link}
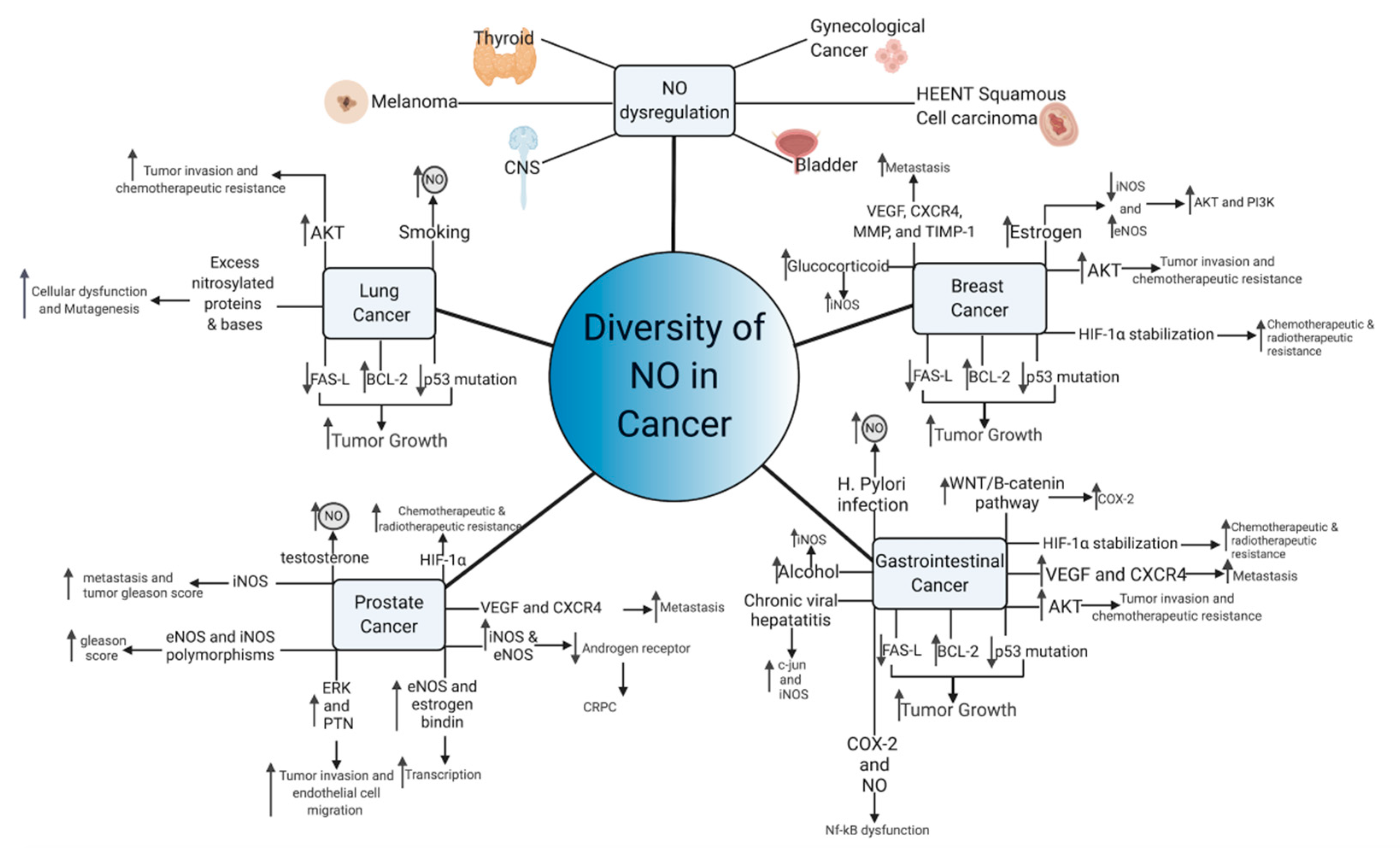{kind=link}
Table 1.
Major classes of NO donors and their examples of their proposed clinical uses.
| Class | Mechanism of Action [207,208] | Examples [207] | Known or Potential Clinical Uses |
|---|---|---|---|
| Endogenous NO precursors | Provide excess reagents or intermediates for NO synthesis. Direct antioxidant. Reversal of NOS inhibition. | L-Arginine, ω-hydroxy-L-arginine (NHA) | Stroke [209], Pulmonary hypetesion [210] |
| Organic Nitrates | Stimulation of guanylyl cyclase pathway, through unknown intermediate. May activate enzymatic NO production through cytochrome P450 or glutathione-S-transferase. Non-enzymatic thiol activity also described, although reaction proceeds at slower rate | Glyceryl trinitrate (GTN), isosorbide mononitrate (ISMN), pentaerythritol tetranitrate (PETN). nicorandil | Cardiac Angina [211], acute coronary syndrome [212], heart failure [213] |
| Organic Nitrites | Activation of NO signaling pathways | Butyl nitrite (BN), isobutyl nitrite (ISBN), tert-butyl nitrite (TBN), amyl nitrite (AMN), isoamyl nitrite (IAMN) | Minimal clinical uses due to cardiotoxicity and cytotoxicity |
| Metal Complexes | Direct release of NO. NO bound to metals such as iron is prone to nucleophilic attack | Sodium Nitroprusside (SNP) | Hypertensive Emergency, cardiac and aortic surgery [214,215], heart failure [216], acute coronary syndrome [217], pheochromocytoma [218] |
| Diazeniumdiolates | Spontaneously decompress to 2 molecules of NO | Methylamine hexa-methylene methylamine NONOate (MAHMA/NO), diethylamine NONOate (DEA/NO), proli NONOate (PROLI/NO) and diethylenetriamine NONOate (DETA/NO) | No current clinical uses |
| Sydnonimines | Spontaneous enzymatic degradation to O2 and NO. Also known as ONOO-donors. cGMP independent activation. Increase in K+ channel activity | 3-morpholinosydnonimine (SIN-1), molsidomine (N-ethoxycarbonyl-3-morpholinosydnonimine) | Minimal clinical trial evidence to support use [207] |
| S-nitrosothiols | Cleavage of S-NO bond releases NO and disulfide. S-nitrosation of cellular components. cGMP stimulation. | S-nitrosoglutathione (GSNO), N-acetylpenicill-amine (SNAP), trityl S-nitrosothiol, (Ph3SNO) and tert-butyl S-nitrosothiol (tButSNO) | Onychomycosis [207], sexual dysfunction [219], antiplatelet agent [220], cardiac surgery [221], cystic fibrosis [222] |
Table 2.
Clinical Studies involving NO donors and Cancer.
| Completed Clinical Studies | |||||
|---|---|---|---|---|---|
| NO Drug | Cancer Type | Combination Treatment | Compared with | Effect | Reference |
| Transdermal Nitroglycerin | Stage IV NSCLC | Paclitaxel, carboplatin, and bevacizumab | Combination treatment minus nitroglycerin | Increased response rate in the nitroglycerin group. Increased overall survival and progression-free survival in nitroglycerin group that did not reach statistical significance | [232] |
| Transdermal Nitroglycerin | Stage III/IV NSCLC | Vinorelbine and cisplatin | Combination treatment minus nitroglycerin | 30% higher response rate in those treated with transdermal nitroglycerin and longer time to disease progression | [226] |
| Transdermal Nitroglycerin | Stage IIIB/IV NSCLC | Vinorelbine and cisplatin | Combination treatment minus nitroglycerin | Higher overall response rate and disease control rate in nitroglycerin arm. Time to progression and overall survival were similar | [233] |
| Transdermal Nitroglycerin | Stages IIIA and IIIB NSCLC | Vinorelbine and cisplatin and radiotherapy | None: Non-randomized | 75% overall response rate after chemotherapy and radiotherapy. Median progression-free survival of 13.5 months (95% CI, 8.8–18.2), while the median overall survival was 26.9 months | [230] |
| Transdermal Nitroglycerin | PSA recurrent prostate cancer after definitive radiotherapy or radical prostatectomy | N/A | N/A | The mean PSA doubling time of the entire cohort increased to 31.8 months from 13.2 months before starting treatment | [228] |
| Transdermal Nitroglycerin | Operable clinical stage T3-4, or T1-4 node-positive, M0 rectal adenocarcinoma | 5-fluorouracil and radiation therapy prior to surgery | None: Phase I Trial | Pathological Complete response of 17%. Only one patient experienced side effects attributed to nitroglycerin | [229] |
| IV Nitroglycerin | Barcelona clinic liver cancer stage A/B hepatocellular carcinoma | Doxorubicin emulsified in Lipiodol followed by Transcatheter arterial embolization (TAE) and transcatheter arterial chemoembolization (TACE) | Combination treatment minus nitroglycerin | Greater change in lesion size from baseline was observed for nitroglycerin group. Nitroglycerin therapy group showed higher concentration of lipiodol (and thus chemotherapeutic) inside the lesion. | [227] |
| Isosorbide mononitrate | Stage IIIB/IV NSCLC | Irinotecan plus cisplatin and Irinotecan plus capecitabine | Combination treatment minus Isosorbide mononitrate | Isosorbide mononitrate addition did not improve outcomes to either treatment group | [234] |
| Isosorbide mononitrate | T1-T4 Oral Squamous Cell Carcinoma | Surgery after drug administration | Surgery alone | No difference in Ki-67 tumor staining was noted | [235] |
| Ongoing Clinical Studies without Posted Results | |||||
| Drug | Cancer Type | Combination Treatment | Compared with | Completed | Clinical Trial Number |
| Transdermal Nitroglycerin | NSCLC with brain metastases | Whole brain radiation | Combination treatment minus nitroglycerin | Yes | NCT04338867 |
| IV Nitroglycerin | Pediatric Retinoblastoma | Intra-arterial chemotherapy | Normal Saline and combination treatment | No | NCT04564521 |
| Dietary Arginine | Colon Cancer | Nutritional Supplement Prior to Surgery | Surgery alone | Yes | NCT04564521 |
| Dietary Arginine | Stage III/IV Head and Neck Cancer | omega-3 fatty acids and nucleotides oral supplement with cisplatin and radiation therapy | Cisplatin and radiation alone | Yes | NCT00559156 |
| Dietary Arginine | Bladder Cancer | Radical cystoscopy | Radical cystoscopy alone | Yes | NCT02655081 |
| Dietary Arginine | unresectable metastatic brain tumors | Radiation | Radiation alone | Yes | NCT02844387 |
| Nicorandil | stage II—IV NSCLC | Radiation | Radiation alone | Unknown | NCT02809456 |
N/A: No additional treatment.
Table 3.
In vivo and in vitro preclinical studies involving NO donors.
| In Vivo Preclinical Studies | ||||
|---|---|---|---|---|
| NO Drug | Cancer Type | Additional Treatment | Effect | Reference |
| Isosorbide mononitrate (ISMN) and isosorbide dinitrate (ISDN) | Lewis Lungcarcinoma in mice | None | Inhibited angiogenesis and tumor growth | [241] |
| Nicorandil | Sprague-Dawley rats without malignancy | Bleomycin | Reduced lung inflammation and fibrosis | [242] |
| DETA/NO | BALB/c female mice with mammary adenocarcinoma in a experimental metastasis model | CORM-A1 | Inhibited the EMT | [243] |
| PROLI/NO | Sprague–Dawley rats with Brain Gliomas | Carboplatin | Increased blood brain barrier permeability and survival | [239] |
| S-nitrosoglutathione | Mouse xenograft model with head and neck squamous cell carcinoma | Radiation and cisplatin | Decreased tumor growth and enhanced therapySTAT3 inhibition | [244] |
| S-nitrosoglutathione | C57BL/6J mice with castration resistant prostate cancer | None | Decreased tumor burdenIncreased the expression of LH, FSH, and testosteroneDecreased M2 TAM and increased T1 TAMAndrogen receptor downregulation | [182] |
| In Vitro Preclinical Studies | ||||
| NO Drug | Cancer Type | Additional Treatment | Effect | Reference |
| ω-hydroxy-L-arginine | MDA-MB-468 Breast Cancer Cells | None | Decreased cellular proliferative and Increased apoptosis | [245] |
| isosorbide mononitrate | HCT116 and SW620 colon cancer cells | Aspirin | Synergistic effect of therapy on inhibition of cell growth | [246] |
| Sodium Nitroprusside/L-arginine | AGS gastric cancer cell line | None | Inhibition of Epidermal growth factorActivation of type II cGMP-dependent protein kinase | [247] |
| Sodium Nitroprusside | SGC-7901, AGS, MKN45 and MKN28 gastric cancer cells | None | Increased apoptosis through TRAIL cytotoxicity | [248] |
| Sodium Nitroprusside | TSCCa tongue oral squamous cell carcinoma | None | Concentration-dependent cytotoxicity and increased apoptosis | [249] |
| Sodium Nitroprusside | N1E-115 neuroblastoma cells | Cycloheximide | Induction of cell death | [250] |
| Sodium Nitroprusside | HeLa cervical cancer cells | GS28 siRNA (siGS28) transfection | Inhibited cytotoxic responseIncreased ERK | [251] |
| Sodium Nitroprusside | HepG2 and Hep3B Hepatocellular carcinoma cells | Deferoxamine | Induced apoptosisApoptoic effect inhibited by deferoxamine pretreatment | [252] |
| Sodium Nitroprusside | Eight human pancreatic tumour cell lines | Radiation | Increased sensitivity to radiotherapy | [253] |
| Sodium Nitroprusside | SK-MEL-28 and WM793 Melanoma Cell Lines | Arginine Deprivation | Increased therapeutic effect | [254] |
| Sodium Nitroprusside | SH-SY5Y neuroblastoma cells | 2-day light-exhausted compound SNP(ex) | Increased apoptosisIncrease in p53 activation of both SNP and SNP(ex) | [255] |
| S-nitroso-N-acetylpenicillamine and sodium nitroprusside | CHP212 neuroblastoma cells | Deferoxamine | Induced apoptosis, although with a different time to inhibition. | [256] |
| ruthenium nitrosyl complex trans-[Ru(NO)(NH3)4(py)](PF6)3] | HepG2 Liver cancer cells | None | Induced apoptosis | [239] |
| Spermine nitric oxide complex hydrate (SPER/NO)/ diethylenetriamine nitric oxide adduct (DETA/NO) | SK-OV-3 and OVCAR-3 ovarian cancer cell | None | Enhanced cytotoxicity and inhibited apoptosisDownregulation of STAT3 and AKT | [249] |
| DETA-NONOate | MDA-MB-231 breast cancer cells | None | Induced G1 phase growth arrestDownregulation of cyclin D1Hyperphosphorylation of RB | [257] |
| DETA-NONOate | MDA-MB-468 breast cancer cells | Farnesyltransferase inhibitor | Induced apoptosisCytochrome-c release and caspase 3/9 activation | [236] |
| DETA-NONOate | MDA-MB-231, MDA-MB-157, MDA-MB-436, HCC-1806, HCC-70, MDA-MB-468, HCC-1395 and BT-549 breast cancer cell | None | Increased mitochondrial induced apoptosis in African American cancer cell lines but not caucasian cells lines | [258] |
| DETANONOate | DU145 and PC-3 Prostate cancer cells | None | Prevented the EMTSNAIL/EMK inhibition | [240] |
| SNAP and DETA-NONOate | Myeloid derived suppressor cells | None | Inhibited cancer antigen presentation to CD4+ T cells | [259] |
| Sodium nitroprusside, S-nitroso-N-acetylpenicilamine, S-nitrosoglutathione, (+/−)-(E)-methyl-2-[(E)-hydroxyimino]-5-nitro-6-methoxy-3-hexeneamide, and iNOS transfection | PC-3MM2, LNCaP, and DU145 prostate cancer cells | NO scavengers | Inhibited TGF-B productionSodium nitroprusside and iNOS transfection were ablated with the NO scavengers | [260] |
| S-nitrosoglutathione | HCT116 and SW620 colon cancer cells | None | Increased apoptosisActivation of ERK1/2 and p38 kinase | [261] |
| S-nitrosoglutathione | MIAPaCa-2, HCT-116, Panc-1, MCF-7, HT-29 cell lines and AGS cells | U0126 MEK Inhibitor | Growth inhibition through EGFR, IGF-1, and AKT signallingGrowth inhibition increased with U0126 MEK inhibitor | [262] |
| S-nitrosoglutathione | A549 and NCI-H1299 lung cancer cells | None | Growth inhibition via Prdx2 and AMPK | [263] |
Table 4.
Clinical Trials Involving PDE-is. Studies that tested the efficacy of PDE-is at preventing side effects of therapy (i.e., ED after radical prostatectomy) are not included.
Table 4.
Clinical Trials Involving PDE-is. Studies that tested the efficacy of PDE-is at preventing side effects of therapy (i.e., ED after radical prostatectomy) are not included.
| Completed Clinical Studies | |||||
|---|---|---|---|---|---|
| Phosphodiesterase Inhibitor | Cancer Type | Combination Treatment | Compared with | Effect | Reference |
| Tadalafil | T1-T4 Oral Squamous Cell Carcinoma | Surgery | Surgery Alone | Myeloid-derived suppressor cells and regulatory T cells were reduced in the blood and tumor, although effect was maximized at the intermediate dose | [281]. |
| Tadalafil | Primary or Secondary Stage III or IV Head and Neck Squamous Cell Carcinoma | Anti-MUC1 Vaccine/ Anti-Influenza Vaccine | Vaccine Alone/Tadalafil Alone | There were no significant adverse effects of combination therapy. Immunohistochemical analysis shows decreased immune cell exclusion from inside the tumor | NCT02544880 (Active, non-recruiting) [282] |
| Tadalafil | Invasive head and neck squamous cell carcinoma | None | Placebo | An increase of ex vivo T cell expression and reduction in myeloid-derived suppressor cells were noted, suggesting reversal of tumor mediated immune suppression | [283] |
| Tadalafil | Invasive head and neck squamous cell carcinoma | None | Placebo | One patient in the tadalafil arm died, although no other adverse events were reported in either treatment group | NCT01697800 |
| Exisulind (PDE-2/5i) | Stage IIIB/IV or recurrent non-squamous cell lung cancer | Carboplatin and Gemcitabine | None | Median progression-free survival was 4.7 months while median overall survival was 9.0 months. Combination therapy was well tolerated with the goal of the primary endpoint being met | [284] |
| Exisulind (PDE-2/5i) | Metastatic breast cancer | Capecitabine | Capecitabine alone | In pretreated patients with other therapies, the addition of exisulind is similar to that of capecitabine alone | [285] |
| Exisulind (PDE-2/5i) | Solid Malignancy | Docetaxel | None | Combination therapy was well tolerated | [285,286] |
| Exisulind (PDE-2/5i) | Castration-resistant prostate cancer | Docetaxel | None | Overall survival and progression-free survival were similar to other studies of chemotherapy alone | [287] |
| Exisulind (PDE-2/5i) | Metastatic castration-resistant prostate cancer | Docetaxel | None | Low likelihood of benefit of exisulind to docetaxel therapy | [288] |
| CP-461 (PDE-2/5i) | Advanced Solid Malignancy | Usual Treatment | None | Four of 21 patients displayed stable disease and CP461 was well tolerated | [288,289] |
| Roflumilast (PDE-4i) | Advanced B-cell malignancy | prednisone | None | PI3K activity was suppressed in over 75% of patients, with 66% exhibited partial response or disease stability | [290] |
| Theophylline (non-selective PDE-i) | Metastatic castration-resistant prostate cancer | Abiraterone/prednisone | Combination Therapy with Dextromethorphan | One subject in the theophylline group experienced a grade 3 increase in alkaline phosphatase | NCT01017939 |
| RA-233( Dipyridamole derivative) | Non-small cell lung carcinoma, small cell lung carcinoma, extensive colon adenocarcinoma | Multiple | Combination Therapy Alone | RA-233 significantly extended median survival in only this with NSCLC limited to 1 hemithorax | [291] |
| Dipyridamole ( platelet PDE-5/6i | stage II or III unresectable adenocarcinoma of the pancreas | 5-Fluorouracil, leucovorin, mitomycin C | None | Median Survival of 13.8 months and an overall response rate of 26%. Six patients underwent curative resection, 2 of whom did not experience disease recurrence | [292] |
| Aminophylline/ theophylline | B Cell Chronic Lymphocytic Leukemia | None | None | Dose-dependent and time-dependent apoptosis was noted in 45% of patients, who experienced a longer progression-free survival time | [293] |
| Ongoing Clinical Studies without Posted Results | |||||
| Drug | Cancer Type | Combination Treatment | Compared with | Completed | Clinical Trial Number |
| Sildenafil | IIIB or IV non-small cell lung cancer | paclitaxel/carboplatin | Combination Therapy Alone | Yes | NCT00752115 |
| Sildenafil | Advanced solid tumors | Regorafenib | None | Yes | NCT02466802 |
| Sildenafil | Kidney Cancer | Surgery after treatment | Placebo and Surgery | Yes | NCT01950923 |
| Sildenafil | Waldenstrom’s Macroglobulinemia | None | None | Yes | NCT00165295 |
| Sildenafil | WHO Grade III or IV Brain Glioma | Valproic Acid/sorafenib | None | Active-Not Recruiting | NCT01817751 |
| Tadalafil | Refractory hepatocellular carcinoma and pancreatic/colorectal cancer with liver metastasis | Oral vancomycin/nivolumab | None | Recruiting | NCT03785210 |
| Tadalafil | Recurrent or Metastatic Head and Neck Cancer | Pembrolizumab | None | Recruiting | NCT03993353 |
| Tadalafil | Resectable Head and Neck Cancer | Nivolumab/surgery | Combination Therapy Alone | Active-Not Recruiting | NCT03238365 |
| Aminophylline (non-selective PDE-i) | Bladder Cancer | BCG Vaccine | None | Yes | NCT01240824 |
| pentoxifylline (non-selective PDE-i) | grade IV astrocytoma/glioblastoma multiforme | hydroxyurea/radiotherapy | Combination Therapy Alone | Yes | NCT00019058 |
| Dipyridamole | Stage III/IV ovarian carcinoma refractory to platinum chemotherapy | Intraperitoneal methotrexate | Unknown | Unknown | NCT00002487 |
| Thymoquinone (PDE-1i) | Premalignant oral Lesions | None | Placebo | Yes | NCT03208790 |
| Vesnarinone (PDE-3i) | Kaposi Sarcoma | None | Unknown | Yes | NCT00002131 |
| PBF-999 (PDE-10i) | Advanced metastatic solid tumor | Usual Treatment | None | Recruiting | NCT03786484 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mintz, J.; Vedenko, A.; Rosete, O.; Shah, K.; Goldstein, G.; Hare, J.M.; Ramasamy, R.; Arora, H. Current Advances of Nitric Oxide in Cancer and Anticancer Therapeutics. Vaccines 2021, 9, 94. https://doi.org/10.3390/vaccines9020094
AMA Style
Mintz J, Vedenko A, Rosete O, Shah K, Goldstein G, Hare JM, Ramasamy R, Arora H. Current Advances of Nitric Oxide in Cancer and Anticancer Therapeutics. Vaccines. 2021; 9(2):94. https://doi.org/10.3390/vaccines9020094
Chicago/Turabian StyleMintz, Joel, Anastasia Vedenko, Omar Rosete, Khushi Shah, Gabriella Goldstein, Joshua M. Hare, Ranjith Ramasamy, and Himanshu Arora. 2021. "Current Advances of Nitric Oxide in Cancer and Anticancer Therapeutics" Vaccines 9, no. 2: 94. https://doi.org/10.3390/vaccines9020094
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.